

Abstract
Afflicting hundreds of millions of individuals globally, diabetes mellitus is a chronic disorder of energy metabolism characterized by hyperglycemia and other metabolic derangements that result in significant individual morbidity and mortality as well as substantial healthcare costs. Importantly, the impact of diabetes in the United States is not uniform across the population; rather, communities of color and those with low income are disproportionately affected. While excessive caloric intake, physical inactivity, and genetic susceptibility are undoubted contributors to diabetes risk, these factors alone fail to fully explain the rapid global rise in diabetes rates. Recently, environmental contaminants acting as endocrine-disrupting chemicals (EDCs) have been implicated in the pathogenesis of diabetes. Indeed, burgeoning data from cell-based, animal, population, and even clinical studies now indicate that a variety of structurally distinct EDCs of both natural and synthetic origin have the capacity to alter insulin secretion and action as well as global glucose homeostasis. This chapter reviews the evidence linking EDCs to diabetes risk across this spectrum of evidence. It is hoped that improving our understanding of the environmental drivers of diabetes development will illuminate novel individual-level and policy interventions to mitigate the impact of this devastating condition on vulnerable communities and the population at large.
1. Introduction
Diabetes mellitus is a common, chronic disease that is defined phenotypically by elevations in blood glucose levels (i.e., hyperglycemia). Diabetes is estimated to afflict over 463 million individuals worldwide, including over 34 million people in the United States; significantly, disease rates continue to increase across the globe (CDC, 2020; Saeedi et al., 2019). Importantly, diabetes is associated with significant burdens on individuals and society at large. Indeed, diabetes is associated with various micro- and macrovascular complications, and is a leading cause of kidney failure, blindness, and non-traumatic amputations. Diabetes can quadruple a person’s risk of heart disease, which is the leading cause of death in individuals with diabetes (Agarwal et al., 2009). In addition to the toll of the disease on the individual, diabetes imposes a significant economic burden. In the United States alone, the total economic costs of diabetes, including lost productivity, is estimated to be $327 billion annually (American Diabetes Association, 2018).
Unfortunately, the impact of diabetes across the population is not uniform, and diabetes is a source of significant health disparities. Certain racial and ethnic groups are more likely to suffer from the disease as well as its complications. People of Hispanic/Latino ancestry have 80% higher rates of diabetes compared to non-Hispanic Whites (Aguayo-Mazzucato et al., 2019), while non-Hispanic Blacks are almost twice as likely to be diagnosed with diabetes compared to non-Hispanic Whites (CDC, 2020). Importantly, these same communities of color are also disproportionately afflicted by the complications of diabetes, including diabetes-associated death (Aguayo-Mazzucato et al., 2019; Marshall, 2005). Additionally, indigenous populations, including Native American and American Indian peoples, are almost three times more likely to be diagnosed with diabetes and have a 2.5-fold greater likelihood of diabetes-related complications and associated death (Kochanek, Murphy, Xu, & Arias, 2019). While many factors have been posited to account for these health disparities, their origins remain incompletely understood, and the potential contribution of environmental injustice is likely underappreciated.
There are many underlying risk factors for diabetes that contribute to the development and progression of disease. These include lifestyle factors such as excess energy intake via poor diets, physical inactivity, and insufficient or disrupted sleep as well as genetic predisposition. While these risk factors for diabetes are unquestioned, they fail to fully account for the dramatic rise and spread of diabetes over the last decades, raising the likelihood that other factors are at play. This includes the possibility that exposures to endocrine-disrupting chemicals (EDCs) may be an underappreciated risk factor for diabetes and many other chronic metabolic disorders (Heindel et al., 2017). This chapter will review the current literature linking environmental exposures with the pathogenesis of hyperglycemia and the development of diabetes, with a specific focus on type 2 diabetes mellitus (T2DM), the most common form of the disease.
2. Pathogenesis of type 2 diabetes mellitus
Type 2 diabetes mellitus (T2DM) is defined clinically by elevations in blood glucose levels that can arise from various disruptions in the molecular physiology that regulates glucose homeostasis. Specifically, T2DM is diagnosed with evidence of hyperglycemia defined by a hemoglobin A1c (HbA1c) ≥6.5%; fasting plasma glucose ≥126mg/dL; a 2-h plasma glucose of ≥200 mg/dL after a 75-g oral glucose load; or a random plasma glucose of ≥200mg/dL with associated symptoms of hyperglycemia (American Diabetes Association, 2021). Importantly, the pathophysiology of hyperglycemia underlying T2DM development stems from various disruptions in molecular physiology across the multiple metabolic tissues regulating energy homeostasis; however, the common physiological characteristics of diabetes development are impairments in insulin secretion, insulin action, or both.
Pancreatic islets of Langerhans are composed of a variety of cell types, including α-cells that secrete glucagon, β-cells that secrete insulin, and δ-cells that secrete somatostatin, among others. The majority of the islet is composed of α-cells and β-cells, the latter of which couple insulin secretion to glucose metabolism. Whole-body glucose homeostasis is a dynamic process that ensures the economic use of energy; it is dependent upon β-cell insulin secretion and insulin signaling in peripheral tissues (e.g., liver, muscle, and adipose tissue). Insulin secretion from pancreatic β-cells is primarily induced by rising plasma glucose concentrations. Glucose is taken up by glucose transporters 1 and 3 (GLUT1 and GLUT3) on the surface of human β-cells (GLUT2 in rodent β-cells), which subsequently undergoes glycolytic metabolism resulting in the production of adenosine triphosphate (ATP) and an increase in the ATP-to-ADP ratio. Increases in the ATP-to-ADP ratio result in the closure of ATP-dependent potassium (KATP) channels and cellular depolarization. This results in the activation of voltage-sensitive calcium channels that increase cytosolic calcium, which in turn triggers release of insulin secretory granules (Fu, Gilbert, & Liu, 2013). Although glucose is the primary trigger for insulin secretion, pancreatic β-cells also respond to amino acids, other monosaccharides, and fatty acids. Furthermore, various endocrine hormones can influence β-cell function and insulin secretion. For example, stimulation of estrogen receptors by 17-β-estradiol augments insulin secretion (Sutter-Dub, 2002), while gut-derived incretin hormones such as glucagon-like peptide-1 (GLP-1) augment glucose-dependent insulin secretion via G-protein coupled receptors (GPCRs) (Orskov, 1992). Conversely, leptin and growth hormone (acting via insulin-like growth factor-1) can inhibit insulin secretion (Fu et al., 2013; Sonksen, 2001). Finally, β-cell development and physiology are influenced by other hormonal signaling pathways, including those responsible for glucocorticoid receptor and prolactin signaling (Arumugam et al., 2008). Thus, multiple signaling pathways modulate insulin secretion, some of which may be targets of metabolic toxicants.
Upon secretion from pancreatic β-cells, insulin enters the circulation where it acts on insulin-responsive tissues that express the insulin receptor, a tyrosine kinase receptor; these include liver, muscle, and adipose tissue. Through a complex intracellular signaling cascade, insulin stimulation of the insulin receptor leads to glucose uptake in insulin-responsive tissues. Specifically, insulin receptor autophosphorylation results in kinase activation and the phosphorylation of insulin receptor substrate (IRS) proteins. A subsequent cascade of phosphorylation events and protein activation follows that ultimately results in activation of Akt and mTORC, inhibition of FoxO signaling, and GLUT4 translocation to the cell membrane. The sum of these effects is the promotion of glucose uptake and a shift toward anabolic metabolism. In skeletal muscle, insulin receptor activation stimulates the expression and membrane translocation of GLUT4 glucose transporters to the myocyte cell membrane to facilitate glucose uptake and clearance, with glucose storage in muscle principally as glycogen (Stump, Short, Bigelow, Schimke, & Nair, 2003). In adipose tissue, insulin stimulation triggers increased expression and membrane translocation of GLUT4 for glucose uptake and storage as triglycerides, as well as increased lipoprotein lipase (LPL) expression for clearance of plasma triglycerides and storage in fat. In the liver, insulin promotes de novo lipogenesis and glucose storage through incorporation into glycogen while suppressing gluconeogenesis and hepatic glucose output. Thus, insulin signaling in insulin-responsive tissues results in clearance of plasma glucose for storage and later energy mobilization during periods of fasting or exercise. Critically, impairments in insulin signaling (i.e., insulin resistance) result in a reduced ability for insulin to clear glucose and triglycerides from the circulation, resulting in higher insulin requirements to maintain euglycemia. As insulin secretion becomes insufficient to overcome insulin resistance, hyperglycemia develops.
Thus, T2DM is a disruption in normal glucose homeostasis that arises from insulin resistance and β-cell dysfunction. Together they result in chronically elevated plasma glucose levels that wreak havoc through the induction of multiple stress pathways, including oxidative stress, inflammation, osmotic stress, and non-enzymatic glycosylation. Activation of these pathways in various tissues leads to the micro- and macrovascular complications of diabetes (i.e., nephropathy, neuropathy, and retinopathy) as well as accelerated atherosclerosis and other derangements. A main driver of insulin resistance is obesity, with excess accumulation of lipids and lipid metabolites being an important mechanism driving impaired insulin action (Samuel & Shulman, 2012). Chronic insulin resistance necessitates increased demand for insulin secretion, resulting in β-cell exhaustion and ultimate failure (Esser, Utzschneider, & Kahn, 2020). Additionally, chronic exposure to elevated glucose and fatty acids is toxic to β-cells (Chang-Chen, Mullur, & Bernal-Mizrachi, 2008). As discussed, when glucose enters β-cells it enters the glycolytic pathway to produce ATP. Excess oxidative phosphorylation can disrupt β-cell mitochondrial function while increasing production of reactive oxygen species (ROS) (Robertson, Harmon, Tran, Tanaka, & Takahashi, 2003). It is suggested that pancreatic β-cells are ill-equipped to handle oxidative stress due to a dearth of antioxidant capacity. In order to compensate, the cells reduce the efficiency of oxidative phosphorylation, thus throttling down ATP generation and ultimately reducing insulin production and cell mass (Prentki & Nolan, 2006). Reductions in insulin secretory capacity arising from β-cell dysfunction results in insufficient insulin delivery to peripheral tissues to effectively clear glucose from the circulation resulting in hyperglycemia and ultimately frank T2DM. Importantly, factors that promote insulin resistance and/or β-cell dysfunction can amplify metabolic deterioration and increase the risk of T2DM. While many such factors have been implicated in this metabolic deterioration, burgeoning evidence now implicates various environmental contaminants acting as endocrine-disrupting chemicals (EDCs) as novel, underappreciated diabetes risk factors.
3. Endocrine-disrupting chemicals (EDCs)
The Endocrine Society defines EDCs as “endogenous chemicals, or mixture of chemicals, that interfere with any aspect of hormone action” (Gore et al., 2015). EDCs have classically been implicated in the disruption of sex steroid and thyroid hormone signaling; however, increasing data across the spectrum of evidence from cells to populations now indicate that various EDCs can disrupt other signaling cascades and may play a role in many chronic diseases, including metabolic disorders such as obesity, diabetes, and non-alcoholic fatty liver disease (Heindel et al., 2017). To date, hundreds of chemicals have been identified as having hormone-disrupting properties, and this list continues to grow with new chemicals and pathways of disruption emerging (Diamanti-Kandarakis et al., 2009; La Merrill et al., 2020). Critically, but often poorly recognized, the nature of endocrine signaling as well as the organizational effects of hormones on development help explain how EDCs can exert adverse effects at both low concentrations and with non-linear dose-response relationships (Vandenberg et al., 2012). The ultimate impact is adverse effects on health at the time of exposure as well as later in life. Notable EDCs that have been extensively studied and will be reviewed herein, including arsenic, bisphenol A (BPA), the organochlorine pesticide dichlorodiphenyltrichloroethane (DDT) and its metabolite dichlorodiphenyldichlorethylene (DDE), tolylfluanid, polychlorinated biphenyls (PCBs), and multiple air pollutants. Importantly, other EDCs associated with diabetes, its pathological origins, and its clinical manifestations continue to emerge, increasingly supporting the contention that environmental health contributes to metabolic wellness.
Potential exposure to EDCs can occur through various routes and sources (Fig. 1). Arsenic is a common naturally occurring element that has also been used in pesticides, preservatives, and for other industrial purposes (Chung, Yu, & Hong, 2014). BPA exposure can occur through water contamination, soil absorption, paper receipts, and food contamination due to incorporation into plastics as well as the BPA coating on the surfaces of metals and other storage containers (Kang, Kondo, & Katayama, 2006; La Merrill et al., 2020). DDT/DDE are among the organochlorine (OC) pesticides and their metabolites that bioaccumulate and are persistent in the environment. OC pesticides have been implicated in the harming of wildlife, and many have been banned; however, humans continue to be exposed through contaminated foods due to the chemicals’ persistence and bioaccumulation in the food chain as well the continued direct handling of some of these agents (Jaga & Dharmani, 2003). Tolylfluanid is a phenylsulfamide fungicide that can contaminate groundwater, and humans can be exposed occupationally in the agriculture industry (Regnier et al., 2015). Finally, PCBs were historically used industrially in electrical and building equipment. They have long half-lives in soil and in the fatty tissue of animals, which results in persistence in the environment and continued human exposure primarily via consumption of contaminated food (Faroon & Ruiz, 2016).
Fig. 1.
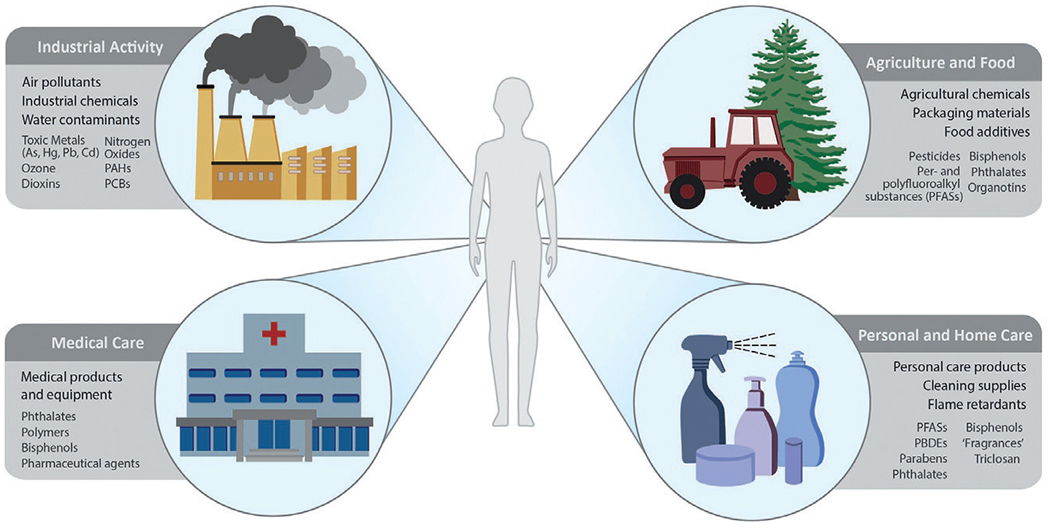
Sources of environmental endocrine-disrupting chemicals. PAH: polycyclic aromatic hydrocarbon, PCB: polychlorinated biphenyl, PBDE: polybrominated diphenyl ethers.
In addition to exposures occurring via contaminated food and water, inhalational exposure to air pollutants has also been implicated in endocrine disruption (Darbre, 2018). Exposure to air pollutants can occur in both indoor and outdoor environments, with specific pollutants being of either natural or synthetic origin. Non-gaseous EDCs can be vaporized into gaseous forms through aerosols, combustion, exhaust, and other routes that can then be absorbed through the respiratory tract (Annamalai & Namasivayam, 2015). Intriguingly, additional sources of EDC exposures include personal and home care products as well as even some medications and medical devices (Encarnacao, Pais, Campos, & Burrows, 2019; Genco, Anderson-Shaw, & Sargis, 2020). While many exposures are inadvertent and need to be addressed through policy, the latter suggest that some EDCs may be avoided through education and improved practice as well as via regulatory policies.
4. EDCs and pathways to environmentally-mediated diabetes
As T2DM can be viewed as arising from a combination of insulin resistance and insulin insufficiency, exposure to EDCs from various sources that affect insulin physiology may play a role in diabetes development. Herein we review evidence linking various EDCs to disruptions in insulin secretion and action across the spectrum of evidence from cellular assays to population-based and even clinical studies. These data are summarized in Table 1, and taken together, indicate that EDCs represent an underappreciated threat to metabolic health.
Table 1.
Example endocrine disruptors and implications in diabetes pathogenesis.
| EDC | Insulin secretion | Insulin action | References |
|---|---|---|---|
| Arsenic | • Impaired glucose sensing • Beta cell mitochondrial dysfunction and oxidative stress |
• Increased HOMA-IR and reduced skeletal muscle mass • Adipocyte, hepatic, and skeletal inflammation • Impairment of insulin signaling |
Zhang et al. (2020) Mondal et al. (2020) Jia et al. (2020); S. Wei et al. (2020) Dover, Patel, and Styblo (2018) |
| Air pollution | • Increased HOMA-IR • Inflammation and increased expression of inflammatory markers in adipose |
Li et al. (2021)
Lin et al. (2020) Kim and Lee (2014) |
|
| Bisphenol A | • Reduced GIIS and downregulation of insulin related genes • Mitochondrial dysfunction and apoptosis of beta cells |
• Increased HOMA-IR • Hepatic DNA methylation and reduction of hepatic glucokinase and overall glucose sensing |
Wang et al. (2012)
Cimmino et al. (2020) Wei, Ding, Wang, Liu, and Lin (2017) Ma et al. (2013) |
| DDT/DDE | • Altered beta cell protein expression and toxicity • Impaired GIIS • Reduced insulin mRNA, proinsulin, an insulin content |
• Oxidative stress with reduced insulin signaling in skeletal muscle • Adipocyte inflammation |
Pavlikova, Sramek, Jelinek, Halada, and Kovar (2020)
Singh, Sarkar, Saxena, and Koner (2019) Park, Kim, and Rhyu (2020) Pavlikova, Smetana, Halada, and Kovar (2015) Mangum et al. (2016) |
| Polychlorinated biphenyls | • Increased insulin release and reduced cellular insulin • Pancreatic atrophy, steatosis, and fibrosis |
• Increased adipocyte droplet size and reduced insulin response • Adipocyte inflammation |
Fischer, Zhou, and Wagner (1996)
Shi et al. (2019) Kim et al. (2017) Baker et al. (2015) |
| Per/polyfluoroalkyl compounds | • Increased HOMA-IR • Activation of PPAR-α and hepatic steatosis |
Cardenas et al. (2017)
Shipley et al. (2004) Lv et al. (2013) |
|
| Organophosphates | • Pancreatitis and pancreatic necrosis • Oxidative stress • Mitochondrial dysfunction |
• Reduced expression of glucose transporters and insulin induced proteins |
Farkhondeh et al. (2020) Leonel Javeres et al. (2020) Karami-Mohajeri and Abdollahi (2013), Nurulain, Szegi, Tekes, and Naqvi (2013) Hou et al. (2019) Wang et al. (2018) |
| Polycyclic aromatic hydrocarbons (PAHs) | • β-cell oxidative stress and toxicity | • Suppression of insulin signaling pathway • Inhibition of beta-adrenergic receptors • Oxidative stress |
Fang et al. (2020) Irigaray et al. (2006), Smiljevska-Ristovska et al. (2020) Hu, Kan, Kearney, and Xu (2015) |
| Neonicotinoid insecticides | • DNA damage • Glutathione reduction |
• Increased weight gain, adiposity, and insulin resistance thorough inhibition of downstream signaling molecules and phosphorylation |
Kara, Ozta, and Ozhan (2020)
Sun et al. (2017) |
| Phthalates | • Reduced β-cell mass and insulin content • Altered expression of genes involved insulin production and pancreatic development • Induce apoptosis |
• Epigenetic modifications in skeletal muscle with increased oxidative stress |
Lin et al. (2011)
Li et al. (2021) Wei et al. (2020) |
| Heavy metals (cadmium, mercury, etc.) | • Cytotoxicity due to decreased ATP • Inhibit GIIS • Mitochondrial swelling and permeation of membranes • Oxidative stress |
• Increased HOMA-IR • Mitochondrial dysfunction |
Elmorsy, Al-Ghafari, Al Doghaither, and Ghulam (2021)
Buha et al. (2020) Wang et al. (2020) Dover et al. (2018) Kim and Lee (2014) |
| 2,3,7,8-Tetrachlorodibenzodioxin | • Disruption in calcium signaling | • Inflammatory cytokine activation |
Ruiz, Perlina, Mumtaz, and Fowler (2016)
Rasinger, Carroll, Lundebye, and Hogstrand (2014) |
| Vacor | • Inhibition of mitochondrial oxidative phosphorylation • Islet necrosis and inflammation |
Miller, Stokes, and Silpipat (1978), Taniguchi et al. (1989) | |
| Triphenyltin | • Induces β-cell apoptosis • Reduced ATP production • Reduced calcium signaling |
• Reduced insulin signaling in adipose, skeletal, and hepatic tissue | Li et al. (2017), Miura et al. (2012), Zuo et al. (2014) |
| Alloxan | • Inhibits glucose sensing, glucokinase, and GIIS • Oxidative stress • β-cell necrosis |
• Inhibition of insulin signaling |
Lenzen (2008)
Ader, Richey, and Bergman (1998) |
4.1. Evidence linking EDCs to alterations in insulin secretion
4.1.1. Evidence from cell-based studies
A plethora of in vitro studies have demonstrated the capacity for EDCs to disrupt endocrine pancreatic function while illuminating mechanisms of toxicity that result in insulin secretory defects (Fig. 2). For example, in murine and human β-cells, short-term BPA exposure increased insulin secretion via an estrogen receptor α (ERα) mechanism (Alonso-Magdalena et al., 2008); however, longer term exposure resulted in reduced glucose-induced insulin secretion (GIIS) and downregulated expression of insulin-related genes with impaired β-cell capacity for compensation (Wei et al., 2017). In earlier studies, PCBs have also been shown to augment insulin release while depleting insulin content (Fischer et al., 1996). Cellular DDT exposure has been shown to impair GIIS (Park et al., 2020; Yau & Mennear, 1977). Human pancreatic β-cells exposed to high concentrations of DDT showed reduced expression of proteins involved in endoplasmic reticulum stress responses, mitochondrial function, and cell morphology (Pavlikova et al., 2020). Also, lower, non-lethal concentrations of DDT have been shown to reduce expression of insulin and proinsulin in the NES2Y human pancreatic β-cell model (Pavlikova et al., 2015). In addition to organic pollutants, inorganic chemicals have also been shown to impair β-cell function as well. For example, arsenic in both its organic and inorganic sub-species has been shown to disrupt β-cell function (Carmean et al., 2019; Dover et al., 2018; Huang, Douillet, & Styblo, 2019; Li, Douillet, et al., 2020). Other EDCs linked to disruptions in insulin secretion and β-cell function in cellular assays include alloxan, cadmium, mercury, phthalates, PBDEs, TCDD, triphenyltin, and other bisphenols such as BPS and BPF. In addition to β-cell disruptions, data also suggest that EDCs, such as BPA, may also disrupt the function of glucagon-secreting pancreatic islet α-cells that function to raise glucose levels (Alonso-Magdalena et al., 2005). Because α-cells make up a larger fraction of cell distribution in human islets, the metabolic impacts of α-cell-disrupting EDCs on human physiology may be more significant than suggested by rodent models.
Fig. 2.
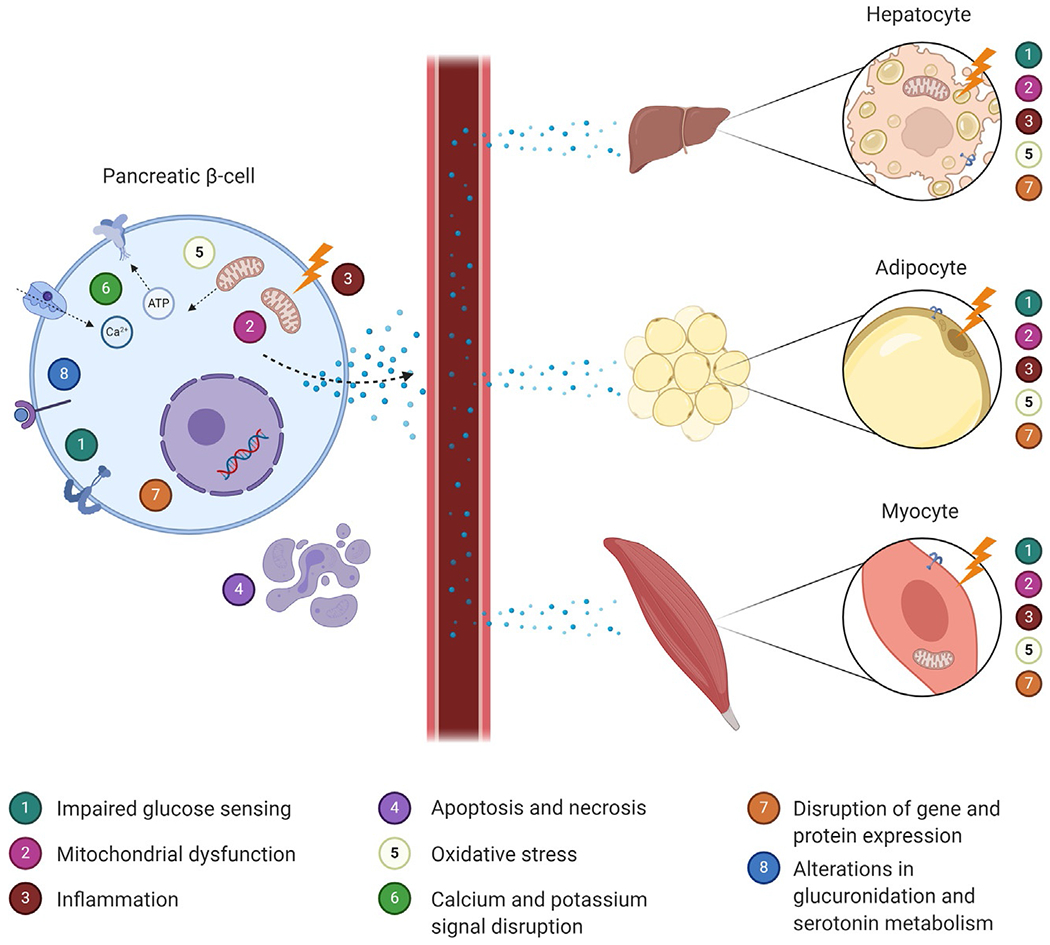
Mechanisms by which environmental endocrine-disrupting chemicals have been linked to dysfunction in metabolic tissues. Environmental EDCs may disrupt insulin signaling by damaging both insulin-releasing pancreatic beta cells (left) and insulin-responsive target tissues such as liver, adipose, and skeletal muscle (right). Created with BioRender.com.
Intriguingly, EDCs linked to disrupted β-cell function and insulin secretion include structurally diverse compounds of diverse origins. While all result in alterations in β-cell physiology, the mechanisms responsible for these effects vary across EDCs, with multiple mechanisms implicated. For example, arsenic has been shown to disrupt β-cell function through multiple mechanisms, including the induction of mitochondrial dysfunction and oxidative stress, as well as through alterations in serotonin metabolism (Carmean et al., 2019; Dover et al., 2018; Huang et al., 2019; Li et al., 2020). Similar mechanisms have been proposed for other EDCs. In addition, EDCs have been shown to alter other key signaling pathways that regulate β-cell function, including oxidative and endoplasmic reticulum (ER) stress; cellular apoptosis, necrosis, and decreased cellular proliferation; mitochondrial dysfunction and reduced ATP synthesis; disruptions of the cytoskeleton and insulin granule exocytosis; altered insulin gene expression and DNA damage; disturbed incretin signaling and calcium flux; disruptions in estrogen receptor signaling; altered epigenetics; disruption in ion channel expression and electrical membrane potential; and increased inflammation among others. Importantly, the cellular disruptions induced by EDCs are consistent with known pathways linked to the pathogenesis of diabetes, providing molecular linkage between EDCs and disease development. Furthermore, the fact that human exposure to environmental toxicants is best characterized as being that of polychemical mixtures, real-world scenarios likely reflect multiple pathways altered simultaneously.
4.1.2. Evidence from animal studies
Because in vivo glucose homeostasis and metabolic physiology require the coordinated interplay of various tissues, our understanding of the impact of EDCs on metabolic disease risk is markedly augmented by physiological studies conducted using intact animal models. Indeed, extensive in vivo studies using rodent models have demonstrated the impact of EDCs on insulin secretion, insulin sensitivity, and global glucose homeostasis. Evidence linking environmental toxicants to disruptions in pancreatic islets dates to the early 1940s when alloxan was shown to induce islet necrosis (Dunn, Duffy, Gilmour, Kirkpatrick, & McLetchie, 1944). Since that time, an increasing body of evidence has implicated environmental toxicants with disruptions in insulin secretion. Adult rats exposed to arsenic displayed altered DNA methylation of the GLUT2 transporter gene in pancreatic islets, which impairs glucose sensing and subsequent insulin release (Khan et al., 2020). Consistent with these findings, another study found that arsenic exposure induces glucose intolerance after 8 weeks of exposure in adult male mice with concordant reductions in early insulin release after a glucose load (Kirkley et al., 2018). Furthermore, arsenic exposure in adult male rats promoted iron-dependent oxidation of pancreatic β-cells in mice resulting in membrane lipid peroxidation and cell death (Wei, Qiu, et al., 2020). Adult male mice exposed to a dioxin-like and non-dioxin-like PCB mixture suffered acinar cell atrophy, steatosis, and fibrosis of pancreatic tissue, in addition to reduced expression of insulin and insulin-regulating genes (Shi et al., 2019). Consistent with in vitro models, in vivo exposure to BPA initially augmented insulin release and increased cellular insulin content in an estrogen receptor-β-dependent manner in adult mice, but ultimately induced β-cell apoptosis due to mitochondrial dysfunction in pregnant mouse offspring (Alonso-Magdalena, Morimoto, Ripoll, Fuentes, & Nadal, 2006; Lin et al., 2013; Manukyan, Dunder, Lind, Bergsten, & Lejonklou, 2019). Lastly, developmental BPA exposure resulted in β-cell dysfunction with hyperglycemia in mouse offspring during adulthood (Garcia-Arevalo et al., 2014; Mao et al., 2015). In addition to these models, in vivo exposure to other EDCs has been shown to disrupt insulin secretion, including cadmium, mercury, Vacor, and alloxan as well as DDT and its metabolite DDE (El Muayed et al., 2012; Iavicoli, Fontana, & Bergamaschi, 2009; Lenzen, 2008; Taniguchi et al., 1989). Collectively, these animal models reinforce cellular data indicating that EDC exposure has the capacity to disrupt insulin secretion, with attendant threats to glucose homeostasis and metabolic physiology more broadly.
4.2. Evidence linking EDCs to alterations in insulin action
Central to the pathophysiology of T2DM are impairments in insulin action, often arising from obesity-induced disruptions in lipid metabolism, enhanced inflammation, and other molecular disruptions. This induction of insulin resistance increases secretory demands on pancreatic β-cells, ultimately leading to their failure. Importantly, while genetic predisposition, excess energy intake, physical inactivity, and sleep disturbances among other factors can promote insulin resistance, data now indicate that exposure to EDCs may also promote insulin resistance and subsequent diabetes risk.
4.2.1. Evidence from cell-based studies
Evidence from cell-based studies demonstrate how EDCs can disrupt insulin action and induce cellular insulin resistance (Fig. 2). Arsenic has been shown to specifically impair insulin signaling and response in cultured 3T3-L1 adipocytes (Paul, Harmon, Devesa, Thomas, & Styblo, 2007). Arsenite also reduces insulin-mediated glucose uptake and induces chronic inflammation in these same cells (Xue et al., 2011). Lastly, arsenic exposure both induces myocyte apoptosis and inhibits myoblast proliferation, resulting in reduced muscle mass that can contribute to insulin resistance (Liu et al., 2015; Yen et al., 2012). In cultured rat L6 myoblast-derived myotubes, DDT exposure resulted in oxidative stress that impaired insulin action (Singh et al., 2019). BPA impairs adipocyte functioning and differentiation, ultimately resulting in inhibition of glucose uptake following insulin stimulation as well as increased expression of inflammatory mediators (Ariemma et al., 2016; Valentino et al., 2013). The phenylsulfamide fungicide tolylfluanid has also been shown to impair cellular insulin signaling. In primary murine and human adipocytes, tolylfluanid disrupts insulin signaling through a specific reduction in the expression of insulin receptor substrate-1 (IRS-1) (Sargis et al., 2012). In C2C12 skeletal myocytes, tolylfluanid reduced insulin-dependent downstream protein phosphorylation and reduced mitochondrial oxygen consumption and membrane potential (Davis, Thomas, Shorter, Brown, & Baumgarner, 2018; Sargis et al., 2012). In studies of primary adipose tissue, tolylfluanid-mediated induction of insulin resistance via reduced IRS-1 expression appears to result from the activation of glucocorticoid receptor signaling (Neel, Brady, & Sargis, 2013). Also, in adipocytes, exposure to PCBs promoted differentiation, increased lipid droplet size, and induced insulin resistance (Kim et al., 2017). The prototypical obesogen, tributyltin (TBT), has been shown to promote adipocyte differentiation via activation of signaling through the retinoid X receptor (RXR) and the peroxisome proliferator-activated receptor-γ (PPARγ) (Shoucri, Hung, Chamorro-Garcia, Shioda, & Blumberg, 2018). Importantly, these adipocytes appear to be physiologically distinct, with characteristics predicted to promote metabolic dysfunction (Regnier et al., 2015).
Other EDCs linked to disruptions in insulin action in cell-based studies include: 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), phthalates, cadmium, and perfluoroalkyl substances (PFASs) among others. In addition to inappropriate activation of nuclear hormone signaling, reductions in the expression of insulin signaling intermediates, and mitochondrial dysfunction, multiple other cellular pathways have been implicated in the pathophysiology of EDC-induced insulin resistance. These include: increased expression of inflammatory mediators, altered lipid metabolism, disrupted adipokine expression, oxidative stress, enhanced cellular senescence, impaired glucose transport, and disruptions in gluconeogenesis and glycogen handling. In summary, the increase in evidence linking EDCs with altered cellular function in tissues regulating metabolic physiology clearly delineate potential connections between diverse toxicant exposures and molecular pathways linked to the development of diabetes.
4.2.2. Evidence from animal studies
Animal models remain essential tools for assessing the impact of EDCs on metabolic physiology, including insulin action. This is because insulin exerts biological effects on multiple tissues that communicate with one another. Thus, in vivo studies remain necessary tools for assessing the impacts of EDCs on energy metabolism, and they have provided intriguing insights regarding the capacity of these pollutants to promote metabolic dysfunction. For example, adult male mice exposed to tolylfluanid exhibited increased body weight that was characterized by increased fat mass, and these changes were accompanied by impaired glucose tolerance resulting from global insulin resistance (Regnier, Kirkley, et al., 2015). Importantly, adipose from these mice also showed increased glucocorticoid receptor signaling, consistent with cell-based assays and the predicted effects of inappropriate signaling through this important nuclear hormone receptor (Neel et al., 2013). Interestingly, the precise effects of tolylfluanid on global energy metabolism appear to be influenced by dietary macronutrient composition (Regnier et al., 2018). In addition to its effects on insulin secretion, BPA has also been linked to impairments in insulin action through alterations in hepatic glucose sensing as well as impairments in glucokinase activity and the glycolytic pathway when administered to adult male mice (Perreault et al., 2013). In agreement with cellular studies, animal models of arsenic exposure have demonstrated hepatic inflammation that may play a role in arsenic-induced insulin resistance (Jia et al., 2020). Exposure to DDE and other organochlorine pesticides in 6-week-old mice resulted in macrophage recruitment and inflammation of adipocytes, which may contribute to impaired insulin action and resistance (Mangum et al., 2016). Lastly, adult mice exposed to PCBs exhibited insulin resistance in conjunction with adipocyte inflammation (Baker et al., 2015). Other EDCs linked to alterations in insulin sensitivity include: TCDD, phthalates, particulate matter air pollution, cadmium, PFASs, malathion, atrazine, polybrominated diphenyl ethers (PBDEs), mercury, and other persistent organic pollutants (POPs).
In addition to adult exposure paradigms, developmental exposure to EDCs has also been linked to metabolic perturbations in the offspring. For example, perinatal DDT exposure during development resulted in glucose intolerance and hyperinsulinemia in adult female offspring (La Merrill et al., 2014). Mice exposed to tolylfluanid throughout gestation and lactation demonstrated sex-dependent effects on glucose tolerance and insulin sensitivity during adulthood (Ruiz et al., 2019). In adult exposure models, tributyltin exposure has been shown to induce insulin resistance (Xu et al., 2019). Intriguingly, developmental exposure to tributyltin has been shown to induce transgenerational disruptions in metabolic regulation (Chamorro-Garcia et al., 2017). Mechanisms responsible for these heritable changes include epigenetic alterations and changes in chromatin modeling (Rattan & Flaws, 2019; Skinner, 2016). These findings are in line with the Developmental Origins of Health and Disease Hypothesis (Dover, 2009) and raise the intriguing possibility that EDC-induced disruptions in metabolic homeostasis may not only reflect current exposure but may result from historical exposure during sensitive windows of development or even from prior generations.
4.3. Evidence from epidemiological and population-based studies
While cell- and animal-based studies facilitate assessment of the mechanisms of toxicant induced β-cell dysfunction and impaired insulin action, human epidemiological and population-based studies are vital in determining the consequence of EDC exposure on diabetes risk, measures of glycemic control, and diabetes-related outcomes. Unfortunately, many population studies that examine the associations between EDC exposure and diabetes-related traits do not inform the pathophysiological origins of those links, i.e. whether associations are mediated by impaired insulin secretion, insulin resistance, or both. Nevertheless, these studies are critical to link real-life exposures to human disease. Importantly, the potential for environmental toxicants to promote diabetes development has been known for decades. In 1978, a case report described a patient who ingested large amounts of the rodenticide Vacor and subsequently developed diabetic ketoacidosis, a metabolic condition characterized by hyperglycemia resulting from marked insulin deficiency (Miller et al., 1978). While this poisoning event is not characteristic of most environmental exposures, it provides proof-of-principle that agents disseminated into the environment may be linked to diabetes pathogenesis.
Arsenic exposure is associated with an increased risk for diabetes, and the metabolism of the metalloid into its dimethylated sub-species is associated with a further increase in risk (Zhang et al., 2020). In recent studies, even low levels of blood arsenic have been associated with a higher prevalence of diabetes (Dai et al., 2020). Furthermore, arsenic exposure, evaluated by hair and nail samples, was positively associated with increased fasting blood glucose, serum insulin, and increased homeostatic model assessment of insulin resistance (HOMA-IR). Arsenic was also associated with reduced lean body mass, which could be a mechanism for the insulin resistance (Mondal et al., 2020). BPA has been heavily studied in human populations, and the results support the in vitro and in vivo evidence. BPA is associated with increased overall risk for diabetes in a dose-dependent manner (Lee et al., 2020). Furthermore, other studies have established a dose-response relationship between urinary BPA levels and increased diabetes prevalence (Shankar & Teppala, 2011), and BPA exposure was found to be associated with an increased odds ratio for insulin resistance (Wang et al., 2012). Serum BPA levels were higher in patients with T2DM compared to healthy controls and were associated with poorer glycemic control, increased insulin resistance, and overall worse clinical outcomes (Soundararajan, Prabu, Mohan, Gibert, & Balasubramanyam, 2019). Large case-control and cohort studies found that exposure to organochlorine pesticides was associated with increased incidence of diabetes, and patients with diabetes had higher DDT and DDE serum concentrations than healthy individuals (Mansouri & Reggabi, 2020; Rignell-Hydbom et al., 2009; Salihovic et al., 2016; Turyk, Anderson, Knobeloch, Imm, & Persky, 2009). PCB exposure in a Taiwanese population was linked to increased diabetes risk in women (Wang, Tsai, Yang, & Guo, 2008). Both PCB and organochlorine pesticide exposure are associated with increased HOMA-IR, HbA1c, and reduced insulin sensitivity (Lind & Lind, 2018).
The primary route for studying the relationship between air pollution and diabetes is through epidemiological cohort and ecological studies. Recent studies have found very compelling and interesting impacts of air pollution on metabolic health, with most studies focusing on particulate matter, nitrogen dioxide, and ozone. Particulate matter is a mixture of particles in the air that vary in size and can penetrate the lungs (Brunekreef & Holgate, 2002). PM2.5 is particulate matter that is less than 2.5 μm in diameter. Firstly, overall air pollution was found to be associated with reduced insulin sensitivity and β-cell function (Zhang, Mwiberi, et al., 2021). Ozone exposure was positively associated with increased diabetes risk, elevated fasting plasma glucose, fasting insulin, insulin resistance, and β-cell function in non-diabetic adults in China (Li, Mei, et al., 2021; Lin et al., 2020). Additionally, individuals exposed to ambient nitrogen dioxide had an increased risk for diabetes, increased HbA1c, and increased fasting plasma glucose (Zhang, Liu, et al., 2021). Furthermore, PM2.5 is associated with increased incidence and prevalence of diabetes and worsened glycemic control among those previously diagnosed with diabetes (Elbarbary et al., 2020; Hwang, Kim, Koo, Yun, & Cheong, 2020). Importantly, the impact of air pollution, particularly particulate matter, appears to be enhanced by warm, humid weather, which raises important concerns about the amplifying impact of climate change on diabetes risk (Jabbari et al., 2020; Lin et al., 2020).
In addition to these EDCs, population-based studies have also linked other EDCs to diabetes and diabetes-related traits, including: TCDD, phthalates, cadmium, PFASs, malathion, PBDEs, polychlorinated dibenzo-p-dioxins and furans, and mercury. In addition to these data from epidemiological studies, data from clinical studies have also emerged. In a cross-over study of male and female subjects intentionally given a single low dose of BPA, exposure to this EDC altered insulin secretion during an oral glucose tolerance test and a hyperglycemic clamp (Stahlhut et al., 2018). While further randomized clinical trials have been proposed to illuminate the effects of BPA on metabolic physiology have been proposed, evidence of toxicity in cells, animal models, and epidemiological studies raise significant ethical concerns regarding the justification of such proposed studies (Hagobian et al., 2020). While the failure of regulatory agencies to address the threats posed by BPA is deeply problematic, deliberately exposing research subjects to EDCs with substantial evidentiary support for adverse health effects is ethically dubious and raises significant concerns not just of subject harm, but also of creating an evidence threshold that cannot be met for more toxic EDCs. Despite this, it is important to note that for BPA, we have evidence of adverse metabolic effects across an incredibly wide spectrum of scientific proof. For other EDCs, the strength of evidence varies; however, the totality of the data suggests that environmental exposures are an important-yet-underappreciated contributor to diabetes rates.
5. EDCs and type 1 diabetes
While T2DM is characterized by insulin resistance and relative insulin insufficiency, type 1 diabetes mellitus (T1DM) is a hyperglycemic disorder arising from absolute insulin insufficiency, often arising from autoimmune destruction of pancreatic β-cells. Importantly, data indicate that rates of T1DM are also increasing, with notable geographic variation in rates (Liu et al., 2020). Because of its distinct pathophysiology, understanding the linkage between environmental toxicant exposures and T1DM is important for better understanding how environments modulate the pathogenesis of this devastating autoimmune disease. Unfortunately, this realm of environmental diabetes has been relatively understudied compared to studies examining linkage between EDCs and T2DM. Despite this, there is some evidence to suggest that EDCs may be linked to increasing rates of T1DM (Howard, 2018). Certainly, EDCs have been shown to promote β-cell apoptosis or necrosis consistent with T1DM development, and exposure to pollutants can induce hypersensitivity reactions, suggesting immune system disruption (Ritz, 2010). Indeed, the early data from Vacor poisoning and alloxan exposure are consistent with these findings (Lenzen, 2008; Miller et al., 1978). Other experimental studies have shown that arsenic and other EDCs directly target pancreatic β-cells resulting in insulin deficient diabetes (Ramdas, Sharma, Kaul, & Bhatia, 2018). Additionally, DDT exposure in rats during development not only resulted in pancreatic dysfunction later in life but in two subsequent generations as well (Song & Yang, 2017). Animal studies have found that exposure to both BPA and its replacement chemical bisphenol S worsens inflammation and glycemia in a non-obese diabetic (NOD) mouse model, which is frequently used to evaluate type 1 diabetes development (Xu, Huang, & Guo, 2019; Xu, Huang, Nagy, & Guo, 2019). Some epidemiological studies have also been consistent with these findings, showing EDCs may be implicated in T1DM pathogenesis. For example, exposure to air pollution during pregnancy is associated with increased risk for T1DM in children (Elten et al., 2020; Malmqvist et al., 2015). Overall, current evidence linking EDCs to T1DM and its pathogenesis is intriguing; however, more work is needed to better understand how environmental exposures contribute to T1DM risk.
6. EDCs and gestational diabetes
Gestational diabetes (GDM) is a physiological condition characterized by hyperglycemia during pregnancy that is associated with adverse maternal and fetal outcomes (American Diabetes Association, 2004). In addition, a prior history of GDM is associated with a greater risk of T2DM in both mother and child (Ben-Haroush, Yogev, & Hod, 2004). Somewhat akin to T2DM, the hyperglycemia of GDM results from insufficient insulin production to meet the demands imposed by gestational insulin resistance. As such, EDCs that augment insulin resistance or impair β-cell compensation, essential for maintaining euglycemia during pregnancy, are likely to augment GDM risk. This area, however, remains relatively understudied. Studies have found that phthalate and BPA exposure are associated with increased plasma glucose and reduced glucose tolerance in pregnant women (Filardi, Panimolle, Lenzi, & Morano, 2020). Other epidemiological studies have found associations between arsenic and cadmium exposure and increased risk for GDM (Farzan et al., 2016; Salmeri et al., 2020; Xing et al., 2018). Other EDCs, including PFASs and POPs have also been implicated in increased GDM risk (Xu et al., 2020). Animal studies have corroborated this epidemiological evidence. Rats, mice, and sheep exposed to gestational BPA exhibited elevated free fatty acid levels, deleterious adipose distribution, and disruption of glucose homeostasis (Veiga-Lopez et al., 2015). Other experimental studies have shown that EDCs mediate GDM through gene disruption in trophoblastic tissue (Ehrlich et al., 2016). Lastly, studies have shown that glycemic control during pregnancy is worsened by climate change and warming temperatures (Preston, Eberle, Brown, & James-Todd, 2020). Altogether, there is some evidence for the role of EDCs in GDM, but further study is needed to better quantify this risk and to illuminate its underlying mechanisms.
7. EDCs, diabetes disparities, and health justice
A particularly disturbing aspect of the diabetes pandemic is its disproportionate impact on communities of color and those with low income. In the U.S., diabetes rates are higher among Blacks, Hispanics/Latinos, and Native Americans than among non-Hispanic Whites (Aguayo-Mazzucato et al., 2019). Furthermore, these same populations are more likely to suffer the consequences of diabetes, including the microvascular complications of nephropathy, neuropathy, and retinopathy and even diabetes-associated death (Clements et al., 2020). In addition to the greater impact of diabetes on these population groups, communities of color are also more likely to have undiagnosed and untreated diabetes (Hsueh et al., 2020). While many factors have been proposed to account for these disparities, too often neglected in this discussion is the contribution of environmental injustice (Ruiz, Becerra, Jagai, Ard, & Sargis, 2018). Indeed, there is evidence that persons of color are disproportionately exposed to EDCs compared to the general population. Importantly, various social and structural determinants of health contribute to EDC exposure and toxicity, including diet, consumer products, neighborhood characteristics, and geographical area (Hicken et al., 2012; James-Todd, Chiu, & Zota, 2016). People of non-white ethnicity are also more likely to work in occupational settings that increase their risk for EDC exposure. Furthermore, EDC exposure in non-white populations tend to be chronic rather than acute, with the health consequences of these exposures disproportionately impacting non-white populations financially (Attina, Malits, Naidu, & Trasande, 2019). These health disparities are deeply concerning. Understanding the contribution of environmental health to diabetes risk is essential for illuminating the sources of health injustice and identifying opportunities to develop and implement countermeasures. Importantly, because environments are modifiable, addressing the disproportionate burden of diabetogenic exposures on vulnerable communities likely represents an underutilized tool for addressing diabetes-associated health injustice.
8. Crosstalk between clinical care and EDC exposure
While clinical management of diabetes rests upon a foundation of dietary changes and increased physical activity, most individuals with diabetes will ultimately require the use of medications and medical devices (e.g. insulin pumps and continuous glucose monitoring systems) to improve glycemic control. Furthermore, the clinical management of diabetes includes pharmacological and lifestyle interventions to address common diabetes-associated comorbidities and factors that accelerate the development of diabetes-associated complications such as atherosclerosis (e.g., dyslipidemia, hypertension). As such, patients with diabetes have significant contact with the healthcare system, which helps explain the significant and rising health costs associated with diabetes (American Diabetes, 2018). Importantly, however, this engagement may lead to treatments that themselves expose patients with diabetes to EDCs (Genco et al., 2020). This creates a unique ethical challenge as health care providers become purveyors of exposure largely unbeknownst to the patient or the clinician. As such, addressing healthcare-associated EDC exposure and its ethical implications on non-maleficence, informed consent, and patient autonomy is a significant concern that warrants interventions at the levels of the patient and provider, pharmaceutical and device manufactures, and government policy makers. Finally, we must begin efforts to instruct patients on ways that they can reduce their exposures to metabolically deleterious EDCs. Some evidence-based advice exists on this topic (Sargis, Heindel, & Padmanabhan, 2019; Trasande, 2019), and new data continue to emerge (Mengozzi et al., 2021). Despite this, more work is needed to demonstrate which interventions are most useful for addressing environmental drivers of diabetes risk.
9. Bridging the gap to address ecosystems of diabetes risk
Based on the proliferation of data indicating that environmental exposures represent underappreciated risks to metabolic health that disproportionately affect already vulnerable populations, it is incumbent upon society to use all available tools to reduce exposures to diabetogenic chemicals in order to promote public metabolic health and to address environmental contributors to health injustice. This includes policy- and individual-level action. Unfortunately, while some chemicals linked to diabetes have been regulated, a recent analysis of federal environmental policy showed that diabetes and metabolic disease risk have not been considered in public policy (Shaikh, Jagai, Ashley, Zhou, & Sargis, 2018). Closing this gap through improved environmental policies as well as improved urban planning and development offer novel tools to address the devastating rise of diabetes rates and its disproportionate impact on vulnerable communities.
10. Data gaps and the need for further studies
Despite the proliferation of data linking various EDCs with diabetes risk across cell, animal, and human studies, there remain important gaps in our knowledge. Significant among these include the lack of data on the vast majority of the tens of thousands of chemicals used in consumer products as well as a dearth of studies examining the effects of chemical mixtures on health outcomes. Indeed, evaluating mixture exposure is vital for understanding real-world EDC exposures and their consequences. With regard to diabetes-related outcomes, there have been a few studies that have begun to examine the effects of chemical mixtures on metabolic outcomes. For example, in one study a mixture of persistent organic pollutants (POPs) found in fish oil was shown to promote insulin resistance in rats (Ruzzin et al., 2010). Indeed, many studies examining PCB effects on metabolic outcomes have used PCB mixtures (Liu et al., 2021). Additionally, epidemiological advancements have allowed for the study of metal and metalloid mixtures in relation to diabetes risk, status, and control. Specifically, one study evaluated diabetes risk and β-cell function for a mixture of 15 urinary metals and found an increased incidence of diabetes, higher HOMA-IR, and reduced HOMA-B (Wang et al., 2020; Wang, Karvonen-Gutierrez, et al., 2020). Another human population study found metal mixtures to be associated with elevated oxidative stress biomarkers (Domingo-Relloso et al., 2019). Advancements in statistical techniques to evaluate these complex exposures continues to evolve, and these advancements will help better illuminate the impact of complex exposures on metabolic health. Given the plethora of chemicals to which humans are exposed, as well as the possibility that these mixtures might simultaneously impact both insulin secretion and action, the threat of mixtures of diabetogenic chemicals to metabolic health is real and needs to be better understood.
In addition to EDC mixtures, understanding how EDCs interact with traditional diabetes risk factors is essential. Some studies have begun to examine how EDCs modulate the effects of metabolically deleterious diets (Carmean et al., 2020; Ishikawa, Graham, Stanhope, Havel, & La Merrill, 2015; Regnier et al., 2018); however, more work is required in this area, especially studies examining the dual impact of EDCs and the high sucrose and fructose intake that characterizes the secular dietary trends of the last several decades that have coincided with exploding rates of diabetes. Similarly, much more work is needed to understand the impact of physical inactivity on the metabolic toxicity of EDCs; moreover, there is a significant need for additional studies examining effects of EDCs on muscle physiology that may amplify the effect of or contribute to physical inactivity.
Fascinating studies have begun to elucidate the impact of underlying genetics on the impact of EDCs. For example, polymorphisms in the arsenic methyltransferase gene modulate the biological effects of arsenic (Douillet et al., 2017; Valenzuela et al., 2009). Such knowledge has the potential to help us identify those with increased EDC susceptibility. In addition, improved understanding of genetic risk has important implications for the translation of animal models into human risk. For example, mouse versus human PPARs may behave differently regarding responses to EDCs (Casals-Casas, Feige, & Desvergne, 2008). In epidemiological studies, some work has begun to investigate the differential impact of EDCs on diabetes risk and diabetes-related traits in relation to underlying polygenic risk scores that are associated with disease development (McCarthy, 2010; Qi et al., 2017). Ultimately, this knowledge of gene-by-environment interactions may become an essential component of personalized medicine in which clinical care can be tailored to provide individualized advice regarding environmental health threats.
In addition to diet, activity, and genetics, other individual level conditions may influence the risk of diabetes related to EDC exposures. These include factors such as underlying medical conditions and therapies that may influence the metabolism and toxicity of EDCs. This also includes diseases that modulate detoxification and clearance pathways such as liver and kidney dysfunction. Similarly, underlying lung disease may predispose to the toxicity of air pollution, which evidence has shown is associated with diabetes risk. Moreover, medications may themselves disrupt endocrine function or may serve as an addition source of EDC exposure, as discussed. Furthermore, various medications influence hepatic and renal function; thus, they may augment or inhibit the adverse effects of EDCs on metabolic health. Indeed, incorporating environmental health into clinical care has the capacity to empower personalized medicine (Sargis, 2015). The identification of individuals with a heightened risk of pollutant exposure requires clinicians and researchers to consider these various factors, and in doing so empowers us to provide better care for our patients.
11. Conclusion
The consequences of diabetes are enormous for both individuals and society at large. While genetics and lifestyle factors clearly promote risk, burgeoning evidence over the last three decades has emerged to suggest that exposure to EDCs represent a novel yet underappreciated diabetes risk factor. Indeed, evidence from cells to human populations shows that various EDCs have the capacity to disrupt the production and release of insulin and/or interfere with insulin’s action in insulin-responsive tissues, promoting the development of hyperglycemia and diabetes risk. Furthermore, studies suggest that differential exposure to EDCs may underlie diabetes disparities. While further work is needed to clarify the risk posed by EDCs to metabolic health, the current state of the evidence suggests that individualized clinical care as well as public policy must include environmental health as critical tools for understanding and addressing the global diabetes pandemic.
Acknowledgments
This work is supported by the National Institutes of Health (R01 ES028879, R21 ES030884, and P30 ES027792 supporting RMS) and the University of Illinois at Chicago’s Medical-Scientist Training Program (T32 GM079086 supporting MCS). The authors wish to express their sincere thanks to Michelle K. Sheena for her assistance with the illustrations.
Abbreviations
- ATP
adenosine triphosphate
- BPA
bisphenol A
- DDE
dichlorodiphenyldichlorethylene
- DDT
dichlorodiphenyltrichloroethane
- GDM
gestational diabetes mellitus
- GIIS
glucose-induced insulin secretion
- GLP-1
glucagon-like peptide-1
- GPCR
G-protein coupled receptor
- HbA1c
hemoglobin A1c
- HOMA-β
homeostatic model assessment for β-cell function
- HOMA-IR
homeostatic model assessment for insulin resistance
- LPL
lipoprotein lipase
- OC
organochlorine
- PAH
polycyclic aromatic hydrocarbon
- PBDE
polybrominated diphenyl ethers
- PCB
polychlorinated biphenyl
- PFAS
perfluoroalkyl substances
- PPARγ
peroxisome proliferator-activated receptor-γ
- POP
persistent organic pollutants
- ROS
reactive oxygen species
- RXR
retinoid X receptor
- TBT
tributyltin
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
Footnotes
Conflict of interest statement
RMS declares he has received honoraria from the American Medical Forum and CVS/Health. Neither of these relationships pertain the content of this chapter.
References
- Ader M, Richey JM, & Bergman RN (1998). Evidence for direct action of alloxan to induce insulin resistance at the cellular level. Diabetologia, 41(11), 1327–1336. 10.1007/s001250051073. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Singla S, Singla S, Singla R, Lal A, Wardhan H, et al. (2009). Prevalence of coronary risk factors in type 2 diabetics without manifestations of overt coronary heart disease. The Journal of the Association of Physicians of India, 57, 135–142. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/19582981. [PubMed] [Google Scholar]
- Aguayo-Mazzucato C, Diaque P, Hernandez S, Rosas S, Kostic A, & Caballero AE (2019). Understanding the growing epidemic of type 2 diabetes in the Hispanic population living in the United States. Diabetes/Metabolism Research and Reviews, 35(2), e3097. 10.1002/dmrr.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Laribi O, Ropero AB, Fuentes E, Ripoll C, Soria B, et al. (2005). Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environmental Health Perspectives, 113(8), 969–977. 10.1289/ehp.8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, & Nadal A (2006). The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environmental Health Perspectives, 114(1), 106–112. 10.1289/ehp.8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Ropero AB, Carrera MP, Cederroth CR, Baquie M, Gauthier BR, et al. (2008). Pancreatic insulin content regulation by the estrogen receptor ER alpha. PLoS One, 3(4), e2069. 10.1371/journal.pone.0002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes A (2018). Economic costs of diabetes in the U.S. in 2017. Diabetes Care, 41(5), 917–928. 10.2337/dci18-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. (2004). Gestational diabetes mellitus. Diabetes Care, 27(Suppl. 1), S88–S90. 10.2337/diacare.27.2007.s88. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. (2021). Summary of revisions: Standards of medical care in diabetes-2021. Diabetes Care, 44(Suppl. 1), S4–S6. 10.2337/dc21-Srev. [DOI] [PubMed] [Google Scholar]
- Annamalai J, & Namasivayam V (2015). Endocrine disrupting chemicals in the atmosphere: Their effects on humans and wildlife. Environment International, 76, 78–97. 10.1016/j.envint.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Ariemma F, D’Esposito V, Liguoro D, Oriente F, Cabaro S, Liotti A, et al. (2016). Low-dose bisphenol-A impairs adipogenesis and generates dysfunctional 3T3-L1 adipocytes. PLoS One, 11(3), e0150762. 10.1371/journal.pone.0150762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam R, Horowitz E, Lu D, Collier JJ, Ronnebaum S, Fleenor D, et al. (2008). The interplay of prolactin and the glucocorticoids in the regulation of beta-cell gene expression, fatty acid oxidation, and glucose-stimulated insulin secretion: Implications for carbohydrate metabolism in pregnancy. Endocrinology, 149(11), 5401–5414. 10.1210/en.2008-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attina TM, Malits J, Naidu M, & Trasande L (2019). Racial/ethnic disparities in disease burden and costs related to exposure to endocrine-disrupting chemicals in the United States: An exploratory analysis. Journal of Clinical Epidemiology, 108, 34–43. 10.1016/j.jclinepi.2018.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Shoemaker R, English V, Larian N, Sunkara M, Morris AJ, et al. (2015). Effects of adipocyte aryl hydrocarbon receptor deficiency on PCB-induced disruption of glucose homeostasis in lean and obese mice. Environmental Health Perspectives, 123(10), 944–950. 10.1289/ehp.1408594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Haroush A, Yogev Y, & Hod M (2004). Epidemiology of gestational diabetes mellitus and its association with type 2 diabetes. Diabetic Medicine, 21(2), 103–113. 10.1046/j.1464-5491.2003.00985.x. [DOI] [PubMed] [Google Scholar]
- Brunekreef B, & Holgate ST (2002). Air pollution and health. Lancet, 360(9341), 1233–1242. 10.1016/S0140-6736(02)11274-8. [DOI] [PubMed] [Google Scholar]
- Buha A, Dukic-Cosic D, Curcic M, Bulat Z, Antonijevic B, Moulis JM, et al. (2020). Emerging links between cadmium exposure and insulin resistance: Human, animal, and cell study data. Toxics, 8(3), 63. 10.3390/toxics8030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas A, Gold DR, Hauser R, Kleinman KP, Hivert MF, Calafat AM, et al. (2017). Plasma concentrations of per- and polyfluoroalkyl substances at baseline and associations with glycemic indicators and diabetes incidence among high-risk adults in the diabetes prevention program trial. Environmental Health Perspectives, 125(10), 107001. 10.1289/EHP1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmean CM, Kirkley AG, Landeche M, Ye H, Chellan B, Aldirawi H, et al. (2020). Arsenic exposure decreases adiposity during high-fat feeding. Obesity (Silver Spring), 28(5), 932–941. 10.1002/oby.22770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmean CM, Yokoi N, Takahashi H, Oduori OS, Kang C, Kanagawa A, et al. (2019). Arsenic modifies serotonin metabolism through glucuronidation in pancreatic beta-cells. American Journal of Physiology. Endocrinology and Metabolism, 316(3), E464–E474. 10.1152/ajpendo.00302.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals-Casas C, Feige JN, & Desvergne B (2008). Interference of pollutants with PPARs: Endocrine disruption meets metabolism. International Journal of Obesity, 32(Suppl. 6), S53–S61. 10.1038/ijo.2008.207. [DOI] [PubMed] [Google Scholar]
- CDC. (2020). Centers for disease control and prevention National Diabetes Statistics Report, 2020. Retrieved from https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf.
- Chamorro-Garcia R, Diaz-Castillo C, Shoucri BM, Kach H, Leavitt R, Shioda T, et al. (2017). Ancestral perinatal obesogen exposure results in a transgenerational thrifty phenotype in mice. Nature Communications, 8(1), 2012. 10.1038/s41467-017-01944-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Chen KJ, Mullur R, & Bernal-Mizrachi E (2008). Beta-cell failure as a complication of diabetes. Reviews in Endocrine & Metabolic Disorders, 9(4), 329–343. 10.1007/s11154-008-9101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JY, Yu SD, & Hong YS (2014). Environmental source of arsenic exposure. Journal of Preventive Medicine and Public Health, 47(5), 253–257. 10.3961/jpmph.14.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino I, Fiory F, Perruolo G, Miele C, Beguinot F, Formisano P, et al. (2020). Potential mechanisms of bisphenol A (BPA) contributing to human disease. International Journal of Molecular Sciences, 21(16), 5761. 10.3390/ijms21165761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JM, West BT, Yaker Z, Lauinger B, McCullers D, Haubert J, et al. (2020). Disparities in diabetes-related multiple chronic conditions and mortality: The influence of race. Diabetes Research and Clinical Practice, 159, 107984. 10.1016/j.diabres.2019.107984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Lv X, Chen Z, Huang Z, Li B, Xie Y, et al. (2020). Elevated whole blood arsenic level is associated with type 2 diabetes in coal-burning areas in Guizhou. Toxicology and Applied Pharmacology, 403, 115135. 10.1016/j.taap.2020.115135. [DOI] [PubMed] [Google Scholar]
- Darbre PD (2018). Overview of air pollution and endocrine disorders. International Journal of General Medicine, 11, 191–207. 10.2147/IJGM.S102230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Thomas AA, Shorter KS, Brown SL, & Baumgarner BL (2018). Cellular fatty acid level regulates the effect of tolylfluanid on mitochondrial dysfunction and insulin sensitivity in C2C12 skeletal myotubes. Biochemical and Biophysical Research Communications, 505(2), 392–398. 10.1016/j.bbrc.2018.09.131. [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. (2009). Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocrine Reviews, 30(4), 293–342. 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo-Relloso A, Grau-Perez M, Galan-Chilet I, Garrido-Martinez MJ, Tormos C, Navas-Acien A, et al. (2019). Urinary metals and metal mixtures and oxidative stress biomarkers in an adult population from Spain: The Hortega study. Environment International, 123, 171–180. 10.1016/j.envint.2018.11.055. [DOI] [PubMed] [Google Scholar]
- Douillet C, Huang MC, Saunders RJ, Dover EN, Zhang C, & Styblo M (2017). Knockout of arsenic (+3 oxidation state) methyltransferase is associated with adverse metabolic phenotype in mice: The role of sex and arsenic exposure. Archives of Toxicology, 91(7), 2617–2627. 10.1007/s00204-016-1890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover GJ (2009). The barker hypothesis: How pediatricans will diagnose and prevent common adult-onset diseases. Transactions of the American Clinical and Climatological Association, 120, 199–207. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/19768178. [PMC free article] [PubMed] [Google Scholar]
- Dover EN, Patel NY, & Styblo M (2018). Impact of in vitro heavy metal exposure on pancreatic beta-cell function. Toxicology Letters, 299, 137–144. 10.1016/j.toxlet.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JS, Duffy E, Gilmour MK, Kirkpatrick J, & McLetchie NG (1944). Further observations on the effects of alloxan on the pancreatic islets. The Journal of Physiology, 103(2), 233–243. 10.1113/jphysiol.1944.sp004072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich S, Lambers D, Baccarelli A, Khoury J, Macaluso M, & Ho SM (2016). Endocrine disruptors: A potential risk factor for gestational diabetes mellitus. American Journal of Perinatology, 33(13), 1313–1318. 10.1055/s-0036-1586500. [DOI] [PubMed] [Google Scholar]
- El Muayed M, Raja MR, Zhang X, MacRenaris KW, Bhatt S, Chen X, et al. (2012). Accumulation of cadmium in insulin-producing beta cells. Islets, 4(6), 405–416. 10.4161/isl.23101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary M, Honda T, Morgan G, Kelly P, Guo Y, & Negin J (2020). Ambient air pollution exposure association with diabetes prevalence and glycosylated hemoglobin (HbA1c) levels in China. Cross-sectional analysis from the WHO study of AGEing and adult health wave 1. Journal of Environmental Science and Health. Part A, Toxic/Hazardous Substances & Environmental Engineering, 55(10), 1149–1162. 10.1080/10934529.2020.1787011. [DOI] [PubMed] [Google Scholar]
- Elmorsy E, Al-Ghafari A, Al Doghaither H, & Ghulam J (2021). Effects of environmental metals on mitochondrial bioenergetics of the CD-1 mice pancreatic beta-cells. Toxicology In Vitro, 70, 105015. 10.1016/j.tiv.2020.105015. [DOI] [PubMed] [Google Scholar]
- Elten M, Donelle J, Lima I, Burnett RT, Weichenthal S, Stieb DM, et al. (2020). Ambient air pollution and incidence of early-onset paediatric type 1 diabetes: A retrospective population-based cohort study. Environmental Research, 184, 109291. 10.1016/j.envres.2020.109291. [DOI] [PubMed] [Google Scholar]
- Encarnacao T, Pais AA, Campos MG, & Burrows HD (2019). Endocrine disrupting chemicals: Impact on human health, wildlife and the environment. Science Progress, 102(1), 3–42. 10.1177/0036850419826802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser N, Utzschneider KM, & Kahn SE (2020). Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia, 63(10), 2007–2021. 10.1007/s00125-020-05245-x. [DOI] [PubMed] [Google Scholar]
- Fang L, Guo J, Wang Q, Ou K, Zou M, Lv L, et al. (2020). Chronic exposure to environmental level phenanthrene induces non-obesity-dependent insulin resistance in male mice. Environmental Science & Technology, 54(23), 15225–15234. 10.1021/acs.est.0c04171. [DOI] [PubMed] [Google Scholar]
- Farkhondeh T, Aschner M, Sadeghi M, Mehrpour O, Naseri K, Amirabadizadeh A, et al. (2020). The effect of diazinon on blood glucose homeostasis: A systematic and meta-analysis study. Environmental Science and Pollution Research International, 28, 4007–4018. 10.1007/s11356-020-11364-0. [DOI] [PubMed] [Google Scholar]
- Faroon O, & Ruiz P (2016). Polychlorinated biphenyls: New evidence from the last decade. Toxicology and Industrial Health, 32(11), 1825–1847. 10.1177/0748233715587849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan SF, Gossai A, Chen Y, Chasan-Taber L, Baker E, & Karagas M (2016). Maternal arsenic exposure and gestational diabetes and glucose intolerance in the New Hampshire birth cohort study. Environmental Health, 15(1), 106. 10.1186/s12940-016-0194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardi T, Panimolle F, Lenzi A,& Morano S (2020). Bisphenol A and phthalates in diet: An emerging link with pregnancy complications. Nutrients, 12(2), 525. 10.3390/nu12020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LJ, Zhou HR, & Wagner MA (1996). Polychlorinated biphenyls release insulin from RINm5F cells. Life Sciences, 59(24), 2041–2049. 10.1016/s0024-3205(96)00557-7. [DOI] [PubMed] [Google Scholar]
- Fu Z, Gilbert ER, & Liu D (2013). Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Current Diabetes Reviews, 9(1), 25–53. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/22974359. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Arevalo M, Alonso-Magdalena P, Rebelo Dos Santos J, Quesada I, Carneiro EM, & Nadal A (2014). Exposure to bisphenol-A during pregnancy partially mimics the effects of a high-fat diet altering glucose homeostasis and gene expression in adult male mice. PLoS One, 9(6), e100214. 10.1371/journal.pone.0100214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco M, Anderson-Shaw L, & Sargis RM (2020). Unwitting accomplices: Endocrine disruptors confounding clinical care. The Journal of Clinical Endocrinology and Metabolism, 105(10), e3822–e3827. 10.1210/clinem/dgaa358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, et al. (2015). Executive summary to EDC-2: The Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocrine Reviews, 36(6), 593–602. 10.1210/er.2015-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagobian TA, Brunner-Gaydos H, Seal A, Schaffner A, Kitts C, Hubbard R, et al. (2020). Rationale and design of a randomized controlled trial examining oral administration of bisphenol A on hepatic glucose production and skeletal muscle insulin sensitivity in adults. Contemporary Clinical Trials Communications, 17, 100549. 10.1016/j.conctc.2020.100549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. (2017). Metabolism disrupting chemicals and metabolic disorders. Reproductive Toxicology, 68, 3–33. 10.1016/j.reprotox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicken MT, Gee GC, Morenoff J, Connell CM, Snow RC, & Hu H (2012). A novel look at racial health disparities: The interaction between social disadvantage and environmental health. American Journal of Public Health, 102(12), 2344–2351. 10.2105/AJPH.2012.300774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou R, Zhang H, Chen H, Zhou Y, Long Y, & Liu D (2019). Total pancreatic necrosis after organophosphate intoxication. Frontiers in Medicine, 13(2), 285–288. 10.1007/s11684-018-0626-z. [DOI] [PubMed] [Google Scholar]
- Howard SG (2018). Developmental exposure to endocrine disrupting chemicals and type 1 diabetes mellitus. Frontiers in Endocrinology, 9, 513. 10.3389/fendo.2018.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh L, Wu W, Hirsh AT, de Groot M, Mather KJ, & Stewart JC (2020). Undiagnosed diabetes among immigrant and racial/ethnic minority adults in the United States: National Health and Nutrition Examination Survey 2011-2018. Annals of Epidemiology, 51, 14–19. 10.1016/j.annepidem.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Kan H, Kearney GD, & Xu X (2015). Associations between exposure to polycyclic aromatic hydrocarbons and glucose homeostasis as well as metabolic syndrome in non-diabetic adults. The Science of the Total Environment, 505, 56–64. 10.1016/j.scitotenv.2014.09.085. [DOI] [PubMed] [Google Scholar]
- Huang M, Douillet C, & Styblo M (2019). Arsenite and its trivalent methylated metabolites inhibit glucose-stimulated calcium influx and insulin secretion in murine pancreatic islets. Archives of Toxicology, 93(9), 2525–2533. 10.1007/s00204-019-02526-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang MJ, Kim JH, Koo YS, Yun HY, & Cheong HK (2020). Impacts of ambient air pollution on glucose metabolism in Korean adults: A Korea National Health and Nutrition Examination Survey study. Environmental Health, 19(1), 70. 10.1186/s12940-020-00623-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavicoli I, Fontana L, & Bergamaschi A (2009). The effects of metals as endocrine disruptors. Journal of Toxicology and Environmental Health. Part B, Critical Reviews, 12(3), 206–223. 10.1080/10937400902902062. [DOI] [PubMed] [Google Scholar]
- Irigaray P, Ogier V, Jacquenet S, Notet V, Sibille P, Mejean L, et al. (2006). Benzo[a] pyrene impairs beta-adrenergic stimulation of adipose tissue lipolysis and causes weight gain in mice. A novel molecular mechanism of toxicity for a common food pollutant. The FEBS Journal, 273(7), 1362–1372. 10.1111/j.1742-4658.2006.05159.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Graham JL, Stanhope KL, Havel PJ, & La Merrill MA (2015). Effect of DDT exposure on lipids and energy balance in obese Sprague-Dawley rats before and after weight loss. Toxicology Reports, 2, 990–995. 10.1016/j.toxrep.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbari F, Mohseni Bandpei A, Daneshpour MS, Shahsavani A, Hashemi Nazari SS, Faraji Sabokbar H, et al. (2020). Role of air pollution and rs10830963 polymorphism on the incidence of type 2 diabetes: Tehran Cardiometabolic Genetic Study. Journal Diabetes Research, 2020, 2928618. 10.1155/2020/2928618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaga K, & Dharmani C (2003). Global surveillance of DDT and DDE levels in human tissues. International Journal of Occupational Medicine and Environmental Health, 16(1), 7–20. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/12705713. [PubMed] [Google Scholar]
- James-Todd TM, Chiu YH, & Zota AR (2016). Racial/ethnic disparities in environmental endocrine disrupting chemicals and women’s reproductive health outcomes: Epidemiological examples across the life course. Current Epidemiology Reports, 3(2), 161–180. 10.1007/s40471-016-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Qiu T, Yao X, Jiang L, Wang N, Wei S, et al. (2020). Arsenic induces hepatic insulin resistance via mtROS-NLRP3 inflammasome pathway. Journal of Hazardous Materials, 399, 123034. 10.1016/j.jhazmat.2020.123034. [DOI] [PubMed] [Google Scholar]
- Kang JH, Kondo F, & Katayama Y (2006). Human exposure to bisphenol A. Toxicology, 226(2–3), 79–89. 10.1016/j.tox.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Kara M, Ozta SE, & Ozhan G (2020). Acetamiprid-induced Cyto- and genotoxicity in the AR42J pancreatic cell line. Turkish Journal of Pharmaceutical Sciences, 17(5), 474–479. 10.4274/tjps.galenos.2019.89719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karami-Mohajeri S, & Abdollahi M (2013). Mitochondrial dysfunction and organophosphorus compounds. Toxicology and Applied Pharmacology, 270(1), 39–44. 10.1016/j.taap.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Khan F, Hodjat M, Rahimifard M, Nigjeh MN, Azizi M, Baeeri M, et al. (2020). Assessment of arsenic-induced modifications in the DNA methylation of insulin-related genes in rat pancreatic islets. Ecotoxicology and Environmental Safety, 201, 110802. 10.1016/j.ecoenv.2020.110802. [DOI] [PubMed] [Google Scholar]
- Kim HY, Kwon WY, Kim YA, Oh YJ, Yoo SH, Lee MH, et al. (2017). Polychlorinated biphenyls exposure-induced insulin resistance is mediated by lipid droplet enlargement through Fsp27. Archives of Toxicology, 91(6), 2353–2363. 10.1007/s00204-016-1889-2. [DOI] [PubMed] [Google Scholar]
- Kim JT, & Lee HK (2014). Metabolic syndrome and the environmental pollutants from mitochondrial perspectives. Reviews in Endocrine & Metabolic Disorders, 15(4), 253–262. 10.1007/s11154-014-9297-5. [DOI] [PubMed] [Google Scholar]
- Kirkley AG, Carmean CM, Ruiz D, Ye H, Regnier SM, Poudel A, et al. (2018). Arsenic exposure induces glucose intolerance and alters global energy metabolism. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 314(2), R294–R303. 10.1152/ajpregu.00522.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek KD, Murphy SL, Xu J, & Arias E (2019). Deaths: Final data for 2017. National Vital Statistics Reports, 68(9), 1–77. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/32501199. [PubMed] [Google Scholar]
- La Merrill M, Karey E, Moshier E, Lindtner C, La Frano MR, Newman JW, et al. (2014). Perinatal exposure of mice to the pesticide DDT impairs energy expenditure and metabolism in adult female offspring. PLoS One, 9(7). 10.1371/journal.pone.0103337, e103337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Merrill MA, Vandenberg LN, Smith MT, Goodson W, Browne P, Patisaul HB, et al. (2020). Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nature Reviews. Endocrinology, 16(1), 45–57. 10.1038/s41574-019-0273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Park YJ, Kim MJ, Kim S, Choi S, Park J, et al. (2020). Associations of urinary concentrations of phthalate metabolites, bisphenol A, and parabens with obesity and diabetes mellitus in a Korean adult population: Korean National Environmental Health Survey (KoNEHS) 2015-2017. Environment International, 146, 106227. 10.1016/j.envint.2020.106227. [DOI] [PubMed] [Google Scholar]
- Lenzen S (2008). The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia, 51(2), 216–226. 10.1007/s00125-007-0886-7. [DOI] [PubMed] [Google Scholar]
- Leonel Javeres MN, Raza S, Judith N, Anwar F, Habib R, Batool S, et al. (2020). Mixture of organophosphates chronic exposure and pancreatic dysregulations in two different population samples. Frontiers in Public Health, 8, 534902. 10.3389/fpubh.2020.534902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Douillet C, Huang M, Beck R, Sumner SJ, & Styblo M (2020). Exposure to inorganic arsenic and its methylated metabolites alters metabolomics profiles in INS-1832/13 insulinoma cells and isolated pancreatic islets. Archives of Toxicology, 94(6), 1955–1972. 10.1007/s00204-020-02729-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Guo J, Xi Z, Xu J, Zuo Z, & Wang C (2017). Tributyltin in male mice disrupts glucose homeostasis as well as recovery after exposure: Mechanism analysis. Archives of Toxicology, 91(10), 3261–3269. 10.1007/s00204-017-1961-6. [DOI] [PubMed] [Google Scholar]
- Li A, Mei Y, Zhao M, Xu J, Seery S, Li R, et al. (2021). The effect of ambient ozone on glucose-homoeostasis: A prospective study of non-diabetic older adults in Beijing. Science of the Total Environment, 761, 143308. 10.1016/j.scitotenv.2020.143308. [DOI] [PubMed] [Google Scholar]
- Li L, Wang F, Zhang J, Wang K, De X, Li L, et al. (2021). Typical phthalic acid esters induce apoptosis by regulating the PI3K/Akt/Bcl-2 signaling pathway in rat insulinoma cells. Ecotoxicology and Environmental Safety, 208, 111461. 10.1016/j.ecoenv.2020.111461. [DOI] [PubMed] [Google Scholar]
- Lin Y, Sun X, Qiu L, Wei J, Huang Q, Fang C, et al. (2013). Exposure to bisphenol A induces dysfunction of insulin secretion and apoptosis through the damage of mitochondria in rat insulinoma (INS-1) cells. Cell Death & Disease, 4, e460. 10.1038/cddis.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wei J, Li Y, Chen J, Zhou Z, Song L, et al. (2011). Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. American Journal of Physiology. Endocrinology and Metabolism, 301(3), E527–E538. 10.1152/ajpendo.00233.2011. [DOI] [PubMed] [Google Scholar]
- Lin Y, Zhou S, Liu H, Cui Z, Hou F, Feng S, et al. (2020). Risk analysis of air pollution and meteorological factors affecting the incidence of diabetes in the elderly population in Northern China. Journal Diabetes Research, 2020, 3673980. 10.1155/2020/3673980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PM, & Lind L (2018). Endocrine-disrupting chemicals and risk of diabetes: An evidence-based review. Diabetologia, 61(7), 1495–1502. 10.1007/s00125-018-4621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ren ZH, Qiang H, Wu J, Shen M, Zhang L, et al. (2020). Trends in the incidence of diabetes mellitus: Results from the Global Burden of Disease Study 2017 and implications for diabetes mellitus prevention. BMC Public Health, 20(1), 1415. 10.1186/s12889-020-09502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SH, Yang RS, Yen YP, Chiu CY, Tsai KS, & Lan KC (2015). Low-concentration arsenic trioxide inhibits skeletal myoblast cell proliferation via a reactive oxygen species-independent pathway. PLoS One, 10(9), e0137907. 10.1371/journal.pone.0137907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang L, Chen L, Li J, Wang J, Zhao Y, et al. (2021). Identification and prioritization of the potent components for combined exposure of multiple persistent organic pollutants associated with gestational diabetes mellitus. Journal of Hazardous Materials, 409, 124905. 10.1016/j.jhazmat.2020.124905. [DOI] [PubMed] [Google Scholar]
- Lv Z, Li G, Li Y, Ying C, Chen J, Chen T, et al. (2013). Glucose and lipid homeostasis in adult rat is impaired by early-life exposure to perfluorooctane sulfonate. Environmental Toxicology, 28(9), 532–542. 10.1002/tox.20747. [DOI] [PubMed] [Google Scholar]
- Ma Y, Xia W, Wang DQ, Wan YJ, Xu B, Chen X, et al. (2013). Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia, 56(9), 2059–2067. 10.1007/s00125-013-2944-7. [DOI] [PubMed] [Google Scholar]
- Malmqvist E, Larsson HE, Jonsson I, Rignell-Hydbom A, Ivarsson SA, Tinnerberg H, et al. (2015). Maternal exposure to air pollution and type 1 diabetes—Accounting for genetic factors. Environmental Research, 140, 268–274. 10.1016/j.envres.2015.03.024. [DOI] [PubMed] [Google Scholar]
- Mangum LH, Crow JA, Stokes JV, Howell GE 3rd, Ross MK, Pruett SB, et al. (2016). Exposure to p,p’-DDE alters macrophage reactivity and increases macrophage numbers in adipose stromal vascular fraction. Toxicological Sciences, 150(1), 169–177. 10.1093/toxsci/kfv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri EH, & Reggabi M (2020). Association between type 2 diabetes and exposure to chlorinated persistent organic pollutants in Algeria: A case-control study. Chemosphere, 264(Pt. 2), 128596. 10.1016/j.chemosphere.2020.128596. [DOI] [PubMed] [Google Scholar]
- Manukyan L, Dunder L, Lind PM, Bergsten P, & Lejonklou MH (2019). Developmental exposure to a very low dose of bisphenol A induces persistent islet insulin hypersecretion in Fischer 344 rat offspring. Environmental Research, 172, 127–136. 10.1016/j.envres.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Mao Z, Xia W, Chang H, Huo W, Li Y, & Xu S (2015). Paternal BPA exposure in early life alters Igf2 epigenetic status in sperm and induces pancreatic impairment in rat off-spring. Toxicology Letters, 238(3), 30–38. 10.1016/j.toxlet.2015.08.009. [DOI] [PubMed] [Google Scholar]
- Marshall MC Jr. (2005). Diabetes in African Americans. Postgraduate Medical Journal, 81(962), 734–740. 10.1136/pgmj.2004.028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI (2010). Genomics, type 2 diabetes, and obesity. The New England Journal of Medicine, 363(24), 2339–2350. 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- Mengozzi A, Carli F, Guiducci L, Parolini F, Biancalana E, Gastaldelli A, et al. (2021). SGLT2 inhibitors and thiazide enhance excretion of DEHP toxic metabolites in subjects with type 2 diabetes: A randomized clinical trial. Environmental Research, 192, 110316. 10.1016/j.envres.2020.110316. [DOI] [PubMed] [Google Scholar]
- Miller LV, Stokes JD, & Silpipat C (1978). Diabetes mellitus and autonomic dysfunction after vacor rodenticide ingestion. Diabetes Care, 1(2), 73–76. 10.2337/diacare.1.2.73. [DOI] [PubMed] [Google Scholar]
- Miura Y, Hori Y, Kimura S, Hachiya H, Sakurai Y, Inoue K, et al. (2012). Triphenyltin impairs insulin secretion by decreasing glucose-induced NADP(H) and ATP production in hamster pancreatic beta-cells. Toxicology, 299(2–3), 165–171. 10.1016/j.tox.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Mondal V, Hosen Z, Hossen F, Siddique AE, Tony SR, Islam Z, et al. (2020). Arsenic exposure-related hyperglycemia is linked to insulin resistance with concomitant reduction of skeletal muscle mass. Environment International, 143, 105890. 10.1016/j.envint.2020.105890. [DOI] [PubMed] [Google Scholar]
- Neel BA, Brady MJ, & Sargis RM (2013). The endocrine disrupting chemical tolylfluanid alters adipocyte metabolism via glucocorticoid receptor activation. Molecular Endocrinology, 27(3), 394–406. 10.1210/me.2012-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurulain SM, Szegi P, Tekes K, & Naqvi SN (2013). Antioxidants in organophosphorus compounds poisoning. Arhiv za Higijenu Rada i Toksikologiju, 64(1), 169–177. 10.2478/10004-1254-64-2013-2294. [DOI] [PubMed] [Google Scholar]
- Orskov C (1992). Glucagon-like peptide-1, a new hormone of the entero-insular axis. Diabetologia, 35(8), 701–711. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/1324859. [PubMed] [Google Scholar]
- Park CM, Kim KT, & Rhyu DY (2020). Low-concentration exposure to organochlorine pesticides (OCPs) in L6 myotubes and RIN-m5F pancreatic beta cells induces disorders of glucose metabolism. Toxicology In Vitro, 65, 104767. 10.1016/j.tiv.2020.104767. [DOI] [PubMed] [Google Scholar]
- Paul DS, Harmon AW, Devesa V, Thomas DJ, & Styblo M (2007). Molecular mechanisms of the diabetogenic effects of arsenic: Inhibition of insulin signaling by arsenite and methylarsonous acid. Environmental Health Perspectives, 115(5), 734–742. 10.1289/ehp.9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlikova N, Smetana P, Halada P, & Kovar J (2015). Effect of prolonged exposure to sublethal concentrations of DDT and DDE on protein expression in human pancreatic beta cells. Environmental Research, 142, 257–263. 10.1016/j.envres.2015.06.046. [DOI] [PubMed] [Google Scholar]
- Pavlikova N, Sramek J, Jelinek M, Halada P, & Kovar J (2020). Markers of acute toxicity of DDT exposure in pancreatic beta-cells determined by a proteomic approach. PLoS One, 15(10), e0229430. 10.1371/journal.pone.0229430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault L, McCurdy C, Kerege AA, Houck J, Faerch K, & Bergman BC (2013). Bisphenol A impairs hepatic glucose sensing in C57BL/6 male mice. PLoS One, 8(7), e69991. 10.1371/journal.pone.0069991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, & Nolan CJ (2006). Islet beta cell failure in type 2 diabetes. The Journal of Clinical Investigation, 116(7), 1802–1812. 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston EV, Eberle C, Brown FM, & James-Todd T (2020). Climate factors and gestational diabetes mellitus risk—A systematic review. Environmental Health, 19(1), 112. 10.1186/s12940-020-00668-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Stilp AM, Sofer T, Moon JY, Hidalgo B, Szpiro AA, et al. (2017). Genetics of type 2 diabetes in U.S. Hispanic/Latino individuals: Results from the Hispanic Community Health Study/Study of Latinos (HCHS/SOL). Diabetes, 66(5), 1419–1425. 10.2337/db16-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdas M, Sharma S, Kaul D, & Bhatia A (2018). Possible role of miR-2909 RNomics in arsenic mediated pancreatic beta-cell dysfunction. Journal of Trace Elements in Medicine and Biology, 50, 263–267. 10.1016/j.jtemb.2018.07.006. [DOI] [PubMed] [Google Scholar]
- Rasinger JD, Carroll TS, Lundebye AK, & Hogstrand C (2014). Cross-omics gene and protein expression profiling in juvenile female mice highlights disruption of calcium and zinc signalling in the brain following dietary exposure to CB-153, BDE-47, HBCD or TCDD. Toxicology, 321, 1–12. 10.1016/j.tox.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Rattan S, & Flaws JA (2019). The epigenetic impacts of endocrine disruptors on female reproduction across generationsdagger. Biology of Reproduction, 101(3), 635–644. 10.1093/biolre/ioz081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier SM, El-Hashani E, Kamau W, Zhang X, Massad NL, & Sargis RM (2015). Tributyltin differentially promotes development of a phenotypically distinct adipocyte. Obesity (Silver Spring), 23(9), 1864–1871. 10.1002/oby.21174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier SM, Kirkley AG, Ruiz D, Kamau W, Wu Q, Kannan K, et al. (2018). Diet-dependence of metabolic perturbations mediated by the endocrine disruptor tolylfluanid. Endocrine Connections, 7(1), 159–168. 10.1530/EC-17-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier SM, Kirkley AG,Ye H, El-Hashani E, Zhang X, Neel BA,et al. (2015). Dietary exposure to the endocrine disruptor tolylfluanid promotes global metabolic dysfunction in male mice. Endocrinology, 156(3), 896–910. 10.1210/en.2014-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rignell-Hydbom A, Lidfeldt J, Kiviranta H, Rantakokko P, Samsioe G, Agardh CD, et al. (2009). Exposure to p,p’-DDE: A risk factor for type 2 diabetes. PLoS One, 4(10), e7503. 10.1371/journal.pone.0007503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz SA (2010). Air pollution as a potential contributor to the ‘epidemic’ of autoimmune disease. Medical Hypotheses, 74(1), 110–117. 10.1016/j.mehy.2009.07.033. [DOI] [PubMed] [Google Scholar]
- Robertson RP, Harmon J, Tran PO, Tanaka Y, & Takahashi H (2003). Glucose toxicity in beta-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes, 52(3), 581–587. 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- Ruiz D, Becerra M, Jagai JS, Ard K, & Sargis RM (2018). Disparities in environmental exposures to endocrine-disrupting chemicals and diabetes risk in vulnerable populations. Diabetes Care, 41(1), 193–205. 10.2337/dc16-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz P, Perlina A, Mumtaz M, & Fowler BA (2016). A systems biology approach reveals converging molecular mechanisms that link different POPs to common metabolic diseases. Environmental Health Perspectives, 124(7), 1034–1041. 10.1289/ehp.1510308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz D, Regnier SM, Kirkley AG, Hara M, Haro F, Aldirawi H, et al. (2019). Developmental exposure to the endocrine disruptor tolylfluanid induces sex-specific later-life metabolic dysfunction. Reproductive Toxicology, 89, 74–82. 10.1016/j.reprotox.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J, Petersen R, Meugnier E, Madsen L, Lock EJ, Lillefosse H, et al. (2010). Persistent organic pollutant exposure leads to insulin resistance syndrome. Environmental Health Perspectives, 118(4), 465–471. 10.1289/ehp.0901321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Research and Clinical Practice, 157, 107843. 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- Salihovic S, Ganna A, Fall T, Broeckling CD, Prenni JE, van Bavel B, et al. (2016). The metabolic fingerprint of p,p’-DDE and HCB exposure in humans. Environment International, 88, 60–66. 10.1016/j.envint.2015.12.015. [DOI] [PubMed] [Google Scholar]
- Salmeri N, Villanacci R, Ottolina J, Bartiromo L, Cavoretto P, Dolci C, et al. (2020). Maternal arsenic exposure and gestational diabetes: A systematic review and meta-analysis. Nutrients, 12(10), 3094. 10.3390/nu12103094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, & Shulman GI (2012). Mechanisms for insulin resistance: Common threads and missing links. Cell, 148(5), 852–871. 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargis RM (2015). Metabolic disruption in context: Clinical avenues for synergistic perturbations in energy homeostasis by endocrine disrupting chemicals. Endocrine Disruptors, 3(1), e1080788. 10.1080/23273747.2015.1080788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargis RM, Heindel JJ, & Padmanabhan V (2019). Interventions to address environmental metabolism-disrupting chemicals: Changing the narrative to empower action to restore metabolic health. Frontiers in Endocrinology, 10, 33. 10.3389/fendo.2019.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargis RM, Neel BA,Brock CO, Lin Y, Hickey AT, Carlton DA, et al. (2012). The novel endocrine disruptor tolylfluanid impairs insulin signaling in primary rodent and human adipocytes through a reduction in insulin receptor substrate-1 levels. Biochimica et Biophysica Acta, 1822(6), 952–960. 10.1016/j.bbadis.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh S,Jagai JS, Ashley C, Zhou S, & Sargis RM (2018). Underutilized andunder threat: Environmental policy as a tool to address diabetes risk. Current Diabetes Reports, 18(5), 25. 10.1007/s11892-018-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar A, & Teppala S (2011). Relationship between urinary bisphenol A levels and diabetes mellitus. The Journal of Clinical Endocrinology and Metabolism, 96(12), 3822–3826. 10.1210/jc.2011-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Jan J, Hardesty JE, Falkner KC, Prough RA, Balamurugan AN, et al. (2019). Polychlorinated biphenyl exposures differentially regulate hepatic metabolism and pancreatic function: Implications for non-alcoholic steatohepatitis and diabetes. Toxicology and Applied Pharmacology, 363, 22–33. 10.1016/j.taap.2018.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley JM, Hurst CH, Tanaka SS, DeRoos FL, Butenhoff JL, Seacat AM, et al. (2004). trans-activation of PPARalpha and induction of PPARalpha target genes by perfluorooctane-based chemicals. Toxicological Sciences, 80(1), 151–160. 10.1093/toxsci/kfh130. [DOI] [PubMed] [Google Scholar]
- Shoucri BM, Hung VT, Chamorro-Garcia R, Shioda T, & Blumberg B (2018). Retinoid X receptor activation during adipogenesis of female mesenchymal stem cells programs a dysfunctional adipocyte. Endocrinology, 159(8), 2863–2883. 10.1210/en.2018-00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh VK, Sarkar SK, Saxena A, & Koner BC (2019). Effect of subtoxic DDT exposure on glucose uptake and insulin signaling in rat L6 myoblast-derived myotubes. International Journal of Toxicology, 38(4), 303–311. 10.1177/1091581819850577. [DOI] [PubMed] [Google Scholar]
- Skinner MK (2016). Endocrine disruptors in 2015: Epigenetic transgenerational inheritance. Nature Reviews. Endocrinology, 12(2), 68–70. 10.1038/nrendo.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiljevska-Ristovska V, Sabriu-Haxhijaha A, Ristoski T, Kosharkoska-Spasovska F, Krstanoski L, & Dimitrova-Shumkovska J (2020). Markers involved in proinflammatory effects by environmental toxicants. Toxicology Mechanisms and Methods, 30(8), 570–579. 10.1080/15376516.2020.1791293. [DOI] [PubMed] [Google Scholar]
- Song Y, & Yang L (2017). Transgenerational pancreatic impairment with Igf2/H19 epigenetic alteration induced by p,p’-DDE exposure in early life. Toxicology Letters, 280, 222–231. 10.1016/j.toxlet.2017.08.083. [DOI] [PubMed] [Google Scholar]
- Sonksen PH (2001). Insulin, growth hormone and sport. The Journal of Endocrinology, 170(1), 13–25. 10.1677/joe.0.1700013. [DOI] [PubMed] [Google Scholar]
- Soundararajan A, Prabu P, Mohan V, Gibert Y, & Balasubramanyam M (2019). Novel insights of elevated systemic levels of bisphenol-A (BPA) linked to poor glycemic control, accelerated cellular senescence and insulin resistance in patients with type 2 diabetes. Molecular and Cellular Biochemistry, 458(1–2), 171–183. 10.1007/s11010-019-03540-9. [DOI] [PubMed] [Google Scholar]
- Stahlhut RW, Myers JP, Taylor JA, Nadal A, Dyer JA, & Vom Saal FS (2018). Experimental BPA exposure and glucose-stimulated insulin response in adult men and women. Journal of the Endocrine Society, 2(10), 1173–1187. 10.1210/js.2018-00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump CS, Short KR, Bigelow ML, Schimke JM, & Nair KS (2003). Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proceedings of the National Academy of Sciences of the United States of America, 100(13), 7996–8001. 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Qi W, Xiao X, Yang SH, Kim D, Yoon KS, et al. (2017). Imidacloprid promotes high fat diet-induced adiposity in female C57BL/6J mice and enhances adipogenesis in 3T3-L1 adipocytes via the AMPKalpha-mediated pathway. Journal of Agricultural and Food Chemistry, 65(31), 6572–6581. 10.1021/acs.jafc.7b02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter-Dub MT (2002). Rapid non-genomic and genomic responses to progestogens, estrogens, and glucocorticoids in the endocrine pancreatic B cell, the adipocyte and other cell types. Steroids, 67(2), 77–93. 10.1016/s0039-128x(01)00142-8. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Yamashiro Y, Chung MY, Hara Y, Ishihara K, Ejiri K, et al. (1989). Vacor inhibits insulin release from islets in vitro. Journal of Endocrinological Investigation, 12(4), 273–275. 10.1007/BF03349985. [DOI] [PubMed] [Google Scholar]
- Trasande L (2019). Sicker, fatter, poorer: the urgent threat of hormone-disrupting chemicals on our health and future … and what we can do about it (p. 1). online resource.
- Turyk M, Anderson H, Knobeloch L, Imm P, & Persky V (2009). Organochlorine exposure and incidence of diabetes in a cohort of Great Lakes sport fish consumers. Environmental Health Perspectives, 117(7), 1076–1082. 10.1289/ehp.0800281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino R, D’Esposito V, Passaretti F, Liotti A, Cabaro S, Longo M, et al. (2013). Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS One, 8(12), e82099. 10.1371/journal.pone.0082099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela OL, Drobna Z, Hernandez-Castellanos E, Sanchez-Pena LC, Garcia-Vargas GG, Borja-Aburto VH, et al. (2009). Association of AS3MT polymorphisms and the risk of premalignant arsenic skin lesions. Toxicology and Applied Pharmacology, 239(2), 200–207. 10.1016/j.taap.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr., Lee DH, et al. (2012). Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocrine Reviews, 33(3), 378–455. 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Pennathur S, Kannan K, Patisaul HB, Dolinoy DC, Zeng L, et al. (2015). Impact of gestational bisphenol a on oxidative stress and free fatty acids: Human association and interspecies animal testing studies. Endocrinology, 156(3), 911–922. 10.1210/en.2014-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Karvonen-Gutierrez CA, Herman WH, Mukherjee B, Harlow SD, & Park SK (2020). Urinary metals and incident diabetes in midlife women: Study of Women’s Health Across the Nation (SWAN). BMJ Open Diabetes Research & Care, 8(1), e001233. 10.1136/bmjdrc-2020-001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Li M, Chen B, Xu M, Xu Y, Huang Y, et al. (2012). Urinary bisphenol A (BPA) concentration associates with obesity and insulin resistance. The Journal of Clinical Endocrinology and Metabolism, 97(2), E223–E227. 10.1210/jc.2011-1989. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li YL, Meng FZ, Hou BL, Zhuang CN, Xiong SH, et al. (2018). Effects of omethoate on liver insulin signaling in mice. Biomedical and Environmental Sciences, 31(8), 627–631. 10.3967/bes2018.086. [DOI] [PubMed] [Google Scholar]
- Wang X, Mukherjee B, Karvonen-Gutierrez CA, Herman WH, Batterman S, Harlow SD, et al. (2020). Urinary metal mixtures and longitudinal changes in glucose homeostasis: The Study of Women’s Health Across the Nation (SWAN). Environment International, 145, 106109. 10.1016/j.envint.2020.106109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SL, Tsai PC, Yang CY, & Guo YL (2008). Increased risk of diabetes and polychlorinated biphenyls and dioxins: A 24-year follow-up study of the Yucheng cohort. Diabetes Care, 31(8), 1574–1579. 10.2337/dc07-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Ding D, Wang T, Liu Q, & Lin Y (2017). MiR-338 controls BPA-triggered pancreatic islet insulin secretory dysfunction from compensation to decompensation by targeting Pdx-1. The FASEB Journal, 31(12), 5184–5195. 10.1096/fj.201700282R. [DOI] [PubMed] [Google Scholar]
- Wei J, Hao Q, Chen C, Li J, Han X, Lei Z, et al. (2020). Epigenetic repression of miR-17 contributed to di(2-ethylhexyl) phthalate-triggered insulin resistance by targeting Keap1-Nrf2/miR-200a axis in skeletal muscle. Theranostics, 10(20), 9230–9248. 10.7150/thno.45253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia X, et al. (2020). Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. Journal of Hazardous Materials, 384, 121390. 10.1016/j.jhazmat.2019.121390. [DOI] [PubMed] [Google Scholar]
- Xing Y, Xia W, Zhang B, Zhou A, Huang Z, Zhang H, et al. (2018). Relation between cadmium exposure and gestational diabetes mellitus. Environment International, 113, 300–305. 10.1016/j.envint.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Xu J, Huang G, & Guo TL (2019). Bisphenol S modulates type 1 diabetes development in non-obese diabetic (NOD) mice with diet- and sex-related effects. Toxics, 7(2), 35. 10.3390/toxics7020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Huang G, Nagy T, & Guo TL (2019). Bisphenol A alteration of type 1 diabetes in non-obese diabetic (NOD) female mice is dependent on window of exposure. Archives of Toxicology, 93(4), 1083–1093. 10.1007/s00204-019-02419-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ou K, Chen C, Li B, Guo J, Zuo Z, et al. (2019). Tributyltin exposure disturbs hepatic glucose metabolism in male mice. Toxicology, 425, 152242. 10.1016/j.tox.2019.152242. [DOI] [PubMed] [Google Scholar]
- Xu H, Zhou Q, Zhang J, Chen X, Zhao H, Lu H, et al. (2020). Exposure to elevated per- and polyfluoroalkyl substances in early pregnancy is related to increased risk of gestational diabetes mellitus: A nested case-control study in Shanghai, China. Environment International, 143, 105952. 10.1016/j.envint.2020.105952. [DOI] [PubMed] [Google Scholar]
- Xue P, Hou Y, Zhang Q, Woods CG, Yarborough K, Liu H, et al. (2011). Prolonged inorganic arsenite exposure suppresses insulin-stimulated AKT S473 phosphorylation and glucose uptake in 3T3-L1 adipocytes: Involvement of the adaptive antioxidant response. Biochemical and Biophysical Research Communications, 407(2), 360–365. 10.1016/j.bbrc.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau DT, & Mennear JH (1977). The inhibitory effect of DDT on insulin secretion in mice. Toxicology and Applied Pharmacology, 39(1), 81–88. 10.1016/0041-008x(77)90179-x. [DOI] [PubMed] [Google Scholar]
- Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, & Liu SH (2012). Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Archives of Toxicology, 86(6), 923–933. 10.1007/s00204-012-0864-9. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Hou Y, Wang D, Xu Y, Wang H, Liu J, et al. (2020). Interactions of arsenic metabolism with arsenic exposure and individual factors on diabetes occurrence: Baseline findings from Arsenic and Non-Communicable disease cohort (AsNCD) in China. Environmental Pollution, 265(Pt. A), 114968. 10.1016/j.envpol.2020.114968. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Liu C, Wang Y, Gong J, Wang G, Ge W, et al. (2021). Associations of long-term exposure to ambient nitrogen dioxide with indicators of diabetes and dyslipidemia in China: A nationwide analysis. Chemosphere, 269, 128724. 10.1016/j.chemosphere.2020.128724. Epub 2020 Oct 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mwiberi S, Pickford R, Breitner S, Huth C, Koenig W, et al. (2021). Longitudinal associations between ambient air pollution and insulin sensitivity: Results from the KORA cohort study. The Lancet. Planetary Health, 5(1), e39–e49. 10.1016/S2542-5196(20)30275-8. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Wu T, Lin M, Zhang S, Yan F, Yang Z, et al. (2014). Chronic exposure to tributyltin chloride induces pancreatic islet cell apoptosis and disrupts glucose homeostasis in male mice. Environmental Science & Technology, 48(9), 5179–5186. 10.1021/es404729p. [DOI] [PubMed] [Google Scholar]