

Abstract
Histone deacetylases (HDACs) inhibit the acetylation of crucial autophagy genes, thereby deregulating autophagy and autophagic cell death (ACD) and facilitating cancer cell survival. Vorinostat, a broad-spectrum pan-HDAC inhibitor, inhibits the deacetylation of key autophagic markers and thus interferes with ACD. Vorinostat-regulated ACD can have an autophagy-mediated, -associated or -dependent mechanism depending on the involvement of apoptosis. Molecular insights reveal that hyperactivation of the PIK3C3/VPS34–BECN1 complex increased lysosomal disparity and enhanced mitophagy. These changes were followed by reduced mitochondrial biogenesis and by secondary signals that enabled superactivated, nonselective or bulk autophagy, leading to ACD. Although the evidence is limited, this review focuses on molecular insights into vorinostat-regulated ACD and describes critical concepts for clinical translation.
Keywords: autophagy, cancer, cell death, histone deacetylases, vorinostat
Teaser:
Vorinostat activates excessive autophagy through hyperactivation of the PIK3C3/VPS34–BECN1 complex, resulting in disrupted lysosomal homeostasis, altering the coordination of mitophagy-mitochondrial biogenesis and leading to superactivated, nonselective/bulk autophagy that could act as an alternative cell death mechanism in potential cancer therapy.
Introduction
Disruption of the ‘epigenetic code’ as the result of legitimate epimutations and post-translational modifications drives the aberrant gene expression responsible for malignant transformation, cancer initiation and progression. Histone deacetylation, an antagonistic post-translational epigenetic co-repressor mechanism, promotes carcinogenesis and tumor progression through deacetylation of tumor suppressor genes and transcription factors.1, 2 By removing acetylated lysine residues, histone deacetylases (HDACs) favor a non-permissive chromatin conformation that aids transcriptional repression. HDACs mediate transcriptional repression that provokes the proliferation of cancer cells, cell cycle progression and inhibition of apoptosis and autophagy.2 HDAC inhibitors (HDACis) reverse the HDAC-mediated transcriptional repression, and thus activate tumor-suppressor and cell-death-associated genes.3 In apoptosis-deficient cancer cells specifically, HDACis target autophagy-associated molecular markers to mediate autophagic cell death (ACD; formerly called type II programmed cell death on the basis of its morphological characteristics) and thus these inhibitors possess immense potential for cancer prevention.4
Autophagy, a catabolic cellular self-degradative mechanism, has been under investigation in recent times as an alternative cell death mechanism. Under normal conditions, autophagy serves as a cytoprotective quality control mechanism that eliminates redundant cargos and damaged cellular organelles. Under stress conditions, autophagy provides a continuous source of energy required for cell survival and energy homeostasis.5 However, the Nomenclature Committee on Cell Death has denoted ACD as a separate ‘cell death subroutine’, with a mechanism that is distinct from that of apoptosis.6 In certain circumstances, autophagy without the involvement of alternative cell death pathways can be lethal, as a result of failed or excessive cargo degradation.7 ACD has been established as a genuine cell death mechanism that has distinct autophagic morphologies, along with the participation of unique molecular components and excessive autophagic flux.8 In fact, ACD can have an autophagy-mediated, -associated or -dependent mechanism , depending on its direct role in the cell death process and its association with apoptosis.9 Several ACD-mediating drugs have been under clinical investigation, but the HDACis have garnered more interest in recent times because of the principal involvement of HDACs in cancer progression.
Among all of the broad spectrum and selective HDACis, vorinostat, a hydroxamate-class Zn2+-dependent pan-HDACi, has been studied extensively for its involvement in the induction of ACD.10 With limited reports available, however, this review focuses on how vorinostat mediates ACD, with particular emphasis on autophagy-mediated, -associated and -dependent cell death. In addition, we emphasize the molecular mechanisms that are associated with vorinostat-induced ACD, such as superactivated autophagy, mitophagy, disrupted lysosomal homeostasis , and the involvement of secondary signals. We try to focus especially on the clinical landscape for vorinostat in different cancer subtypes, looking at autophagy as the principal regulatory mechanism that determines the pharmacokinetics and pharmacodynamics of the drug.
HDACs, HDACis and autophagy: an intricate interplay
HDACs mediate the transcriptional regulation of several proteins and play a key role in cancer initiation and progression. Through dynamic post-translational modifications and non-permissive chromatin conformations, HDACs target key proteins and transcription factors that are associated with cell proliferation, cell differentiation, cell survival, cell cycle progression, apoptosis and autophagy.1, 2 The deacetylation of crucial molecular components that are associated with tumor suppressor genes and apoptotic genes promotes the survival of cancer cells. Conversely, HDACis oppose the survival and proliferation of cancer cells by causing dynamic alterations in the acetylation and deacetylation status of key target proteins and transcription factors.1 For example, HDACis target the acetylation of apoptotic genes for cancer regulation. In apoptosis-deficient cells, however, HDACis target autophagy regulatory genes to induce ACD.10 Autophagy, a catabolic cellular self-degradative pathway, has been under investigation in recent times as an alternative mechanism of cell death.5 Several reports have indicated that HDACs deregulate key autophagic genes to sustain cancer cell survival.3 In this context, HDACis are effective yet selective cytostatic agents that target autophagy to inhibit cancer cell survival.
HDACs mediate the deregulation of cytostatic autophagy in cancer progression, and the HDAC inhibitor vorinostat acts as an autophagy modulator
Deacetylation of key autophagic genes promotes cytoprotective autophagy that encourages cancer cell proliferation, survival and drug resistance.11 The application of broad-spectrum multipharmacological HDACi in cancer therapy has emerged as a prominent therapeutic strategy in cancer treatment.3, 12 Hence, understanding the molecular mechanisms and critical molecules that are associated with cancer cell survival will provide insight into the best opportunities for therapeutic applications.
Deacetylation of key molecular markers during the stage-specific progression of autophagy for cancer progression
Cancer cells preferentially activate cytoprotective autophagy as a survival mechanism to overcome apoptosis-mediated cell death.5 From this perspective, regulation of the deacetylation status of selective autophagy regulatory genes drives cytoprotective autophagy and promotes cancer cell survival and progression.1 Cancer cell survival and progression, in lieu of the deacetylation of stage-specific autophagic regulatory genes, are described below.
Deacetylation of autophagic marker proteins during autophagy initiation
In MCF-7 breast cancer cells, downregulation of ESR1/estrogen receptor α is transcriptionally regulated by HDAC1 and RPS6KB/p70S6K. The phosphorylation of HDAC1 and activation of the PI3K–MTOR signaling pathway is regulated by RPS6KB/p70S6K, which recruits HDAC1 to the ESR1 promoter and enhances drug resistance.13 In triple-negative breast cancer cells, activators of HDACs and MTORC1 enhance the expression of EIF4EBP1 to promote cell viability.14 In endometrial cancer, HDAC6 supports cancer cell proliferation, metastasis and invasion by modulating the PTEN–AKT–MTOR signaling pathway.15, 16 Although the direct involvement of autophagy was not reported in the study by Zheng et al.,15 the dynamic regulation of AKT-MTOR is expected to play a cytoprotective role in nullifying a MIR206-mediated carcinostatic effect. In human multiple myeloma cell lines, dynamic co-regulation of HDAC and MTOR promotes the stability of the oncogenic protein MYC, which promotes cancer cell survival and progression.17
Deacetylation of autophagic marker proteins during phagophore membrane expansion
In hepatocellular carcinoma cells, HDAC1 overexpression regulates enhanced LC3-II lipidation to induce cytoprotective autophagy and thus to sustain hepatocarcinogenesis.18, 19 In Waldenstrom macroglobulinemia cells, HDAC4 and HDAC5 induce cytoprotective autophagy by enhancing the expression of LC3-II and RAB7, which abrogates apoptosis regulated by LBH589 (a specific HDAC4 and HDAC5 inhibitor) and MIR9 (microRNA 9).20, 21 In human cervical cancer (HeLa) cells, HDAC6 deacetylates LC3-II, promoting autophagosome formation.22 Tubacin, a specific HDAC6 inhibitor, results in partial acetylation of LC3-II, suggesting the involvement of other HDACs in the deacetylation process. Moreover, tubacin-induced LC3-II acetylation further impairs the autophagic flux by promoting the accumulation of SQSTM1/p62.23 In gastric cancer cells, class III HDACs deacetylate BECN1, ATG5, ATG7 and Atg8-family proteins, which preferentially support cell survival, proliferation, drug resistance, invasion and metastasis. Moreover, deacetylation of BECN1 by class III HDACs promotes epithelial-to-mesenchymal transition (EMT) in hepatocellular carcinoma via autophagic degradation of CDH1/E-cadherin.24
Deacetylation of autophagic marker proteins during flux in autophagy
In metastatic prostate cancer (DU145) cells, a dynamic co-regulation of SQSTM1 and HDAC6 directs the acetylation of TUBA/α-tubulin, thereby reducing the stability and motility of this complex . The less stable and motile acetylated form of TUBA/α-tubulin impairs the autophagy flux, and subsequently promotes invasion, metastasis and EMT.25 In MYC/v-Myc myelocytomatosis-overexpressing neuroblastoma cells (BE[2]-C, Kelly and IMR32 cells), HDAC10 facilitates the onset of cytoprotective autophagy against doxorubicin-induced cytotoxicity via deacetylation of HSPA/heat shock protein 70 and HSPA8/HSC70. Mechanistically, HDAC10-mediated deacetylation of HSPA8 results in the selective transport of cargos to lysosomes in a LAMP2-dependent molecular cascade, whereas HSPA maintains lysosomal membrane integrity for sustained autophagic flux. Hence, the autophagosome formation that results from HDAC10-mediated deacetylation induces cytoprotective autophagy in neuroblastoma cells.11
Vorinostat as a broad-spectrum pan-HDACi
As HDACs promote the survival and progression of cancer cells, there is a dire need for strategies to inhibit HDAC and thus prevent cancer.3 Several HDACi, such as vorinostat, panobinostat, romidepsin, Trichostatin A, tubacin and tubastatin, mediate ACD in several cancer subtypes. Among all the pan-HDACi, vorinostat and romidepsin have been approved by the US Food and Drug Administration (FDA). Panobinostat has been used experimentally in several solid cancers and has shown the ability to induce ACD at a lower dosage than vorinostat. Panobinostat has been under clinical trial in recent times.1, 26 In the ACD context, vorinostat (suberanilohydroxamic acid (SAHA)), a member of the hydroxamate class of broad-spectrum pan-HDACI, has been studied extensively and approved by the FDA for the treatment of cutaneous T-cell lymphoma. Vorinostat inhibits class I, II and IV HDACs that display Zn2+-dependent deacetylase activity. Chemically, vorinostat binds to the Zn2+ ion present in the active site of HDACs, thereby restricting their enzymatic activity.27 Several reports have implied the potential of vorinostat as an anticancer drug in various cancer subtypes because of its abilities to induce cell cycle arrest, apoptosis, and DNA-damage repair, thereby inhibiting metastasis and angiogenesis, and because of its autophagy-modulating activities.27 From a cancer treatment perspective, the ability of cancer cells to escape apoptosis suggests autophagy as an alternative cell death mechanism that could be modulated by therapeutics.5, 8 Various reports suggest that vorinostat-mediated autophagy can function in this capacity, and this broad-spectrum pan-HDACi has become highly prevalent in cancer therapeutics. Moreover, vorinostat can act on multipharmacological targets (EGFR, RTKs and synergistic agents), making it a more effective drug against heterogeneous tumors.28
Modes of ACD induced by vorinostat
The Nomenclature Committee on Cell Death has denoted ACD as a separate ‘cell death subroutine’ with distinct morphological characteristics, including rounded cell structure, detachment from the substratum, deposition of small vesicles, excessive accumulation of cytoplasmic vacuoles, and double-membrane compartments that resemble autophagosomes containing damaged cellular aggregates, damaged organelles and cytoplasmic cargos.6, 8 ACD can further be divided into autophagy-mediated (autophagic facilitation of apoptotic cell death), autophagy-associated (co-existence of autophagic and apoptotic cell death) and autophagy-dependent (autophagy as an explicit cell death mechanism that is independent of apoptosis) modes.9 With limited evidence available, we summarize all three modes of ACD in the context of nomenclature, mode and molecular mechanism of action (Figure 1). Table 1 lists the modes of ACD seen after vorinostat treatment in several in vivo and in vitro models of numerous cancer subtypes.
Figure 1. Intricate role of vorinostat-regulated autophagy in cell death.
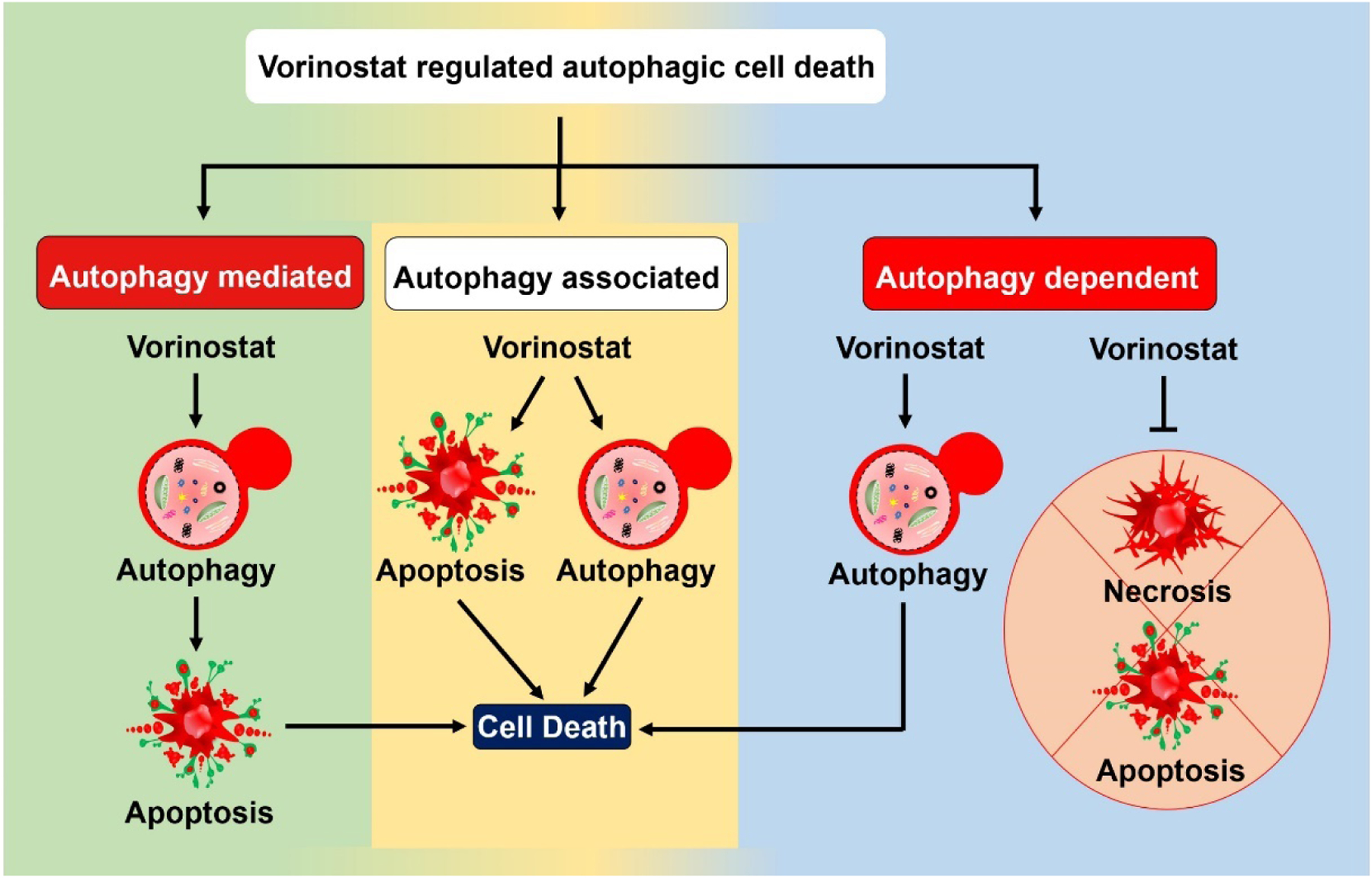
Vorinostat-regulated autophagic cell death (ACD) can be grouped into autophagy-mediated, -associated and -dependent modes depending on the involvement of apoptosis. In autophagy-mediated death (autophagic facilitation of apoptotic cell death) cell, the autophagy pathway activates the apoptotic cell death pathways. In autophagy-associated cell death (in which autophagic and apoptotic cell death co-exist), the induction of autophagy accompanies apoptotic cell death. Autophagy-dependent (autophagy as an explicit cell death mechanism) cell death occurs independently of the apoptotic and necrotic cell-death pathways.
Table 1:
Different modes of vorinostat regulated autophagic cell death (ACD).
| Types of ACD | Drug entity | Cancer type or model | Molecular mechanisms of action | Reference |
|---|---|---|---|---|
| Autophagy-mediated cell death | Synergism of temozolomide and vorinostat with chloroquine | Glioblastoma (C6 and U251MG) cells | Temozolomide induces cyto-protective autophagy via sustained upregulation of AMPK–ULK1 and inhibition of AKT–mTOR. Vorinostat inhibits mTOR signaling to aid cyto-protective autophagy. Inhibition of temozolomide-vorinostat regulated cyto-protective autophagy by chloroquine facilitates the onset of apoptotic cell death pathways. | 61 |
| GL261 glioma tumor-bearing C57BL/6 mice | Temozolomide (10 mg/kg dose, every 3 days for 10 days) vorinostat (i.p. 25 mg/kg/day for 10 days) and chloroquine (pH 7; 20 mg/kg/day for 10 days) inhibits cyto-protective autophagy to aid apoptosis. | |||
| Vorinostat in synergism with chloroquine | Glioblastoma (U87MG) cells | Autophagic flux inhibition by chloroquine facilitates vorinostat-mediated apoptosis via hyperaccumulation of autophagosomes, prevention of the elimination of damaged mitochondria and increased lysosomal pH with an eventual generation of reactive oxygen species (ROS). | 60 | |
| Vorinostat in combination with quinacrine | T-cell ALL (Jurkat and Molt-4) cells | Quinacrine in synergism with vorinostat facilitates the inhibition of mitophagy, enhances the K-63 associated ubiquitination of mitochondrial proteins, enhances the accumulation of mitochondrial aggresomes, enhances ROS production, decreases mitochondrial membrane potential, enhances the expression of Bax and downregulates the expression of Bcl-2. | 62 | |
| Jurkat-cell-bearing SCID mice | Vorinostat (i.p. 70 mg/kg/d for 12 d) in synergism with quinacrine (i.p. 50 mg/kg/d for 12 d) mediates the onset of apoptosis, resulting in reduced tumor cell proliferation. | |||
| Vorinostat in synergism with chloroquine | Acute myeloid leukemia (AML) t(8;21) cells | Inhibition of vorinostat-mediated pro-survival autophagy by chloroquine results in excessive accumulation of ubiquitinated proteins (AML1-ETO) and aggresomes, leading to cell death. | 63 | |
| Synergism of gefitinib-vorinostat with metformin and spautin-1 | EGFR-TKI resistant PC-9GR and H1975 non small cell lung cancer (NSCLC) | Metformin and spautin-1 reduce the lipidation of LC3, reduces the phosphorylation of Beclin-1 and enhances the expression of P62, leading to chemosensitization of the resistant NSCLC towards apoptosis (evidenced by upregulation in the expression of BIM and Bax and downregulation of antiapoptotic proteins Bcl-2, Bcl-XL and Mcl-1). | 64 | |
| Vorinostat | Neuroblastoma (SK-N-SH and QDDQ-NM) | Decreased LC3 lipidation and decreased expression of ATG5 and Beclin-1 , leading to chemosensitization of neuroblastoma cells towards apoptosis. | 65 | |
| Vorinostat in synergism with simvastatin | Triple negative breast cancer (TNBC) (MDA-MB-468, MDA-MB-231 and DA-MB-453) cells | Inhibition of autophagic flux, leading to accumulation of LC3II, P62, NDP52 and NBR1, reduced the prenylation of Rab7 that is required for autolysosome formation. Eventual onset of apoptosis through caspase-dependent signaling pathway. | 66 | |
| Synergism of bortezomib and vorinostat with chloroquine | Colon cancer (HT-29 and HCT-8) cell | Disruption of autophagic flux, bulk generation of superoxide as chief regulator of ubiquitinated protein accumulation, and subsequent onset of apoptosis. | 67 | |
| Vorinostat in synergism with chloroquine | HCT-8 colon cancer-bearing female BALB/c xenograft mouse model | Chloroquine (60mg/kg/d for 21 d) and vorinostat (100mg/kg/d for 21 d) treatment results in the inhibition of autophagy, leading to ubiquitinated protein accumulation, greater superoxide generation and the subsequent onset of apoptosis. | 68 | |
| Vorinostat in combination with sorafenib | Hepatoma (HepG2, Hep3B and PLC/PRF/5) cells | Inhibition of autophagy, via genetic and pharmacological inhibitors, elicits the acetylation of p53 and caspase-dependent apoptosis. | ||
| Vorinostat and pharmacological inhibitors of autophagy | Glioblastoma stem cells (GSCs) and GSC-bearing mouse xenograft model | Inhibition of autophagy by pharmacological agents (3MA, wortmannin, BafA1 and chloroquine) and genetic agents (ShLC3, shBeclin-1 and shATG5) results in the onset of apoptotic cell death (evidenced by cleaved Caspase 3 and PARP). | 69 | |
| Autophagy-associated cell death | Vorinostat in combination with olaparib | TNBC (MDA-MB-231 and MDA-MB-2468) cells | Vorinostat mediates the onset of autophagy-associated cell death, which in turn enhances the cytotoxic effect of olaparib (a PARP inhibitor) through PTEN modulation to sustain tumor cell proliferation and to induce apoptotic cell death. | 70 |
| Vorinostat in combination with BIBW2992/WZ4002 | EGFR-TKI NSCLC (PC-9 and NCI-H1975) cells | Enhanced expression of LC3II and Beclin-1 and downregulation of p62 dynamically regulates the onset of caspase-dependent intrinsic apoptosis. | 71 | |
| NCI-H1975-bearing mouse xenograft model | Vorinostat (25mg/kg) in combination with BIBW2992 (10mg/kg) and WZ4002 (25mg/kg) induces autophagy (enhanced LC3 lipidation and Beclin-1 expression) and apoptosis (evidenced through cleaved PARP and Caspase 3, and by downregulation of Bcl-XL). | |||
| Vorinostat in combination with tamoxifen | TAMR/MCF-7 and TAMR/T47D cells | Induces both apoptotic cells death (evidenced by cleaved Caspase 3, 7, 9 and PARP, upregulation of Bax, and downregulation of Bcl-2 together with ER-associated gene transactivation) and autophagic cell death (evidenced by enhanced LC3 lipidation and expression of Beclin-1, ATG 5 and ATG 7). | 34 | |
| Vorinostat in combination with ionizing radiation | MDA-MB-231/4T1 cells and 4T1-bearing Balb/c in vivo mouse model | Co-existence of apoptotic cell death (evidenced by annexin-v-positive cells, nuclear pyknosis and chromatin condensation) and autophagic cell death (evidenced by enhanced expression of LC3II and Beclin-1). | 72 | |
| Vorinostat in combination with decitabine | Ovarian cancer (SKOv3 and Hey) cells | Co-existence of apoptotic (evidenced by enhanced expression of Caspase 3/7) and autophagic cell death (evidenced by enhanced LC3 lipidation and overexpression of ARH1 and PEG3). | 73 | |
| Autophagy-dependent cell death | Vorinostat in combination with BIBW2992/WZ4002 | EGFR-TKI NSCLC (PC-9) cells | Induction of autophagic cell death independent of apoptosis: treatment with the pan caspase inhibitor Z-VED-FMK does not abrogate cell death. | 71 |
| Vorinostat | TAMR/MCF-7-bearing athymic mouse xenograft model | Administration of vorinostat (i.p. 50mg/kg, every 2 d for 21 d for 10 treatment cycles) induces autophagic cell death (evidenced through enhanced expression of LC3, Beclin-1, ATG5 and ATG7). | 34 | |
| Vorinostat | Bcl-XL overexpressing cervical cancer (HeLa) cells | Induction of non-apoptotic cell death despite inhibition of cytoplasmic shuttling of cytochrome C and caspase activation with distinct autophagic cellular morphologies. | 10 | |
| Vorinostat in combination with pazopanib | Fibrosarcoma (HT1080), uterine sarcoma (MES) and osteosarcoma (Saos-2) cells | Autophagic cell death with an evident increase in LC3 lipidation and Beclin-1 expression. Inhibition of the mTOR-p70S6K pathway for the induction of lethal autophagy. | 74 | |
| Vorinostat and pharmacological inhibitors of autophagy | Gastric cancer (AGS and KATO-III) cells | Induction of autophagy (enhanced lipidation of LC3 and decreased expression of p62) followed by autophagy flux inhibition facilitates autophagy-dependent cell death. | 75 | |
| Vorinostat | Hepatocellular carcinoma (Huh7, Hep3B and HepG2) | Enhances LC3 lipidation, Beclin-1 expression, downregulation of p62 and inhibition of the PI3K-AKT-mTOR signaling axis. Phosphorylation of ER stress regulates markers such as PERK and eIF2α, leading to autophagy-dependent cell death. | 76 | |
| Vorinostat | Endometrial stomal sarcoma (ESS-1) cells | Downregulation of p-mTOR, mTOR and pS6K expression, leading to induced autophagy-dependent cell death that was independent of activation of Caspase 3. | 77 |
Where and how does ACD occur after vorinostat treatment
How autophagy, the catabolic process that helps in cell survival under stress conditions, becomes lethal by over-consuming cellular components is not fully understood because of the lack of molecular evidence. Nevertheless, recent investigations have provided multiple indications of autophagy being lethal in the context of cellular dependency.8 A few of the suggested molecular mechanisms are discussed below (Figure 2).
Figure 2. Mechanisms through which vorinostat-regulated autophagy promotes cell death.
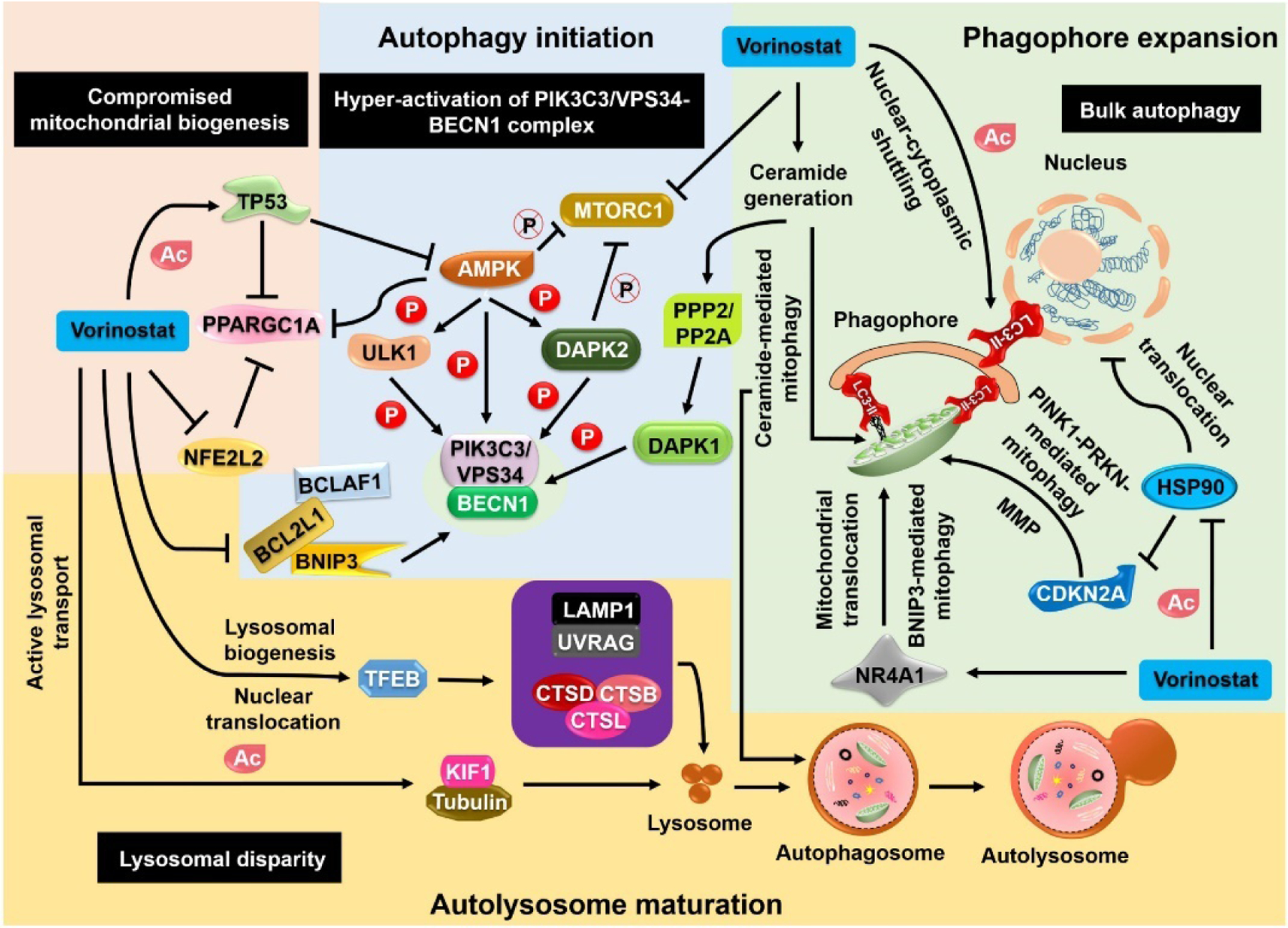
Lethal autophagy can be introduced via hyperactivation of the PIK3C3/VPS34–BECN1 complex in the autophagy initiation stage. Apart from direct inhibition of mTOR (MTORC1), vorinostat activates AMP-activated protein kinase (AMPK) and causes the phosphorylation of particular substrates (ULK1, DAPK2), which inhibits mTOR expression. Further modulation of the phosphorylation status of BECN1 by PPP2/PP2A–DAPK1 inhibits its interaction with BCL2 homology domain proteins, leading to the sequestration of BNIP3 and BCLAF1. Despite bypassing canonical initiation of autophagy, the noncanonical pathways drive the formation of autophagosomes. Secondary signals, such as ceramide generation, that recruit mitochondria to the phagophore membranes initiate mitophagy by interacting with LC3-II. Moreover, vorinostat regulates the acetylation of LC3-II to mediate nuclear–cytoplasmic shuttling, which has a substantial role in enhancing autophagosome formation. In addition, vorinostat inhibits the acetylation of HSP90, preventing its nuclear translocation. The inhibition of HSP90 translocation inhibits CDKN2A degradation and facilitates PINK1–PRKN-mediated mitophagy. Furthermore, vorinostat mediates the mitochondrial translocation of NR4A1/Nur77, thereby aiding BNIP3-mediated mitophagy. During the autolysosome maturation stage, vorinostat causes nuclear translocation of Transcription Factor EB (TFEB), leading to further activation of downstream lysosomal targets such as LAMP1, UVRAG, CTSB, CTSD and CTSL and to enhanced lysosomal biogenesis. In addition to lysosomal biogenesis, vorinostat mediates the acetylation of tubulin and KIF1, resulting in enhanced lysosomal transport. During the compromised mitochondrial biogenesis, vorinostat acetylates TP53, which in turn inhibits AMPK and PPARGC1A. Moreover, vorinostat-mediated inhibition of NFE2L2 also inhibits PPARGC1A-mediated mitochondrial biogenesis.
Hyperactivation of the PIK3C3/VPS34–BECN1 complex induces superactivated autophagy
The PIK3C3/VPS34–BECN1 complex plays a significant role in the onset of hyperactivated autophagy, which drives autophagic cell death.8 Previous reports have demonstrated that the activation of BECN1 after vorinostat treatment induces ACD in several cancer subtypes.22 Moreover, it has also been reported that vorinostat downregulates BCL2 and BCL2L1/Bcl-xL protein expression during autophagy associated with cell death, with subsequent activation of BECN1.22 In a molecular context, DAPK1 mediates the kinase cascades that are involved in the phosphorylation of protein kinase D (PRKD), which then phosphorylates and activates PIK3C3/VPS34 .29 Second, the phosphorylation of BECN1 is mediated by DAPK1, which subsequently dissociates BECN1 from BCL2 and promotes its association with PIK3C3/VPS34. In acute myeloid leukemia, vorinostat, in synergism with tyrosine kinase inhibitors and FLT3 (fms-related receptor tyrosine kinase 3) inhibitors, mediates the derepression of DAPK1.30 Furthermore, a cyclic synergistic administration of sorafenib and vorinostat in acute myeloid leukemia mediates DAPK1 derepression.31 Although the reports are limited, it appears that the functional association of vorinostat and DAPK1 activation might drive hyperactivation of the PIK3C3/VPS34–BECN1 complex and the subsequent induction of ACD. Interestingly, another pan-HDACi panobinostat, LBH589, induces DAPK1-dependent ACD via enhanced LC3 lipidation. In autophagy-deficient cells, LBH589 promotes DAPK1-independent apoptotic cell death.32
Sustained autophagic flux to restrain bulk autophagy for autophagic cellular morphologies
Progressive autophagic flux overwhelms the cellular cytoplasm with the accumulation of non-cargo-containing autophagic vacuoles and empty autophagosomes. In the later stages of bulk autophagy, the autophagic vacuoles fill the cytoplasmic volume , which is presumably devoid of intracellular organelles such as mitochondria, Golgi and endoplasmic reticulum (ER).8, 33 In addition to overconsumption of intracellular organelles, rerouting of cellular membranes to facilitate the production of autophagosomes disrupts cellular membrane homeostasis .8 The mechanistic observations in TAMR/MCF-7, MDA-MB-231 and 4T1 cells, as well as those in apaf1-knockout mouse embryonic fibroblasts (MEFs) and BCL2L1-overexpressing HeLa cells, have suggested the onset of ACD with prominent autophagic cellular morphologies following vorinostat treatment.10, 34
Death from disrupted lysosomal homeostasis
Lysosomal homeostasis acts as a key component for maintaining bulk autophagy for subsequent activation of autophagic cell death. Conservation of the lysosomal pool during hyperactivated autophagy induction promotes enhanced autophagic flux.35 In this context, it has been reported that vorinostat treatment improves lysosomal function (lysosomal acidification), number and enzymatic activity; particularly proteolytic including CTSB (cathepsin B), CTSD (cathepsin D) and CTSL (cathepsin L).36 Moreover, vorinostat activates lysosomal biogenesis through acetylation of TFEB and its nuclear translocation. Besides TFEB, vorinostat enhances the expression of TFEB target genes such as UVRAG and LAMP1, which are important during the stage-specific progression of autophagy.36 By contrast, a recent finding from a drug screening using the lysosomal-METRIQ probe has demonstrated vorinostat functions as a downregulator of lysosomal membrane trafficking and activity.37 It has been expected that vorinostat-mediated downregulation of lysosomal activity might inhibit autophagic flux leading to autophagosome accumulation and subsequent activation of autophagy-mediated cell death.
Although Transcription Factor EB (TFEB) acts as a master regulator of autophagy and lysosomal biogenesis, selective cargo sorting and degradation via chaperone-mediated autophagy with the help of HSP90 and LAMP2 also play a key roles in ACD .38 A combination of vorinostat with doxorubicin and paclitaxel enhances the acetylation of HSP90 and reduces the ectopic expression of HSP90 client proteins. This reduction in the expression of HSP90 client proteins, together with reduced expression of LAMP2A, prevents the lysosomal degradation of CDKN2A, which in turn elicits ACD.38, 39 Moreover, acetylated microtubules facilitate autophagosome–lysosome fusion, which is critical for hyperactivated autophagy.40 Acetylation of tubulin (at Lys40) is enhanced in both the labile and stable populations of microtubules, and results in the recruitment of dyneins and KIF1 that deliver autophagosomes centripetally towards lysosomes along microtubules.40, 41 In this context, vorinostat-mediated microtubule acetylation might expedite the enhanced autophagosome–lysosome fusion required for ACD.
Death from excessive mitophagy followed by compromised mitochondrial biogenesis
Excessive removal of mitochondria through autophagy creates a problem for mitochondria-depleted cells in generating the energy required for cellular homeostasis. The association of autophagy markers with mitochondria leads to lethal autophagy of these organelles.42 A mechanistic investigation revealed the role of smARF/CDKN2A (CDKN2A is the translational precursor of smARF) in mitophagic cell death. smARF/CDKN2A overexpression leads to autophagy-dependent cell death independent of caspase-mediated apoptosis.8, 43 Moreover, under hypoxia or external stimuli, enhanced expression of smARF/CDKN2A sustains bulk autophagy via SQSTM1 degradation. Subsequently, smARF/CDKN2A deregulation induces PINK1–PRKN/Parkin mitophagy.8, 44 In cutaneous T-cell lymphoma, the deregulated expression of CDKN2A that is induced by vorinostat treatment reduces tumor proliferation, in a process that might involve autophagy-dependent cell death resulting from excessive mitophagy.45
In another investigation, mitochondrial translocation of orphan nuclear receptor NR4A1/TR3/Nur77 increased mitochondrial membrane permeability through the permeability transition pore ANT1–VDAC1, leading to mitophagic cell death. Mechanistically, the mitochondrial translocation of NR4A1/TR3/Nur77 occurs via the interaction of BNIP3L/Nix with the outer mitochondrial membrane proteins TOMM20 and TOMM70.46 It has been shown that vorinostat upregulates the expression of NR4A1/TR3/Nur77 and its associated signaling mediators such as CDKN1A/p21.47 Limited evidence suggests that the dynamic interaction of vorinostat and NR4A1/TR3/Nur77 might be associated with cell death as the result of mitophagy. In addition to NR4A1/TR3/Nur77, BNIP3 also mediates mitophagy-dependent cell death. In the context of autophagy-associated cell death, a synergistic combination of several drugs with vorinostat regulates the expression of BNIP3, which might follow the induction of mitophagic cell death in close coordination with apoptosis.48 Moreover, the extensive interplay between DAPK1 and BNIP3 illustrates the possibility of mitophagic cell death.8
Mitochondrial energy homeostasis post mitophagy is thought to be maintained by mitochondrial biogenesis. PPARGC1A/PGC-1α is a critical regulator of mitochondrial biogenesis, working alongside several transcriptional and signaling cascades.49 In osteosarcoma cells, vorinostat downregulates the expression of PPARGC1A, which might be responsible for diminishing the metastatic behavior of these cancer cells.50 In addition to this, vorinostat also suppresses the MYC/c-MYC-regulated expression of NFE2L2/NRF2 (a transcription factor associated with PPARGC1A), and restores the function of KEAP1, which might promote mitochondrial biogenesis.51 Moreover, acetylated TP53/p53 inhibits the expression of AMPK, a critical regulator of cellular energy metabolism via mitochondrial biogenesis. Vorinostat treatment results in the acetylation of TP53 and, subsequently, AMPK, potentially inhibiting mitochondrial biogenesis.52
Secondary signals and autophagy-associated signaling cascades elicit cell death
Ceramides are critical for the induction of apoptosis and necrosis-independent cell death.8 Furthermore, several reports have demonstrated the essential involvement of ceramides in the induction of autophagy-dependent cell death. In gastrointestinal tumor cells, vorinostat in synergism with sorafenib resulted in enhanced ceramide generation, which in turn mediates PPP2/PP2A-DAPK1-dependent cell death.53 Furthermore, vorinostat directly causes the acetylation of the HIF1A/HIF1-α-associated chaperone HSP90, subsequently hindering the nuclear translocation of HSP90 and consequently inhibiting the transcriptional activity that might regulate HIF1A-dependent ACD.54 In addition to this, vorinostat-mediated acetylation deregulates the expression of oncogenic RAS, leading to PMAIP1/Noxa- and BECN1-dependent ACD.55 More importantly, the acetylation of cytoplasmic LC3, which might be achieved through vorinostat therapy, is critically involved in the regulation of superactivated autophagy that leads to cell death.56
Vorinostat-mediated regulation of autophagy for cancer therapy
The clinical efficacy of vorinostat in terms of regulating autophagy, specifically the expression of key autophagic markers in tumors, has been demonstrated in several cancer subtypes. In addition to its ability to regulate autophagy, however, the clinical pharmacology (i.e., pharmacokinetics and pharmacodynamics) of vorinostat, used either as a mono-chemopreventive agent or in combination with autophagy activators or inhibitors, is immensely important.57–59 A number of pharmacological parameters have been estimated in different phases of randomized and non-randomized clinical trials (Table 2). These include the dose and periodicity of treatment, drug synergism, drug efficacy, drug-related adverse toxicity, pharmacodynamics targets and biomarkers of primary tumor tissues, pharmacokinetic drift, the area under curve, median half-life, and drug concentrations in tumor, blood and plasma.57 With limited clinical trials involving an assessment of autophagy regulation available, we have summarized clinical pharmacology results for various vorinostat combinations in Table 2.
Table 2:
Detailed evaluation of the clinical pharmacology of vorinostat in the regulation of autophagy.
| Details of clinical trial | Drug entity | Cancer type | Clinical pharmacology properties and their relevance | Autophagy-associated molecular mechanisms of action | Reference |
|---|---|---|---|---|---|
| Phase I clinical trial (NCT01023737*) | Hydroxychloroquine (HCQ) (400–1000) and vorinostat (300) mg/m2/day | Patients with advanced or malignant solid tumors (colorectal and renal) | Evaluation of safety, tolerability, drug response rate, progression-free survival and anti-tumor activity. | Inhibition of autophagy by HCQ in combination with vorinostat critically regulated autophagy, resulting in inhibition of cancer progression with minimal toxicity. | – |
| Phase I clinical trial | HCQ and oral ingestion of vorinostat (400 mg/m2, once a day for 21 d) | Colorectal, soft tissue sarcoma, ovarian cancer, melanoma, NSCLC, renal cell carcinoma, prostate, breast and bladder cancers | Evaluation of minimal dose-related (MDR) toxicity at maximum tolerated dose (MTD), Cmax of 768 and 786 μg/L, AUC of 3387 and 2410 μg × hr/L, median half time of 2.06 h and 1.3 h and median Vd/f of 304–309 L. | Enhanced expression of cathepsin D (CTSD), which is responsible for the onset of HCQ-vorinostat-regulated autophagy-mediated apoptosis. Accumulation of autophagic vacuoles (AVs). Enhanced expression of CDKN1A, SQSTM1 (p62) and MAP1LC3B. | 57 |
| Early phase clinical trial | HCQ (600) and oral ingestion of vorinostat (400) mg/m2, once a day for 21 d) | Advanced and refractory metastatic colorectal cancer | Evaluation of median overall survival (6.7 months), median progression-free survival (2.8 months) and minimal drug-related adverse toxicity. | Enhanced CTSD and p62 level. | 58 |
| Phase II randomized, therapeutic clinical trial (NCT02316340*) | HCQ (600 mg/m2) and oral ingestion of vorinostat (400 mg/m2, once a day for 28 days) | Chemorefractory metastatic colorectal cancer | Evaluation of enhanced anti-tumor efficacy, overall survival and median progression-free survival | Autophagy inhibition leading to enhanced tumor immunity (decreased expression of IL-1b, IL-6, IL-10, IL-2, IFN-γ and TNF-α). | – |
| Phase II clinical trial (NCT00882206*) | Vorinostat (230 mg/m2, oral gavage, twice a day), intravenous injection of decitabine (15 mg/m2), prednisone (40 mg/m2, twice a day for 8 days), vincristine (1.5 mg/m2), doxorubicin (60 mg/m2), Pegasparaginase (2,500 IU/m2) and intrathecal cytarabine (70 mg for 14 d) | Relapsed and refractory acute lymphoblastic leukemia (ALL) | Evaluation of enhanced chemo-sensitization and identification of 32 differentially methylated genes in the responding groups as compared to non-responding ALL patients | Hypomethylation of cell death (apoptosis and autophagy) and transcription-regulating genes, such as SQSTM1, MAP3K5, SIRT1, DNAJB6, BTAF1 and UB2E1. | – |
The NCT reference codes identify the clinical trials, and details of these clinical trials can be found in ClinicalTrials.gov.
Challenges for clinical translation and scope for future expansion
After the approval of vorinostat by the FDA in 2006 for the treatment of cutaneous T-cell lymphoma, several clinical trials in different phases have looked at the use of vorinostat as a mono-therapeutic agent or in combination with other drugs as a treatment for hematological and solid tumors.27 The clinical outcomes achieved by vorinostat as a mono-therapeutic agent against solid tumors have been disappointing. The restricted pharmacokinetic properties of the drug, which affect its absorption, distribution, excretion, metabolism, targeted drug delivery, and short half-life, hinder the clinical translation of vorinostat. Moreover, adverse side effects and toxicity during early-phase clinical trials have further restricted its clinical translation.57 In addition to this, the complexity of the autophagic response, which ranges from pro-survival to lethal for cancer cells , makes the use of vorinostat more challenging. The drug synergism of vorinostat with specific autophagy mediators may provide a way to overcome these challenges for translation, ultimately delivering valuable outcomes in personalized and precision cancer therapy. The combination of chloroquine (CQ) or hydroxychloroquine (HCQ) with vorinostat has shown promising clinical results in several cancer subtypes.57, 58, 60 Hence, the design of stage-specific autophagy-targeting drugs and drug combinations will offer improved clinical efficacy over that provided by vorinostat alone , owing to their improved pharmacokinetics and pharmacodynamics. Importantly, the identification of drug targets and an understanding of the mechanistic regulation of ACD will make it easier to use vorinostat to provide an enhanced carcinostatic effect in apoptosis-deficient or -resistant cancer cells.
Conclusions
Deacetylation of key molecular markers that are involved in apoptosis and autophagy plays a crucial role in the initiation and progression of cancer. With the dire need to identify and establish autophagy as an alternative cell death mechanism, apoptosis-resistant and -deficient cancer cells deregulate autophagy-related genes through deacetylation. By reversing such deregulation, HDACis impart a critical response in cancer cells and facilitate ACD. Vorinostat, a broad-spectrum HDACi, enables autophagy-mediated, -associated and -dependent death of cancer cells. By acetylation of several key components of the autophagy machinery, vorinostat promotes superactivated autophagy through hyperactivation of PIK3C3/VPS34-BECN1. In addition, vorinostat mediates nonselective autophagy via lysosomal disparity and mitophagy, with subsequent inhibition of mitochondrial biogenesis, that expedites ACD. Notably, an understanding of the clinical pharmacology of ACD regulation allows vorinostat to be implemented as a mono-therapeutic or as a synergistic drug for cancer prevention. More investigation is warranted to delineate the mechanistic involvement of vorinostat-mediated inhibition of acetylation in ACD induction. Further elucidation of the vorinostat specificity of novel molecular ACD targets will afford enhanced therapeutic value for personalized cancer therapy.
Highlights.
HDACs mediate deregulation of cytostatic autophagy for cancer progression.
Vorinostat mediates autophagy-mediated, associated and dependent cell death.
Hyperactivation of PIK3C3/VPS34-BECN1 complex aids vorinsostat regulated cell death.
Lysosomal disparity; biogenesis and functionality facilitates cell death.
Excessive mitophagy followed by compact mitochondrial biogenesis provokes cell death.
Acknowledgments
We convey our thanks to the National Institute of Technology, Rourkela. SP acknowledges DST INSPIRE (Award reference number IF180167), Department of Science and Technology, Government of India for providing a fellowship. DJK was supported by NIH grant GM131919.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
The authors declare that they have no conflicts of interest.
References
- 1.Patra S, Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Mishra SR, et al. Dysregulation of histone deacetylases in carcinogenesis and tumor progression: a possible link to apoptosis and autophagy. Cell Mol Life Sci 2019; 76: 3263–82. doi: 10.1007/s00018-019-03098-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26: 5420–32. doi: 10.1038/sj.onc.1210610 [DOI] [PubMed] [Google Scholar]
- 3.Chueh AC, Tse JW, Tögel L, Mariadason JM. Mechanisms of histone deacetylase inhibitor-regulated gene expression in cancer cells. Antioxid Redox Signal 2015; 23: 66–84. doi: 10.1089/ars.2014.5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A 2012; 109: 6561–5. doi: 10.1073/pnas.1204429109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patra S, Mishra SR, Behera BP, Mahapatra KK, Panigrahi DP, Bhol CS, et al. Autophagy-modulating phytochemicals in cancer therapeutics: current evidences and future perspectives. Semin Cancer Biol 2020; S1044-579X(20)30104-8. doi: 10.1016/j.semcancer.2020.05.008 [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018; 25: 486–541. doi: 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fulda S, Kögel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015; 34: 5105–13. doi: 10.1038/onc.2014.458 [DOI] [PubMed] [Google Scholar]
- 8.Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death—where, how and why a cell eats itself to death. J Cell Sci 2018; 131: jcs.215152. doi: 10.1242/jcs.215152 [DOI] [PubMed] [Google Scholar]
- 9.Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ 2019; 26: 605–16. doi: 10.1038/s41418-018-0252-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A 2004; 101: 18030–5. doi: 10.1073/pnas.0408345102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oehme I, Linke JP, Böck BC, Milde T, Lodrini M, Hartenstein B, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A 2013; 110: E2592–601. doi: 10.1073/pnas.1300113110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci 2017; 18: 1414. doi: 10.3390/ijms18071414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Citro S, Miccolo C, Meloni L, Chiocca S. PI3K/mTOR mediate mitogen-dependent HDAC1 phosphorylation in breast cancer: a novel regulation of estrogen receptor expression. J Mol Cell Biol 2015; 7: 132–42. doi: 10.1093/jmcb/mjv021 [DOI] [PubMed] [Google Scholar]
- 14.Sulaiman A, McGarry S, Lam KM, El-Sahli S, Chambers J, Kaczmarek S, et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis 2018; 9: 815. doi: 10.1038/s41419-018-0811-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Y, Yang X, Wang C, Zhang S, Wang Z, Li M, et al. HDAC6, modulated by miR-206, promotes endometrial cancer progression through the PTEN/AKT/mTOR pathway. Sci Rep 2020; 10: 3576. doi: 10.1038/s41598-020-60271-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh BN, Zhou H, Li J, Tipton T, Wang B, Shao G, et al. Preclinical studies on histone deacetylase inhibitors as therapeutic reagents for endometrial and ovarian cancers. Future Oncol 2011; 7: 1415–28. doi: 10.2217/fon.11.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simmons JK, Michalowski AM, Gamache BJ, DuBois W, Patel J, Zhang K, et al. Cooperative targets of combined mTOR/HDAC inhibition promote MYC degradation. Mol Cancer Ther 2017; 16: 2008–21. doi: 10.1158/1535-7163.mct-17-0171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie HJ, Noh JH, Kim JK, Jung KH, Eun JW, Bae HJ, et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS One 2012; 7: e34265. doi: 10.1371/journal.pone.0034265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 2016; 6: a026831. doi: 10.1101/cshperspect.a026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koeneke E, Witt O, Oehme I. HDAC family members intertwined in the regulation of autophagy: a druggable vulnerability in aggressive tumor entities. Cells 2015; 4: 135–68. doi: 10.3390/cells4020135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roccaro AM, Sacco A, Jia X, Azab AK, Maiso P, Ngo HT, et al. microRNA-dependent modulation of histone acetylation in Waldenstrom macroglobulinemia. Blood 2010; 116: 1506–14. doi: 10.1182/blood-2010-01-265686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rikiishi H Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol 2011; 2011: 830260. doi: 10.1155/2011/830260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaliszczak M, van Hechanova E, Li Y, Alsadah H, Parzych K, Auner HW, Aboagye EO. The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br J Cancer 2018; 119: 1278–87. doi: 10.1038/s41416-018-0232-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peixoto P, Grandvallet C, Feugeas JP, Guittaut M, Hervouet E. Epigenetic control of autophagy in cancer cells: a key process for cancer-related phenotypes. Cells 2019; 8: 1656. doi: 10.3390/cells8121656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang X, Huang Y, Liang X, Jiang F, He Y, Li T, et al. Metastatic prostate cancer-associated P62 inhibits autophagy flux and promotes epithelial to mesenchymal transition by sustaining the level of HDAC6. Prostate 2018; 78: 426–34. doi: 10.1002/pros.23487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Fazio P, Waldegger P, Jabari S, Lingelbach S, Montalbano R, Ocker M, et al. Autophagy-related cell death by pan-histone deacetylase inhibition in liver cancer. Oncotarget 2016; 7: 28998–9010. doi: 10.18632/oncotarget.8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richon VM. Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br J Cancer 2006; 95: S2–6. doi: 10.1038/sj.bjc.6603463 [DOI] [Google Scholar]
- 28.Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM, et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol 2009; 2: 31. doi: 10.1186/1756-8722-2-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H, Luo W, Zeng C, Zhang Y, Wang L, Yao W, Nie C. PP2A mediates apoptosis or autophagic cell death in multiple myeloma cell lines. Oncotarget 2017; 8: 80770–89. doi: 10.18632/oncotarget.20415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shanmugam R, Gade P, Wilson-Weekes A, Sayar H, Suvannasankha A, Goswami C, et al. A noncanonical Flt3ITD/NF-κB signaling pathway represses DAPK1 in acute myeloid leukemia. Clin Cancer Res 2012; 18: 360–9. doi: 10.1158/1078-0432.ccr-10-3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pratz K, Levis M. Incorporating FLT3 inhibitors into acute myeloid leukemia treatment regimens. Leuk Lymphoma 2008; 49: 852–63. doi: 10.1080/10428190801895352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gandesiri M, Chakilam S, Ivanovska J, Benderska N, Ocker M, Di Fazio P, et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012; 17: 1300–15. doi: 10.1007/s10495-012-0757-7 [DOI] [PubMed] [Google Scholar]
- 33.Dasari SK, Bialik S, Levin-Zaidman S, Levin-Salomon V, Merrill AH Jr, Futerman AH, Kimchi A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ 2017; 24: 1288–302. doi: 10.1038/cdd.2017.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YJ, Won AJ, Lee J, Jung JH, Yoon S, Lee BM, Kim HS. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int J Med Sci 2012; 9: 881–93. doi: 10.7150/ijms.5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discov 2020; 6: 6. doi: 10.1038/s41421-020-0141-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Wang J, Zhou Z, Park JE, Wang L, Wu S, et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy 2018; 14: 1043–59. doi: 10.1080/15548627.2018.1447290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishii S, Matsuura A, Itakura E. Identification of a factor controlling lysosomal homeostasis using a novel lysosomal trafficking probe. Sci Rep 2019; 9: 11635. doi: 10.1038/s41598-019-48131-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robert G, Jacquel A, Auberger P. Chaperone-mediated autophagy and its emerging role in hematological malignancies. Cells 2019; 8: 1260. doi: 10.3390/cells8101260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hui KF, Leung YY, Yeung PL, Middeldorp JM, Chiang AK. Combination of SAHA and bortezomib upregulates CDKN2A and CDKN1A and induces apoptosis of Epstein-Barr virus-positive Wp-restricted Burkitt lymphoma and lymphoblastoid cell lines. Br J Haematol 2014; 167: 639–50. doi: 10.1111/bjh.13089 [DOI] [PubMed] [Google Scholar]
- 40.Xie R, Nguyen S, McKeehan WL, Liu L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol 2010; 11: 89. doi: 10.1186/1471-2121-11-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bánréti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013; 9: 819–29. doi: 10.4161/auto.23908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012; 111: 1208–21. doi: 10.1161/circresaha.112.265819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell 2006; 22: 463–75. doi: 10.1016/j.molcel.2006.04.014 [DOI] [PubMed] [Google Scholar]
- 44.Grenier K, Kontogiannea M, Fon EA. Short mitochondrial ARF triggers Parkin/PINK1-dependent mitophagy. J Biol Chem 2014; 289: 29519–30. doi: 10.1074/jbc.M114.607150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martínez Leal JF, de La Cueva P, et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica 2010; 95: 613–21. doi: 10.3324/haematol.2009.013870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell 2017; 66: 141–153.e6. doi: 10.1016/j.molcel.2017.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou L, Ruvolo VR, McQueen T, Chen W, Samudio IJ, Conneely O, et al. HDAC inhibition by SNDX-275 (Entinostat) restores expression of silenced leukemia-associated transcription factors Nur77 and Nor1 and of key pro-apoptotic proteins in AML. Leukemia 2013; 27: 1358–68. doi: 10.1038/leu.2012.366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiu HW, Yeh YL, Wang YC, Huang WJ, Ho SY, Lin P, Wang YJ. Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol Cancer 2016; 15: 46. doi: 10.1186/s12943-016-0531-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patra S, Mahapatra KK, Praharaj PP, Panigrahi DP, Bhol CS, Mishra SR, et al. Intricate role of mitochondrial calcium signalling in mitochondrial quality control for regulation of cancer cell fate. Mitochondrion 2021; 57: 230–40. doi: 10.1016/j.mito.2021.01.002 [DOI] [PubMed] [Google Scholar]
- 50.Mu X, Brynien D, Weiss KR. The HDAC inhibitor vorinostat diminishes the in vitro metastatic behavior of osteosarcoma cells. Biomed Res Int 2015; 2015: 290368. doi: 10.1155/2015/290368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leone A, Roca MS, Ciardiello C, Terranova-Barberio M, Vitagliano C, Ciliberto G, et al. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic Biol Med 2015; 89: 287–99. doi: 10.1016/j.freeradbiomed.2015.07.155 [DOI] [PubMed] [Google Scholar]
- 52.Mrakovcic M, Kleinheinz J, Fröhlich LF. p53 at the crossroads between different types of HDAC inhibitor-mediated cancer cell death. Int J Mol Sci 2019; 20: 2415. doi: 10.3390/ijms20102415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Häussinger D, et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res 2010; 70: 6313–24. doi: 10.1158/0008-5472.can-10-0999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang C, Yang C, Feldman MJ, Wang H, Pang Y, Maggio DM, et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017; 8: 56110–25. doi: 10.18632/oncotarget.18125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 2019; 4: 62. doi: 10.1038/s41392-019-0095-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang R, Xu Y, Wan W, Xin Shou X, Qian J, You Z, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell 2015; 57: 456–66. doi: 10.1016/j.molcel.2014.12.013 [DOI] [PubMed] [Google Scholar]
- 57.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10: 1403–14. doi: 10.4161/auto.29231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel S, Hurez V, Nawrocki ST, Goros M, Michalek J, Sarantopoulos J, et al. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 2016; 7: 59087–97. doi: 10.18632/oncotarget.10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burke MJ, Lamba JK, Pounds S, Cao X, Ghodke-Puranik Y, Lindgren BR, et al. A therapeutic trial of decitabine and vorinostat in combination with chemotherapy for relapsed/refractory acute lymphoblastic leukemia. Am J Hematol 2014; 89: 889–95. doi: 10.1002/ajh.23778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lohitesh K, Saini H, Srivastava A, Mukherjee S, Roy A, Chowdhury R. Autophagy inhibition potentiates SAHA-mediated apoptosis in glioblastoma cells by accumulation of damaged mitochondria. Oncology Rep 2018; 39: 2787–96. doi: 10.3892/or.2018.6373 [DOI] [PubMed] [Google Scholar]
- 61.Gonçalves RM, Agnes JP, Delgobo M, de Souza PO, Thomé MP, Heimfarth L, et al. Late autophagy inhibitor chloroquine improves efficacy of the histone deacetylase inhibitor SAHA and temozolomide in gliomas. Biochem Pharmacol 2019; 163: 440–50. doi: 10.1016/j.bcp.2019.03.015 [DOI] [PubMed] [Google Scholar]
- 62.Jing B, Jin J, Xiang R, Liu M, Yang L, Tong Y, et al. Vorinostat and quinacrine have synergistic effects in T-cell acute lymphoblastic leukemia through reactive oxygen species increase and mitophagy inhibition. Cell Death Dis 2018; 9: 589. doi: 10.1038/s41419-018-0679-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Torgersen ML, Engedal N, Bøe SO, Hokland P, Simonsen A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood 2013; 122: 2467–76. doi: 10.1182/blood-2013-05-500629 [DOI] [PubMed] [Google Scholar]
- 64.Chen H, Wang Y, Lin C, Lu C, Han R, Jiao L, et al. Vorinostat and metformin sensitize EGFR-TKI resistant NSCLC cells via BIM-dependent apoptosis induction. Oncotarget 2017; 8: 93825–38. doi: 10.18632/oncotarget.21225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhen Z, Yang K, Ye L, You Z, Chen R, Liu Y, He Y. Suberoylanilide hydroxamic acid sensitizes neuroblastoma to paclitaxel by inhibiting thioredoxin-related protein 14-mediated autophagy. Cancer Sci 2017; 108: 1485–92. doi: 10.1111/cas.13279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kou X, Yang Y, Jiang X, Liu H, Sun F, Wang X, et al. Vorinostat and Simvastatin have synergistic effects on triple-negative breast cancer cells via abrogating Rab7 prenylation. Eur J Pharmacol 2017; 813, 161–71. doi: 10.1016/j.ejphar.2017.08.022 [DOI] [PubMed] [Google Scholar]
- 67.Carew JS, Medina EC, Esquivel JA 2nd, Mahalingam D, Swords R, Kelly K, et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J Cell Mol Med 2010; 14: 2448–59. doi: 10.1111/j.1582-4934.2009.00832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L, Wu MC, et al. Inhibition of autophagy significantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J Gastroenterol 2014; 20: 4953–62. doi: 10.3748/wjg.v20.i17.4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chiao MT, Cheng WY, Yang YC, Shen CC, Ko JL. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013; 9: 1509–26. doi: 10.4161/auto.25664 [DOI] [PubMed] [Google Scholar]
- 70.Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res 2015; 17: 33. doi: 10.1186/s13058-015-0534-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee TG, Jeong EH, Kim SY, Kim HR, Kim CH. The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790M-mutated lung cancer. Int J Cancer 2015; 136: 2717–29. doi: 10.1002/ijc.29320 [DOI] [PubMed] [Google Scholar]
- 72.Chiu HW, Yeh YL, Wang YC, Huang WJ, Chen YA, Chiou YS. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS One 2013; 8: e76340. doi: 10.1371/journal.pone.0076340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen MY, Liao WSL, Lu Z, Bornmann WG, Hennessey V, Washington MN, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011; 117: 4424–38. doi: 10.1002/cncr.26073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tavallai S, Hamed HA, Grant S, Poklepovic A, Dent P. Pazopanib and HDAC inhibitors interact to kill sarcoma cells. Cancer Biol Ther 2014; 15: 578–85. doi: 10.4161/cbt.28163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Claerhout S, Lim JY, Choi W, Park YY, Kim K, Kim SB, et al. Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS One 2011; 6: e24662. doi: 10.1371/journal.pone.0024662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6: 1057–65. doi: 10.4161/auto.6.8.13365 [DOI] [PubMed] [Google Scholar]
- 77.Hrzenjak A, Kremser ML, Strohmeier B, Moinfar F, Zatloukal K, Denk H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J Pathol 2008; 216: 495–504. doi: 10.1002/path.2434 [DOI] [PubMed] [Google Scholar]