

Abstract
We hypothesize that intrauterine hypoxia (HPX) alters the mitochondrial phenotype in fetal hearts contributing to developmental programming. Pregnant guinea pigs were exposed to normoxia (NMX) or hypoxia (HPX, 10.5% O2), starting at early [25 days (25d), 39d duration] or late gestation (50d, 14d duration). Near-term (64d) male and female fetuses were delivered by hysterotomy from anesthetized sows, and body/organ weights were measured. Left ventricles of fetal hearts were excised and frozen for measurement of expression of complex (I–V) subunits, fusion (Mfn2/OPA1) and fission (DRP1/Fis1) proteins, and enzymatic rates of I and IV from isolated mitochondrial proteins. Chronic HPX decreased fetal body weight and increased relative placenta weight regardless of timing. Early-onset HPX increased I, III, and V subunit levels, increased complex I but decreased IV activities in males but not females (all P < 0.05). Late-onset HPX decreased (P < 0.05) I, III, and V levels in both sexes but increased I and decreased IV activities in males only. Both HPX conditions decreased cardiac mitochondrial DNA content in males only. Neither early- nor late-onset HPX had any effect on Mfn2 levels but increased OPA1 in both sexes. Both HPX treatments increased DRP1/Fis1 levels in males. In females, early-onset HPX increased DRP1 with no effect on Fis1, whereas late-onset HPX increased Fis1 with no effect on DRP1. We conclude that both early- and late-onset HPX disrupts the expression/activities of select complexes that could reduce respiratory efficiency and shifts dynamics toward fission in fetal hearts. Thus, intrauterine HPX disrupts the mitochondrial phenotype predominantly in male fetal hearts, potentially altering cardiac metabolism and predisposing the offspring to heart dysfunction.
Keywords: cardiac, fetal, hypoxia, mitochondria, programming
INTRODUCTION
Chronic fetal hypoxia is a major obstetric problem that leads to maternal, placental, and fetal complications, resulting in asymmetric fetal growth restriction (1). Fetal growth restriction is associated with increased risk of cardiovascular disease and other disorders (2–4) in the adult offspring through programming (3–8) of future organ dysfunction such as the fetal heart (9). In several animal models, fetal hearts exposed to chronic hypoxia exhibit hypertrophic growth, altered cardiac function, decreased systolic function (reviewed in Ref. 42), and decreased cardiomyocyte proliferation (10–14), illustrating a deleterious effect of hypoxia on cardiac function and development. Offspring hearts exposed to hypoxia during gestation exhibit an abnormal phenotype that includes cardiac hypertrophy (15), reduced cardiac efficiency (16), diastolic and systolic dysfunction (15), decreased contractile and mitochondrial respiratory function (17), and an increased susceptibility to ischemia/reperfusion injury (7, 16, 18).
The fetal heart is a critical organ for survival. It undergoes a profound process of development and maturation over the course of gestation in both cardiac cell morphology and energy metabolism. Several aspects of heart development follow a temporal profile that is regulated by both its genome and environment (19, 20). Thus, understanding how an adverse intrauterine environment, such as chronic hypoxia, impacts fetal heart maturation is important for assessing not only fetal health but also the risk of the offspring to adult disease (3).
Maturation of the fetal heart relies on processes that impact cell number and size, as well as the organelles within the cardiac cell. The early embryonic heart increases its size by cell proliferation (21, 22). The mature fetal heart continues to grow by increasing cell size, during which mononucleated cells fuse with neighboring cells and become multinucleated (23, 24). In addition, cardiac cells undergo changes in the organization of cytoplasmic ultrastructure with a progressive increase in mitochondrial and myofibrillar content before birth (8, 20, 25, 26). In the fetal guinea pig, heart growth includes a transition from hyperplasia to hypertrophy (25) and ultrastructural organization of mitochondria and myofibrils that increases the efficiency of energy transfer to the muscle (20, 25, 27). This optimizes a tight coupling between mitochondrial mechanisms and ATP synthesis along with generating well-regulated pathways of mitochondrial biogenesis, protein import and assembly, cristae formation, respiratory chain function, and mitochondrial dynamics to assure a healthy mitochondrial population (28). The timing of these processes varies depending on the animal species, with guinea pigs (29), sheep (30), and rabbits (31) maturing before birth, and mice and rats being highly immature at birth (32). Because of its maturity, the ultrastructure of the fetal guinea pig heart is comparable to fetal humans (8), sharing similar characteristics in myofibril density and alignment, compartmentation of metabolic pathways, reliance on transarcolemmal calcium, and development of sarcoplasmic reticulum (reviewed in Ref. 8). Thus, the impact of intrauterine HPX on fetal heart function and ultrastructure organization may depend on the relative maturational state of the fetus at the time of exposure.
Cellular metabolism of the fetal heart relies primarily on anaerobic glycolysis for its energy supply (20, 33). Because of the reduced O2 levels, hypoxia inducible factor (HIF-1), a heterodimeric transcription factor, plays a critical role in oxygen sensing in cells, induces the expression of glycolytic enzymes (34), as well as inhibits mitochondrial fusion (35). Yet, the mature fetal heart exhibits significant oxidative capacities generating ∼50% of total ATP via oxidative phosphorylation (OXPHOS) (36–40). Thus, despite a high reliance of the fetal heart on anaerobic glycolysis, the development of OXPHOS mechanisms in the mature fetal heart is critical for the metabolic switch to aerobic metabolism at the time of birth.
Cellular hypoxia alters the redox status by the generation of reactive oxygen species (ROS) (41). The HPX fetal heart exhibits oxidative stress (5, 15, 42) as evidenced by increased ROS generation and lipid peroxides (43) and by the protective effects of antioxidant treatments (42–44). In rat offspring, chronic HPX has been reported to alter the cardiac mitochondrial phenotype by reducing respiratory complex expression, increasing H2O2 production, and reducing the respiratory capacity (45). Thus, cardiac mitochondria play a central role in mediating cardiac cell injury in response to chronic HPX. Although focus has been on programming effects of intrauterine HPX on heart function in the offspring, limited study has focused on the initiating causes of mitochondrial dysfunction in the fetal heart.
This study investigated the effects of intrauterine hypoxia on mitochondrial proteins, central to oxidative phosphorylation and energy supply in the fetal guinea pig heart. Since mitochondria are signaling organelles capable of adapting to the cell’s changes in bioenergetic needs through mitochondrial biogenesis and dynamics (8, 21), we propose that chronic exposure to hypoxia disrupts mitochondrial protein expression as a mechanism for disrupting normal cardiomyocyte maturation. We studied the effects of hypoxia initiated at early and late gestation to determine if the effects of hypoxia vary with duration and/or timing of exposure. Having previously shown that chronic intrauterine hypoxia in late gestation reduces mitochondrial respiratory function in the offspring in a sex-related manner (43, 46), we hypothesized that these postnatal consequences are initiated in utero by disrupting mitochondrial pathways important in heart cell function and/or cardiomyocyte maturation.
METHODS
All animal procedures using guinea pigs were approved by the University of Maryland Institutional Animal Care and Use Committee in accordance with AAALAC International accreditation (Animal Welfare Assurance no. A3200-01).
Animal Model and Fetal Extraction
Pregnant guinea pigs were exposed to normoxia for the duration of pregnancy or hypoxia (10.5% O2, HPX) starting at either early [25 days (25d)] or late gestation (50d, respectively) resulting in differences in both timing and duration of 39d and 14d, respectively. This level of hypoxia is equivalent to oxygen levels at high altitude (∼18,200 ft) generating both maternal (60% maternal blood O2 saturation) and fetal hypoxia (increased cardiac HIF1α protein, hematocrit, and reticulocyte count) in the guinea pig as previously documented (43, 47). Maternal body weight and water and food intake rates were measured for each treatment group. All near-term fetuses were extracted at 64d gestation (term = ∼65d gestation). The timing of early-onset HPX occurs at the early stages of trophoblast invasion (∼21 days gestation and placental development) (48). The rationale is based on testing the effects of HPX on an immature fetus after placental development has been established. This also results in a longer duration of HPX exposure, which could give rise to compensatory responses. The timing of late-onset HPX corresponds to the period of rapid fetal growth, postorganogenesis, and placental maturation (8, 49). Exposure to late-onset HPX occurs at a gestational age of high fetal oxygen utilization and exponential growth. Differences between early- versus late-onset HPX could reflect differences in the mitochondrial capacity to respond to HPX stress.
Near-term male and female fetuses and placentas were delivered by hysterotomy after sows were anesthetized with ketamine (80 mg/kg, sc) and xylazine (6 mg/kg, sc). Lidocaine (2%) was administered subcutaneously before making an abdominal incision. Fetal body, placenta, and organ weights were measured from excised fetuses. Left ventricles of fetal hearts were excised and frozen in liquid N2 and stored at −80°C until assayed. Fetuses were sexed by identifying reproductive organs following euthanasia.
Male and female sibling fetuses were analyzed to compare sex differences. In the NMX group, 11 male and 10 female siblings were obtained from 11 pregnant sows (∼3–4 fetuses/litter). For early- and late-onset HPX, 8 male and 8 female siblings were obtained from eight pregnant sows for each group. Fetal selection was based on those closest to the cervix in the uterine horn.
Mitochondrial Isolation
Mitochondrial proteins of left ventricles were obtained from hearts of near-term NMX and early- and late-onset HPX fetal guinea pigs. For determining protein abundance of complex subunits (I–V) by Western immunoblotting and enzymatic activity rates of complex I and IV, a mitochondrial fraction of each sample was obtained using a standard differential centrifugation protocol (43, 50). Mitochondrial isolation methods were followed as described in Thompson et al. (46). Briefly, the frozen heart tissues (20–30 mg) were ground to a fine powder in liquid N2. Tissues were then separately resuspended in 1 mL of ice-cold homogenization buffer (0.25 M sucrose, 5 mM HEPES, 1 mM EDTA, pH 7.2) and homogenized for 10 min at 4°C. Samples were centrifuged twice at 600 g for 10 min at 4°C to remove cellular debris. The supernatant was recentrifuged at 12,500 g for 10 min to generate an enriched mitochondrial fraction. The pellet containing the mitochondrial fraction was resuspended in 1× RIPA Lysis buffer supplemented with a protease inhibitor (Bio-Rad, Hercules, CA) for Western blot or was solubilized with 0.1 mM N-dodecyl β-d-maltoside (Sigma, St. Louis, MO) for complex activity assays. Total protein concentration of each sample was determined by the Bio-Rad Protein Assay (Bio-Rad).
Western Blot Analysis
Enriched mitochondrial fractions from left ventricles were obtained using the differential centrifugation protocol described above for Western immunoblot analysis of complex (I–V) subunits and mitochondrial fusion/fission proteins. As described in Thompson et al. (46), purified mitochondrial proteins were separated on 4%–15% precast SDS-PAGE gels (Bio-Rad) and then transferred to PVDF membranes with a Trans-Blot Turbo Transfer System (Bio-Rad). The membrane was blocked with 5% nonfat milk in TBST buffer for 2 h and then incubated overnight at 4°C with primary antibody. After washing with TBST, the membrane was incubated with an appropriate peroxidase-conjugated secondary antibody for 1 h and visualized by ChemiDoc Touch Imaging System (Bio-Rad). Protein bands of CI–V subunits (I:NDUFB8, 20 kDa MW; II:SDHB, 30 kDa MW; III:UQRC2, 48 kDa MW; IV:MITCO1, 40 kDa MW; V:ATP5a, 55 kDa MW) were detected by a total OXPHOS cocktail antibody (1:500, Abcam, Cambridge, MA). Fusion proteins were detected by anti-Mfn2 (1:500, Sigma, St. Louis, MO) or anti-OPA1 (1:1,000, Proteintech, Rosemont, IL) and fission proteins by anti-DRP1 (1:1,000, Abcam, Cambridge, MA) or anti-Fis1 (1:1,000, Proteintech). Voltage-dependent anion channel (VDAC) protein was used as a loading control and probed with anti-VDAC/Porin antibody (1:2,000, Boster Biological Technology Co., Pleasanton, CA). Band density was quantified by Bio-Rad Image Lab System, normalized to the loading control, VDAC, and expressed as relative expression.
Quantitative Real-Time PCR of Mitochondrial DNA
Mitochondrial:nuclear DNA ratio (mtDNA) was measured as an index of mitochondrial copy number as described by Thompson et al. (46). Total genomic DNA was isolated from heart tissue of NMX and early- and late-onset HPX fetal guinea pigs using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA concentration was determined by Nanodrop (Thermo Fisher Scientific, Waltham, MA). Relative quantification of mtDNA content for each group was determined by quantitative real-time PCR (qRT-PCR) using primers for a mitochondrial gene (mt-ND1, forward 5′-CTAAAAACCCTTGCGCTCAC-3′; reverse 5′- TGGGAAGGGAAATGTGTCAT-3′) and a nuclear gene (β-actin, forward 5′- ACTCTCCACCTTCCAGCAGA-3′; reverse 5′- AAAGCCATGCCAATCTCATC-3′). qPCR was performed with a two-step cycling program by using SYBR Green ROX qPCR Mastermix (Qiagen) and read on QuantStudio 3 Real-Time PCR System (Thermo Fisher). Gene expression was quantified by using the 2−ΔΔCt method (51).
Complex Enzyme Activity Assays
Respiratory complexes I and IV are key enzymes in the electron transport chain. These complexes were selected because of their importance as the first (I) and last (IV) complexes in the respiratory chain. Altered enzymatic activity would be expected to affect electron flux along the chain.
Complex enzyme activity rates were measured as previously described in Thompson et al. (46) using mitochondrial protein fractions of heart tissue from NMX and early- and late-onset HPX fetal guinea pigs. Complex I (NADH:ubiquinone oxidoreductase) enzyme activity was measured as the oxidation of NADH to NAD+ with a complex I enzyme activity microplate assay kit (Abcam, Cambridge, MA). This assay measures the diaphorase-type activity of complex I, which is independent of ubiquinone and rotenone sensitivity. Briefly, mitochondrial proteins were isolated as previously described above, and 4 µg were added to the microplate wells precoated with complex I-specific antibody. After 3 h of incubation at 25°C, substrates (NADH and dye) were added to the wells and OD values were measured at 450 nm. Enzyme activity was expressed as the change in OD values per minute per mg protein.
Complex IV (cytochrome c oxidase) activity is responsible for reduction of O2 to H2O and is a measure of the oxidative capacity of the respiratory chain (52). Cytochrome c oxidase activity was measured colorimetrically following the oxidation of reduced cytochrome c (ferrocytochrome c). As described in Thompson et al. (46), mitochondrial protein amount (4 µg) was selected for an optimal reaction rate and added to a 96-well plate containing the assay buffer (10 mM Tris·HCl, pH 7.0, and 120 mM KCl) plus 0.04 mM reduced cytochrome c (Sigma-Aldrich, St. Louis, MO) following reduction by 3 mM dithiothreitol. The OD values generated by oxidation of the reduced cytochrome c were measured as a decrease in absorbance at 550 nm in a 96-well plate reader (BioTek, Winooski, VT) at 10-s intervals. The reaction rates of each sample were directly determined from a tangent drawn on the reaction curve at the 3-min time interval. The observed reaction kinetics reflect the complex interaction of the effect of decreasing substrate concentration and increasing product inhibition on cytochrome c oxidase (53). Cytochrome c oxidase activity is measured as units/mg protein derived from the following equation, ΔOD/time (Δt)/ε·protein (mg), where ε = 7.04 mM−1·cm−1.
Statistical Analysis
Systat software (San Jose, CA) was used for statistical analyses. Data are expressed as means ± SE. Biochemical assays of enzymatic activities were performed on samples in triplicate with a variance of <5%. Comparisons of fetal body and organ weights and complex I and IV enzyme activities between treatments (NMX vs. early-onset HPX and NMX vs. late-onset HPX) and sex (male vs. female) were made using two-way ANOVA using the same NMX group as control. If significant main effects were detected (P < 0.05), post hoc analysis using the Holm–Sidak method was performed to identify differences between groups. Comparisons of protein expression (complexes I–V, Mfn2, OPA1, DRP1, Fis1) between NMX and each HPX group were made using Student’s t test because only two groups were run on a single gel and comparisons were not made between gels. Each N value indicates a single fetus. Male and female fetuses were matched from the same litters. Because of tissues being used up in the multiple assays, three male and two female NMX fetuses were obtained from separate litters to match equal n values for either HPX comparisons. Total number of fetuses include 21 NMX (11 males, 10 females), 16 early-onset HPX (8 males, 8 females), and 16 late-onset HPX (8 males, 8 males).
RESULTS
Fetal Organ Responses to Early- and Late-Onset HPX
The effects of intrauterine HPX on fetal body and organ weights are listed in Tables 1 and 2. With early-onset HPX (Table 1), fetal body weight was significantly reduced (P < 0.05) in males but not females. Absolute fetal heart weight was reduced in females only, and fetal kidney weight was reduced in both males and females (all P < 0.05). Early-onset HPX significantly (P < 0.05) increased relative brain (organ weight/fetal body weight ratio) and placental weight in both males and females but increased relative kidney weight in females only. There was no effect of early-onset HPX on relative fetal heart in either sex.
Table 1.
Effects of early-onset hypoxia on fetal and placental weights of males and females
| Early-Onset HPX | ||||
|---|---|---|---|---|
| Males |
Females |
|||
| NMX | EO-HPX | NMX | EO-HPX | |
| Fetal parameters | ||||
| n values | 11 | 8 | 10 | 8 |
| Absolute Wt | ||||
| FBW, g | 89.0 ± 3.8 | 76.7 ± 4.0* | 92.5 ± 4.5 | 79.4 ± 5.1 |
| FHt, g | 0.54 ± 0.03 | 0.49 ± 0.03 | 0.54 ± 0.02 | 0.45 ± 0.03* |
| FBr, g | 2.60 ± 0.04 | 2.62 ± 0.03 | 2.58 ± 0.05 | 2.59 ± 0.06 |
| FLiv, g | 4.51 ± 0.46 | 3.65 ± 0.43 | 4.66 ± 0.44 | 3.70 ± 0.33 |
| FKid, g | 0.42 ± 0.02 | 0.34 ± 0.02* | 0.47 ± 0.03 | 0.33 ± 0.02* |
| Plac Wt, g | 4.42 ± 0.32 | 5.06 ± 0.21 | 4.62 ± 0.26 | 4.78 ± 0.35 |
| Relative Wt | ||||
| FHt/FBW | 0.0060 ± 0.0002 | 0.0064 ± 0.0002 | 0.0059 ± 0.0002 | 0.0057 ± 0.0003 |
| FBr/FBW | 0.0295 ± 0.0012 | 0.0347 ± 0.0002* | 0.0283 ± 0.0012 | 0.0334 ± 0.0019* |
| FLiv/FBW | 0.0499 ± 0.003 | 0.0468 ± 0.0030 | 0.0501 ± 0.003 | 0.0462 ± 0.0014 |
| FKid/FBW | 0.0047 ± 0.0001 | 0.0044 ± 0.0002 | 0.0051 ± 0.0003 | 0.0042 ± 0.0001* |
| Plac/FBW | 0.049 ± 0.002 | 0.067 ± 0.003* | 0.050 ± 0.002 | 0.060 ± 0.002* |
Comparisons between treatment (NMX vs. HPX) in males and females were made using two-way ANOVA with statistical significance (*P < 0.05) indicated by an asterisk to indicate difference from its respective NMX control. EO-HPX, early-onset hypoxia; FBr, fetal brain; FBW, fetal body weight; FHt, fetal heart; FKid, fetal kidney; FLiv, fetal liver; NMX, normoxia; plac, placenta; Wt, weight.
Table 2.
Effects of late-onset hypoxia on fetal and placental weights of males and females
| Late-Onset HPX | ||||
|---|---|---|---|---|
| Males |
Females |
|||
| NMX | LO-HPX | NMX | LO-HPX | |
| Fetal parameters | ||||
| n values | 11 | 8 | 10 | 8 |
| Absolute Wt | ||||
| FBW, g | 89.0 ± 3.8 | 79.8 ± 4.1 | 92.5 ± 4.5 | 77.6 ± 4.6* |
| FHt, g | 0.54 ± 0.03 | 0.43 ± 0.02* | 0.54 ± 0.02 | 0.44 ± 0.03* |
| FBr, g | 2.60 ± 0.04 | 2.46 ± 0.07 | 2.58 ± 0.05 | 2.43 ± 0.06* |
| FLiv, g | 4.51 ± 0.46 | 4.02 ± 0.30 | 4.66 ± 0.44 | 4.08 ± 0.33 |
| FKid, g | 0.42 ± 0.02 | 0.40 ± 0.02 | 0.47 ± 0.03 | 0.39 ± 0.02* |
| Plac Wt, g | 4.42 ± 0.32 | 4.88 ± 0.46 | 4.62 ± 0.26 | 4.52 ± 0.39 |
| Relative Wt | ||||
| FHt/FBW | 0.0060 ± 0.0002 | 0.0054 ± 0.0002* | 0.0059 ± 0.0002 | 0.0057 ± 0.0002 |
| FBr/FBW | 0.0295 ± 0.0012 | 0.0313 ± 0.0002 | 0.0283 ± 0.0012 | 0.0318 ± 0.0013 |
| FLiv/FBW | 0.0499 ± 0.003 | 0.0501 ± 0.0020 | 0.0501 ± 0.003 | 0.0524 ± 0.0024 |
| FKid/FBW | 0.0047 ± 0.0001 | 0.005 ± 0.0002 | 0.0051 ± 0.0003 | 0.005 ± 0.0001 |
| Plac/FBW | 0.049 ± 0.002 | 0.061 ± 0.004* | 0.050 ± 0.002 | 0.058 ± 0.002* |
Comparisons between treatment (NMX vs. HPX) in males and females were made using two-way ANOVA with statistical significance (*P < 0.05) indicated by an asterisk to indicate difference from its respective NMX control. FBr, fetal brain; FBW, fetal body weight; FHt, fetal heart; FKid, fetal kidney; FLiv, fetal liver; LO-HPX, late-onset hypoxia; NMX, normoxia; plac, placenta; Wt, weight.
With late-onset exposure (Table 2), HPX significantly reduced fetal body weight, brain weight, and kidney weight in females only but reduced fetal heart weight in both sexes. Similar to early-onset HPX, late-onset HPX significantly increased (P < 0.05) relative placental weight and relative brain weight in both males and females and decreased relative fetal heart weight in males only.
Maternal body weight (MBW, g) at term was 1,064 ± 12.4 (NMX), 992 ± 14.3 (early-onset HPX), and 957 ± 9.3 (late-onset HPX), which corresponded to a weight reduction of 6.8% and 10.0%, respectively, from NMX control (P < 0.05). This is unlikely to account for nutritional deficiency since food and water intake was similar with treatment. The average maternal food intake rate (g/day/kg MBW), measured over the last 14 days of gestation, was increased (P < 0.05) with early-onset HPX (55.1 ± 2.5) but similar with late-onset HPX (42.0 ± 1.5) to NMX controls (45.2 ± 0.7). The average maternal water intake rate (mL/day/kg MBW) for the same gestational period as measured for food intake was similar between NMX and late-onset HPX groups (123.7 ± 8.3 vs. 128.8 ± 6.1) but increased with early-onset HPX (199.8 ± 10.2; P < 0.05 vs. NMX). These results indicate a hypoxic pregnant guinea pig model that generates fetal growth restriction independent of reduced nutrient intake.
Effects of HPX on Respiratory Complex Subunit Expression (I–V)
The protein expression levels of mitochondrial respiratory complexes I–V subunits from fetal heart tissue homogenates were measured in males and females (Fig. 1) by Western blot. A representative immunoblot from male fetal hearts is included to illustrate the variable abundance of selective subunits associated with complexes I–V. Expression of complexes I, III, and V was increased with early-onset HPX (P < 0.05) and decreased with late-onset HPX in male hearts. In female hearts, late- but not early-onset HPX significantly decreased (P < 0.05) expression of complexes I, III, and V subunits compared with NMX controls.
Figure 1.
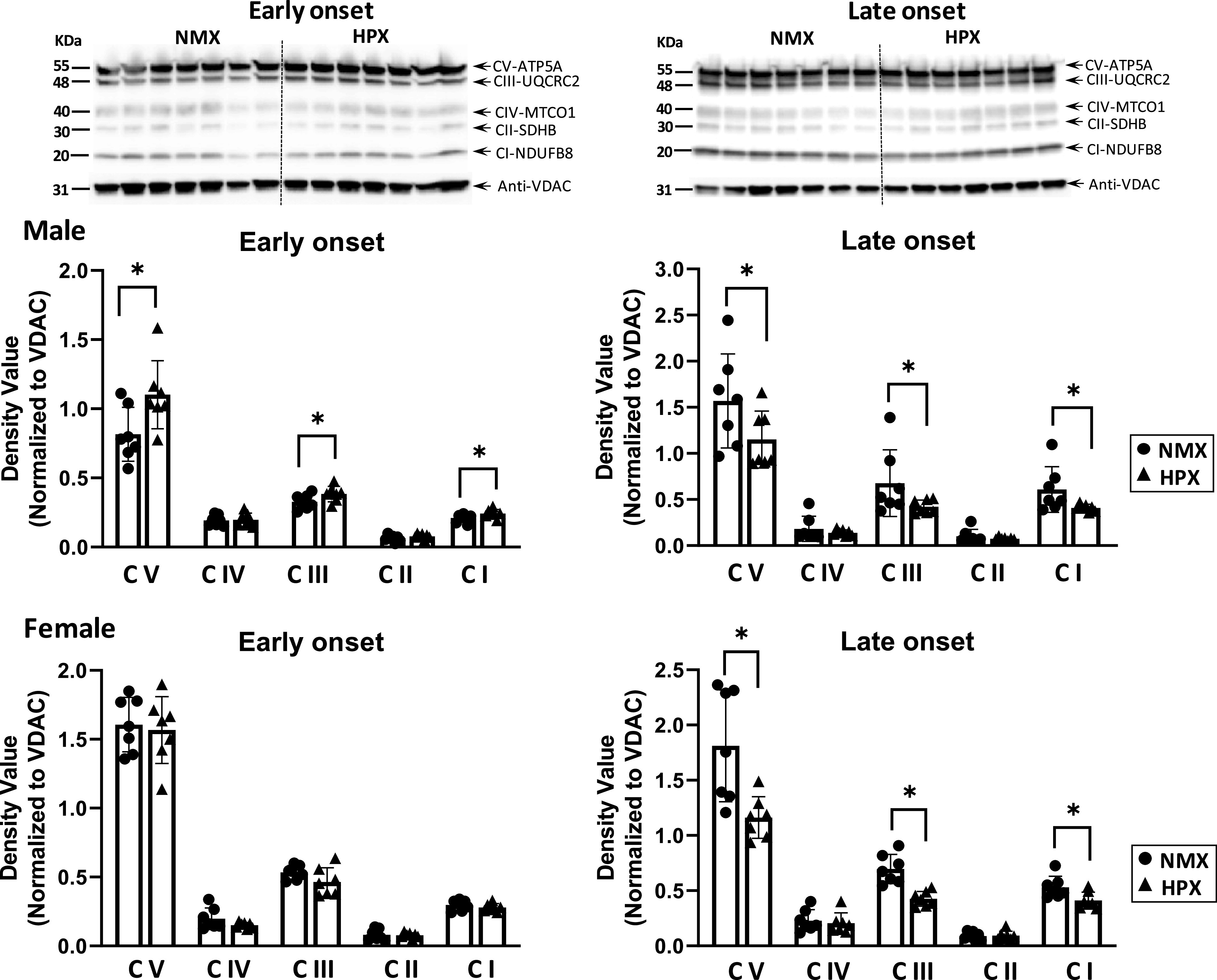
Western immunoblot (top, male fetal hearts) and analysis (bottom) of mitochondrial complex (CI–V) subunit expression of isolated cardiac mitochondria of left ventricles. Male and female fetuses were exposed to normoxia (NMX, circles) and early-onset [25 days (25d) gestation, 39d duration] and late-onset (50d gestation, 14d duration) hypoxia (HPX, triangles). Protein expression was measured in fetal heart ventricles obtained at term (64d gestation). Density values are target band values normalized to VDAC loading controls. *Significant differences at P < 0.05 vs. NMX; n = 7 animals in each group. Representative blots identified Complex I–V subunits are NFUFB8, SDHB, UQCRC2, MITCO1, and ATP5a, respectively. VDAC, voltage-dependent anion channel.
Effects of Hypoxia on Respiratory Complex I and IV Enzyme Activities
Intrauterine HPX differentially altered enzyme activities of complex I and IV in fetal heart ventricles (Fig. 2). Both early- and late-onset HPX significantly (P < 0.05) increased complex I and decreased complex IV activities in heart tissue of males compared with their NMX controls. In contrast, neither early- nor late-onset HPX affected complex I or IV activity in female left ventricles.
Figure 2.

Enzyme activities of respiratory complexes I (CI) and IV (CIV) in isolated cardiac mitochondria of left ventricles. Male and female fetuses were exposed to normoxia (NMX, circles) and early-onset [25 days (25d) gestation, 39d duration] and late-onset (50d gestation, 14d duration) hypoxia (HPX, triangles). Activity rates were measured in tissues obtained at term (64d gestation) as oxidation rates of substrate per minute per milligram mitochondrial protein (CI: oxidation of NADH; CIV: oxidation of ferricytochrome c). *Significant differences at P < 0.05 vs. NMX controls; n = 7 or 8 animals in each group.
Effects of Hypoxia on Mitochondrial Fusion and Fission Proteins
Mitochondrial dynamics are dictated in part by the relative expression of fusion/fission proteins within the cell. Representative immunoblots of male and female fetal hearts illustrate expression relative to VDAC of both fusion (Fig. 3) and fission proteins (Fig. 4). Both early- and late-onset HPX had similar effects on Mfn2 and OPA1 expression in fetal hearts. For example, neither early- nor late-onset HPX had any effect on expression levels of the fusion protein, Mfn2, in either male or female hearts. However, both treatments significantly increased (P < 0.05) OPA1 levels in both sexes compared with NMX (Fig. 3). Regarding fission proteins, both early- and late-onset HPX significantly increased (P < 0.05) expression levels of DRP1 and Fis1 in males (Fig. 4). In females, there were variable effects of HPX exposure on expression of fission proteins. For example, early-onset HPX significantly increased (P < 0.05) DRP1 levels whereas late-onset HPX increased (P < 0.05) Fis1 levels (Fig. 4).
Figure 3.

Western immunoblot (top, male fetal hearts) and analysis (bottom) of fusion proteins (Mfn-2 and OPA-1) in isolated cardiac mitochondria of left ventricles. Male and female fetuses were exposed to normoxia (NMX, circles) and early-onset [25 days (25d) gestation, 39d duration] and late-onset (50d gestation, 14 duration) hypoxia (HPX, triangles). Protein expression was measured in fetal heart ventricles obtained at term (64d gestation). Density values are ratios of target band values normalized to VDAC as a loading control. *Significant differences from NMX controls; n = 7 animals in each group (*P < 0.05, **P < 0.01, ****P < 0.0001). VDAC, voltage-dependent anion channel.
Figure 4.

Western immunoblot (top, male fetal hearts) and analysis (bottom) of fission proteins (DRP1 and Fis1) in isolated cardiac mitochondria of left ventricles. Male and female fetuses were exposed to normoxia (NMX, circles) and early-onset [25 days (25d) gestation, 39d duration] and late-onset (50d gestation, 14d duration) hypoxia (HPX, triangles). Protein expression was measured in fetal heart ventricles obtained at term (65d gestation). Density values are ratios of target band values normalized to VDAC as a loading control. *Significant differences from NMX controls; n = 7 animals in each group (*P < 0.05, **P < 0.01). VDAC, voltage-dependent anion channel.
Mitochondrial DNA Content
mtDNA content was measured by qPCR as the mitochondrial DNA:nuclear DNA ratio (Fig. 5). Both early- and late-onset HPX significantly reduced (P < 0.05) mtDNA content in male but not female heart ventricles.
Figure 5.

Effects of early-onset [25 days (25d) gestation, 39 duration] and late-onset (50d gestation, 14d duration) hypoxia (HPX, 10.5% O2, triangles) compared with normoxic controls (NMX, circles) on mitochondrial content of heart left ventricles of male and female fetuses obtained at term (64d gestation). Mitochondrial (mito) content was measured as the ratio of mito DNA/nuclear DNA ratios. *P < 0.05 from NMX controls; n = 6–8 animals in each group.
DISCUSSION
Fetal heart cell metabolism is linked to both mitochondrial function and cytosolic glycolysis depending on the aerobic conditions and the cell’s relative maturity. During fetal heart maturation, the ultrastructure of the cardiac cell (i.e., myofibrils, sarcoplasmic reticulum) and its mitochondria (i.e., cristae morphology, network formation) progresses concomitantly to provide an efficient energy supply for contractile function. At term, the fetal heart undergoes a metabolic switch to OXPHOS in response to changes in substrate availability, oxygenation of the tissue, and HIF1 signaling (20). This study demonstrated that chronic intrauterine HPX alters expression and activities of select mitochondrial complex proteins that could disrupt its respiratory efficiency and cardiac metabolism before birth. This is supported by altered abundance of complex proteins in both sexes and sex-dependent changes in complex enzyme activity rates and mitochondrial fission in male hearts.
Effects of Hypoxia on Respiratory Chain Complexes
Mitochondrial respiratory protein expression links biogenesis to efficient energy metabolism and OXPHOS. Complexes I, III, and IV exist in the respiratory chain as respirosomes and increased expression of these supercomplex subunits has been shown to increase stability and efficiency of the respiratory chain (54). In the current study, early-onset HPX increased the expression of complexes I, III, and V in male hearts, suggesting a mitochondrial response toward respiratory chain stability. In late-onset HPX, expression of fetal cardiac complex I, III, and V subunits was reduced in both males and females, suggesting that timing of HPX exposure and/or heart maturity at the time of exposure differentially affects the regulation of mitochondrial protein expression. In early-onset HPX, the increase in complex expression may be compensatory, and the decrease, in late-onset HPX, may be decompensatory, both of which may alter electron flux along the respiratory chain. These changes in protein expression, differing with duration of HPX exposure and/or maturation, reflect the developmental plasticity of mitochondrial biogenesis in response to hypoxic stress (55).
Mitochondrial biogenesis is the process of expression and assembly of protein subunits into the mitochondrial membranes (56). Peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1-α) is a transcriptional coactivator considered the master regulator of mitochondrial biogenesis. The mechanisms underlying subunit expression can be regulated by HPX with increased activation/expression of nuclear PGC1-α via phosphorylation and deacetylation, inducing nuclear-encoded mitochondrial proteins that are transcribed in the nucleus and transported to the mitochondria. Hypoxia increases PGC1-α expression and activity, and subsequently, biogenesis, in cardiac cell culture via AMPK pathway (57) and SIRT activation (58), respectively. HPX-induced changes in PGC1-α expression/activation may be a possible underlying mechanism mediating altered complex expression. Evidence for differences in timing/duration of HPX exposure on PGC1-α expression is needed to account for the differential expression of complex proteins in fetal hearts.
Epigenetic mechanisms may also contribute to changes in gene expression of respiratory complex subunits in response to chronic HPX. Prenatal HPX has been shown to differentially alter DNA methylation patterns in rat hearts of fetal versus adult offspring in opposing directions (59–61). For example, chronic HPX induces hypermethylation of the promoter region of PKCε in fetal rat hearts, resulting in decreased PKCε gene expression (7, 62). In addition, chronic HPX reduces expression of the cold shock protein, cold-inducible RNA binding protein (CIRBP), in neonatal rat cardiomyocytes via Cirbp hypermethylation (63). The changes in redox status occurring with chronic HPX are directly linked to epigenetic mechanisms during development (64). Thus, the conditions of HPX and/or oxidative stress may mediate DNA methylation patterns, contributing to altered cardiac gene expression (59, 60, 62, 63). Furthermore, sex differences in DNA methyltransferases and DNA methylation patterns have been identified in early development and influenced by sex hormones and nutrient restriction (65), although unknown for fetal hearts. Therefore, identifying the effects of chronic HPX on methylation patterns of specific promoter regions encoding for mitochondrial proteins is key for understanding its role in mitochondrial biogenesis and how it varies with sex in the fetal heart.
Effects of Hypoxia on Complex I and IV Enzyme Activities
Efficiency of cellular respiration is determined by the kinetic properties of the respiratory complexes, as well as their abundance. Both early- and late-onset HPX increased complex I and decreased complex IV activity in male but not female hearts. The decrease in complex IV activity along with enhanced complex I activity would both be expected to significantly disrupt electron flux, resulting in excessive electron accumulation and generation of ROS (66, 67).
Prenatal HPX is associated with oxidative stress and reported in a variety of HPX animal models (reviewed in Ref. 68). Excess superoxide anions generated at complexes I, II, and IV (67) form ROS that can have direct effects on enzyme activity. We have shown that maternal administration of the antioxidant, N-acetylcysteine, reverses the HPX-induced decrease in cytochrome c oxidase (a complex IV subunit) activity in heart ventricles of male fetal guinea pigs (43), along with preventing the increase in the lipid peroxide, malondialdehyde (43), and peroxynitrite (69), both of which are inhibitory adducts of cytochrome c oxidase (70, 71). Others have reported that maternal antioxidant treatment reverses the HPX-induced effects in placenta and fetal hearts (15, 43, 72, 73) and the programming effects of cardiovascular dysfunction in the offspring (5, 15, 18). Mitochondrial-targeted antioxidants have increasingly demonstrated the role of inhibiting oxidative stress in the prevention of programming of cardiovascular dysfunction in the offspring (74–76).
In addition, excess ROS generation can induce mitochondrial injury (e.g., DNA degradation and altered membrane integrity) and alter cardiomyocyte proliferation/differentiation (77). Differences in enzyme activities may generate different levels of ROS since ROS production is related to electron flux in the respiratory chain (67). Female mitochondria generate less ROS and have a higher antioxidant capacity as a result of higher reduced glutathione, glutathione peroxidase, and MnSOD levels compared with male mitochondria (78, 79). Furthermore, in adult offspring exposed to developmental hypoxia, female hearts had higher oxygen consumption rates and produced less H2O2 than their male cohorts (45). Higher estradiol levels in females compared with males (59, 80) confer greater cardioprotection in females via ERα/β-induced gene expression (81). However, since estradiol levels are similar between male and female fetuses, the role of sex hormones, particularly estradiol, likely differs in determining sex differences in the fetus.
Mitochondrial complex enzyme activity is also regulated by acetylation of complex subunits by acetyl CoA (82). Mitochondrial sirtuins such as SIRT3 are deacetylases, which can affect activity by removal of acetyl groups. In the heart, studies have shown that complex I and IV can be differentially acetylated (61, 83) depending on the levels of NAD+ and NADH with complex I subunit (NDUFS4) hyperacetylated in the presence of SIRT3 inhibition and complexes III and IV, each having different levels of acetylation (61). Since SIRT activity is regulated by NAD+/NADH ratios (58), the effect of HPX on complex I and IV enzyme activities along with the differences in response between males and females may depend on the relative amount of protein acetylation. Thus, HPX-induced oxidative stress can have both direct effects on enzyme activity and indirect effects through posttranscriptional modification of protein expression.
Effects of Hypoxia on Mitochondrial Dynamics
Mitochondrial dynamics plays a significant role in respiratory efficiency by balancing the generation of mitochondrial network formation via fusion and removing defective mitochondria via fission coupled to mitophagy (84). In adult heart cells, a highly organized ultrastructure ensures an efficient energy transfer within the cell to support cellular function (32, 84, 85). Since fetal heart cells exhibit less organized ultrastructure compared with mature adult cells, the progressive formation of new mitochondria is important for establishing the organizational arrangement of mitochondria, myofibrils, and sarcoplasmic reticulum (37, 86) in preparation for the metabolic switch that occurs at birth (28).
Studies demonstrate that altered fusion/fission protein expression alters mitochondrial metabolism (87–90), along with changes in mitochondrial calcium uptake and release (91), an important function in overall calcium homeostasis. Further, the effects of decreased Mfn2 or OPA1 and increased DRP1 levels in the heart are generally harmful and lead to altered cardiac cell metabolism (92). Mfn2, along with other proteins (e.g., Mfn1), governs the fusion of outer mitochondrial membranes (93), whereas OPA1 proteins regulate the fusion of inner mitochondrial membranes. The lack of change in Mfn2 protein levels with either early- or late-onset HPX suggests that HPX had little effect on mitochondrial fusion and generation of new mitochondria.
The increase in OPA1 may reflect a change in cristae formation or remodeling in response to HPX because of its regulatory role in cristae morphology (94–96). Acute hypoxia (1% O2, 0–12 h) has been shown to stimulate cristae restructuring in mitochondria of neuronal cell cultures as a metabolic response to maintain ATP generation (97). OPA1 is a master regulator of cristae remodeling that increases folding of the inner mitochondrial membrane, localizes OXPHOS complexes closer together, and enhances respiration (98). Overexpression of OPA1 levels maintains cristae integrity and enhances respiratory complex assembly and stability (98). Both early- and late-onset HPX increased expression of OPA1 while having no effect on Mfn2 levels in the fetal heart, which may indicate a compensatory change to HPX in the ultrastructure of the inner mitochondrial membrane.
DRP1 and Fis1, as well as other proteins (e.g., Mff, MiD49, MiD51), are the major proteins that regulate mitochondrial fission (99). Both early- and late-onset HPX increased DRP1 and Fis1 expression in males, whereas the expression of fission proteins differed with HPX treatment in females. The shift toward fission may limit overall mitochondrial function, remove mitochondria, and/or reduce generation of new mitochondria under HPX stress. Both early- and late-onset HPX decreased mitochondrial DNA content in males but not females, consistent with reduced mitochondrial number in HPX hearts. Since mitochondrial DNA copy number increases at birth (77), a decrease in the normal trajectory of mitochondrial formation due to HPX during fetal maturation could limit biogenic and bioenergetic pathways in males. Thus, the HPX-induced changes toward fission may unfavorably alter the cell’s oxidative capacity by reducing mitochondrial number, along with expression of complex subunits and their kinetic properties (88, 100–102).
Cardiac mitochondria have been shown to differ in size between males and females. Female mitochondria are larger and elongated, a phenotype indicative of fusion, whereas male mitochondria have a round, fragmented phenotype, typical of fission (78). Although there were no sex differences in NMX, HPX increased fission in male mitochondria to a greater extent than female mitochondria and further reduced mitochondrial density in male compared with female hearts. Thus, mitochondria of male hearts may be more susceptible to chronic HPX stress than mitochondria of female hearts in maintaining a stable population.
In summary, both conditions of early- and late-onset HPX alter respiratory complex I and IV activities despite compensation of complex expression with early-onset HPX and decompensation with late-onset HPX, suggesting late-gestation exposure may be more deleterious. Thus, intrauterine hypoxia disrupts expression and activity of select respiratory chain components that likely contribute to inefficient electron transport. Since maturation of mitochondrial ultrastructure is a driving force for cardiomyocyte maturation (37), the disruptive effects of HPX on mitochondrial complex subunit expression and activities, along with altered dynamics processes, are likely to alter the metabolic efficiency of the cell. The mismatch between mitochondrial complex expression and kinetics induced by hypoxia may reduce the cell’s bioenergetic function, thereby reducing the efficiency of electron flux along the respiratory chain (103). Furthermore, this study identified sex differences in the mitochondrial phenotype of fetal hearts that suggest males are more vulnerable than females to HPX, which we have shown to persist in the adult offspring exposed to prenatal HPX (17).
Notably, there are limitations of the study that include a lack of measurement of oxygen consumption rate and/or ATP generation of fetal cardiomyocytes as an index of mitochondrial respiratory function. Furthermore, lacking is an ability to distinguish the effects of duration of HPX exposure from its timing of gestational age on mitochondrial biology.
Perspectives and Significance
Altered fetal heart mitochondrial function may be an underlying cause of programming of the offspring. We previously reported that late-onset HPX, identical to the current study, reduces contractile function of the offspring guinea pig heart concomitant with reduced mitochondrial function, as measured by reduced complex IV activity, reduced complex IV subunit expression, and reduced maximal respiratory and reserve capacity of isolated cardiac cells (17). Thus, the programming effect of prenatal hypoxia may be initiated in utero by targeting cardiac mitochondria, causing dysregulation of respiratory chain subunits, enzyme activities, and mitochondrial dynamics. The gestational age of HPX exposure also plays an important role in mitochondrial function, given the temporal relationship with the relative maturity of the fetal heart cell and its spatial organization with regards to ultrastructure (40). Finally, chronic HPX may generate defective fetal cardiac mitochondria that predispose the offspring heart to an increased risk of mitochondrial dysfunction.
GRANTS
The project described is supported, in part, by a National Institutes of Health (NIH HL126859, to L.P.T.) grant.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.P.T. conceived and designed research; H.S. performed experiments; H.S. analyzed data; H.S., B.M.P., and L.P.T. interpreted results of experiments; H.S. prepared figures; L.P.T. drafted manuscript; H.S., B.M.P., and L.P.T. edited and revised manuscript; H.S., B.M.P., and L.P.T. approved final version of manuscript.
ACKNOWLEDGMENTS
Data from this study was presented in part at the 2020 SRI Annual Scientific Meeting. Repro. Sci. 27: Supplement 1, 323A, 2020.
REFERENCES
- 1.Giussani DA. The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol 594: 1215–1230, 2016. doi: 10.1113/JP271099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodríguez-Rodríguez P, Ramiro-Cortijo D, Reyes-Hernández CG, López de Pablo AL, González MC, Arribas SM. Implication of oxidative stress in fetal programming of cardiovascular disease. Front Physiol 9: 602, 2018. doi: 10.3389/fphys.2018.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet 341: 938–941, 1993. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 4.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73, 2008. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giussani DA, Davidge ST. Developmental programming of cardiovascular disease by prenatal hypoxia. J Dev Orig Health Dis 4: 328–337, 2013. doi: 10.1017/S204017441300010X. [DOI] [PubMed] [Google Scholar]
- 6.Nuyt AM, Alexander BT. Developmental programming and hypertension. Curr Opin Nephrol Hypertens 18: 144–152, 2009. doi: 10.1097/MNH.0b013e328326092c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue Q, Zhang L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase Cε. J Pharmacol Exp Ther 330: 624–632, 2009. doi: 10.1124/jpet.109.153239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison JL, Botting KJ, Darby JRT, David AL, Dyson RM, Gatford KL, Gray C, Herrera EA, Hirst JJ, Kim B, Kind KL, Krause BJ, Matthews SG, Palliser HK, Regnault TRH, Richardson BS, Sasaki A, Thompson LP, Berry MJ. Guinea pig models for translation of the developmental origins of health and disease hypothesis into the clinic. J Physiol 596: 5535–5569, 2018. doi: 10.1113/JP274948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Y, Dasgupta C, Huang L, Song R, Zhang Z, Zhang L. Multi-omics integration reveals short and long-term effects of gestational hypoxia on the heart development. Cells 8: 1608, 2019. doi: 10.3390/cells8121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Botting KJ, Wang KC, Padhee M, McMillen IC, Summers-Pearce B, Rattanatray L, Cutri N, Posterino GS, Brooks DA, Morrison JL. Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clin Exp Pharmacol Physiol 39: 814–823, 2012. doi: 10.1111/j.1440-1681.2011.05649.x. [DOI] [PubMed] [Google Scholar]
- 11.Botting KJ, McMillen IC, Forbes H, Nyengaard JR, Morrison JL. Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia-responsive genes. J Am Heart Assoc 3: e000531, 2014. doi: 10.1161/JAHA.113.000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Botting KJ, Loke XY, Zhang S, Andersen JB, Nyengaard JR, Morrison JL. IUGR decreases cardiomyocyte endowment and alters cardiac metabolism in a sex- and cause-of-IUGR-specific manner. Am J Physiol Regul Integr Comp Physiol 315: R48–R67, 2018. doi: 10.1152/ajpregu.00180.2017. [DOI] [PubMed] [Google Scholar]
- 13.Bubb KJ, Cock ML, Black MJ, Dodic M, Boon WM, Parkington HC, Harding R, Tare M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J. Physiol 578: 871–881, 2007. doi: 10.1113/jphysiol.2006.121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol 580: 639–648, 2007. doi: 10.1113/jphysiol.2006.122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, Blake EZ, Horder KA, Thakor AS, Hansell JA, Kane AD, Wooding FBP, Cross CM, Herrera EA. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PloS One 7: e31017, 2012. doi: 10.1371/journal.pone.0031017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rueda-Clausen CF, Morton JS, Lopaschuk GD, Davidge ST. Long-term effects of intrauterine growth restriction on cardiac metabolism and susceptibility to ischaemia/reperfusion. Cardiovasc Res 90: 285–294, 2011. doi: 10.1093/cvr/cvq363. [DOI] [PubMed] [Google Scholar]
- 17.Thompson LP, Chen L, Polster BM, Pinkas G, Song H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex-related manner. Am J Physiol Regul Integr Comp Physiol 315: R1232–R1241, 2018. doi: 10.1152/ajpregu.00224.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Invest 10: 265–274, 2003. doi: 10.1016/S1071-5576(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 19.Sánchez-Díaz M, Nicolás-Ávila JA, Cordero MD, Hidalgo A. Mitochondrial adaptations in the growing heart. Trends Endocrinol Metab 31: 308–319, 2020. doi: 10.1016/j.tem.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Piquereau J, Ventura-Clapier R. Maturation of cardiac energy metabolism during perinatal development. Front Physiol 9: 959, 2018. doi: 10.3389/fphys.2018.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christoffels VM, Habets PE, Franco D, Campione M, de Jong F, Lamers WH, Bao ZZ, Palmer S, Biben C, Harvey RP, Moorman AF. Chamber formation and morphogenesis in the developing mammalian heart. Dev Biol 223: 266–278, 2000. [Erratum in Dev Biol 225: 266, 2000]. doi: 10.1006/dbio.2000.9753. [DOI] [PubMed] [Google Scholar]
- 22.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464, 2011. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 23.Paradis AN, Gay MS, Zhang L. Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state. Drug Discov Today 19: 602–609, 2014. doi: 10.1016/j.drudis.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zak R. Cell proliferation during cardiac growth. Am J Cardiol 31: 211–219, 1973. doi: 10.1016/0002-9149(73)91034-5. [DOI] [PubMed] [Google Scholar]
- 25.Hoerter JA, Ventura-Clapier R, Kuznetsov A. Compartmentation of creatine kinases during perinatal development of mammalian heart. Mol Cell Biochem 133: 277–286, 1994. doi: 10.1007/BF01267960. [DOI] [PubMed] [Google Scholar]
- 26.Rolph TP, Jones CT, Parry D. Ultrastructural and enzymatic development of fetal guinea pig heart. Am J Physiol Heart Circ Physiol 243: H87–H93, 1982. doi: 10.1152/ajpheart.1982.243.1.H87. [DOI] [PubMed] [Google Scholar]
- 27.Sano HI, Toki T, Naito Y, Tomita M. Developmental changes in the balance of glycolytic ATP production and oxidative phosphorylation in ventricular cells: a simulation study. J Theor Bio 419: 269–277, 2017. doi: 10.1016/j.jtbi.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 28.Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol 4: 102, 2013. doi: 10.3389/fphys.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirakow R, Gotoh T. Quantitative studies on the ultrastructural differentiation and growth of mammalian cardiac muscle cells. II. The atria and ventricles of the guinea pig. Acta Anat (Basel) 108: 230–237, 1980. doi: 10.1159/000145304. [DOI] [PubMed] [Google Scholar]
- 30.Sheldon CA, Friedman WF, Sybers HD. Scanning electron microscopy of fetal and neonatal lamb cardiac cells. J Mol Cell Cardiol 8: 853–862, 1976. doi: 10.1016/0022-2828(76)90068-7. [DOI] [PubMed] [Google Scholar]
- 31.Smith HE, Page E. Ultrastructural changes in rabbit heart mitochondria during the perinatal period: neonatal transition to aerobic metabolism. Dev Biol 57: 109–117, 1977. doi: 10.1016/0012-1606(77)90358-x. [DOI] [PubMed] [Google Scholar]
- 32.Piquereau J, Novotova M, Fortin D, Garnier A, Ventura-Clapier R, Veksler V, Joubert F. Postnatal development of mouse heart: formation of energetic microdomains. J Physiol 588: 2443–2454, 2010. doi: 10.1113/jphysiol.2010.189670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol Heart Circ Physiol 261: H1698–H1705, 1991. doi: 10.1152/ajpheart.1991.261.6.H1698. [DOI] [PubMed] [Google Scholar]
- 34.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol 76: 347–353, 2011. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 35.Neary MT, Ng KE, Ludtmann MH, Hall AR, Piotrowska I, Ong SB, Hausenloy DJ, Mohun TJ, Abramov AY, Breckenridge RA. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J Mol Cell Cardiol 74: 340–352, 2014. doi: 10.1016/j.yjmcc.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warshaw JB. Cellular energy metabolism during fetal development. I. Oxidative phosphorylation in the fetal heart. J Cell Biol 41: 651–657, 1969. doi: 10.1083/jcb.41.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol 56: 130–140, 2010. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 38.Mühlfeld C, Singer D, Engelhardt N, Richter J, Schmiedl A. Electron microscopy and microcalorimetry of the postnatal rat heart (Rattus norvegicus). Comp Biochem Physiol A Mol Integr Physiol 141: 310–318, 2005. doi: 10.1016/j.cbpb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A, Chung S. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med 4 Suppl 1: S60–S67, 2007. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Carvalho AETS, Bassaneze V, Forni MF, Keusseyan AA, Kowaltowski AJ, Krieger JE. Early postnatal cardiomyocyte proliferation requires high oxidative energy metabolism. Sci Rep 7: 15434, 2017. doi: 10.1038/sr1598-017-15656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: mechanisms of O2 sensing. J Biol Chem 275: 25130–25138, 2000. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 42.Giussani DA, Niu Y, Herrera EA, Richter HG, Camm EJ, Thakor AS, Kaen AD, Hansel JA, Brain KL, Skeffingtonn KL, Itani N, Wooding FBP, Cross CM, Allison BJ. Heart disease link to fetal hypoxia and oxidative stress. In: Advances in Fetal and Neonatal Physiology, Advances in Experimental Medicine and Biology, edited by Zhang L, Ducsay C.. New York, NY: Springer, 2014, Vol. 814. doi: 10.1007/978-1-4939-1031-1_7. [DOI] [PubMed] [Google Scholar]
- 43.Al-Hasan YM, Evans LC, Pinkas GA, Dabkowski ER, Stanley WC, Thompson LP. Chronic hypoxia impairs cytochrome oxidase activity via oxidative stress in selected fetal guinea pig organs. Reprod Sci 20: 299–307, 2013. doi: 10.1177/1933719112453509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aljunaidy MM, Morton JS, Cooke C-LM, Davidge ST. Prenatal hypoxia and placental oxidative stress: linkages to developmental origins of cardiovascular disease. Am J Physiol Regul Integr Comp Physiol 313: R395–R399, 2017. doi: 10.1152/ajpregu.00245.2017. [DOI] [PubMed] [Google Scholar]
- 45.Hellgren KT, Premanandhan H, Quinn CJ, Trafford AW, Galli GLJ. Sex-dependent effects of developmental hypoxia on cardiac mitochondria from adult murine offspring. Free Radic Biol Med 162: 490–499, 2021. doi: 10.1016/j.freeradbiomed.2020.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Thompson LP, Song H, Polster BM. Fetal programming and sexual dimorphism of mitochondrial protein expression and activity of hearts of prenatally hypoxic guinea pig offspring. Oxid Med Cell Longev 2019: 7210249–7210211, 2019. doi: 10.1155/2019/7210249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson L, Dong Y, Evans L. Chronic hypoxia increases inducible NOS-derived nitric oxide in fetal guinea pig hearts. Pediatr Res 65: 188–192, 2009. doi: 10.1203/PDR.0b013e31818d6ad0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaufmann P, Davidoff M. The guinea-pig placenta. Adv Anat Embryol Cell Biol 53: 5–91, 1977. doi: 10.1007/978-3-642-66618-6. [DOI] [PubMed] [Google Scholar]
- 49.Valdés G, Erices R, Chacón C, Corthorn J. Angiogenic, hyperpermeability and vasodilator network in utero-placental units along pregnancy in the guinea-pig (Cavia porcellus). Reprod Biol Endocrinol 6: 13, 2008. doi: 10.1186/1477-7827-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gostimskaya I, Galkin A. Preparation of highly coupled rat heart mitochondrial. J Vis Exp 43: e2202, 2010. doi: 10.3791/2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Kadenbach B, Hüttemann M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion. 24: 64–76, 2015. doi: 10.1016/j.mito.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 53.Yonetani T, Ray GS. Studies on cytochrome oxidase. VI. Kinetics of the aerobic oxidation of ferrocytochrome C by cytochrome oxidase. J Biol Chem 240: 3392–3398, 1965. [PubMed] [Google Scholar]
- 54.Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20: 267–284, 2019. [Erratum in Nat Rev Mol Cell Biol 22: 367, 2021]. doi: 10.1038/s41580-018-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gyllenhammer LE, Entringer S, Buss C, Wadhwa PD. Developmental programming of mitochondrial biology: a conceptual framework and review. Proc Biol Sci 287: 20192713, 2020. doi: 10.1098/rspb.2019.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dorn GW 2nd, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29: 1981–1991, 2015. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, Gu Z, Zhang J, Wang J, Zen K, Xiang Y, Wang D, Zhang C-Y. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res 20: 676–687, 2010. doi: 10.1038/cr.2010.46. [DOI] [PubMed] [Google Scholar]
- 58.Brenmoehl J, Hoeflich A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 13: 755–761, 2013. doi: 10.1016/j.mito.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 59.Chen M, Zhang L. Epigenetic mechanisms in developmental programming of adult disease. Drug Discov Today 16: 1007–1018, 2011. doi: 10.1016/j.drudis.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen X, Zhang L, Wang C. Prenatal hypoxia-induced epigenomic and transcriptomic reprogramming in rat fetal and adult offspring hearts. Sci Data 6: 238, 2019. doi: 10.1038/s41597-019-0253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, Meyer-Steenbuck M, Cenkerova K, Hoffmann MM, Jaeger C, Odening KE, Kammerer B, Hein L, Bode C, Bugger H. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol 110: 36, 2015. doi: 10.1007/s00395-015-0493-6. [DOI] [PubMed] [Google Scholar]
- 62.Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKC{epsilon} gene repression in rat heart. Circ Res 107: 365–373, 2010. doi: 10.1161/CIRCRESAHA.110.221259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Y, Xing J, Li Y, Luo Q, Su Z, Zhang X, Zhang H. Chronic hypoxia-induced Cirbp hypermethylation attenuates hypothermic cardioprotection via down regulation of ubiquinone biosynthesis. Sci Transl Med 11: eaat8406, 2019. doi: 10.1126/scitranslmed.aat8406. [DOI] [PubMed] [Google Scholar]
- 64.Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med 43: 1023–1036, 2007. doi: 10.1016/j.freeradbiomed.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dearden L, Bouret SG, Ozanne SE. Sex and gender differences in developmental programming of metabolism. Mol Metab 15: 8–19, 2018. doi: 10.1016/j.molmet.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scialò F, Fernández-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol 8: 428, 2017. doi: 10.3389/fphys.2017.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson LP, Al-Hasan Y. Impact of oxidative stress in fetal programming. J Pregnancy 2012: 582748, 2012. doi: 10.1155/2012/582748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evans LC, Liu H, Pinkas GA, Thompson LP. Chronic hypoxia increases peroxynitrite, MMP9 expression, and collagen accumulation in fetal guinea pig hearts. Pediatr Res 71: 25–31, 2012. doi: 10.1038/pr.2011.10. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, Petersen DR, Schenker S, Henderson GI. Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcohol Clin Exp Res 24: 544–552, 2000. [PubMed] [Google Scholar]
- 71.Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J Biol Chem 278: 37223–37230, 2003. doi: 10.1074/jbc.M305694200. [DOI] [PubMed] [Google Scholar]
- 72.Kane AD, Herrera EA, Camm EJ, Giussani DA. Vitamin C prevents intrauterine programming of in vivo cardiovascular dysfunction in the rat. Circ J 77: 2604–2611, 2013. doi: 10.1253/circj.cj-13-0311. [DOI] [PubMed] [Google Scholar]
- 73.Botting KJ, Skeffington KL, Niu Y, Allison BJ, Brain KL, Itani N, Beck C, Logan A, Murray AJ, Murphy MP, Giussani DA. Translatable mitochondria-targeted protection against programmed cardiovascular dysfunction. Sci Adv 6: eabb1929, 2020. doi: 10.1126/sciadv.abb1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spiroski A-M, Niu Y, Nicholas LM, Austin-Williams S, Camm EJ, Sutherland MR, Ashmore TJ, Skeffington KL, Logan A, Ozanne SE, Murphy MP, Giussani DA. Mitochondria antioxidant protection against cardiovascular dysfunction programmed by early-onset gestational hypoxia. FASEB J 35: e21446, 2021. doi: 10.1096/fj.202002705R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aljunaidy MM, Morton JS, Kirschenman R, Phillips T, Case CP, Cooke C-LM, Davidge ST. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol Res 134: 332–342, 2018. doi: 10.1016/j.phrs.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 76.Ganguly E, Aljunaidy MM, Kirschenman R, Spaans F, Morton JS, Phillips TEJ, Case CP, Cooke C-LM, Davidge ST. Sex-specific effects of nanoparticle-encapsulated MitoQ (nMitoQ) delivery to the placenta in a rat model of fetal hypoxia. Front Physiol 10: 562, 2019. doi: 10.3389/fphys.2019.00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao Q, Sun Q, Zhou L, Liu K, Jiao K. Complex regulation of mitochondrial function during cardiac development. J Am Heart Assoc 8: e012731, 2019. doi: 10.1161/JAHA.119.012731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khalifa ARM, Abdel-Rahman EA, Mahmoud AM, Ali MH, Noureldin M, Saber SH, Mohsen M, Ali SS. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol Rep 5: e13125, 2017. doi: 10.14814/phy2.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Demarest TG, McCarthy MM. Sex differences in mitochondrial (dys)function: implications for neuroprotection. J Bioenerg Biomembr 47: 173–188, 2015. doi: 10.1007/s10863-014-9683-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of cardiovascular disease. Gend Med 5, Suppl A: S121–S132, 2008. doi: 10.1016/j.genm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen M, Xiong F, Zhang L. Promoter methylation of Egr-1 site contributes to fetal hypoxia-mediated PKCε gene repression in the developing heart. Am J Physiol Regul Integr Comp Physiol 304: R683–R689, 2013. doi: 10.1152/ajpregu.00461.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parodi-Rullán RM, Chapa-Dubocq XR, Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front Physiol 9: 1094, 2018. doi: 10.3389/fphys.2018.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250, 2013. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dorn GW 2nd. Evolving concepts of mitochondrial dynamics. Annu Rev Physiol 81: 1–17, 2019. doi: 10.1146/annurev-physiol-020518-114358. [DOI] [PubMed] [Google Scholar]
- 85.Wilding JR, Joubert F, de Araujo C, Fortin D, Novotova M, Veksler V, Ventura-Clapier R, Wilding JR. Altered energy transfer from mitochondria to sarcoplasmic reticulum after cytoarchitectural perturbations in mice hearts. J. Physiol 575: 191–200, 2006. doi: 10.1113/jphysiol.2006.114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Papanicolaou KN, Ngoh GA, Dabkowski ER, O'Connell KA, Ribeiro RF Jr, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol 302: H167–H179, 2012. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278: 7743–7746, 2003. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 88.Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet 14: R283–R289, 2005. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 89.Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V, Cortopassi G, Chiamvimonvat N, Bers DM, Votruba M, Knowlton AA. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc 1: e003012, 2012. doi: 10.1161/JAHA.112.003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci 120: 838–848, 2007. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 91.Kowaltowski AJ, Menezes-Filho SL, Assali EA, Gonçalves IG, Cabral-Costa JV, Abreu P, Miller N, Nolasco P, Laurindo FRM, Bruni-Cardoso A, Shirihai OS. Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. FASEB J 33: 13176–13188, 2019. doi: 10.1096/fj.201901136R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parra V, Verdejo H, del Campo A, Pennanen C, Kuzmicic J, Iglewski M, Hill JA, Rothermel BA, Lavandero S. The complex interplay between mitochondrial dynamics and cardiac metabolism. J Bioenerg Biomembr 43: 47–51, 2011. doi: 10.1007/s10863-011-9332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B, Alexander C. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215, 2000. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 94.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189, 2006. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 95.Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabò R, Costa V, Civiletto G, Pesce P, Viscomi C, Zeviani M, Lisa FD, Mongillo M, Sandri M, Scorrano L. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21: 834–844, 2015. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Shackelford DB, Liesa M, Shirihai OS. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J 38: e101056, 2019. doi: 10.15252/embj.2018101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, Bergeron R, Cregan SP, Harper M-E, Park DS, Slack RS. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun 5: 3550, 2014. doi: 10.1038/ncomms4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci 41: 261–273, 2016. doi: 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 99.Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci 117: 4389–4400, 2004. doi: 10.1242/jcs.01299. [DOI] [PubMed] [Google Scholar]
- 100.Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacín M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 14: 1405–1415, 2005. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 101.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241–1252, 2006. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 102.Liesa M, Palacín M, Zorzano A, Liesa M. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 103.Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the “gluc” back in glucocorticoids. Nat Rev Endocrinol 10: 303–310, 2014. doi: 10.1038/nrendo.2014.22. [DOI] [PubMed] [Google Scholar]