

Abstract
With numerous infections and fatalities, COVID-19 has wreaked havoc around the globe. The main protease (Mpro), which cleaves the polyprotein to form non-structural proteins, thereby helping in the replication of SARS-CoV-2, appears as an attractive target for antiviral therapeutics. As FDA-approved drugs have shown effectiveness in targeting Mpro in previous SARS-CoV(s), molecular docking and virtual screening of existing antiviral, antimalarial, and protease inhibitor drugs were carried out against SARS-CoV-2 Mpro. Among 53 shortlisted drugs with binding energies lower than that of the crystal-bound inhibitor α-ketoamide 13 b (−6.7 kcal/mol), velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, danoprevir, nelfinavir, and indinavir (−9.1 to −7.5 kcal/mol) were the most significant on the list (hereafter referred to as the 53-list). Molecular dynamics (MD) simulations confirmed the stability of their Mpro complexes, with the MMPBSA binding free energy (ΔGbind) ranging between −124 kJ/mol (glecaprevir) and −28.2 kJ/mol (velpatasvir). Despite having the lowest initial binding energy, velpatasvir exhibited the highest ΔGbind value for escaping the catalytic site during the MD simulations, indicating its reduced efficacy, as observed experimentally. Available inhibition assay data adequately substantiated the computational forecast. Glecaprevir and nelfinavir (ΔGbind = −95.4 kJ/mol) appear to be the most effective antiviral drugs against Mpro. Furthermore, the remaining FDA drugs on the 53-list can be worth considering, since some have already demonstrated antiviral activity against SARS-CoV-2. Hence, theoretical pKi (Ki = inhibitor constant) values for all 53 drugs were provided. Notably, ΔGbind directly correlates with the average distance of the drugs from the His41–Cys145 catalytic dyad of Mpro, providing a roadmap for rapid screening and improving the inhibitor design against SARS-CoV-2 Mpro.
Keywords: SARS-CoV-2, Main protease, FDA-Approved drugs, Molecular dynamics, Binding free energy
Graphical abstract

1. Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), which emerged in Wuhan, China, in late 2019, has drawn considerable attention from the scientific community worldwide due to the severity and rapid spread of the disease [1,2]. An increasing number of cases and deaths caused by the novel coronavirus (COVID-19) worldwide have notably had an adverse impact on health and economics [1]. Thus, the identification of antiviral agents against SARS-CoV-2 for the treatment of COVID-19 infections is a matter of the utmost urgency. Since the identification, development, and human clinical trials of any new drug require a considerable amount of time, the repurposing of already available FDA-approved drugs seems to be the fastest route to combat the current novel COVID-19 pandemic. The entry of SARS-CoV-2 is mediated by the host cellular receptor angiotensin-converting enzyme 2 (ACE-2), causing infection in the lungs, heart, gastrointestinal tract, or central nervous system (CNS) [3].
To date, among all the coronaviruses, SARS-CoV-2 is the largest in size, approximately 27–32 kb, and it belongs to a family of positive-sense, single-stranded RNA viruses. Its replicase genome encodes two polyproteins, pp1a and pp1ab, overlapped by ribosomal frameshifting [4]. Proteolytic cleavage produces 16 non-structural proteins that form the replicase transcriptase complex. Non-structural protein 5 (NSP5) or the main protease (Mpro), also known as 3CLpro, exists in polyprotein pp1a [5].
Mpro consists of three domains with amino acid sequences 8–101 (domain I), 102–184 (domain II), and 185–306 (domain III). The substrate-binding site is located between domains I and II, whereas domain III is a globular cluster that helps the dimerization of Mpro [6]. A loop with residues 189–191 connects domains II and III. The active site comprises a His41–Cys145 catalytic dyad at the catalytic cleavage site. Other key substrate-binding residues within the S1 to S5 subsites are Thr25, Met49, Phe140, Leu 141, Asn142, Gly143, Ser144, Cys145, His163, Met165, Glu166, His172, Gln189, and Thr190 [7,8] (see Fig. 1 in Section 3).
Fig. 1.

Three domains of the SARS-CoV-2 main protease (Mpro). Displayed separately are the conserved catalytic dyad His41–Cys145, active-site residues, and the co-crystallized α-ketoamide 13 b ligand.
Among all the therapeutic targets of SARS-CoV-2, Mpro is the most vital and best-characterized drug target for rational drug design [9,10]. The protease has a key role in infecting host cells [11] and plays an important role in the processing of polyproteins that are translated from the viral RNA [12]. Thus, inhibiting the activity of this enzyme would block viral replication. Moreover, since no proteases with similar cleavage specificity are present in humans, such inhibitors are unlikely to produce any side effects/toxicity [[12], [13], [14]]. Therefore, the replication process of the virus can be inhibited by searching for a potential inhibitor against SARS-CoV-2 Mpro.
While drug repurposing can offer the fastest solution to prevent the rapid spread of COVID-19 infections [15,16], the recently solved high-resolution crystal structure of SARS-CoV-2 Mpro, complexed with a modified α-ketoamide inhibitor (α-ketoamide 13 b) [17], can be used for the screening of potential inhibitors from existing FDA-approved drugs. By employing molecular docking and virtual screening, the best candidates from the FDA-approved drugs were identified, which were further validated by molecular dynamics (MD) simulations and Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA)-based binding free energy calculations. Existing biological data testifies to the usefulness of the screening. The study underlines the inefficacy of some drugs arising from their affinity for an allosteric site [18]. The shorter the distance from the catalytic dyad, the greater the efficacy of a drug. Finally, the findings of this study demonstrate the significance of the shortlisted candidates, which may direct experiments and improve drug design at a rapid pace against the novel coronavirus.
2. Materials and methods
2.1. Protein structure preparation
The crystal structure of SARS-CoV-2 Mpro with PDB_ID: 6Y2F [17] at 1.95 Å resolution was retrieved from the RCSB (Protein Data Bank). The missing hydrogens and residues Glu47, Asp48, and Gln 306 were added using MODELLER [19].
2.2. Binding cleft identification
To determine a protein's binding site, (i) the shape of the receptor in different cavities and (ii) the volume occupied by the known ligand poses already located in an active site are useful [20,21]. In this study, the residues within the 5 Å radius of the α-ketoamide 13 b inhibitor were selected as active residues for molecular docking studies. These were found to be amino acids 24–27, 41, 49, 54, 140–145, 163–168, 172, and 186–192. These residues are highly conserved and form a catalytic His41–Cys145 dyad, as mentioned earlier. Cysteine and histidine are the essential amino acids that initiate the enzymatic process [7,8]. Additionally, there was a substrate-binding subsite positioned in the active-site groove of the protease. The specific subsites located in the enzyme active site are S1 and S2 (buried subsites) and S3, S4, and S5 (shallow subsites), categorized according to their positions relative to the cleavage site [7]. In the Mpro active-site region of SARS-CoV-2, the S1 subsite consists of Phe140, His163, Glu166, Cys145, Gly143, and His172, which also serve as the oxyanion (or hydroxyl group) hole of serine protease [8]. The Cys145, His41, and Thr25 residues are located at the S2 subsite, involved in hydrophobic and electrostatic interactions. The shallow subsites, S3 to S5, contain the Met49, His41, Met165, Glu166, Pro168, and Gln189 amino acid residues [8]. Structural details and interactions of SARS-CoV-2 Mpro with the co-ligand α-ketoamide 13 b are shown in Fig. 1 (Section 3).
2.3. Ligand preparation
All available FDA-approved antiviral, antimalarial, and protease inhibitor drugs (a total of 200) were retrieved from DrugBank 5.0 [22] and SuperDrug2 [23]. The ligands (drugs) were prepared by adding the missing hydrogens. Thereafter, energy minimization of all the ligands was carried out using the semiempirical PM6 method.
2.4. Molecular docking
The optimized ligands, as well as the co-ligand, α-ketoamide 13 b, were docked in the active site of SARS-CoV-2 Mpro after assigning the Gasteiger charges with AutoDock Vina [24], which uses the Lamarckian genetic algorithm (GA) in combination with grid-based energy estimation incorporated in PyRx [25]. The docking protocol adopted herein is the same as that used in other peer-reviewed works [26,27]. Due to the significant accuracy of AutoDock Vina, demonstrated in a recent benchmark study [28], we also used it for molecular docking. Based on docking scores and binding interactions, the top 53 ligands out of 200 were selected. To improve the screening and docking scores, these selected ligands were re-optimized by using density functional theory (DFT) at the B3LYP/6-311G (d, p) level. From these, seven ligands – namely, velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, danoprevir, nelfinavir, and indinavir – had the best docking scores and were selected for further MD simulations and binding free energy calculations.
2.5. Molecular dynamics (MD) simulations
MD simulations were carried out for Mpro complexed with velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, nelfinavir, danoprevir, and indinavir for a period of 100 ns using GROMACS (Groningen Machine for Chemical Simulations) v2020 [29]. The unit cell, defined as a cubical box with a minimum distance of 10 Å from the protein surface to the edges of the box, was solvated using the Simple Point Charge (SPC) water model; the topologies of Mpro and the selected drugs were created using the GROMOS96 53a6 force field [30]. Counterions were added to make every system electrically neutral at a salt concentration of 0.15 mol/L. Before the MD simulations, each system was subjected to energy minimization by employing the steepest descent integrator for 5000 steps with a force convergence of <1000 kcal mol−1 nm−1.
Thereafter, each protein–ligand complex was equilibrated for 5 ns using the canonical (NVT) and the isothermal–isobaric (NPT) ensembles. During equilibration, each system was coupled with the Berendsen temperature and the Parrinello-Rahman pressure controllers to maintain a temperature of 300 K and a pressure of 1 bar, respectively. The particle mesh Ewald (PME) algorithm [31] was employed to compute the long-range Coulomb interactions with a Fourier grid spacing of 0.12 nm. The short-range van der Waals interactions were modeled using the Lennard-Jones potential with a cut-off distance of 1 nm. All bond lengths were constrained by the linear constraint solver (LINCS) method [32].
Subsequently, 100-ns production runs were performed under the microcanonical ensemble by relaxing the coupling with the thermostats. In principle, the same protocol was applied to all the protein–ligand systems. A time step of 2 fs was used, and the coordinates were saved every 10 ps during the production runs. For the structural analyses of every system, the resultant MD trajectories were analyzed using the built-in modules of GROMACS and visual molecular dynamics (VMD 1.9.1) [33]. The 2D plots depicting the intrinsic dynamical stabilities captured by the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), hydrogen bond (HB), and principal component analysis (PCA) of the complexes were generated with the Origin v2021 program, as in our previous studies [34,35]. The RMSD was obtained using Equation (1):
| (1) |
where and r i (t) is the position of atom i at time t after a least-squares fitting of the structure to the reference structure. The RMSF was obtained as in Equation (2):
| (2) |
where T is the time over which one wants to average, and is the reference position of particle i. Rg was obtained using Equation (3):
| (3) |
where m i is the mass of atom i, and r i is the position of atom i with respect to the center of mass of the molecule.
Proteins control their functions by changing their shape. The collective movements of the atoms in a protein control the overall conformational change. The relative conformational dynamics and atomic fluctuations of the functionally relevant substructures in the native and ligand-bound forms were studied using PCA [36,37]. The trajectories generated during the 100-ns MD simulations were used for PCA analysis. A cross-correlation matrix was developed by removing the translational and rotational movements and monitoring the relative Cα-backbone atomic fluctuations. The generated eigenvalues represented the energetic contribution from the corresponding principal components (PCs), while the eigenvectors represented the direction of motion. The projected eigenvalues and eigenvectors were analyzed to determine the overall flexibility of the protein. PCA was carried out by using the formula in Equation (4) [38]:
| (4) |
where are the coordinates of the ith and jth atoms of the protein, respectively, and < > and <> are the ensemble averages.
2.6. Binding free energy calculations
The MMPBSA method is widely used for free energy calculations from MD trajectories [39]. A benchmark study by Wang et al. in 2019 showed that MMPBSA outperformed the Glide SP scoring function (with a success rate of 58.6%) and MMGBSA in most cases, with an overall success rate of approximately 74% [40]. The binding free energy (ΔGbind) in a solvent medium was calculated as in Equation (5):
| ΔGbind = Gcomplex - (Gprotein + Gligand), | (5) |
where Gcomplex is the total free energy of the protein–ligand complex, and Gprotein and Gligand are the free energies of the protein and ligand alone in a solvent, respectively.
Each free energy Gp (p = protein, ligand, or complex) is a combination of the average molecular mechanics potential energy (EMM) in vacuum and the solvation free energy (Gsolv), as given by Equation (6):
| Gp = EMM + Gsolv. | (6) |
EMM in vacuum was calculated as in Equation (7):
| EMM = Ebonded + Enon-bonded = Ebonded (Eint) + Evdw + Eelec, | (7) |
where Ebonded (or Eint) is the total bonded interaction, which includes bond, angle, dihedral, and improper interactions; and Enon-bonded is the total non-bonded interaction, consisting of both van der Waals (Evdw) and electrostatic (Eelec) interactions. Ebonded is always taken as zero.
Gsolv was estimated as the sum of the electrostatic/polar solvation free energy (Gpolar) and nonpolar solvation free energy (Gnon-polar), as given in Equation (8):
| Gsolv = Gpolar + Gnon-polar, | (8) |
where Gpolar was determined using the Poisson–Boltzmann (PB) linear equation, and the non-polar contribution, Gnon-polar, was estimated from the solvent-accessible surface area (SASA), as per Equation (9):
| Gnon-polar = γSASA + b, | (9) |
where γ (a coefficient related to the surface tension of a solvent) = 0.02267 kJ/mol/Å2, and b = 3.849 kJ/mol.
The binding free energies for all seven complexes were calculated based on 10,000 snapshots taken at equal time intervals during the 100-ns MD simulations.
3. Results and discussion
3.1. Molecular docking and binding mode analysis
All the candidates from the prepared drug library were docked at the same active-site region where the α-ketoamide 13 b ligand had been co-crystallized with SARS-CoV-2 Mpro. Table 1 presents the best screened (the top seven) FDA-approved drugs from this study along with the co-crystallized ligand α-ketoamide 13 b. In total, 53 drugs with binding energies lower than that of the co-crystallized ligand (≤−7.0 kcal/mol) were initially shortlisted (hereafter referred to as the 53-list; Table S1).
Table 1.
The binding affinities of the seven FDA-approved drugs with the best docking scores along with that of the co-crystallized ligand, α-ketoamide 13 b, before running MD simulations.
| SI. No. | Repurposed FDA-approved Drugs | 2D Structures | Binding Affinity (kcal/mol) | Mode of Action |
|---|---|---|---|---|
| 1 | Velpatasvir | 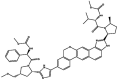 |
−9.1 | HCV NS5B and NS5A protease inhibitor |
| 2 | Glecaprevir | 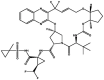 |
−8.7 | HCV NS3/4 A protease inhibitor |
| 3 | Grazoprevir | 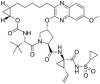 |
−8.7 | HCV protease inhibitor |
| 4 | Baloxavir marboxil | 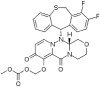 |
−8.4 | Influenza A and influenza B inhibitor |
| 5 | Nelfinavir | 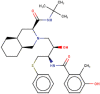 |
−7.9 | Protease inhibitor |
| 6 | Danoprevir | 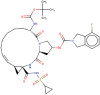 |
−8.4 | NS3/4 A HCV protease inhibitor |
| 7 | Indinavir (reference) | 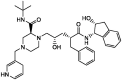 |
−7.5 | Protease inhibitor |
| 8 | α-ketoamide 13 b (co-crystallized ligand) | 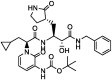 |
−6.7 | Broad-spectrum inhibitor |
In the 53-list, some drugs – such as danoprevir, nelfinavir, indinavir, paritaprevir, daclatasvir, faldaprevir, lonafarniv, voxilaprevir, asunaprevir, etc. – have been found to be effective against SARS-CoV-2 in biological assays/clinical trials [41,42]. Among them, including the α-ketoamide 13 b compound reported in Ref. [17], the top four compounds in the list – velpatasvir, glecaprevir, grazoprevir, and baloxavir marboxil – have respective binding energies of −9.1, −8.7, −8.7, and −8.4 kcal/mol for SARS-CoV-2 Mpro. Danoprevir, nelfinavir, and indinavir, which have shown in-vitro efficacy against SARS-CoV-2, have binding affinities of −8.4, −7.9, and −7.5 kcal/mol, respectively.
Velpatasvir is an antiviral agent as well as an NSP5A inhibitor used to treat chronic liver infection caused by the Hepatitis C virus [43]. NSP5A is a viral protein that plays a key role in the replication of the Hepatitis C virus, its assembly, and the modulation of the host immune response [44]. Likewise, glecaprevir acts as a protease inhibitor of the NS3/4 A Hepatitis C virus by hijacking the viral replication machinery [45]. Grazoprevir is also an inhibitor of the Hepatitis C virus used to inhibit the function of the NS3/4 A protease protein [46]. Baloxavir marboxil, an antiviral drug marketed under the brand name xofluza, is used to treat influenza A and influenza B [47]. Danoprevir is a 15-membered macrocyclic peptidomimetic inhibitor of NS3/4 A HCV protease [48,49] and is suggested to be a viable treatment plan as compared to lopinavir/ritonavir, owing to a significant shortening of the period of hospital stay and the time required to achieve a negative nucleic acid test for COVID-19 patients. Nelfinavir belongs to the class of protease inhibitors and is used to treat the human immunodeficiency virus (HIV). In the case of SARS-CoV-2, it has been reported to inhibit the replication of the virus with effective concentrations of 1.13 and 1.76 μM, respectively, for 50% and 90% inhibitions [50].
Fig. 1 shows the three domains of SARS-CoV-2 Mpro, and the docking poses and 2D interaction plots of velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, danoprevir, nelfinavir, and indinavir with Mpro are shown in Fig. 2 and S1, respectively. Table S2 provides details on the various interactions present for all the drugs along with the co-crystallized ligand given in Table 1. Velpatasvir showed four C–H … X (X = O, N, etc.)-type non-classical hydrogen bonds with Gly143 (3.89 Å), Asn 119 (4.41 Å), and Thr45 (two, at 4.29 and 4.56 Å). It also formed three A-H•••B (A and B = N, O, F, etc.)-type classical hydrogen bonds with Ser46 (two, at 3.78 and 4.14 Å) and Thr190 (4.55 Å). The Met49 and Met165 residues formed two π–sulfur bonds with velpatasvir within 5.71 and 7.36 Å, respectively. The side chains of Cys145 were also involved in π–alkyl interactions with velpatasvir within distances of 5.44 and 7.64 Å. These interactions gave rise to a binding energy of −9.1 kcal/mol, as mentioned earlier.
Fig. 2.

The docking poses of (A) velpatasvir, (B) glecaprevir, (C) grazoprevir, (D) baloxavir marboxil, (E) danoprevir, (F) nelfinavir, and (G) indinavir with Mpro before (t = 0 ns) and after (t = 100 ns) the MD runs of corresponding complexes. For every complex, the interacting amino acids are also denoted.
Glecaprevir bonded strongly to the target site of Mpro with three classical hydrogen bonds to the following residues: Ser46 (two, at 2.40 and 2.99 Å) and Asn142 (2.51 Å), as shown in Figures 2B and S1 (B). There were non-classical hydrogen bonds with Ser46 (2.71 Å), Asn142 (2.60 Å), Gln189 (2.98 Å), and His164 (two, at 3.06 and 3 Å). The Glu166 residue interacted with two aromatic rings within distances of 4.59 and 4.05 Å. Moreover, residues such as His41 (4.77 Å), Met49 (5.22 Å), Cys145 (4.32 Å), and Met165 (5.07 Å) were involved in hydrophobic interactions with glecaprevir, one of the halogen atoms of which interacted with Ser46 (2.99 Å) and Glu166 (3.37 Å), as seen in Fig. S1. All these interactions account for the binding energy of −8.7 kcal/mol between glecaprevir and Mpro, as given in Table 1. As depicted in Fig. 2C and S1 (C), grazoprevir had one classical hydrogen-bond interaction with residue Glu166 (4.71 Å) and three non-classical hydrogen-bond interactions with Asn142 (4.82 Å), Met165 (5.30 Å), and Glu166 (3.65 Å). It also had hydrophobic interactions with Cys145 (4.18 Å) and Pro168 (two, at 3.92 and 5.42 Å). In Fig. 2D and S1 (D), baloxavir marboxil showed classical hydrogen-bond interactions with residue Gly143 (two, at 3.91 and 3.36 Å) and non-classical hydrogen-bond interactions with His41 (two, at 5.35 and 4.02 Å), His164 (two, at 5.97 and 6.51 Å), and Gln189 (4.69 Å). Hydrophobic interactions were present with residues Leu27 (5.65 Å), His41 (two, at 4.95 and 6.64 Å), Met49 (6.80 Å), Asn142 (5.41 Å), Met165 (4.35 Å), and Glu166 (4.04 Å).
In the case of another FDA-approved drug, danoprevir (Fig. 2E and S1 [E]), the residues involved in classical hydrogen bonding were His41, Thr25 (2), Asp48, Cys145, and Gln 192 within distances of 4.80, 4.11, 4.49, 4.86, 5.05, and 3.20 Å, respectively. Residues involved in carbon–hydrogen bond interactions were His41 (two, at 4.61 and 3.93 Å) and Glu47 (4.39 Å). Further, a halogen interaction was exhibited by Thr190 (4.85 Å). There were also hydrophobic interactions with Pro168 (6.82 Å).
Fig. 2F and S1 (F) show the docking pose and interacting residues of Mpro with the effective in-vitro drug, nelfinavir. The residues Thr26 (3.44 Å), Ser144 (4.62 Å), Gly143 (3.56 Å), and Glu166 (4.23 Å) were involved in classical hydrogen bonding. Glu166 (2.79 Å) and Asn142 (4.70 Å) formed non-classical hydrogen bonds with nelfinavir. There were π–π and π–sulfur interactions with His41 (5.08 Å) and Met49 (5.16 Å), respectively. Further, residues such as Leu27 (4.41 Å) and Cys145 (4.14 Å) were also involved in hydrophobic interactions.
Fig. 2G and S1 (G) represent the docking pose and interaction plot for Mpro and another effective in-vitro drug, indinavir. Indinavir interacted with Mpro through three conventional hydrogen bonds involving Asn142 (3.36 Å), Glu166 (4.04 Å), and Gln189 (3.64 Å). Similarly, it also had non-classical hydrogen bonds with Thr24 (5.78 Å), Ser46 (4.00 Å), Met165 (4.75 Å), Glu166 (two, at 4.53 and 5.52 Å), and Gln189 (two, at 3.76 and 6.03 Å). Residues involved in π–π and π–sulfur interactions were His41 (5.32 Å) and Met49 (5.93 Å), respectively. A residue, Gln 198 (3.79 Å), also exhibited a π–amide interaction with indinavir. Altogether, these interactions resulted in a binding energy of −7.8 kcal/mol between indinavir and Mpro, as mentioned earlier (see Table 1).
Thus, initial docking results distinguish velpatasvir as the best candidate followed by glecaprevir, grazoprevir, baloxavir marboxil, danoprevir, nelfinavir, and indinavir. A strong affinity essentially stems from the cumulative interactions of a large number of residues.
3.2. MD trajectory analysis
MD simulations are indispensable for scrutinizing the internal motions, conformational changes, stability, etc. of protein–ligand complexes pertaining to inhibitor design and mutational analysis [34,35,51]. Using the MD trajectories of the complexes produced, the RMSD, RMSF, Rg, and hydrogen bonds were computed and PCA conducted, and the results are discussed as follows.
In addition to assessing the equilibration and convergence of an MD run, the RMSD is an essential structural and dynamical parameter to investigate conformational stability [34]. Larger RMSD fluctuations are indicative of lower conformational stability of a protein–ligand complex and vice versa. Fig. 3 displays RMSD plots of the Mpro–velpatasvir, Mpro–glecaprevir, Mpro–grazoprevir, Mpro–baloxavir marboxil, Mpro–danoprevir, Mpro–nelfinavir, and Mpro–indinavir complexes computed for the Cα atomic positions over the entire MD simulation period. Surprisingly, although its docking score was low, Mpro–velpatasvir had larger RMSD values, with an average value of 0.36 nm. On the contrary, the Mpro–glecaprevir, Mpro–grazoprevir, Mpro–baloxavir marboxil, Mpro–danoprevir, Mpro–nelfinavir, and Mpro–indinavir complexes had average RMSD values of approximately 0.22, 0.21, 0.30, 0.26, 0.22, and 0.21 nm, respectively. Initially, the Mpro–velpatasvir complex had a smooth variation in RMSD, but after 30 ns, it increased sharply and fluctuated violently. A similar pattern was observed for Mpro–baloxavir marboxil, although the fluctuations were less pronounced here. All other systems had a stable RMSD plot throughout the entire simulation period. During the trajectory analysis, the velpatasvir molecule escaped from the catalytic cavity for a different binding site, that is, an allosteric site in Mpro, at approximately 60 ns, as shown in Fig. 2A. The RMSD fluctuations and Fig. 2A concurrently indicate the instability of the Mpro–velpatasvir complex despite initially having the smallest binding energy, which accords well with earlier experimental observations [52].
Fig. 3.

Plots of RMSD as a function of simulation time for the Mpro–velpatasvir (black), Mpro–glecaprevir (red), Mpro–grazoprevir (magenta), Mpro–baloxavir marboxil (cyan), Mpro–danoprevir (gray), Mpro–nelfinavir (blue), and Mpro–indinavir (green) complexes.
The RMSF is a useful parameter that assesses residue flexibility during dynamic simulations with respect to the Cα backbone atoms of each amino acid residue of Mpro and is plotted in Fig. 4 . In the case of the Mpro–velpatasvir complex, the average RMSF was approximately 0.20 nm. The highest degree of fluctuation in this complex is reminiscent of its instability. The remaining complexes – that is, Mpro–glecaprevir, Mpro–grazoprevir, Mpro–baloxavir marboxil, Mpro–danoprevir, Mpro–nelfinavir, and Mpro–indinavir – had an average RMSF of 0.10, 0.11, 0.10, 0.09, 0.13, and 0.11 nm, respectively. When compared to velpatasvir, these complexes exhibited fewer fluctuations. This complies with the RMSD analysis in inferring the stability of these complexes.
Fig. 4.

Plots of RMSF as a function of simulation time for the Mpro–velpatasvir (black), Mpro–glecaprevir (red), Mpro–grazoprevir (magenta), Mpro–baloxavir marboxil (cyan), Mpro–danoprevir (gray), Mpro–nelfinavir (blue), and Mpro–indinavir (green) complexes.
In the case of Mpro–velpatasvir, RMSF fluctuations occurred in almost all areas of the complex. In other complexes, however, the loop regions were responsible for larger fluctuations. The specific amino acids that showed more fluctuations were as follows: Mpro–glecaprevir – 1, 52, 120, 142, 302, and 304–306; Mpro–grazoprevir – 47 and 216; Mpro–baloxavir marboxil – 47, 155, and 302; Mpro–danoprevir – 1–3 and 47–50; Mpro–nelfinavir – 1, 2, 46, 47, 300, and 301; and Mpro–indinavir – 1–3, 94, 155, 232, 302, 305, and 306. The active site did not include these residues.
Fig. S2 depicts the minimum distance between Mpro and the ligands. This distance remained almost constant, with negligible variations over the simulation period, for all except baloxavir marboxil, which displayed spikes corresponding to large sharp increases. The baloxavir marboxil molecule moved out of the active site pocket approximately between 55 and 70 ns. Despite velpatasvir having the largest RMSD, its minimum distance was unexpectedly similar to that of other drug molecules. To remove this contradiction, the minimum distance was estimated with respect to the catalytic dyad residues (His41 and Cys145), which is shown in Fig. 5 A and B. In addition, frames of the MD trajectory at different times categorically showed that the velpatasvir molecule escaped from the active site to another binding location (allosteric site) situated between domains II and III of Mpro during the simulation (see Fig. 2A). Similarly, baloxavir marboxil moved away from the catalytic dyad site but never left the region permanently for a different location (see Fig. 2, Fig. 5B). On the contrary, all other molecules consistently remained in the vicinity of the catalytic dyad. Glecaprevir had the shortest minimum distance from the catalytic dyad residue His41, followed by nelfinavir, indinavir, danoprevir, and grazoprevir. Similarly, nelfinavir had the shortest average minimum distance from the other catalytic dyad residue, Cys145, followed by indinavir, glecaprevir, danoprevir, and grazoprevir. With the exception of these two drugs, the results of the minimum distance, RMSD, and RMSF, therefore, predict the large stability and tight binding of other drugs to the catalytic site of Mpro.
Fig. 5.

The minimum distance of velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, danoprevir, nelfinavir, and indinavir from the catalytic dyad residues (A) His41 and (B) Cys145.
Rg describes the level of compaction of a protein and is defined as the mass-weighted root-mean-square distance for a collection of atoms from their common center of mass. Hence, Rg depicts the evolution of the overall dimension of a protein during a dynamic simulation. This is shown in Fig. 6 . In the Mpro–velpatasvir, Mpro–glecaprevir, Mpro–grazoprevir, Mpro–baloxavir marboxil, Mpro–danoprevir, Mpro–nelfinavir, and Mpro–indinavir complexes, the average values of Rg for the Cα atoms of Mpro were 2.22, 2.19, 2.20, 2.20, 2.15, 2.21, and 2.19 nm, respectively. In the Rg plots, the fluctuations were larger for velpatasvir and nelfinavir as compared to grazoprevir and baloxavir marboxil, followed by glecaprevir, indinavir, and danoprevir. Hence, the complexes of danoprevir, glecaprevir, and indinavir are more compact. While velpatasvir and baloxavir marboxil escaped Mpro's active site, all other drug molecules showed a stable conformation at the site (Fig. 2, Fig. 5). The 2D interaction plots after the 100-ns dynamic simulations (Fig. S1) demonstrate this, with all molecules except velpatasvir and baloxavir marboxil showing substantial binding with active residues.
Fig. 6.

Plots of the radius of gyration (Rg) of the Mpro–velpatasvir (black), Mpro–glecaprevir (red), Mpro–grazoprevir (magenta), Mpro–baloxavir marboxil (cyan), Mpro–danoprevir (gray), Mpro–nelfinavir (blue), and Mpro–indinavir (green) complexes.
Glecaprevir retained its interaction with His41 and Glu166 in which the former takes part in catalytic dyad formation. The His41, Glu166, Cys145, Met165, and Met49 residues sustained their interactions with nelfinavir to the end of the simulation. Danoprevir also retained interactions with His41 and Cys145. Indinavir continued its interaction with Glu166, forming an additional hydrogen bond with His41. While the number of interactions of glecaprevir, nelfinavir, and danoprevir remained almost intact in the MD simulations, it was reduced to half or less for velpatasvir and indinavir, which accounts for the weaker binding affinity of the latter two drugs. Interestingly, we observed that the Glu166 residue in the Mpro complexes of glecaprevir, nelfinavir, and indinavir retained its interaction throughout the simulation, which indicates that it may play an important role in the catalytic site [53]. Table 2 presents the computed binding free energies and decompositions for all complexes.
Table 2.
The binding free energy (ΔGbind) and contributory van der Waals (ΔEvdw), electrostatic (ΔEelec), polar solvation, and solvent accessible surface area (SASA) energies, reported in kJ/mol.
| SI. No. | SARS-CoV-2 complex | ΔEvdw | ΔEelec | Polar Solvation Energy | SASA Energy | ΔGbind |
|---|---|---|---|---|---|---|
| 1 | Mpro–velpatasvir | −63.8 ± 22.8 | −12.6 ± 4.9 | 55.0 ± 15.9 | −5.9 ± 2.2 | −28.2 ± 16.0 |
| 2 | Mpro–glecaprevir | −205.7 ± 2.0 | −50.2 ± 1.5 | 150.0 ± 6.5 | −18.2 ± 0.3 | −124.0 ± 6.6 |
| 3 | Mpro–grazoprevir | −128.7 ± 11.7 | −29.1 ± 3.9 | 108.5 ± 10.3 | −15.7 ± 1.4 | −65.6 ± 8.2 |
| 4 | Mpro–baloxavir marboxil | −36.4 ± 9.6 | −5.5 ± 3.5 | 31.4 ± 8.4 | −4.4 ± 1.0 | −15 ± 7.2 |
| 6 | Mpro–danoprevir | −150.1 ± 15.2 | −37.9 ± 8.2 | 136.6 ± 15.4 | −15.7 ± 1.7 | −66.3 ± 9.8 |
| 5 | Mpro–nelfinavir | −144.6 ± 14.4 | −24.8 ± 2.8 | 89.2 ± 9.4 | −15.2 ± 1.5 | −95.4 ± 9.5 |
| 7 | Mpro–indinavir | −96.2 ± 19.0 | −21.0 ± 4.4 | 5.2 ± 14.9 | −10.6 ± 2.1 | −42.9 ± 14.0 |
In Table 2, the ΔGbind values were computed with MMPBSA [39], which has been widely used to study crucial molecular recognition processes. ΔGbind of the Mpro–velpatasvir, Mpro–glecaprevir, Mpro–grazoprevir, Mpro–baloxavir marboxil, Mpro–danoprevir, Mpro–nelfinavir, and Mpro–indinavir complexes were, respectively, −28.2, −124.0, −65.6, −15, −66.3, −95.4, and −42.9 kJ/mol. From the different interactions or contributions to ΔGbind, the polar solvation energy was the most positive contributor, amounting to 55.0, 150.0, 108.5, 31.4, 136.6, 89.2, and 85.2 kJ/mol for the seven complexes, which opposes binding and, thus, reduces the absolute value of the total binding free energy. The remaining van der Waals (ΔEvdw), electrostatic (ΔEelec), and solvent-accessible surface area (SASA) energies were negative, favorably contributing to binding in all the seven complexes (Table 2).
Noticeably, the lowest ΔEvdw (−205.7 kJ/mol) and ΔEelec (−50.2 kJ/mol) led to the lowest ΔGbind and, hence, the strongest binding between Mpro and glecaprevir among all complexes. Fig. 7 shows the correlation between the average distance of the molecules from the catalytic dyad and their binding free energies.
Fig. 7.

The correlation between the average distance of the best screened molecules from the catalytic dyad and their binding free energies.
The His41–Cys145 dyad in the active site of Mpro is critically important for catalytic activity [17]. Thus, it is instructive to unearth any correlation existing between the average distance of drug molecules from the catalytic dyad and the binding free energy. Notably, we found a good linear relationship between the two, having a Pearson R-value of 0.76. The shorter the distance to the catalytic dyad, the stronger the binding and, hence, the inhibitory potential (Table S3 and Fig. 7). This would serve as a strategy for rapid screening as well as improving inhibitor design against SARS-CoV-2 Mpro. The validity of this correlation can be verified with large datasets. Table 3 presents biological data for the studied drug molecules from literature sources. Fig. 8 presents the PCA results of the complexes.
Table 3.
Biological data for velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, nelfinavir, danoprevir, and indinavir against SARS-CoV-2 from the literature.
Fig. 8.

The principal component analysis (PCA) of the Mpro–velpatasvir (black), Mpro–glecaprevir (red), Mpro–grazoprevir (magenta), Mpro–baloxavir marboxil (cyan), Mpro–danoprevir (gray), Mpro–nelfinavir (blue), and Mpro–indinavir (green) complexes: (A) the 2D projection plot of the first two principal eigenvectors and (B) the plot of eigenvalues versus the corresponding eigenvector indices from the Cα covariance matrix during the MD simulations.
The MD trajectories were further used for PCA to observe the conformational motions relevant to protein functions. Concerning the first two PCs, the simulation results revealed the smallest subspace dimension for Mpro–glecaprevir, followed by Mpro–indinavir, Mpro–nelfinavir, Mpro–danoprevir, Mpro–baloxavir marboxil, Mpro–grazoprevir, and Mpro–velpatasvir, in ascending order (Fig. 8A). The same trend was also reflected in their 2D projection plots of the trajectories, with the lowest trace values of the covariance matrix for the Mpro–glecaprevir complex. This again signifies that the Mpro–glecaprevir complex has the greatest stability among all for having the lowest degree of conformational changes due to decreased collective motions, as reflected in the trends of other dynamical parameters.
Eigenvalues were used to calculate the extent of conformational changes due to the movement of atoms, which were generated by diagonalizing the covariance matrix of Cα atomic fluctuations against the equivalent eigenvector's (EV's) indices. Taking the first 12 modes into consideration in the analysis of the essential subspace as they cover >95% variance of the protein, an exponentially decaying curve of eigenvalues against the EVs was obtained (Fig. 8B). In this study, PC1 and PC2, which dominated the protein conformational fluctuations, were used for the analysis of the seven complexes. The plot of the first 12 eigenvalues against the EV indices produced by the diagonalization of the covariance matrix showed that the eigenvalues of the seven complexes, in ascending order, were Mpro–glecaprevir, Mpro–indinavir, Mpro–danoprevir, Mpro–nelfinavir, Mpro–baloxavir marboxil, Mpro–grazoprevir, and Mpro–velpatasvir. When compared to other drugs, Mpro–glecaprevir had the lowest eigenvalue, indicating that it is structurally more stable and energetically more favorable. The first eight EVs account for 90% of the protein motion. The first eigenvector (EV1) accounts for 61%, 35%, 40%, 30%, 28%, 47%, and 52%, and the second eigenvector (EV2) corresponds to 24%, 20%, 18%, 22%, 17%, 20%, and 18% for Mpro–velpatasvir, Mpro–glecaprevir, Mpro–grazoprevir, Mpro–baloxavir marboxil, Mpro–danoprevir, Mpro–nelfinavir, and Mpro–indinavir, respectively.
Overall, owing to lower degrees of fluctuations in the RMSD, RMSF, and Rg; smaller subspace dimensions (PCA); and the presence of more interacting residues or hydrogen bonds, glecaprevir and nelfinavir are revealed to be more effective in inhibiting Mpro due to the notable stability of their complexes as compared to the others. More importantly, the lowest ΔGbind value of glecaprevir emphasizes the importance of performing experimental and clinical investigations on it.
Table 3 summarizes experimental EC50 or IC50, CC50, and SI data for the top seven selected drugs against SARS-CoV-2 that are currently available. The half-maximal effective concentration (EC50) is the drug concentration that gives the half-maximal response. The lower the EC50 value, the more potent the drug. The 50% cytotoxic concentration (CC50) is defined as the concentration of a drug that reduces cell viability by 50%. Drugs with a lower CC50 value are less potent. The selectivity index (SI) is simply the ratio of CC50 to IC50. Larger SI values indicate better drug efficacy [52]. This quantity provides valuable information about a drug that inhibits viral replication without killing the host cell. A drug with an SI value greater than 10 is a good candidate. Velpatasvir has been reported to have no direct antiviral activity against SARS-CoV-2 as its cell viability is very low (CC50 = 17 μM) [52]. In addition, experimental studies have demonstrated that nelfinavir has potential SARS-CoV-2 antiviral activity with EC50 = 1.13 μM, followed by indinavir with EC50 = 59.14 μM (Table 3) [50]. Thus far, however, experimental data is lacking regarding glecaprevir's activity against SARS-CoV-2. Danoprevir also displays good antiviral activity against SARS-CoV-2, with a CC50 > 50 μM [52]. From the screening of FDA-approved drugs, Chang et al. recently reported indinavir and remdesivir as the best drug candidates against SARS-CoV-2 [56]. In phase III clinical trials, remdesivir demonstrated prophylactic action against the RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2 by not only reducing the risk of death but also improving pulmonary activity in infected patients [57]. Being an RdRp inhibitor, remdesivir is excluded from consideration here. The experimental data accumulated in Table 3 accords well with our prediction of the inhibitory potential of the selected drugs from the 53-list. Even velpatasvir's reduced efficacy, as reflected by its lower CC50 value, has been correctly captured in our simulation as having a fragile interaction with the catalytic site. This demonstrates the high accuracy of the screening protocol adopted herein, and it suggests that the other identified drugs in the 53-list (Table S1) may be worth considering as many of them, including paritaprevir, daclatasvir, faldaprevir, lonafarnib, voxilaprevir, and asunaprevir, have been reported to have antiviral activity against SARS-CoV-2 [58,59]. Therefore, this study provides sufficient insight into the importance of the enlisted compounds in combating SARS-CoV-2, where glecaprevir and nelfinavir may show great promise. Nonetheless, the negative logarithm of the inhibitor constant (Ki), which is approximately the concentration needed to achieve half-maximal inhibition, has also been predicted for the drugs in Table S1.
4. Conclusions
The present study has addressed the screening of existing FDA-approved antiviral, antimalarial, and protease inhibitor drugs against a crucial molecular target of SARS-CoV-2, the main protease Mpro, in order to offer a faster remedy to the ever-spreading infection of COVID-19. Molecular docking and virtual screening revealed 53 drugs with binding energies below −7.0 kcal/mol (i.e., having a stronger affinity than the co-crystallized ligand α-ketoamide 13 b) with Mpro. The top candidates of the list included velpatasvir, glecaprevir, grazoprevir, baloxavir marboxil, danoprevir, nelfinavir, and indinavir, some of which – such as danoprevir, nelfinavir, indinavir, etc. – have already been reported to be biologically effective against SARS-CoV-2. After a complete analysis based on docking, screening, and MD simulations, glecaprevir and nelfinavir were the most significant, exhibiting the strongest affinities to SARS-CoV-2 Mpro with ΔG bind = −124 and −95.4 kJ/mol, respectively. In addition, RMSD, RMSF, Rg, and PCA analysis corresponding to lower values indicated the significant stability of their complexes. An excellent agreement between the predicted and experimental efficacies accentuates the high accuracy of the computational protocol adopted herein and the reliability of the results. Some significant insights were gained that could help improve drug design targeting Mpro, such as the existence of different binding site(s) (allosteric) responsible for lower efficacy (e.g., for velpatasvir and baloxavir marboxil) and closer proximity to the catalytic dyad, indicative of the increased inhibitory potential of a drug. Moreover, the study implies that the candidates on the 53-list that were not considered in this study may also be worth considering in future evaluations since some of them have already been reported to possess antiviral activity against the virus. Hence, the present work can serve as a basis to perform biological assays and clinical trials of glecaprevir and nelfinavir along with many other notable candidates for antiviral activity against SARS-CoV-2 Mpro.
Declaration of competing interest
The authors report no conflicts of interest.
Acknowledgments
The authors acknowledge IISER Berhampur for computational support. P.S.S.G. also sincerely acknowledges IISER Berhampur for providing him the Institute Postdoc Fellowship to carry out this work. The authors also acknowledge AMD COVID-19 High-Performance Computing (HPC) for supporting us with a remote supercomputing facility. The AMD EPYC processors accelerated the computing.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.105183.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muralidharan N., Sakthivel R., Velmurugan D., Gromiha M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn. 2020:1–6. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- 3.Olsen M., Cook S.E., Huang V., Pedersen N., Murphy B.G. Perspectives: potential therapeutic options for SARS-CoV-2 patients based on feline infectious peritonitis strategies: central nervous system invasion and drug coverage. Int. J. Antimicrob. Agents. 2020;55(6):105964. doi: 10.1016/j.ijantimicag.2020.105964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adegbola P.I., Fadahunsi O.S., Adegbola A.E., Semire B. In silico studies of Potency and safety assessment of selected trial drugs for the treatment of COVID-19. In Silico Pharmacol. 2021;9(1):45. doi: 10.1007/s40203-021-00105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J., Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273(5):1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., et al. 2020. Structure of M Pro from COVID-19 Virus and Discovery of its Inhibitors. [Google Scholar]
- 8.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020;63(9):4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 9.Xia B., Kang X. Activation and maturation of SARS-CoV main protease. Protein Cell. 2011;2(4):282–290. doi: 10.1007/s13238-011-1034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu I.-L., Mahindroo N., Liang P.-H., Peng Y.-H., Kuo C.-J., Tsai K.-C., et al. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe Acute respiratory Syndrome coronavirus main protease. J. Med. Chem. 2006;49(17):5154–5161. doi: 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- 11.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020;94(7) doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mengist H.M., Dilnessa T., Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021;9:622898. doi: 10.3389/fchem.2021.622898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adegbola P.I., Semire B., Fadahunsi O.S., Adegoke A.E. Molecular docking and ADMET studies of Allium cepa, Azadirachta indica and Xylopia aethiopica isolates as potential anti-viral drugs for Covid-19. Virusdisease. 2021:1–13. doi: 10.1007/s13337-021-00682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004;3(8):673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 16.Xu J., Shi P.-Y., Li H., Zhou J. Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect. Dis. 2020;6(5):909–915. doi: 10.1021/acsinfecdis.0c00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Baba T.J., Lutomski C.A., Kantsadi A.L., Malla T.R., John T., Mikhailov V., et al. Allosteric inhibition of the SARS-CoV-2 main protease: insights from mass spectrometry based assays. Angew Chem. Int. Ed. Engl. 2020;59(52):23544–23548. doi: 10.1002/anie.202010316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Šali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 20.Sakkiah S., Thangapandian S., Park C., Son M., Lee K.W. Molecular docking and dynamics simulation, receptor-based hypothesis: application to identify novel sirtuin 2 inhibitors. Chem. Biol. Drug Des. 2012;80(2):315–327. doi: 10.1111/j.1747-0285.2012.01406.x. [DOI] [PubMed] [Google Scholar]
- 21.Panda S.K., Saxena S., Guruprasad L. Homology modeling, docking and structure-based virtual screening for new inhibitor identification of Klebsiella pneumoniae heptosyltransferase-III. J. Biomol. Struct. Dyn. 2020;38(7):1887–1902. doi: 10.1080/07391102.2019.1624296. [DOI] [PubMed] [Google Scholar]
- 22.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siramshetty V.B., Eckert O.A., Gohlke B.-O., Goede A., Chen Q., Devarakonda P., et al. SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res. 2018;46(D1):D1137–D1143. doi: 10.1093/nar/gkx1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009 doi: 10.1002/jcc.21334. NA-NA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dallakyan S., Olson A.J. In: Chemical Biology. Hempel J.E., Williams C.H., Hong C.C., editors. Springer New York; New York, NY: 2015. Small-molecule library screening by docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 26.Gupta P.S.S., Bhat H.R., Biswal S., Rana M.K. Computer-aided discovery of bis-indole derivatives as multi-target drugs against cancer and bacterial infections: DFT, docking, virtual screening, and molecular dynamics studies. J. Mol. Liq. 2020;320:114375. doi: 10.1016/j.molliq.2020.114375. [DOI] [Google Scholar]
- 27.Panda S.K., Saxena S., Gupta P.S.S., Rana M.K. Inhibitors of Plasmepsin X Plasmodium falciparum: structure-based pharmacophore generation and molecular dynamics simulation. J. Mol. Liq. 2021:116851. doi: 10.1016/j.molliq.2021.116851. [DOI] [Google Scholar]
- 28.Nguyen N.T., Nguyen T.H., Pham T.N.H., Huy N.T., van Bay M., Pham M.Q., et al. Autodock Vina adopts more accurate binding poses but Autodock 4 forms better binding affinity. J. Chem. Inf. Model. 2020;60(1):204–211. doi: 10.1021/acs.jcim.9b00778. [DOI] [PubMed] [Google Scholar]
- 29.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 30.Oostenbrink C., Villa A., Mark A.E., van Gunsteren W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004;25(13):1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 31.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103(19):8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 32.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18(12):1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463:AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 33.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 34.Sen Gupta P.S., Islam R.N.U., Banerjee S., Nayek A., Rana M.K., Bandyopadhyay A.K. Screening and molecular characterization of lethal mutations of human homogentisate 1, 2 dioxigenase. J. Biomol. Struct. Dyn. 2020:1–11. doi: 10.1080/07391102.2020.1736158. [DOI] [PubMed] [Google Scholar]
- 35.Singh V.K., Srivastava R., Gupta P.S.S., Naaz F., Chaurasia H., Mishra R., et al. Anti-HIV potential of diarylpyrimidine derivatives as non-nucleoside reverse transcriptase inhibitors: design, synthesis, docking, TOPKAT analysis and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020:1–17. doi: 10.1080/07391102.2020.1748111. [DOI] [PubMed] [Google Scholar]
- 36.Pearson K. LIII. On lines and planes of closest fit to systems of points in space. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science. 1901;2(11):559–572. doi: 10.1080/14786440109462720. [DOI] [Google Scholar]
- 37.Hotelling H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933;24(6):417–441. doi: 10.1037/h0071325. [DOI] [Google Scholar]
- 38.Altis A., Otten M., Nguyen P.H., Hegger R., Stock G. Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. J. Chem. Phys. 2008;128(24):245102. doi: 10.1063/1.2945165. [DOI] [PubMed] [Google Scholar]
- 39.Kumari R., Kumar R., Lynn A. g_mmpbsa —a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z., Wang X., Li Y., Lei T., Wang E., Li D., et al. farPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics. 2019;35(10):1777–1779. doi: 10.1093/bioinformatics/bty879. [DOI] [PubMed] [Google Scholar]
- 41.Gammeltoft K.A., Zhou Y., Galli A., Offersgaard A., Pham L.V., Fahnøe U., et al. 2020. Hepatitis C Virus Protease Inhibitors Show Differential Efficacy and Interactions with Remdesivir for Treatment of SARS-CoV-2 in Vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sacramento C.Q., Fintelman-Rodrigues N., Temerozo J.R., Paula Dias Da Silva A de, da Silva Gomes Dias S., dos Santos da Silva C., et al. 2020. The in Vitro Antiviral Activity of the Anti-hepatitis C Virus (HCV) Drugs Daclatasvir and Sofosbuvir against SARS-CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee R., Kottilil S., Wilson E. Sofosbuvir/velpatasvir: a pangenotypic drug to simplify HCV therapy. Hepatol Int. 2017;11(2):161–170. doi: 10.1007/s12072-016-9776-8. [DOI] [PubMed] [Google Scholar]
- 44.Mogalian E., German P., Kearney B.P., Yang C.Y., Brainard D., Link J., et al. Preclinical pharmacokinetics and first-in-human pharmacokinetics, safety, and tolerability of velpatasvir, a pangenotypic hepatitis C virus NS5A inhibitor, in healthy subjects. Antimicrob. Agents Chemother. 2017;61(5) doi: 10.1128/AAC.02084-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salam K.A., Akimitsu N. Hepatitis C virus NS3 inhibitors: current and future perspectives. BioMed Res. Int. 2013;2013:1–9. doi: 10.1155/2013/467869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawitz E., Gane E., Pearlman B., Tam E., Ghesquiere W., Guyader D., et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1075–1086. doi: 10.1016/S0140-6736(14)61795-5. [DOI] [PubMed] [Google Scholar]
- 47.O'Hanlon R., Shaw M.L. Baloxavir marboxil: the new influenza drug on the market. Curr Opin Virol. 2019;35:14–18. doi: 10.1016/j.coviro.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Deutsch M., Papatheodoridis G.V. Danoprevir, a small-molecule NS3/4A protease inhibitor for the potential oral treatment of HCV infection. Curr. Opin. Invest. Drugs. 2010;11(8):951–963. [PubMed] [Google Scholar]
- 49.Ascher D.B., Wielens J., Nero T.L., Doughty L., Morton C.J., Parker M.W. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci. Rep. 2015;4(1) doi: 10.1038/srep04765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto N., Matsuyama S., Hoshino T., Yamamoto N. 2020. Nelfinavir Inhibits Replication of Severe Acute Respiratory Syndrome Coronavirus 2 in Vitro. [Google Scholar]
- 51.Sen Gupta P.S., Biswal S., Panda S.K., Ray A.K., Rana M.K. Binding mechanism and structural insights into the identified protein target of COVID-19 and importin-α with in-vitro effective drug ivermectin. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1839564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu S., Lien C.Z., Selvaraj P., Wang T.T. 2020. Evaluation of 19 Antiviral Drugs against SARS-CoV-2 Infection. [Google Scholar]
- 53.Ngo S.T., Quynh Anh Pham N., Thi Le L., Pham D.-H., van Vu V. Computational determination of potential inhibitors of SARS-CoV-2 main protease. J. Chem. Inf. Model. 2020;60(12):5771–5780. doi: 10.1021/acs.jcim.0c00491. [DOI] [PubMed] [Google Scholar]
- 54.Milani M., Donalisio M., Bonotto R.M., Schneider E., Arduino I., Boni F., et al. Combined in silico and in vitro approaches identified the antipsychotic drug lurasidone and the antiviral drug elbasvir as SARS-CoV2 and HCoV-OC43 inhibitors. Antivir. Res. 2021;189:105055. doi: 10.1016/j.antiviral.2021.105055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lou Y., Liu L., Yao H., Hu X., Su J., Xu K., et al. Clinical outcomes and plasma concentrations of baloxavir marboxil and favipiravir in COVID-19 patients: an exploratory randomized, controlled trial. Eur. J. Pharmaceut. Sci. 2021;157:105631. doi: 10.1016/j.ejps.2020.105631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang Y.-C., Tung Y.-A., Lee K.-H., Chen T.-F., Hsiao Y.-C., Chang H.-C., et al. 2020. Potential Therapeutic Agents for COVID-19 Based on the Analysis of Protease and RNA Polymerase Docking. [Google Scholar]
- 57.Zhai P., Ding Y., Wu X., Long J., Zhong Y., Li Y. The epidemiology, diagnosis and treatment of COVID-19. Int. J. Antimicrob. Agents. 2020;55(5):105955. doi: 10.1016/j.ijantimicag.2020.105955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gammeltoft K.A., Zhou Y., Duarte Hernandez C.R., Galli A., Offersgaard A., Costa R., et al. Hepatitis C virus protease inhibitors show differential efficacy and interactions with remdesivir for treatment of SARS-CoV-2 in vitro. Antimicrob. Agents Chemother. 2021:AAC0268020. doi: 10.1128/AAC.02680-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sacramento C.Q., Fintelman-Rodrigues N., Temerozo J.R., Da Silva AdPD., Da Dias S.S.G., Da Silva C.D.S., et al. In vitro antiviral activity of the anti-HCV drugs daclatasvir and sofosbuvir against SARS-CoV-2, the aetiological agent of COVID-19. J. Antimicrob. Chemother. 2021;76(7):1874–1885. doi: 10.1093/jac/dkab072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.