

Abstract
One of the hallmarks of cancer cells is their exceptional ability to migrate within the extracellular matrix (ECM) for gaining access to the circulatory system, a critical step of cancer metastasis. RhoA, a small GTPase, is known to be a key molecular switch that toggles between actomyosin contractility and lamellipodial protrusion during cell migration. Current understanding of RhoA activity in cell migration has been largely derived from studies of cells plated on a two-dimensional (2D) substrate using a FRET biosensor. There has been increasing evidence that cells behave differently in a more physiologically relevant three-dimensional (3D) environment. However, studies of RhoA activities in 3D have been hindered by low signal-to-noise ratio in fluorescence imaging. In this paper, we present a FRET technique in conjunction with a machine learning-assisted cell segmentation method to follow the spatiotemporal dynamics of RhoA activities of single breast tumor cells (MDA-MB-231) migrating in a 3D as well as a 2D environment using a RhoA biosensor. We found that RhoA activity is more polarized along the long axis of the cell for single cells migrating on 2D fibronectin-coated glass versus those embedded in 3D collagen matrices. In particular, RhoA activities of cells in 2D exhibit a distinct front-to-back and back-to-front movement during migration in contrast to those in 3D. Finally, regardless of dimensionality, RhoA polarization is found to be moderately correlated with cell shape.
Keywords: RhoA, Polarization, Cell migration, FRET, Motility, ECM
INTRODUCTION
Cell migration within a three-dimensional (3D) extracellular matrix plays a critical role in many physiological processes (e.g. immune response and wound healing) as well as pathological processes (e.g. cancer metastasis and fibrosis)[1–2]. To metastasize, cancer cells need to move away from the primary site, into interstitial space and gain access to the circulatory system[3–4]. Traditionally, cell migration studies have been performed when cells are plated on a 2D substrate[5]. With the advancement of biomaterials and microfabrication technology, we now know that what we have learnt of cells in 2D does not always apply to those in 3D[6]. In 2D, cells are adhered to a flat substrate and mechanically supported on one side. In 3D, cells are surrounded by extracellular matrices and are supported architecturally on all sides[7–8]. 2D cell migration has been studied extensively in the existing literature, and is mostly governed by actin protrusion in the front and actomyosin contraction in the back and it is critically regulated by integrin-based adhesion[5]. 3D cell motility, however, is more diverse due to its interactions with ECM. Broadly speaking, mammalian cell migration within a 3D ECM can be grouped into three categories: (i) an integrin-independent amoeboid motility, where cells migrate in a path-seeking manner with diffuse, short lived actin protrusions, and squeezing through the ECM pores[9]; (ii) an integrin-dependent and matrix metalloproteinases (MMPs)-independent mesenchymal motility where cells use integrins to adhere to and pull on collagen fibers, and contract to move through the matrix when the pore size is large enough for passage of the nucleus[9–10]; (iii) an integrin- and MMPs-dependent path creating migration where cells use MMPs to digest the matrix in the front and migrate through the self-dug micro-channels using adhesion and contraction[6,11–15]. Central to all types of cell motility is cell contractility.
For cell contractility, mammalian cells often use RhoA GTPase, one of the canonical Rho-family p21 small GTPases, which acts as a crucial molecular switch that controls downstream cytoskeletal effectors by cycling between its GTP-bound (active) state and GDP-bound (inactive) state under the regulation of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs)[15]. While its counterparts Rac1 and Cdc42 control protusion formation, RhoA triggers actomyosin rearrangement, morphological changes, and cell contractility through activation of mammalian Diaphanous (mDia)-related formins and Rho-associated protein kinases (ROCKs), and interactions with focal adhesion kinase (FAK)[16–19]. In lamellipodium-driven migration, while Rac1 predominantly acts to form protrusions, its counterpart RhoA is activated also at the leading edge and the lateral edges of lamellipodia which may be important for protrusion shaping[20–21]. On the opposite end of the cell, RhoA can be activated by RhoGEFs leading to rear retraction[22]. RhoGTPases shuttle between the plasma membrane when activated and the cytosol in a complex with the guanine nucleotide dissociation inhibitor (GDI) when inactivated[23]. Moreover, the localization and spatial distribution of RhoGTPases plays a key role during directed cell migration[24–29]. On one hand, local activation of RhoGTPases can drive directional cell migration, but, on the other hand, disruption of RhoGTPase localization can lead to general defects in motility[21,29–31].
To study the dynamics of RhoA and other RhoGTPases, the Förster Resonance Energy Transfer (FRET) biosensors have played important roles in our ability to visualize the localization of their active and inactive forms within living cells[21,25,32–36]. While traditional bioassays such as western blot and ELISA have been successful in providing many biological insights on cellular responses, these analyses are limited to studies at a populational level and not suitable for revealing spatiotemporal dynamics of RhoA GTPases important for single cell migration. A critical step of the ratiometric FRET calculation is that the FRET/CFP image pairs need to be accurately aligned and segmented. Previously, these image pairs had been segmented manually, which is time consuming and limits the robustness of tracking fine cellular features like protrusions. Although automated approaches including Otsu’s thresholding and k-means clustering have gained popularity in rapid image segmentation, they are often challenged by a continuous gradient of fluorescence intensities within a cell and near cell edges close to background. To overcome these difficulties, we used a neural network assisted algorithm (U-Net, University of Freiberg)[37] to trace the cell outlines of FRET/CFP image pairs. This technique enabled a robust way to obtain cell outlines precisely, and allowed us to visualize spatial and temporal dynamics of RhoA activities of cells in 3D.
MATERIAL AND METHODS
Cell Line and RhoA Biosensor
Cell line
Triple-negative breast tumor cells (MDA-MB-231) with the RhoA FRET biosensor was produced following previously established protocols[38]. Briefly, cells were transduced with retrovirus harboring the tetOFF tTA (pQCXIN-tetOFF tetracycline-TransActivator, second generation; Clontech) and selected for stable genomic integration using G418 at 1mg/mL in the culture media, as previously described[38–39]. This biosensor is based on RhoA FLARE.sc[25,40], in the pRetro-X-Puro backbone (Clontech). This biosensor design has been described previously by Pertz et al, and consists of a Rho binding domain from Rhotekin (RBD), enhanced cyan fluorescent protein (ECFP), an unstructured linker, Citrine yellow fluorescent protein (YFP), and the full length RhoA GTPase, in one single chain[25]. The cells were selected for stable genomic integration using 10μg/mL puromycin dihydrochloride (A1113802, Gibco) in culture media, together with 2ug/mL doxycycline hyclate (D9891, Sigma) to repress the biosensor expression under normal culture[34]. Cells were maintained at 37°C and 100% humidity in a 5% CO2 incubator and were cultured in T75 flasks (10062–860, Corning) using Dulbecco’s Modified Eagle’s Medium (DMEM) (15–013-CV, Corning) with 4.5 g/L glucose, sodium pyruvate supplemented with 1:100 GlutaMAX (35050061, Gibco), 10% fetal bovine serum (FBS) (S11150, Atlanta Biologicals), 1% penicillin/streptomycin (15140122, Gibco), and 1 mg/mL G418 (10131027, ThermoFisher). Culture medium was changed every 2 days to maintain a reasonable level of doxycycline[34,41]. Cells were cultured in phenol red-free complete DMEM (17–205-CVR, Corning) 1 day prior to the experiment to reduce the background autofluorescence from the media during imaging. Cells were passaged when they reached 70–90% confluency. Cells with 19–20 passages were used for the experiments.
Biosensor expression
For induction of biosensor expression, 48hrs prior to experiments, cells were trypsinized and doxycycline was removed to allow expression of the biosensor by replacing medium with doxycycline-free complete medium. Cells were then plated at a density of 100,000 cells/T25 flask and were incubated for 48h. The cells were checked for YFP signal before experiments to ensure the expression of biosensor.
We note that a possible concern in any FRET-based detection approach is the molecular crowding effect, which may produce inter-molecular FRET in the case of these single-chain biosensors when they are overexpressed. However, previous expression analysis of this class of single-chain FRET biosensors and expression control revealed that the fundamental protrusive leading edge dynamics and the associated kinetics and kinematics of the FRET biosensor readouts did not change within a 3–7.5 : 1 dynamic range of expression levels[42–43].
Cell Preparation
In both 2D and 3D experiments, cells were serum starved overnight (~16h) and epidermal growth factor (EGF) was added for EGF-dependent studies. While serum starvation can decrease the basal activity of many cellular pathways before any drug treatment, the removal of serum can therefore eliminate serum-dependent confounding factors[44–45]. Serum-starved cells were resuspended in serum-free medium, and plated at a density of ~4280 cells/cm2. The plated cells were incubated at 37°C and 5% CO2 for 1hr for attachment. Medium was then replaced with fresh medium with 20 ng/mL EGF (AF-100–15, PeproTech) and cells were settled for 2hrs prior to imaging.
For 3D experiments, type I collagen extracted from rat tail tendon (354249, Corning) in 0.1% acetic acid (9.41 mg/mL) was used. On ice, 31.9 μL collagen stock (9.41 mg/mL), 20ug fibronectin (F0556, Sigma-Aldrich), and 0.2 μL 1N NaOH were mixed with the cell suspension in complete medium to reach a final volume of 200 μL. The final collagen concentration was 1.0 mg/mL and cell density was 150 cells/μL. 5 μL of the cell embedded collagen mixture was placed into the each of the three PDMS microwells (2mm diameter and 300 μm in depth) for polymerization in a 37°C and 5% CO2 incubator for collagen polymerization. To prevent cells from sinking to the bottom of the glass bottom dish during polymerization, the device was first placed upside-down for 5 mins. Subsequently, the dish was flipped twice at time points 15 min and 30 min. Note that we lifted the glass above any incubator surfaces to adopt a slow warming procedure for polymerization to obtain long and thick collagen fibers in contrast to the fast warming process described in our previous publication that yields thin and dense collagen fibers[46]. Throughout the process, the glass bottom dish was contained within a second moist petri dish to prevent collagen dryout. After polymerization, cells were serum-starved overnight, and 1.5mL complete medium with 20 ng/mL EGF (AF-100–15, PeproTech) was added to each glass bottom dish after starvation. Cells were settled for 2hrs prior to imaging.
Device Preparation
For both 2D and 3D experiments, 50mm uncoated glass bottom dishes with No. 0 glass (D50-14-0-U, Matsunami) were used. For 2D experiments, cover glasses were coated with 1μg/cm2 fibronectin[7,47–49] (F0556, Sigma-Aldrich) by incubating for 1hr at room temperature according to manufacturer’s protocol. Note that this coating density is beyond the theoretical saturation density which ensures a complete coverage of fibronectin on the substrate. Although a fibronectin study conducted by DiMilla et al. recommended an optimal coating density of 0.073μg/cm2 for migration of human smooth muscle cells (HSMCs)[49], we found that, under 1μg/cm2 fibronectin, the velocity of MDA-MB-231 in the presence of 20 ng/mL EGF was comparable to the peak velocity (~60μm hr−1) demonstrated by DiMilla et al[49]. Following incubation, the fibronectin solution was aspirated and rinsed with media. The coated dish was used the same day after the coating process. For 3D experiments, polydimethylsiloxane (PDMS) microwells were prepared by mixing 10g silicone elastomer with 1g curing agent thoroughly. The mixture was degassed in vacuum for 20mins to remove air bubbles, followed by incubation at 60°C overnight for polymerization. Three through holes with a diameter of 2 mm were created using a biopsy punch on the PDMS sheet with a thickness of 300 μm. PDMS sheet and cover glass were bonded following plasma treatment with an oxygen plasma oven (PDC-001, Harrick Plasma), followed by autoclave sterilization. To enhance attachment of collagen matrices to the glass/PDMS surfaces, cover glasses were treated with 5 μL of 1% polyethyleneimine (PEI) for 10 mins and then with 5 μL of 0.1% glutaraldehyde for 30mins. The dishes were left in biohood overnight in sterile distilled water and were washed with phosphate buffered saline (PBS) and aspirated before experiments.
FRET Technique - Imaging
Image acquisition
A dual channel image acquisition system is used to acquire the FRET and CFP image pair as shown in Fig. S2. This setup minimizes motion artifacts confounded in sequential image acquisitions[49]. Excitation is via CFP, and images from the YFP channel are denoted as “FRET” images, and images from the CFP emission filter as “CFP” images, with the final ratiometric FRET images denoted as “rFRET”. All CFP-FRET image pairs were taken with a 60x magnification water immersion objective lens (NA = 1.2; UPLSAPO60XW, Olympus America) installed on an epifluorescence microscope (IX81, Olympus America) and a 16-bit sCMOS camera (ORCA Flash 4.0 V3, Hamamatsu Photonics). Immersol W 2020 (η = 1.3339; 444969, Carl Zeiss), a non-evaporative immersion medium, was used to match optical indices between the sample and objective lens. The light source for fluorescence imaging was a xenon arc lamp (Lambda LB-LS/30, Sutter Instruments). An absorptive neutral density filter with OD = 0.3 (NE03B-A, ThorLabs) was used to attenuate light to minimize phototoxicity. To obtain CFP and FRET signals simultaneously for ratiometric FRET calculation, the W-View Gemini beam splitter was used (A12801–01, Hamamatsu Photonics). The optical filters used were as follows: ET436/20x (CFP excitation), ET480/40m (CFP emission); ET500/20x (YFP excitation), ET535/30m (YFP emission); T455lp (dichroic mirror for reflecting excitation light and relaying emission from cells), T505lpxr (dichroic mirror for splitting CFP and FRET signals), T515lp (longpass filter for checking sensor expression by YFP signals). All filters and dichroic mirrors were purchased from Chroma Technology. Prior to each experiment, the x-y positions of CFP and FRET channels were coarsely adjusted using the W-VIEW Adjustment software (Hamamatsu Photonics) to achieve a reasonable alignment for easier image registration and processing. The epifluorescence microscope was surrounded by an incubator with a temperature of 37°C, 5% CO2, and humidity of ~70%.
For each experimental run, images were taken using an automatic x-y microscope stage (MS-2000, Applied Scientific Instrumentation). At each position, images were taken every 7 min for 133 mins using Metamorph (version 7.7.7.0) (Molecular Devices) with an exposure time of 800ms. We constrained both the exposure time and experiment time to limit phototoxic effects on cells. To effectively maintain a consistent focal plane at this high magnification (60x) setting, between each image acquisition, an image-based automatic step focusing procedure in MetaMorph (Search type: Step; Algorithm: Standard; Range +/−: 1 μm; Max. step size: 0.2μm) was carried out with a minimal exposure time of 50ms to avoid photobleaching. In both 2D and 3D setting, cells were tracked in the focal plane and the measured speed is the actual speed of cells in the focal plane.
Field alignment
To align the CFP/FRET image channels from misalignments caused by the difference in optical paths (Fig. S3A,B), we conducted the following calibration procedures. First, a target pattern provided by Olympus America was imaged under brightfield as a guideline for rough cropping (Fig. S2B). The center of the target served as a reference for aligning the two channels. To further align and map the channels, a glass slide coated with 4-μm fluorescence marker beads of 4 different colors, blue, green, orange, and dark red (T14792, Invitrogen) was imaged in fluorescence mode to produce reference images from the CFP and FRET channels. Then, the bead image was cropped and split into two images according to the reference point calculated earlier (Fig. S2B). The split bead images were then used to generate registration parameters using the affine transformation algorithm from MatLab to achieve subpixel accuracy in field alignment. This feature-based algorithm corrects for x-y-translation, rotation, scaling, and shear. We were able to reduce misalignment down to less than 1 pixel (Fig. S3C).
Flatfield correction and background subtraction
Flatfield correction and dark current (DC) subtraction described by Spiering et al[34] were performed after field alignment. For dark current subtraction, 10 DC images were taken from the field of view with no illumination and a closed shutter. To compensate for spatial unevenness in illumination of the field of view, 10 shade images of a dish of medium with the same optical setup as the experiments were taken. Both DC images and shade images were averaged respectively for calibration of the two channels. The following formula was used to calculate the corrected image[34]:
Since we were taking a ratiometric calculation, the choice of scaling factor did not affect our final calculation as long as it was consistent for both channels. We used a scaling factor of 3000 to bring images to visualization under a 16-bit domain.
After flatfield correction, we selected a blank area away from the cell and any cell debris to measure its average intensity value. This was done for each individual frame as the background intensity can change in a time series movie. The average intensity of the blank area was then subtracted from the flatfield corrected image.
FRET Technique - Image analysis
Cell segmentation and tracking
A convolutional neural network package, U-Net, developed in the University of Freiburg[37] was adopted to segment the epi-fluorescence images of the cells. Details of this segmentation method are illustrated in Results and Discussion section. To track cell movements, the centroids of the binary masks of the segmented cells were defined as the center of cells.
Ratiometric FRET calculation and RhoA biosensor validation
The FRET channel was divided by the CFP channel, then multiplied by a scaling factor to bring the image back to the 16-bit dynamic range for visualization purposes. The ratiometric FRET image, rFRET, was calculated using the following equation:
where [FRET]corr and [CFP]corr are the corrected images of FRET and CFP channels respectively.
The final images were then corrected for photobleaching using the biexponential intensity decay model described in Spiering et al[34,36].
To validate the cell line with the FRET biosensor, we modulated RhoA activity by treating cells with CN03, a constitutive RhoA activator (CN03, Cytoskeleton, Inc.)[51], and Rhosin (Fig. S4), a RhoA inhibitor[52]. CN03 activates RhoA GTPase by deamidating glutamine-63, which is located at the Switch II region[52]. This allows constitutive activation of RhoA without altering the availability of Switch I region for binding with Rho-Binding Domain (RBD) within the biosensor.
On the other hand, Rhosin (555460-M, Millipore) is an RhoA inhibitor which targets the RhoGEF binding domain of RhoA[52]. We found that Rhosin treatment reduced the average FRET ratio in MDA-MB-231, while cells treated with CN03 had a higher average FRET ratio compared to those treated with Rhosin (Fig. S4).
Data Analysis
RhoA polarization, cell shape, and motility characterization
We used the first spatial moment of the rFRET signal, , to represent the polarization of RhoA activities, analogous to the definition of electric dipole moment in physics[53], which is commonly used to quantify the polarization of an electric charge distribution. As shown below, RhoA polarization , is the sum of the product of rFRET intensity at a given point and the displacement with respect to the centroid of the cell (Fig. S5A):
where Ii is the rFRET intensity at position , is position vector of the centroid of the cell extrapolated from elliptic fit using ImageJ. n is the total number of pixels within the cell. Polarization strength is represented by .
After an elliptic fit was performed, component vectors of velocity and polarization were calculated with reference to the cell’s long (L)-axis and short (S)-axis (Fig. S5B). Here, the angle between and is defined as the circular distance between two vectors in counterclockwise direction. Finally, to distinguish between amoeboid and mesenchymal motility, a cell is considered mesenchymal when its aspect ratio (i.e. ) is greater than or equal to two, to be consistent with previous work[54–55]. Aspect ratios of cells were generated from the elliptical fit using ImageJ (NIH). Cell speed is calculated by the distance travelled by a cell (i.e. centroid shift) divided by time. Cell velocity is calculated by the displacement of a cell divided by time. Polarization of RhoA activities, cell speed and velocity, and cell trajectories were computed using in-house MatLab programs. Codes have been made available on Github: https://github.com/chunbrian16/FRET-image-analysis.
Statistical analysis
All statistical analyses were performed using Prism 8 (GraphPad) and in-house Matlab codes. Student’s t tests were performed on data sets which had similar variances, and Welch’s t tests were performed on data sets with unequal variances. Spearman’s ranking correlation analyses were performed in all correlation studies. To test for significance between correlation studies, we compared the coefficient ρ with null hypotheses that ρ0 = 0. For analyses between velocity or speed versus polarization, we used a polarization value one time-point before velocity or speed, i.e. P at t = 0min was paired with V at t = 7min.
For angular data, the Rao’s spacing test and resultant vector length were used to study the uniformity of vector orientations[56]. To define the resultant vector length in directional data analysis, we assign unit vectors to corresponding angles θi, i = 1, ⋯, n. The mean direction of θ1, ⋯, θn is the direction of the resultant vector from , which is also the direction of the center of mass of . The Cartesian coordinates of the center of mass are therefore . The resultant vector length is given by .
RESULTS AND DISCUSSION
FRET signal computation using a machine learning-assisted cell segmentation method
Ratiometric computation of the FRET signal involves dividing the fluorescence cell image of the FRET channel by that of the CFP channel (Fig. 1). An important component of this computation is the accurate segmentation of cell images with weak fluorescence signal. We adopted a machine learning cell segmentation method previously developed for brightfield cell images to accurately identify the outlines of the cells in CFP-FRET image pairs[37]. Fig. 1 illustrates the procedures that we used to segment an image pair. (i) 10 images from the FRET channel (after alignment and correction) with manually drawn outlines together with a pre-trained model provided by the U-Net package were used as input to the U-Net finetuning module, and the outcome was an adapted model (Fig. 1A). (ii) An original FRET cell image along with the adapted model was used as the input to the U-Net segmentation model, and the output was the binary mask of the cell (Fig. 1B). (iii) Finally, the ratiometric FRET image was generated by taking a pixel-wise division of the intensity matrices of FRET and CFP images, and multiplied by the binary mask (Fig. 1C). We note that FRET images, in contrast to the CFP images, were used for model training and segmentation because of their higher signal-to-noise ratios. While this process has been used successfully to automatically analyze live cells from time series of images as well as slices of images for 3D reconstruction, this workflow can be potentially be applied on other biological FRET imaging tools that require simultaneous image acquisition of two fluorophores[57–58]. Note that this segmentation method does not work if cells are overlapping.
Figure 1. Illustration of a machine learning-assisted FRET technique.
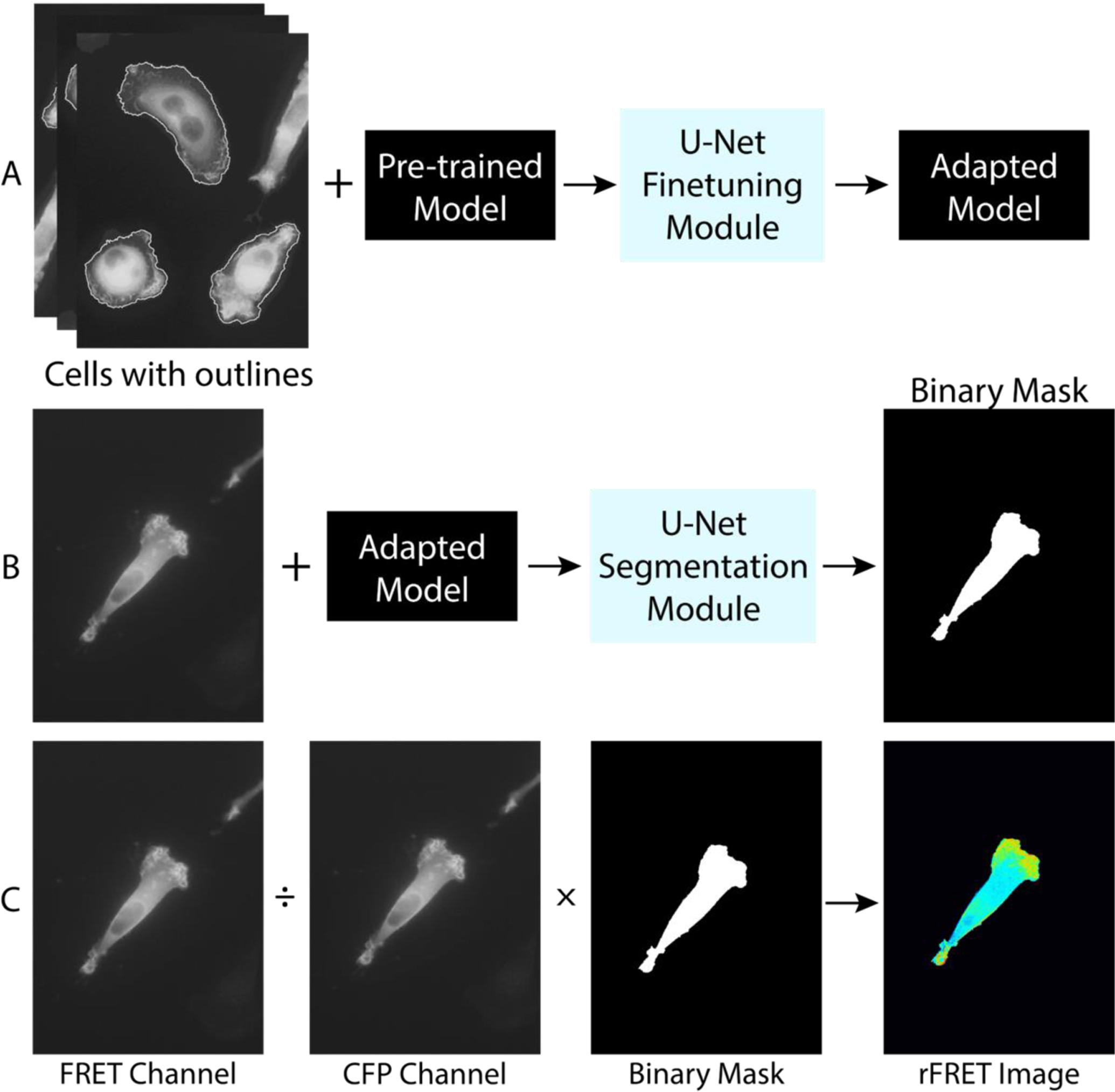
A machine learning approach is adopted in cell segmentation prior to the ratiometric FRET calculation. A. Model training using a convolutional neural network (U-Net) package. Original images of cells with manually drawn outlines together with pre-trained model are input to the U-Net finetuning module. The output is the trained adapted model. B. Cell binary mask generation using the adapted model. The cell image from FRET channel and the adapted model are fed into the U-Net segmentation module. The output is a binary mask of the cell. Here, cell images from the FRET channel are used. C. Ratiometric FRET calculation. The ratiometric FRET image is obtained by dividing the image of the FRET channel from that of the CFP channel, and then segmented by the binary mask. The final image is then corrected for photobleaching.
Traditionally, segmentation of a cell image is done manually, which leads to person-to-person variability and misinterpretation of fine features such as cell protrusions which are critical for cell migration studies. For example, with a generally weak fluorescence signal and a histogram with no apparent bimodality, the Otsu method cannot accurately search for an optimal threshold that minimizes intra-class variance[59]. Despite requiring less computational power, the other commonly used segmentation method, k-means clustering, has also been challenged by low contrast situations where centroids of intensity clusters become proximal to each other[60]. The machine learning-assisted cell segmentation method has enabled us to successfully identify the outlines of cells in an automatic and consistent way. We also note that an additional important feature of our FRET analysis is the alignment of cell images. The two images (CFP and FRET) are not always perfectly superimposable due to physical factors such as optical path differences introduced by the beam splitter or slight mechanical misalignment between filter cubes and curvature differences between dichroic mirrors (Fig. S3A). This misalignment could cause extreme ratios at the cell edge and miscalculated ratiometric FRET signal (Fig. S3B). Therefore, it is important to align images in a reproducible way. Here, we used multi-fluorescent beads to generate reference images in CFP and FRET channels and aligned image fields by affine transformation, which has enabled us to achieve image alignment at a subpixel resolution (Fig. S3C).
RhoA activities are more polarized in cells plated on 2D surfaces than those embedded within a 3D collagen matrix
To follow the spatial and temporal dynamics of RhoA activities of malignant breast tumor cells (MDA-MB-231), we took time series of CFP-FRET image pairs of MDA-MB-231 cells with the RhoA biosensor in both 2D and 3D environments. RhoA activities were more polarized in 2D than those in 3D as seen in the ratiometric FRET images (Fig. 2A, Supplementary Video 1). In addition, RhoA activities of cells had a clear oscillation movement in a front-to-back and back-to-front fashion in 2D in contrast to that of in 3D (Supplementary Video 1–2).
Figure 2. Spatial and temporal dynamics of RhoA activities of MDA-MB-231 Cells in 2D versus 3D.
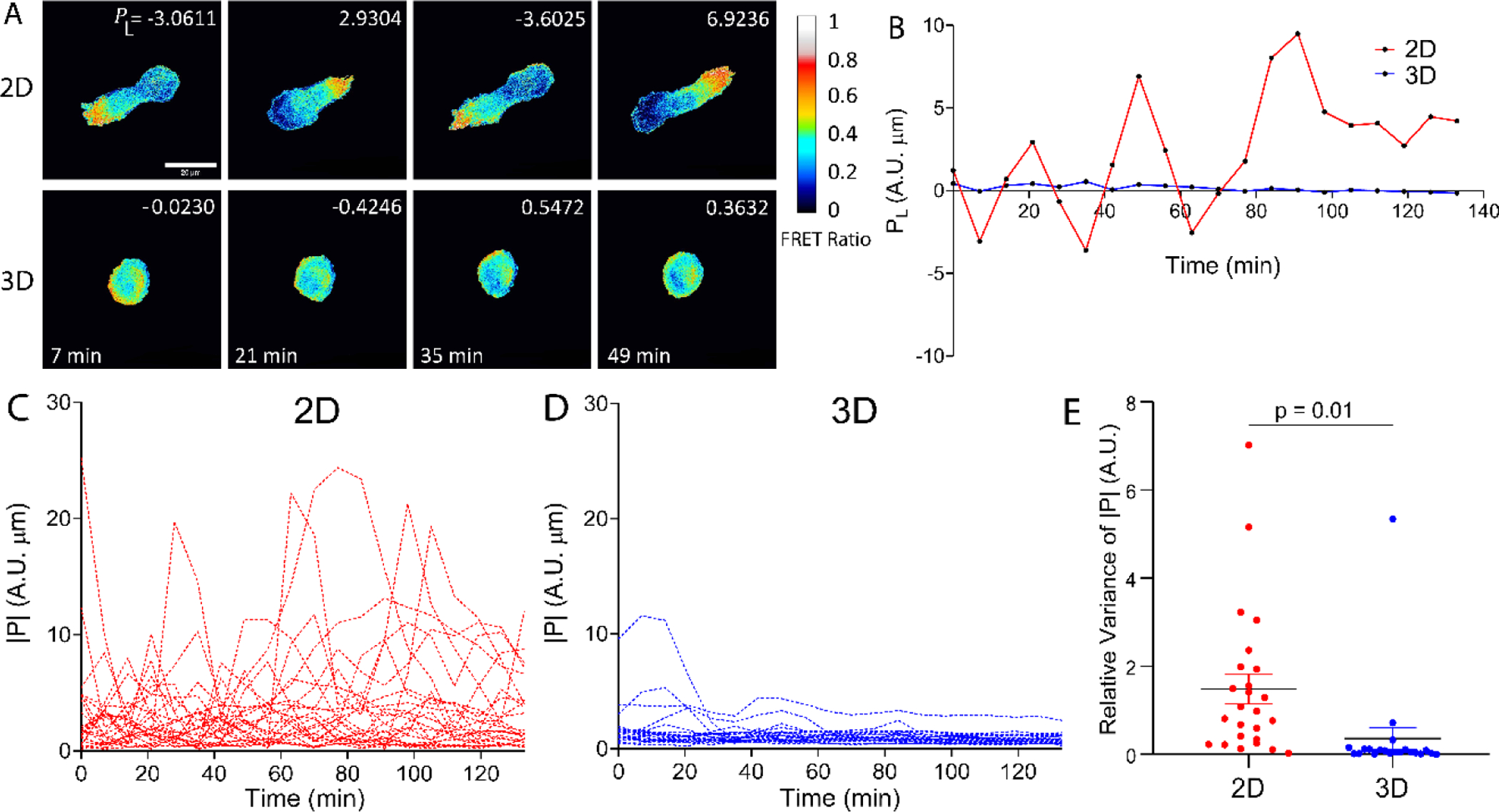
A. RhoA activities represented by the ratiometric FRET signal of the cells seeded in 2D versus in 3D. The color indicates the FRET ratio. Cells in 2D showed strong RhoA polarization activities and elongated morphologies, whereas cells in 3D remained rounded and polarization was not as pronounced. The upper right legend in each image is the polarization magnitude along the long axis of the cell, PL. The scale bar represents 20 μm. Images were taken at every 7 mins, and t=0 is defined as the starting point of imaging, which is about 2h after EGF treatment for motility enhancement. B. Computed polarization along the long axis of the cell, PL, demonstrates a more pronounced oscillating movement in a front-to-back and back-to-front fashion during migration in 2D, in contrast to that of 3D. C-D. Temporal dynamics of RhoA activities of cells in 2D and 3D. Each line is time evolution of the absolute value of the polarization from one cell (n = 25 for 2D and n = 21 for 3D). E. While RhoA polarization oscillated in both 2D and 3D, the relative variance of polarization strength in 2D was significantly higher than that of 3D (Student’s t test, p = 0.01) (n = 25 for 2D and n = 21 for 3D). Relative variance is defined as the variance of polarization strength normalized by the mean polarization strength of individual cells.
To gain a quantitative understanding on how RhoA polarization is correlated with cell migration, we computed the polarization of RhoA activity, , using the rFRET images shown in Fig. 2A. We define RhoA polarization, , to be the first moment of the rFRET signals with respect to the centroid of the cell (Fig. S5). PL and PS are the component of along the long (L)- and short (S)-axis of the cell. Fig. 2B illustrates an example of the time evolution of PL of a cell in 2D versus 3D. It shows that the cell in 2D had a larger PL oscillation magnitude than that in 3D. This observation is consistent with the computation of P for multiple cells in 2D versus 3D (Fig. 2C–D) where the oscillation magnitude of RhoA activity for cells in 2D was significantly higher than that in 3D (Fig. 2C–E). Fourier transforms of |P| in 2D and 3D are consistent with Fig. 2C–D, in that a few cells in 2D showed clear periodic movement with a period of ~30 minutes while no discernible peak was observed from the cells in 3D (Fig. S6A and B).
It is interesting to note that, in 2D, cells were mostly elongated and adopted mesenchymal motility; while in 3D, cells were more rounded and exhibited an amoeboid motility (Fig. 2A). With a mesenchymal phenotype, the RhoA activities of the cell in 2D exhibited an apparent oscillating behavior where cell extension and lamellipodia formation was locally associated with weak RhoA activity, and cell contraction appeared to be accompanied by strong local RhoA activity (Fig. 2A). This behavior is less evident in cells in 3D (Fig. 2A). While Costigliola et al suggested that RhoA regulates calcium-independent periodic contractions of the cell cortex[61], it will be interesting to explore whether this behavior is applicable to all mesenchymal and amoeboid motility in the future.
Shape and RhoA polarization of cells in 2D and 3D
We calculated the cell aspect ratio (long axis/short axis) using cell images from the FRET channel. Fig. 3A and B show that cells in 2D had larger aspect ratio and polarization strength than those in 3D. Also, cells in 3D became more rounded when treated with EGF, but this was not evident in 2D (Fig. S7). To answer the question whether elongated cells are more polarized, we computed the Spearman’s ranking correlation coefficient between |P| and aspect ratio (Fig. 3C,D). Here, ρ = 0 means no association, ρ = +1 indicates a perfect positive monotonic relationship, and ρ = −1 indicates a perfect negative monotonic relationship. The results show that, in both 2D and 3D, cells with high RhoA polarization tend to be more elongated (Fig. 3C and D). This observation is consistent with Zemel et al, in which elongated cells tend to exhibit aligned stress fibers which are largely regulated by RhoA during actin polymerization[62–63]. To answer the question whether RhoA polarization follows the long axis of the cell, we computed the angle, , between polarization vector and the long axis (Fig. S8). A plot of shows that polarization vectors in 2D were significantly more aligned with the long axes, compared to 3D (Fig. S8).
Figure 3. Cell shape and the polarization of RhoA activities of the cell in 2D versus 3D.
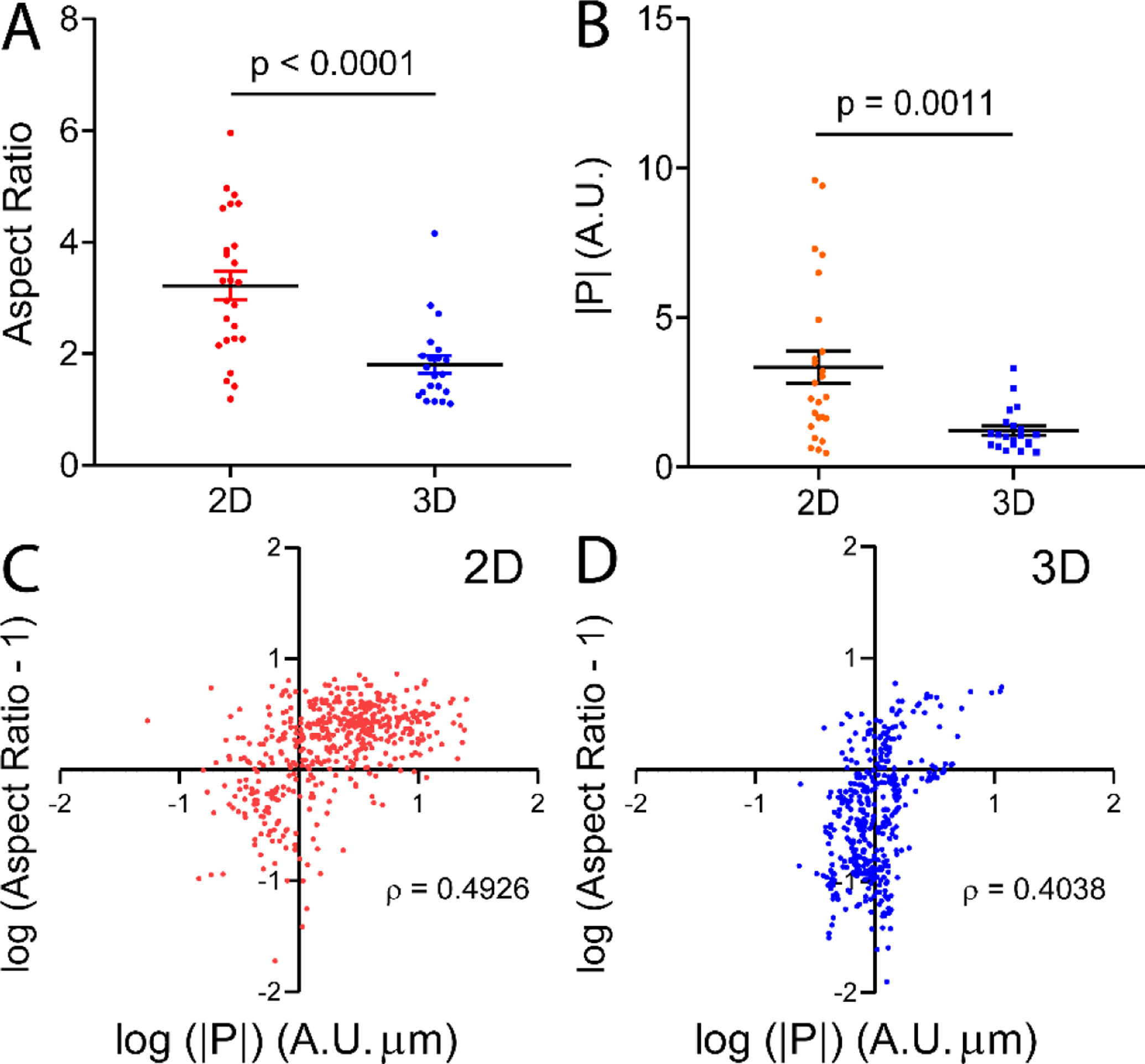
A. Aspect ratio of individual cells in 2D versus 3D. Cells are more elongated in 2D than 3D. B. Magnitude of PL of cells in 2D versus 3D. Cells in 2D are significantly more polarized than those in 3D. Each dot represents the polarization strength |P| of a cell. Error bar = SEM in both A, B. p is the statistical significance of Student’s t test. C, D. Scatter plots of log of (aspect ratio-1) versus log of polarization strength |P| for cells in 2D (C) and 3D (D). Spearman’s ranking correlation coefficient, ρ, between |P| and aspect ratio at all instances during cell migration was computed. Here, r = 0 means no correlation, r = +1 indicates a perfect positive monotonic relationship, and r = −1 indicates a perfect negative monotonic relationship. Each dot represents a time point of a cell, with n = 500 for 2D, and n = 420 for 3D. 25 cells were followed for 2D, and 21 cells were followed for 3D. ρ is the Spearman’s ranking correlation coefficient. Cells in both 2D and 3D exhibited a positive relationship between polarization strength and cell elongation.
In summary, we see that RhoA activities were more polarized and cells were more elongated in 2D than 3D. This differential behavior may be explained by the distinct cell-environment interactions for cells in 2D versus 3D. In 2D, cells are plated on a rigid glass substrate where cells adhere to the fibronectin coated substrate via focal adhesions[64–65]. This allows greater cellular traction force which orients and aligns stress fibers, and subsequently leads to a polarized and an elongated phenotype[65–66]. In 3D, cells experience less adhesion to the matrix which means less traction force and less stress fiber alignment. This leads to a less polarized and more rounded cell morphology. Nonetheless, we note that the nature of the 3D matrix and cell type can impact cell shape and mode of migration[66–67]. For instance, migration among elongated cells can be driven by small lateral blebs called lobopodia which, consistent with other contractility-based motility, are governed by RhoA, ROCK, and myosin II[66,68].
RhoA polarization occurs at the rear-end of the cell during migration in 2D but not evident in 3D
To delineate the relationship between migration direction and RhoA polarization of cells in 2D and 3D, we measured θVP, the angle between velocity vector and polarization vector as defined in Fig. S5A–B. The polar plot for the distribution of θVP peaks at 180° for cells in 2D, which means cells had a tendency to move towards the direction that was opposite to its RhoA polarization. Meanwhile, the distribution of θVP was uniform in all directions for cells in 3D. We also computed the resultant vector length to evaluate whether had a preferred direction relative to . Here, a resultant vector length of 0 would indicate non-directionality, and a length of 1 represents a strong preference of direction. As revealed by the resultant vector length and Rao’s spacing test, polarization vectors are oriented away from the direction of cell migration in 2D, but not in 3D.
These findings suggest that RhoA is mostly activated at the rear-end of the cell in 2D in a directional manner, but not in 3D. As cells are more capable of forming stable adhesions and protrusions with a rigid substrate in 2D, it is conceivable that polarization and actin alignment are more favorable for cells in 2D than in 3D. In breast cancer, short filopodium-like protrusions (FLPs), controlled upstream by RhoA-ROCK signaling, have been shown to be more abundant in the metastasis-competent MDA-MB-231 cell line[69]. While these RhoA-ROCK-driven short FLPs are responsible for initiating migration direction[70], we speculate that, in 3D, as cell adhesions are less stable due to a soft fibrous matrix, the regulation of RhoA activity via adhesion molecules such as integrins becomes less stable, which may result in less actin alignment. We also suspect that the requirement for RhoA polarization in 3D may indeed vary with matrix fiber density, as Lammermann et al reported that myosin II-inhibited dentritic cells can still migrate and drag the rear cell body in low density-collagen gels[11], even when contraction is blocked. It will be interesting to study how tumor cells dynamically orchestrate their RhoA activity at different fiber densities as they often migrate in highly heterogenous metastatic niches.
Cells migrate faster in 2D than 3D, and cell speed and RhoA polarization strength are poorly correlated
We measured the speed of cells in 2D versus 3D, and investigated its correlation with RhoA polarization strengths. Fig. 5A shows the maximum speed of cells in 2D versus 3D. In agreement with our previous work[71], cells in 3D migrated at a significantly lower speed than in 2D (Fig. 5A), and the speed was consistent with previous work using collagen matrices with similar concentrations (1.0 mg/mL)[71]. The difference in speed between 2D and 3D can be explained by that, in a collagen matrix, cells have to overcome steric resistance from fibers and undergo nuclear deformation before they can migrate to a new position, whereas cells can form stable focal adhesions to exert force on the planar substrate and migrate in a 2D environment[72–74].
Figure 5. Cells move faster in 2D than in 3D, cell speed and polarization strength are not or poorly correlated in 2D or 3D.
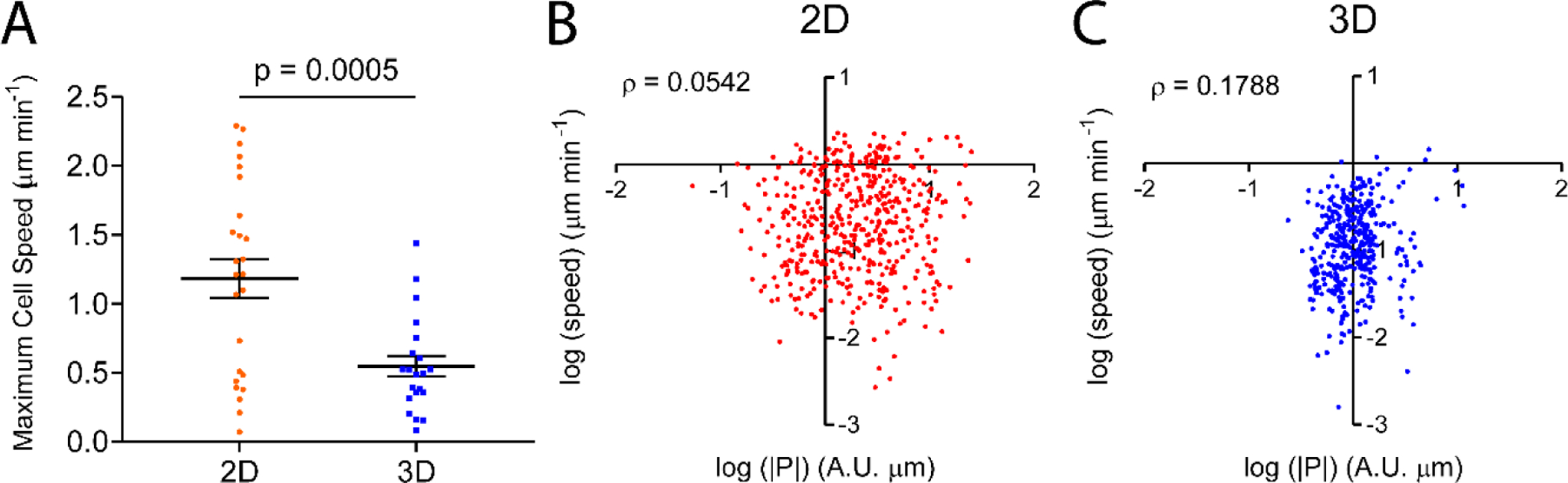
A. Cells in 2D migrate at a significantly higher speed than those in 3D. Error bar: +/−SEM. p is the statistical significance of Student’s t test. B, C. Scatter plots of speed versus polarization magnitude |P| for cells in 2D (C) and 3D (D). Speed and polarization strength are very weakly correlated (ρ=0.1788) in 3D, but not in 2D (ρ=0.0542). ρ is the Spearman’s ranking correlation coefficient. n = 475 for 2D and n = 399 for 3D.
To test whether migration speed is associated with RhoA polarization, we performed Spearman’s ranking coefficient analysis between speed and polarization. Interestingly, weak (3D) or no correlation (2D) were found between these two parameters (Fig. 5B, C). We conjecture that, polarization strength becomes less important for cell migration in 2D as cells use integrin dependent adhesion forces to push forward on a stiff and stable substrate, in contrast to a soft and dynamic 3D fibrous network. Together with low aspect ratios, these findings are consistent with cells in 3D undergoing amoeboid motility[68,75].
CONCLUSIONS
In this study, we utilized a machine learning-assisted FRET analytical technique for studies of RhoA dynamics of EGF stimulated MDA-MB-231 cells in both 2D and 3D platforms. This technique can potentially facilitate consistent and efficient analytical work on a high volume of FRET images, in particular, 3D reconstruction of cells embedded in collagen matrices to gain biophysical insights on how RhoGTPases like RhoA are modulated and generate contractile forces on the matrix during migration. In our experiments, we found that MDA-MB-231 cells were mostly elongated on a 2D glass substrate while cells in a 3D collagen matrix had a rounded morphology. During cell migration, compared to 3D, stronger oscillations of RhoA polarization activities were observed in 2D. We showed that while polarization is associated with cell elongation in both 2D and 3D, RhoA activities tend to align better with the long axis of cells in 2D, compared to 3D. Finally, cell tracking results show that cells moved faster in 2D than in 3D. While RhoA polarization is found to be weakly correlated with cell speed in 3D and not in 2D, further molecular studies are needed to delineate the mechanism by which RhoA polarization differentially affects cell migration speed and direction in 2D and 3D.
Supplementary Material
Figure 4A-B. Directionality of RhoA polarization revealed by the Rao’s spacing test between polarization and velocity.
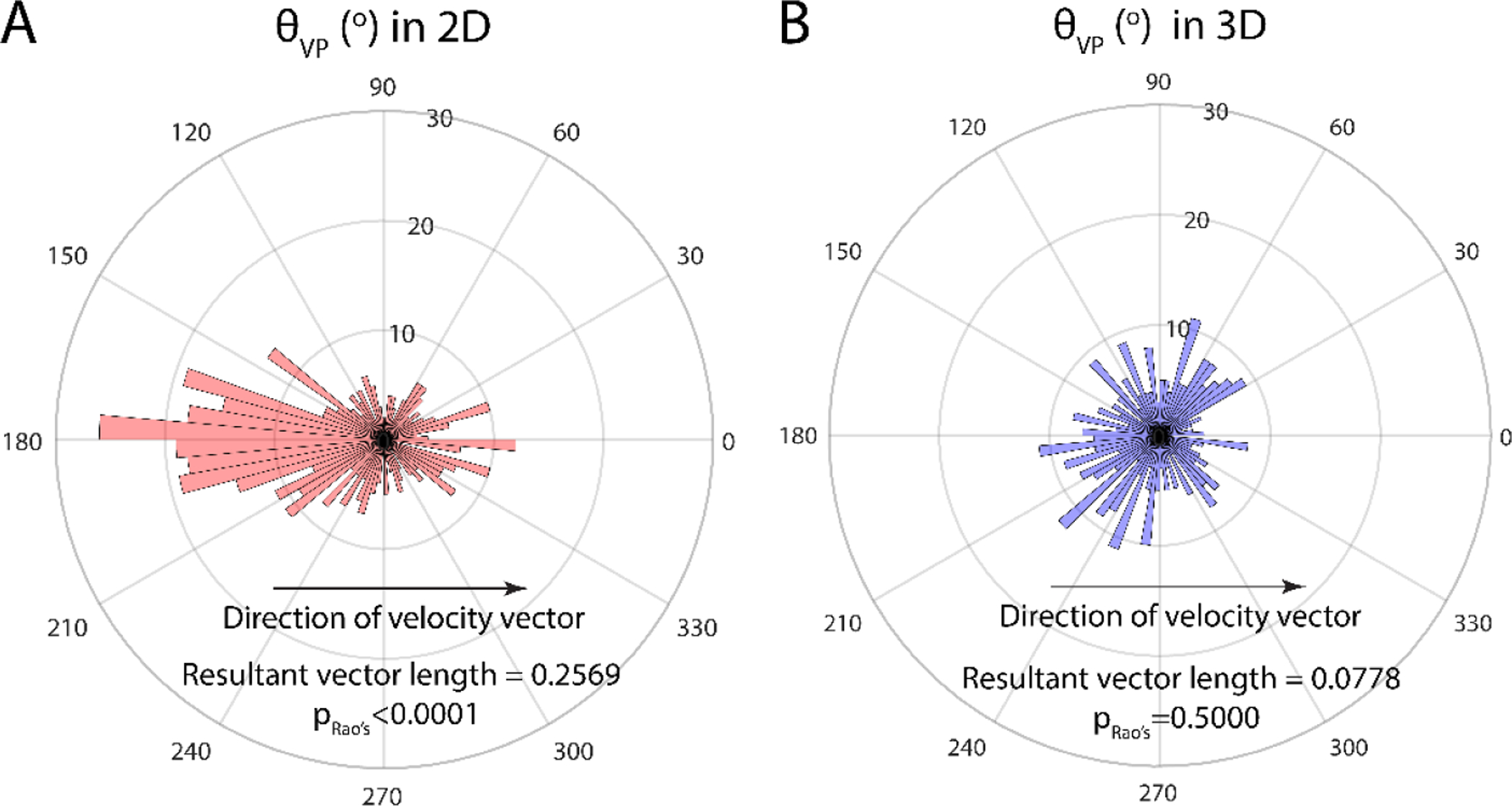
Polar plot of the distribution of , the angle between velocity and polarization vector in 2D (A) and 3D (B). In 2D, RhoA polarization peaks around 180° with respect to the direction of velocity (p < 0.0001, Rao’s spacing test), indicating that cells tend to move against the direction of polarization. RhoA polarization in 3D exhibits no preferred direction (p = 0.5000, Rao’s spacing test). The axis in the radial direction shows the number of data points in a given direction (n = 475 for 2D, and n = 399 for 3D).
HIGHLIGHTS.
Robust machine learning approach for FRET image preparation and analysis
Revealed spatial and temporal dynamics of RhoA activities of single cells in 2D and 3D
Polarization strength and cell shape are correlated for cells in 2D and 3D
ACKNOWLEDGEMENT
This work was supported by grants from the National Cancer Institute [Grant No. R01CA221346 (MW and JES)], National Institute of General Medical Sciences [Grant No. R35GM136226 (LH)]; and by the Cornell Center on the Microenvironment & Metastasis [Award No. U54CA143876 from the National Cancer Institute]; the Cornell NanoScale Science and Technology, and the Cornell BRC imaging facility. JES is the Betty and Sheldon Feinberg Senior Faculty Scholar in Cancer Research. LH is a Irma T. Hirschl Career Scientist. We thank Dr. Yu Ling Huang and Young Joon Suh for their assistance in experiments. We also thank Dr. Thorsten Falk from the University of Freiburg for advice on the U-net software package. All Matlab codes and MetaMorph routines are available upon request.
ABBREVIATIONS
- 2D
Two-dimensional
- 3D
Three-dimensional
- CFP
Cyan fluorescent protein
- DC
Dark current
- ECM
Extracellular matrix
- EGF
Epidermal growth factor
- FAK
Focal adhesion kinase
- FLP
Filopodium-like protrusions
- FRET
Förster resonance energy transfer
- GAP
GTPase activation protein
- GDI
Rho GDP-dissociation inhibitor
- GEF
Guanine nucleotide exchange factors
- mDia
Mammalian diaphanous-related formin
- MMP
Matrix metalloproteinase
- PBS
Phosphate buffered saline
- PDMS
Polydimethylsiloxane
- PEI
Polyethylenimine
- PIP3
Phosphatidylinositol (3,4,5)-trisphosphate
- RBD
Rho-binding domain
- RhoA
Ras homolog gene family, member A
- RhoGTPase
Rho family guanosine triphosphatase
- ROCK
Rho-associated protein kinase
- YFP
Yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCE
- 1.Sahai E Mechanisms of Cancer Cell Invasion. Current Opinion in Genetics & Development. 2005;15(1):87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Sacchi M, Bansal R, Rouwkema, J. Bioengineered 3D Models to Recapitulate Tissue Fibrosis. Trends in Biotechnology. 2020;38(6):623–636. doi: 10.1016/j.tibtech.2019.12.010. [DOI] [PubMed] [Google Scholar]
- 3.Reymond N, d’Agua BB, Ridley AJ. Crossing the Endothelial Barrier During Metastasis. Nature Reviews Cancer. 2013;13(12):858–870. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 4.Chambers AF, Groom AC, MacDonald IC. Dissemination and Growth of Cancer Cells in Metastatic Sites. Nature Reviews Cancer. 2002;2(8):563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 5.Yamada KM, Sixt M. Mechanisms of 3D Cell Migration. Nature Reviews Molecular Cell Biology. 2019;20:738–752. doi: 10.1038/s41580-019-0172-9. [DOI] [PubMed] [Google Scholar]
- 6.Friedl P, Wolf K. Plasticity of Cell Migration: A Multiscale Tuning Model. Journal of Cell Biology. 2010;188(1):11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffith LG, Swartz MA. Capturing Complex 3D Tissue Physiology in vitro. Nature Reviews Molecular Cell Biology. 2006;7(3):211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 8.Kim BJ, Zhao S, Bunaciu RP, Yen A, Wu M. A 3D In Situ Cell Counter Reveals That Breast Tumor Cell (MDA-MB-231) Proliferation Rate is Reduced by the Collagen Matrix Density. Biotechnology Progress. 2015;31:990–996. doi: 10.1002/btpr.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu PH, Gilkes DM, Wirtz D. The Biophysics of 3D Cell Migration. Annual Review of Biophysics. 2018;47:549–67. doi: 10.1146/annurev-biophys-070816-033854. [DOI] [Google Scholar]
- 10.Wolf K, te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Physical Limits of Cell Migration: Control by ECM Space and Nuclear Deformation and Tuning by Proteolysis and Traction Force. Journal of Cell Biology. 2013;201:1069–84. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, Sixt M. Rapid Leukocyte Migration by Integrin-independent Flowing and Squeezing. Nature. 2008;453:51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 12.Hall MS, Alisafaei F, Ban E, Feng X, Hui CY, Shenoy VB, Wu M. Fibrous Nonlinear Elasticity Enables Positive Mechanical Feedback Between Cells and ECMs. PNAS. 2016;113(49):14043–14048. doi: 10.1073/pnas.1613058113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pathak A, Kumar S. Biophysical Regulation of Tumor Cell Invasion: Moving Beyond Matrix Stiffness. Integrative Biology. 2011;3(4):267–278. doi: 10.1039/c0ib00095g. [DOI] [PubMed] [Google Scholar]
- 14.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. Multi-step Pericellular Proteolysis Controls The Transition from Individual to Collective Cancer Cell Invasion. Nature Cell Biology. 2007;9(8):893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 15.Schaefer A, Reinhard NR, Hordijk PL. Toward Understanding RhoGTPase Specificity: Structure, Function and Local Activation. Small GTPases. 2014;5(2):6. doi: 10.4161/21541248.2014.968004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Connor K, Chen M. Dynamic Functions of RhoA in Tumor Cell Migration and Invasion. Small GTPases. 2013;4(3):141–147. doi:/ 10.4161/sgtp.25131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ridley AJ, Hall A. The Small GTP-binding Protein Rho Regulates the Assembly of Focal Adhesions and Actin Stress Fibers in Response to Growth Factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 18.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated Contractility Drives the Formation of Stress Fibers and Focal Adhesions. Journal of Cell Biology. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nobes CD, Hall A. Rho, Rac, and Cdc42 GTPases Regulate the Assembly of Multimolecular Focal Complexes Associated with Actin Stress Fibers, Lamellipodia, and Filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 20.Zawistowski JS, Sabouri-Ghomi M, Danuser G, Hahn KM, Hodgson L. A RhoC Biosensor Reveals Differences in the Activation Kinetics of RhoA and RhoC in Migrating Cells. PLoS ONE. 2013;8(11):e79877. doi: 10.1371/journal.pone.0079877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shcherbakova DM, Cammer NC, Huisman TM, Verkhusha VV, Hodgson L. Direct Mutiplex Imaging and Optogenetics of Rho GTPases Enabled by Near-infrared FRET. Nature Chemical Biology. 2018;14:591–600. doi: 10.1038/s41589-018-0044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hetmanski JHR, de Belly H, Busnelli I, Waring T, Nair RV, Sokleva V, Dobre O, Cameron A, Gauthier N, Lamaze C, Swift J, del Campo A, Starborg T, Zech T, Goetz JG, Paluch EK, Schwartz JM, Caswell PT. Membrane Tension Orchestrates Rear Retraction in Matrixdirected Cell Migration. Developmental Cell. 2019;51(4):460–470. doi: 10.1016/j.devcel.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chuang TH, Xu X, Knaus UG, Hart MJ, Bokoch GM. GDP Dissociation Inhibitor Prevents Intrinsic and GTPase Activating Protein-stimulated GTP Hydrolysis by The Rac GTP-binding Protein. Journal of Biological Chemistry. 1993;268(2):775–8. doi: 10.1016/S0021-9258(18)54000-X. [DOI] [PubMed] [Google Scholar]
- 24.Lauffenburger DA, Horwitz AF. Cell Migration: A Physically Integrated Molecular Process. Cell. 1996;84:359–369. doi: 10.16/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 25.Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal Dynamics of RhoA Activity in Migrating Cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- 26.Lin M, van Golen KL. Rho-regulatory Proteins in Breast Cancer Cell Motility and Invasion. Breast Cancer Research and Treatment. 2004;84:49–60. doi: 10.1023/B:BREA.0000018424.43445.f3. [DOI] [PubMed] [Google Scholar]
- 27.Haugh JM, Codazzi F, Teruel M, Meyer T. Spatial Sensing in Fibroblasts Mediated by 3’ Phosphoinositides. Journal of Cell Biology. 2000;151:1269–1280. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuoka S, Ueda M. Mutual Inhibition Between PTEN and PIP3 Generates Bistability for Polarity in Motile Cells. Nature Communications. 2018;9:4481. doi: 10.1038/s41467-018-06856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valon L, Marín-Llauradó A, Wyatt T, Charras G, Trepat X. Optogenetic Control of Cellular Forces and Mechanotransduction. Nature Communications. 2017;8:14396. doi: 10.1038/ncomms14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolado-Carrancio A, Rukhlenko OS, Nikonova E, Tsyganov MA, Wheeler A, Garcia-Munoz A, Kolch W, von Kriegsheim A, Kholodenko BN. Periodic Propagating Waves Coordinate RhoGTPase Network Dynamics At the Leading and Trailing Edges During Cell Migration. eLife. 2020;9:58165. doi: 10.7554/eLife.58165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bass MD, Roach KA, Morgan MR, Mostafavi-Pour Z, Schoen T, Muramatsu T, Mayer U, Ballestrem C, Spatz JP, Humphries MJ. Syndecan-4-dependent Rac1 Regulation Determines Directional Migration In Response to The Extracellular Matrix. Journal of Cell Biology. 2017;177(3):527–38. doi: 10.1083/jcb.200610076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nobis M, Herrmann D, Warren SC, Kadir S, Leung W, Killen M, Magenau A, Stevenson D, Lucas MC, Reischmann N, Vennin C, Conway JRW, Boulghourjian A, Zaratzian A, Law AM, Gallego-Ortega D, Ormandy CJ, Walters SN, Grey ST, Bailey J, Chtanova T, Quinn JMW, Baldock PA, Croucher PI, Schwarz JP, Mrowinska A, Zhang L, Herzog H, Masedunskas A, Hardeman EC, Gunning PW, Monte-Nieto GD, Harvey RP, Samuel MS, Pajic M, McGhee EJ, Johnsson AKE, Sansom OJ, Welch HCE, Morton JP, Strathdee D, Anderson KI, Timpson P. A RhoA-FRET Biosensor Mouse for Intravital Imaging in Normal Tissue Homeostasis and Disease Contexts. Cell Report. 2017;21(1):274–288. doi: 10.1016/j.celrep.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 33.Lakhani VV, Hinde E, Gratton E, Elston TC. Spatio-Temporal Regulation of Rac1 Mobility by Actin Islands. PLoS One 2015;10(11):e0143753. doi: 10.1371/journal.pone.0143753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spiering D, Bravo-Cordero JJ, Moshfegh Y, Miskolci V, Hodgson L. Quantitative Ratiometric Imaging of FRET-biosensors in Living Cells. Methods in cell biology. 2013;114:593–609. doi: 10.1016/B978-0-12-407761-4.00025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell GR, Natwick DE, Collins SR. Parallel High-Resolution Imaging of Leukocyte Chemotaxis Under Agarose with Rho-Family GTPase Biosensors. Methods in Molecular Biology. 2018;1821:71–85. doi: 10.1007/978-1-4939-8612-5_6. [DOI] [PubMed] [Google Scholar]
- 36.Hodgson L, Nalbant P, Shen F, Hahn K. Imaging and Photobleach Correction of Mero-CBD, Sensor of Endogenous Cdc42 Activation. Methods Enzymology. 2006;406:140–56. doi: 10.1016/S0076-6879(06)06012-5. [DOI] [PubMed] [Google Scholar]
- 37.Falk T, Mai D, Bensch R, Çiçek Ö, Abdulkadir A, Marrakchi Y, Böhm A, Deubner J, Jäckel Z, Seiwald K, Dovzhenko A, Tietz O, Bosco CD, Walsh S, Saltukoglu D, Tay TL, Prinz M, Palme K, Simons M, Diester I, Brox T, Ronneberger O. U-Net – Deep Learning for Cell Counting, Detection, and Morphometry. Nature Methods. 2019;16:67–70. doi: 10.1038/s41592-018-0261-2. [DOI] [PubMed] [Google Scholar]
- 38.Bravo-Cordero JJ, Moshfegh Y, Condeelis J, Hodgson L. Live Cell Imaging of RhoGTPase Biosensors in Tumor Cells. 2013;1046:359–370. doi: 10.1007/978-1-62703-538-5_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miskolci V, Hodgson L, Cox D. Using Fluorescence Resonance Energy Transfer-Based Biosensors to Probe Rho GTPase Activation during Phagocytosis. Methods in Molecular Biology. 2017;1519:125–143. doi: 10.1007/978-1-4939-6581-6_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu B, Miskolci V, Sato H, Tutucci E, Kenworthy CA, Donnelly SK, Yoon YJ, Cox D, Singer RH, Hodgson L. Synonymous Modification Results in High-fidelity Gene Expression of Repetitive Protein and Nucleotide Sequences. Genes Development. 2015;29(8):876–86. doi: 10.1101/gad.259358.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gossen M, Bujard H. Tight Control of Gene Expression in Mammalian Cells by Tetracycline-responsive Promoters. Proceedings of the National Academy of Sciences. 1992;89(12):5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodgson L, Spiering D, Sabouri-Ghomi M, Dagliyan O, DerMardirossian C, Danuser G, Hahn KM. FRET Binding Antenna Reports Spatiotemporal Dynamics of GDI-Cdc42 GTPase Interactions. Nature Chemical Biology. 2016;12(10):802–809. doi: 10.1038/nchembio.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodgson L, Shen F, Hahn K. Biosensors for Characterizing the Dynamics of Rho Family GTPases in Living Cells. Current Protocols in Cell Biology. 2010;Chapter 14:Unit-14.11.26. doi: 10.1002/0471143030.cb1411s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price JT, Tiganis T, Agarwal A, Djakiew D, Thompson EW. Epidermal Growth Factor Promotes MDA-MB-231 Breast Cancer Cell Migration through a Phosphatidylinositol 3’-Kinase and Phospholipase C-dependent Mechanism. Cancer Research. 1999;59(21):5475–5478. [PubMed] [Google Scholar]
- 45.Pirkmajer S, Chibalin AV. Serum Starvation: caveat emptor. American Journal of Cell Physiology. 2011;301(2):C272–C279. doi: 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- 46.Hall MS, Long R, Feng X, Huang YL, Hui CY, Wu M. Towards Single Cell Traction Microscopy Within 3D Collagen Matrices. Experimental Cell Research. 2013;319(16):2396–2408. doi: 10.1016/j.yexcr.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grinnell F, Feld MK. Adsoption Characteristics of Plasma Fibronectin in Relationship to Biological Activity. Journal of Biomedical Materials Research. 1981;15:363–381. doi: 10.1002/jbm.820150308. [DOI] [PubMed] [Google Scholar]
- 48.Kowalczyńska HM, Nowak- Wyrzykowska M, Dobkowski J, Kołos R, Kamiński J, Makowska-Cynka A, Marciniak E. Adsoption Characteristics of Human Plasma Fibronectin in Relationship to Cell Adhesion. Journal of Biomedical Materials Research. 2002;61(2):260–9. doi: 10.1002/jbm.10151. [DOI] [PubMed] [Google Scholar]
- 49.DiMilla PA, Stone JA, Quinn JA, Albelda SM, Lauffenburger DA. Maximal Migration of Human Smooth Muscle Cells on Fibronectin and Type IV Collagen Occurs at an Intermediate Attachment Strength. Journal of Cell Biology. 1993;122(3):729–737. doi: 10.1083/jcb.122.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM. Activation of Endogenous Cdc42 Visualized in Living Cells. Science. 2004;305(5690):1615–9. doi: 10.1126/science.1100367 [DOI] [PubMed] [Google Scholar]
- 51.Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is Deamidated by Escherichia Coli Cytotoxic Necrotizing Factor-1. Nature. 1997;387:725–729. doi: 10.1038/42735. [DOI] [PubMed] [Google Scholar]
- 52.Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, Duhr S, Seibel W, Wortman M, Zheng Y. Rational Design of Small Molecule Inhibitors Targeting RhoA Subfamily Rho GTPases. Chemical Biology. 2012;19:699–710. doi: 10.1016/j.chembiol.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dugdale DE. Essentials of Electromagnetism. New York: American Institute of Physics, 1993:80–81. [Google Scholar]
- 54.Petrie RJ, Gavara N, Chadwick RS, Yamada KM. Nonpolarized Signaling Reveals Two Distinct Modes of 3D Cell Migration. Journal of Cell Biology. 2012;197(3):439–455. doi: 10.1083/jcb.201201124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell. 2008;135(3):510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 56.Mardia KV and Jupp PE. Directional Statistics. J. Wiley, 2000: 15–18. doi: 10.1002/9780470316979. [DOI] [Google Scholar]
- 57.Zhou Z, Sohn YS, Nechushtai R, Willner I. DNA Tetrahedra Modules as Versatile Optical Sensing Platforms for Multiplexed Analysis of miRNAs, Endonucleases, and Aptamer–Ligand Complexes. ACS Nano. 2020;14(7):9021–9031. doi: 10.1021/acsnano.0c04031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma F, Zhang Q, Zhang C. Catalytic Self-Assembly of Quantum-Dot-Based MicroRNA Nanosensor Directed by Toehold-Mediated Strand Displacement Cascade. Nano Letter. 2019;19(9):6370–6376. doi: 10.1021/acs.nanolett.9b0. [DOI] [PubMed] [Google Scholar]
- 59.Liu D, Yu J. Otsu Method and K-means. 2009 Ninth International Conference on Hybrid Intelligent Systems. 2009;1:344–349. doi: 10.1109/HIS.2009.74. [DOI] [Google Scholar]
- 60.Dhanachandra N, Manglem K, Chanu YJ. Image Segmentation Using K -means Clustering Algorithm. Procedia Computer Science. 2015;54:764–771. doi: 10.1016/j.procs.2015.06.090. [DOI] [Google Scholar]
- 61.Costigliola N, Kapustina MT, Weinreb GE, Monteith A, Rajfur Z, Elston TC, Jacobson K. RhoA Regulates Calcium-Independent Periodic Contractions of the Cell Cortex. Biophysical Journal. 2010;99(4):1053–1063. doi: 10.1016/j.bpj.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zemel A, Rehfeldt F, Brown AEX, Discher DE, Safran SA. Optimal Matrix Rigidity for Stress-fibre Polarization in Stem Cells. Nature Physics. 2010;6:468–473. doi: 10.1038/nphys1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohashi K, Fujiwara S, Mizuno K. Roles of the Cytoskeleton, Cell Adhesion and Rho Signaling in Mechanosensing and Mechanotransduction. The Journal of Biochemistry. 2017;161(3):245–254. doi: 10.1093/jb/mvw082. [DOI] [PubMed] [Google Scholar]
- 64.Califano JP, Reinhart-King CA. Substrate Stiffness and Cell Area Predict Cellular Traction Stresses in Single Cells and Cells in Contact. Cellular and Molecular Bioengineering. 2010;3:68–75. doi: 10.1007/s12195-010-0102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deshpande VS, Mrksich M, McMeeking RM, Evans AG. A Bio-mechanical Model for Coupling Cell Contractility with Focal Adhesion Formation. Journal of the Mechanics and Physics of Solids. 2008;56:1484–1510. doi: 10.1016/j.jmps.2007.08.006. [DOI] [Google Scholar]
- 66.Kusters R, Simon C, Dos Santos RL, Caorsi V, Wu S, Joanny JF, Sens P, Sykes C. Actin Shells Control Buckling and Wrinkling of Biomembranes. Soft Matter. 2019;15:9647–9653. doi: 10.1039/C9SM01902B. [DOI] [PubMed] [Google Scholar]
- 67.Stolarska MA, Rammohan AR. Center or Periphery? Modeling The Effects of Focal Adhesion Placement During Cell Spreading. PLoS One. 2017;12:1–22. doi: 10.1371/journal.pone.0171430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petrie RJ, Gavara N, Chadwick RS, Yamada KM. Nonpolarized Signaling Reveals Two Distinct Modes of 3D Cell Migration. Journal of Cell Biology. 2012;197(3):439–455. doi: 10.1083/jcb.201201124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The Outgrowth of Micrometastases is Enabled by the Formation of Filopodium-like Protrusions. Cancer Discovery. 2013;2(8):706–721. doi: 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paul NR, Allen JL, Chapman A, Morlan-Mairal M, Zindy E, Jacquemet G, del Ama LF, Ferizovic N, Green DM, Howe JD, Ehler E, Hurlstone A, Caswell PT. α5β1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. Journal of Cell Biology. 2015;210(6):1013–1031. doi: 10.1083/jcb.201502040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geum DT, Kim BJ, Hall MS, Wu M. Epidermal Growth Factor Promotes A Mesenchymal Over An Amoeboid Motility of MDA-MB-231 Cells Embedded Within A 3D Collagen Matrix. The European Physical Journal Plus. 2016;131(8). doi: 10.1140/epjp/i2016-16008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu PH, Gilkes DM, Wirtz D. The Biophysics of 3D Cell Migration. Annual Review of Biophysics. 2018;47:529–567. doi: 10.1146/annurev-biophys-070816-033854. [DOI] [Google Scholar]
- 73.Doyle AD, Petrie RJ, Kutys ML, Yamada KM. Dimensions in Cell Migration. Current Opinion in Cell Biology. 2013;25:642–649. doi: 10.1016/j.ceb.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lammerding J, Wolf K. Nuclear Envelope Rupture: Actin Fibers Are Putting the Squeeze on the Nucleus. Journal of Cell Biology. 2016;215(1):5. doi: 10.1083/jcb.201609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petrie RJ, Yamada KM. At the Leading Edge of Three-dimensional Cell Migration. Journal of Cell Science. 2012;125:5917–5926. doi: 10.1242/jcs.093732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.