

Keywords: aryl hydrocarbon receptor, colonic epithelial cells, IL22 signaling, SOCS3
Abstract
IL22 signaling plays an important role in maintaining gastrointestinal epithelial barrier function, cell proliferation, and protection of intestinal stem cells from genotoxicants. Emerging studies indicate that the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, promotes production of IL22 in gut immune cells. However, it remains to be determined if AhR signaling can also affect the responsiveness of colonic epithelial cells to IL22. Here, we show that IL22 treatment induces the phosphorylation of STAT3, inhibits colonic organoid growth, and promotes colonic cell proliferation in vivo. Notably, intestinal cell-specific AhR knockout (KO) reduces responsiveness to IL22 and compromises DNA damage response after exposure to carcinogen, in part due to the enhancement of suppressor of cytokine signaling 3 (SOCS3) expression. Deletion of SOCS3 increases levels of pSTAT3 in AhR KO organoids, and phenocopies the effects of IL22 treatment on wild-type (WT) organoid growth. In addition, pSTAT3 levels are inversely associated with increased azoxymethane/dextran sulfate sodium (AOM/DSS)-induced colon tumorigenesis in AhR KO mice. These findings indicate that AhR function is required for optimal IL22 signaling in colonic epithelial cells and provide rationale for targeting AhR as a means of reducing colon cancer risk.
NEW & NOTEWORTHY AhR is a key transcription factor controlling expression of IL22 in gut immune cells. In this study, we show for the first time that AhR signaling also regulates IL22 response in colonic epithelial cells by modulating SOCS3 expression.
INTRODUCTION
Interleukin (IL)-22 is a member of the IL-10 family of cytokines that regulate cell proliferation, host defense, inflammation, and tissue repair (1). Human IL22, encoded by the IL22 gene located on chromosome 12, contains 146 amino acids in its secreted form and signals through a heterodimeric receptor consisting of two different subunits IL22R1 and IL-10R2. IL22R1 is exclusively expressed in some subsets of tissues, such as skin, small intestine, liver, colon, lung, kidney, and pancreas, but not in immune cells, thymus, and spleen, whereas IL-10R2 is ubiquitously expressed (1, 2). Therefore, unlike most interleukins, IL22 signaling does not directly regulate the function of immune cells, but targets epithelial cells in the skin, gastrointestinal tract, respiratory system, and other organs (1, 2).
In the gastrointestinal tract, IL22 signaling plays an important role in maintaining gut-barrier function, resolution of inflammation, wound-associated regeneration, and genotoxin-induced DNA damage response (DDR) (3–6). Many different types of immune cells including innate lymphoid cells, natural killer (NK) T cells, ɣδ T cells, Th17 cells, neutrophils, and CD4+ T cells are capable of producing IL22 (4, 5, 7–11). In patients with inflammatory bowel disease (IBD), IL22 mRNA expression is upregulated in the colon, which is also observed in murine dextran sulfate sodium (DSS)-induced colitis (12, 13). Antagonism of IL22 signaling worsens clinical disease scores and wound recovery in DSS-treated mice (4, 13, 14), implying that IL22 signaling plays a protective role with regard to intestinal inflammation.
The IL22 receptor is heterogeneously expressed across different intestinal cell types, and slightly enriched in transit-amplifying (TA) progenitor cells in the small intestine (15). IL22 has a high binding affinity to the extracellular domain of IL22R1 (16). Upon binding to IL22R1, IL22 undergoes a conformational change which supports the interaction between the IL22-IL22R1 complex and IL-10R2. The intracellular moieties of IL22R1 and IL-10R2 form a complex with the kinases Janus kinase 1 (JNK1) and tyrosine kinase 2 (TYK2), respectively. Generation of the tripartite complex induces the phosphorylation of JAK1 and TYK2, which further phosphorylate IL22R1 in its cytoplasmic domain, thereby facilitating the docking of STAT3. Upon STAT3 recruitment to IL22R1, JAK1 then phosphorylates STAT3 predominately at Tyr705. The phosphorylation of STAT3 enables its dimerization and translocation into the nucleus, where it regulates the expression of downstream targets (1, 16). In addition, IL22 signaling can promote STAT1 and STAT5 phosphorylation (6, 17).
Emerging studies have shown that the aryl hydrocarbon receptor (AhR), a ligand-activated basic helix-loop-helix transcription factor, plays a pivotal role in IL22 production in immune cells. In the absence of ligands, the AhR is bound to several chaperone proteins in the cytoplasm, including Hsp90, p23, and XAP2. Upon ligand binding, AhR shuttles into the nucleus and heterodimerizes with AhR nuclear translocator (ARNT), affecting downstream gene expression. The major sources of naturally occurring AhR ligands include plant-derived dietary ingredients, such as polyphenolics and indole-containing glucobrassicin, gut microbiota, and host metabolites of tryptophan (18). It has been demonstrated that AhR can directly bind to the promoter of the IL22 gene in mouse CD4+ T cells and RORɣt(+) innate lymphoid cells (ILCs), thereby promoting IL22 transcription (19, 20). In addition, AhR signaling affects immune cell fate programming, favoring IL22-induced ILCs and FoxP3+ Treg cell differentiation (21, 22). Global AhR-deficient mice exhibit reduced IL22 expression and RORɣt(+) ILC3 development as well as increased intestinal Th17 cell numbers (22). In contrast, AhR activation by AhR ligand 6-formylindolo[3,2-b]carbazole (FICZ) treatment ameliorates 2,4,6-trinitrobenzene sulfonic acid (TNBS)-, dextran sulfate sodium (DSS)-, and T cell transfer-induced colitis in part by upregulating IL22 and downregulating the expression of proinflammatory cytokines, such as IFN-ɣ, IL-17α, and TNF-α (23). In addition, IL22 signaling is required for the effective initiation of DNA damage-induced repair, and thus maintenance of genome integrity in intestinal stem cells (ISCs) (5). However, the effect of AhR signaling on the response to IL22 in ISCs is not fully understood. To this end, we used intestinal epithelial cell-specific AhR KO mice to determine the response of colonic stem/progenitor cells to IL22 signaling. We demonstrate that AhR KO impaired the response of colonic stem cells to IL22 signaling by upregulating the expression of the suppressor of cytokine signaling 3 (SOCS3), a negative regulator of STAT3. These findings provide novel insight into the specific role of AhR signaling in mediating colonic epithelial cell responsiveness to IL22.
METHODS
Mice
Animals were housed under conventional conditions, adhering to the guidelines approved by the Institutional Animal Care and Use Committee at Texas A&M University. Lgr5-EGFP-IRES-CreERT2, CDX2P-CreERT2, and AhRf/f mouse strains have all been previously described. Specifically, Lgr5-EGFP-IRES-CreERT2 mice express EGFP and CreERT2 in crypt base columnar cells in the intestine under control of the Lgr5 promoter (24). CDX2P-CreERT2 mice express CreERT2 throughout the entire intestinal epithelium under the control of the CDX2 promoter (25, 26). AhRf/f mice carry loxP sites bordering exon 2 of the AhR gene, with recombination resulting in the generation of a premature stop codon at codon 29 (27). Mice were intraperitoneally injected with 2.5 mg of tamoxifen (Sigma, T5648) dissolved in corn oil (25 mg/mL) once per day for 4 consecutive days. Mice were maintained on an AIN-76A semipurified diet (Research Diets, D12450B), fed ad libitum and housed on a 12-h light-dark cycle. For all experiments, littermate controls were cohoused with the knockout mice, unless specifically indicated.
For genotyping analyses, DNA was extracted from tails using DNeasy Blood and Tissue Kit (Qiagen, 69506). PCR was subsequently performed using the following primer sets: AhR (5′- CAGTGGGAATAAGGCAAGAGTGA-3′ and 5′- GGTACAAGTGCACATGCCTGC-3′), CDX2P-Cre (5′- GGACTTGAGCAGCTAGCTGTGCAACTT-3′ and 5′- TGTCTCGTGCCTGGAATGACCTT-3′), and Lgr5-EGFP (5′- CACTGCATTCTAGTTGTGG-3′ and 5′- CGGTGCCCGCAGCGAG-3′). AhR recombination in vivo has been previously described (28).
Recombinant murine IL22 (BioLegend, 576206) was reconstituted in 3% BSA containing phosphate-buffered saline (PBS) at a concentration of 40 μg/mL. Mice were treated daily via intraperitoneal injection with either 100 μL of PBS or with 100 μL of PBS containing 4 μg IL22 as previously described (6). To induce DNA damage, mice were administrated a single subcutaneous injection of azoxymethane (AOM; 10 mg/kg body wt; Sigma A5486).
Crypt Single Cell Isolation and Cell Sorting
Colons were removed, washed with cold PBS without calcium and magnesium (PBS−/−), everted on a disposable mouse gavage needle (Instech Laboratories, Inc.), and incubated in 15 mM EDTA in PBS−/− at 37°C for 35 min as previously described (29). Subsequently, after transfer to chilled PBS−/−, crypts were mechanically separated from the connective tissue by vigorous vortexing. Crypts were embedded in Matrigel (Corning 356231) and overlaid with crypt culture media as previously described (29). For intestinal stem cell (ISC) isolation, crypt suspensions were dissociated to single cells with 0.25% trypsin-EDTA containing 200 U/mL DNase. Cell suspensions were then filtered through a 40-µm mesh and GFPhi and GFPlow-expressing cells were collected using a Bio-Rad S3e Cell Sorter. Dead cells were excluded by staining with propidium iodide or 7-aminoactinomycin D. Sorted cells were collected in RNA lysis buffer (for RNA isolation).
Organoid Culture
Mouse colonic organoid cultures were maintained in crypt culture medium, containing advanced DMEM/F12 (Gibco) supplemented with 2 mM GlutaMAX, penicillin-streptomycin, 10 mM HEPES, 50 ng/mL EGF (Life Technologies), 100 ng/mL Noggin (PeproTech), 10% R-spondin conditioned medium, 1 µM N-acetyl-l-cysteine (Sigma), 1X N2 (Life Technologies), 1X B27 (Life Technologies), and 50% Wnt-conditioned medium as described previously (30). Crypt media was changed every 2 days. Organoids were quantified on day 5 of culture, unless otherwise specified. For AhR-related treatments, DMSO or 2,3,7,8-tetrachlorodibenzodioxin (TCDD; 10 nM; gifted from Dr. Stephen Safe) were added to cultures for 3 days. In some experiments, colonic organoids were cultured in 50% WRN-conditioned medium derived from L-WRN cells (ATCC, CRL-3276), and 10% FBS (WRN medium; 31).
To measure organoid viability, CellTiter-Blue reagent (Promega G8081) was used according to manufacturer’s instructions. Fluorescence was measured on a CLARIOstar microplate reader. For experiments evaluating the effect of recombinant mouse IL22 (BioLegend, 576202) on mouse organoid growth in the absence or presence of AhR, 1 or 5 ng/mL IL22 was added to the culture medium after passaging. To probe phosphorylated STAT3 in response to IL22 treatment, organoids were harvested for protein extraction 30 min after the initiation of IL22 treatment. To measure organoid growth in response to IL22 treatment, organoids were cultured in medium containing IL22 for 2 or 5 days. Images of organoids were captured using an upright AZ100 Nikon microscope or all-in-one BZ-X800 Keyence fluorescence microscope.
Secondary Murine Organoid Assay
For secondary organoid assays, organoids were treated with PBS or 5 ng/mL IL22 for 2 days followed by dissociation for 8 min in 0.25% trypsin-EDTA at 37°C. Cell suspensions were filtered through a 20-µm mesh, centrifuged for 3 min at 500 g, and resuspended in cold ADF+ medium. Live cell density was counted. Approximately, 5,000 live cells were seeded into 30 µL Matrigel in flat-bottomed 24-well plates. After Matrigel polymerization, cells were overlaid with 300 µL of crypt culture medium supplemented with 10 µM Y-27632 (Sigma Y0503), 1 µM Jagged-1, and 2.5 µM CHIR99021 (Stemgent, 04-0004). Y-27632, Jagged-1, and CHIR99021 were withdrawn from crypt culture medium 2 days after plating. The crypt media was changed every 2 days and organoids were quantified on day 5 of culture, unless otherwise specified.
Gene Editing
Organoid lipofection was performed as previously described (32). Briefly, mouse organoids were dissociated into single or small cell clusters using 0.25% trypsin-EDTA at 37°C, filtered through a 20-μm cell strainer and washed twice with cold advanced DMEM/F12. Cells were then resuspended in 1.5 mL of crypt culture medium containing 10 μM Y-27632 and plated in a six-well plate at high density (80%–90% confluent). Nucleic acid-Lipofectamine 3000 complexes were prepared according to the standard Lipofectamine 3000 Reagent protocol. For this purpose, 7.5 μL of Lipofectamine 3000 Reagent (Life Technologies, L3000008) was diluted in 125 μL of Opti-MEM medium. pSpCas9(BB)-2A-GFP (10 μg) with gRNA targeting Socs3 ( TTCTACTGGAGCGCCGTGAC or CGAGCTGTCGCGGATAAGAA) was diluted into 125 μL of Opti-MEM medium and then mixed with 20 μL of P3000 Reagent. Diluted DNA was then added into diluted Lipofectamine 3000 Reagent, mixed well, incubated for 15 min at room temperature, and added to the cells (250 μL per well). Plates were centrifuged at 600 g at 32°C for 1 h and incubated for 4 h at 37°C before single cells were replated in Matrigel. Cells were subsequently collected, centrifuged, resuspended with Matrigel, and plated into the center of each well of prewarmed 24-well plates. Crypt culture medium supplemented with 1 µM Jagged-1, 5 μM CHIR99021, and 10 μM Y-27632 was added after cell-Matrigel suspension drops had solidified. Two days after lipofection, cells were dissociated into single cells using 0.25% trypsin-EDTA at 37°C and filtered through a 20-μm cell strainer. Dead cells were excluded by staining with 7-AAD. The top (highest) 30% green fluorescent protein (GFP)-expressing cells were collected using a Bio-Rad S3e Cell Sorter. Sorted GFP-expressing cells were mixed with an appropriate volume of Matrigel (∼30 cells per 30 µL of Matrigel) and seeded into the center of each well of prewarmed 24-well plates. Subsequently, 10–14 days after being plated, single organoids were picked and trypsinized into small cell clusters. Cells were resuspended with Matrigel and plated as described earlier.
RNA Isolation and Quantitative Real-Time PCR
RNA from sorted cells or organoids was isolated using the Quick-RNA Microprep Kit (Zymo, R1050) and further processed using a DNA removal kit (DNA Free, Ambion, AM1906). RNA integrity was assessed using a Bioanalyzer 2100 (Agilent Technologies), quantified by Nanodrop, and stored at −80°C. Real-time PCR was performed on a QuantStudio 3 System (Applied Biosystems) using TaqMan Universal PCR Master Mix (Applied Biosystems). Specific primer/probe mix for each gene was obtained from Applied Biosystems: Reg3b (Mm00440616_g1), Reg3g (Mm00441127_m1), Lgr5 (Mm00438890_m1), Ascl2 (Mm01268891_g1), Atm (Mm01177457_m1), p21 (Mm04205640_g1), Puma (Mm00519268_m1), Socs3 (Mm00545913_s1), and Gapdh (Mm99999915_g1).
Immunohistochemistry
To assess cell proliferation in the colon, mice were intraperitoneally injected with EdU 2 h before termination as previously described (33). Colonic cell proliferation was measured using the Click-IT EdU kit (Life Technologies, C10340). Antigen retrieval was performed by subboiling in 10 mmol/L sodium citrate (pH 6.0) for 20 min. Antibodies used include: rabbit polyclonal antibody to SOCS3 (Abcam, ab16030) and rabbit monoclonal antibody to pSTAT3 (Cell Signaling Technology, 9145S), followed by Alexa-568 donkey anti-rabbit secondary antibody (Life Technologies, A-10042). Prolong Gold antifade with DAPI (Life Technologies, P36935) was used to coverslip the slides. Images of colonic crypts were captured on a Leica DMi8 TCS SPE spectral confocal microscope. Images were processed using ImageJ software (ImageJ 1.51n version).
Western Blotting
Organoids were treated in a lysis buffer [50 mM Tris-HCl (pH = 7.2), 250 mM sucrose, 2 mM EDTA (pH = 7.6), 1 mM EGTA (pH = 7.5), 1% Triton X-100, and 10 mM β-mercaptoethanol] supplemented with protease inhibitor cocktail (Sigma) and 1× phosphatase inhibitor (Life Technologies). Lysates were subjected to standard SDS-PAGE and Western blotting procedures using primary antibodies against STAT3 (1:2,000, 4904S, Cell Signaling Technology), STAT3 (1:1,000, Abcam 119352), phospho STAT3 (Tyr705; 1:2,000, 9145S, Cell Signaling Technology), β-actin (1:5,000, Abcam ab8227), and SOCS3 (1:1,000, Abcam, ab16030). Secondary anti-mouse or rabbit conjugated to horseradish peroxidase or secondary StarBright Blue 700 goat anti-mouse IgG (Bio-Rad, no. 12004159) was used to detect primary antibodies. Signal was detected using enhanced chemiluminescence substrate (Bio-Rad), imaged with the Bio-Rad Chemidoc System and protein bands quantified using Image Lab 6.0 (Bio-Rad).
Statistics
Two-tailed Student’s t tests were used to assess statistical significance of the differences between means across experimental groups. Paired Student’s t tests were used to examine the statistical significance of the differences between treatments within genotypes in organoids, unless otherwise specified. One-way ANOVA with Tukey’s multiple-comparisons test were used to compare more than two groups. All data are presented as means ± SE (standard error), and all analyses were conducted using Prism 8 statistical software (GraphPad Software, Inc.).
RESULTS
IL22 Responsiveness Is Impaired in AhR KO Colonic Organoids
The IL22 receptor is heterogeneously expressed in the intestinal stroma and intestinal epithelial cells (6, 15, 34). Therefore, we initially determined the IL22 concentration-dependent effects on mouse colonic AhR wild-type (WT) and knockout (KO) organoids (100% AhR deletion), which have been characterized previously (28). IL22 treatment (5 ng/mL) robustly increased phosphorylation of STAT3 within 30 min in a dose-dependent manner both in AhR WT and KO organoids and the total protein level of STAT3 remained unchanged (Supplemental Fig. S1, A–C; see https://doi.org/10.6084/m9.figshare.16641667), indicating that IL22 signaling was functional in AhR WT and KO organoids. Next, we determined the kinetic response of AhR WT and KO organoids to 5 ng/mL IL22, which robustly induced pSTAT3, indicating that pSTAT3 levels were gradually decreased in a time-dependent manner (Supplemental Fig. S1, D–E). Interestingly, reduced pSTAT3 was observed in AhR KO organoids at 4, 8, and 24 h after IL22 treatment (Supplemental Fig. S1, D–E; see https://doi.org/10.6084/m9.figshare.16641667). Consistent with a previous study (6), we also found that IL22 induced phosphorylation of STAT1, but AhR KO had no effect on pSTAT1 (Supplemental Fig. S2; see https://doi.org/10.6084/m9.figshare.16683535).
Next, we determined the effect of AhR KO on ISC-dependent organoid growth in response to IL22 treatment. For this purpose, we incubated mouse colonic organoids with IL-22 (1 or 5 ng/mL) for 5 consecutive days as previously described (6). Organoids exceeding a diameter of 50 µm were quantified 5 days after plating based on previous study (28). As expected, AhR KO enhanced organoid growth relative to WT control (Fig. 1, A and B), since AhR deletion increases ISC proliferation (28). Interestingly, IL22 supplementation decreased the number of organoids and organoid size in a dose-dependent manner in the WT group, resulting in a decrease in total viable cells in organoids (Fig. 1, A–C). Increased cleaved caspase 3 and cell death was observed in WT organoids treated with IL22 after 5 days (Supplemental Fig. S3; see https://doi.org/10.6084/m9.figshare.16683667). In contrast, AhR KO abrogated the inhibitory effects of IL22 on organoid growth (Fig. 1, A–C). Immunoblot analysis also revealed that the phosphorylation of STAT3 was attenuated by AhR depletion 5 days after IL22 treatment, and the total protein level of STAT3 was not affected either by AhR status or IL22 treatment (Fig. 1, D–F). We also examined the expression level of STAT3 downstream targets, antimicrobial Reg3b and Reg3g, and found that IL22 treatment robustly induced the expression of Reg3b and Reg3g in WT organoids in a dose-dependent manner, and AhR KO reduced the effect of IL22 on Reg3b and Reg3g expression (Fig. 1, G and H). Since IL22 inhibited colonic organoid growth, we hypothesized that IL22 signaling had an impact on colonic stem cells. Interestingly, IL22 treatment suppressed the expression of colonic stem cell markers, Lgr5 and Ascl2 (Fig. 1, I and J), and decreased the growth of secondary organoids (Fig. 1K). This indicates that IL22 inhibited colonic stem cell expansion, whereas AhR KO reduced the effect of IL22 signaling in colonic stem cells (Fig. 1, I and J). Moreover, IL22 treatment decreased the expression of goblet cell marker Muc2 and enhanced the expression of absorptive enterocyte marker Slc26a3 (Supplemental Fig. S4; see https://doi.org/10.6084/m9.figshare.16683877). Collectively, these findings indicate that IL22 treatment suppressed colonic ISC-dependent organoid growth and AhR KO desensitized colonic organoid responsiveness to IL22 signaling.
Figure 1.

Loss of AhR attenuates the effects of IL22 signaling. A: representative brightfield images of colonic organoids treated with IL22 for 5 days. Scale bar 200 μm. B: colonic organoid diameter. Organoids with a diameter >50 μm were quantified. Data were analyzed by one-way ANOVA without Fisher’s LSD correction. C: organoid growth assessed by CellTiter Blue assay, n = 3 mice per group. D: representative immunoblots for pSTAT3 (Tyr705) and STAT3 in AhR WT and KO organoids treated with IL22 for 5 days, n = 3 mice. E and F: quantification of pSTAT3 (Tyr705) and STAT3 protein levels. Expression of antimicrobial peptides Reg3b (G) and Reg3g (H) and colonic stem cell markers, Lgr5 (I) and Ascl2 (J) in response to IL22 treatment, n = 3 mice. K: representative images of secondary organoid growth for WT colonic organoids pretreated with IL22 for 2 days. Scale bar 200 μm. *P < 0.05, **P < 0.01. AhR, aryl hydrocarbon receptor; KO, knockout; LSD, least significant difference; n.s, not significant; WT, wild type.
Loss of AhR Inhibits IL22-Induced Cell Proliferation in Vivo
Since previous studies reported that IL22 promotes colonic cell proliferation, for example, progenitor cell proliferation (6, 15, 35), we further investigated the link between IL22 signaling, AhR, and gastrointestinal (GI) cell proliferation. Lgr5-GFP-IRES-CreERT2 reporter mice were used to mark colonic stem (GFPhi) and progenitor (GFPlow) cells as previously described (28). To more broadly delete AhR in colonic epithelial cells, mice expressing the AhRf/f alleles were crossed with mice expressing a CreERT2 transgene driven by Lgr5 and CDX2 promoters. AhR KO efficiency has been characterized previously (28), with ∼85% KO efficiency in sorted GFP-expressing cells. Fourteen days after the final tamoxifen injection, mice were administrated an intraperitoneal PBS (control) or 4 μg IL22 per day for 2 consecutive days and cell proliferation was assessed in vivo. In this context, IL22 treatment induced the expression of Reg3b, which was decreased in AhR KO colonic stem and progenitor cells (Fig. 2, A and B). In addition, IL22 treatment dramatically increased cell proliferation in vivo, including stem cell proliferation in the AhR WT group to a level similar to AhR KO alone. However, IL22 did not further increase cell proliferation in AhR KO cells (Fig. 2, C–E). We also analyzed the number of goblet cells upon IL22 treatment, since IL22 can promote goblet cell hyperplasia (36). Surprisingly, the number of goblet cells was not affected by IL22 (Supplemental Fig. S5; see https://doi.org/10.6084/m9.figshare.16683949.v1).
Figure 2.

AhR KO suppresses IL22-induced colonic cell proliferation in vivo. Expression of Reg3b in sorted colonic stem cells (A) and sorted colonic progenitor cells (B), n = 6–8 mice per group. C: representative images of cell proliferation marked by EdU+ in the distal colon of n = 3 or 4 mice treated with IL22 or vehicle (control) for 2 days. GFP+ crypts were quantified; green, GFP; blue, DAPI; white, EdU. Scale bar 50 μm. Quantification of EdU+ cells (D) or EdU+ stem cells (E) per crypt. n = 64–102 from n = 3 or 4 mice. Data were analyzed using a one-way ANOVA with Tukey’s multiple-comparisons test in D and E. *P < 0.05, ***P < 0.001, ****P < 0.0001. AhR, aryl hydrocarbon receptor; GFP, green fluorescent protein; KO, knockout; WT, wild type.
Loss of AhR Suppresses Carcinogen-Induced DNA Damage Response
It has been recently demonstrated that IL22 signaling is required to initiate efficient DNA damage response after genotoxic stress, for example, AOM exposure, to maintain ISC genome integrity (5). Thus, we determined the effect of AhR KO on AOM-induced DNA damage response after IL22 treatment. Lgr5-GFP-IRES-CreERT2 reporter mice were administrated 4 μg IL22 twice before AOM injection and terminated 12 h after AOM injection (Fig. 3A). We found that AhR KO decreased the expression of p21 and Puma in sorted colonic GFPhi stem and GFPlow progenitor cells, compared with WT counterparts, but no effect on Atm expression (Fig. 3, B–G). In addition, we also examined the induction of γH2AX, which is required for assembly of DNA repair complex at DNA damage foci (37, 38), and cell apoptosis 12 h after AOM exposure in response to IL22 treatment. Consistently, AhR KO significantly decreased the number of γH2AX+ cells and γH2AX+ colonic stem cells per crypt, compared with WT mice (Fig. 3, H–J). However, AhR KO had no effect on AOM-induced apoptosis (cCaspase3+ cells per crypt and cCaspase3+ stem cells per crypt) (Fig. 3, K and L). Collectively, our findings suggest that AhR KO compromised the ability of colonic stem and progenitor cells to repair damaged cells, potentially leading to the accumulation of DNA lesions.
Figure 3.

AhR KO compromises IL22-mediated DNA damage response induced by AOM exposure. A: schematic illustration of experimental design. AhR WT or KO mice were injected intraperitoneally with 4 μg IL22 daily for 2 consecutive days. Expression of DNA damage response genes Atm (B), p21 (C), and Puma (D) in sorted colonic stem cells and in sorted colonic progenitor cells (E–G), n = 7 mice per group. H: ɣH2AX formation in distal colonic epithelial cells 12 h after AOM exposure. Insets are representative magnified GFP+ crypts. GFP+ crypts were quantified. Green, GFP; gray, DAPI; blue, ɣH2AX; red, cleaved caspase 3. Arrowhead indicates cCaspase 3 positive cells. n = 7 mice per group. Scale bar 50 μm. Quantification of ɣH2AX+ colon cells (I) and ɣH2AX+ colonic stem cells (J) per crypt, n = 107–144 from 7 mice per group. Quantification of cCaspase3+ colon cells (K) and Caspase3+ colonic stem cells (L) per crypt, n = 107–123 from 7 mice per group. *P < 0.05, **P < 0.01. AhR, aryl hydrocarbon receptor; AOM, azoxymethane; GFP, green fluorescent protein; KO, knockout; WT, wild type.
AhR KO Enhances the Expression of SOCS3
What is the molecular mechanism by which AhR KO suppresses the response of colonic epithelial cells to IL22? To address this question, we examined the expression of each member of the canonical IL22 signaling pathway based on RNAseq data from sorted colonic GFPhi stem and GFPlow progenitor cells isolated from WT and AhR KO mice in our previous study (28). These data revealed that Socs3 expression was upregulated in AhR KO mice, whereas the expression of other members remained unaltered (data not shown). qPCR was performed to confirm the expression of Socs3 expression in sorted colonic stem/progenitor cells. As expected, Socs3 expression was upregulated in AhR KO mouse colonic stem and progenitor cells, respectively (Fig. 4, A and B). In addition, AhR activation by TCDD decreased the expression of Socs3 in AhR WT organoids, whereas AhR KO organoids increased the expression of Socs3, and TCDD had no effect on Socs3 expression in AhR KO organoids (Fig. 4C). Effects on the protein level of SOCS3 were consistent with mRNA expression in organoids (Fig. 4, D and E). Immunostaining of SOCS3 revealed that SOCS3 was widely expressed in all colonic epithelial cells, and AhR KO upregulated SOCS3 expression in vivo (Fig. 4, F and G).
Figure 4.

SOCS3 expression is upregulated in AhR-deficient organoids. mRNA expression of Socs3 in sorted GFPhi colonic stem cells (A), sorted GFPlow colonic progenitor cells (B), and colonic organoids treated with DMSO or 10 nM TCDD (C), n = 4 mice per group. D: representative immunoblots for SOCS3 in AhR WT and KO colonic organoids treated with DMSO or TCDD, n = 3 mice per group. E: quantification of SOCS3 protein level. Data were analyzed using a paired Student’s t test within the same genotype. F: representative images of SOCS3 expression in colonic crypts. GFP+ crypts were quantified; magenta, SOCS3; green, GFP; blue, DAPI. Scale bar 50 μm. G: quantification of mean fluorescence intensity of SOCS3 per crypt, n = 3 mice per group. *P < 0.05, **P < 0.01, ****P < 0.0001. AhR, aryl hydrocarbon receptor; GFP, green fluorescent protein; KO, knockout; SOCS3, suppressor of cytokine signaling 3; TCDD, tetrachlorodibenzodioxin; WT, wild type.
Loss of SOCS3 Potentiates pSTAT3 Levels and Inhibits Organoid Growth
Next, we determined whether the enhanced expression of SOCS3 was attributed to diminished IL22 responsiveness in the AhR KO colonic cells. For this purpose, CRISPR-Cas9 was used to delete SOCS3 expression in AhR KO organoids. SOCS3 KO was validated by immunoblotting, and SOCS3 expression was also increased by IL22 treatment (Fig. 5A). SOCS3 KO dramatically enhanced the basal phosphorylation of STAT3 (without IL22 treatment), and additional IL22 treatment had no effect on pSTAT3 in SOCS3 KO and AhR KO (DKO) organoids (Fig. 5, A and B). SOCS3 KO or IL22 treatment did not affect total levels of STAT3 (Fig. 5B). Consistent with pSTAT3 levels in DKO organoids, SOCS3 KO increased the basal expression of Reg3b and Reg3g, and IL22 treatment slightly enhanced their expression in DKO organoids (Fig. 5, C and D). SOCS3 KO also inhibited colonic stem cell expansion, marked by decreased Lgr5 and Ascl2 expression (Fig. 5, E and F). We also assessed the effects of SOCS3 KO on organoid growth in the presence or absence of IL22 treatment. As expected, IL22 treatment decreased WT organoid growth, but had no obvious effect on AhR KO organoids (Fig. 5, G and H). Importantly, SOCS3 KO inhibited AhR KO organoid growth under basal conditions, including organoid number and size, and additional IL22 treatment did not exhibit notable changes in DKO organoids (Fig. 5, G and H). In addition, the passage efficiency of DKO organoids was very low, compared with AhR KO organoids. Overall, these findings underscore the fact that SOCS3 plays an important role in suppressing the phosphorylation of STAT3, and diminished responsiveness to IL22 in AhR KO organoids is at least partly attributed to increased SOCS3 expression.
Figure 5.

SOCS3 deletion increases basal pSTAT3 and reduces organoid growth. A: representative immunoblot for pSTAT3 at Tyr705, STAT3, and SOCS3. Colonic organoids were incubated with IL22 (5 ng/mL) for 2 days. DKO: AhR and SOCS3 double KO. B: quantification of pSTAT3 and total STAT3 protein levels, n = 3 from pooled independent experiments. mRNA expression of Reg3b (C), Reg3g (D), Lgr5 (E), and Ascl2 (F) in AhR KO organoids with or without SOCS3 treated with IL22 (5 ng/mL) for 2 days, n = 4 from 2 independent experiments. G: representative brightfield images of organoid growth 5 days after IL22 (5 ng/mL) treatment. Activated STAT3 reduced organoid growth. Scale bar 200 μm. H: quantification of organoid size. Organoids with a diameter >50 μm were quantified. Data were analyzed using a one-way ANOVA with Tukey’s multiple-comparisons test in C–F and H. *P < 0.05, **P < 0.01, ****P < 0.0001; n.s, not significant. AhR, aryl hydrocarbon receptor; DKO, double knockout; KO, knockout; SOCS3, suppressor of cytokine signaling 3; WT, wild type.
Decreased pSTAT3 Is Associated with Increased Colon Tumorigenesis
Since AhR deficiency compromised IL22-mediated DNA damage response in colonic epithelial cells and we previously demonstrated that intestinal-specific AhR KO (AhRf/f; Villin-Cre) promotes AOM/DSS-induced colon tumorigenesis (28), we tested our hypothesis that the decreased pSTAT3 level due to upregulated SOCS3 modulates colitis-associated colon tumorigenesis in intestinal-specific AhR KO mice. To this end, we analyzed the expression level of SOCS3 and a key mediator of IL22 signaling, pSTAT3, in uninvolved colonic crypts from AOM/DSS-treated mice. Consistently, AhR KO promoted SOCS3 expression both at the mRNA and protein level (Fig. 6, A–C) and decreased the amount of pSTAT3 in uninvolved colonic crypts (Fig. 6, D and E). Importantly, we previously showed that the expression of IL22 was not changed between AhR WT and KO mice after DSS treatment (28), indicating that the decreased pSTAT3 was unlikely to result from changes in IL22 production in intestinal-specific AhR KO mice. This was also corroborated by similar staining of pSTAT3 levels in nonepithelial cells in those mice. In comparison, the expression of antimicrobial peptides Reg3b and Reg3g was not significantly altered in uninvolved colonic crypts from AhR KO mice, compared with WT control (Fig. 6F). In addition, SOCS3 expression was comparable in colonic tumors between AhR WT and KO mice (Fig. 6, G–I). Interestingly, Socs3 expression and pSTAT3 were remarkably upregulated in tumor tissues compared with matched tumor uninvolved tissues (Fig. 6, J and K). However, the expression of Reg3b and Reg3g were decreased in tumor tissues (Fig. 6L), and thus, were not positively correlated with pSTAT3 level in colon following tumor progression. This is in contrast to IL22 administration in vitro and in vivo, and could be related to altered cell composition and programming in the tumor niche. For example, Paneth cells are highly responsive to IL22 signaling (34). Collectively, these findings indicate that AhR deletion decreased pSTAT3 by promoting SOCS3 expression, which was associated with carcinogen-mediated colon tumor initiation.
Figure 6.

Decreased pSTAT3 levels mediated by AhR KO is associated with the promotion of colon tumorigenesis. WT and intestinal-specific AhR KO (AhRf/f; Villin-Cre) mice were treated with a single dose of AOM (10 mg/kg) by subcutaneous injection, followed by three cycles of 2% DSS for 5 days. Mice were terminated 6 wk after the third cycle of DSS. A: relative Socs3 mRNA expression in uninvolved colonic mucosa, n = 10 mice per group. Immunohistochemistry (B) and quantification (C) of mean fluorescence intensity (MFI) of SOCS3 in uninvolved colonic crypts, n = 4 or 5 mice per group. Immunohistochemistry (D) and quantification (E) of MFI of pSTAT3 in uninvolved colonic crypts, n = 5–7 mice per group. F: relative Reg3b and Reg3g mRNA expression in uninvolved colonic crypts, n = 9 mice per group. G: relative Socs3 mRNA expression in colon tumors from WT and AhR KO mice, n = 3 or 4 mice per group. Immunohistochemistry (H) and quantification (I) of MFI of SOCS3 in colon tumors from WT and AhR KO mice, n = 15–21 fields of view from 2 to 3 mice per group. J: upregulated Socs3 mRNA in colon tumors relative to matched uninvolved mucosa, n = 11 mice. K: quantification of MFI of pSTAT3 between tumor uninvolved and tumor tissues, n = 16–22 fields of view from 5 mice per group. L: decreased Reg3b and Reg3g mRNA expression in colon tumors relative to matched uninvolved mucosa, n = 11 mice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. AhR, aryl hydrocarbon receptor; AOM, azoxymethane; DSS, dextran sulfate sodium; KO, knockout; SOCS3, suppressor of cytokine signaling 3; WT, wild type.
DISCUSSION
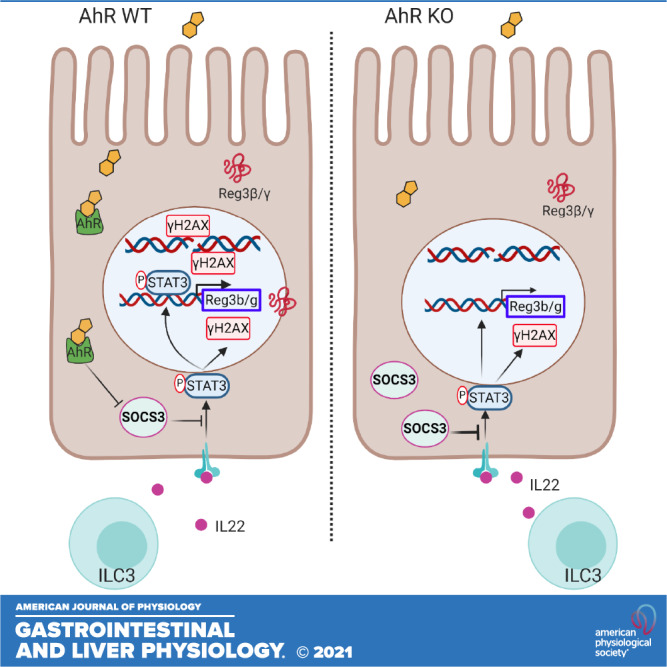
Previous studies have focused on the ability of AhR signaling to regulate IL22 production in intestinal immune cells, for example, group 3 innate lymphoid cells (5, 10, 20, 22). This supports the hypothesis that AhR signaling ensures the on-demand production of IL22 by immune cells, thereby indirectly affecting intestinal epithelial cells (IECs), the ultimate effector cells of IL22 signaling (39, 40). In this study, we provide evidence demonstrating that loss of mucosal epithelial AhR signaling leads to an impaired response of colonic stem/progenitor cells to IL22 signaling. The impaired response by IECs to IL22 was in part mediated via upregulation of SOCS3 expression, an important negative regulator of IL22 receptor-mediated STAT3 activation (41, 42). These data reveal a novel homeostatic control circuit by which AhR signaling regulates the response of colonic stem cells to IL22 (Fig. 7).
Figure 7.

Illustrative summary of IL22 signal modulation by AhR. IL22 is produced by gut immune cells, such as group 3 innate lymphoid cells (ILC3s), binds to the IL22 receptor in colonic stem/progenitor cells, and induces the phosphorylation of STAT3. pSTAT3 induces the expression of antimicrobial peptides Reg3β/γ. In addition, pSTAT3 enhances γH2AX levels to initiate DNA damage response. Increased expression of SOCS3 by AhR KO inhibits phosphorylation of STAT3 as a negative regulator, thereby attenuating IL22-pSTAT3-mediated effects. AhR, aryl hydrocarbon receptor; KO, knockout; SOCS3, suppressor of cytokine signaling 3; WT, wild type.
IL22 along with IL-10 play an important role in mitigating intestinal inflammation, promoting pathogen defense, and tissue regeneration. One of the hallmarks of IL22 signaling is to induce the expression of antimicrobial peptides, such as Reg3b, Reg3g, β-defensin, and serum amyloid A, to enable the host to defend against pathogen invasion (43, 44). Interestingly, intestinal-specific AhR KO mice exhibit increased susceptibility to Citrobacter rodentium infection (45), and IL22 plays a crucial role in the early phase of host defense against C. rodentium (46), which is consistent with our findings that intestinal-specific AhR KO suppresses IEC responsiveness to IL22 signaling. However, IL22 and Reg3g levels were elevated in intestinal-specific AhR KO mice (45), suggesting the contribution of a secondary response to impaired IL22 signaling due to increased bacterial loads in the host. In addition, Lindemans et al. (6) reported that IL22 directly promoted ISC expansion and epithelial regeneration after allogeneic bone marrow transplantation or graft-versus-host disease. They found that IL22 induced STAT3 phosphorylation in ISCs and promoted cell proliferation and organoid growth in vitro. Collectively, these reports suggest that IL22 is an intestinal stem cell growth factor (47). In contrast, our results in healthy mice suggest that in certain contexts, IL22 suppresses colonic stem cells and secondary organoid formation and inhibits organoid growth in a dose-dependent manner by increasing stem cell differentiation and cell apoptosis. Specifically, IL22 treatment favorably promoted absorptive enterocytes differentiation, and suppressed goblet cell differentiation ex vivo, even though IL22 treatment had no effect on the number of goblet cells in vivo. These apparent differences may be attributed to the use of organoids from distinct anatomical sites, for example, small intestine versus colon, duration and dose of IL22 treatment between in vitro and in vivo contexts, and the involvement of nonepithelial cells in vivo. Interestingly, recent studies indicate that IL22 downregulates Wnt and Notch signaling pathways, consequently decreasing intestinal stem cell pools (15, 35), even though IL22 signaling is not required for cell lineage commitment and turnover (34). IL22 signaling can also promote ER stress and induce intestinal epithelial cell apoptosis (48). Moreover, organoid culture conditions can also modify the effect of IL22 on organoid growth. For example, Zha et al. (35) found that IL22 promotes jejunal- and ileal-derived organoids in medium without Wnt3a, while suppressing jejunal derived organoid growth in medium containing Wnt3a, which was utilized to culture colonic organoids in our study. In addition, emerging studies indicate that IL22 does not promote ISC proliferation but spares progenitor cell proliferation (15, 35). However, we observed that IL22 treatment promotes ISC cell proliferation in vivo. Thus, it is possible that IL22 can affect the ISC niche in vivo, thereby indirectly promoting ISC cell proliferation. Additional investigation is required to address the apparent discrepancies with respect to IL22 signaling in intestinal epithelial cells.
IL22 was recently implicated in the initiation of the DDR induced by genotoxic stress, such as radiation and carcinogen (5). Since global AhR KO mice exhibit an increase in DNA adducts and colitis-associated colon tumorigenesis after exposure to carcinogen AOM (49), we hypothesized that this enhanced susceptibility may be explained by the impairment of IL22 signaling after the loss of AhR KO. Indeed, we found that intestinal-specific AhR KO desensitized the response of colonic epithelial cells to IL22 signaling, thereby leading to the undesirable accumulation of damaged cells due to defective DNA damage response. Moreover, AhR KO upregulated FoxM1 signaling to promote cell proliferation (28), which further promoted the propagation of DNA lesions following carcinogen. It is possible that the regulation of cell proliferation and DNA damage response by AhR are evolutionarily selected to safeguard colonic epithelial cells. Consequently, our laboratory and other groups showed that intestinal-specific AhR KO promoted carcinogen and colitis-associated colon tumor growth (28, 45, 50), even though the immune system was not affected. It is noteworthy, that loss of Apc function can also render intestinal epithelial cells resistant to IL22 signaling (51). Furthermore, Gronke et al. (5) found that serine/threonine kinase (ATM) was the main downstream target of IL22-pSTAT3 after radiation or carcinogen exposure. Interestingly, the IL22 receptor is heterogeneously expressed in the intestinal stroma and intestinal epithelial cells (6, 15, 34), and different cell types respond differently to IL22 treatment. For instance, IL22 signaling regulates Paneth cell maturation and antimicrobial effector function (34). These findings lay the foundation for future studies aiming to determine how AhR status, biological context, and cell type shape the response to IL22.
In summary, our study provides mechanistic insight into the AhR-SOCS3-pSTAT3 signaling axis in colonic Lgr5+ stem cells (Fig. 7). SOCS3 constitutes one of the most important negative feedback pathways to regulate IL22 signaling strength and duration. SOCS3 itself is a STAT3 downstream target, and thus provides transcription-dependent negative feedback regulation. Consistent with previous studies showing that increased pSTAT3 levels are detected in the colonic epithelium of intestinal cell-specific SOCS3 KO mice (52), whereas SOCS3 overexpression decreased pSTAT3 (53, 54), we found that loss of SOCS3 increased pSTAT3 levels even in the absence of exogenous IL22 in AhR KO organoids. In addition, restoring pSTAT3 levels in AhR KO organoids by deleting SOCS3 rescued AhR KO-mediated organoid growth. The regulation of the AhR-SOCS3-pSTAT3 axis has also been identified in other tissues, such as liver and kidney (55, 56). Interestingly, SOCS3 is a direct transcriptional target of AhR in the liver (56), where AhR activation increased the expression of SOCS3, in contrast with our findings. This suggests that AhR-SOCS3 regulation is cell context dependent. In addition to inhibition of pSTAT3, SOCS3 is also involved in other signaling pathways, including inhibition of pSTAT1, degradation of indoleamine dioxygenase, NF-κB, insulin receptor, insulin receptor substrate-1, and inhibition of Smad3 nucleus translocation (42, 57). It is, therefore, possible that AhR signaling could affect multiple SOCS3-regulated signaling pathways. In addition to IL22, several other cytokines can induce the phosphorylation of STAT3, including IL-6, IL-11, Glycoprotein 130, IL-27, and leukemia inhibitory factor (LIF) (42). Whether defective AhR signaling decreases the response of colonic epithelial cells to these cytokines via the upregulation of SOCS3 expression remains to be determined. This is noteworthy because AhR signaling is compromised in individuals with inflammatory bowel diseases (IBD), for example, decreased mucosal AhR expression level and its gut microbiota-derived ligands in blood and feces have been reported (23, 58). In addition, an array of cytokines including IL-6, IL-10, IL-17, IL22, and TNF-α, are elevated in subjects with IBD (59). In conclusion, data from our preclinical orthogonal models indicate that AhR signaling modulates the response of colonic epithelial cells to IL22, resulting in the suppression of carcinogen-associated colon tumorigenesis. These findings provide rationale for targeting AhR as a means of ameliorating IBD and reducing colon cancer risk.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16641667;
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.16683535;
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.16683667;
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.16683877;
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.16683949.v1.
GRANTS
Funding was provided by Texas AgriLife Research, the Sid Kyle Chair Endowment, the Allen Endowed Chair in Nutrition & Chronic Disease Prevention, the Cancer Prevention Research Institute of Texas (RP160589), and the National Institutes of Health (R01-ES025713, R01-CA202697, and R35-CA197707).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.H. and R.S.C. conceived and designed research; H.H., Y.-Y.F., and K.K.L. performed experiments; H.H. analyzed data; H.H. and R.S.C. interpreted results of experiments; H.H. prepared figures; H.H. and R.S.C. drafted manuscript; H.H., L.A.D., A.J., S.H.S., and R.S.C. edited and revised manuscript; H.H., S.H.S., and R.S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Hans Clevers and Eric Fearon for providing the Lgr5CreERT2 reporter and CDX2PCreERT2 mice, respectively. The illustrative figure was created with BioRender.com.
REFERENCES
- 1.Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov 13: 21–38, 2014. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 2.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity 21: 241–254, 2004. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol 53: 465–474, 2018. doi: 10.1007/s00535-017-1401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zindl CL, Lai J-F, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. IL-22–producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA 110: 12768–12773, 2013. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gronke K, Hernández PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, Witkowski M, Tizian C, Amann L, Schumacher F, Glatt H, Triantafyllopoulou A, Diefenbach A. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566: 249–253, 2019. doi: 10.1038/s41586-019-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu YY, Takashima S, Hua G, Martin ML, O'Rourke KP, Lo YH, Mokry M, Romera-Hernandez M, Cupedo T, Dow LE, Nieuwenhuis EE, Shroyer NF, Liu C, Kolesnick R, van den Brink MR, Hanash AM. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528: 560–564, 2015. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity 31: 15–23, 2009. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, Tardif MR, Sathaliyawala T, Kubota M, Farber DL, Collman RG, Shaked A, Fouser LA, Weiner DB, Tessier PA, Friedman JR, Kiyono H, Bushman FD, Chang KM, Artis D. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336: 1321–1325, 2012. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol 30: 447–454, 2009. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol 13: 144–151, 2011. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, Bracken AP, Mills KHG. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med 210: 1117–1124, 2013. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte J-M, Diepolder H, Marquardt A, Jagla W, Popp A, Leclair S, Herrmann K, Seiderer J, Ochsenkühn T, Göke B, Auernhammer CJ, Dambacher J. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 290: G827–G838, 2006. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 13.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118: 534–544, 2008. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29: 947–957, 2008. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zwarycz B, Gracz AD, Rivera KR, Williamson IA, Samsa LA, Starmer J, Daniele MA, Salter-Cid L, Zhao Q, Magness ST. IL22 Inhibits Epithelial Stem Cell Expansion in an Ileal Organoid Model. Cell Mol Gastroenterol Hepatol 7: 1–17, 2019. doi: 10.1016/j.jcmgh.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudakov JA, Hanash AM, van den Brink MR. . Interleukin-22: immunobiology and pathology. Annu Rev Immunol 33: 747–785, 2015. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld J-C. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line: pathways that are shared with and distinct from IL-10. J Biol Chem 277: 33676–33682, 2002. doi: 10.1074/jbc.M204204200. [DOI] [PubMed] [Google Scholar]
- 18.Han H, Jayaraman A, Safe S, Chapkin RS. Targeting the aryl hydrocarbon receptor in stem cells to improve the use of food as medicine. Curr Stem Cell Rep 6: 109–118, 2020. doi: 10.1007/s40778-020-00184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah A-M, Santiago A, Wu C, Patel B, Kumar D, Quintana FJ. IL-21 induces IL-22 production in CD4+ T cells. Nat Commun 5: 3753, 2014. doi: 10.1038/ncomms4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36: 92–104, 2012. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453: 65–71, 2008. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 22.Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, Fu YX, Zhou L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39: 386–399, 2013. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, MacDonald TT, Pallone F, Monteleone G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 141: P237–P248.e1, 2011. doi: 10.1053/j.gastro.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007, 2007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 25.Hardiman KM, Liu J, Feng Y, Greenson JK, Fearon ER. Rapamycin inhibition of polyposis and progression to dysplasia in a mouse model. PLoS One 9: e96023, 2014. doi: 10.1371/journal.pone.0096023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, Cho KR, Fearon ER. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am J Pathol 183: 493–503, 2013. doi: 10.1016/j.ajpath.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci USA 102: 17858–17863, 2005. doi: 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han H, Davidson LA, Fan Y-Y, Goldsby JS, Yoon G, Jin U-H, Wright GA, Landrock KK, Weeks BR, Wright RC, Allred CD, Jayaraman A, Ivanov I, Roper J, Safe SH, Chapkin RS. Loss of aryl hydrocarbon receptor potentiates FoxM1 signaling to enhance self-renewal of colonic stem and progenitor cells. EMBO J 39: e104319, 2020., doi: 10.15252/embj.2019104319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan YY, Davidson LA, Chapkin RS. Murine colonic organoid culture system and downstream assay applications. Methods Mol Biol 1576: 171–181, 2016. doi: 10.1007/7651_2016_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265, 2009. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 31.Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 8: 2471–2482, 2013. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13: 653–658, 2013. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Turk HF, Monk JM, Fan YY, Callaway ES, Weeks B, Chapkin RS. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing. Am J Physiol Cell Physiol 304: C905–C917, 2013. doi: 10.1152/ajpcell.00379.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaudino SJ, Beaupre M, Lin X, Joshi P, Rathi S, McLaughlin PA, Kempen C, Mehta N, Eskiocak O, Yueh B, Blumberg RS, van der Velden AWM, Shroyer KR, Bialkowska AB, Beyaz S, Kumar P. IL-22 receptor signaling in Paneth cells is critical for their maturation, microbiota colonization, Th17-related immune responses, and anti-Salmonella immunity. Mucosal Immunol 14: 389–401, 2021. doi: 10.1038/s41385-020-00348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zha J-M, Li H-S, Lin Q, Kuo W-T, Jiang Z-H, Tsai P-Y, Ding N, Wu J, Xu S-F, Wang Y-T, Pan J, Zhou X-M, Chen K, Tao M, Odenwald MA, Tamura A, Tsukita S, Turner JR, He W-Q. Interleukin 22 expands transit-amplifying cells while depleting Lgr5+ stem cells via inhibition of Wnt and notch signaling. Cell Mol Gastroenterol Hepatol 7: 255–274, 2019. doi: 10.1016/j.jcmgh.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turner J-E, Stockinger B, Helmby H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog 9: e1003698, 2013. doi: 10.1371/journal.ppat.1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mah LJ, El-Osta A, Karagiannis TC. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24: 679–686, 2010. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 38.Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids 2010: 920161, 2010. doi: 10.4061/2010/920161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zenewicz LA. IL-22: there is a gap in our knowledge. ImmunoHorizons 2: 198–207, 2018. doi: 10.4049/immunohorizons.1800006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perusina Lanfranca M, Lin Y, Fang J, Zou W, Frankel T. Biological and pathological activities of interleukin-22. J Mol Med (Berl) 94: 523–534, 2016. doi: 10.1007/s00109-016-1391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer 19: 82–96, 2019. doi: 10.1038/s41568-018-0090-8. [DOI] [PubMed] [Google Scholar]
- 42.Carow B, Rottenberg ME. SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol 5: 58–58, 2014. doi: 10.3389/fimmu.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moyat M, Bouzourene H, Ouyang W, Iovanna J, Renauld JC, Velin D. IL-22-induced antimicrobial peptides are key determinants of mucosal vaccine-induced protection against H. pylori in mice. Mucosal Immunol 10: 271–281, 2017. doi: 10.1038/mi.2016.38. [DOI] [PubMed] [Google Scholar]
- 44.Mulcahy ME, Leech JM, Renauld JC, Mills KH, McLoughlin RM. Interleukin-22 regulates antimicrobial peptide expression and keratinocyte differentiation to control Staphylococcus aureus colonization of the nasal mucosa. Mucosal Immunol 9: 1429–1441, 2016. doi: 10.1038/mi.2016.24. [DOI] [PubMed] [Google Scholar]
- 45.Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E, Li V, Maradana MR, Schiering C, Stockinger B. The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49: 353–362.e5, 2018. doi: 10.1016/j.immuni.2019.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14: 282–289, 2008. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 47.Lindemans CA, Mertelsmann A, O'Connor MH, Dudakov JA, Jenq R, Velardi E, Young LF, Smith OM, Lawrence G, Luo N, Ivanov J, Hua G, Martin ML, Liu C, Kolesnick R, Van Den Brink M, Hanash AM. IL-22 is an intestinal stem cell growth factor, and IL-22 administration in vivo reduces morbidity and mortality in murine GvHD. Blood 124: 651–651, 2014. doi: 10.1182/blood.V124.21.651.651. [DOI] [Google Scholar]
- 48.Powell N, Pantazi E, Pavlidis P, Tsakmaki A, Li K, Yang F, Parker A, Pin C, Cozzetto D, Minns D, Stolarczyk E, Saveljeva S, Mohamed R, Lavender P, Afzali B, Digby-Bell J, Tjir-Li T, Kaser A, Friedman J, MacDonald TT, Bewick GA, Lord GM. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut 69: 578–590, 2020. doi: 10.1136/gutjnl-2019-318483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diaz-Diaz CJ, Ronnekleiv-Kelly Sm Nukaya M, Geiger PG, Balbo S, Dator R, Megna BW, Carney PR, Bradfield CA, Kennedy GD. The Aryl hydrocarbon receptor is a repressor of inflammation-associated colorectal tumorigenesis in mouse. Ann Surg 264: 429–436, 2016. doi: 10.1097/SLA.0000000000001874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Villatoro EL, DeLuca JAA, Callaway ES, Allred KF, Davidson LA, Hensel ME, Menon R, Ivanov I, Safe SH, Jayaraman A, Chapkin RS, Allred CD. Effects of high-fat diet and intestinal aryl hydrocarbon receptor deletion on colon carcinogenesis. Am J Physiol Gastrointest Liver Physiol 318: G451–G463, 2020. doi: 10.1152/ajpgi.00268.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, Vandereyken M, Newton IP, Moraga I, Näthke IS, Swamy M. Loss of adenomatous polyposis coli function renders intestinal epithelial cells resistant to the cytokine IL-22. PLoS Biol 17: e3000540, 2019. doi: 10.1371/journal.pbio.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene 26: 4833–4841, 2007. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- 53.Ortiz-Muñoz G, Lopez-Parra V, Lopez-Franco O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J, Mezzano S, Ortiz A, Egido J, Gomez-Guerrero C. Suppressors of cytokine signaling abrogate diabetic nephropathy. J Am Soc Nephrol 21: 763–772, 2010. doi: 10.1681/ASN.2009060625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwahori K, Serada S, Fujimoto M, Nomura S, Osaki T, Lee CM, Mizuguchi H, Takahashi T, Ripley B, Okumura M, Kawase I, Kishimoto T, Naka T. Overexpression of SOCS3 exhibits preclinical antitumor activity against malignant pleural mesothelioma. Int J Cancer 129: 1005–1005, 2011. doi: 10.1002/ijc.25716. [DOI] [PubMed] [Google Scholar]
- 55.Tsai C-H, Lee Y, Li C-H, Cheng Y-W, Kang J-J. Down-regulation of aryl hydrocarbon receptor intensifies carcinogen-induced retinal lesion via SOCS3-STAT3 signaling. Cell Biol Toxicol 36: 223–242, 2020. doi: 10.1007/s10565-019-09499-z. [DOI] [PubMed] [Google Scholar]
- 56.Wada T, Sunaga H, Miyata K, Shirasaki H, Uchiyama Y, Shimba S. Aryl hydrocarbon receptor plays protective roles against high fat diet (HFD)-induced hepatic steatosis and the subsequent lipotoxicity via direct transcriptional regulation of Socs3 gene expression. J Biol Chem 291: 7004–7016, 2016. doi: 10.1074/jbc.M115.693655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sood V, Lata S, Ramachandran VG, Banerjea AC. Suppressor of cytokine signaling 3 (SOCS3) degrades p65 and regulate HIV-1 replication. Front Microbiol 10: 114, 2019. doi: 10.3389/fmicb.2019.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Larmurier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay JM, Langella P, Xavier RJ, Sokol H. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 22: 598–605, 2016. doi: 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140: 1756–1767, 2011. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16641667;
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.16683535;
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.16683667;
Supplemental Fig. S4: https://doi.org/10.6084/m9.figshare.16683877;
Supplemental Fig. S5: https://doi.org/10.6084/m9.figshare.16683949.v1.