

Abstract
Plasmacytoid dendritic cells (pDCs) produce type I interferons (IFNs) and promote pathogenesis of multiple autoimmune diseases. Autoimmune Sjögren’s syndrome (SS) primarily affects salivary and lacrimal gland, causing their inflammation, destruction and dysfunction. pDCs and type I IFN activity are elevated in salivary glands of SS patients, and this study seeks to elucidate the in vivo actions of pDCs in SS pathogenesis using the non-obese diabetic (NOD) mouse model. We confirmed the type I IFN-dependency of SS development in female NOD mice and elevation of pDC-type I IFN in their submandibular glands (SMGs). We administered a pDC-depleting anti-BST2/CD317 antibody to female NOD mice from 4 to 7 weeks of age, the early stage of SS, and assessed SS pathologies at age 10 weeks, the time of disease onset. Depletion of pDCs impeded the development of SMG inflammation and secretory dysfunction. It drastically reduced the amount of type I IFN mRNA and that of total leukocytes, and T- and B lymphocytes in SMGs. Gene expression analyses showed that pDC depletion markedly diminished SMG expression of IL-7, BAFF, TNF-α, IFN-γ, CXCL9, CXCL11, CD40, CD40L, Lt-α, Lt-β and NOS2. Hence, pDCs critically contribute to the development and onset of SS-like salivary gland exocrinopathy.
Keywords: Sialadenitis, type I interferons, salivary gland, hyposalivation, autoimmune inflammation, innate immune cells
1. Introduction
Sjӧgren’s syndrome (SS) is a systemic autoimmune disease affecting an approximate 4 million Americans [1, 2]. It is characterized by the autoimmune inflammation, damage and dysfunction of exocrine glands, primarily salivary and lacrimal glands, frequently accompanied with abnormal production of autoantibodies [1–4]. The most common clinical manifestations of SS are dry mouth and dry eyes, which are often accompanied by a variety of systemic complications [1, 2]. Although the etiology of SS is still largely unknown, a key pathogenic role of type I interferons (IFNs) in this disease has been well characterized [5–8]. In patients with SS, activation of type I IFN pathways has been detected in the salivary gland biopsy specimens, with the severity of salivary gland inflammation and secretory dysfunction positively correlated with the type I IFN activity [6–11]. In the C57BL/6.NOD-Aec1Aec2 mouse, a spontaneous model of SS, IFNα receptor deficiency that obliterates type I IFN signaling prevents the leukocyte infiltration of salivary glands and enhances salivary secretion [8]. Therefore, understanding the function of major type I IFN-producing cells in SS pathogenesis and unraveling the underlying cellular and molecular mechanisms could facilitate the development of effective cell-targeted therapeutic strategies against SS disease, for which no effective treatment is currently available.
Plasmacytoid dendritic cells (pDCs) are the most potent cellular producers of type I IFNs and have the capacity to produce up to 1,000-fold more type I IFNs than other cell types upon activation [12–14]. In autoimmune diseases such as psoriasis, systemic lupus erythematous and type I diabetes, pDCs infiltrate into the disease target organs and cause excessive production of type I IFNs by recognition of self-derived nucleic acids. These early events have been demonstrated to critically contribute to the initiation and induction of the autoimmune inflammatory response and immunopathology [9, 15–20], as in vivo ablation of pDCs during the early stage of inflammatory responses leads to amelioration of these autoimmune disorders [17, 20, 21]. In addition to robust type I IFN production, pDCs also can secrete various other inflammatory cytokines, such as TNFα and IL-6, produce chemokines and express costimulatory molecules on their surfaces, to influence both innate and adaptive immune responses through different mechanisms in a disease context-dependent manner [9, 19, 22]. In SS patients, pDCs are detected in salivary glands, along with activated type I IFN signaling pathways [9–11, 13]. Furthermore, a recent transcriptome study has demonstrated that pDCs from SS patients have an more activated transcriptional profile and are more poised to produce high-levels of pro-inflammatory cytokines [23]. Collectively, these findings strongly support a potential pathogenic role of pDCs in SS disease.
In the present study, we first confirmed the type I IFN-dependency of SS pathogenesis and the correlation between the presence of pDCs and activation of type I IFN pathways in submandibular glands (SMGs) in the female non-obese diabetic (NOD) mouse strain, a well-characterized and commonly used SS disease model. We next sought to determine the specific role of pDCs in SS pathogenesis using the NOD mouse model by using an anti-BST2/CD317 antibody-mediated pDC depletion approach, which reveals that pDCs make indispensable contributions to the development and onset of SS and provides a rationale for therapeutically targeting pDCs to combat this disease.
2. Materials and Methods
2.1. Mice.
Female NOD/ShiLtJ mice (Cat# 001976) and BALB/cJ mice (Cat# 000651) were purchased from the Jackson Laboratory and kept under the specific pathogen-free condition at the Forsyth Institute. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Forsyth Institute (protocol number: 19-002; approval date: 02/12/2019), which is fully accredited by the AAALAC. All experiments and assays were carried out in compliance with the “Guide for the Care and Use of Laboratory Animals” of the National Institutes of Health and the ARRIVE guidelines.
2.2. Antibodies.
For injections, purified monoclonal anti-mouse IFNAR1 (MAR1–5A3), isotype mouse IgG1 and rat IgG2b were obtained from BioXCell, and purified monoclonal anti-mouse BST2/CD317 antibody (JF05–1C2.4.1) was obtained from Miltenyi Biotec. For flow cytometric staining, fluorescence conjugated anti-CD45 (30-F11), anti-CD4 (GK-1.5), anti-CD8 (53–5.8), anti-B220 (RA3–6B2), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD86 (GL-1), anti-BST2/CD317, anti-CD11b (M1/70), anti-CD11c (N418), anti-Siglec-H (clone 551) and anti-CD16/32 antibodies were all purchased from BioLegend. For immunohistochemical staining, rat anti-BST2/CD317 antibody (clone 927) was obtained from BioXCell, and biotin conjugated anti-rat IgG was purchased from Vector Laboratories. For immunofluorescence staining, Alexa Fluor568-conjugated anti-mouse IgG was purchased from Abcam.
2.3. In vivo administration of antibodies.
For IFNAR1 blockade, 1 mg of anti-mouse IFNAR1 antibody or its mouse IgG1, dissolved in 100 μl PBS, was intraperitoneal (i.p.) administered into 4-week-old female NOD mice, once every 5 days for a total of 4 times. For pDC depletion, 500 μg pDC-depleting anti-BST2/CD317 antibody or its isotype control, rat IgG2b, dissolved in 100 μl PBS, was i.p. administered into 9-week-old female NOD mice on day 0 and day 2, or into 4-week-old female NOD mice, 3 times weekly for 3 weeks, as indicated in the specific experiments.
2.4. Histological and immunohistochemical staining.
The procedures were performed as previously described [24–28]. SMGs were fixed in 4 % paraformaldehyde, embedded in paraffin and sectioned to 5μm thickness. The sections were stained with hematoxylin and eosin (H&E) to quantify the leukocyte aggregates in SMG tissues. Leukocytic foci containing at least 50 leukocytes in each of the three non-consecutive sections, separated by 100 microns, of an SMG sample were enumerated, and the average number was used for subsequent statistical analysis. For immunohistochemical staining, the SMG sections were incubated with anti-mouse BST2/CD317 antibody overnight at 4°C and the procedures were carried out using a VECTASTAIN Elite ABC Kit (Vector Laboratories) following the instructions of the kit. The stained sections were then imaged with a light microscope at 200× or 400× magnification.
2.5. Detection of serum autoantibodies.
The procedures were performed as previously described [24–28]. Mouse sera were serial diluted by 2-fold from 1:40 to 1:1280 in PBS and mounted onto the HEp-2 human epithelial cell substrate slides (INOVA Diagnostics), which were then incubated at room temperature for 1 hour. After washing with PBS, the slides stained with 1:100-diluted Alexa Fluor568–conjugated goat anti-mouse IgG (Invitrogen) for 1 hour at room temperature, and washed with PBS. The stained slides were imaged using a Zeiss wide-field fluorescence microscope (Zeiss) at 400× magnification and analyzed using the corresponding Zeiss software. The fluorescence staining intensity was measured using the ImageJ 1.50i software, and the titer of the autoantibody was determined based on the negative staining control with PBS.
2.6. ELISA of serum anti-M3 muscarinic acetylcholine receptor (M3R).
A peptide (AILFWQYFVGKRTVP) that contains 15 amino acids of the second extracellular loop of the M3R protein (synthesized by Biomatic Corporation, kindly provided by Dr. Kawai, Nova Southeastern University) was dissolved in PBS, and further diluted in 1×BioLegend ELISA coating buffer. The resulting M3R peptide solution (2 μg/ml) was used to coat Nunc™ MaxiSorp™ flat-bottom 96 well plates. Non-specific binding sites on the plates were blocked with the ELISA assay diluent buffer (BioLegend), and 1:6-diluted serum samples were added to the plates and incubated overnight at 4 °C. After washing, the plates were incubated with 1:300-diluted biotinylated goat anti-mouse IgG (Vector Laboratories) for 1 h, washed, and incubated with the avidin-HRP solution for 30 min. The plates were washed and then incubated with the TMB substrate to detect the antibodies bound to the plates. The absorbance was measured using a BioTek microplate reader at 450 nm.
2.7. Measurement of salivary flow rate.
Mice were i.p. injected with 100 μl PBS-based secretagogue solution containing isoproterenol (1 mg/ml) and pilocarpine (2 mg/ml). One min after the secretagogue injection, saliva was collected continuously for 5 min from the oral cavity of mice with a micropipette. The volume of saliva was measured and normalized to the body weight and presented in the unit of μl/g.
2.8. Preparation of single cells from SMGs.
SMGs were harvested from mice, minced into small pieces in cold Dulbecco’s Modified Eagle Medium, and digested in the presence of collagenase type II (0.63 mg/ml; Gibco), hyaluronidase (0.5 mg/ml; Sigma-Aldrich), and CaCl2 (6.25 mM; Sigma-Aldrich) at 37°C for 20 min. The tissue pieces were pipetted up and down repeatedly to further break up cell clumps and filtered sequentially through 100 um- and 50um pore sized nylon meshes to remove any cell aggregates and yield single cells.
2.9. Flow cytometry.
Freshly prepared SMG single cells were incubated with anti-CD16/32 antibody to block non-specific Fc receptor binding. The cells were subsequently stained with a combination of fluorescence-conjugated antibodies against surface markers for pDCs, CD4 and cD8 T cells, activated effector CD4 and CD8 T cell populations, B cells and activated B cell population, at 4°C for 30 min, rinsed twice with cold PBS. The stained cells were then analyzed with a flow cytometer (Invitrogen Attune) and the FlowJo V10 software. Dead cells and cell debris were excluded by gating based on both forward- and side scatters.
2.10. Real-time RT-PCR.
Total RNA was isolated using RNeasy Micro kit (Qiagen) and reverse transcribed into cDNA by MLV reverse transcriptase (Promega). SYBR Green-based real-time PCR amplification (Qiagen) was performed for 40 cycles with annealing and extension temperature at 60°C, on a LightCycler 480 Real-Time PCR System (Roche). Primer sequences for mouse gene analysis are: β-actin, forward, 5’- TGG ATG ACG ATA TCG CTG CG −3’; reverse, 5’- AGG GTC AGG ATA CCT CTC TT −3’. IFNα, forward, 5’- ACC TCA GGA ACA AGA GAG CC −3’; reverse, 5’- CTG CGG GAA TCC AAA GTC CT −3’. IFN β, forward, 5’- TAA GCA GCT CCA GCT CCA AG-3’; reverse, 5’- CCC TGT AGG TGA GGT TGA TC-3’. IL-7, forward, 5’- GGA ACT GAT AGT AAT TGC CCG −3’; reverse, 5’- TTC AAC TTG CGA GCA GCA CG −3’. BAFF, forward, 5’- AGT TGG CTG CCT TGC AAG CA −3’; reverse, 5’- TCA GGA GTT TGA CTC CAG CG −3’. IRF-1, forward, 5’- GGG TCA GGA CTT GGA TAT GG −3’; reverse, 5’- GAT CAG TGG TGC TAT CTG GT −3’. IRF-7, forward, 5’- GGT CGT AGG GAT CTG GAT GA −3’; reverse, 5’- GGA AGT TGG TCT TCC AGC CT −3’. All transcript levels were normalized to that of β-actin.
2.11. PCR array analysis.
RT2 Profiler PCR Array (Cat no. PAMM-077Z; Qiagen) analysis for 84 key genes involved in mouse inflammatory response and autoimmunity was performed following the manufacturer’s instructions. The data were interpreted with the web-based PCR Array Data Analysis tool (Qiagen). The examined genes with at least two-fold (up or down) differences between the treatment and control groups were considered as differentially expressed genes.
2.12. Statistical analysis.
The normal distribution of all the data was confirmed by SPSS analysis, and the statistical significance was determined by the Student’s t-test (two-tailed, two sample equal variance). P values equal to or smaller than 0.05 were considered as statistically significant.
3. Results
3.1. Blockade of IFNAR1 (IFN-α/β receptor subunit 1) impedes the development of SS-like salivary gland disorder in NOD mice
Elevated type I IFN activity has been detected in a large proportion of SS patients, and its functional importance has been demonstrated in a mouse model of this disease, the C57BL/6.NOD-Aec1.Aec2 mouse strain [5–8]. In this study, we used female NOD/ShiLtJ (referred to as NOD) mice that spontaneously develop SS-like sialadenitis and exhibit initial disease onset at around 10 weeks of age, based on characteristic SS pathologies and impaired salivary secretion [26, 29, 30]. It has been shown that the onset of clinical diabetes in female NOD mice generally starts around 12–16 weeks of age [31], and we also previously confirmed that none of the female NOD mice used in our studies became diabetic up to 13 weeks, when the mice had established SS disease, as indicated by normal urine glucose levels [27]. Hence, the onset of SS is earlier than that of diabetes and hyperglycemia.
To examine the role of type I IFNs in the early pathogenic and pathological events of SS prior to clinical disease onset in the NOD mouse model, we i.p. administered 1 mg of monoclonal blocking antibody against IFNAR1 or its isotype IgG, to 4-week-old female NOD mice, once every 5 days for a total of 4 times, and analyzed the SS disease profile in these mice at 10 weeks of age, the time of initial disease onset. Anti-IFNAR1 antibody treatment significantly suppressed the leukocyte infiltration of SMGs based on histological analyses and increased the salivary flow rate (Fig. 1A–B). Moreover, real-time PCR results showed that the gene expression of two key SS-promoting cytokines, IL-7 and BAFF, were substantially downregulated in the SMGs by anti-IFNAR1 treatment (Fig. 1C). In addition, anti-IFNAR1 treatment markedly decreased the relative level of anti-M3 muscarinic acetylcholine receptor (M3R) antibody, an autoantibody closely associated with SS with the ability to impair salivary secretion triggered by neurotransmitters, in the sera as determined by ELISA using a specific M3R peptide (Fig. 1D). In accordance, indirect immunofluorescence staining analysis unequivocally revealed that anti-IFNAR1-treatment also significantly reduced serum autoantibody levels, and the titers of autoantibodies (Fig. 1E). Taken together, these results strongly support the notion that endogenous type I IFN signaling is crucial for the development of SS-like salivary gland disorder in the NOD model of this disease.
Figure 1. Blockade of IFNAR1 impedes the development of SS-like salivary gland pathologies in NOD mice.
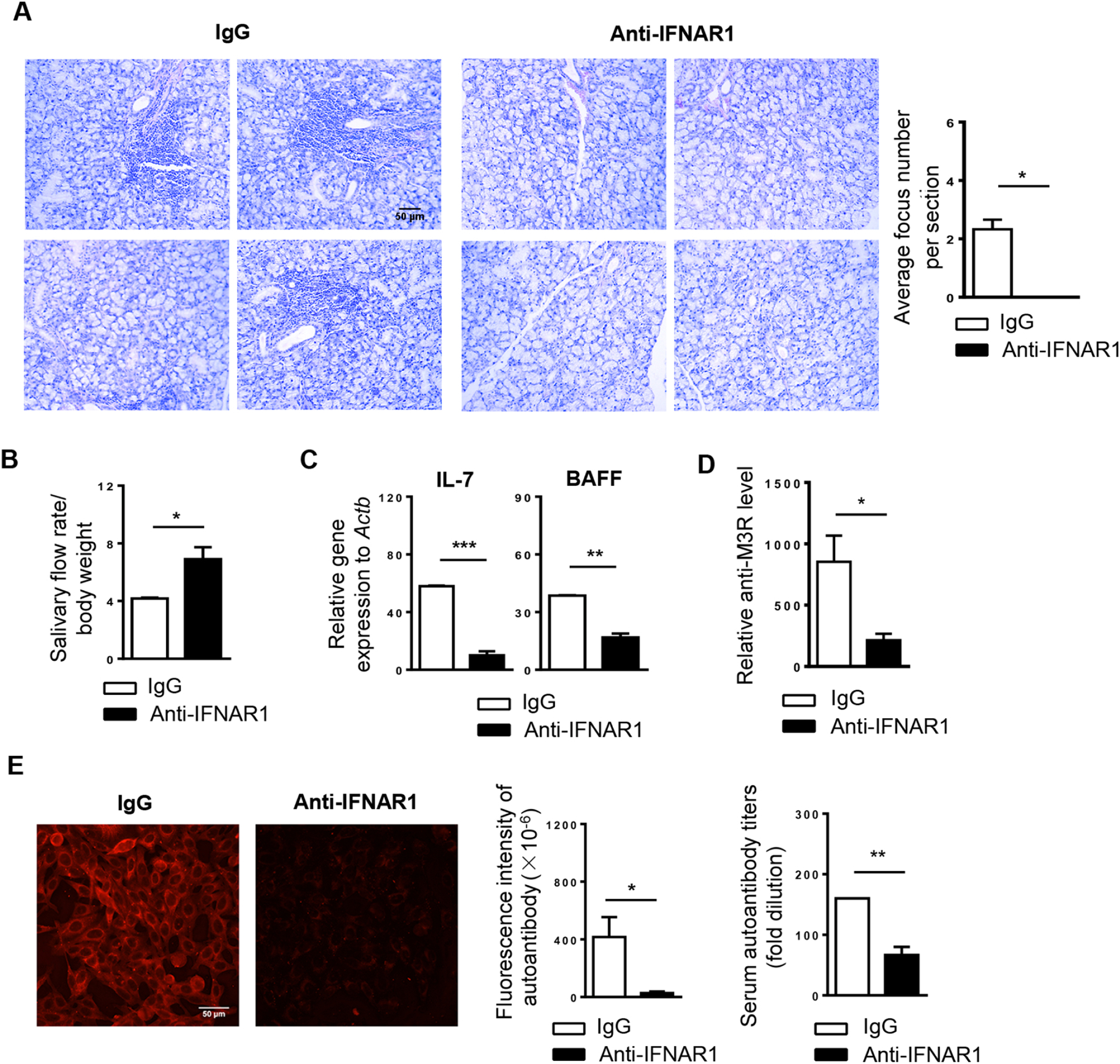
Anti-IFNAR1 antibody or IgG was i.p. administered to 4-week-old female NOD mice, once every 5 days for a total of 4 times. All the analyses were performed when these mice were 10 weeks of age. (A) Tile images of H&E stained areas of one representative SMG sample each treatment group (scale Bar=100 μm). Bar graph shows the average number of leukocyte foci per SMG section. (B) Stimulated saliva flow rate normalized to body weight. (C) Real-time PCR analysis of IL-7 and BAFF mRNA levels in SMGs. Gene expression was presented relative to that of β-actin. (D) Relative level of anti-M3R in the sera as determined by ELISA. (E) Detection of serum autoantibodies (scale bar=50 μm). Bar graphs show the fluorescence intensity of autoantibody staining of the 1:40-diluted sera, left, and the autoantibody titers, right. All data are the representative or average of analyses of 6 mice for each group. Error bars represent the standard error of mean (SEM). *P< 0.05, ** P<0.01, ***P<0.001.
3.2. pDCs are detected in the SMGs of female NOD mice accompanied by a local upregulation of type I IFN-responsive genes
pDCs and activated type I IFN pathways are readily detected in the salivary glands of SS patients compared to the control subjects [9–11, 13]. To examine the presence of pDCs in the SMGs of NOD mice, we analyzed SMG single cells by antibody staining and multicolor flow cytometry. The results revealed a significantly higher percentage of pDCs, defined as CD11b−CD11cmidB220+Siglec-H+BST2+ cells, in total SMG cells of 13-week-old female NOD mice, which had established SS disease, compared to that of age- and gender-matched BALB/c control mice (Fig. 2A). Immunohistochemical staining with an antibody specific for BST2 showed the presence of BST2+ cells, enriched of pDCs, in the SMGs of NOD mice at 7- and 10 weeks of age, corresponding respectively to the pre-disease autoimmune phase and the initial disease onset. In comparison, almost no BST2+ cells were detected in SMGs of 4-week-old, young NOD mice (Fig. 2B). Furthermore, the presence of pDCs was accompanied by a local upregulation of IRF-1 and IRF-7, two type I IFN-responsive genes, in the SMGs based on real-time PCR analysis (Fig. 2C). Hence, pDCs and activated type I IFN pathways are detected in the SMGs of NOD mice, which is consistent with the observation in SS patients [7, 10, 11] and suggests a vital participation of pDCs in SS disease pathogenesis.
Figure 2. pDCs are present in the SMGs of female NOD mice, accompanied by local upregulation of type I IFN-responsive genes.
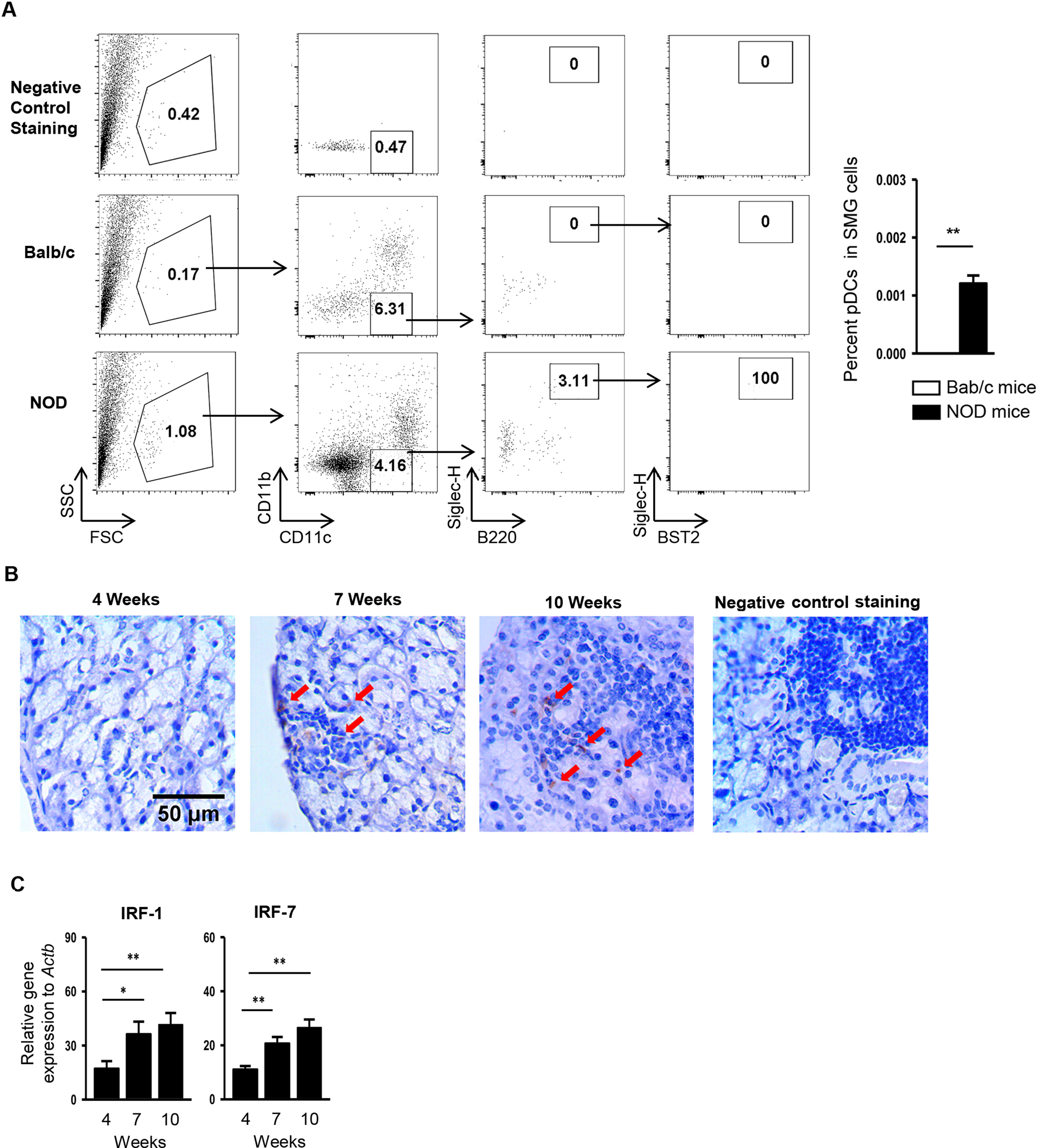
(A) Flow cytometric analysis of the percentage of pDCs (CD11b−CD11cmidB220+Siglec-H+BST2+) in total SMG cells of BALB/c mice and NOD mice aged 13 weeks. The top panels show the negative control staining profile of the cells (a mixture of BALB/c and NOD SMG cells at the 1:1 ratio) under each gating. Bar graph shows the mean percentage of pDCs in total SMG cells, calculated as: Percentage of pDCs in the total SMG cells (%) = % mononuclear cells in SMG cells × % CD11b−CD11cmid cells in mononuclear cells × % B220+Siglec-H+ cells among CD11b−CD11cmid cells × % B220+BST2+ cells among B220+Siglec-H+ cells × 100. (B) Immunohistochemical staining with anti-BST2 antibody of SMG sections from female NOD mice aged 4, 7, and 10 weeks (scale bar = 50 μm). Red arrows indicate the representative areas stained positive for BST2. Negative staining control: SMG sections from female NOD mice aged 10 weeks stained without the without the primary antibody. (C) Real-time PCR analysis of IRF-1 and IRF-7 levels in the SMGs of female NOD mice. The results are presented relative to that of β-actin. All data are representative or the average of analyses of 4–7 mice for each group.
3.3. Anti-BST2 antibody-mediated pDC depletion rapidly reduces expression levels of type I IFNs, type I IFN-responsive genes and SS-promoting cytokines in the SMGs of NOD mice
Many lines of studies have identified pDCs as the most potent cellular producers of type I IFNs [12–14]. To confirm the efficacy of antibody-mediated pDC-depletion and to test whether pDCs are the key type I IFN-producing cells in the SMGs in the SS setting, we ablated pDCs by i.p. administration of 500 μg pDC-depleting anti-BST2 antibody into 9-week-old female NOD mice during the pre-disease autoimmune phase on day 0 and day 2, and analyzed the mice for SS disease pathologies 24 hours after the last injection. Almost all pDCs were effectively depleted with a high efficiency in SMGs, SMG-draining lymph nodes (smLNs) and spleens after this short course of anti-BST2 treatment based on flow cytometric analysis (Fig. 3A). Real time PCR revealed a significant reduction in mRNA levels of type I IFNs, including IFNα and IFNβ, and those of IRF-1 and IRF-7 in the SMGs resulting from anti-BST2 administration (Fig. 3B). In addition, anti-BST2-mediated pDC-depletion also led to a marked downregulation of IL-7 and BAFF gene expression in the SMGs (Fig. 3B). These results have validated the high efficiency of anti-BST2-mediated pDC depletion in the NOD mice using this regimen, and strongly suggested pDCs as the major cellular sources of type I IFNs in the SMGs in the NOD mice with a disease-promoting effect in SS.
Figure 3. Administration of anti-BST2 antibody efficiently depletes pDCs and reduces expression of type I IFNs, type I IFN-responsive genes and SS-promoting cytokines in SMGs.
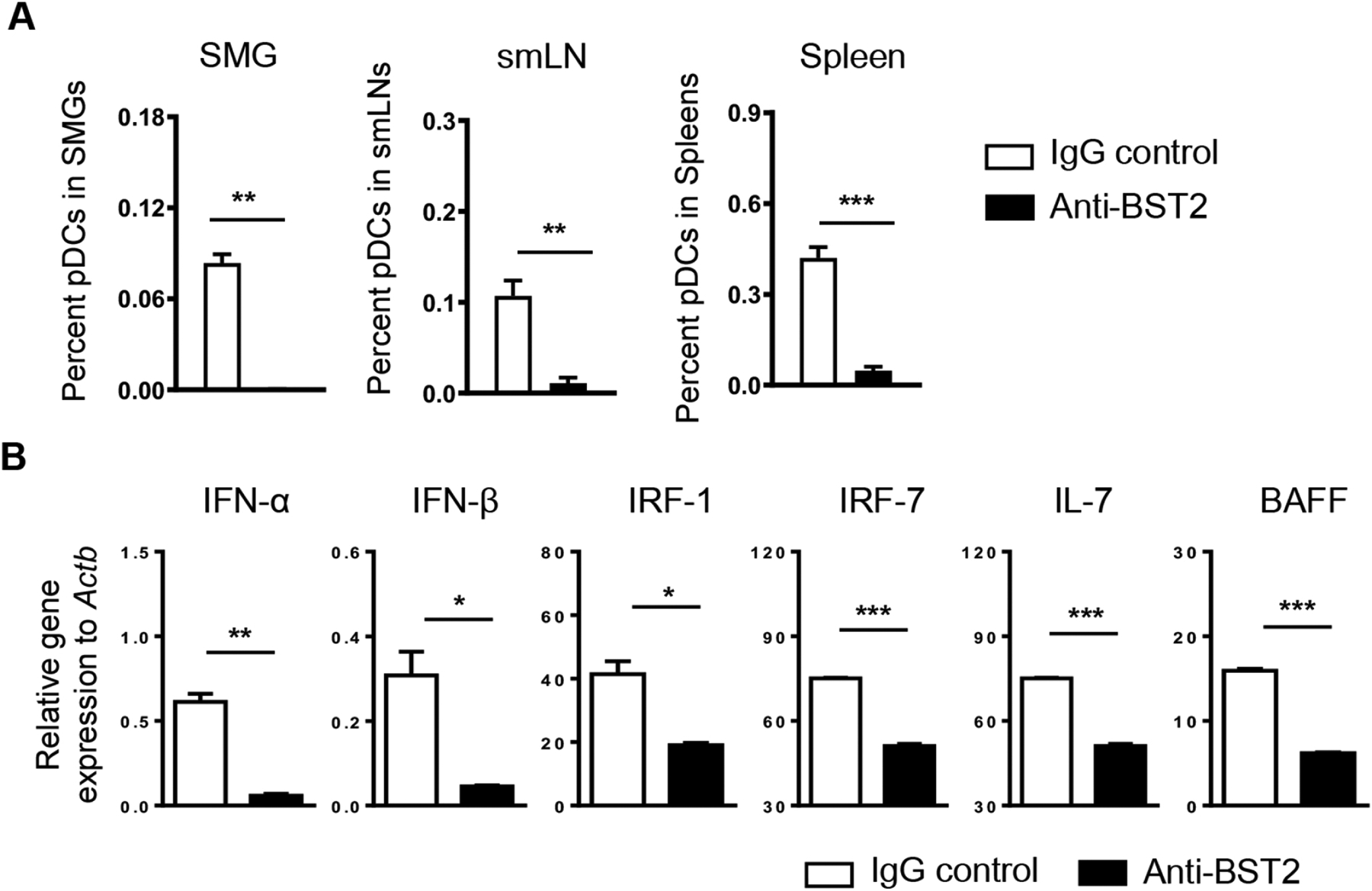
Anti-BST2 or isotype rat IgG was i.p. administered into 9-week-old female NOD mice on day 0 and day 2. The analyses were performed 24 hours after the last injection. (A) Percentages of pDCs (defined as CD11b−CD11cmidB220+Siglec-H+BST2+) among total SMG cells, SMG-draining lymph node, and spleen cells based on flow cytometric analysis. (B) Real-time PCR analysis of the mRNA levels of type I IFNs, type I IFN-responsive genes and SS-promoting cytokines in the SMGs. The results are presented relative to that of β-actin. All data are representative or the average of analyses of 3 mice for each group.
3.4. Anti-BST2-mediated pDC depletion over a 3-week period improves salivary gland secretory function and attenuates SMG inflammation in NOD mice
To determine whether a longer duration anti-BST2 treatment can impede the early pathogenic and pathological events and hinder the onset of SS disease, we i.p. administered 500 ug of anti-BST2 antibody or its isotype control IgG into 4-week-old female NOD mice, 3 times weekly for a consecutive 3 weeks. The characteristic pathologies of SS were analyzed at 10 weeks of age, the time of normal clinical disease onset in these mice. We first evaluated the effect of anti-BST2 treatment on hyposalivation, one of the hallmark manifestations of SS, and found that anti-BST2-treated NOD mice exhibited markedly higher salivary flow rate than the control mice (Fig. 4A), suggesting an improvement of salivary gland secretory function and alleviation of hyposalivation. H&E staining of SMG sections showed markedly fewer leukocyte infiltrates in anti-BST2 antibody-treated mice than the control IgG-treated mice (Fig. 4B, left). Accordingly, quantification of leukocyte infiltration revealed that the number of leukocyte foci was significantly lowered by anti-BST2 treatment (Fig. 4B, right). Therefore, these findings suggest that pDC depletion at the early disease development stage hinders the initiation and onset of SS-associated salivary gland pathology and dysfunction in the NOD model of this disease.
Figure 4. Administration of anti-BST2 antibody over a 3-week period reduces leukocyte infiltration of SMGs and improves salivary secretion in female NOD mice.
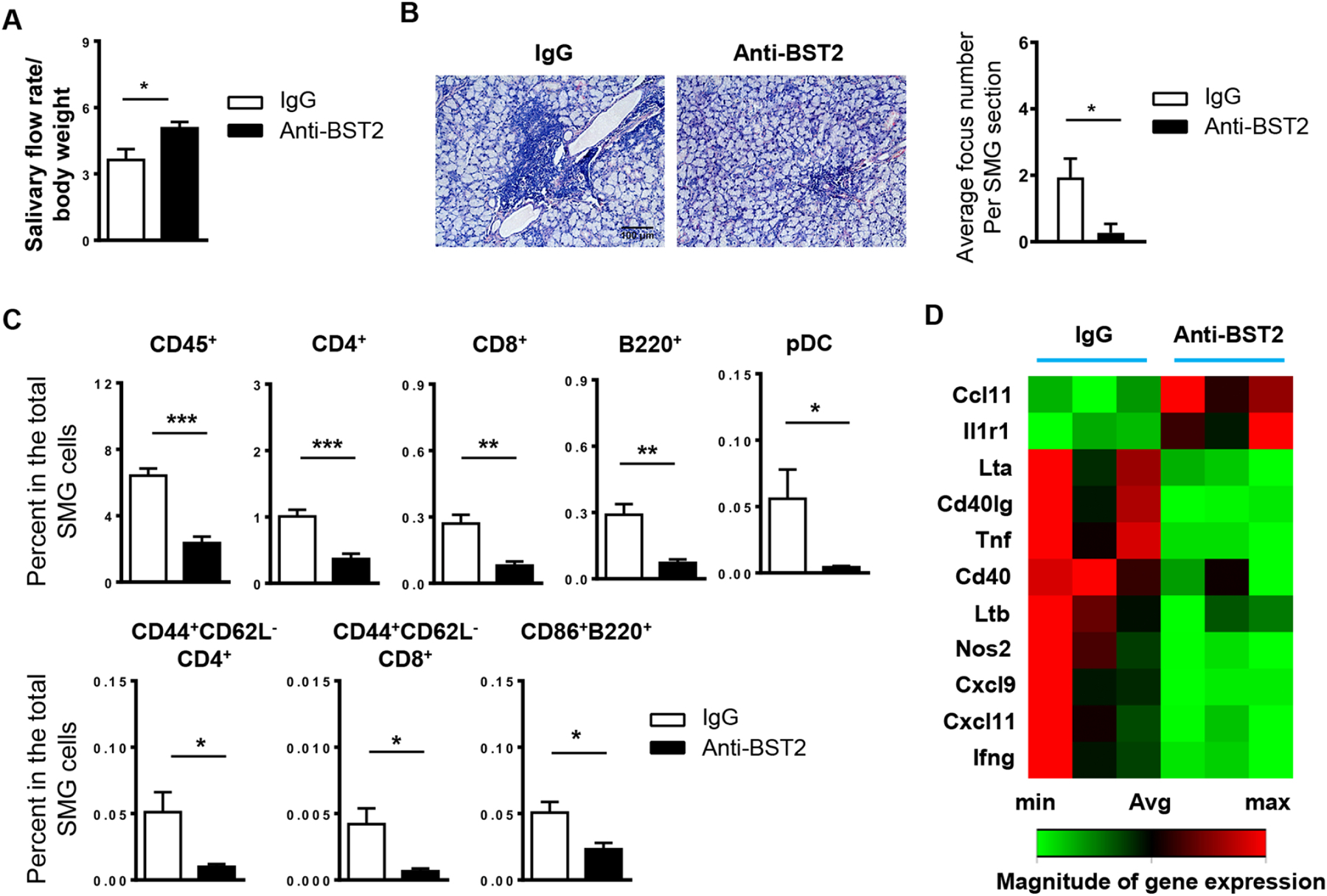
Anti-BST2 or isotype rat IgG was i.p. administered into 4-week-old female NOD mice, 3 times weekly for 3 weeks. All the analyses were performed when mice were 10 weeks of age. (A) Stimulated saliva flow rate normalized to body weight. (B) Images of H&E staining of SMG sections (scale Bar=100 μm). Bar graph shows the average number of leukocyte foci per SMG section. (C) Flow cytometric analysis of lymphocyte populations in the SMGs. (D) The clustergram of RT2 Profiler PCR Array results displaying gene expression levels with statistically significant changes upon anti-BST2 treatment. All data are representative or the average of analyses of 6 mice for each group.
We next determined the impact of anti-BST2 treatment on SMG leukocyte compositions. Consistent with a reduced leukocyte focus number, flow cytometry analysis demonstrated that the frequency of total leukocytes, defined by surface expression of CD45, a leukocyte common antigen, was dramatically lowered by anti-BST2 treatment. Moreover, administration of anti-BST2 antibody decreased the proportion of CD4 and CD8 T cells, B220+ B cells and pDCs (CD11b−CD11cmidB220+Siglec-H+BST2+), in the total SMG cells (Fig. 4C). In accordance, the frequency of activated effector CD4 T cells (CD44+CD62L−CD4+) and CD8 T cells (CD44+CD62L−CD8+), and that of activated B cells (CD86+B220+) in the total SMG cells were also significantly lowered by anti-BST2 treatment (Fig. 4C).
We next comprehensively assessed the impact of anti-BST2-mediated pDC depletion on an array of inflammatory cytokines, chemokines and their receptors known to be critically involved in autoimmune and inflammatory responses, in the SMGs. The RT2 Profiler Inflammatory Response & Autoimmunity PCR Array analysis (Qiagen) showed that the mRNA level of TNF-α, IFN-γ, NOS2, CD40, CD40lg, Ltα, Ltβ, CXCL9 and CXCL11 in the SMGs of NOD mice with anti-BST2 treatment was markedly lower than that of control mice (Fig. 4D).
In summary, these results indicate that pDCs play an indispensable role at the early stage of the development of SS-like sialadenitis and hyposalivation, which is associated with an increased leukocyte accumulation, and an upregulated expression of multiple critical pro-inflammatory factors and chemokines in the SMGs.
3.5. Anti-BST2-mediated pDC depletion does not alter autoantibody production
Elevation in serum autoantibody levels is another characteristic manifestation of SS disease in addition to salivary gland inflammation and hypofunction. Indirect immunofluorescence staining with human Hep-2 epithelial cells as substrates showed that anti-BST2-treated mice exhibited varying levels of autoantibodies in the sera. On average, anti-BST2 treatment did not significantly alter the levels of autoantibodies based on the fluorescence intensity of the staining or the titers of autoantibodies (Fig. 5A), compared to the IgG-treated control group (p>0.05) (Fig. 5A). In addition, anti-BST2 treatment did not significantly change the relative levels of serum anti-M3R antibodies, as determined by ELISA with a specific M3R peptide (Fig. 5B). Hence, anti-BST2-mediated pDC depletion at the early stage of SS development does not significantly alter the production of these autoantibodies.
Figure 5. Administration of anti-BST2 antibody does not significantly affect serum autoantibody levels.
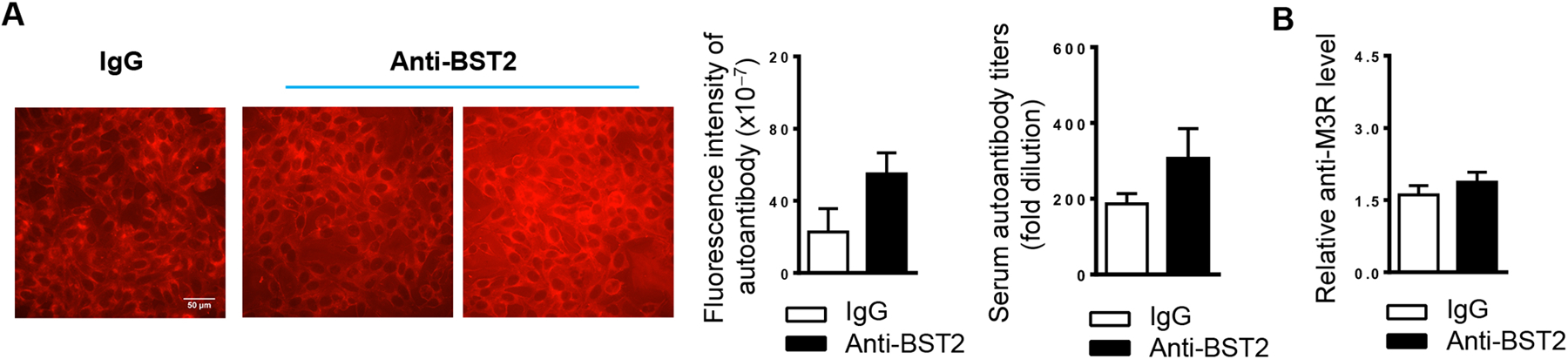
(A) Anti-BST2 or the isotype rat IgG was i.p. administered into 4-week-old female NOD mice, 3 times weekly for 3 weeks. Immunofluorescence staining images show the detection of serum autoantibodies, measured by the HEp-2 human epithelial cell substrate slides, in mice aged 10 weeks (scale bar = 50 μm). The two images shown for the anti-BST2 treatment group represent serum samples from two of the NOD mice in this group. Bar graphs show the fluorescence intensity of autoantibody staining of the 1:40-diluted sera, left, and the autoantibody titers, right. (B) Relative level of anti-M3R in the sera as determined by ELISA. All data are representative or the average of analyses of 6 mice for each group.
4. Discussion
The present study was undertaken to elucidate the role of pDCs in the development of SS using the NOD model of this disease, and the results showed that pDCs critically contribute to the initiation and development of SS-like salivary gland disorder, including sialadenitis and hyposalivation, in these mice. The results also provide several potential cellular and molecular mechanisms underlying the SS-promoting effect of pDCs, which include production of type 1 IFNs as the major cellular sources and upregulation of a number of pro-inflammatory factors and chemoattractants in the target SMG tissues.
Elevated amounts of pDCs and activated type I IFN signatures have been detected in the salivary glands of SS patients, with the severity of glandular inflammation and hyposalivation closely associated with type I IFN activity [9–11, 13]. Moreover, IFNAR1-deficiency impedes the full development of SS pathologies in C57BL/6.NOD-Aec1Aec2 mice, another spontaneous mouse model of SS [8]. pDCs are the main cellular sources of type I IFNs in multiple inflammatory and autoimmune conditions [13, 14]. Our study confirmed type I IFN pathways as key pathogenic factors and pDCs as major producers of type I IFNs in SS disease using the NOD mouse model and the anti-BST2 antibody-mediated pDCs depletion approach.
The present study showed that pDC depletion and IFNR1-ablation both cause a down-regulation of SMG expression of IL-7 and BAFF, two critical pathogenic factors in SS by enhancing T and B cell responses, respectively, as we and other groups previously reported [24, 32–35]. These results also further support and strengthen the previous findings that type I IFN-signaling enhances IL-7 and BAFF production in salivary gland cells that we and others have illustrated [25, 36].
Importantly, our study demonstrated that depletion of pDCs by repeated administration of an anti-BST2 antibody during the early stage of SS significantly impedes the development and onset of salivary gland inflammation and dysfunction in NOD mice, accompanied by reduced local T and B cell responses in SMGs, which was similar to the outcome of IFNAR1 blockade. Together with the finding that pDCs are the major cellular sources of type 1 IFNs in the SMGs of the NOD mice, these results strongly suggest that pDCs promote the pathogenic SS, to a large degree, through production of type 1 IFNs.
This study also uncovered a panel of immunological players that are down-regulated in the target SMG tissues by pDC-ablation, including costimulatory factors, proinflammatory cytokines and mediators, and T cell chemoattractants [28, 29, 32, 37–41]. All these molecules have been shown to contribute to SS pathogenesis by controlling different pathological events and processes, suggesting that they are the mediators of pDC actions in this disease. Interestingly, the expression of CCL11 and IL-1R1, two immunological factors that bear complex context-dependent pathophysiological effects, was markedly upregulated by pDC-depletion. The functional significance of these changes and the previously unexplored role of CCL11 and IL-1R1 in SS require further investigation and delineation using both in vitro and in vivo approaches.
Antibodies against BST2 are commonly used to deplete pDCs in vivo [21, 42, 43]. Our study also confirms a high efficiency of pDC ablation in the SMGs of NOD mice by administration of the JF05–1C2.4.1 clone of anti-BST2 antibody. In addition to being expressed in pDCs, BST2 can also be induced in several other cell types following exposure to type I and II IFNs [44]. However, it has been reported that administration of the same clone of anti-BST2 antibody to a mouse model of experimental autoimmune encephalomyelitis specifically depletes pDCs without affecting other immune cell populations including NK cells, NKT cells, B cells, and macrophages in peripheral lymphoid organs [18], supporting the specificity and validity of this approach for pDC depletion. It is also important to note that this study excluded the interference from the clinical type-1 diabetes, since the onset of SS in female NOD mice, which was also our time of endpoint analysis, is earlier than that of clinical diabetes and hyperglycemia that typically starts around 12–16 weeks of age [26, 29, 30] [31]. We also previously confirmed that none of the NOD mice used in our studies became diabetic up to 13 weeks of age based on the urine glucose level [27]. Hence, the measurements of SS pathologies and assessment of the effects of pDC depletion at 10 weeks of age in this study were done in the absence of clinical diabetes.
Elevation in serum autoantibody concentrations is a characteristic manifestation in autoimmune rheumatic diseases including SS [45]. It has been reported that depletion of pDCs in lupus-prone mice prior to the disease onset reduces serum autoantibody levels [20]. In this study, we did not observe a significant impact of pDC-depletion on serum autoantibodies in NOD mice. The difference could be attributed to the distinct disease contexts and genetic backgrounds of the mouse models used. In addition, it is well documented that anti-TNF-α therapies in the patients with inflammatory diseases such as rheumatic arthritis and inflammatory bowel disease induce a lupus-like condition characterized by elevated serum autoantibody levels [46, 47]. In accordance, our previous study showed that TNF-α blockade in NOD mice prior to SS onset impedes the development of salivary gland inflammation and hypofunction but enhances serum autoantibody levels [29]. In the present study, we observed a decrease in serum autoantibody concentrations by blockade of type I IFN pathways. Whereas pDC-depletion caused a dramatic downregulation of type I IFNs, it did not achieve the similar suppression of autoantibody production. One possible reason is that anti-BST2-treatment suppressed the production of both type I IFNs and TNF-α that have opposing effects on autoantibody production. Nonetheless, we are able to conclude that anti-BST2-mediated pDC-depletion impedes the development of SS without significantly affecting autoantibody production.
Finally, the present study elucidates the critical role of the pDC-type I IFN axis in the early development and pathogenesis of SS. It will be important to elucidate the in vivo role of this axis in the persistence of the established SS in future studies by depleting pDCs or blocking type I IFNs in mouse models with established, ongoing disease, and evaluate the therapeutic effects of these strategies. Indeed, several phase I clinical trials have demonstrated that pDC-depletion by a monoclonal antibody significantly reduce type I IFN levels and ameliorating the severity of cutaneous lupus [48]. Moreover, a humanized monoclonal antibody that inhibit pDC production of type I IFNs show efficacy in reducing skin inflammation and disease activity in cutaneous lupus patients in a randomized, double-blind and placebo-controlled clinical trial [49]. These promising results suggest that ablation of the pDC-type I IFN pathway may have similar effect on SS, which is also an type I IFN-dependent autoimmune rheumatic disease, and this line of studies could greatly facilitate the development of pDC-type I IFN-targeted effective new therapies for SS disease to improve the oral and systemic health of millions of SS patients.
Highlights.
Depletion of plasmacytoid dendritic cells prevents Sjögren’s syndrome in NOD mice
Ablation of plasmacytoid dendritic cell dramatically reduces type I interferon levels
Plasmacytoid dendritic cell-ablation curbs early salivary gland inflammatory events
Depletion of plasmacytoid dendritic cells dampens T helper 1- and B cell responses
Acknowledgement
We thank Dr. BoRa You for her contribution to the preliminary study of this project. We thank the staff at the Forsyth Institute animal facility for their work.
Funding
This study was supported by grants from NIH/NIDCR (R03 DE028033, R01 DE030646) to JZ, and NIH/NIDCR (R01 DE023838, R56 DE023838) and NIH/NIAID (R03 AI142273) to QY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
The authors have no competing financial interests.
Credit Author Statement
Jing Zhou: Conceptualization, Methodology, Investigation, Formal analysis, Writing - Original Draft, Visualization, Project administration, Funding acquisition
Xiaofeng Zhang: Investigation, Formal analysis
Qing Yu: Conceptualization, Methodology, Formal analysis, Writing - Review & Editing, Visualization, Supervision, Funding acquisition
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Fox PC, Autoimmune diseases and Sjogren’s syndrome: an autoimmune exocrinopathy, Ann N Y Acad Sci, 1098 (2007) 15–21. [DOI] [PubMed] [Google Scholar]
- [2].Jin JO, Yu Q, T Cell-Associated Cytokines in the Pathogenesis of Sjogren’s Syndrome, Journal of clinical & cellular immunology, S! (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lee BH, Tudares MA, Nguyen CQ, Sjogren’s syndrome: an old tale with a new twist, Arch Immunol Ther Exp (Warsz), 57 (2009) 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Voulgarelis M, Tzioufas AG, Pathogenetic mechanisms in the initiation and perpetuation of Sjogren’s syndrome, Nat Rev Rheumatol, 6 (2010) 529–537. [DOI] [PubMed] [Google Scholar]
- [5].Di Domizio J, Cao W, Fueling autoimmunity: type I interferon in autoimmune diseases, Expert review of clinical immunology, 9 (2013) 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yao Y, Liu Z, Jallal B, Shen N, Ronnblom L, Type I interferons in Sjogren’s syndrome, Autoimmun Rev, 12 (2013) 558–566. [DOI] [PubMed] [Google Scholar]
- [7].Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, Alm GV, Ronnblom L, Activation of the type I interferon system in primary Sjogren’s syndrome: a possible etiopathogenic mechanism, Arthritis and rheumatism, 52 (2005) 1185–1195. [DOI] [PubMed] [Google Scholar]
- [8].Szczerba BM, Rybakowska PD, Dey P, Payerhin KM, Peck AB, Bagavant H, Deshmukh US, Type I interferon receptor deficiency prevents murine Sjogren’s syndrome, Journal of dental research, 92 (2013) 444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cao W, Pivotal Functions of Plasmacytoid Dendritic Cells in Systemic Autoimmune Pathogenesis, Journal of clinical & cellular immunology, 5 (2014) 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, Jacques S, Ba N, Ittah M, Lepajolec C, Labetoulle M, Ardizzone M, Sibilia J, Fournier C, Chiocchia G, Mariette X, Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome, Proc Natl Acad Sci U S A, 103 (2006) 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wildenberg ME, van Helden-Meeuwsen CG, van de Merwe JP, Drexhage HA, Versnel MA, Systemic increase in type I interferon activity in Sjogren’s syndrome: a putative role for plasmacytoid dendritic cells, European journal of immunology, 38 (2008) 2024–2033. [DOI] [PubMed] [Google Scholar]
- [12].Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ, The nature of the principal type 1 interferon-producing cells in human blood, Science, 284 (1999) 1835–1837. [DOI] [PubMed] [Google Scholar]
- [13].Guery L, Hugues S, Tolerogenic and activatory plasmacytoid dendritic cells in autoimmunity, Frontiers in immunology, 4 (2013) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fitzgerald-Bocarsly P, Dai J, Singh S, Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history, Cytokine Growth Factor Rev, 19 (2008) 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Panda SK, Kolbeck R, Sanjuan MA, Plasmacytoid dendritic cells in autoimmunity, Current opinion in immunology, 44 (2017) 20–25. [DOI] [PubMed] [Google Scholar]
- [16].Takagi H, Arimura K, Uto T, Fukaya T, Nakamura T, Choijookhuu N, Hishikawa Y, Sato K, Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation, Scientific reports, 6 (2016) 24477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu YJ, Gilliet M, Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production, The Journal of experimental medicine, 202 (2005) 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ioannou M, Alissafi T, Boon L, Boumpas D, Verginis P, In vivo ablation of plasmacytoid dendritic cells inhibits autoimmunity through expansion of myeloid-derived suppressor cells, J Immunol, 190 (2013) 2631–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Swiecki M, Colonna M, The multifaceted biology of plasmacytoid dendritic cells, Nature reviews. Immunology, 15 (2015) 471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, Colonna M, Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model, The Journal of experimental medicine, 211 (2014) 1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li Q, McDevitt HO, The role of interferon alpha in initiation of type I diabetes in the NOD mouse, Clinical immunology, 140 (2011) 3–7. [DOI] [PubMed] [Google Scholar]
- [22].Swiecki M, Colonna M, Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance, Immunological reviews, 234 (2010) 142–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hillen MR, Pandit A, Blokland SLM, Hartgring SAY, Bekker CPJ, van der Heijden EHM, Servaas NH, Rossato M, Kruize AA, van Roon JAG, Radstake T, Plasmacytoid DCs From Patients With Sjogren’s Syndrome Are Transcriptionally Primed for Enhanced Pro-inflammatory Cytokine Production, Frontiers in immunology, 10 (2019) 2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jin JO, Kawai T, Cha S, Yu Q, Interleukin-7 enhances the Th1 response to promote the development of Sjogren’s syndrome-like autoimmune exocrinopathy in mice, Arthritis Rheum, 65 (2013) 2132–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jin JO, Shinohara Y, Yu Q, Innate immune signaling induces interleukin-7 production from salivary gland cells and accelerates the development of primary Sjogren’s syndrome in a mouse model, PloS one, 8 (2013) e77605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou J, Jin JO, Kawai T, Yu Q, Endogenous programmed death ligand-1 restrains the development and onset of Sjgren’s syndrome in non-obese diabetic mice, Scientific reports, 6 (2016) 39105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhou J, Yu Q, Anti-IL-7 receptor-alpha treatment ameliorates newly established Sjogren’s-like exocrinopathy in non-obese diabetic mice, Biochimica et biophysica acta, 1864 (2018) 2438–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou J, Yu Q, Disruption of CXCR3 function impedes the development of Sjogren’s syndrome-like xerostomia in non-obese diabetic mice, Lab Invest, 98 (2018) 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou J, Kawai T, Yu Q, Pathogenic role of endogenous TNF-alpha in the development of Sjogren’s-like sialadenitis and secretory dysfunction in non-obese diabetic mice, Lab Invest, 97 (2017) 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lavoie TN, Lee BH, Nguyen CQ, Current concepts: mouse models of Sjogren’s syndrome, J Biomed Biotechnol, 2011 (2011) 549107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Giarratana N, Penna G, Adorini L, Animal models of spontaneous autoimmune disease: type 1 diabetes in the nonobese diabetic mouse, Methods in molecular biology, 380 (2007) 285–311. [DOI] [PubMed] [Google Scholar]
- [32].Zhou J, Yu Q, Anti-IL-7 receptor-alpha treatment ameliorates newly established Sjogren’s-like exocrinopathy in non-obese diabetic mice, Biochimica et biophysica acta. Molecular basis of disease, 1864 (2018) 2438–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou J, Jin JO, Du J, Yu Q, Innate Immune Signaling Induces IL-7 Production, Early Inflammatory Responses, and Sjogren’s-Like Dacryoadenitis in C57BL/6 Mice, Investigative ophthalmology & visual science, 56 (2015) 7831–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, Tschopp J, Cachero TG, Batten M, Wheway J, Mauri D, Cavill D, Gordon TP, Mackay CR, Mackay F, Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren’s syndrome, The Journal of clinical investigation, 109 (2002) 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thompson N, Isenberg DA, Jury EC, Ciurtin C, Exploring BAFF: its expression, receptors and contribution to the immunopathogenesis of Sjogren’s syndrome, Rheumatology, 55 (2016) 1548–1555. [DOI] [PubMed] [Google Scholar]
- [36].Ittah M, Miceli-Richard C, Gottenberg JE, Sellam J, Eid P, Lebon P, Pallier C, Lepajolec C, Mariette X, Viruses induce high expression of BAFF by salivary gland epithelial cells through TLR- and type-I IFN-dependent and -independent pathways, European journal of immunology, 38 (2008) 1058–1064. [DOI] [PubMed] [Google Scholar]
- [37].Wieczorek G, Bigaud M, Pfister S, Ceci M, McMichael K, Afatsawo C, Hamburger M, Texier C, Henry M, Cojean C, Erard M, Mamber N, Rush JS, Blockade of CD40-CD154 pathway interactions suppresses ectopic lymphoid structures and inhibits pathology in the NOD/ShiLtJ mouse model of Sjogren’s syndrome, Annals of the rheumatic diseases, 78 (2019) 974–978. [DOI] [PubMed] [Google Scholar]
- [38].Shen L, Suresh L, Wu J, Xuan J, Li H, Zhang C, Pankewycz O, Ambrus JL Jr., A role for lymphotoxin in primary Sjogren’s disease, Journal of immunology, 185 (2010) 6355–6363. [DOI] [PubMed] [Google Scholar]
- [39].Cha S, Brayer J, Gao J, Brown V, Killedar S, Yasunari U, Peck AB, A dual role for interferon-gamma in the pathogenesis of Sjogren’s syndrome-like autoimmune exocrinopathy in the nonobese diabetic mouse, Scand J Immunol, 60 (2004) 552–565. [DOI] [PubMed] [Google Scholar]
- [40].Benchabane S, Boudjelida A, Toumi R, Belguendouz H, Youinou P, Touil-Boukoffa C, A case for IL-6, IL-17A, and nitric oxide in the pathophysiology of Sjogren’s syndrome, International journal of immunopathology and pharmacology, 29 (2016) 386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Blokland SLM, Flessa CM, van Roon JAG, Mavragani CP, Emerging roles for chemokines and cytokines as orchestrators of immunopathology in Sjogren’s syndrome, Rheumatology, (2019). [DOI] [PubMed] [Google Scholar]
- [42].Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD, The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse, Journal of immunology, 179 (2007) 5041–5053. [DOI] [PubMed] [Google Scholar]
- [43].Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A, Ponnazhagan S, Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells, Journal of immunology, 189 (2012) 4258–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M, Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation, Journal of immunology, 177 (2006) 3260–3265. [DOI] [PubMed] [Google Scholar]
- [45].Fayyaz A, Kurien BT, Scofield RH, Autoantibodies in Sjogren’s Syndrome, Rheumatic diseases clinics of North America, 42 (2016) 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhu LJ, Yang X, Yu XQ, Anti-TNF-alpha therapies in systemic lupus erythematosus, Journal of biomedicine & biotechnology, 2010 (2010) 465898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Almoallim H, Al-Ghamdi Y, Almaghrabi H, Alyasi O, Anti-Tumor Necrosis Factor-alpha Induced Systemic Lupus Erythematosus(), The open rheumatology journal, 6 (2012) 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, Casey KA, Henault J, Riggs JM, Nicholson SM, Sanjuan MA, Vousden KA, Werth VP, Drappa J, Illei GG, Rees WA, Ratchford JN, Investigators VIBT, Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus, Science translational medicine, 13 (2021). [DOI] [PubMed] [Google Scholar]
- [49].Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, Wang W, Christmann R, Gardet A, Pellerin A, Hamann S, Auluck P, Barbey C, Gulati P, Rabah D, Franchimont N, Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus, The Journal of clinical investigation, 129 (2019) 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]