

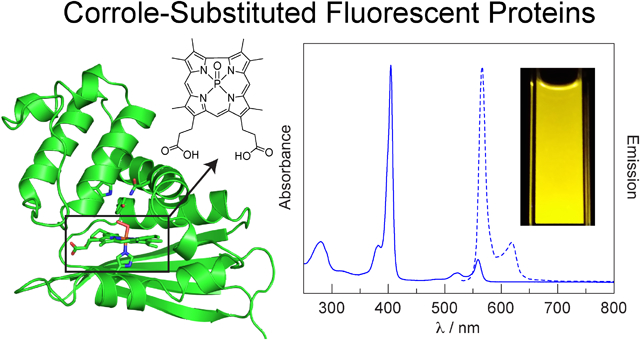
Abstract
Although fluorescent proteins have been utilized for a variety of biological applications, they have several optical limitations, namely weak red and NIR emission and exceptionally broad (> 200 nm) emission profiles. The photophysical properties of fluorescent proteins can be enhanced through the incorporation of novel cofactors with the desired properties into a stable protein scaffold. To this end, a fluorescent phosphorus corrole that is structurally similar to the native heme cofactor is incorporated into two exceptionally stable heme proteins: H-NOX from Caldanaerobacter subterraneus and HasA from Pseudomonas aeruginosa. These yellow-orange emitting protein conjugates are examined by steady-state and time-resolved optical spectroscopy. The HasA conjugate exhibits enhanced fluorescence, whereas emission from the H-NOX conjugate is quenched relative to the free corrole. Despite the low fluorescence quantum yields, these corrole-substituted proteins exhibit more intense fluorescence in a narrower spectral profile than traditional fluorescent proteins that emit in the same spectral window. This study demonstrates that fluorescent corrole complexes are readily incorporated into heme proteins and provides an inroad for the development of novel fluorescent proteins.
Graphical Abstract
Introduction
Since the discovery of green fluorescent protein (GFP)1 and its subsequent demonstration as a fluorescent tag for in vivo labeling,2 the field of fluorescent proteins has greatly expanded. Significant effort has been dedicated to tune the optical properties of fluorescent proteins, spanning UV, visible, and NIR wavelengths. Moreover, fluorescent proteins have been utilized for a variety of applications as sensitizers, labels, and sensors.3 Despite advances to extend the emission of fluorescent proteins into the red and NIR, the emission intensity of these proteins is quite weak, exhibiting quantum yields (ϕ) < 20%.4 Since tissue is transparent to red and NIR light in the 600–1100 nm range (i.e., the “tissue transparency window”),5 the weak emission of traditional fluorescent proteins in this spectral range greatly limits in vivo utility. Indeed, the quantum yields of traditional red fluorescent proteins are significantly lower than typical GFPs and other fluorescent proteins that emit outside the tissue transparency window. Another limitation of traditional fluorescent proteins is their broad emission profiles, which can span over 200 nm. This limits the utility of fluorescent proteins for ratiometric sensing applications. In this sensing modality, two fluorophores are incorporated into the sensor and the analyte is quantified by measuring the emission intensity ratio of the two components. This is a robust sensing methodology that is independent of sensor concentration and is functional in scattering media, such as the biological milieu.6–8
One strategy to improve upon the optical properties of traditional fluorescent proteins is to incorporate a highly fluorescent cofactor into a stable protein scaffold. One example of such a scaffold is the heme nitric oxide/oxygen binding (H-NOX) protein from the thermophilic bacterium Caldanaerobacter subterraneus (Cs) (Figure 1a). H-NOX proteins are a family of bacterial gas sensing proteins that are homologous to the heme domain of the mammalian nitric oxide receptor soluble guanylate cyclase (sGC).9–15 They have a buried, hydrophobic heme pocket comprised of a heme-ligating histidine residue and a conserved Y–S–R motif that hydrogen bonds with the propionate chains of the heme cofactor. In oxygen sensing H-NOXs, such as Cs H-NOX, the protein also contains a distal hydrogen bonding network that stabilizes the O2 adduct. The iron center of the heme cofactor binds diatomic gases, such as NO or O2, resulting in a conformational change of the protein. This triggers a signal cascade that culminates in a physiological response to the gaseous signal, such as biofilm formation or flagellar motion.15 The robust Cs H-NOX protein is readily expressed in E. coli, variants are easily made, and it readily binds unnatural heme cofactors. Together, these properties render Cs H-NOX an ideal platform to construct novel sensing and imaging agents.16 To this end, Cs H-NOX has been tailored for a variety of applications, including NO sensing,17 O2 sensing,18 magnetic resonance imaging (MRI),19 and fluorescent labeling.20
Figure 1.
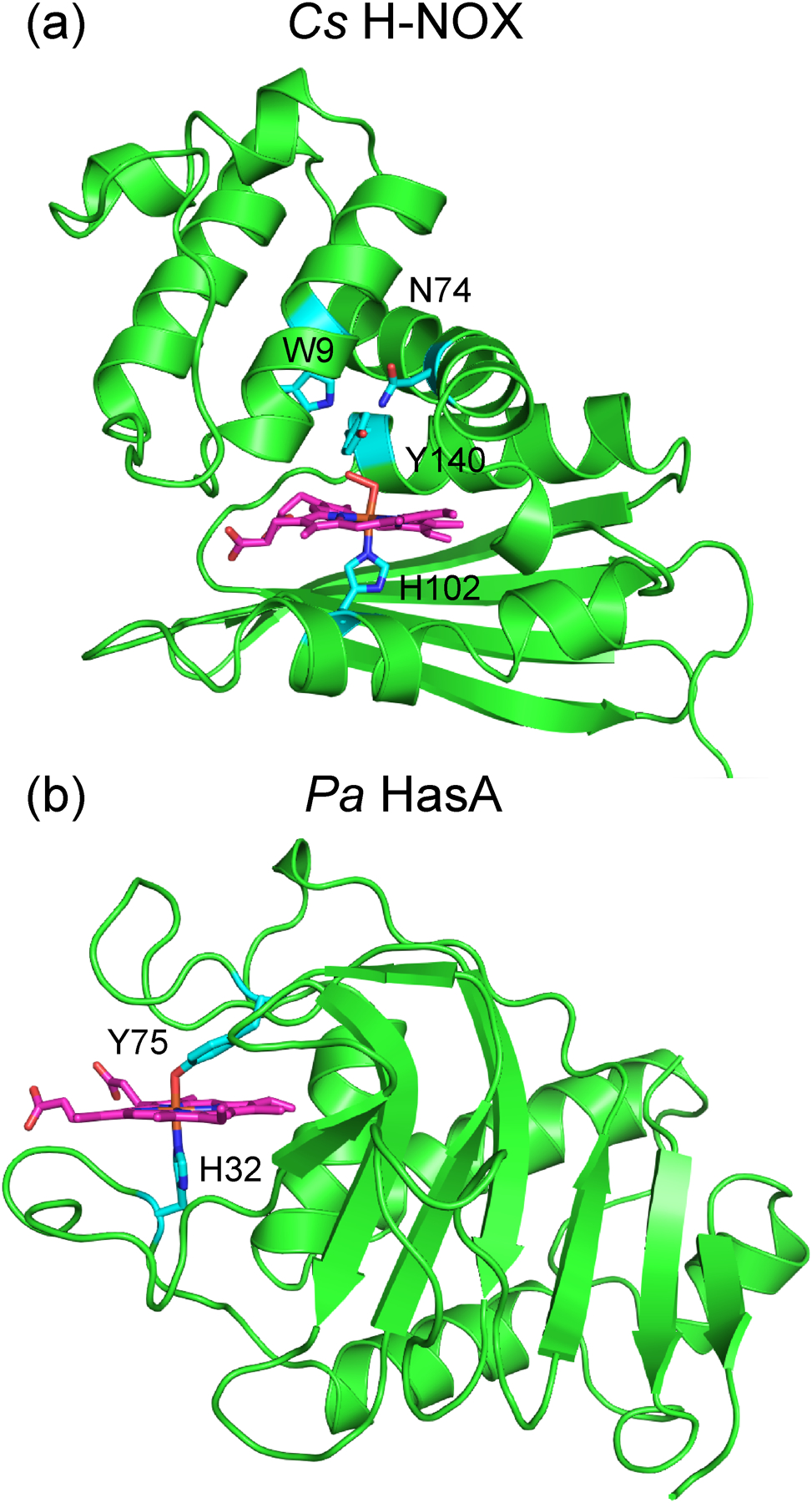
(a) X-ray crystal structure of the Fe(II)–O2 complex of Cs H-NOX (PDB ID: 3TF0), depicting the heme-ligating histidine (H102 in cyan) and the hydrogen bonding network that stabilizes the O2 adduct (W9, Y140, and N74 in cyan). (b) X-ray crystal structure of Pa HasA (PDB ID: 3ELL), illustrating the heme-ligating residues (Y75 and H32 in cyan).
Another stable protein scaffold is the heme acquisition system protein A (HasA) from the pathogenic bacterium Pseudomonas aeruginosa (Pa) (Figure 1b). Under iron-depleted stress conditions, these bacteria secrete HasA to scavenge heme from the host and subsequently import it through a membrane receptor HasR.21–24 Unlike H-NOX proteins, HasA has a surface-exposed heme binding pocket. The secreted apoprotein exhibits an extended conformation; two flexible loops sequester the heme cofactor, leading to high affinity binding (Ka = 5.3 × 1010 M−1)25 and a large conformational change.26 The bound heme is ligated by a tyrosine and a histidine residue, but it has been demonstrated that either one of these two residues alone is sufficient to bind heme.27,28 In addition to the native heme cofactor, Pa HasA can also bind variety of β-alkyl and meso-aryl iron porphyrins, as well as related iron phthalocyanine and salophen complexes.29,30 It has also been demonstrated that HasA can bind gallium complexes of protoporphyrin IX31,32 and phthalocyanine.33
Since both H-NOX and HasA bind artificial cofactors, a strategy to incorporate fluorescent analogs of protoporphyrin IX (Chart 1) was designed to serve as an alternative to traditional fluorescent proteins. Although emissive in the red region of the spectrum, free-base and metalloporphyrins are not exceptionally bright fluorophores. For example, Zn(II) porphyrin complexes exhibit ϕ = 4–5%,34 which is not an improvement over extant fluorescent proteins. Corrole is a closely related tetrapyrrole macrocycle with a contracted 23-atom core due to the presence of a direct pyrrole–pyrrole linkage.35 As a result, corrole is a trianionic ligand when fully deprotonated, rather than dianionic like porphyrin, thereby stabilizing different metal complexes and accessing different molecular properties.36,37 While corroles have not been studied as extensively as their porphyrin congeners, it has been demonstrated that they exhibit optical properties superior to related porphyrin complexes.38 In general, corroles have higher fluorescent quantum yields than related porphyrin complexes.39,40 Specifically, light main group corrole complexes of gallium,39,41 phosphorus,42–44 aluminum,40,41,45,46 and silicon47,48 are highly fluorescent and have been utilized for biological imaging applications.43,49–51 Therefore, main group corrole complexes are ideal heme analogs for fluorescent protein development.
Chart 1.
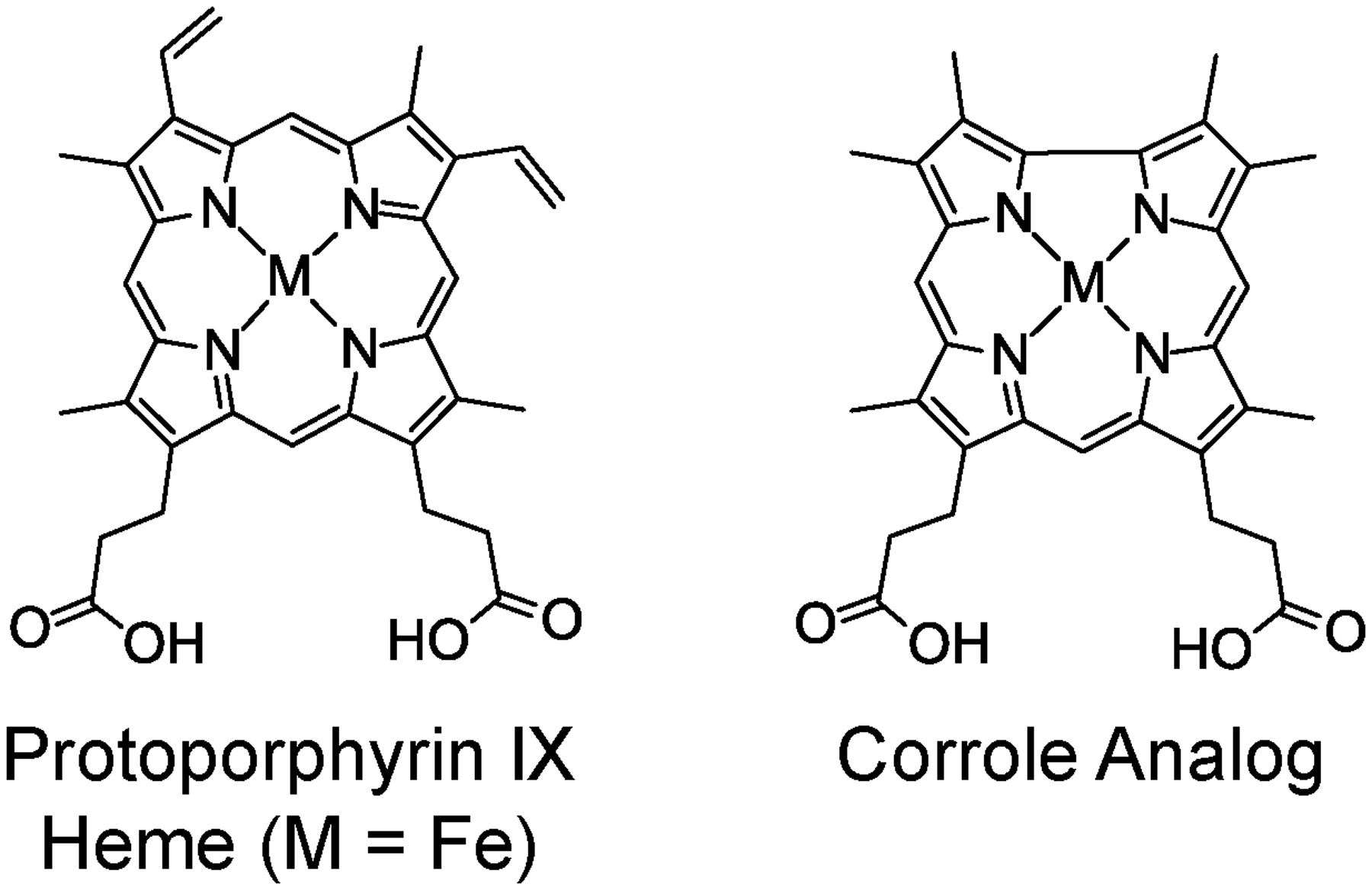
Comparison of the native heme cofactor protoporphyrin IX and the corrole analog utilized in this study.
To this end, the synthesis of a corrole analog that is structurally similar to the native heme cofactor was targeted (Chart 1) for ultimate reconstitution into the heme pocket of the apoprotein to furnish a novel fluorescent protein. Previous studies have examined corrole–protein interactions with a variety of proteins, including serum albumin,52,53 viral capsids,54 and transferrin.55 Additionally, nanoparticle formulations have been prepared with corrole conjugates of these proteins.56,57 In all of these examples, the corroles are amphiphilic meso-aryl corroles with β-sulfonate substituents. These conjugates, which exploit non-specific hydrophobic and charge interactions, have been utilized for a variety of biomedical applications,51 including drug delivery,54 in vitro cancer cell therapy,54 cell imaging,56 and sonodynamic therapy.57 Conversely, only two studies have used heme-like (meso-unsubstituted, β-octaalkyl) corrole complexes to replace the native porphyrin cofactor of heme proteins: an iron corrole in myoglobin and horseradish peroxidase as artificial peroxidases,58 and a copper corrole in myoglobin.59 Here, this approach has been extended by developing fluorescent H-NOX and HasA proteins using a phosphorus corrole as the fluorescent cofactor.
Experimental
Materials for Chemical Synthesis and Characterization.
The following chemicals were used as received: dichloromethane (CH2Cl2), ethyl acetate (EtOAc), hexanes, 4-(dimethylamino)pyridine (DMAP), 3-nitro-2-butanol (mixture of isomers), acetic anhydride (Ac2O), sodium bicarbonate (NaHCO3), sodium sulfate (Na2SO4), ethyl isocyanoacetate, benzyl acetoacetate, methyl 4-acetyl-5-oxohexanoate, zinc powder (Zn), triethylamine (NEt3), trimethyl orthoformate, L-histidine hydrochloride monohydrate, palladium on carbon with 10 wt% loading (Pd/C), silica gel (60 Å pore size, 230–400 mesh particle size), potassium hydroxide (KOH), concentrated hydrobromic acid (HBr), pyridine, phosphorus trichloride (PCl3), rhodamine 101, and Celite from Sigma-Aldrich; trifluoroacetic acid (TFA), concentrated hydrochloric acid (HCl), sodium hydroxide (NaOH), and tetrahydrofuran (THF) from EMD Millipore; methanol (MeOH), chloroform (CHCl3), sodium nitrite (NaNO2), and 85% phosphoric acid (H3PO4) from Fisher Scientific; acetic acid (HOAc) from Macron; tetrachloro-1,4-benzoquinone (p-chloranil) from TCI Chemicals; chloroform-d (CDCl3) and dimethylsulfoxide-d6 (DMSO-d6) from Cambridge Isotope Labs; hydrogen gas (H2) from Praxair; lead(IV) acetate (Pb(OAc)4) from Alfa Aesar; and potassium carbonate (K2CO3) from J. T. Baker Chemicals. Nitrogen gas (N2) from Praxair was passed over a Drierite column prior to use. The phosphorus complex of 5,10,15-tris(4-methoxyphenyl)-corrole was prepared as previously reported.42
Materials for Protein Expression, Purification, and Reconstitution.
The following chemicals were used as received: potassium phosphate dibasic trihydrate (K2HPO4), deoxyribonuclease I from bovine pancreas (DNase), benzamidine, D-(+)-glucose, triethanolamine (TEA), 2-morpholinoethanesulfonic acid (MES), and antifoam SE-15 from Sigma-Aldrich; Luria Broth (LB), Terrific Broth (TB), ampicillin, and 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) from Research Products International; agarose, dimethylsulfoxide (DMSO), BugBuster Master Mix, and Immobilon Classico Western HRP substrate from EMD Millipore; hemin chloride from Frontier Scientific; PrimeSTAR Max DNA polymerase and His60 Ni Superflow resin from Takara Bio; mouse anti-His6 antibody and goat anti-mouse HRP conjugate antibody from Life Technologies; Bacto Agar from BD; glycerol from VWR; lysozyme (from egg white) from Amresco; triethanolamine hydrochloride (TEA•HCl) from Spectrum Chemical; imidazole from Oakwood Chemical; sodium chloride (NaCl) from Macron; Gibson Assembly Master Mix from New England BioLabs; nitrogen gas (N2) from Praxair; isopropyl β-D-1-thiogalactopyranoside (IPTG) and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) from Fisher Scientific; and PD-10 desalting columns (Sephadex G-25 medium) from GE Healthcare. Argon gas (Ar) from Praxair was passed over an Oxiclear gas purifier prior to use. The DH5α and XL1-Blue E. coli competent cells were obtained from the UC Berkeley Macrolab, while the RP523(DE3) competent cells was obtained from lab-generated stocks.13
8,12-Bis(2-methoxycarbonylethyl)-2,3,7,13,17,18-hexamethyl corrole (H3-2).
In a 20 mL glass vial, 60 mg (83 μmol) of a,c-biladiene 1, which was prepared as described in the Supplemental Information, was suspended in 10 mL of MeOH. The mixture was briefly sonicated and 65 mg (264 μmol) of p-chloranil was added. The mixture was briefly sonicated and then stirred at room temperature for 1.5 hours. Two parallel reactions were run and then combined for work-up and purification. The reaction mixtures were transferred to a separatory funnel and CH2Cl2 was added. The red-brown solution was washed with a saturated solution of NaHCO3 and then with water. The organics were dried over Na2SO4 and brought to dryness. The crude material was loaded onto a silica gel column packed with CH2Cl2 and the product was eluted with 4:1 CH2Cl2:EtOAc as a magenta solution. Solvent was removed by rotary evaporation to afford 77 mg (83% yield for two parallel reactions, or 120 mg of 1) of the title compound as a purple solid. 1H NMR (400 MHz, CDCl3, 25 °C) δ 3.19 (t, J = 7.7 Hz, 4H), 3.35 (s, 6H), 3.45 (s, 6H), 3.53 (s, 6H), 3.71 (s, 6H), 4.23 (t, J = 7.7 Hz, 4H), 9.19 (s, 1H), 9.35 (s, 2H). Anal. Calcd for (M + H)+, M = C33H38N4O4: 555.2966; Found ESI-MS: 555.2960. UV–vis (CHCl3), λ in nm (ε in 103 M−1 cm−1): 398 (130.), 409 (97.8), 501 (7.0), 538 (16.3), 550 (16.6), 593 (17.5).
8,12-Bis(2-methoxycarbonylethyl)-2,3,7,13,17,18-hexamethyl-corrolato-(oxo)phosphorus(V) (P-2).
In a 20 mL glass vial, 21 mg (38 μmol) of free-base corrole H3-2 was dissolved in 5 mL of dry pyridine and 100 μL of PCl3 (0.16 g, 1.1 mmol) was added and the resultant mixture was stirred at room temperature for 10 min. An additional 200 μL of PCl3 (0.31 g, 2.3 mmol) was added and the mixture was stirred at room temperature for 10 minutes. Then 200 μL of PCl3 (0.31 g, 2.3 mmol) was added and the mixture was stirred at room temperature for 10 minutes. The reaction mixture was dissolved in CH2Cl2 and transferred to a separatory funnel; water was carefully added to react with the residual PCl3. The product was extracted into CH2Cl2 and the organic layer was washed with a saturated solution of NaCl. The organics were dried over Na2SO4 and brought to dryness. The crude material was loaded onto a silica gel column packed with EtOAc and the product was eluted with 5% MeOH in EtOAc as a pink-orange solution. Solvent was removed by rotary evaporation to afford 18 mg (77% yield) of the title compound as a red-orange solid. 1H NMR (400 MHz, CDCl3, 25 °C) δ 3.25 (m, 4H), 3.57 (s, 6H), 3.63 (s, 6H), 3.70 (s, 6H), 3.77 (s, 6H), 4.38 (t, J = 7.8 Hz, 4H), 9.87 (s, 2H), 9.96 (s, 1H). 31P NMR (162 MHz, CDCl3, 25 °C) δ −98.76. Anal. Calcd for (M + H)+, M = C33H35N4O5P: 599.2418; Found ESI-MS: 599.2410. UV–vis (CHCl3), λ in nm (ε in 103 M−1 cm−1): 274 (16.9), 280 (16.2), 313 (12.3), 399 (215), 403 (238), 485 (3.4), 517 (11.6), 522 (11.2), 558 (25.3).
8,12-Bis(2-carboxyethyl)-2,3,7,13,17,18-hexamethylcorrolato-(oxo)phosphorus(V) (P-3).
In a 50 mL round bottom flask, 29 mg (47 μmol) of corrole ester P-2 was dissolved in 10 mL of THF and 10 mL of 3 M NaOH was added. The biphasic mixture was stirred vigorously and refluxed overnight, protected from light. After this reaction, the corrole had transferred from the organic layer to the aqueous layer. The reaction mixture was cooled in an ice bath and acidified by slowly adding 1 M HCl to reach pH ~0. The solution became cloudy, indicating precipitation of the protonated carboxylic acid corrole. The red-orange suspension was transferred to a 50 mL conical tube and was centrifuged at 4300 g for 20 minutes. The supernatant was discarded and the pellet was resuspended in 20 mL of H2O and the suspension was centrifuged at 4300 g for 20 minutes. This process was repeated until the supernatant had a pH of ~ 5. The pellet was resuspended in a minimal amount of H2O and transferred to a round bottom flask. Solvent was removed by rotary evaporation and then dried under vacuum for several days to afford 20 mg (76% yield) of the title compound as a dark red solid. 1H NMR (400 MHz, DMSO-d6, 25 °C) δ 3.13 (t, J = 7.6 Hz, 4H), 3.54 (s, 6H), 3.62 (s, 6H), 3.63 (s, 6H), 4.32 (m, 4H), 10.09 (s, 2H), 10.17 (s, 1H), 12.39 (bs, 2H). 31P NMR (162 MHz, DMSO-d6, 25 °C) δ −97.66. Anal. Calcd for (M + H)+, M = C31H31N4O5P: 571.2105; Found ESI-MS: 571.2110.
Protein Expression and Purification.
The protein expression plasmids were prepared as described in the Supplementary Information. The plasmids were transformed into the RP523(DE3)13 strain of E. coli then grown on an LB agar plate (1.5% (w/v) agar) supplemented with ampicillin (100 μg/mL) and hemin (20 μg/mL). Ten colonies were selected to test protein expression and were grown in 3 mL of TB supplemented with ampicillin (100 μg/mL) and hemin (20 μg/mL) at 37 °C overnight. Glycerol stocks were prepared for each culture. The cells were then sub-cultured (1:200 dilution) in 50 mL of TB supplemented with ampicillin (100 μg/mL), hemin (20 μg/mL), and 0.2% (w/v) glucose and grown at 37 °C until the OD600 reached ~0.6. IPTG was added (100 μM final concentration) and the cultures were then transferred to an 18 °C incubator and grown overnight. H-NOX expression levels were determined by preparing cell density-matched samples of each culture, as well as a pre-induction control. The cells were pelleted (2300 g for 5 minutes) then lysed using BugBuster® and the whole-cell lysate was analyzed by SDS-PAGE and Western blot to identify His-tagged proteins. Western blotting was performed using a mouse anti-His6 primary antibody, a goat anti-mouse–HRP conjugate secondary antibody, and the Classico HRP substrate. Colonies with the highest level of heme expression were then selected for large-scale protein expression. For large-scale expression of the holoprotein, a 5 mL overnight culture (prepared as described above) was inoculated directly from a frozen glycerol stock and then grown at 37 °C overnight. This culture was then used to inoculate 1 L of TB (containing 100 mg of ampicillin, 20 mg of hemin, and 0.2% (w/v) glucose) and was grown at 37 °C until the OD600 reached ~0.6. IPTG was added (100 μM final concentration) and the cultures were then transferred to an 18 °C incubator and grown for 24 hours. Cells were harvested by spinning the cultures at 4300 g for 20 min and the resultant pellets were snap-frozen in liquid nitrogen and stored at −80 °C.
For anaerobic protein expression to obtain the apoprotein, a starter culture was prepared in a serum bottle containing 60 mL of TB, supplemented with ampicillin (100 μg/mL) and 0.2% (w/v) glucose, then inoculated directly from a frozen RP523 glycerol stock. The bottle was sealed with a rubber septum and purged with Ar for 1 hour, and then grown at 37 °C for at least 20 hours. A freshly autoclaved 10 L fermenter60 was charged with 7 L of hot, sterilized TB and 1 L of hot, sterilized H2O (to account for the ~1 L volume loss due to overnight purging). After cooling to 37 °C, 2 mL of antifoam and 400 mg of ampicillin were added, and the mixture was sparged with N2 overnight. The following day, ampicillin (500 mg) and glucose (0.2% (w/v) final concentration) were added to the media, which was then inoculated with the 60 mL anaerobic starter culture. The resultant culture was grown at 37 °C until the OD600 reached ~0.45, and was then cooled to 18 °C prior to inducing protein expression. Once the OD600 reached ~0.7 and the temperature reached 18 °C, IPTG was added (100 μM final concentration) and the culture was grown at 18 °C for 24 hours to obtain the apo protein. Cells were harvested by spinning the cultures at 4300 g for 20 min and the resultant pellets were snap-frozen in liquid nitrogen and stored at −80 °C.
Cell pellets of Cs H-NOX were thawed in a water bath at room temperature then resuspended in Buffer A (50 mM K2HPO4, 300 mM NaCl, 20 mM imidazole, and 5% (v/v) glycerol at pH 8.0) supplemented with 110 mM benzamidine, 0.4 mM AEBSF, 0.1 mg/mL lysozyme, and 0.3 mg/mL DNase. Similarly, Pa HasA cell pellets were resuspended in Buffer B (20 mM K2HPO4, 250 mM NaCl, and 30 mM imidazole at pH 7.4) supplemented with 110 mM benzamidine, 0.4 mM AEBSF, 0.1 mg/mL lysozyme, and 0.3 mg/mL DNase. For both proteins, cells were lysed using a high-pressure homogenizer (Avestin Emulsiflex-C5). In the case of Cs H-NOX, the lysate was heated in a water bath at 60 °C for 1 hour. The lysate was clarified by spinning at 42,000 g and the supernatant was loaded onto a column containing His60 resin (3 mL for 1 L aerobic cultures, or 5 mL for ~7 L anaerobic cultures) equilibrated with Buffer A for H-NOX or Buffer B for HasA. The resin was then washed with 20 column volumes (CV) of Buffer A for H-NOX or Buffer B for HasA. The protein was then eluted with Buffer C for H-NOX (50 mM K2HPO4, 300 mM NaCl, 300 mM imidazole, and 5% (v/v) glycerol at pH 8.0) or Buffer D for HasA (20 mM K2HPO4, 250 mM NaCl, and 300 mM imidazole at pH 7.4). The eluent was collected in five, 2–3 mL fractions. Fractions were analyzed by SDS-PAGE and UV-vis absorption spectroscopy. Typically, the first fraction contained a significant amount of nucleic acids. The main protein-containing fractions were the second and third fractions. Later fractions contained little protein and typically exhibited a shoulder on the red side of the 280 nm protein absorption peak. Fractions with comparable purity were combined and then dialyzed overnight into Buffer E (20 mM K2HPO4, 250 mM NaCl, and 1 mM EDTA at pH 7.4) using snakeskin dialysis tubing (3500 MW cutoff). The protein was then filtered with a 0.22 μm syringe filter and stored in sterile tubes at 4 °C.
Apoprotein Reconstitution.
A 1 mg/mL stock solution of corrole P-3 was prepared in DMSO. An aliquot of the H-NOX or HasA protein (~1 mg) was diluted to 1 mL with Buffer E in a 1.7 mL microcentrifuge tube. Then, 100 μL of the P-3 corrole solution was slowly added dropwise to the protein; the tube was gently inverted after each addition of two drops of the corrole stock solution. The reaction mixture was gently rocked at room temperature for 2 hours, protected from ambient light. After incubation, the reaction mixture was spun at 21,000 g for 1 minute to pellet any solids. The solution was then applied to a PD-10 column (Sephadex G-25 medium), pre-equilibrated with Buffer F (20 mM TEA and 1 mM EDTA at pH 6.5). The pink eluent was collected in 1 mL fractions. In order to remove any traces of unbound P-3, the protein-containing fractions were concentrated to <1 mL (using a 5000 MW cutoff filter) and a second PD-10 column was run using Buffer F.
Physical Measurements.
NMR spectra were recorded on a Bruker Avance 400 NMR spectrometer at the UC Berkeley College of Chemistry NMR Facility. 1H NMR spectra were internally referenced to the residual solvent signal (δ = 7.26 for CDCl3 or δ = 2.50 for DMSO-d6),61 and 31P NMR were internally referenced to 85% H3PO4 (δ = 0) using a capillary tube containing the standard. Mass spectra were recorded on a Finnigan LTQ FT-ICR mass spectrometer (Thermo Fisher Scientific) equipped with an electrospray ionization source in positive ion mode at the QB3/Chemistry Mass Spectrometry Facility at UC Berkeley. UV–vis absorption spectra were acquired using a Cary 300 spectrometer (Agilent) or a NanoDrop 2000C (Thermo Scientific). Steady-state emission spectra were recorded on a Horiba Scientific FluoroMax-4 spectrofluorometer. Time-resolved fluorescence was measured using a PicoQuant FluoTime 300 fluorometer and a 520 nm PicoQuant pulsed diode laser with a PDL 820 driver. Signal was collected at the emission maximum with a 2 nm bandwidth and detected using a TimeHarp 260 time-correlated single photon counting board. Relative quantum yields of corroles in chloroform (η = 1.4459)62 or proteins in TEA buffer (η = 1.3329, interpolated from published data)63 were calculated using Rhodamine 101 in basic EtOH (η = 1.3611)62 as the reference (Φref = 0.96)64 according to the following equation:
| (1) |
where ∇ is the slope of the plot of integrated fluorescence intensity versus absorbance (constructed with 5 points) and η is the refractive index of the solvent.
Computational Details.
Density functional theory (DFT) calculations were performed with the hybrid functional Becke-3 parameter exchange functional65–67 and the Lee–Yang–Parr nonlocal correlation functional (B3LYP),68 as implemented in the Gaussian 16, Revision A.03 software package.69 A polarized split-valence triple-ζ basis set that includes p functions on hydrogen atoms and d functions on other atoms (i.e., the 6–311G(d,p) or 6–311G** basis set) was used. Calculations were performed with a polarizable continuum (PCM) solvation model in chloroform using a polarizable conductor calculation model (CPCM).70–71 Geometries were confirmed as local minima structures by calculating the Hessian matrix and ensuring that no imaginary eigenvalues were present. Excited state calculations were performed using time-dependent DFT (TD-DFT)72–76 with the same functionals, basis sets, and solvation details as the ground state, but with the inclusion of diffuse functions on all atoms (i.e., the 6–311++G(d,p) or the 6–311++G** basis set). Excited state energies were computed for the 20 lowest singlet and triplet excited states. Optimized geometries were rendered in Gauss View 5. Simulated UV–vis spectra were generated in the program Gauss View 5 by broadening transition lines with Gaussian functions that have a half width of 0.03 eV.
Results
Corrole Synthesis and Characterization.
In order to prepare a corrole analog with approximately the same size and shape of the native heme cofactor, the synthesis of corrole H3-2 was targeted. In order to simplify the synthesis, vinyl groups were substituted for more synthetically tractable methyl groups. Although the 2,3,17,18-tetraethyl derivative of corrole H3-2 has previously been incorporated into heme proteins,58,59 smaller methyl groups would minimize the bulk of the corrole and facilitate binding in the H-NOX heme binding pocket. The free-base corrole H3-2 was prepared via oxidative cyclization of the linear tetrapyrrole a,c-biladiene 1 (see SI for synthesis), using p-chloranil as the oxidant (Scheme 1).77 The scale of the cyclization is critical and the highest yields (>80%) were obtained when the reaction was run on a 60 mg scale. For convenience, two parallel reactions were performed then combined for work-up and purification.
Scheme 1.
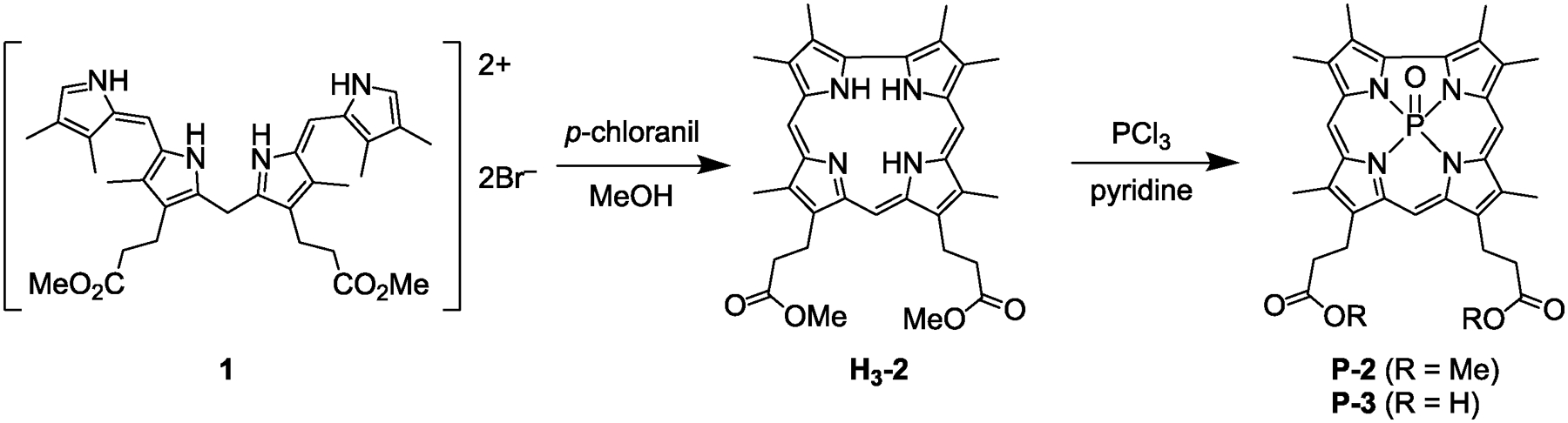
Synthesis of free-base corrole H3-2 and subsequent phosphorus insertion to yield P-2.
Metallation of the free-base corrole was attempted with a variety of light main-group elements that are known to yield highly fluorescent complexes. First, gallium coordination was performed with GaCl3 in pyridine, following literature procedures.39 A stark color change from the magenta free-base to red-orange indicated formation of the complex. Purification of Ga-2 on silica resulted in decomposition of the molecule to the corresponding biliverdin. Presumably, the gallium complex acted as a photosensitizer for the generation of singlet oxygen, which subsequently reacted with the corrole across the pyrrole–pyrrole linkage (Scheme S1). This reactivity is consistent with previous reports of photochemical decomposition of highly electron rich β-octaalkyl corroles.78,79
To circumvent this deleterious reactivity, the preparation of complexes with the lighter main group elements aluminum and phosphorus was targeted. Following published methods,40 aluminum coordination was performed using AlMe3 in toluene; a drastic color change was consistent with complex formation. While the primary method for purification of aluminum corroles is recrystallization, attempts to purify Al-2 in this way resulted in decomposition of the corrole after ~12 hours. It is likely that this complex undergoes photochemical degradation akin to the gallium analog. Attempts to purify the aluminum corrole by column chromatography were unsuccessful. The compound stuck to silica, neutral alumina, and Florisil and could not be eluted with polar solvent mixtures, including 100% methanol.
It should be noted that previously reported Ga39 and Al40 complexes utilize 5,10,15-tris(pentafluorophenyl)corrole (TPFC). Since the corrole core is inherently more electron-rich than porphyrin,80 the strongly electron-withdrawing pentafluorophenyl substituents reduce the electron density of the tetrapyrrole core. Consequently, H3TPFC is highly resistant to decomposition and is one of the most stable corrole ligands.81 Conversely, H3-2 has electron-donating alkyl substituents, which increase the density of the corrole core. This ligand and other β-octaalkyl corroles are prone to oxidative degradation.78
Fortunately, the phosphorus complex of corrole 2 was readily prepared via literature methods using PCl344 (Scheme 1) and the compound was obtained in 77% yield. In order to enhance water solubility and enable hydrogen bonding with the Y–S–R motif in Cs H-NOX, the methyl esters where hydrolyzed under basic conditions. A biphasic mixture of the corrole ester P-2 in THF and 3 M NaOH was refluxed overnight. Acidic workup precipitated the carboxylic acid derivative P-3 in high yield.
The phosphorus center of P-2 and P-3 could exist as one of three possible species: a six-coordinate di-hydroxide, a five-coordinate terminal oxo,82 or a five-coordinate cationic mono-hydroxide83 (Chart 2). Mass spectrometry is consistent with the five-coordinate formulations. It should be noted that these two scenarios are indistinguishable, as [P=O] + H+ and [P–OH]+ have identical masses. The coordination number of the P(V) center can be determined by 31P NMR. Chemical shifts in the −90 to −100 ppm region correspond to five-coordinate species, whereas six-coordinate complexes have resonances between −180 and −200 ppm.37,42 P-2 and P-3 exhibit a single resonance at −98.76 and −97.66 ppm, respectively, confirming both are five-coordinate species. These values are similar to the chemical shift reported for P(V)=O octaethylcorrole (−99.40 ppm).82 Finally, there is no evidence of axial –OH signals in the 1H NMR spectrum, as evidenced by the lack of signals at < −2 ppm. Together, these data indicate that P-2 and P-3 are best formulated as five-coordinate P=O complexes.
Chart 2.
Speciation of phosphorus corroles.
The electronic absorption spectra of H3-2, P-2 (Figure 2), and P-3 (Figure S1) exhibit intense Soret (or B) bands in the near-UV and weaker Q bands in the visible region of the spectrum (Table 1). The free-base corrole (Cs symmetry) exhibits a split Soret band, which arises from distinct x and y polarizations of this transition.84 Out-of-plane coordination of the P=O unit renders P-2 and P-3 Cs symmetric. Consequently, it is expected that the phosphorus complexes would also exhibit a split Soret band, consistent with previously reported symmetry-dependent observations.44,85,86 However, the splitting is poorly resolved and the spectrum is more similar to pseudo-octahedral complexes with C2v symmetry,85 and is consistent with previously reported six-coordinate phosphorus corroles.87–89 The addition of pyridine to a CH2Cl2 solution of P-2 does not change the absorption spectrum (Figure S2a). Similarly, the absorption spectrum of a P-3 solution in TEA buffer does not change upon the addition of histidine (Figure S2b). This result suggests that, unlike meso-triaryl P=O corroles (Figure S2c and d), P-2 and P-3 do not bind nitrogenous bases and remain five-coordinate complexes.
Figure 2.
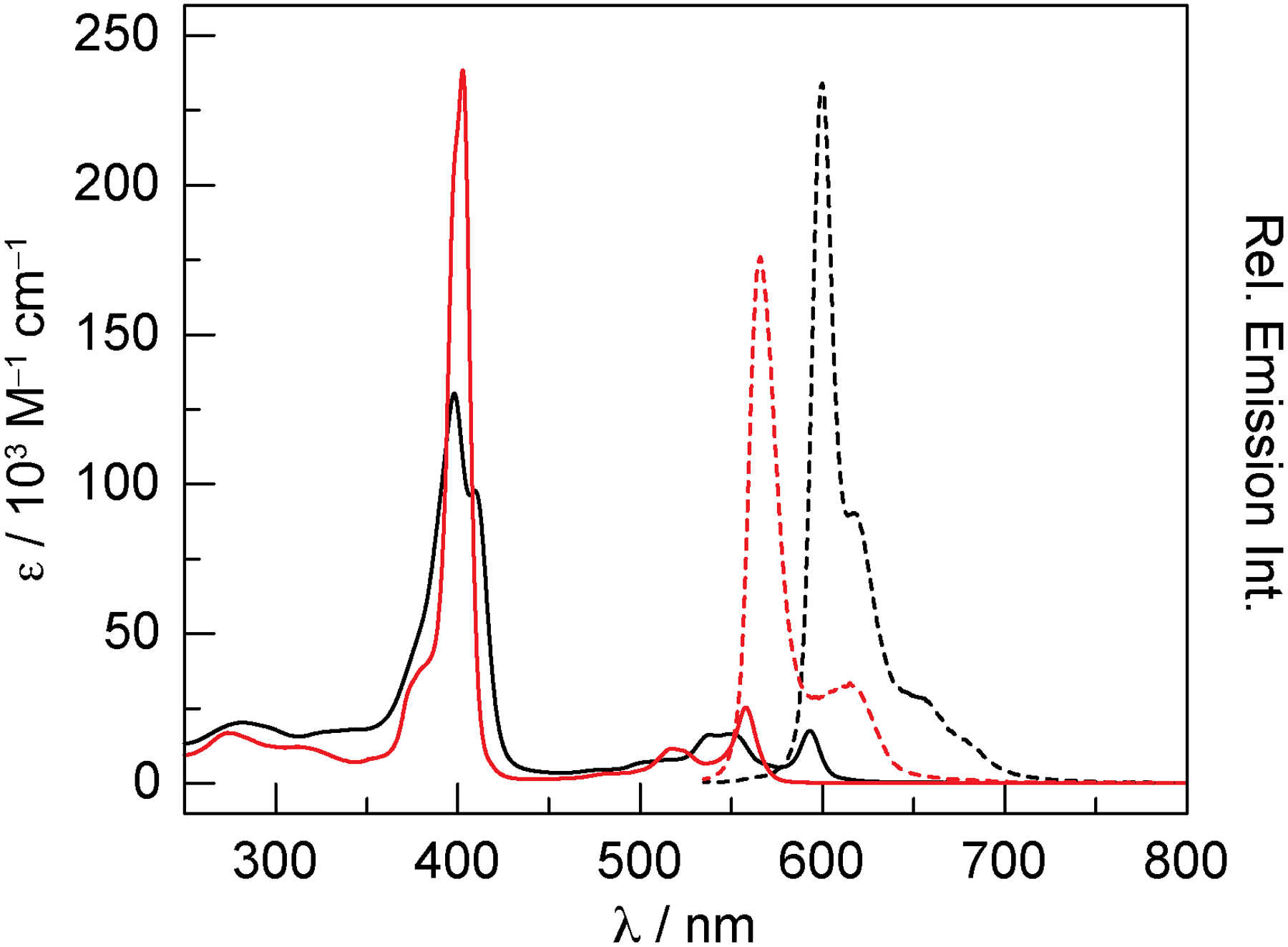
Absorption (solid lines) and emission (dashed lines) spectra of H3-2 (▬) and P-2 (▬) in CHCl3. For the emission spectra, the absorbance of the samples was matched at the excitation wavelength: A(525) = 0.1000 ± 0.0003.
Table 1.
Absorption of P Corroles and Conjugatesa
| B1 | B2 | Q(1,0) | Q(0,0) | |
|---|---|---|---|---|
| P-2 b | 399 (sh)c | 403 | 517 | 558 |
| P-3 d | – | 380 | 520 | 558 |
| P-3 H-NOXd | 399 | 403 | 519 | 561 |
| P-3 HasAd | – | 404 | 523 | 559 |
Transition wavelengths are in units of nm;
Measured in CHCl3;
Shoulder;
Measured in TEA buffer at pH 6.5.
The emission spectra of H3-2, P-2 (Figure 2), and P-3 (Figure S1) exhibit a sharp fluorescent transition with weaker vibrational overtones at longer wavelengths. Figure 2 illustrates the emission intensity for absorbance-matched samples of H3-2 and P-2, indicating that the emission from the free-base corrole is more intense than the phosphorus complex. Indeed, the fluorescence quantum yields for H3-2 and P-2 are 5.9% and 3.9%, respectively (Table 2). Emission from P-3 is extremely weak in aqueous buffer, exhibiting a quantum yield of only 1.2%. Time-resolved measurements (Figure S3) indicate that the excited-state lifetime for P-2 is extremely short (1.2 ns), whereas the lifetime of H3-2 is considerably longer (5.7 ns), and this value is consistent with previously reported data for free-base corroles.90 Despite the low quantum yield, P-3 exhibits a longer lifetime of 3.8 ns when fit to a monoexponential decay. We note that a biexponential decay function nominally improves the fit of the data (τ1 = 1.5 ns, 84%; τ2 = 4.9 ns, 16%), but the physical significance of this two-component fit is unclear. Using the lifetime and quantum yield, the radiative (kr) and nonradiative (knr) rate constants can be determined using the following equation:
| (2) |
where ϕ is the fluorescence quantum yield and τ is the excited-state lifetime. While kr is similar for H3-2 and P-2, this value is an order of magnitude slower for P-3. Conversely, knr is similar for both H3-2 and P-3, but this rate constant is nearly four times higher for P-2.
Table 2.
Summary of Emission Properties of Corroles and Protein Conjugates
DFT Calculations.
In order to gain insight into the electronic structure of the corrole complexes, density functional theory (DFT) calculations were performed using the B3LYP functional. Ground-state geometry optimization calculations were performed for H3-2 and P-2, both as a terminal oxo and a dihydroxide (P(OH)2-2), using the 6–311G** basis set and a CPCM solvation model in chloroform. The structures were verified as local minima by performing frequency calculations and ensuring that there were no imaginary frequencies. The optimized structures (Figures S4–S6, Tables S1–S3) are qualitatively similar to previously reported corrole structures determined by X-ray crystallography.44,83,91,92
Since it appears that P-2 does not bind pyridine (Figure S2), calculations were also performed for a six-coordinate complex of P-2 with an axial imidazole ligand (P(O)(Im)-2). This structure would mimic the histidine–corrole interaction in the H-NOX and HasA heme binding pockets. As a starting point for the geometry optimization, one of the axial hydroxide ligands of P(OH)2-2 was replaced with an imidazole ligand while the other was converted to an oxo. Interestingly, the imidazole ligand dissociates from the phosphorus center in the optimized structure (Figure S7, Tables S4 and S5), resulting in a long P–N distance of 4.43 Å. The presence of imidazole destabilizes the structure by 7.2 kcal/mol (relative to the five-coordinate complex and imidazole): ΔGrxn = +7.2 kcal/mol for P-2 + imidazole → P(O)(Im)-2. This supports the experimental observation that P-2 does not bind nitrogenous bases, demonstrating that the phosphorus center prefers to remain 5-coordinate.
The four frontier molecular orbitals (HOMO, HOMO–1, LUMO, and LUMO+1) are energetically well-separated from the rest of the orbital manifold (Figure S8, Table S6). Based on the Gouterman four orbital model for porphyrins,93–95 which has also been shown to hold for corroles,84,96,97 it is expected that the Soret and Q bands arise due to transitions between these four frontier orbitals. To more rigorously characterize the optical properties of these corroles, time-dependent DFT (TD-DFT) calculations were performed using the same methods as for the geometry optimization, but with the inclusion of diffuse functions on all atoms (i.e., the 6–311++G** basis set). Single-point excited-state energy calculations were performed for the 20 lowest singlet and triplet states (Tables S7–S12) of H3-2, P-2, and P(OH)2-2. The four lowest-energy singlet states (S1–S4) give rise to two Q-like and two Soret-like transitions. These states exclusively involve the four frontier orbitals, with the exception of a minor (10%) contribution to S3 of H3-2 that involves HOMO–2. The deviation between the calculated and experimental transitions ranges from 0.01 to 0.18 eV, although S1 for H3-2 deviates by 0.24 eV. Nevertheless, these differences are substantially smaller than the absolute mean error of 0.3–0.5 eV determined from TD-DFT benchmark studies on test sets of organic molecules.98–100
Although the predicted transitions do not precisely align with the data, the calculated spectra (Figures S9–S11) qualitatively reflect experimental observations. The two Q bands and the two Soret bands are orthogonally polarized; this data is summarized in Tables S13–S15, which list the electronic dipole moment vectors for states S1–S4. This demonstrates that both the Q and B bands are split into x and y polarizations due to the low symmetry of the corrole, consistent with previous reports.84 Calculations indicate that H3-2 has a substantial energy difference between the Soret transitions (202 cm−1), which results in clear separation of bands (676 cm−1) in the experimental spectrum. It should be noted that these TD-DFT calculations have underestimated the experimental split in the Soret transitions. Given the out-of-plane coordination of the P=O unit in P-2, the calculated splitting of the Bx and By transitions is 519 cm−1, which is larger than the experimental splitting of 249 cm−1. Conversely, calculations for P(OH)2-2 show a very small 28 cm−1 separation between the Bx and By transitions. The experimental absorption spectrum of P-2 exhibits poor resolution of the Soret transitions, which is more similar to the calculated UV-vis spectrum of P(OH)2-2 rather than the terminal oxo P-2.
Protein Expression.
Expression-based methods were initially explored to incorporate the carboxylic acid derivative P-3 into Cs H-NOX, as this has been a successful methodology to replace the native cofactor of heme proteins with a variety of unnatural porphyrin complexes.60 This approach requires the RP523(DE3) strain of E. coli, which has a disrupted gene in the heme biosynthesis pathway, and also harbors a mutation that renders the cell well permeable to heme and heme-like porphyrins (e.g., mesoporphyrin IX).101 High levels of artificial porphyrin incorporation are achieved by supplementing the media with the cofactor under anaerobic conditions. Given the structural similarity of the corrole analog to the native heme cofactor, extension of this approach to prepare corrole-substituted heme proteins was reasonable.
Small-scale (1 L) anaerobic protein expression of Cs H-NOX was performed in a three-neck bottle sealed with silicone septa and the media was supplemented with 7 mg of P-3. Surprisingly, the bacterial cell pellet was not pink, as would be expected if the corrole diffused across the bacterial cell wall. Although the purified protein appeared colorless, a minimal amount of P-3 was incorporated into the H-NOX, as evidenced by a small Soret band in the absorption spectrum (Figure S12). It is possible that the speciation of P-3 is different in water than it is in organic solvents; additional charge (e.g., [P–OH]+) may prevent the molecule from crossing the bacterial cell wall. The absorption spectrum of P-3 in buffer is pH-dependent, which likely reflects changes in the protonation state of the molecule (Figure S13); such events are more likely for a hydroxide ligand rather than an oxo.
Encouraged by the successful heme reconstitution of an sGC from Chlamydomonas reinhardtii to yield a functional enzyme,102 we then turned to reconstitution methods. Both Pa HasA and Cs H-NOX were expressed using the RP523(DE3) strain of E. coli. When RP523 cells are grown anaerobically, the bacteria can grow in the absence of heme, enabling apoprotein isolation. This methodology circumvents the harsh, denaturing conditions necessary to remove the heme.19,29 The apoprotein was isolated in moderate yield: 2 mg/L for Cs H-NOX and 5 mg/L for Pa HasA. While Pa HasA is stable for over 1 year in imidazole-free buffer at 4 °C, Cs H-NOX undergoes complete degradation over the course of several weeks at 4 °C (or several hours at room temperature), as evidenced by SDS-PAGE. If the H-NOX protein is instead stored at 4 °C in buffer containing 300 mM imidazole, then the protein remains intact for over 1 year. This observation is consistent with a contaminating metalloprotease; imidazole coordination to the active-site Zn center inhibits activity. Leveraging the thermal stability of Cs H-NOX, heat denaturation can be utilized to inactivate any protease contaminants. Although there are several thermostable (> 49 °C) E. coli proteins, none are proteases.103 After heating the crude Cs H-NOX lysate at 60 °C for 1 hour, the IMAC-purified protein is stable for at least four months in imidazole-free buffer when stored at 4 °C. It was determined that 60 °C was the optimal temperature because the majority of E. coli proteins denatured at this temperature while preserving the apo H-NOX, which significantly degrades above 66 °C (Figure S14).
Conjugate Preparation and Characterization.
The conditions used for protein reconstitution were surveyed using apo H-NOX. A DMSO solution of P-3 was added dropwise to a solution of the protein and the resultant mixture was gently rocked at room temperature for 2 hours. The reconstituted protein was separated from excess corrole on a PD-10 G-25 desalting column. The eluent was collected in 1 mL fractions and analyzed by UV-vis absorption spectroscopy (Figure S15). The first fraction exhibited a split Soret band (~400 nm) that is significantly red shifted relative to the free corrole (~380 nm), indicating incorporation in the heme binding pocket. Later fractions have a Soret band comparable to the corrole in buffer, suggestive of contamination from the unbound corrole. While this spectral feature does not decrease after dialyzing the sample overnight, it is readily removed by passage through a second PD-10 desalting column (Figure S16). Perhaps the excess corrole can non-specifically bind to the H-NOX surface, if given a sufficient amount of time.
In order to optimize P-3 incorporation in H-NOX, the effects of buffer identity and salt concentration were surveyed for the buffer used for both the reconstitution reaction and the desalting column (Figure S17). The buffer (phosphate at pH 7.4 vs. triethanolamine (TEA) at pH 7.5) used for the reconstitution reaction had a nominal effect on P-3 incorporation. The inclusion of 250 mM NaCl in the desalting buffer eliminated nonspecific binding to the protein surface, but drastically decreased the level of corrole incorporation. Next, the pH dependence of the TEA desalting buffer was examined over the 6.5 to 8.5 range, and it was found that P-3 incorporation decreased monotonically with increasing pH (Figure S18). A less dramatic effect was observed for the pH dependence of the reconstitution reaction, although pH 7.5 was optimal (Figure S19). Additional experiments were performed using triethanolamine (pKa = 7.8) (Figure S20), TEA•HCl at pH 4.8 (Figure S21), and MES buffer (pKa = 6.1) (Figure S22), but these conditions did not improve P-3 incorporation.
P-3 incorporation in Cs H-NOX is maximized when the reconstitution reaction is performed in phosphate buffer at pH 7.5 and the PD-10 desalting column is run using TEA buffer at pH 6.5 (Figure S23a). These optimized conditions were then utilized to incorporate P-3 into Pa HasA and led to high levels of corrole incorporation (Figure S23b). Importantly, these samples are quite stable when stored at 4 °C protected from light, resulting in nominal corrole loss after nearly two months (Figure S24). In the case of P-3 H-NOX, a 13% loss of corrole was observed after 56 days, while a 5% loss was observed for P-3 HasA after 44 days.
Figures 3a and 3b compare the absorption spectra of P-3 with the H-NOX and HasA conjugates. A significant red shift of the Soret band is observed upon protein binding and the spectrum is comparable to P-2 in CHCl3 (Figure 2, Table 1). Another distinction in the absorption spectra of free and protein-bound P-3 is the width of the Q(0,0) transition ~560 nm. While the full width at half maximum (FWHM) of this feature is 25 nm for P-3, the values are 13 nm and 14 nm for the H-NOX and HasA conjugates, respectively, which is similar to the 15 nm FWHM for P-2 in CHCl3. These results suggest that P-2 in CHCl3 is a better standard for comparison than P-3 in aqueous buffer. Indeed, the extinction coefficients for P-2 were measured in CHCl3 because this solvent has a similar dielectric constant (ε = 4.806)62 to the hydrophobic interior of proteins (ε = 6–7).104 For the H-NOX conjugate (Figure 3a), the observed Soret to protein (280 nm) ratio is 4.88. Based on the extinction coefficients of P-2 and H-NOX (and accounting for corrole absorbance at 280 nm), the expected ratio is 5.06, indicating that >96% of the protein is in the holo form. Similarly, the observed Soret to 280 nm ratio for the HasA conjugate (Figure 3b) is 5.33, which is similar to the expected ratio of 5.18. These nominal deviations from the expected value could reflect differences in the extinction coefficient in the protein environment versus organic solvent. Nevertheless, the observed ratios indicate a high extent of P-3 incorporation in both proteins.
Figure 3.
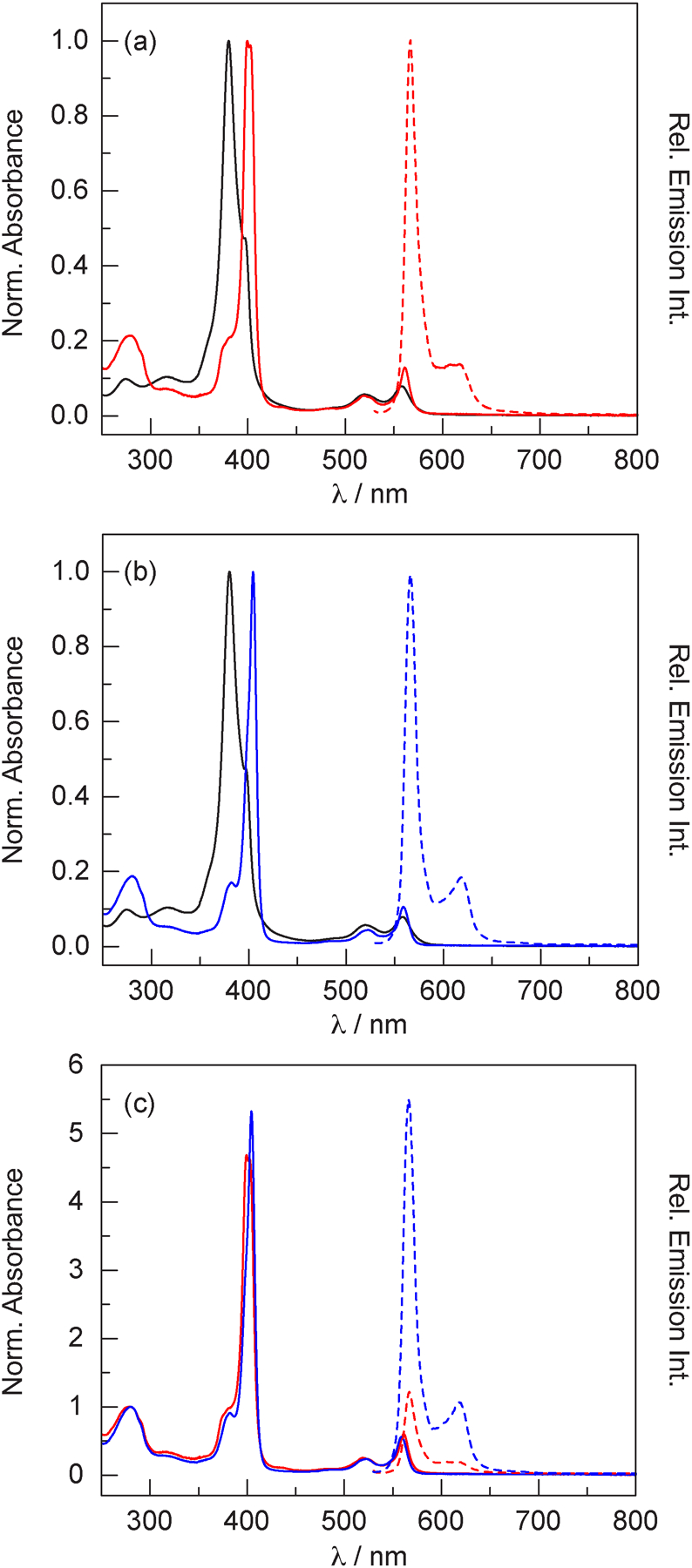
(a) Absorption (solid lines) and emission (dashed lines) spectra of P-3 (▬) and the H-NOX conjugate (▬) in TEA buffer at pH 6.5. (b) Absorption (solid lines) and emission (dashed lines) spectra of P-3 (▬) and the HasA conjugate (▬) in TEA buffer at pH 6.5. (c) Comparison of the absorption (solid lines) and emission (dashed lines) spectra of the H-NOX (▬) and HasA (▬) conjugates of P-3. For the emission spectra, the absorbance of the samples was matched at the excitation wavelength: A(520) = 0.0735 ± 0.0005.
Figure 3c compares the absorption and emission spectra of the H-NOX and HasA conjugates. The H-NOX conjugate displays a split Soret band, while the HasA conjugate displays a single transition. This result suggests that there are distinct corrole–protein interactions in HasA and H-NOX, which could account for the difference between the observed and expected Soret to 280 nm ratios. Unexpectedly, the emission intensity of the H-NOX conjugate is weak, exhibiting a signal that is 4.7 times weaker than the emission from the HasA conjugate (Figure 3c). This is more clearly illustrated in Figure 4, which compares the emission of absorbance-matched samples of P-2, P-3, as well as the protein conjugates.
Figure 4.
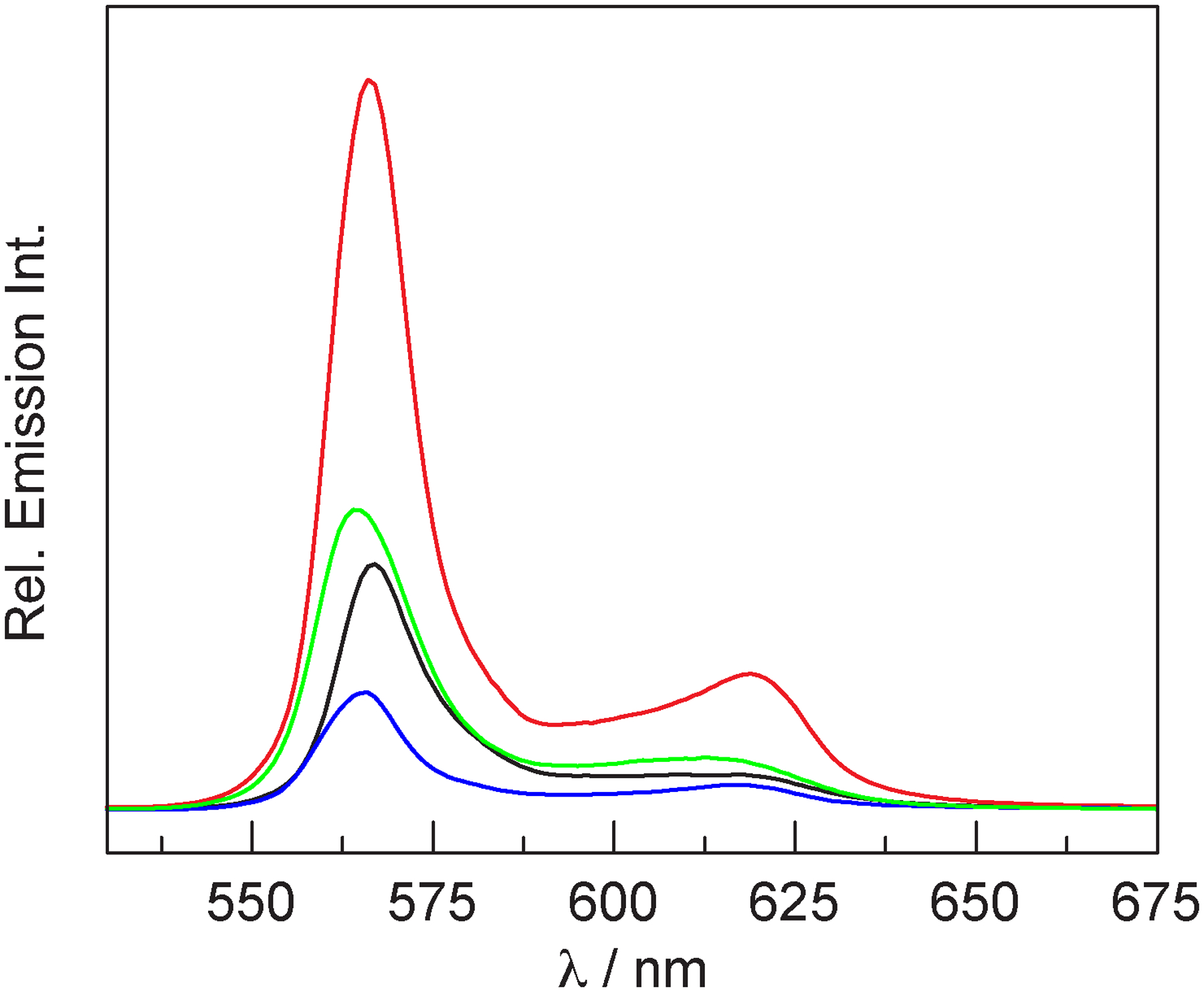
Emission spectra of absorbance-matched samples (A(520) = 0.0575 ± 0.0005) of P-3-substituted H-NOX (▬), HasA (▬), and P-3 (▬) in TEA buffer at pH 6.5, as well as P-2 (▬) in CH2Cl2.
It was our expectation that conjugate formation would enhance the fluorescence intensity (and quantum yield) relative to the free molecule because binding in the heme pocket would restrict the conformational flexibility of the molecule, decreasing non-radiative deactivation of the excited state. We also expected that the fluorescence enhancement would be greater for H-NOX because hydrogen bonding of the propionate chains to the Y–S–R motif would greatly restrict the conformational flexibility of the corrole, whereas the carboxylate groups of the propionate chains are solvent exposed in HasA. Thus, we predicted that ϕ(P-2) < ϕ(P-3 HasA) < ϕ(P-3 H-NOX). It should be noted that P-2 is a better comparison for the conjugates than P-3 (vide supra). However, we find that ϕ(P-3 H-NOX) = 2.5% < ϕ(P-2) = 3.9% < ϕ(P-3 HasA) = 7.3% (Table 2), and this is corroborated by the emission spectra of Figure 4. Time-resolved measurements (Figure S25) indicate that the excited-state lifetime for the H-NOX conjugate is 1.5 ns, which is similar to that of P-2 (1.2 ns). Conversely, the lifetime of the HasA conjugate is longer (3.1 ns) and quite similar to that of P-3 (3.8 ns). As in the case of P-3, a fit of the HasA data to a biexponential decay function nominally improves the fit (τ1 = 1.9 ns, 89%; τ2 = 5.0 ns, 11%), but the physical significance of this two-component fit is unclear. Using this data and Eq. 2, we find that the non-radiative rate constant knr decreases upon protein conjugation: 1.2-fold decrease for H-NOX and 2.7-fold decrease for HasA (relative to P-2). While this result confirms our hypothesis, it is counter to the expectation that H-NOX would restrict the conformation of the corrole more than HasA and result in a lower knr. It should be noted that the relative intensity of the Q(0,1) vibrational shoulder (~620 nm) decreases upon protein binding and this feature is highly suppressed for the H-NOX conjugate. The Q(0,0) to Q(0,1) intensity ratios are as follows: 4.61 for P-3 < 5.27 for P-2 < 5.49 for HasA < 7.41 for H-NOX. This result suggests that the corrole conformation is more restricted in H-NOX than in HasA. Since the Q(0,0) to Q(0,1) intensity ratio does not align with the observed values of knr, there are likely additional factors that dictate the photophysics of these conjugates.
Discussion
Phosphorus meso-triaryl corroles exhibit red-orange emission profiles (λmax > 590 nm) with high fluorescence quantum yields (ϕf = 20–50%).42,44,105 We hoped to translate these favorable optical properties into a protein environment with P-3, thus generating fluorescent proteins with optical properties that are superior to traditional fluorescent proteins. However, P-2 and P-3 exhibit orange emission with a maximum at 566 nm, which is significantly blue-shifted relative to phosphorus meso-triaryl corroles. This observation can readily be rationalized with a qualitative molecular orbital diagram (Figure 5). The b1 HOMO is significantly more destabilized by an electron-donating meso-substituent than a β-substituent because this orbital has greater density at the meso positions relative to the β positions. As a result, the HOMO–LUMO gap (ΔE) for meso-substitution is smaller than β-substitution (ΔEmeso < ΔEβ), which manifests in lower energy (longer wavelength) emission for meso-triaryl corroles relative to β-octaalkyl corroles.
Figure 5.
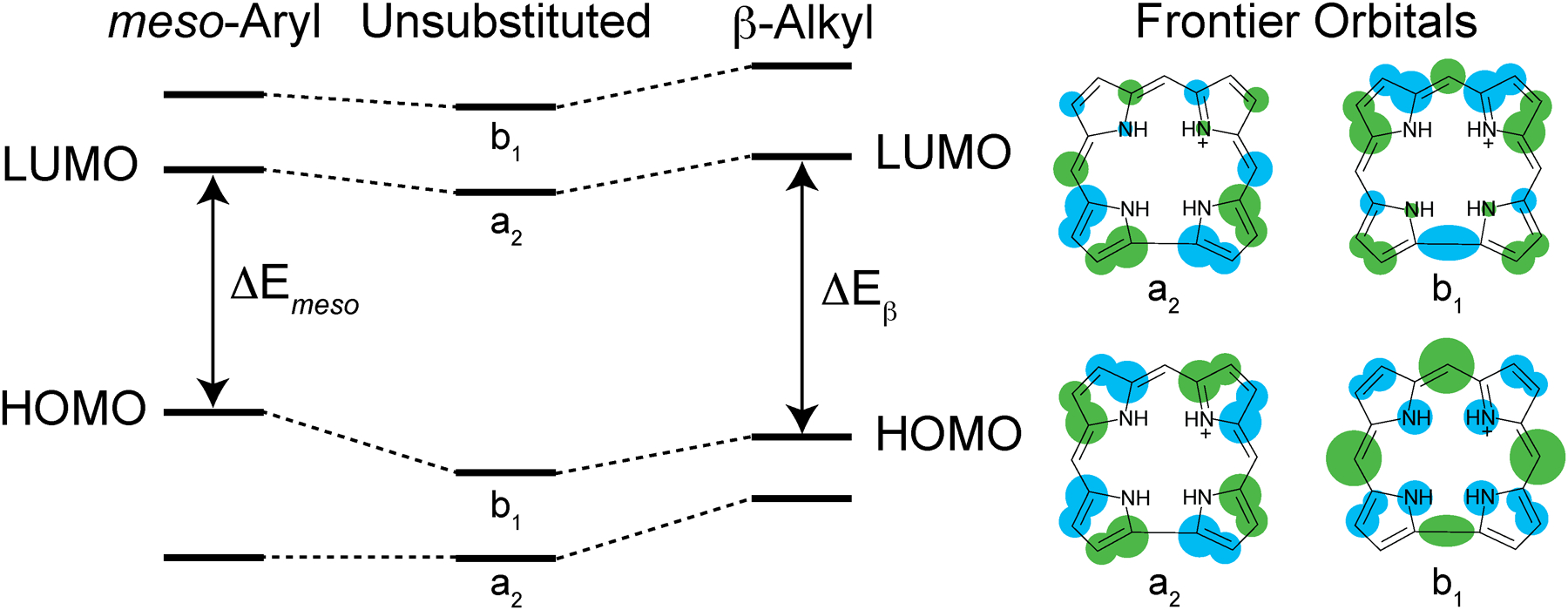
Qualitative molecular orbital diagram illustrating the perturbation of the four frontier orbitals for an unsubstituted corrole core upon meso-aryl substitution and β-alkyl substitution.
While the majority of P(V) meso-triaryl corroles are six-coordinate species, P-2, P-3, and other β-octaalkyl corroles are five-coordinate. It seems that the coordination number correlates with the nature of the peripheral substituents. Phosphorus triaryl corroles exhibit an equilibrium between five- and six-coordinate species, as observed by solution 31P NMR. Electron donating substituents give rise to a 1:1 mixture of five- and six-coordinate complexes, whereas electron-withdrawing groups (e.g., 4-NO2-phenyl, 4-CN-phenyl) yield primarily six-coordinate species.42 Similarly, P corroles with fluorinated aryl groups are exclusively isolated as six coordinate complexes.44,87 Perhaps the reduced electron density of macrocycle increases the effective positive charge on the P center. This leads to an increase in the ionic character of the bonding situation, which could readily be offset with two anionic ligands. Conversely, β-substituted corroles with electron donating alkyl substituents are exclusively isolated as five-coordinate species.82,83 Perhaps this higher electron density at the P center increases the covalent character of the bonds; this may be efficiently utilized for π-bonding to result in the isolation of terminal oxo (P=O) derivatives.
Although phosphorus complexes of β-octaalkyl corroles have been previously characterized,82,83 the emission properties of these molecules have not yet been reported. The fluorescence quantum yield of P-2 is unexpectedly low. This can partially be explained by the conformational flexibility of the propionate chains, which would engender molecular motions that readily deactivate the excited state. In general, six-coordinate P(OH)2 corrole complexes exhibit higher fluorescence quantum yields than the corresponding free-base. For example, ϕf = 31% for the P(OH)2 complex of 5,10,15-tris(pentafluorophenyl)corrole,44 whereas ϕf = 13% for the free-base.90 In the case of corrole 2, the free-base has a higher ϕf than the phosphorus complex: 5.9% vs. 3.9% (Table 1). It is unlikely that the difference in substitution pattern (meso vs. β) accounts for this observation. Indeed, the fluorescence lifetime of H3-2 is comparable to free-base meso-triaryl corroles (4–6 ns),90 and the 2–4 fold decrease in kr likely reflects the molecular motions of the propionate chains. Since the excited-state dynamics of H3-2 is consistent with free-base meso-triaryl corroles, it is expected that P-2 should behave similarly to phosphorus meso-triaryl corroles. The weak fluorescence and short 1.2 ns lifetime of P-2 may be due to solvent interactions that result in the formation of non-radiative decay channels. Since the excited singlet state of P(V) triaryl corroles is a potent oxidant,106 charge-transfer to solvent could occur, thereby enhancing non-radiative deactivation of the excided state. If this were the case, it would be expected that this is a general phenomenon for all P(V) corroles. However, six-coordinate P corroles and related analogs exhibit longer singlet excited state lifetimes (3–4 ns).106–109 Alternatively, the weak fluorescence of P-2 likely reflects differences in phosphorus speciation. Five-coordinate meso-triaryl corroles can be obtained by treating the six-coordinate complex with TFA and it was shown that this five-coordinate species exhibits a lower fluorescence quantum yield (27%) and shorter excited state lifetime (1.61 ns) than the analogous six-coordinate complex (ϕ = 44%, τ = 3.01 ns).91 The short lifetime (1.2 ns) and low quantum yield of five-coordinate P-2 are consistent with these observations. While the speciation accounts for the short lifetime, the underlying chemical explanation for this observation remains unknown.
The short lifetime (1–2 ns) of the phosphorous corrole is consistent for P-2, P-3, and the protein conjugates. Although the excited state of P-3 and the HasA conjugate exhibits biexponental kinetics, the major contribution to the fit (~85%) falls in this range. Despite the similarity in lifetime, the fluorescence quantum yields are quite variable, reflecting differences in the non-radiative decay of the excited state. The protein environment surrounding the corrole likely dictates the photophysical properties of P-3. Both Cs H-NOX and Pa HasA have a tyrosine reside at the heme binding pocket; the phenol side chain is redox active, albeit at high potentials (1.46 V vs. NHE).110 However, since P(V) corroles are potent photo-oxidants (~1.2 V vs. Ag/AgCl or 1.4 V vs. NHE),106 tyrosine oxidation could contribute to the non-radiative deactivation of P-3 in the protein conjugates. Using electrochemical and spectroscopic data, the reduction potential of the singlet excited state can be estimated. Given the structural and spectral similarity of P-2 and the P(V)=O complex of octaethylcorrole (OEC),82 it is expected that the electrochemical properties will also be similar. Using the one-electron reduction potential of PO(OEC) (−1.58 V vs. SCE or −1.34 V vs. NHE)82 and the energy of the Q(0,0) transition of P-2 (2.19 V), the estimated excited state reduction potential is 0.85 V vs. NHE. Consequently, these five-coordinate phosphorus corroles are considerably weaker photo-oxidants than six-coordinate derivatives by 0.55 V. While proton-coupled electron transfer (PCET) lowers the barrier for tyrosyl radical generation,110 it seems unlikely that this process is operative for P-3. A more likely contribution to non-radiative decay of P-3 is histidine residues in the heme binding pocket. Amines are well-known fluorescence quenchers. Since P-3 does not bind histidine, the nitrogen lone-pair of the imidazole side chain can quench corrole fluorescence through photo-induced electron transfer (PET). Both Cs H-NOX and Pa HasA have heme-ligating histidine residues, which are not participating in a bonding interaction with P-3, that are sufficiently close to the corrole cofactor to quench fluorescence. These hypotheses can be tested through site-direct mutagenesis, but such experiments are beyond the scope of this study.
The absorption spectrum of P-3 in buffer is significantly different from that of the protein-bound corrole (Figure 3). Additional evidence of protein-induced changes include a decrease in the FWHM of the Soret and Q bands, as well as the suppression of the Q(0,1) emission band. One interesting observation is the splitting of the Soret band for the H-NOX protein. Protein binding sharpens the Soret band to enable resolution of the x and y polarizations of this transition. DFT calculations demonstrate the presence of two orthogonally polarized transitions (vide supra), but they are rarely resolved experimentally.84 It is likely that hydrogen bonding of the propionate chains to the Y–S–R motif in H-NOX helps to differentiate the dipole moments along the x and y axes of the corrole, enabling the resolution of the polarized Soret transitions. Conversely, HasA lacks this Y–S–R motif and the propionate chains are solvent exposed; the lack of a hydrogen bonding partner does not enable sufficient differentiation of the x and y dipole moments, resulting in the experimental observation of a single Soret transition.
Despite the low fluorescence intensity, the emission profile is concentrated in a relatively sharp band (FWHM = 13–14 nm). This is in contrast to traditional fluorescent proteins,111 which exhibit broad emission profiles that can span over 200 nm, as in the case of mHoneydew, mBanana, and mTangerine (Figure 6a, Figure S26). Of the examples illustrated in Figures 6a and S26, mKO (Kusabira Orange) and mKO2 have the narrowest peak for the main emission transition (FWHM = 27–30 nm), but this is nearly twice as broad as the H-NOX and HasA conjugates. As a result, the protein conjugates of P-3 are better suited for FRET-based and ratiometric applications because the emission intensity is concentrated in a significantly smaller spectral window. This attribute results in enhanced emission intensity, despite their low fluorescence quantum yields. Figure 6b shows the emission profiles of the fluorescent proteins in Figure 6a that have been scaled to the quantum yield of the protein. In this way, the emission intensity of the proteins is directly comparable. Although the HasA conjugate exhibits a 8.5-fold lower fluorescence quantum yield than mKO2, the corrole conjugate only exhibits a 5-fold decrease in relative fluorescence intensity at the emission maximum. Similarly, the HasA conjugate exhibits only a 1.7-fold decrease in emission intensity relative to mTangerine, despite having a 4-fold lower fluorescence quantum yield. The most striking example is a comparison of the HasA conjugate and mBanana; while the quantum yield is nearly 10 times less, the emission intensity of HasA is only 4-fold lower than mBanana. Finally, the H-NOX conjugate and mHoneydew display equal emission intensity, but the HasA conjugate has a 5-fold lower quantum yield. These comparisons illustrate that, despite their low fluorescence quantum yield, the P-3 conjugates of H-NOX and HasA offer several advantages over traditional fluorescent proteins.
Figure 6.
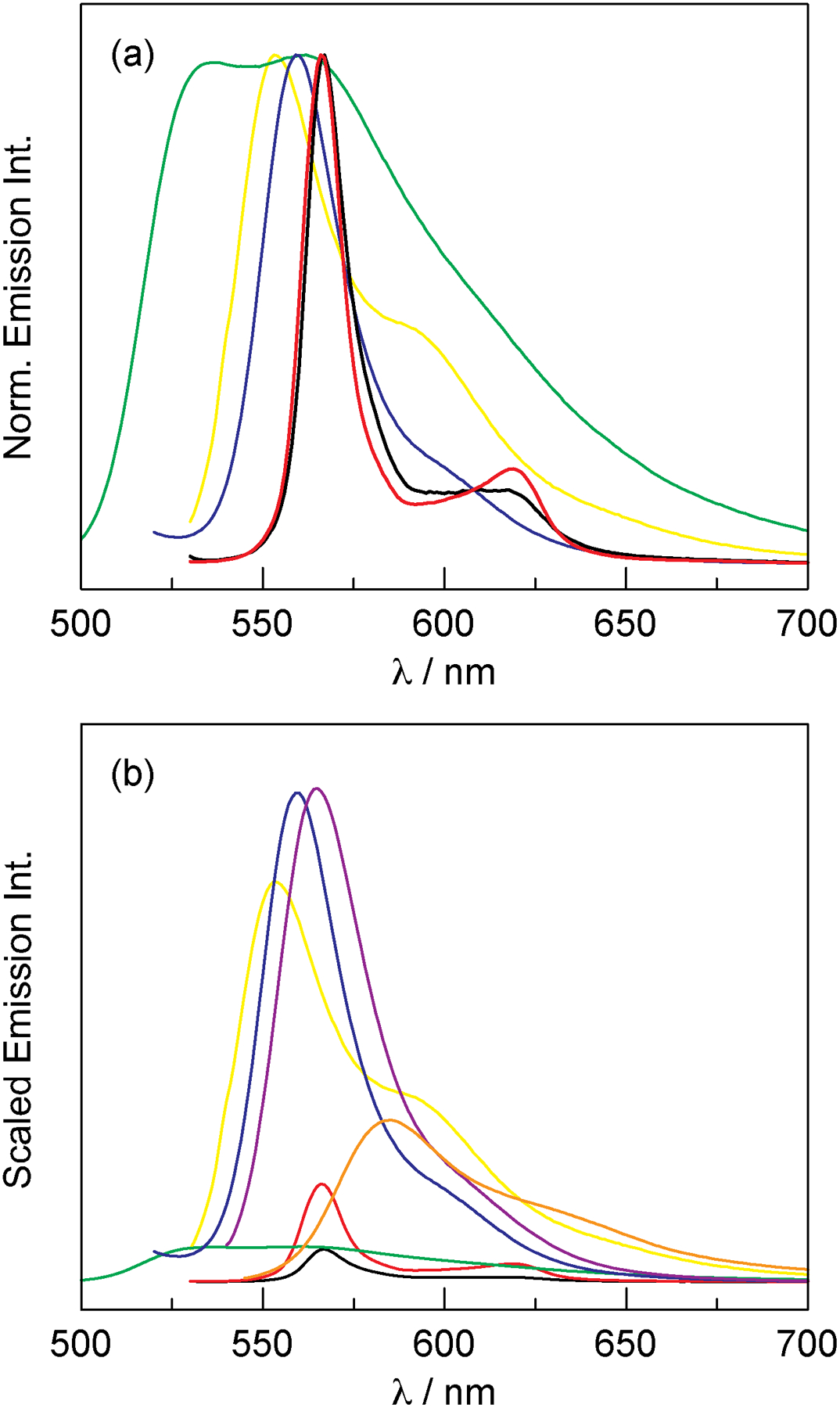
(a) Normalized emission spectra of the P-3 conjugates and traditional fluorescent proteins that emit in a similar spectral window: P-3 H-NOX (▬), P-3 HasA (▬), mBanana (▬), mHoneydew (▬), and mKO (▬). Spectral data for the fluorescent proteins was obtained from the online fluorescent protein database FPbase (fpbase.org).111 (b) Emission spectra of fluorescent proteins that is scaled by quantum yield: P-3 H-NOX ϕ = 2.5% (▬), P-3 HasA ϕ = 7.3% (▬), mBanana ϕ = 70% (▬), mHoneydew ϕ = 12% (▬), mKO ϕ = 60% (▬), mKO2 ϕ = 62% (▬), and mTangerine ϕ = 30% (▬). Quantum yield data was obtained from FPbase (fpbase.org).111
Conclusions
We have presented a proof-of-principle study demonstrating that fluorescent phosphorus corroles can be incorporated into the scaffolds of Cs H-NOX and Pa HasA. Distinctive changes in the absorption and emission spectra confirm corrole binding in the heme pocket of the proteins. Corrole emission is enhanced in the HasA conjugate, while it is quenched in H-NOX, relative to the free molecule. This observation is counter to the expectation that corrole emission would be most enhanced in H-NOX because of the buried heme pocket and hydrogen bonding with the Y–S–R motif, which would limit the conformational flexibility of the corrole. Although vibrational motions are more restricted in H-NOX relative to HasA (i.e., suppression of the Q(0,1) transition), the non-radiative rate constant is slower in HasA than H-HOX. This disparity suggests that environmental factors in the heme binding pocket are playing a significant role in dictating the photophysical properties of these conjugates.
We are currently working to improve the optical properties of the conjugates by modifying both the corrole and the heme binding pocket of the proteins. Conversion of five-coordinate P-2 to a six-coordinate species (as a difluoride (PF2),44 dialkyl (PR2),82 or dialkoxide (P[OR]2)42,89) should enhance the fluorescence of the corrole. However, it is unclear if or how such a six-coordinate species would bind to the H-NOX and HasA proteins. Alternatively, modification of the corrole by β-fluorination or β-trifluoromethylation would reduce the electron density of the macrocycle and could enable the isolation of stable Al and Ga complexes. These derivatives are expected to exhibit more red-shifted emission profiles and higher fluorescence quantum yields than the corresponding P complex. So far, attempts to adapt the synthesis of 1 using 3,4-difluoropyrrole instead of 3,4-dimethylpyrrole have been unsuccessful. Additionally, it is unclear exactly how the corrole complex is interacting with the protein, especially since P-2 prefers to remain five-coordinate in the presence of a nitrogenous base (e.g., pyridine or histidine). X-ray crystal structures of the protein–corrole conjugates would aid in the structure-guided design of these unnatural cofactors. With a detailed understanding of the corrole–protein interaction, selected variants can be generated to enhance the optical properties of these fluorescent proteins. Together, modification of the corrole cofactor and heme binding pocket of the protein will result in enhanced conjugates with higher fluorescent quantum yields.
Supplementary Material
Synopsis:
The native heme cofactor in H-NOX and HasA is replaced with a phosphorus corrole to yield fluorescent protein derivatives. Upon binding to HasA, the corrole exhibits enhanced emission relative to the free corrole, whereas H-NOX binding quenches fluorescence. Despite the low quantum yield, these corrole-substituted proteins exhibit more intense fluorescence in a narrower emission profiles than traditional fluorescence proteins.
ACKNOWLEDGMENTS
C.M.L. acknowledges the Miller Institute at UC Berkeley for a Postdoctoral Fellowship. This research was supported by a grant from the National Institutes of Health (NIH): R01GM127854 (to M.A.M). Calculations were performed using the Molecular Graphics and Computation Facility at UC Berkeley, which is partially supported by NIH S10OD023532. We are deeply indebted to Dr. Benjamin Guthrie for his guidance with cloning and protein expression. We thank Dr. Sho Takatori for providing a sample of Pseudomonas aeruginosa PAO1 and Sarah Chen for assisting with the expression of HasA. We thank Trevor Roberts for acquiring time-resolved emission data as well as Professor Naomi Ginsberg for allowing us to access the instrument. The members of the Marletta lab are graciously thanked for critically evaluating this manuscript.
Footnotes
Supporting Information
NMR spectra, results of DFT and TD-DFT calculations, as well as additional experimental details and results are presented in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Prasher DC; Eckenrode VK; Ward WW; Prendergast FG; Cormier MJ Primary Structure of the Aequorea victoria Green-Fluorescent Protein. Gene 1992, 111, 229–233. [DOI] [PubMed] [Google Scholar]
- (2).Chalfie M; Tu Y; Euskirchen G; Ward WW; Prasher DC Green Fluorescent Protein as a Marker for Gene Expression. Science 1994, 263, 802–805. [DOI] [PubMed] [Google Scholar]
- (3).Chudakov DM; Matz MV; Lukyanov S; Lukyanov KA Fluorescent Proteins and their Applications in Imaging Living Cells and Tissues. Physiol. Rev 2010, 90, 1103–1163. [DOI] [PubMed] [Google Scholar]
- (4).Chu J; Xing Y; Lin M Far-Red and Near-Infrared Fluorescent proteins. In The Fluorescent Protein Revolution; Day RN, Davidson MW, Eds.; CRC Press: Boca Raton, FL, 2014; pp 157–174. [Google Scholar]
- (5).König K Multiphoton Microscopy in the Life Sciences. J. Microsc 2000, 200, 83–104. [DOI] [PubMed] [Google Scholar]
- (6).Lemon CM; Curtin PN; Somers RC; Greytak AB; Lanning RM; Jain RK; Bawendi MG; Nocera DG Metabolic Tumor Profiling with pH, Oxygen, and Glucose Chemosensors on a Quantum Dot Scaffold. Inorg. Chem 2014, 53, 1900–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xu H; Aylott JW; Kopelman R; Miller TJ Philbert MA A Real-Time Ratiometric Method for the Determination of Molecular Oxygen Inside Living Cells Using Sol–Gel-Based Spherical Optical Nanosensors with Applications to Rat C6 Glioma. Anal. Chem 2001, 73, 4124–4133. [DOI] [PubMed] [Google Scholar]
- (8).Sun H; Scharff-Poulsen AM; Gu H; Almdal K Synthesis and Characterization of Ratiometric, pH Sensing Nanoparticles with Covalently Attached Fluorescent Dyes. Chem. Mater 2006, 18, 3381–3384. [Google Scholar]
- (9).Pellicena P; Karow DS; Boon EM; Marletta MA; Kuriyan J Crystal Structure of an Oxygen-Binding Heme Domain Related to Soluble Guanylate Cyclases. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Boon EM; Huang SH; Marletta MA A Molecular Basis for NO Selectivity in Soluble Guanylate Cyclase. Nat. Chem. Biol 2005, 1, 53–59. [DOI] [PubMed] [Google Scholar]
- (11).Erbil WK; Price MS; Wemmer DE; Marletta MA A Structural Basis for H-NOX Signaling in Shewanella oneidensis by Trapping a Histidine Kinase Inhibitory Conformation. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 19753–19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Weinert EE; Plate L; Whited CA; Olea C; Marletta MA Determinants of Ligand Affinity and Heme Reactivity in H-NOX Domains. Angew. Chem. Int. Ed 2010, 49, 720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Herzik MA; Jonnalagadda R; Kuriyan J; Marletta MA Structural Insights into the Role of Iron-Histidine Bond Cleavage in Nitric Oxide-Induced Activation of H-NOX Gas Sensor Proteins. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E4156–E4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Horst BG; Marletta MA Physiological Activation and Deactivation of Soluble Guanylate Cyclase. Nitric Oxide 2018, 77, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Guo Y; Marletta MA Structural Insight into H-NOX Gas Sensing and Cognate Signaling Protein Regulation. ChemBioChem 2019, 20, 7–19. [DOI] [PubMed] [Google Scholar]
- (16).Senge MO; M cGow n SA; O’B ien JM Con or-mation Control of Cofactors in Nature – The Influence of Protein-Induced Macrocycle Distortion on the Biological Function of Tetrapyrroles. Chem. Commun 2015, 51, 17031–17063. [DOI] [PubMed] [Google Scholar]
- (17).Boon EM; Marletta MA Sensitive and Selective Detection of Nitric Oxide Using an H-NOX Domain. J. Am. Chem. Soc 2006, 128, 10022–10023. [DOI] [PubMed] [Google Scholar]
- (18).Winter MB; McLaurin EJ; Reece SY; Olea C; Nocera DG; Marletta MA Ru-Porphyrin Protein Scaffolds for Sensing O2. J. Am. Chem. Soc 2010, 132, 5582–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Winter MB; Klemm PJ; Phillips-Piro CM; Raymond KN; Marletta MA Porphyrin-Substituted H-NOX Proteins as High-relaxivity MRI Contrast Agents. Inorg. Chem 2013, 52, 2277–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nierth A; Marletta MA Direct meso-Alkynylation of Metalloporphyrins through Gold Catalysis for Hemoprotein Engineering. Angew. Chem. Int. Ed 2014, 53, 2611–2614. [DOI] [PubMed] [Google Scholar]
- (21).Binet R; Létoffé S; Ghigo JM; Delepelaire P; Wandersman C Protein Secretion by Gram-Negative Bacterial ABC Exporters – A Review. Gene 1997, 192, 7–11. [DOI] [PubMed] [Google Scholar]
- (22).Izadi N; Henry Y; Haladjian J; Goldberg ME; Wandersman C; Delepierre M; Lecroisey A Purification and Characterization of an Extracellular Heme-Binding Protein, HasA, Involved in Heme Iron Acquisition. Biochemistry 1997, 36, 7050–7057. [DOI] [PubMed] [Google Scholar]
- (23).Létoffé S; Nato F; Goldberg ME; Wandersman C Interactions of HasA, a Bacterial Haemophore, with Haemoglobin and with Its Outer Membrane Receptor HasR. Mol. Microbol 1999, 33, 546–555. [DOI] [PubMed] [Google Scholar]
- (24).Contreras H; Chim N; Credali A; Goulding CW Heme Uptake in Bacterial Pathogens. Curr. Opin. Chem. Biol 2014, 19, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Deniau C; Gilli R; Izadi-Pruneyre N; Létoffé S; Delepierre M; Wandersman C; Briand C; Lecroisey A Thermody-namics of Heme Binding to the HasASM Hemophore: Effect of Mutations at Three Key Residues for Heme Uptake. Biochemistry 2003, 42, 10627–10633. [DOI] [PubMed] [Google Scholar]
- (26).Jepkorir G; Rodríguez JC; Rui H; Im W; Lovell S; Battaile KP; Alontaga AY; Yukl ET; Moënne-Loccoz P; Rivera M Structural, NMR Spectroscopic, and Computational Inves-tigation of Hemin Loading in the Hemophore HasAp from Pseudomonas aeruginosa. J. Am. Chem. Soc 2010, 132, 9857–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kumar R; Lovell S; Matsumura H; Battaile KP; Moënne-Loccoz P; Rivera M The Hemophore HasA from Yersinia pestis (HasAyp) Coordinates Hemin with a Single Residue, Tyr75, and with Minimal Conformational Change. Biochemistry 2013, 52, 2705–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kumar R; Matsumura H; Lovell S; Yao H; Rodríguez JC; Battaile KP; Moënne-Loccoz P; Rivera M Replacing the Axial Ligand Tyrosine 75 or Its Hydrogen Bond Partner Histidine 83 Minimally Affects Hemin Acquisition by the Hemophore HasAp from Pseudomonas aeruginosa. Biochemistry 2014, 53, 2112–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shirataki C; Shoji O; Terada M; Ozaki S; Sugimoto H; Shiro Y; Watanabe Y Inhibition of Heme Uptake in Pseudomonas aeruginosa by its Hemophore (HasAp) Bound to Synthetic Metal Complexes. Angew. Chem. Int. Ed 2014, 53, 2862–2866. [DOI] [PubMed] [Google Scholar]
- (30).Uehara H; Shisaka Y; Nishimura T; Sugiomoto H; Shiro Y; Miyake Y; Shinokubo H; Watanabe Y; Shoji O Structures of the Heme Acquisition Protein HasA with Itron(III)-5,15-Diphenylporphyrin and Derivatives Thereof as an Artificial Prosthetic Group. Angew. Chem. Int. Ed 2017, 56, 15279–15283. [DOI] [PubMed] [Google Scholar]
- (31).Deniau C; Couprie J; Simenel C; Kumar V; Stojiljkovic I; Wandersman C; Delepierre M; Lecroisey A 1H, 15N and 13C Resonance Assignments for the Gallium Protoporphyrin IX-HasAsm Hemophore Complex. J. Biomol. NMR 2001, 21, 189–199. [DOI] [PubMed] [Google Scholar]
- (32).Wolff N; Deniau C; Létoffé S; Simenel C; Kumar V; Stojiljkovic I; Wandersman C; Delepierre M; Lecroisey A Histidine pKa Shifts and Changes of Tautomeric States Induced by the Binding of Gallium-Protoporphyrin IX in the Hemophore HasASM. Protein Sci. 2002, 11, 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shisaka Y; Iwai Y; Yamada S; Uehara H; Tosha T; Sugimoto H; Shiro Y; Stanfield JK; Ogawa K; Watanabe Y; Shoji O Hijacking the Heme Acquisition System of Pseudomonas aeruginosa for the Delivery of Phthalocyanine as an Antimicrobial. ACS Chem. Biol 2019, 14, 1637–1642. [DOI] [PubMed] [Google Scholar]
- (34).Gouterman M Optical Spectra and Electronic Structure of Porphyrins and Related Rings. In The Porphyrins; Dolphin D, Ed.; Academic Press: New York, 1978; Vol. 3; pp 1–165. [Google Scholar]
- (35).O łowski R; G yko D; G yko DT Synthesis o Co oles and Their Heteroanalogs. Chem. Rev 2017, 117, 3102–3137. [DOI] [PubMed] [Google Scholar]
- (36).Barata JFB; Neves MGPMS; Faustino MAF; Tomé AC; Cavaleiro JAS Strategies for Corrole Functionalization. Chem. Rev 2017, 117, 3192–3253. [DOI] [PubMed] [Google Scholar]
- (37).Ghosh A Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations. Chem. Rev 2017, 117, 3798–3881. [DOI] [PubMed] [Google Scholar]
- (38).Aviv-Harel I; Gross Z Coordination Chemistry of Corroles with Focus on Main Group Elements. Coord. Chem. Rev 2011, 255, 717–736. [Google Scholar]
- (39).Bendix J; Dmochowski IJ; Gray HB; Mahammed A; Simkhovich L; Gross Z Structural, Electrochemical, and Photophysical Properties of Gallium(III) 5,10,15-Tris(pentafluorophenyl)-corrole. Angew. Chem. Int. Ed 2000, 39, 4048–4051. [DOI] [PubMed] [Google Scholar]
- (40).Mahammed A; Gross ZJ Inorg. Biochem Aluminum Corrolin, a Novel Chlorophyll Analogue. 2002, 88, 305–309. [DOI] [PubMed] [Google Scholar]
- (41).Kowalska D; Liu X; Tripathy U; Mahammed A; Gross Z; Hirayama S; Steer RP Ground- and Excited-State Dynamics of Aluminum and Gallium Corroles. Inorg. Chem 2009, 48, 2670–2676. [DOI] [PubMed] [Google Scholar]
- (42).Ghosh A; Ravikanth M Synthesis, Structure, Spectroscopic, and Electrochemical Properties of Highly Fluorescent Phospho-rus(V)–meso-Triarylcorroles. Chem. Eur. J 2012, 18, 6386–6396. [DOI] [PubMed] [Google Scholar]
- (43).Liang X; Mack J; Zheng LM; Shen Z; Kobayashi N Phosphorus(V)-Corrole: Synthesis, Spectroscopic Properties, Theoret-ical Calculations, and Potential Utility for in vivo Applications in Living Cells. Inorg. Chem 2014, 53, 2797–2802. [DOI] [PubMed] [Google Scholar]
- (44).Vestfrid J; Kothari R; Kostenko A; Goldberg I; Tumanskii B; Gross Z Intriguing Physical and Chemical Properties of Phosphorus Corroles. Inorg. Chem 2016, 55, 6061–6067. [DOI] [PubMed] [Google Scholar]
- (45).Vestfrid J; Botohansky M; Palmer JH; Durrell AC; Gray HB; Gross Z Iodinated Aluminum(III) Corroles with Long-Lived Triplet Excited States. J. Am. Chem. Soc 2011, 133, 12899–12901. [DOI] [PubMed] [Google Scholar]
- (46).Yang Y; Jones D; von Haimberger T; Linke M; Wagnert L; Berg A; Levanon H; Zacarias A; Mahammed A; Gross Z; Heyne K Assignment of Aluminum Corroles Absorption Bands to Electronic Transitions by Femtosecond Polarization Resolved VIS-Pump IR-Probe Spectroscopy. J. Phys. Chem. A 2012, 116, 1023–1029. [DOI] [PubMed] [Google Scholar]
- (47).Ueta K; Fukuda M; Kim G; Shimizu S; Tanaka T; Kim D; Osuka A The First Silicon(IV) Corrole Complexes: Synthesis, Structures, Properties, and Formation of a μ-Oxo Dimer. Chem. Eur. J 2018, 24, 7637–7646. [DOI] [PubMed] [Google Scholar]
- (48).Pomarico G; Monti D; Bischetti M; Savoldelli A; Fronczek FR; Smith KM; Genovese D; Prodi L; Paolesse R Silicon(IV) Corroles. Chem. Eur. J 2018, 24, 8438–8446. [DOI] [PubMed] [Google Scholar]
- (49).Agadjanian H; Ma J; Rentsendorj A; Valluripalli V; Hwang JY; Mahammed A; Farkas DL; Gray HB; Gross Z; Medina-Kauwe LK Tumor Detection and Elimination by a Target-ed Gallium Corrole. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 6105–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hwang JY; Gross Z; Gray HB; Medina-Kauwe LK; Farkas DL Ratiometric Spectral Imaging for Fast Tumor Detection and Chemotherapy Monitoring in vivo. J. Biomed. Opt 2011, 16, 066007/1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Teo RD; Hwang JY; Termini J; Gross Z; Gray HB Fighting Cancer with Corroles. Chem. Rev 2017, 117, 2711–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mahammed A; Gray HB; Weaver JJ; Sorasaenee K; Gross Z Amphiphilic Corroles Bind Tightly to Human Serum Albumin. Bioconjugate Chem. 2004, 15, 738–746. [DOI] [PubMed] [Google Scholar]
- (53).Acunha TV; Chaves OA; Iglesias BA Fluorescent Pyrene Moiety in Fluorinated C6F5-Corroles Increases the Interaction with HAS and CT-DNA. J. Porphyrins Phthalocyanines 2021, 25, DOI: 10.1142/S1088424620500534. [DOI] [Google Scholar]
- (54).Agadjanian H; Weaver JJ; Mahammed A; Rentsendorj A; Bass S; Kim J; Dmochowski IJ; Margalit R; Gray HB; Gross Z; Medina-Kauwe LK Specific Delivery of Corroles to Cells via Noncovalent Conjugates with Viral Proteins. Pharm. Res 2006, 23, 367–377. [DOI] [PubMed] [Google Scholar]
- (55).Haber A; Agadjanian H; Medina-Kauwe LK; Gross Z Corroles that Bind with High Affinity to Both Apo and Holo Transferrin. J. Inorg. Biochem 2008, 102, 446–457. [DOI] [PubMed] [Google Scholar]
- (56).Soll M; Goswami TK; Chen QC; Saltsman I; Teo RD; Shahgholi M; Lim P; Di Billo AJ; Cohen S; Termini J; Gray HB; Gross Z Cell-Penetrating Protein/Corrole Nanoparticles. Sci. Rep 2019, 9, 2294/1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sharma VK; Mahammed A; Soll M; Tumanskii B; Gross Z Corroles and Corrole/Transferrin Nanoconjugates as Candi-dates for Sonodynamic Therapy. Chem. Commun 2019, 55, 12789–12792. [DOI] [PubMed] [Google Scholar]
- (58).Matsuo T; Hayashi A; Abe M; Matsuda T; Hisaeda Y; Hayashi Y Meso-Unsubstituted Iron Corrole in Hemoproteins: Re-markable Differences in Effects on Peroxidase Activities between Myoglobin and Horseradish Peroxidase. J. Am. Chem. Soc 2009, 131, 15124–15125. [DOI] [PubMed] [Google Scholar]
- (59).Bröring M; Brégier F; Burghaus O; Kleeberg C A Biomimetic Copper Corrole – Preparation, Characterization, and Reconstitution with Horse Heart Apomyoglobin. Z. Anorg. Alle. Chem 2010, 636, 1760–1766. [Google Scholar]
- (60).Winter MB; Woodward JJ; Marletta MA An Escherichia coli Expression-Based Approach for Porphyrin Substitution in Heme Proteins. In Cytochrome P450 Protocols, Methods in Molecular Biology; Phillips IR, Shepard EA, Ortiz de Montellano PR, Eds,; Humana Press: Totowa, NJ, 2013; Vol. 987; pp 95–106. [DOI] [PubMed] [Google Scholar]
- (61).Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar]
- (62).CRC Handbook of Chemistry and Physics, 84th ed.; CRC Press: Boca Raton, FL, 2003. [Google Scholar]
- (63).Tseng Y-M; Thompson AR Densities and Refractive Indices of Aqueous Monoethanolamine, Diethanolamine, Triethanolamine. J. Chem. Eng. Data 1964, 9, 264–267. [Google Scholar]
- (64).Kubin RF; Fletcher AN Fluorescence Quantum Yields of Some Rhodamine Dyes. J. Lumin 1982, 27, 455–462. [Google Scholar]
- (65).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- (66).Becke AD A New Mixing of Hartree–Fock and Local Density-Functional Theories. J. Chem. Phys 1993, 98, 1372–1377. [Google Scholar]
- (67).Becke AD Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (68).Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (69).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Hender-son T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Haseg-awa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogli-aro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, Revision A.03; Gaussian, Inc.: Walling-ford, CT, 2016. [Google Scholar]
- (70).Barone V; Cossi M Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- (71).Cossi M; Rega N; Scalmani G; Barone V Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- (72).Bauernschmitt R; Ahlrichs R Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett 1996, 256, 454–464. [Google Scholar]
- (73).Casida ME; Jamorski C; Casida KC; Salahub DR Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold. J. Chem. Phys 1998, 108, 4439–4449. [Google Scholar]
- (74).Stratmann RE; Scuseria GE; Frisch MJ An Efficient Implementation of Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules. J. Chem. Phys 1998, 109, 8218–8224. [Google Scholar]
- (75).Van Caillie C; Amos RD Geometric Derivatives of Excitation Energies using SCF and DFT. Chem. Phys. Lett 1999, 308, 249–255. [Google Scholar]
- (76).Scalmani G; Frisch MJ; Mennucci B; Tomasi J; Cammi R; Barone V Geometries and Properties of Excited States in the Gas Phase and in Solution: Theory and Application of a Time-Dependent Density Functional Theory Polarizable Continuum Model. J. Chem. Phys 2006, 124, 094107/1–15. [DOI] [PubMed] [Google Scholar]
- (77).Paolesse R; Froiio A; Nardis S; Mastroianni M; Russo M; Nurco DJ; Smith KM Novel Aspects of the Chemistry of 1,19-Diunsubstituted a,c-Biladienes. J. Porphyrins Phthalocyanines 2003, 7, 585–592. [Google Scholar]
- (78).Tardieux C; Gros CP; Guilard R On Corrole Chemistry. An Isomerization Study and Oxidative Cleavage of the Corrole Macroring to a Biliverdin Structure. J. Heterocycl. Chem 1998, 35, 965–970. [Google Scholar]
- (79).Lemon CM Corrole Photochemistry. Pure Appl. Chem 2020, 92, 1901–1919. [Google Scholar]
- (80).Gross Z; Gray HB How Do Corroles Stabilize High Valent Metals? Comments Inorg. Chem 2006, 27, 61–72. [Google Scholar]
- (81).Geier GR; Chick JFB; Callinan JB; Reid CG; Auguscinski WP A Survey of Acid Catalysis and Oxidation Condi-tions in the Two-Step One-Flask Synthesis of Meso-Substituted Corroles via Dipyrromethanedicarbinols and Pyrrole. J. Org. Chem 2004, 69, 4159–4169. [DOI] [PubMed] [Google Scholar]
- (82).Kadish KM; Ou Z; Adamian VA; Guilard R; Gros CP; Erben C; Will S; Vogel E Corroles with Group 15 Ions. 2. Synthesis and Characterization of Octaethylcorroles Containing a Phosphorus Central Atom. Inorg. Chem 2000, 39, 5675–5682. [DOI] [PubMed] [Google Scholar]
- (83).Paolesse R; Boschi T; Licoccia S; Khoury RG; Smith KM Phosphorus Complex of Corrole. Chem. Commun 1998, 1119–1120. [Google Scholar]
- (84).Lemon CM; Halbach RL; Huynh M; Nocera DG Photophysical Properties of β-Substituted Free-Base Corroles. Inorg. Chem 2015, 54, 2713–2725. [DOI] [PubMed] [Google Scholar]
- (85).Lemon CM; Hwang SJ; Maher AG; Powers DC Nocera DG Halogen Photoelimination from SbV Dihalide Corroles. Inorg. Chem 2018, 57, 5333–5342. [DOI] [PubMed] [Google Scholar]
- (86).Lemon CM; Maher AG; Mazzotti AR; Powers DC; Gonzalez MI; Nocera DG Multielectron C–H Photoactivation with an Sb(V) Oxo Corrole. Chem. Commun 2020, 56, 5247–5250. [DOI] [PubMed] [Google Scholar]
- (87).Simkhovich L; Mahammed A; Goldberg I; Gross Zeev. Synthesis and Characterization of Germanium, Tin, Phosphorus, Iron, and Rhodium Complexes of Tris(pentafluorophenyl)corrole, and the Utilization of the Iron and Rhodium Corroles as Cyclopropanation Catalysts. Chem. Eur. J 2001, 7, 1041–1055. [DOI] [PubMed] [Google Scholar]
- (88).Pomarico G; Tortora L; Fronczek FR; Smith KM; Paolesse R Selective Nitration and Bromination of Surprisingly Ruffled Phosphorus Corroles. J. Inorg. Biochem 2016, 158, 17–23. [DOI] [PubMed] [Google Scholar]
- (89).Gao D; Azarias C; D’Aléo A; Giorgi M; Siri O; Balaban TS; Jacquemin D; Canard G Synthesis and Characterization of Ruffled Phosphorus meso-Ester Corroles. Eur. J. Inorg. Chem 2017, 780–788. [Google Scholar]
- (90).Ventura B; Esposti AD; Koszarna B; Gryko DT; Flamigni L Photophysical Characterization of Free-Base Corroles, Promising Chromophores for Light Energy Conversion and Singlet Oxygen Generation. New J. Chem 2005, 29, 1559–1566. [Google Scholar]
- (91).Ghosh A; Lee W-Z; Ravikanth M Synthesis, Structure and Properties of a Five-Coordinate Oxophosphorus(V) meso-Triphenylcorrole. Eur. J. Inorg. Chem 2012, 4231–4239. [Google Scholar]
- (92).Harrison HR; Hodder OJR; Hodgkin DC Crystal and Molecular Structure of 8,12-Diethyl-2,3,7,13,17,18-hexamethylcorrole. J. Chem. Soc. B 1971, 540–645. [Google Scholar]
- (93).Gouterman M Study of the Effects of Substitution on the Absorption Spectra of Porphin. J. Chem. Phys 1959, 30, 1139–1161. [Google Scholar]
- (94).Gouterman M Spectra of Porphyrins. J. Mol. Spectrosc 1961, 6, 138–163. [Google Scholar]
- (95).Gouterman M; Wagnière GH; Snyder LC Spectra of Porphyrins: Part II. Four Orbital Model. J. Mol. Spectrosc 1963, 11, 108–127. [Google Scholar]
- (96).Ghosh A; Wondimagegn T; Parusel ABJ Electronic Structure of Gallium, Copper, and Nickel Complexes of Corrole. High-Valent Transition Metal Centers versus Noninnocent Ligands. J. Am. Chem. Soc 2000, 122, 5100–5104. [Google Scholar]
- (97).Albrett AM; Conradie J; Ghosh A; Brothers PJ DFT Survey of Monoboron and Diboroncorroles: Regio- and Stereochemi-cal Preferences for a Constrained, Low-Symmetry Macrocycle. Dalton Trans. 2008, 4464–4473. [DOI] [PubMed] [Google Scholar]
- (98).Dreuw A; Head-Gordon M Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev 2005, 105, 4009–4037. [DOI] [PubMed] [Google Scholar]
- (99).Jacquemin D; Wathelet V; Perpète EA; Adamo C Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput 2009, 5, 2420–2435. [DOI] [PubMed] [Google Scholar]
- (100).Jacquemin D; Mennucci B; Adamo C Excited-State Calculations with TD-DFT: From Benchmarks to Simulations in Complex Environments. Phys. Chem. Chem. Phys 2011, 13, 16987–16998. [DOI] [PubMed] [Google Scholar]
- (101).Li J-M; Umanoff H; Proenca R; Russell CS; Cosloy SD Cloning of the Escherichia coli K-12 hemB Gene. J. Bacteriol 1988, 170, 1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Horst BG; Stewart EM; Nazarian AA; Marletta MA Characterization of a Carbon Monoxide-Activated Soluble Guanylate Cyclase from Chlamydomonas reinhardtii. Biochemistry 2019, 58, 2250–2259. [DOI] [PubMed] [Google Scholar]
- (103).Kwon S; Jung Y; Lim D Proteomic Analysis of Heat-Stable Proteins in Escherichia coli. BMB Rep. 2008, 41, 108–111. [DOI] [PubMed] [Google Scholar]
- (104).Li L; Li C; Zhang Z; Alexov E On the Dielectric “Const nt” o P oteins: S ooth Dielect ic Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput 2013, 9, 2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Naitana ML; Nardis S; Pomarico G; Raggio M; Caroleo F; Cicero DO; Lentini S; Prodi L; Genovese D; Mitta S; Sgarlata A; Fanfoni M; Persichetti L; Paolesse R A Highly Emissive Water-Soluble Phorphorus Corrole. Chem. Eur. J 2017, 23, 905–916. [DOI] [PubMed] [Google Scholar]
- (106).Mahammed A; Chen K; Vestfrid J; Zhao J; Gross Z Phosphorus Corrole Complexes: From Property Tuning to Applications in Photocatalysis and Triplet–Triplet Annihilation Upconversion. Chem. Sci 2019, 10, 7091–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Chen Q-C; Xiao Z-Y; Fite S; Mizrahi A; Fridman N; Zhan X; Keisar O; Cohen Y; Gross Z Tuning Chemical and Physical Properties of Phosphorus Corroles for Advanced Applications. Chem. Eur. J 2019, 25, 11383–11388. [DOI] [PubMed] [Google Scholar]
- (108).Katturi NK; Balahoju SA; Ramya AR; Biswas C; Raavi SSK; Giribabu L; Soma VR Ultrafast Photophysical and Nonlinear Optical Properties of Novel Free Base and Axially Substituted Phosphorus (V) Corroles. J. Mol. Liq 2020, 311, 113308/1–10. [Google Scholar]
- (109).Lu X; Zhang X-F Phosphorus Tetrabenzocorrolazine from Its Metal-Free Phthalocyanine Precursor: Its Facile Synthesis, High Fluorescence Emission, Efficient Singlet Oxygen Formation, and Promising Hole Transporting Material. Dyes Pigm. 2020, 179, 108421/1–7. [Google Scholar]
- (110).Reece SY; Nocera DG Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem 2009, 78, 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Lambert TJ FPbase: A Community-Editable Fluorescent Protein Database. Nat. Methods 2019, 16, 277–278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.