

Keywords: intestines, liver, manganese, parkinsonism, SLC30A10
Abstract
The essential metal manganese (Mn) induces incurable neurotoxicity at elevated levels that manifests as parkinsonism in adults and fine motor and executive function deficits in children. Studies on Mn neurotoxicity have largely focused on the role and mechanisms of disease induced by elevated Mn exposure from occupational or environmental sources. In contrast, the critical role of excretion in regulating Mn homeostasis and neurotoxicity has received less attention although 1) studies on Mn excretion date back to the 1920s; 2) elegant radiotracer Mn excretion assays in the 1940s to 1960s established the routes of Mn excretion; and 3) studies on patients with liver cirrhosis in the 1990s to 2000s identified an association between decreased Mn excretion and the risk of developing Mn-induced parkinsonism in the absence of elevated Mn exposure. Notably, the last few years have seen renewed interest in Mn excretion largely driven by the discovery that hereditary Mn neurotoxicity due to mutations in SLC30A10 or SLC39A14 is caused, at least in part, by deficits in Mn excretion. Quite remarkably, some of the recent results on SLC30A10 and SLC39A14 provide explanations for observations made ∼40–50 years ago. The goal of the current review is to integrate the historic studies on Mn excretion with more contemporary recent work and provide a comprehensive state-of-the-art overview of Mn excretion and its role in regulating Mn homeostasis and neurotoxicity. A related goal is to discuss the significance of some of the foundational studies on Mn excretion so that these highly consequential earlier studies remain influential in the field.
INTRODUCTION
Manganese (Mn) is an essential trace metal required for the activity of numerous enzymes that regulate diverse physiological processes (e.g., glycosylation, kinases, etc.) (1, 2). As Mn is fairly abundant in plant-based foods (3), deficiency in the absence of genetic mutations is rare (4, 5). In contrast, toxicity induced by elevated levels of Mn is an important public health problem (2). When body levels of Mn increase, the metal accumulates in the brain, primarily in the basal ganglia, and induces neurotoxicity (2, 6–19). Occupationally exposed adults develop a parkinsonian-like movement disorder, whereas environmentally exposed children and adolescents present with fine motor and executive/cognitive function deficits (2, 6–19). Historically, Mn neurotoxicity in adults has been associated with elevated exposure from occupational sources (20). Indeed, the first report of Mn-induced neurotoxicity in 1837 provided a remarkably detailed description of neuromotor dysfunction in factory workers exposed to elevated Mn during bleaching powder manufacture in Glasgow (21). Currently, occupational exposure to Mn remains relevant because Mn is widely used in industry. Occupations with an elevated Mn exposure risk include mining, welding, and battery and steel manufacture (7). In addition, more recent studies indicate that environmental overexposure to Mn, through drinking water, dust, air, or infant formula, poses a risk to the general public, especially to infants, children, and adolescents who may develop lifelong neurological defects (6–19). Overall, the relationship between elevated Mn exposure from environmental or occupational sources and neurotoxicity is a biomedically important issue that is the subject of intense investigation.
However, the relationship between Mn excretion and neurotoxicity has received comparatively little attention. This is surprising because published literature provides clear evidence highlighting the critical role of excretion in regulating body Mn levels. Mn radiotracer excretion studies in the 1940s to 1960s established that Mn is excreted by the liver and intestines (22–26). These studies further revealed that an increase in the body burden of Mn enhances Mn excretion, suggesting that elevated excretion may be a homeostatic control mechanism to regulate body Mn levels (22–26). Separately, clinical studies in the 1990s revealed that liver dysfunction (e.g., cirrhosis) is associated with the retention of Mn in the body and brain and with the onset of neurotoxicity without exposure to elevated Mn (27–36). These findings raised the hypothesis that excretion may be essential to prevent toxic accumulation of Mn under basal physiological conditions. However, despite the above insights, there was limited further progress, and discussion of the importance of excretion in the homeostatic control and toxicity of Mn was lacking in subsequent literature. A primary reason may be that the molecular mechanisms of Mn excretion remained unknown until very recently.
The last few years have seen a resurgence of interest in Mn excretion, largely driven by the discoveries of hereditary forms of Mn neurotoxicity caused by homozygous loss-of-function mutations in SLC30A10 or SLC39A14 (37–41). Study of rare genetic diseases often produces broad leaps in scientific understanding, and a similar development occurred in the Mn biology field. Mechanistic assays in cell culture and Slc30a10 or Slc39a14 single- or double-knockout mice revealed that SLC30A10 and SLC39A14 are critical Mn transporters that mediate Mn excretion—SLC39A14 imports Mn from blood into hepatocytes and enterocytes for subsequent excretion into bile and feces by SLC30A10 (42–55). Furthermore, analyses of tissue-specific Slc30a10 knockout mice revealed that brain Mn levels are regulated primarily by Mn excretion (46). Finally, a very recent study reported that Mn upregulates SLC30A10 expression in the liver via a hypoxia-inducible factor (HIF)-dependent mechanism, and that increasing SLC30A10 expression using HIF-stabilizing compounds protects cells against Mn toxicity and mice against Mn-induced motor dysfunction (56). These new findings provide a mechanistic basis to understand the historical data on Mn excretion and highlight the close interrelationship between Mn excretion, homeostasis, and neurotoxicity.
The renewed recognition of the importance of Mn excretion makes it an ideal time for a comprehensive review on the topic. Here, we integrate prior historical studies with recent molecular findings to provide an overview of the current state of knowledge of Mn excretion and its role in Mn homeostasis and neurotoxicity. In addition, we highlight some of the seminal studies on Mn excretion from the 1940s to 1950s and on patients with liver cirrhosis from the 1990s that provide a foundation for ongoing mechanistic work so that these important findings retain prominence in scientific discussion.
INITIAL INSIGHTS INTO ROUTES OF MANGANESE EXCRETION FROM THE MID-19TH TO THE EARLY 20TH CENTURY
Body Mn is primarily excreted in feces by the liver in bile and directly by the intestines (22–26). Urinary excretion of Mn is minimal (57, 58). Evidence suggestive of the central role of the liver and intestines in Mn excretion dates back to animal studies conducted in the early 20th century, and perhaps even earlier. In the 1920s, analyses of tissue Mn levels in several animal species (e.g., cats, dogs, rabbits, etc.) revealed that the liver contained the highest amount of Mn under basal conditions (Table 1) (58, 60, 61). Moreover, 1 h after intravenous injection of a Mn dioxide suspension, the liver was the primary (61) or major (60) site of Mn accumulation in numerous mammalian species, and there was an associated, rapid clearance of the injected Mn from the blood (Table 1) (60). Tissue Mn distribution analyses over longer periods after similar intravenous injection of Mn dioxide suspension identified a peak in hepatic Mn levels immediately or within a few hours of injection in rabbits and cats, respectively, followed by a steady decline (Table 1) (62). In these studies, Mn was also recovered in bile collected from Mn-injected rabbits (62). In addition, liver tissue had the highest Mn concentrations in measurements of autopsy samples from 14 individuals without a history of Mn overexposure (Table 1) (59). Finally, increasing doses of intraperitoneal Mn chloride injections in monkeys produced histopathologic changes in the liver and locomotor abnormalities (63), and repeated subcutaneous injection of Mn chloride in rabbits produced cirrhosis (Table 1) (64). In totality, these early studies raised the hypothesis that, under basal conditions as well as with elevated exposure, Mn accumulates in the liver for subsequent biliary excretion.
Table 1.
Historical studies on Mn excretion and their main findings
| References | Findings |
|---|---|
| Reiman and Minot, 1920 (59) | Developed colorimetric assay for determining blood and tissue Mn levels. Liver contains the highest amount of Mn in human tissues collected from 14 autopsies. |
| Drinker and Shaw, 1921 (60) | Liver is a major site of Mn accumulation in cats intravenously injected with MnO2. |
| Lund et al., 1921 (61) | Mn accumulates primarily in the liver 1 h after intravenous MnO2 injection in several other animal species. |
| Drinker et al., 1923 (62) | Mn is detected in bile samples collected from rabbits receiving intravenous Mn injections. |
| Mella, 1924 (63) | Observed movement disorders (rigidity, hand tremors) and signs of brain and liver injury in rhesus monkeys receiving intraperitoneal Mn injections. |
| Findlay, 1924 (64) | Repeated Mn injections cause liver cirrhosis in rats and rabbits. |
| Greenberg and Campbell, 1940 (57) | >90% of orally or intraperitoneally administered Mn radioisotopes are excreted in the feces of rats. |
| Greenberg et al., 1943 (26) | 37.3% of injected Mn radiotracer dose is observed in bile after 48 h in rats with artificial gallbladders. |
| Cotzias and Greenough, 1958 (24) | Rate of Mn excretion is affected by levels of dietary Mn, but not by supplement of other metals (Fe, Cr) |
| Bertinchamps et al., 1966 (22) | When hepatobiliary excretory pathway of Mn is saturated, auxiliary excretion occurs through the gut. |
| Papavasiliou et al., 1966 (25) | Biliary obstruction impairs Mn excretion, fecal obstruction abolishes Mn excretion. |
| Mena et al., 1967 (65) | Healthy Mn miners eliminate injected radioisotope Mn faster than miners afflicted with Mn toxicity. |
| Klaassen, 1974 (66) | Mn concentration in bile is ∼150-fold greater than Mn concentration in plasma. |
| Miller et al., 1975 (67) | Neonatal mice show almost complete retention of Mn radiotracer until PND17, when Mn excretion seemingly presents a sudden increase. |
| Ballatori et al., 1987 (68) | Neonatal mice excrete Mn at a lower rate than adults (∼50% lower biliary excretion). Manganese excretory pathways are saturated at relatively low doses of Mn supplement (10 mg Mn/kg). |
Mn, manganese; PND17, postnatal day 17.
In 1935, von Oettingen wrote an excellent review summarizing work on Mn biology until that time (Table 1) (58), which indicates that the importance of biliary and fecal Mn excretion was recognized as early as the mid-19th century. von Oettingen states that Mn was detected in the bile of cats in 1860; and that feces was identified to be the main route of Mn excretion in 1883 and 1884 with subsequent independent confirmation in the early 1900s (58).
Another noteworthy point is that, in the studies in the 1920s, Mn was also detected in the small intestine and colon under basal conditions and after intravenous injection of Mn dioxide (60–62). This raised the possibility that Mn may also be directly excreted by the intestines into feces. Intestinal excretion of Mn was definitively demonstrated in the mid-1960s (22, 25).
LIVER AS A MAJOR ROUTE OF MANGANESE EXCRETION—RADIOTRACER MANGANESE ASSAYS BY GREENBERG AND COLLEAGUES IN THE 1940S
The advent of radioisotope tracers allowed for accurate analyses of in vivo Mn distribution in different organs and quantitative measurements of Mn excretion through different routes. Utilization of this technique in the 1940s substantially advanced understanding of Mn excretion. A major contribution was by Greenberg and colleagues (Table 1). In 1940, Greenberg and Campbell established that radiotracer Mn can be used to study Mn metabolism and further confirmed that Mn is primarily excreted by the feces (57). Subsequently, in a seminal paper in 1943, Greenberg et al. assayed for biliary Mn excretion in rats with an artificial gallbladder fistula exposed to radioactive Mn by intraperitoneal injection or via the oral route (26). After intraperitoneal injection, ∼25%–40% of the administered dose was recovered in the bile within 48 h (Fig. 1) (26). Of the total Mn appearing in the feces after intraperitoneal injection, ∼50%–75% was estimated to be coming from biliary excretion (26). Urinary excretion of radiotracer Mn was minimal (≤5% of the administered dose) (26). These observations were the first to definitively identify the bile as the major route of Mn excretion. Furthermore, since biliary excretion did not account for 100% of radiotracer Mn in feces after intraperitoneal injection, we can also interpret these results to mean that there must be additional routes to introduce Mn into the feces (e.g., direct excretion via the intestines). There are two other noteworthy features of the biliary Mn excretion data. After oral delivery, only ∼1% of the administered dose was recovered in the bile (Fig. 1) (26). As described by the authors, this was likely reflective of limited absorption of Mn from the gastrointestinal tract. Moreover, after intraperitoneal injection, a higher proportion of the administered dose was recovered in the bile with the higher dose of Mn (0.1 mg) (Fig. 1) (26). This result provides an initial glimpse into the regulation of Mn excretion by the intake or body burden of Mn that was later established.
Figure 1.

Biliary excretion of manganese (Mn) in a rat model. Rats with an artificial gall bladder fistula were provided radioactive Mn (curve 5: 0.1 mg Mn ip; curve 6: 0.01 mg Mn ip; curve 7: oral Mn). Graph depicts presence of radiotracer in bile as a percentage of the administered dose. Information about curves 1–4 relating to excretion of iron and cobalt can be found in the original citation and are not discussed here. From Greenberg et al. (26).
COTZIAS’ CONTRIBUTIONS IN THE 1950S–1960S
The next major leap in our understanding of Mn excretion came from a series of studies by George Cotzias and colleagues that also utilized radiotracer Mn to investigate Mn excretion and tissue distribution (Table 1). We describe these findings in two categories—the first demonstrating the relationship between Mn intake and excretion, and the second elaborating on the routes of Mn excretion.
Dependence of Manganese Excretion on Manganese Intake
An initial study in 1958 used mice to assay for the effect of Mn intake on the turnover of radioactive Mn injected intraperitoneally (24). In animals fed regular chow rich in Mn, the whole body retention of Mn followed a smooth elimination curve with an initial fast phase for several hours after the injection followed by a slower phase over several weeks (24) (see Fig. 2 for an example from a different experiment). The retention time was substantially lengthened when animals were fed a reduced Mn diet for a few days before and during the experiment (24). In contrast, the addition of Mn to the diet lowered the retention time of radiotracer Mn in the body (24). To rule out the possibility that the nonradioactive dietary Mn increased radiotracer Mn excretion by interfering with the enterohepatic circulation of radiotracer Mn, a second set of assays were performed where supplemental Mn was provided intraperitoneally. The enhancing effect of supplemental nonradioactive Mn on radiotracer Mn excretion was observed again (24). Supplementation with other metals (e.g., Fe, Cr, etc.) did not enhance Mn elimination, indicating specificity of the Mn excretion system (24).
Figure 2.

Dependence of manganese (Mn) retention on Mn intake. Mice maintained on a low-Mn milk diet were intraperitoneally injected with radiotracer Mn (54Mn), and the diet was immediately supplemented with indicated amounts of stable Mn (55Mn). Graph depicts radioactivity retained in the animal as a measurement of Mn excretion. Increased Mn content in diet increases radiotracer elimination. From Britton and Cotzias (23).
More detailed analyses were then reported by Britton and Cotzias in 1966 (23). In these experiments, mice were fed a basal diet of evaporated milk, which is low in Mn (18 µg Mn/L). Different experimental groups were supplemented with varying amounts of Mn (range 18 µg to 6.9 g Mn/L). Radiotracer Mn was injected intraperitoneally, and retention of Mn in the carcass was assayed. The addition of supplemental Mn to the diet immediately after injection of radiotracer Mn substantially enhanced elimination of the radiotracer (Fig. 2) (23). The enhancing effect was dependent on the amount of Mn added to the diet. A similar effect was noted when the supplemental Mn was provided 2 wk after the injection of radiotracer Mn (23). At the time, these observations were interpreted to mean that the nonradioactive stable isotope of Mn could exchange with the radioactive Mn in tissues, leading to an increase in the excretion of the radiotracer (23). Although this remains an astute explanation, recent findings suggest another mutually nonexclusive possibility—the increase in excretion of the radiotracer by dietary Mn may be reflective of an increase in Mn excretion due to upregulation of hepatic and intestinal SLC30A10. There is another critical observation in this manuscript discussed by the authors that may not be obvious at first glance. Substantial amounts of radioactive Mn were eliminated from animals fed the low-Mn milk diet without supplemental Mn (Fig. 2), suggesting that Mn is obligatorily excreted even when dietary availability is very low (23). Furthermore, there was a linear relationship between the amount of supplemental dietary Mn and the elimination of radiotracer Mn, suggesting that absorption of dietary Mn is not well-regulated even during excess exposure (23). In conjunction with the obligatory excretion of Mn, these findings implicate that excretion is the primary means of controlling body Mn levels. Recent studies on Slc30a10 and/or Slc39a14 knockout mice validate the preeminent role of excretion in controlling body Mn levels (43, 45, 46, 50, 51, 53, 55).
Routes of Manganese Excretion
Two landmark manuscripts in 1966 provided seminal information about the routes of Mn excretion and their interdependence (22, 25). Bertinchamps et al. (22) focused on detailed analyses of biliary and intestinal Mn excretion. The general experimental strategy was to inject radiotracer Mn into rats intravenously and directly assay for biliary or intestinal Mn excretion by ligating and catheterizing the bile duct or the gut. Several important conclusions emerged from these assays. Kinetic analyses of biliary excretion revealed two stages of radiotracer Mn elimination—a sharp and rapidly resolving first peak within minutes of radiotracer injection and a later, slowly resolving second peak (Fig. 3) (22). The first peak represented the initial clearance of the radiotracer into the bile, and the second peak was suggestive of enterohepatic circulation of Mn. Definitive demonstration of enterohepatic circulation came from observing an enhancing effect of stable Mn delivered to the duodenum on total biliary Mn excretion (22). Notably, Bertinchamps et al. was the first study to provide direct experimental evidence for enterohepatic circulation of Mn. In addition, comparisons of intestinal excretion in different segments of the small intestine revealed that the most proximal aspects of the small intestine contained the highest amounts of radioactivity, suggesting that the proximal small intestine is the major site of Mn excretion (22). Direct comparison of the excretory capacity of the small intestinal segments by placement of catheters identified the duodenum and jejunum to be the primary sites of Mn excretion with comparable excretory capacity, whereas the role of the ileum was minor (Fig. 4) (22). Consistent with the above interpretation, both the duodenum and jejunum, but not the ileum, exhibited a robust increase in excretion of the radiotracer Mn when the radiotracer was injected intravenously with supplemental nonradioactive Mn (Fig. 4) (22). Finally, comparison of biliary and intestinal (duodenal) excretion revealed that biliary excretion quantitatively accounted for ∼80%–90% of the total excretion over the experimental period of ∼4 h (Fig. 5) (22). The two phases of radiotracer Mn excretion in the bile, with the second being suggestive of enterohepatic circulation, were again evident (Fig. 5) (22). As expected, the second phase was not evident in the duodenal excretion, which steadily declined (Fig. 5) (22). Interestingly, in this experiment, intravenous injection of stable Mn ∼2.5 h after the initial radiotracer injection led to an increase in both biliary and duodenal excretion, but the duodenal excretion exhibited a sharper peak and a more rapid decline (Fig. 5) (22). When this experiment was conducted on rats on a Mn-poor diet, only an increase in biliary radioactive Mn was observed (22). Put together, the data from the experiments to compare biliary and intestinal excretion imply that although biliary excretion is the major route of Mn elimination, the intestines can play an important secondary role that becomes more prominent when biliary excretion reaches saturation due to high body burden of Mn.
Figure 3.

Kinetics of biliary manganese (Mn) excretion and Mn clearance. Rats with bile duct catheters were intravenously injected with radiotracer Mn (54Mn). Graph depicts radioactivity measured from blood and bile samples as measurement of blood Mn clearance and biliary Mn excretion, respectively. A sharp peak in bile 54Mn is observed within 10 min after radiotracer injection. Whereas blood 54Mn is almost completely cleared 1 h after the injection, bile 54Mn presented a second peak, indicating enterohepatic circulation of 54Mn back into the liver. From Bertinchamps et al. (22). IV, intravenous.
Figure 4.

Intestinal segments involved in manganese (Mn) excretion. Rats with catheters in the ileum, jejunum, and duodenum were intravenously injected with radiotracer Mn (54Mn). Graph depicts radioactivity measured in intestinal segments every 10 min after radiotracer injection for 150 min. Approximately 90% of radioactivity in the intestines is measured in the jejunum and duodenum. Injection of stable Mn after 1 h sharply increases jejunal and duodenal radioactivity. From Bertinchamps et al. (22). cpm, counts per minute.
Figure 5.

Comparison of biliary and intestinal manganese (Mn) excretion. Rats with bile duct and duodenal catheters were intravenously injected with radiotracer Mn (54Mn). Graph depicts radioactivity measured from bile and duodenum samples every 10 min for 4 h after radiotracer injection. Overall, biliary 54Mn radioactivity accounts for 80%–90% of total radioactivity measured in both segments of the body, indicating the bile as the primary route of Mn excretion. Stable Mn injection ∼150 min after the radiotracer induces a sharp increase in duodenal radioactivity and a more stable increase in biliary radioactivity. From Bertinchamps et al. (22). cpm, counts per minute.
Results from Papavasiliou et al. (25), also on rats exposed to radiotracer Mn, complemented those of Bertinchamps et al. (22). This study confirmed the existence of extrahepatic routes of Mn excretion by showing that Mn excretion was abolished with rectal obstruction, but only inhibited with bile duct ligation (25). Ligation of the bile duct, however, led to the substantial retention of radiotracer Mn in the body (Fig. 6), validating the liver as the major route of Mn excretion (25). As expected from studies reported in Bertinchamps et al., supplementation with stable Mn delivered intravenously enhanced the elimination of radiotracer Mn from the body (i.e., increased Mn excretion; Fig. 7) (25). Notably, this enhancement was also evident in bile duct-ligated animals provided with high doses of supplemental stable Mn (Fig. 7), providing strong evidence indicating that the hepatic route of excretion could be bypassed when the body burden of Mn increases (25). Remarkably, similar conclusions have emerged from analyses of tissue-specific Slc30a10 knockout mice in the past few years (46, 50).
Figure 6.

Manganese (Mn) excretion rate is significantly reduced after biliary ligation. Sham-operated or bile duct-ligated rats were intravenously or intraportally injected with radiotracer Mn (54Mn). Graph depicts radioactivity retained in the animal as a measurement of Mn excretion. Biliary ligation causes significant retention of injected Mn regardless of the route of injection (curves A and C). When radiotracer Mn is injected directly to the portal vein, its elimination is more significantly decreased by biliary ligation compared with injection to a peripheral vein. Notably, Mn excretion is inhibited, but not abolished by biliary ligation (n = 3 rats per group). From Papavasiliou et al. (25). I.V., intravenous.
Figure 7.

Dependence of manganese (Mn) retention on Mn intake persists after biliary ligation. Sham-operated or bile duct-ligated rats were first intravenously injected with radiotracer Mn (54Mn) at day 0 and then injected with indicated doses of stable Mn (55Mn) on day 3. Graph depicts radioactivity retained in the animal as a measurement of Mn excretion. Stable Mn injection dose-dependent increase of Mn excretion in sham-operated rats (curves D–F) confirms the findings shown in Fig. 2. Notably, Mn excretion rate is also increased in bile duct-ligated rats provided with high doses of stable Mn (curves B and C). From Papavasiliou et al. (25). I.V., intravenous.
Results in Klaassen (66) provide independent confirmation for some of the observations in Papavasiliou et al. (25) and Bertinchamps et al. (22). In this later study, increasing doses of stable MnCl2 (0.3, 1, 3, or 10 mg Mn/kg) combined with radiotracer Mn were given intravenously to rats and biliary Mn excretion was assayed (66). Excretion of Mn in bile increased in a Mn dose-dependent fashion up to the 3 mg/kg dose, but no further increase was evident after the 10 mg/kg dose, indicating that the hepatic route was saturable (66). Notably, the concentration of Mn in bile was 100- to 150-fold higher than that in plasma for 0.3, 1, and 3 mg Mn/kg doses (66). The high bile-plasma Mn gradient and saturability of transport were interpreted to mean that an active transport mechanism mediated biliary Mn excretion (66). This interpretation was very perceptive as we now know that SLC30A10 is a saturable secondary active Mn transporter (69).
AGE DEPENDENCE OF MANGANESE EXCRETION—1970S–1980S
Work from Cotzias’ group in 1975 revealed that neonatal mice injected with trace amounts of radiotracer Mn on postnatal day (PND) 7 retained nearly all of the radiotracer until ∼PND17 (Fig. 8 and Table 1) (67). This was initially interpreted to mean that neonates lack the capability to excrete Mn (67). However, subsequent work by Ballatori in rats indicated that neonatal animals have reduced, but measurable, Mn excretory capacity (68). In a first set of experiments in this later paper, 8-day-old rats were injected with radiotracer Mn containing varying amounts of stable Mn (0, 0.1, 1, or 10 mg Mn/kg), and total radioactivity retained in the carcass was used to assay for Mn excretion. Little excretion (∼10% of the dose) was evident when the radiotracer was given by itself or with 0.1 mg Mn/kg until PND18 (Fig. 9). However, in animals given the two higher doses of stable Mn, rapid reductions in whole body retention (i.e., rapid excretion) were immediately evident (Fig. 9) (68). Interestingly, the sudden increase in Mn excretion on PND18 was absent among animals receiving the highest Mn dose (10 mg/kg), implying saturation of Mn excretion (Fig. 9) (68). Importantly, Ballatori then directly compared biliary Mn excretion between preweaned 14-day-old and adult rats after intravenous administration of the above doses of stable and radiotracer Mn (68). At each Mn dose, PND14 rats excreted ∼50% less Mn than the adults (Fig. 10) (68). Higher doses of stable Mn (up to 1 mg/kg dose) increased excretion in both the adults and preweaning animals (Fig. 10) (68). Transport was saturated at the 10 mg Mn/kg dose (68); this transport maximum is similar to that reported by Klaassen in 1974 (66). Ballatori provided several insightful interpretations of these results. The reduced capacity of neonates to excrete Mn may be reflective of a greater need to retain Mn at this early life stage because breast milk has exceedingly low Mn content (e.g., 200–300 ng/mL) (47, 68, 70). The increase in excretion around PND18 may be a physiological adaptation as animals move from a milk-only diet to solid food (chow in laboratory setting) with much higher Mn content (e.g., ∼100 µg Mn/g chow) (43, 68). The lack of excretion of tracer doses of radioactive Mn observed may be a consequence of Mn retention (68). Finally, rodents and humans are exceedingly sensitive to Mn neurotoxicity in early life (10–19, 71–73). The decreased excretory capacity of Mn in this vulnerable life stage may be an important contributor to the heightened sensitivity of young animals, including infants and children, to Mn neurotoxicity.
Figure 8.

Manganese (Mn) retention in newborn mice. Seven-day-old mice were intraperitoneally injected with radiotracer Mn (54Mn). Graph depicts radioactivity measurements as percent of the first count after the injection. Tracer Mn is almost completely retained until postnatal day (PND) 17, followed by a sharp increase in Mn excretion (n = 6–8 mice per experiment, experiment repeated 5 times). Animals did not receive supplemental stable Mn in this experiment (i.e., dose of stable Mn was 0 mg/kg). From Miller et al. (67).
Figure 9.

Dose dependence of manganese (Mn) retention rate in newborn rats. Eight-day-old rats were intraperitoneally injected with radiotracer Mn and varying doses (0, 0.1, 1, and 10 mg/kg) of stable Mn. Graph depicts retained whole body Mn radioactivity as a percentage of initial radioactivity measured 4 h after the injection. Dose-dependent increase in Mn excretion rate is observed almost immediately after the injections for 1 mg Mn/kg and 10 mg Mn/kg injected animals. Notably, all animals show an increase in Mn excretion rate after 18 postnatal day (PND). From Ballatori et al. (68).
Figure 10.

Comparison of dose-dependent manganese (Mn) excretion rates in newborn and adult rats. Fourteen-day-old and adult rats were intravenously injected with radiotracer Mn and indicated doses of stable MnCl2. Top: biliary excretion as percent radioactivity of collected bile samples to radioactivity of the administered dose. Bottom: bile flow according to measured bile sample volumes. Bile samples were collected in 1 h intervals for newborn and 30-min intervals for adult rats. Values are means ±SE, n = 4 adult rats, 6 newborn rats for each dose. From Ballatori et al. (68).
LIVER FAILURE AND MANGANESE TOXICITY—1990S–2000S
Subsequent to the radiotracer studies described earlier, the only other notable progress in elucidating mechanisms of Mn excretion and the health impacts of impaired Mn excretion until the last decade came from analyses of patients with liver disease in the 1990s to 2000s (Table 2) (27–36). Neuromotor dysfunction is an outcome commonly associated with liver disease, and the evidence from clinical studies implicates impaired Mn excretion resulting in the accumulation of Mn in the brain, particularly in the basal ganglia, as a contributing factor (Table 2) (27–33, 35). Manganese [as Mn(III)] is a paramagnetic metal, and as such it produces hyperintensities on T1-weighted MRI scans. Several human studies have leveraged this property of Mn to assay for brain Mn accumulation in patients (Table 2) (27–33, 35). In 1991, T1-weighted MRI hyperintensities were reported in the globus pallidus of 30 out of 42 patients with chronic liver failure (Table 2) (34). Subsequently, a correlation between MRI signal hyperintensity and blood Mn concentration was also reported (blood Mn concentrations: 20.6 ± 10.2 µg/L in cirrhotics vs. 7.2 ± 2.7 in controls; r = 0.65 for correlation between blood Mn and MRI signals) (Table 2) (29). Similar T1-weighted pallidal hyperintensities along with increases in blood Mn levels (125 ± 49.5 nmol/L in controls vs. 331 ± 190 nmol/L in patients) were reported by Spahr and colleagues (28) (Table 2). Notably, pallidal hyperintensities were observed in 88% of patients with cirrhosis in this study (28). Consistent with this, rats with induced biliary cirrhosis or portacaval shunts exhibited 27% and 57% increase in pallidal Mn levels, respectively (Table 2) (30). Furthermore, autopsy findings revealed two- to sevenfold increases in pallidal Mn levels of nine patients who died due to complications of cirrhosis compared with 12 age-matched controls (average of 5.4 ± 2.0 SD µg Mn/g dry wt in cirrhotics vs. 1.9 ± 0.4 µg Mn/g in controls) (Table 2) (27). A related study of brain Mn levels in autopsy samples from patients with cirrhosis confirmed and extended the above observations by documenting increases in Mn levels in the pallidus (186%), caudate (54%), putamen (66%), and cortex (<50%; Table 2) (30). Interestingly, a more recent study detected strikingly similar increases in pallidal Mn levels measured by atomic absorption spectrometry and T1-weighted pallidal hyperintensities of rhesus monkeys exposed to Mn by inhalation, suggesting that both exogenous Mn exposure and increased Mn retention due to impaired hepatic Mn excretion can lead to similarly elevated neurotoxic levels of Mn accumulation in the brain (74). Finally, patients with liver disease who received a liver transplant showed improvement of neuromotor symptoms and reduction of MRI hyperintensities in globus pallidus several months after the operation (Table 2) (31, 32). Notably, cirrhosis and chronic liver disease are associated with functional deficits in the intestines (75), raising the possibility that intestinal Mn excretion may also be compromised in patients with cirrhosis. Although causal inferences cannot be drawn from these studies, the data are consistent with the hypothesis that decreased hepatic, and likely intestinal, Mn clearance due to liver damage may be sufficient to lead to Mn retention and neurotoxicity in the absence of elevated Mn exposure. This possibility is supported by more recent observations from tissue-specific Slc30a10 knockout mice showing that simultaneous depletion of SLC30A10 in the liver and the intestines substantially elevates Mn levels in the body, including in the brain, without excessive Mn exposure (46, 50) (see discovery of the molecular mechanisms of manganese excretion—2012 onward below for more details).
Table 2.
Studies on liver damage-related Mn toxicity due to compromised Mn excretion
| References | Findings |
|---|---|
| Brunberg et al., 1991 (34) | MRI T1 hyperintensity in globus pallidus observed in 30 of 42 patients with chronic liver failure. |
| Hauser et al., 1996 (29) | Correlation between blood Mn levels and MRI T1-hyperintensity scores observed in patients with chronic hepatic cirrhosis. |
| Pomier-Layrargues et al., 1995 (27) | 2- to 7-fold higher pallidal Mn detected in the autopsies of patients who died from cirrhosis-related complications. |
| Spahr et al., 1996 (28) | 88% of patients with cirrhosis exhibit pallidal T1 hyperintensity. In addition, shows association between blood Mn levels and T1 pallidal MRI intensity. |
| Maeda et al., 1997 (35) | Increased Mn levels were observed in the pallidus, caudate, putamen, and cortex in autopsy samples from patients with cirrhosis. |
| Rose et al., 1999 (30) | Rats with biliary cirrhosis or portacaval shunt present increased Mn levels in pallidum and caudate. |
| Spahr et al. 2000 (32) | MRI T1 hyperintensity of patients with hepatic encephalopathy correlates with severity of parkinsonian signs. 3 patients showed disappearance of T1 hyperintensity and improvement in parkinsonian signs after liver transplant. |
| Burkhard et al. 2003 (33) | Blood Mn levels of all examined patients with cirrhosis are higher than normal values and comparable with those of patients with occupational Mn toxicity. |
| Long et al. 2009 (31) | Observed correlation between Mn levels in globus pallidus and chronic liver disease severity. Patients with cirrhosis who receive liver transplants have normalized brain Mn levels 3 mo posttransplant. |
Mn, manganese.
DISCOVERY OF THE MOLECULAR MECHANISMS OF MANGANESE EXCRETION—2012 ONWARD
Understanding of the molecular mechanisms of Mn excretion has significantly advanced over the past decade, such that a fairly well-established molecular basis of Mn excretion is now available (42–48, 51–53, 56, 76). This advancement in the field arose out of the discoveries of two hereditary disorders of Mn metabolism caused by homozygous loss-of-function mutations in SLC30A10 or SLC39A14. In addition, homozygous loss-of-function mutations in SLC39A8 were identified to cause Mn deficiency. Several detailed reviews on this topic have recently been written by us and others (42, 76, 77). Therefore, we only summarize the salient features of this line of work below and direct the reader to these other reviews and readily accessible primary papers for more detailed information.
SLC30A10—A Manganese Efflux Transporter Essential for Manganese Excretion
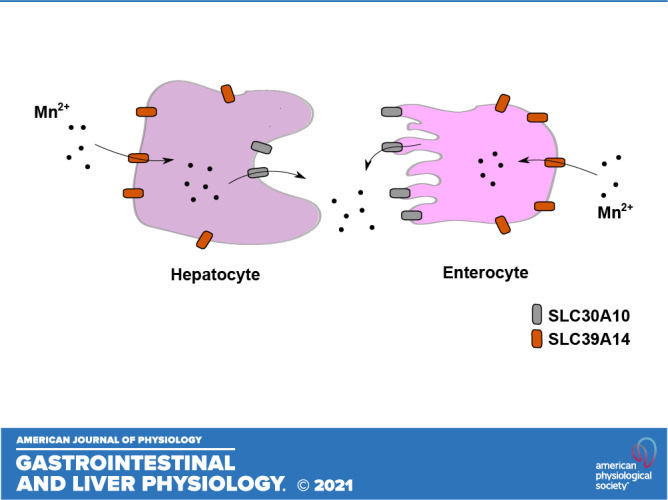
In 2008, Tuschl and colleagues reported that a pediatric patient born to consanguineous parents had clinical features of neuromotor defects, cirrhosis, and elevated blood and brain Mn levels (39). Notably, this patient did not have a history of exposure to elevated Mn (39), suggesting that an inherited disorder of Mn metabolism, likely in Mn excretion, may be the underlying cause of the Mn retention and neurological deficit. In 2012, additional patients with similar clinical features and family history were identified, and whole genome homozygosity mapping revealed that all patients carried homozygous mutations in SLC30A10 gene (37, 40, 41). Our assays in cell culture, including primary neurons and Caenorhabditis elegans revealed that SLC30A10 is a cell surface localized Mn efflux transporter that transports Mn from the cytosol to the cell exterior, reduces cellular Mn levels, and protects against Mn toxicity (44). Several disease-causing SLC30A10 mutants mislocalized to the endoplasmic reticulum and lacked Mn efflux activity (44). Furthermore, SLC30A10 was targeted to the apical domain of differentiated HepG2 cells that model hepatocytes (45) and CaCo2 cells that model enterocytes (46), suggesting that its efflux activity may mediate Mn excretion (Fig. 11) (47). Consistent with this hypothesis, full-body Slc30a10 knockout mice exhibited marked increases in tissue Mn levels by 6 wk of age and developed neuromotor deficits in the absence of elevated Mn exposure (43, 45, 46). This phenotype was rescued by a reduced Mn diet (43, 46). More direct evidence for the role of SLC30A10 in Mn excretion, and the critical function of excretion in regulating body Mn levels, came from the analyses of tissue-specific Slc30a10 knockouts (46). By 6 wk of age, Mn levels in tissues, including the brain, were highly elevated in endoderm-specific Slc30a10 knockouts that lacked SLC30A10 expression in both the liver and intestines (46). However, fecal Mn levels of the endoderm-specific knockouts were lower than controls (46). Put together, the above results imply that activity of SLC30A10 is essential for Mn excretion, and excretion is a primary means to control blood and brain Mn levels (46). Interestingly, liver and brain tissue Mn levels of liver-specific Slc30a10 knockouts were only modestly elevated in our assays, suggesting that the intestines can compensate for loss-of-function of SLC30A10 in the liver, and by extension, for hepatic Mn excretion (46), consistent with Cotzias’ earlier findings (22, 25). A subsequent study confirmed the critical role of SLC30A10 in hepatic and intestinal Mn excretion using radiotracer Mn elimination assays, including biliary Mn excretion, in full-body and tissue-specific Slc30a10 knockout mice (this study also recapitulated the previously reported enhancing effect of elevated dietary Mn on Mn excretion) (50). Finally, additional work by us revealed that brain SLC30A10 plays an essential role in providing neuroprotection that becomes particularly critical during elevated Mn exposure (Fig. 11) (46).
Figure 11.

The mechanisms of manganese (Mn) excretion by SLC30A10 and SLC39A14 in the liver, intestines, and brain. From Taylor et al. (47) with permission.
SLC39A14—A Manganese Importer That Functions Cooperatively with SLC30A10 to Mediate Manganese Excretion
In 2016, Tuschl and colleagues identified a second hereditary disorder of Mn metabolism by demonstrating that individuals with loss-of-function mutations in SLC39A14 also accumulate Mn in the blood and brain and develop neurotoxicity (38). SLC39A14 is a metal importer with established Mn transport activity (78). The lack of a liver phenotype in patients with SLC39A14 mutations suggested that SLC39A14 likely played an important role in importing Mn into hepatocytes (38). Consistent with this, SLC39A14 was detected in the basolateral domain of polarized HepG2 cells (45) and rat hepatocytes (79). Full-body Slc39a14 knockout mice exhibited elevated Mn levels in the blood and brain, but not the liver, and developed neurotoxicity in the absence of elevated Mn exposure (51, 53, 55). Further evidence for the indispensability of SLC39A14 for hepatic Mn excretion came from the comparison of Slc30a10 or Slc39A14 single-knockout mice with Slc30a10/Slc39a14 double knockouts (45). Although blood and brain Mn levels were elevated in all three strains, liver Mn levels were elevated only in the Slc30a10 single knockouts (45). Put together, the available evidence indicates that SLC39A14 transports Mn from blood into hepatocytes for subsequent excretion into bile by SLC30A10 (Fig. 11). Other studies revealed that SLC39A14 is also critical for intestinal Mn excretion by localizing to the basolateral domain of, and transporting Mn into, enterocytes, where it can subsequently be excreted via SLC30A10 into the intestinal lumen (52, 54, 80).
SLC39A8—A Manganese Importer That Reclaims Manganese Excreted in Bile
In 2015, two groups independently reported that individuals harboring homozygous loss-of-function mutations in SLC39A8 develop Mn deficiency (81, 82). SLC39A8 is a metal importer, and subsequent mechanistic studies suggested that a critical function of SLC39A8 is to reclaim Mn excreted in bile (83). Although SLC39A8 does not directly mediate Mn excretion, conceivably, changes in SLC39A8 expression, localization, or activity may indirectly influence Mn turnover by affecting biliary Mn import into hepatocytes. A more comprehensive discussion about SLC39A8 and its role in Mn metabolism is provided in a review by Balachandran et al. (42).
HOMEOSTATIC CONTROL OF MANGANESE EXCRETION DURING MANGANESE TOXICITY
Metal homeostasis is primarily controlled by changes in the expression or activity of specific transporters. Until the discovery of the genetic diseases described earlier, understanding of the responses of mammalian systems to changes in Mn levels was limited by a lack of information about critical Mn transporters. The identification of SLC30A10, SLC39A14, and SLC39A8 as transporters that play a fundamental role in regulating body Mn levels in humans and rodents led us to hypothesize that alterations in the activity of these transporters may underlie a homeostatic response to Mn. Our recent studies identified a Mn-dependent transcriptional elevation of SLC30A10 as a central, protective response to elevated Mn exposure. We discovered that exposure to an environmentally relevant level of elevated Mn in drinking water increased SLC30A10 mRNA levels in liver of 129S4/SvJaeJ or intestines of C57BL/6J mice (mechanisms underlying the strain-specific differences are yet unclear) (56). Assays in HepG2 cells and primary human hepatocytes demonstrated that Mn transcriptionally upregulated SLC30A10 (56). A hypoxia response element in the SLC30A10 promoter and the activity of hypoxia inducible factor (HIF) 1 or HIF2 were required and sufficient for this response (56). The molecular mechanism was a Mn-induced inhibition of prolyl hydroxylase enzymes that target HIF proteins for degradation under normoxic conditions. The Mn-induced elevation of SLC30A10 was homeostatically important because inhibition of the response by genetic attenuation of the HIF cascade significantly increased intracellular Mn levels under elevated Mn exposure conditions and further enhanced sensitivity to Mn-induced cell death (56). Moreover, prolyl hydroxylase inhibitor drugs that have been approved for human use or are in advanced clinical trials for treatment of renal anemia reduced intracellular Mn levels and protected against Mn toxicity in cell culture. Finally, analyses of one prolyl hydroxylase inhibitor in C57BL/6J mice, roxadustat, revealed a strong protective effect against Mn-induced neuromotor deficits. Thus, elevated Mn levels activate HIF1/HIF2 to upregulate hepatic/intestinal SLC30A10 providing a pathway to reduce cellular and organismal Mn levels. These results provide a molecular framework to understand the Mn-induced increases in radiotracer Mn elimination reported previously and raise the exciting possibility of using pharmacological methods to enhance SLC30A10 expression and Mn excretion for the management of Mn overexposure and risk for neurotoxicity.
RECONCILING RECENT MOLECULAR FINDINGS WITH HISTORICAL STUDIES
As described in the preceding sections, results obtained from mechanistic studies on SLC30A10 and SLC39A14 (see discovery of the molecular mechanisms of manganese excretion—2012 onward and homeostatic control of manganese excretion during manganese toxicity) corroborate with the historic radiotracer Mn excretion studies (see liver as a major route of manganese excretion—radiotracer manganese assays by greenberg and colleagues in the 1940s, cotzias’ contributions in the 1950s–1960s, age dependence of manganese excretion—1970s–1980s, and liver failure and manganese toxicity—1990s–2000s). We summarize these consistencies here. Prior studies showed that Mn is primarily excreted by the liver (22–26, 66), but the intestines also have the capability to excrete Mn (22, 25), and that the role of the intestines gains prominence when hepatic excretion is compromised or overwhelmed (22, 25). These findings predict that loss of function of critical Mn exporters in both the liver and intestines, but not just one organ, would be necessary to block Mn excretion. Results obtained from the tissue-specific Slc30a10 knockout mice are consistent with this prediction (46, 50). We also note that liver- or intestine-specific Slc39a14 knockout mice only exhibit modest elevations in tissue Mn levels, unlike the full-body Slc39a14 knockouts (52, 54, 55) (Slc39a14 knockouts with gene deletion in both the liver and intestines have not yet been reported). As cirrhosis also impacts intestinal function, Mn retention inherent to patients with cirrhosis is also broadly consistent with the prior radiotracer Mn studies and phenotypes of tissue-specific Slc30a10 or Slc39a14 knockout mice. Moreover, the historic studies indicated that Mn intake modulates Mn excretion, with elevated Mn levels increasing excretion (23, 24, 26, 68). Identification of the Mn-dependent transcriptional upregulation of SLC30A10 via HIF1/HIF2 provides the mechanistic bases for this observation.
CONCLUSIONS AND FUTURE DIRECTIONS
Summary of Our Current Understanding of Manganese Excretion and Its Role in Neurotoxicity
Research on Mn excretion spans more than a century with periods of rapid progress alternating with years of limited breakthroughs. A notable feature of the historic radiotracer studies is their perceptive nature that helped set the stage for the recent mechanistic analyses. Several important, shared conclusions emerge from the studies presented in this review: 1) under physiological conditions, the liver is the primary organ that excretes Mn, but the intestines play an important role as well; 2) the role of the intestines in Mn excretion becomes more prominent when hepatic excretion is saturated or impaired; 3) SLC30A10 and SLC39A14 are the critical Mn transporters that cooperatively mediate Mn excretion, with SLC39A14 transporting Mn from blood into hepatocytes and enterocytes, and SLC30A10 excreting Mn into bile and feces; 4) body Mn excretion is a major physiological mechanism to control tissue Mn levels, and under basal conditions, brain Mn levels are primarily regulated by Mn excretion; 5) increases in Mn excretion in response to elevated Mn exposure, by the transcriptional upregulation of SLC30A10 in the liver and intestines, provide a homeostatic protective pathway to reduce body Mn levels and protect against Mn toxicity; and 6) it may be possible to leverage the Mn responsiveness of SLC30A10 expression for the development of therapeutic treatments for Mn overexposure and risk for neurotoxicity using prolyl hydroxylase inhibitors.
Emerging Directions for Future Investigations
Several new questions arise from the work on Mn reported to date. We note a few examples of important lines of investigations that have already been, or will likely soon be, initiated. Tissue-specific Slc30a10 knockout mice are being used to determine the influence of age, sex, and Mn exposure on the relative contributions of the liver and intestines in Mn excretion. More detailed analyses of the role of excretion in modulating Mn neurotoxicity are also ongoing. This line of work may complement findings from epidemiological studies indicating that Mn exposure impacts neuromotor function in children in a sex-specific manner (84). In addition, effects of Mn on intestinal physiology are also expected to emerge over the coming years, as recent studies indicate that Mn exposure in mice impacts the gut microbiome (85) as well as the enteric nervous system and gastrointestinal transit time (86). Furthermore, intestinal SLC30A10 is now known to be transcriptionally upregulated by bile acids (87) and vitamin D (88, 89). Elucidating the mechanisms by which diverse physiological ligands (Mn, bile acids, vitamin D, and perhaps others yet to be identified) modulate SLC30A10 expression is a required next step. Additional work on the effects of prolyl hydroxylase inhibitors and the HIF1/HIF2 pathway on Mn excretion and neurotoxicity in animal models is also necessary from a drug development perspective. In this realm, the influence of the route of exposure (e.g., oral vs. inhalation) on the homeostatic control of SLC30A10 transcription and Mn excretion is a critical topic. From a public health perspective, recent epidemiological studies revealed that common single-nucleotide polymorphisms (SNPs) in SLC30A10, prevalent in ∼20%–25% of the population, were associated with increased blood Mn (90, 91), neurological markers of parkinsonism among the elderly (90), neurological performance difficulties among children (91), and for one SNP for which gene expression was tested, decreased SLC30A10 levels (90, 91). SLC30A10 SNPs associated with reduced blood Mn, suggestive of increased Mn excretory capacity, were also identified (90, 91). Potentially disease-relevant SNPs in SLC39A14 and SLC39A8 have also been reported (91–94). Interestingly, a 1967 study had shown that healthy Mn miners presented faster total body Mn turnover compared with miners exhibiting signs of Mn toxicity, implicating the possibility of genetic variations in modulating Mn excretion and protection against Mn toxicity (65). Determining whether common genetic variations in Mn transporters modulate Mn excretion capacity in a manner that influences the risks and outcomes of Mn neurotoxicity, with elevated risk in individuals with lower excretory capacity, is of fundamental importance. Cell- and organ-specific studies on SLC39A14 and SLC39A8 are also ongoing. Since SLC39A14 and SLC39A8 are metal importers with a broader substrate specificity compared with SLC30A10, these studies are expected to provide insights beyond Mn excretion and into areas such as the transport of Mn across the blood-brain barrier (95), as well as into neurons and glia, homeostatic control of other metals (e.g., SLC39A14 was recently reported to play a role in biliary excretion of excess iron; Ref. 96), and biology of other disease conditions (e.g., association of SLC39A8 missense mutation with Crohn’s disease; Ref. 97). As current interest in Mn excretion is high, the rapid progress in this area will likely continue and the questions raised above may soon be answered.
GRANTS
This study was supported by NIH/National Institute of Environmental Health Sciences (NIEHS) Grants R01-ES024812 and R01-ES031574 (to S.M.).
DISCLOSURES
D. R. Smith and S. Mukhopadhyay are inventors on a provisional patent application filed by The University of Texas at Austin on the use of HIF stabilizing compounds for the treatment of Mn-induced neurotoxicity. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
K.C.G. drafted manuscript; K.C.G., M.A., D.R.S., and S.M. edited and revised manuscript; S.M. approved final version of manuscript.
REFERENCES
- 1.Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Mol Aspects Med 26: 353–362, 2005. doi: 10.1016/j.mam.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aschner M, Erikson KM, Herrero Hernández E, Tjalkens R. Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromolecular Med 11: 252–266, 2009. [Erratum in Neuromolecular Med 11: 267, 2009]. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeland-Graves JH, Mousa TY, Kim S. International variability in diet and requirements of manganese: causes and consequences. J Trace Elem Med Biol 38: 24–32, 2016. doi: 10.1016/j.jtemb.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Keen CL, Ensunsa JL, Watson MH, Baly DL, Donovan SM, Monaco MH, Clegg MS. Nutritional aspects of manganese from experimental studies. Neurotoxicology 20: 213–223, 1999. [PubMed] [Google Scholar]
- 5.Friedman BJ, Freeland-Graves JH, Bales CW, Behmardi F, Shorey-Kutschke RL, Willis RA, Crosby JB, Trickett PC, Houston SD. Manganese balance and clinical observations in young men fed a manganese-deficient diet. J Nutr 117: 133–143, 1987. doi: 10.1093/jn/117.1.133. [DOI] [PubMed] [Google Scholar]
- 6.Bowman AB, Aschner M. Considerations on manganese (Mn) treatments for in vitro studies. Neurotoxicology 41: 141–142, 2014. doi: 10.1016/j.neuro.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Racette BA, Criswell SR, Lundin JI, Hobson A, Seixas N, Kotzbauer PT, Evanoff BA, Perlmutter JS, Zhang J, Sheppard L, Checkoway H. Increased risk of parkinsonism associated with welding exposure. Neurotoxicology 33: 1356–1361, 2012. doi: 10.1016/j.neuro.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olanow CW. Manganese-induced parkinsonism and Parkinson’s disease. Ann N Y Acad Sci 1012: 209–223, 2004. doi: 10.1196/annals.1306.018. [DOI] [PubMed] [Google Scholar]
- 9.Perl DP, Olanow CW. The neuropathology of manganese-induced parkinsonism. J Neuropathol Exp Neurol 66: 675–682, 2007. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- 10.Bhang S-Y, Cho S-C, Kim J-W, Hong Y-C, Shin M-S, Yoo HJ, Cho IH, Kim Y, Kim B-N. Relationship between blood manganese levels and children’s attention, cognition, behavior, and academic performance—a nationwide cross-sectional study. Environ Res 126: 9–16, 2013. doi: 10.1016/j.envres.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D. Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ Health Perspect 115: 122–127, 2007. doi: 10.1289/ehp.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouchard MF, Sauvé S, Barbeau B, Legrand M, Brodeur ME, Bouffard T, Limoges E, Bellinger DC, Mergler D. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect 119: 138–143, 2011. doi: 10.1289/ehp.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claus Henn B, Ettinger AS, Schwartz J, Téllez-Rojo MM, Lamadrid-Figueroa H, Hernández-Avila M, Schnaas L, Amarasiriwardena C, Bellinger DC, Hu H, Wright RO. Early postnatal blood manganese levels and children’s neurodevelopment. Epidemiology 21: 433–439, 2010. doi: 10.1097/ede.0b013e3181df8e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan K, Factor-Litvak P, Wasserman GA, Liu X, Ahmed E, Parvez F, Slavkovich V, Levy D, Mey J, van Geen A, Graziano JH. Manganese exposure from drinking water and children’s classroom behavior in Bangladesh. Environ Health Perspect 119: 1501–1506, 2011. doi: 10.1289/ehp.1003397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan K, Wasserman GA, Liu X, Ahmed E, Parvez F, Slavkovich V, Levy D, Mey J, van Geen A, Graziano JH, Factor-Litvak P. Manganese exposure from drinking water and children's academic achievement. Neurotoxicology 33: 91–97, 2012. doi: 10.1016/j.neuro.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucchini RG, Guazzetti S, Zoni S, Donna F, Peter S, Zacco A, Salmistraro M, Bontempi E, Zimmerman NJ, Smith DR. Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology 33: 687–696, 2012. doi: 10.1016/j.neuro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oulhote Y, Mergler D, Barbeau B, Bellinger DC, Bouffard T, Brodeur ME, Saint-Amour D, Legrand M, Sauvé S, Bouchard MF. Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ Health Perspect 122: 1343–1350, 2014. doi: 10.1289/ehp.1307918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riojas-Rodríguez H, Solís-Vivanco R, Schilmann A, Montes S, Rodríguez S, Ríos C, Rodríguez-Agudelo Y. Intellectual function in Mexican children living in a mining area and environmentally exposed to manganese. Environ Health Perspect 118: 1465–1470, 2010. doi: 10.1289/ehp.0901229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wasserman GA, Liu X, Parvez F, Ahsan H, Levy D, Factor-Litvak P, Kline J, van Geen A, Slavkovich V, LoIacono NJ, Cheng Z, Zheng Y, Graziano JH. Water manganese exposure and children’s intellectual function in Araihazar, Bangladesh. Environ Health Perspect 114: 124–129, 2006. doi: 10.1289/ehp.8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanc PD. The early history of manganese and the recognition of its neurotoxicity, 1837-1936. Neurotoxicology 64: 5–11, 2018. doi: 10.1016/j.neuro.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Couper J. On the effects of black oxide of manganese when inhaled into the lungs. Br Ann Med Pharmacol 1: 41–42, 1837. [Google Scholar]
- 22.Bertinchamps AJ, Miller ST, Cotzias GC. Interdependence of routes excreting manganese. Am J Physiol 211: 217–224, 1966. doi: 10.1152/ajplegacy.1966.211.1.217. [DOI] [PubMed] [Google Scholar]
- 23.Britton AA, Cotzias GC. Dependence of manganese turnover on intake. Am J Physiol 211: 203–206, 1966. doi: 10.1152/ajplegacy.1966.211.1.203. [DOI] [PubMed] [Google Scholar]
- 24.Cotzias GC, Greenough JJ. The high specificity of the manganese pathway through the body. J Clin Invest 37: 1298–1305, 1958. doi: 10.1172/JCI103718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papavasiliou PS, Miller ST, Cotzias GC. Role of liver in regulating distribution and excretion of manganese. Am J Physiol 211: 211–216, 1966. doi: 10.1152/ajplegacy.1966.211.1.211. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg DM, Copp DH, Cuthbertson EM. Studies in mineral metabolism with the aid of artificial radioactive isotopes. VII. The distribution and excretion, particularly by way of the bile, of iron, cobalt, and manganese. J Biol Chem 147: 749–756, 1943. [Google Scholar]
- 27.Pomier-Layrargues G, Spahr L, Butterworth RF. Increased manganese concentrations in pallidum of cirrhotic patients. Lancet 345: 735, 1995. doi: 10.1016/s0140-6736(95)90909-5. [DOI] [PubMed] [Google Scholar]
- 28.Spahr L, Butterworth RF, Fontaine S, Bui L, Therrien G, Milette PC, Lebrun LH, Zayed J, Leblanc A, Pomier-Layrargues G. Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensity and neurological symptoms. Hepatology 24: 1116–1120, 1996. doi: 10.1002/hep.510240523. [DOI] [PubMed] [Google Scholar]
- 29.Hauser RA, Zesiewicz TA, Martinez C, Rosemurgy AS, Olanow CW. Blood manganese correlates with brain magnetic resonance imaging changes in patients with liver disease. Can J Neurol Sci 23: 95–98, 1996. doi: 10.1017/s0317167100038786. [DOI] [PubMed] [Google Scholar]
- 30.Rose C, Butterworth RF, Zayed J, Normandin L, Todd K, Michalak A, Spahr L, Huet PM, Pomier-Layrargues G. Manganese deposition in basal ganglia structures results from both portal-systemic shunting and liver dysfunction. Gastroenterology 117: 640–644, 1999. doi: 10.1016/s0016-5085(99)70457-9. [DOI] [PubMed] [Google Scholar]
- 31.Long LL, Li XR, Huang ZK, Jiang YM, Fu SX, Zheng W. Relationship between changes in brain MRI and 1H-MRS, severity of chronic liver damage, and recovery after liver transplantation. Exp Biol Med (Maywood) 234: 1075–1085, 2009. doi: 10.3181/0903-RM-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spahr L, Vingerhoets F, Lazeyras F, Delavelle J, DuPasquier R, Giostra E, Mentha G, Terrier F, Hadengue A. Magnetic resonance imaging and proton spectroscopic alterations correlate with parkinsonian signs in patients with cirrhosis. Gastroenterology 119: 774–781, 2000. doi: 10.1053/gast.2000.17857. [DOI] [PubMed] [Google Scholar]
- 33.Burkhard PR, Delavelle J, Du Pasquier R, Spahr L. Chronic parkinsonism associated with cirrhosis: a distinct subset of acquired hepatocerebral degeneration. Arch Neurol 60: 521–528, 2003. doi: 10.1001/archneur.60.4.521. [DOI] [PubMed] [Google Scholar]
- 34.Brunberg JA, Kanal E, Hirsch W, Van Thiel DH. Chronic acquired hepatic failure: MR imaging of the brain at 1.5 T. AJNR Am J Neuroradiol 12: 909–914, 1991. [PMC free article] [PubMed] [Google Scholar]
- 35.Maeda H, Sato M, Yoshikawa A, Kimura M, Sonomura T, Terada M, Kishi K. Brain MR imaging in patients with hepatic cirrhosis: relationship between high intensity signal in basal ganglia on T1-weighted images and elemental concentrations in brain. Neuroradiology 39: 546–550, 1997. doi: 10.1007/s002340050464. [DOI] [PubMed] [Google Scholar]
- 36.Butterworth RF, Spahr L, Fontaine S, Layrargues GP. Manganese toxicity, dopaminergic dysfunction and hepatic encephalopathy. Metab Brain Dis 10: 259–267, 1995. doi: 10.1007/BF02109357. [DOI] [PubMed] [Google Scholar]
- 37.Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK, Mills PB. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet 90: 457–466, 2012. [Erratum in Am J Hum Genet 99: 521, 2016]. doi: 10.1016/j.ajhg.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun 7: 11601, 2016. doi: 10.1038/ncomms11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, Clayton PT. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia—a new metabolic disorder. J Inherit Metab Dis 31: 151–163, 2008. doi: 10.1007/s10545-008-0813-1. [DOI] [PubMed] [Google Scholar]
- 40.Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM Jr.. Pathology of inherited manganese transporter deficiency. Ann Neurol 75: 608–612, 2014. doi: 10.1002/ana.24131. [DOI] [PubMed] [Google Scholar]
- 41.Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 90: 467–477, 2012. doi: 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balachandran RC, Mukhopadhyay S, McBride D, Veevers J, Harrison FE, Aschner M, Haynes EN, Bowman AB. Brain manganese and the balance between essential roles and neurotoxicity. J Biol Chem 295: 6312–6329, 2020. doi: 10.1074/jbc.REV119.009453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem 292: 9760–9773, 2017. doi: 10.1074/jbc.M117.783605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci 34: 14079–14095, 2014. doi: 10.1523/JNEUROSCI.2329-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem 292: 16605–16615, 2017. doi: 10.1074/jbc.M117.804989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor CA, Hutchens S, Liu C, Jursa T, Shawlot W, Aschner M, Smith DR, Mukhopadhyay S. SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J Biol Chem 294: 1860–1876, 2019. doi: 10.1074/jbc.RA118.005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor CA, Tuschl K, Nicolai MM, Bornhorst J, Gubert P, Varão AM, Aschner M, Smith DR, Mukhopadhyay S. Maintaining translational relevance in animal models of manganese neurotoxicity. J Nutr 150: 1360–1369, 2020. doi: 10.1093/jn/nxaa066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zogzas CE, Aschner M, Mukhopadhyay S. Structural elements in the transmembrane and cytoplasmic domains of the metal transporter SLC30A10 are required for its manganese efflux activity. J Biol Chem 291: 15940–15957, 2016. doi: 10.1074/jbc.M116.726935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zogzas CE, Mukhopadhyay S. Putative metal binding site in the transmembrane domain of the manganese transporter SLC30A10 is different from that of related zinc transporters. Metallomics 10: 1053–1064, 2018. doi: 10.1039/c8mt00115d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercadante CJ, Prajapati M, Conboy HL, Dash ME, Herrera C, Pettiglio MA, Cintron-Rivera L, Salesky MA, Rao DB, Bartnikas TB. Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J Clin Invest 129: 5442–5461, 2019. doi: 10.1172/JCI129710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aydemir TB, Kim MH, Kim J, Colon-Perez LM, Banan G, Mareci TH, Febo M, Cousins RJ. Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J Neurosci 37: 5996–6006, 2017. doi: 10.1523/JNEUROSCI.0285-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aydemir TB, Thorn TL, Ruggiero CH, Pompilus M, Febo M, Cousins RJ. Intestine-specific deletion of metal transporter Zip14 (Slc39a14) causes brain manganese overload and locomotor defects of manganism. Am J Physiol Gastrointest Liver Physiol 318: G673–G681, 2020. [Erratum in Am J Physiol Gastrointest Liver Physiol 320: G557, 2021]. doi: 10.1152/ajpgi.00301.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jenkitkasemwong S, Akinyode A, Paulus E, Weiskirchen R, Hojyo S, Fukada T, Giraldo G, Schrier J, Garcia A, Janus C, Giasson B, Knutson MD. SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc Natl Acad Sci USA 115: E1769–E1778, 2018. [Erratum in Proc Natl Acad Sci USA 115: E4730, 2018]. doi: 10.1073/pnas.1720739115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheiber IF, Wu Y, Morgan SE, Zhao N. The intestinal metal transporter ZIP14 maintains systemic manganese homeostasis. J Biol Chem 294: 9147–9160, 2019. doi: 10.1074/jbc.RA119.008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xin Y, Gao H, Wang J, Qiang Y, Imam MU, Li Y, Wang J, Zhang R, Zhang H, Yu Y, Wang H, Luo H, Shi C, Xu Y, Hojyo S, Fukada T, Min J, Wang F. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov 3: 17025, 2017. doi: 10.1038/celldisc.2017.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C, Jursa T, Aschner M, Smith DR, Mukhopadhyay S. Up-regulation of the manganese transporter SLC30A10 by hypoxia-inducible factors defines a homeostatic response to manganese toxicity. Proc Natl Acad Sci USA 118: e2107673118, 2021. doi: 10.1073/pnas.2107673118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greenberg DM, Campbell WW. Studies in mineral metabolism with the aid of induced radioactive isotopes: IV-manganese. Proc Natl Acad Sci USA 26: 448–452, 1940. doi: 10.1073/pnas.26.7.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.von Oettingen WF. Manganese: its distribution, pharmacology and health hazards. Physiol Rev 15: 175–201, 1935. doi: 10.1152/physrev.1935.15.2.175. [DOI] [Google Scholar]
- 59.Reiman CK, Minot AS. A method for manganese quantitation in biological material together with data on the manganese content of human blood and tissues. J Biol Chem 42: 329–345, 1920. doi: 10.1016/S0021-9258(18)87150-2. [DOI] [Google Scholar]
- 60.Drinker CK, Shaw LA. Quantitative distribution of particulate material (manganese dioxide) administered intravenously to the cat. J Exp Med 33: 77–98, 1921. doi: 10.1084/jem.33.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lund CC, Shaw LA, Drinker CK. Quantitative distribution of particulate material (manganese dioxide) administered intravenously to the dog, rabbit, guinea pig, rat, chicken, and turtle. J Exp Med 33: 231–238, 1921. doi: 10.1084/jem.33.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drinker CK, Shaw LA, Drinker KR. The deposition and subsequent course of particulate material (manganese dioxide and manganese meta-silicate) administered intravenously to cats and to rabbits. J Exp Med 37: 829–850, 1923. doi: 10.1084/jem.37.6.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mella H. The experimental production of basal ganglion symptomatology in macacus rhesus. Arch NeurPsych 11: 405–417, 1924. doi: 10.1001/archneurpsyc.1924.02190340027003. [DOI] [Google Scholar]
- 64.Findlay G. The experimental production of biliary cirrhosis by salts of manganese. Br J Exp Pathol 5: 92–99, 1924. [Google Scholar]
- 65.Mena I, Marin O, Fuenzalida S, Cotzias GC. Chronic manganese poisoning. Clinical picture and manganese turnover. Neurology 17: 128–136, 1967. doi: 10.1212/wnl.17.2.128. [DOI] [PubMed] [Google Scholar]
- 66.Klaassen CD. Biliary excretion of manganese in rats, rabbits, and dogs. Toxicol Appl Pharmacol 29: 458–468, 1974. doi: 10.1016/0041-008x(74)90117-3. [DOI] [PubMed] [Google Scholar]
- 67.Miller ST, Cotzias GC, Evert HA. Control of tissue manganese: initial absence and sudden emergence of excretion in the neonatal mouse. Am J Physiol 229: 1080–1084, 1975. doi: 10.1152/ajplegacy.1975.229.4.1080. [DOI] [PubMed] [Google Scholar]
- 68.Ballatori N, Miles E, Clarkson TW. Homeostatic control of manganese excretion in the neonatal rat. Am J Physiol Regul Integr Comp Physiol 252: R842–R847, 1987. doi: 10.1152/ajpregu.1987.252.5.R842. [DOI] [PubMed] [Google Scholar]
- 69.Levy M, Elkoshi N, Barber-Zucker S, Hoch E, Zarivach R, Hershfinkel M, Sekler I. Zinc transporter 10 (ZnT10)-dependent extrusion of cellular Mn2+ is driven by an active Ca2+-coupled exchange. J Biol Chem 294: 5879–5889, 2019. doi: 10.1074/jbc.RA118.006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keen CL, Lönnerdal B, Clegg M, Hurley LS. Developmental changes in composition of rat milk: trace elements, minerals, protein, carbohydrate and fat. J Nutr 111: 226–236, 1981. [DOI] [PubMed] [Google Scholar]
- 71.Beaudin SA, Strupp BJ, Strawderman M, Smith DR. Early postnatal manganese exposure causes lasting impairment of selective and focused attention and arousal regulation in adult rats. Environ Health Perspect 125: 230–237, 2017. doi: 10.1289/EHP258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beaudin SA, Nisam S, Smith DR. Early life versus lifelong oral manganese exposure differently impairs skilled forelimb performance in adult rats. Neurotoxicol Teratol 38: 36–45, 2013. doi: 10.1016/j.ntt.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lasley SM, Fornal CA, Mandal S, Strupp BJ, Beaudin SA, Smith DR. Early postnatal manganese exposure reduces rat cortical and striatal biogenic amine activity in adulthood. Toxicol Sci 173: 144–155, 2019. doi: 10.1093/toxsci/kfz208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dorman DC, Struve MF, Wong BA, Dye JA, Robertson ID. Correlation of brain magnetic resonance imaging changes with pallidal manganese concentrations in rhesus monkeys following subchronic manganese inhalation. Toxicol Sci 92: 219–227, 2006. doi: 10.1093/toxsci/kfj209. [DOI] [PubMed] [Google Scholar]
- 75.Kalaitzakis E. Gastrointestinal dysfunction in liver cirrhosis. World J Gastroenterol 20: 14686–14695, 2014. doi: 10.3748/wjg.v20.i40.14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mukhopadhyay S. Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14. Neurotoxicology 64: 278–283, 2018. doi: 10.1016/j.neuro.2017.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anagianni S, Tuschl K. Genetic disorders of manganese metabolism. Curr Neurol Neurosci Rep 19: 33, 2019. doi: 10.1007/s11910-019-0942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jeong J, Eide DJ. The SLC39 family of zinc transporters. Mol Aspects Med 34: 612–619, 2013. doi: 10.1016/j.mam.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nam H, Wang CY, Zhang L, Zhang W, Hojyo S, Fukada T, Knutson MD. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica 98: 1049–1057, 2013. doi: 10.3324/haematol.2012.072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guthrie GJ, Aydemir TB, Troche C, Martin AB, Chang SM, Cousins RJ. Influence of ZIP14 (slc39A14) on intestinal zinc processing and barrier function. Am J Physiol Gastrointest Liver Physiol 308: G171–G178, 2015. doi: 10.1152/ajpgi.00021.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, Redl D, Qin W, Hampson S, Küry S, Tetreault M, Puffenberger EG, Scott JN, Bezieau S, Reis A, Uebe S, Schumacher J, Hegele RA, McLeod DR, Gálvez-Peralta M, Majewski J, Ramaekers VT; Care4Rare Canada Consortium, Nebert DW, Innes AM, Parboosingh JS, Abou Jamra R. Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am J Hum Genet 97: 886–893, 2015. doi: 10.1016/j.ajhg.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park JH, Hogrebe M, Grüneberg M, DuChesne I, von der Heiden AL, Reunert J, Schlingmann KP, Boycott KM, Beaulieu CL, Mhanni AA, Innes AM, Hörtnagel K, Biskup S, Gleixner EM, Kurlemann G, Fiedler B, Omran H, Rutsch F, Wada Y, Tsiakas K, Santer R, Nebert DW, Rust S, Marquardt T. SLC39A8 deficiency: a disorder of manganese transport and glycosylation. Am J Hum Genet 97: 894–903, 2015. doi: 10.1016/j.ajhg.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin W, Vann DR, Doulias PT, Wang T, Landesberg G, Li X, Ricciotti E, Scalia R, He M, Hand NJ, Rader DJ. Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J Clin Invest 127: 2407–2417, 2017. doi: 10.1172/JCI90896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiu YM, Claus Henn B, Hsu HL, Pendo MP, Coull BA, Austin C, Cagna G, Fedrighi C, Placidi D, Smith DR, Wright RO, Lucchini RG, Arora M. Sex differences in sensitivity to prenatal and early childhood manganese exposure on neuromotor function in adolescents. Environ Res 159: 458–465, 2017. doi: 10.1016/j.envres.2017.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chi L, Gao B, Bian X, Tu P, Ru H, Lu K. Manganese-induced sex-specific gut microbiome perturbations in C57BL/6 mice. Toxicol Appl Pharmacol 331: 142–153, 2017. doi: 10.1016/j.taap.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ghaisas S, Harischandra DS, Palanisamy B, Proctor A, Jin H, Dutta S, Sarkar S, Langley M, Zenitsky G, Anantharam V, Kanthasamy A, Phillips GJ, Kanthasamy A. Chronic manganese exposure and the enteric nervous system: an in vitro and mouse in vivo study. Environ Health Perspect 129: 87005, 2021. doi: 10.1289/EHP7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahmad TR, Higuchi S, Bertaggia E, Hung A, Shanmugarajah N, Guilz NC, Gamarra JR, Haeusler RA. Bile acid composition regulates the manganese transporter Slc30a10 in intestine. J Biol Chem 295: 12545–12558, 2020. doi: 10.1074/jbc.RA120.012792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Claro da Silva T, Hiller C, Gai Z, Kullak-Ublick GA. Vitamin D3 transactivates the zinc and manganese transporter SLC30A10 via the vitamin D receptor. J Steroid Biochem Mol Biol 163: 77–87, 2016. doi: 10.1016/j.jsbmb.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 89.Li S, De La Cruz J, Hutchens S, Mukhopadhyay S, Criss ZK, Aita R, Pellon-Cardenas O, Hur J, Soteropoulos P, Husain S, Dhawan P, Verlinden L, Carmeliet G, Fleet JC, Shroyer NF, Verzi MP, Christakos S. Analysis of 1,25-dihydroxyvitamin D3 genomic action reveals calcium-regulating and calcium-independent effects in mouse intestine and human enteroids. Mol Cell Biol 41: e00372-20, 2020. doi: 10.1128/MCB.00372-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wahlberg K, Kippler M, Alhamdow A, Rahman SM, Smith DR, Vahter M, Lucchini RG, Broberg K. Common polymorphisms in the solute carrier SLC30A10 are associated with blood manganese and neurological function. Toxicol Sci 149: 473–483, 2016. doi: 10.1093/toxsci/kfv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wahlberg KE, Guazzetti S, Pineda D, Larsson SC, Fedrighi C, Cagna G, Zoni S, Placidi D, Wright RO, Smith DR, Lucchini RG, Broberg K. Polymorphisms in manganese transporters SLC30A10 and SLC39A8 are associated with children’s neurodevelopment by influencing manganese homeostasis. Front Genet 9: 664, 2018. doi: 10.3389/fgene.2018.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rentschler G, Kippler M, Axmon A, Raqib R, Skerfving S, Vahter M, Broberg K. Cadmium concentrations in human blood and urine are associated with polymorphisms in zinc transporter genes. Metallomics 6: 885–891, 2014. doi: 10.1039/c3mt00365e. [DOI] [PubMed] [Google Scholar]
- 93.Wahlberg K, Arora M, Curtin A, Curtin P, Wright RO, Smith DR, Lucchini RG, Broberg K, Austin C. Polymorphisms in manganese transporters show developmental stage and sex specific associations with manganese concentrations in primary teeth. Neurotoxicology 64: 103–109, 2018. doi: 10.1016/j.neuro.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Broberg K, Taj T, Guazzetti S, Peli M, Cagna G, Pineda D, Placidi D, Wright RO, Smith DR, Lucchini RG, Wahlberg K. Manganese transporter genetics and sex modify the association between environmental manganese exposure and neurobehavioral outcomes in children. Environ Int 130: 104908, 2019. doi: 10.1016/j.envint.2019.104908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steimle BL, Smith FM, Kosman DJ. The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels. J Biol Chem 294: 19197–19208, 2019. doi: 10.1074/jbc.RA119.009371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Prajapati M, Conboy HL, Hojyo S, Fukada T, Budnik B, Bartnikas TB. Biliary excretion of excess iron in mice requires hepatocyte iron import by Slc39a14. J Biol Chem 297: 100835, 2021. doi: 10.1016/j.jbc.2021.100835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li D, Achkar JP, Haritunians T, Jacobs JP, Hui KY, D'Amato M, et al. A pleiotropic missense variant in SLC39A8 is associated with Crohn’s disease and human gut microbiome composition. Gastroenterology 151: 724–732, 2016. doi: 10.1053/j.gastro.2016.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]