

Summary
Trajectories of cognitive decline vary considerably among individuals with mild cognitive impairment (MCI). To address this heterogeneity, subtyping approaches have been developed, with the objective of identifying more homogeneous subgroups. To date, subtyping of MCI has been based primarily on cognitive measures, often resulting in indistinct boundaries between subgroups and limited validity. Here, we introduce a subtyping method for MCI based solely upon brain atrophy. We train a deep learning model to differentiate between Alzheimer’s disease (AD) and cognitively normal (CN) subjects based on whole-brain MRI features. We then deploy the trained model to classify MCI subjects based on whole-brain gray matter resemblance to AD-like or CN-like patterns. We subsequently validate the subtyping approach using cognitive, clinical, fluid biomarker, and molecular imaging data. Overall, the results suggest that atrophy patterns in MCI are sufficiently heterogeneous and can thus be used to subtype individuals into biologically and clinically meaningful subgroups.
Keywords: mild cognitive impairment, Alzheimer’s disease, subtyping, deep learning, neurodegeneration, brain atrophy
Graphical abstract
Highlights
-
•
Individuals with mild cognitive impairment (MCI) are subtyped using a deep learning model
-
•
The model is able to subtype MCI based solely on structural brain atrophy patterns
-
•
The model-based subtypes differ in amyloid burden, brain metabolism, and cognition
-
•
The model-based subtyping approach captures marked differences in cognitive decline
Rates of cognitive decline vary considerably among individuals with mild cognitive impairment (MCI). Kwak et al. develop a deep learning model based solely on brain atrophy patterns. This model divides individuals with MCI into distinct subtypes, showing differences in amyloid burden, brain metabolism, and cognition.
Introduction
Mild cognitive impairment (MCI) is often construed as the transitional stage between normal aging-related cognitive decline and Alzheimer’s disease (AD).1,2 However, MCI is associated with marked etiological heterogeneity.3 While the yearly risk of progression from MCI to AD is set at around 10%–12%,4, 5, 6 not all individuals with MCI eventually progress to AD and many demonstrate different outcomes, including the development of non-AD dementia or other neuropsychiatric conditions,7,8 or reversion to cognitively normal (CN) status.9 Despite its ubiquity, the heterogeneity of MCI with respect to rates of cognitive decline and progression to AD remains poorly understood, challenging further progress in research and care.
Attempts to constrain the heterogeneity of MCI via subtyping approaches have been proposed by several groups,10, 11, 12 almost exclusively relying on cognitive subtyping, that is, classification into subtypes, which is based on subjects’ performance in cognitive tests and tasks.11,13 For example, a common subtyping framework defines individuals with MCI as amnestic and non-amnestic, depending on whether or not memory loss is a predominant feature.11 Other common subtyping approaches further classified MCI as being either single or multiple domain as a function of the number of cognitive domains where decline is observed.14 More recently, studies have shown that a more comprehensive subtyping for MCI can be achieved based on similarities in neuropsychological test scores using clustering techniques.15,16
While cognitive subtyping has been instrumental in delineating the various dimensions of cognitive performance that are affected in MCI, cognitive subtypes show heterogeneity in their longitudinal trajectories and clinical outcoms17,18 and may not be sufficiently distinct from one another18 in capturing rates of cognitive decline and progression to AD. In recent years, a shift in the definition of preclinical, prodromal, and clinical AD from a syndromic to a biological construct has been suggested.19 Indeed, the recently proposed “AT(N)” framework for AD research20,21 attempts to provides accurate, biologically centered definitions for AD research based on multi-domain biomarkers for β-amyloid deposition (“A”), pathologic tau (“T”), and neurodegeneration (“N”). Biomarkers for neurodegeneration, however, particularly those based on structural MRI, are not specific to AD, and alterations in their levels may be attributable to various other comorbidities.19 Consequently, the degree to which heterogeneity in atrophy patterns in MCI can be leveraged to subtype individuals into homogeneous subgroups, showing similar clinical outcomes, as was done in AD,22, 23, 24, 25 remains unclear.
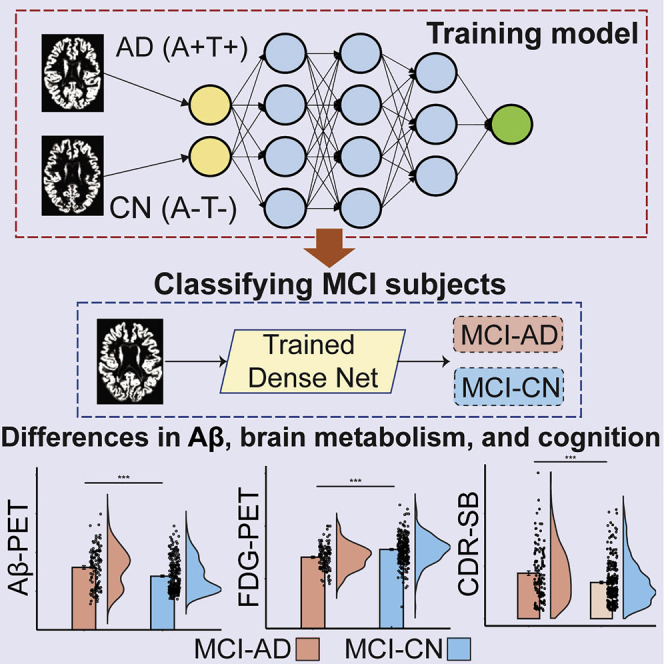
In the current study, we tested whether patterns of brain atrophy, derived from MRI, are sufficient in allowing for the subtyping of MCI into biologically and clinically distinct subgroups. We propose and validate a novel data-driven subtyping approach based upon deep learning. We first train a dense convolutional neuronal network (CNN) to differentiate between A+T+ AD patients (i.e., AD patients with pathologic changes) and A−T− CN subjects (i.e., CN subjects with no pathologic changes) based on whole-brain gray matter (GM) morphometric data. We then deploy the trained CNN to classify MCI subjects into two subgroups, corresponding to MCI subjects with AD-like and CN-like morphometric characteristics,26 respectively. We also identified the major regional atrophy patterns contributing to the differentiation between the two MCI subgroups through occlusion analysis.27 The resulting labels were then validated against cerebrospinal fluid (CSF) biomarkers for β-amyloid (Aβ) and tau, baseline fluorodeoxyglucose (FDG) and Aβ-positron emission tomography (PET), as well as baseline and longitudinal cognitive scores. Finally, we evaluated the degree of overlap between the modeling-based labels and those obtained through cognitive subtyping.
Results
Participant characteristics
We analyzed data from 489 subjects, obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. An additional sample of 78 MCI subjects was obtained from the Open Access Series of Imaging Studies-3 (OASIS-3) database28 and used as an independent validation cohort. Our proposed modeling approach (see STAR Methods) utilized AD and CN data for training, and MCI data for testing. Following the proposed NIA-AA guidelines,19 AD subjects were included in the analysis if they displayed abnormal CSF Aβ42 and p-tau181 levels (denoted henceforth as A+T+). CN subjects were included in the analysis only if they displayed normal CSF Aβ42 and p-tau181 levels (henceforth, A−T−). The demographic characteristics of subjects in all groups are shown in Table 1. When comparing the AD, CN, and MCI groups (Table S2), there were significant difference in Clinical Dementia Rating Sum of Boxes (CDR-SB: F2,596 = 458.2, p < 0.001), Alzheimer’s Disease Assessment Scale (ADAS: F2,596 = 177.90, p < 0.001), and CSF biomarker concentrations (A42: F2,596 = 146.45, p < 0.001, p-tau181: F2,596 = 86.08, p < 0.001). Post hoc comparisons with the Tukey test revealed significant pairwise difference (all p’s < 0.001) between the three groups in all cognitive measures and CSF biomarker concentrations. No significant differences were observed between the groups in age (F2,596 = 2.16, p = 0.12) or gender distribution ( = 3.06, p = 0.22).
Table 1.
Demographics
| ADNI |
|||
|---|---|---|---|
| AD (A+T+) | CN (A−T−) | MCI | |
| n | 110 | 109 | 380 |
| Age | 72.91 (8.03) | 70.98 (5.32) | 72.15 (7.19) |
| Gender, female | 46 (41.82%) | 57 (52.29%) | 166 (43.68%) |
| ADAS | 21.97 (7.03) | 6.97 (3.05) | 9.64 (4.43) |
| CDR-SB | 4.58 (1.72) | 0.0 (0.07) | 1.49 (0.89) |
| MMSE | 21.97 (7.03) | 29.09 (1.06) | 27.98 (1.74) |
| A 42 (pg/mL) | 597.79 (158.44) | 1454.75 (247.64) | 987.22 (425.97) |
| p-tau181 (pg/mL) | 40.41 (14.97) | 16.4 (2.82) | 27.59 (14.91) |
Continuous variables are presented as means, with SDs and categorical variables are presented as percentages in parentheses. AD, Alzheimer’s disease; CN, cognitively normal; MCI, mild cognitive impairment; N, number of subjects; ADAS, Alzheimer’s disease assessment scale; CDR-SB, clinical dementia rating sum of boxes; MMSE, mini-mental state exam; A 42, beta-amyloid 42; p-tau181, phosphorylated-tau 181; SD, standard deviation.
A deep learning model for subtyping MCI subjects
Our major objective in the current study was to develop a deep learning modeling framework for subtyping MCI. To that end, we utilized a dense CNN architecture (Figure 1),29 which relies on whole-brain GM density as input data. The model was first trained to differentiate AD and CN subjects, with the assumption that these two distinct groups would provide the model with an adequate distribution of morphometric features sufficient for subtyping MCI subjects. Data augmentation was applied within the training data to increase its size and improve the performance of the model and its generalizability. As a result, a total of 14,016 images were generated for training the model. We used 5-fold cross-validation within the training set to optimize and fine-tune the model’s performance, finding similar performance across the different folds (Figure S1A). The model with the best performance achieved maximal accuracy of 93.75%, with an area under the curve (AUC) of the receiver operating characteristic (ROC) of 0.983 (Figure S1A). The model achieved better performance than other standard machine learning classification models including support vector machine, random forest, and logistic regression (Figure S1B). We thus subsequently deployed the model on the MCI data (Figure 1), formalized as a binary classification problem with class labels AD and CN. Then, output values less than the default threshold of 0.5 were assigned to the class MCI-CN, and values greater than or equal to 0.5 were assigned to the class MCI-AD. We tested whether the threshold of 0.5 was appropriate, given the distribution of the model’s output, finding that in both the training and testing datasets this threshold value was at a location in the distribution with little variance around it (Figure S1C). We additionally retrained the model with T1 intensity values, replacing GM density. AUC of the ROC was similar to that obtained when using GM density, while the accuracy of the model was lower (Figure S1D).
Figure 1.

Study methods
Illustration of proposed deep learning framework. A dense convolutional neural network is trained to differentiate patients with Alzheimer disease (AD) and cognitively normal (CN) controls based on whole-brain GM morphometric data. Subsequently, the trained model is deployed to classify individuals with mild cognitive impairment (MCI), into two groups, MCI-AD and MCI-CN, based on structural morphometric data. AD, Alzheimer’s disease; CN, cognitively normal; MCI, mild cognitive impairment; GM, gray matter.
Validation of model-based MCI subgroups with CSF biomarker concentrations and cognitive scores
We investigated 380 MCI subjects (57.4% were CSF Aβ42 positive; 56.6% were p-tau181 positive). Our data-driven approach for subtyping MCI resulted in two subgroups, MCI-AD and MCI-CN. We next assessed the validity of these two data-driven labels. We first compared the prevalence of the various biomarker profiles19 in each of the subgroups, based on each subject’s CSF Aβ42 and p-tau181 biomarkers. Each subject was rated as either positive (i.e., abnormal) or negative (i.e., normal) in each biomarker, based on previously published cutoff values19 (see Figure S2). This resulted in four different profiles (A+T+, A+T−, A−T+, and A−T−, where “A” denotes Aβ, and “T” denotes tau) (Figures 2A and 2B). The prevalence of the biomarker profiles differed significantly between the MCI-AD and MCI-CN subgroups ( = 21.40, p < 0.001). In particular, subjects with an abnormal CSF Aβ42 or p-tau181 were more common in the MCI-AD (CSF Aβ42: 71.6%; p-tau181: 69.3%) than in the MCI-CN group (CSF Aβ42: 50.6%; p-tau181: 50.2%). Subjects with an abnormal CSF Aβ42 and p-tau181 were more prominent in the MCI-AD (A+T+: 55.1%) than in the MCI-CN group (A+T+: 32.8%). In contrast, subjects with a normal CSF Aβ42 and p-tau181 were more common in the MCI-CN (A−T−: 32.0%) than in MCI-AD group (A−T−: 14.2%).
Figure 2.

Validation of the MCI subgroups using fluid biomarker and cognitive data
Subjects in the MCI-AD and MCI-CN groups were rated as Amyloid (A) and p-tau (T), positive or negative, based on the CSF A 42, and p-tau181 biomarkers. Pie charts depict the biomarker score combinations in the MCI-AD (A) and MCI-CN (B) subgroups. These score distributions were significantly different between the two subgroups. Bar plots show differences in CDR-SB scores (C), CSF A 42 (D), and CSF p-tau181 (E). Bar graphs depict means SEs. AD, Alzheimer’s disease; CN, cognitively normal; MCI, mild cognitive impairment; CDR-SB, clinical dementia rating sum of boxes. ∗∗∗p < 0.001.
We then investigated baseline differences in demographic characteristics, cognitive scores, and continuous CSF concentrations between the MCI-AD and MCI-CN subgroups. There was a significant difference between the two subgroups in age (t378 = 6.44, p < 0.001), but not in gender distribution ( = 3.49, p = 0.06). Comparing cognitive scores between the MCI-AD and MCI-CN subgroups reveled significant differences in CDR-SB (t378 = 3.46, p < 0.001; Figure 2C) and ADAS (t378 = 6.51, p < 0.001; see Figure S3A) scores. All significant results were retained when controlling for age (all p < 0.01). The comparison of CSF concentrations between the MCI-AD and MCI-CN subgroups revealed significant differences in CSF Aβ42 (t378 = 4.55, p < 0.001; Figure 2D) and CSF p-tau181 (t378 = 3.81, p < 0.001; Figure 2E). The significant results were retained when controlling for age (both p < 0.001). Moreover, group differences between the MCI-CN and MCI-AD groups were retained when varying the threshold used for binary classification (Figures S3B–S3J), again suggesting that the specific value used for classification had little effect on the reported results.
Comparison of PET uptake in subtyped MCI
We next assessed group differences in PET uptake, focusing on Aβ- and FDG-PET. While Aβ-PET and CSF measures of Aβ are generally properly correlated with one another, a certain degree of discordance between the two markers has been consistently reported.30,31 Our analysis above revealed that the MCI-AD and MCI-CN subgroups differed in CSF Aβ42 levels. We thus next aimed to complement this analysis by also comparing the subgroups across Aβ-PET. There were significant differences between the two subgroups in Aβ-PET (t377 = 4.36, p < 0.001; Figure 3A), with lower levels observed in the MCI-AD group, relative to the MCI-CN group. We have additionally examined group differences in FDG-PET uptake. Metabolic imaging studies utilizing FDG-PET for AD diagnosis are common,32 and this modality has been proposed as a reliable and valid marker for neurodegeneration in AD.19 We observed significant group differences in FDG-PET (t377 = 5.53, p < 0.001; Figure 3C), with lower uptake values obtained in the MCI-AD group. Group differences in both Aβ-PET and FDG-PET were retained after adjusting for the effect of age. We have additionally assessed group differences in PET uptake, comparing normal/abnormal uptake values after binarizing the data with established cutoff values.33,34 The prevalence of normal and abnormal Aβ-PET and FDG-PET differed between the two subtyped subgroups (Figures 3B and 3D). These differences were apparent in both Aβ-PET and FDG-PET (Aβ-PET: = 12.35, p < 0.001; FDG-PET: = 14.70, p < 0.001). In particular, there were more subjects with abnormal Aβ-PET in the MCI-AD (Aβ-PET+: 71.4%) than in MCI-CN group (Aβ-PET+: 52.6%). Similarly, abnormal FDG-PET was more common in the MCI-AD (FDG-PET+: 48.4%) than in the MCI-CN group (FDG-PET+: 28.5%).
Figure 3.

Comparison of the MCI subgroups using PET uptake data
Bar plots show the comparison of A -PET (A) and FDG-PET (C) uptake between the MCI-AD and MCI-CN subgroups. In both measurements, group differences were statistically significant. Pie charts depict the proportion of subjects in the MCI-AD and MCI-CN subgroups with normal and abnormal A -PET (B) and FDG-PET (D). The distributions of normal and abnormal PET uptake values were significantly different between the two subgroups (Aβ-PET: = 12.35, p < 0.001; FDG-PET: = 14.70, p < 0.001). Bar graphs depict means SEs. AD, Alzheimer’s disease; CN, cognitively normal; MCI, mild cognitive impairment; A -PET, beta-amyloid positron emission tomography; FDG-PET, fluorodeoxyglucose-positron emission tomography. ∗∗∗p < 0.001
Contribution of regional atrophy to MCI subtyping: Occlusion analysis
The results suggest that patterns of whole-brain GM are sufficient for differentiating MCI subjects into two distinct subgroups. As our approach utilizes whole-brain GM features, the major regional contributors to the model’s output remain unclear. We thus next examined the relative lobar (frontal, parietal, medial temporal, lateral temporal, occipital, and cingulate) contribution to the performance of the model, through occlusion analysis, as proposed previously.27 Briefly, we retested the deep learning model iteratively, occluding a bilateral binary mask composed of each lobe from the model’s test-set input data (Figure 4A). This was achieved by setting the intensity values of each lobe to zero on each iteration. The percentages of change in the model’s output, with respect to the original results for classifying MCI subgroups were ranked and then compared across the different occluded lobes (see Figure 4B). We found that the occlusion of the medial temporal and lateral temporal lobes led to dramatic changes in the model’s output (Figure 4C), relative to the original results. On the other hand, occlusion of the occipital and cingulate lobes had relatively little effect, resulting in model output that resembled the original results (Figure 4C). Thus, the medial and lateral temporal lobes had the largest impact on the performance of the model and on the classification of MCI subjects into two distinct subgroups.
Figure 4.

Identifying the major contributors to atrophy-centered subtyping of MCI via occlusion analysis
(A) Schematic illustration of the occlusion analysis. The testing phase in the deep learning model was repeated, whereby in each step cortical lobes were occluded from the input data (temporal lobes were further divided to medial and lateral). The percentages of change with respect to the original results for classifying MCI subgroups were ranked.
(B) The results of the occlusion analysis are shown, in each tested lobe. Shown are percentages of change with respect to the original intact model.
(C) Percentage of change following occlusion analysis, in each cortical lobe, superimposed on medial and lateral cortical surface models.
MCI, mild cognitive impairment.
Longitudinal analysis of cognitive changes in the MCI subgroups
Our analysis reveals marked baseline differences between the MCI-AD and MCI-CN subgroups. These observed differences, nevertheless, cannot be taken to imply that the two MCI subgroups also differ in their prognostic outcomes. We next set out to evaluate whether the MCI-AD and MCI-CN subgroups also exhibit differences in the progression to AD and in longitudinal cognitive performance. This analysis focused on subjects with data from at least three follow-up visits. Individual trajectories of longitudinal changes in cognitive performance varied between the two MCI subgroups (Figures 5A–5D). Survival analysis35 revealed marked differences between the two subgroups in their progression to AD (log-rank test; = 64.40, p < 0.001), with the MCI-AD subgroup showing faster progression, relative to the MCI-CN subgroup (Figure 5E). We next used repeated-measures analysis of variance (RM-ANOVA) to compare changes in cognitive performance from baseline to the second-year follow-up visit, with group (MCI-AD, MCI-CN) as the between-subjects factor and time (baseline, follow-up) as the within-subject, repeated-measure factor. Focusing on CDR-SB scores (Figure 5F), this analysis revealed a significant interaction (group time; F1,358 = 14.92, p < 0.001), along with significant main effects for group (F1,358 = 27.87, p < 0.001) and time (F1,358 = 38.91, p < 0.001). Thus, the MCI-AD group showed more pronounced changes in CDR-SB scores between the testing sessions. Similarly, in the analysis of ADAS scores (Figure 5G), a significant interaction (group time; F1,358 = 7.56, p < 0.001) was observed, along with significant main effects for group (F1,358 = 5.92, p = 0.02), and time (F1,358 = 87.12, p < 0.001).
Figure 5.

Longitudinal comparison of the MCI subgroups
(A–D) The plots show longitudinal changes in cognition and clinical rating scores, observed at baseline and at the second-year follow-up. Longitudinal changes are shown for CDR-SB scores (A and B) and ADAS scores (C and D).
(E) Kaplan-Meier plots depicting disease-free survival in the MCI-AD and MCI-CN subgroups. The MCI-CN group showed significantly better disease-free survival over time (log-rank test). Shaded areas depict confidence intervals. Longitudinal changes in CDR-SB scores (F) and ADAS scores (G), displayed by the two MCI subgroups, tested with a RM-ANOVA. MCI, mild cognitive impairment; CDR-SB, clinical dementia rating sum of boxes; ADAS, Alzheimer’s disease assessment scale. RM-ANOVA, repeated-measures analysis of variance.
Validation in an independent cohort
We next validated our subtyping approach with data from the OASIS-3 dataset (Table S1). The MCI samples in the ADNI and OASIS-3 datasets did not differ in age (t = 1.4, p = 0.16) or gender distribution ( = 0.04, p = 0.84). We first identified MCI-AD and MCI-CN subjects in the OASIS-3 sample, using the same approach described above. Survival analysis showed that, similar to the original test data, there was a significant difference between the subgroups in their progression to AD (log-rank test; = 22.6, p < 0.001), with the MCI-AD subgroup showing faster progression over time, compared to the MCI-CN group (Figure S4A). Similar to the original testing data, the MCI-AD and MCI-CN subgroups also showed longitudinal differences in cognitive performance (mini-mental state exam [MMSE] scores) observed over a period of 2 years (Figure S4B). Namely, a significant interaction effect was observed between group and time in the validation cohort (F1,76 = 7.34, p < 0.01), along with significant main effects for group (F1,76 = 10.97, p < 0.005) and time (F1,76 = 7.44, p < 0.01).
Different atrophy levels in the MCI subgroups
One possibility that emerges from the findings reported above is that subjects in the MCI-AD and MCI-CN subgroups merely differ in levels of atrophy since they are at different phases of AD. However, a closer comparison of longitudinal data from the two subgroups also reveals marked differences in the rate of atrophy changes they display. Namely, we calculated the annualized rate of hippocampal atrophy, a common marker of neurodegeneration,36 expressed in percentages of volume loss relative to baseline.37 The annual rate of hippocampal atrophy was significantly (p < 0.001) reduced in the MCI-AD (annual rate: 3.77 2.98%) than in the MCI-CN group (annual rate: 1.72 2.67%), while no significant differences (p = 0.50) were observed between MCI-CN and CN subjects (annual rate: 1.52 2.22%).
Concordance between the proposed MCI subtyping approach and cognitive subtyping
The data-driven approach proposed here subtypes MCI subjects solely based on patterns of whole-brain GM volume. As the vast majority of existing subtyping approaches for MCI are based on cognitive profiles,18 we wished to determine the extent of concordance between the current MCI subtyping approach and cognitive subtyping. We focused the comparison on a recently introduced subtyping method where neuropsychological assessments are clustered into distinct subgroups.15 We first clustered subjects (n = 374 subjects who had available data) based on their neuropsychological assessments (Table S3). This resulted in four subgroups: dysnomic (37.43% of subjects), amnestic MCI (36.63%), dysexecutive (6.95%), and cluster-derived normal (18.98%) (Figure 6A). When testing the prediction of disease-free survival observed when using this subtyping approach, we found a large degree of overlap among three of the four subtypes (Figure S5A). The four subgroups showed significant group differences in the six neuropsychological assessments used for clustering (p < 0.001). Performance in none of the cognitive domain scores showed a better predictive performance than that of our model (Figure S5B). Post hoc comparisons with the Tukey multiple comparison test revealed that the dysnomic group performed worse than all other groups in five of six measures of language. The dysexecutive group performed worse than all other groups in assessments of attention/executive function, and the amnestic MCI group performed worse than the dysnomic and cluster-derived normal groups in assessments of memory function. Thus, the clustering of neuropsychological assessments resulted in distinct MCI subtypes, as previously reported.15 We next compared the distributions of the neuropsychological subtypes within the MCI-AD and MCI-CN subgroups. We found significant differences between the two distributions ( = 30.45, p < 0.001), observing more dysnomic and dysexecutive subjects in the MCI-AD (60%) than in the MCI-CN group (36.5%). On the other hand, as expected, the cluster-derived normal profile, which shows the normal range of scores across all neuropsychological measures, was more common in the MCI-CN (24.9%) than in the MCI-AD group (7.2%). Interestingly, the prevalence of the amnestic MCI profile was similar between the MCI-AD (32.8%) and MCI-CN subgroups (38.6%). Altogether, while both the MCI-AD and MCI-CN subgroups displayed impairments in memory function, the former subgroup displayed more significant deficits in attention/executive and language function than the latter subgroup.
Figure 6.

Overlap between atrophy-centered and neuropsychological subtypes
(A) Hierarchical clustering on neuropsychological data was used to define four MCI subtypes within the dataset used here (dysnomic MCI, aMCI, dysexecutive MCI, and cluster-derived normal).
(B and C) Pie charts show the prevalence of these subtypes in the MCI-AD (B) and MCI-CN (C) subgroups. The two distributions were significantly different. AD, Alzheimer’s disease; CN, cognitively normal; MCI, mild cognitive impairment; aMCI, amnestic mild cognitive impairment. ∗∗∗p < 0.001.
Discussion
Subtyping approaches for MCI have been proposed as a remedy for the large etiological heterogeneity characteristic of this elusive stage in cognitive aging. Current subtyping approaches primarily rely on neuropsychological profiles and may often result in blurred boundaries between subgroups and limited validity.18 Here, we propose a novel data-driven subtyping approach, which utilizes CNNs to divide MCI subjects into subgroups based on the extent to which their brain atrophy patterns resemble those observed in AD as opposed to CN subjects. This approach resulted in two subgroups, MCI-AD and MCI-CN, denoting closer correspondence in GM patterns with data from AD and CN subjects, respectively. We then comprehensibly validated the model-based subgroups, findings marked group differences in baseline CSF biomarker concentrations and PET uptake, along with annual rate of hippocampal volume loss, and baseline and longitudinal cognitive performance scores. The latter differences were also validated using an independent dataset. Through occlusion analysis,27 we also investigated lobar contributions to the performance of the deep learning model, reporting that it mostly relied on GM volume from the medial and lateral temporal lobes. Finally, we found a limited degree of overlap between the current subtyping approach and that based on neuropsychological examination.
The purpose of the current study was to test whether distinct subgroups with differing structural brain atrophy patterns could be delineated within a heterogeneous clinical sample of individuals diagnosed with MCI. Rather than providing an AT(N)-like subtyping solution for MCI, our goal was to investigate whether the heterogeneity of MCI as it pertains to rates of cognitive decline and progression to AD can be captured using an imaging biomarker of neurodegeneration. To that end, we utilized a deep learning framework, rather than other machine learning models, such as support vector machine, primarily since there has been a growing body of research demonstrating the utility of deep learning models based on MRI-derived features in various tasks, such as diagnostic prediction,38 image reconstruction39 and segmentation,40 and prognostic prediction of disease progression.41 Our choice to utilize a deep learning framework was further motivated by the assumption that complex and non-linear relationships exist between whole-brain structure and progression of MCI/AD. Our chosen model indeed outperformed other standard machine learning models in the task of differentiating data from AD and CN subjects. Similar to other machine and deep learning models where “transfer learning”42 is applied, we propose a classification framework that is trained on a domain different than the one being tested.43, 44, 45 However, rather than evaluating the performance of the model against clinically defined labels (e.g., progressive and stable MCI, or AD convertors and non-convertors), our approach was to re-label data from individuals with MCI based on its proximity to the model’s trained labels, that is, AD and CN.
We found robust differences between the MCI-AD and MCI-CN subgroups in CSF biomarker concentrations, cognitive scores, annual rate of hippocampal atrophy, and PET uptake, suggesting that our data-driven method subtypes MCI into biologically and clinically distinct subgroups. Moreover, the prevalence of biomarker profiles, defined based on established cutoff thresholds for the CSF Aβ42 and p-tau181 biomarkers differed significantly between the MCI-AD and MCI-CN subgroups. Namely, abnormal CSF Aβ42 (A+) and p-tau181 (T+) were more prevalent in the MCI-AD group. These findings are consistent with earlier studies where it was shown that positive CSF biomarker concentrations can predict conversion from MCI to AD with accuracy larger than 80%.46,47 We additionally found more pronounced cognitive impairment, as assessed with the CDR-SB and ADAS scores, in the MCI-AD subgroup, relative to the MCI-CN subgroup. The two subtyped subgroups also exhibited marked differences in Aβ-PET, a marker of amyloid deposition, and FDG-PET, a commonly used marker of neurodegeneration.19 The topographical distribution of Aβ deposition, assessed with PET is predicative of progression of individuals with MCI to AD,48 with abnormalities appearing long before the onset of clinical symptoms.49 The Aβ-PET abnormalities observed in the MCI-AD subgroup, where longitudinal cognitive outcomes were poorer and progression to AD was quicker, are consistent with these findings. Similarly, the finding that negative FDG-PET was more predominant in the MCI-CN subgroup is consistent with the observation that negative FDG-PET is highly predictive of clinically stable MCI.50
The recently proposed AT(N) framework20,21 provides biologically centered definitions for AD research based on biomarkers for Aβ, tau, and neurodegeneration. Unlike the latter two, however, biomarkers for neurodegeneration lack specificity, often showing overlap with other comorbidities.19 Our results show that patterns of brain atrophy are sufficiently heterogeneous to allow for the subtyping of MCI into biologically and clinically distinct subgroups. Indeed, when considering prediction of progression from MCI to AD, our analysis revealed a marked distinction between the MCI subgroups, when using an atrophy-centered subtyping approach, relative to neuropsychological subtyping, which resulted in an overlap between several of the subtypes. Our findings are consistent with the observation that similarities in patterns of GM topography between subjects can be used as features in a predictive model of MCI progression.51 Our approach also joins other data-driven approaches that explored the heterogeneity of regional neurodegeneration patterns based on structural neuroimaging in AD and related dementias.52
We complemented the initial analysis, which relied on whole-brain GM patterns, with an analysis that combined deep learning classification with occlusion analysis.27 This allowed us to identify the major lobar contributors to the performance of the subtyping model. The results revealed that the medial temporal, lateral temporal, and to a lesser extent the parietal lobe were more central to the model’s performance than other lobes. These findings are consistent with earlier studies on AD pathology, where atrophy was reported in the hippocampus, amygdala, and entorhinal cortex.26,53,54 These findings are also consistent with recent findings on the possible neuroprotective role of redundancy in the hippocampus55 and other large-scale brain networks,56 which may contribute to the observed differences between the subgroups. Our results also suggest that the occipital lobe played a more minor role in the performance of the model, consistent with Braak’s staging scheme,57 where the occipital lobe is shown to be affected only at later stages of AD.58 Although our results highlight the central role of the medial and lateral temporal lobes in the subtyping of MCI, further examination into the possible involvement of other cortical and subcortical regions is warranted.
One question that arises is whether the MCI-AD and MCI-CN subgroups merely reflect different stages along the course of AD, or rather reveal distinct MCI subtypes. Our findings show that the two subgroups differed not only in their underlying patterns of neurodegeneration but rather also in biomarker composition (i.e., combination of A and T biomarkers), PET uptake, and current and subsequent clinical presentation. Moreover, we found that the two subgroups exhibited different annual rates of hippocampal atrophy,59 with the one found in the MCI-CN group being indistinguishable from that seen in CN subjects. Thus, while more data, in particular “omics” data,60 will be needed to determine whether the atrophy-centered subgroups we introduced here are indeed real biological subtypes, our finding suggest that the subgroups do not simply differ in their AD disease stage.
We examined the agreement between our atrophy-centered data-driven subtyping approach and that obtained based on neuropsychological assessments. The latter approach groups subjects based on the similarity of their neuropsychological assessments using clustering techniques, and thus goes beyond the more traditional amnestic/non-amnestic MCI subgrouping studied extensively in the literature.61 However, neuropsychological and biomarker profiles are strongly heterogeneous in subjects who can be classified as having amnestic MCI,62 and empirically derived subtyping approaches depict heterogeneity that is not captured by conventional criteria.15 In our analysis, we found that the cluster-derived normal group, where cognitive function is unimpaired, was more predominant in the MCI-CN than the MCI-AD subgroups (see Figure 6B). Moreover, while the amnestic MCI profile showed a similar distribution in the two subgroups, dysnomic or dysexecutive subtypes were primarily represented in the MCI-AD subgroup. Overall, our findings demonstrate that the matching between the two subtyping approaches is incomplete. Future research could attempt to combine the two approaches, to achieve neuropsychologically distinct subgroups, that show differing patterns of brain atrophy.
In conclusion, the current study demonstrates that patterns of GM atrophy are sufficient for subtyping MCI into biologically and clinically distinct subgroups. These results further highlight the need to consider the heterogeneity of MCI when attempting to understand the pathological mechanisms of dementia, while providing a potential tool for individualized disease prognosis.
Limitations of the study
Several limitations should be noted when considering the current results. First, in this proof-of-concept stage, we used atrophy patterns to subtype MCI into two distinct subgroups. We acknowledge that a larger number of subgroups would be needed to better capture the different pathways of progression from prodromal to clinical AD. In principle, our approach could be easily modified to output more than two subgroups; however, we believe that a larger number of MCI subjects than that used here, and/or the inclusion of data from multiple cohorts would be needed in order to achieve robust and generalizable results. This could hopefully be achieved in future research. Second, as also noted above, we did not attempt to combine neuropsychological and atrophy-centered features in the current study, primarily as our major motivation was to examine whether the latter type of features is sufficient in subtyping MCI. Future research could extend our approach to test whether a combination of cognitive and neurobiological features better captures the heterogeneity of MCI.
STAR★Methods
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Software and algorithms | ||
| Deep learning model for MCI subtypes | current paper | https://github.com/rlckd/MCI-subtype |
| R version 4.0.8 with the following packages: | R Core Team | https://www.R-project.org |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| ggplot2_3.3.3 | R Core Team | https://ggplot2.tidyverse.org/ |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| dplyr_1.0.7 | R Core Team | https://dplyr.tidyverse.org/ |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| tidyr_1.1.2 | R Core Team | https://github.com/atorus-research/Tplyr |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| WRS2_1.1.0 | R Core Team | https://github.com/cran/WRS2 |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| R.matlab_3.6.2 | R Core Team | https://github.com/HenrikBengtsson/R.matlab |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| tidyverse_1.3.0 | R Core Team | https://www.tidyverse.org/ |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| survival_3.1.12 | R Core Team | https://github.com/therneau/survival |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| survminer_0.4.8 | R Core Team | https://rpkgs.datanovia.com/survminer/ |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| ggforce_0.3.2 | R Core Team | https://ggforce.data-imaginist.com/ |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| ggseg_1.5.5 | R Core Team | https://github.com/ggseg/ggseg |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
| R_rainclouds.r | R Core Team | https://github.com/RainCloudPlots |
| R: A Language and Environment for Statistical Computing. | ||
| R Foundation for Statistical Computing, 2016 | ||
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Eran Dayan (eran_dayan@med.unc.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
In this study, we subtyped MCI subjects using a deep learning approach, based on patterns of brain structural atrophy, derived from MRI. We then validated the model’s output (i.e., subgroups) using CSF biomarker, PET and cognitive/clinical data. Specifically, we trained a dense CNN,29 to differentiate data from AD and CN subjects based on whole brain atrophy patterns. To provide the model with adequate well-defined training data, all AD subjects were A+ and T+, that is, they had abnormal levels of CSF A 42 and p-tau181. CN subjects were all A− and T−, in other words, they showed normal levels in the same CSF biomarkers. Previously determined cutoff values63 for abnormal Aβ42 (Aβ42 < 976.6pg/ml) and p-tau181 (p-tau181 > 21.8pg/ml) were used. We reasoned that training the model with well-differentiated AD (A+T+) and CN (A−T−) data would allow the model to learn a more discriminative set of pathological features. We then deployed the trained CNN to classify MCI subjects (n= 380), as either AD-like or CN-like. We subsequently validated the model’s output labels with baseline molecular and metabolic neuroimaging, cognitive scales and tests and CSF biomarkers, given strong associations between atrophy, tau, and amyloid burden and cognitive performance (e.g.,64,65). We additionally examined longitudinal changes in cognitive scores (between baseline and the 2nd year follow-up visit, which was the latest visit where imputation for missing data could be achieved reliably) and calculated disease-free survival in the MCI-AD and MCI-CN subgroups, to assess differences in the progression to AD. To test the generalizability of our subtyping model, we also investigated longitudinal changes in cognitive performance in an independent cohort (n= 78; OASIS-3 dataset), where MCI subtyping was applied as in the original dataset. Finally, we identified the major regions (lobes) contributing to the performance of the model through occlusion analysis27 (see below), and assessed the intersection between the modeling-based labels and those obtained through cognitive subtyping.
Subjects
Data used in the preparation of this study were obtained from the ADNI and OASIS-3 databases. The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial MRI, other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and early AD. For up-to-date information, see http://adni.loni.usc.edu/. The OASIS-3 dataset was used to test the validity of our subtyping model. OASIS-3 is a longitudinal neuroimaging, neuropsychological, clinical and biomarker dataset for normal aging and AD (https://www.oasis-brains.org/). The dataset includes participants enrolled through Washington University in St. Louis’ Knight Alzheimer Research Center. We used AD (n= 110) and CN (n= 109) data from ADNI to train the proposed deep learning model. Then, we subtyped MCI (n= 380) into subgroups, validating the model’s output with cognitive scores, CSF biomarker levels and PET uptake available in the same subjects. We excluded MCI subjects who reverted to CN status during follow-up. Additionally, one subject had no FDG-PET uptake data, while 20 subjects were excluded from the longitudinal analysis of cognitive performance since they had data in less than 3 testing time points. Missing data at the 2nd year follow-up was imputed using linear interpolation/extrapolation. Data from the OASIS-3 dataset included a sample of 78 subjects with MCI. MMSE scores were extracted for each of the subjects in this dataset. Linear interpolation/extrapolation was used based on at least 2 time points to estimate MMSE scores 2 years after baseline (and thus match the time frame considered for subjects in the ADNI dataset). All subjects provided written informed consent and the studies’ protocols were approved by the local Institutional Review Boards.
Method details
Image acquisition
Structural MRI data used in the deep learning model were acquired at ADNI sites using 3T scanners and were based on either an inversion recovery-fast spoiled gradient recalled (IR-SPGR) or magnetization-prepared rapid gradient-echo (MP-RAGE) sequences.66 Aβ-PET data were acquired in a 20-min dynamic emission scan, composed of four 5-min frames. The data was acquired 50–70 min after the injection of 10.0 mCi of [18F]-AV45. FDG-PET data were acquired in a 30-min dynamic emission scan, composed of six 5-min frames. The data was acquired 30–60 min after the injection of 5.0 mCi of [18F]-FDG. PET data ran through a strict quality control procedure to assess image quality. Standardized image preprocessing correction steps were applied to produce uniform data across the ADNI PET cores. These steps included frame co-registration, averaging across the dynamic range, and standardization with respect to orientation, voxel size, and intensity. Full details of the T1 and PET acquisition parameters and imaging processing steps are listed on the ADNI website (http://adni.loni.usc.edu/methods/). In the independent validation dataset (OASIS-3), structural MRI data were collected on 2 different Siemens 3T scanner, using MP-RAGE sequences (TR= 2.4ms; TE= 3.2ms; flip angle= 8 ; voxel size= 1 1 1 mm3).
CSF collection
CSF collection, shipping, aliquoting, storage as well as analysis followed ADNI’s standardized procedures (http://adni.loni.usc.edu/).67,68 Collected CSF samples were frozen on dry ice right after collection (within 1 hour). The samples were then shipped overnight, also on dry ice, to the ADNI Biomarker Core laboratory at the University of Pennsylvania. Aliquots (0.5mL) were prepared from the CSF samples and were then stored at −80 in barcode-labeled polypropylene vials. Samples for Aβ42 and p-tau181 were then measured using Elecsys immunoassays. The lower and upper technical limits for the Elecsys Aβ42 CSF immunoassay were 200 to 1700pg/mL. The limits for the Elecsys p-tau181 CSF immunoassay were 8 to 120pg/mL.
Image processing
The deep learning model used for subtyping MCI data utilized whole brain GM data. MRI data were analyzed using Statistical Parametric Mapping 12 (SPM12; Wellcome Department of Imaging Neuroscience, Institute of Neurology, London, UK; https://www.fil.ion.ucl.ac.uk/spm) running on MATLAB 9.8.0 (Math-Works, Natick, MA, USA). GM morphometric data was chosen as input data in our model due to its wide use in studies focusing on the pathological and clinical progression of AD.69, 70, 71, 72, 73, 74 Briefly, all MR images were aligned to the anterior commissure and segmented into GM, white matter and CSF using the unified segmentation procedure,75 implemented in SPM12. To improve the registration of the GM maps, we used the diffeomorphic anatomic registration through an exponentiated lie algebra algorithm (DARTEL).76 This resulted in more precise spatial normalization to the template. The DARTEL used subject-specific deformation fields to warp the GM map into subject-specific space, resampled at 2mm isotropic voxels. Then the warped GM maps were affine transformed into Montreal Neurological Institute (MNI) space. In addition to using whole brain GM volume data, we evaluated lobar contribution to the performance of the deep learning model through occlusion analysis (see below).
PET imaging data was used to validate the MCI subgroups outputted by the deep learning model. Detailed acquisition and standardized pre-processing procedures used with the PET images are available at the ADNI website (http://adni.loni.usc.edu/methods/). Aβ-PET uptake was calculated by averaging across 4 cortical regions (frontal, anterior cingulate, precuneus, and parietal cortex) relative to the whole cerebellum region.33 Similarly, FDG-PET uptake was calculated by averaging across a set of pre-defined regions (angular gyrus, posterior cingulate, inferior temporal gyrus) relative to pons/vermis reference regions.77
Deep learning model architecture
Model architecture: The deep learning model used for subtyping of MCI (See Figure 1) was based on the DenseNet architecture.29 It consisted of a convolutional layer, 4 dense blocks, 3 transition layers, a global averaging pooling layer and a fully-connected layer. This convolutional neural network architecture was chosen as it shows excellent feature propagation and classification performance while alleviating the vanishing gradient problem and significantly reducing the number of parameters used by the model.29 First, the whole brain image with dimensions of 91 109 91 was passed through a stack of convolutional layers, where the filters were of size 5 5 5. The convolution stride was set to 1 voxel, while the size of the max-pooling layer was 2 2 2, with a kernel size set at 2 2 2. The dense block consisted of multiple convolution units, which were equipped with a batch normalization layer, leaky rectified linear unit, a 1 1 1 convolutional layer, a 5 5 5 convolutional layer and a dropout layer. Every convolutional unit was connected to all the previous layers via shortcut connections. Dimensionality reduction of feature maps between dense blocks was achieved through the transition layer. The transition layer included a batch normalization layer, a leaky rectified linear unit, a convolutional layer of size 1 1 1, and an averaging pooling layer of size 2 2 2. The global averaging pooling layer was then concatenated and connected through a fully-connected layer. The model’s output values were processed by the fully-connected layer which used a sigmoid activation function. The output layer mapped all values greater than 0.5 as 1 (positive class: MCI-AD) and all values less than or equal to 0.5 as 0 (negative class: MCI-CN), while testing MCI data based on the pre-trained model (Figure 1).
Implementation
The Keras application programming interface in TensorFlow 2.0 was used for building the deep learning model. Model training and testing were performed in a parallelized manner with an Ubuntu 18.04.3 operating system, utilizing two Nvidia Tesla V100 graphic cards with 16GB memory each. The model was trained with a mini-batch size of 24 and 200 epochs, and optimized using stochastic gradient descent based on adaptive estimation of first- and second-order moments78 and an exponentially decaying learning rate. The initial learning rate was set at 0.0001 and decayed by 0.9 after every 10000 steps. A dropout layer was added to the dense block, with the dropout rate set to 0.2. In the batch normalization step, beta and gamma weights were initialized with L2 regularization set at 1 10−4 and epsilon set to 1.1 10−5. In the fully-connected layer, the L2 regularization penalty coefficient was set at 0.01. We observed stability in the model after an iteration of 150 epochs.
Model comparison
We compared the performance of the deep learning model to the performance of 3 widely-used machine learning models, including support vector machine,79 logistic regression,80 and random forest,81 all available in the scikit-learn library. Similar to the training of the deep learning model, whole-brain GM maps were used as features, and 5-fold cross-validation was used during training. Support vector machine classification was performed, adopting a soft margin with a Gaussian radial basis function. Logistic regression was initiated by setting the penalty to “L2” for regularization, and other hyper-parameters were set at default settings. Random forest classification was used with default settings as well.
Occlusion analysis
To identify the more specific regional contribution to the performance of the deep learning model and the differentiation between the two MCI subgroups, we integrated occlusion analysis (e.g.,82) into the classification framework. Cortical regions were first segmented with an automated segmentation tool available in FreeSurfer v6.0 (https://surfer.nmr.mgh.harvard.edu/), resulting in a parcellation of the cerebral cortex into 34 sulcal and gyral regions of interest (ROI) per hemisphere, according to the Desikan-Killiany protocol.83,84 We then merged individual ROIs to 6 lobes: frontal, parietal, medial temporal, lateral temporal, occipital, and cingulate.85 These ROIs were then masked out (setting their voxels to zero) from the testing phase’s input data (Figure 4A). We evaluated the performance of the different models (i.e., with each occluded lobe), relative to the intact model, quantifying the percentage of change in the model’s accuracy in reference to the original results.
Clustering based on neuropsychological assessments
Neuropsychological testing was comprised of six measures assessing three different cognitive domains: (1) Memory: Rey auditory verbal learning test (RAVLT),86 30-minute delayed free recall and the RAVLT recognition test. (2) Language: Animal fluency87 and the 30-item Boston naming test total score.88 (3) Attention/Executive: Trail making test (TMT),89 part A and part B. Similar to previous research15 raw scores in these neuropsychological measures were used to cluster MCI subjects into subgroups. Raw scores were first transformed into age- and education-adjusted z-scores based on means and standard deviations in each measure, calculated in the CN group. Then, an agglomerative hierarchical clustering analysis was performed on the z-scores using Ward’s method.90 The clustering analysis resulted in four distinct subgroups15: amnestic MCI, dysnomic MCI, dysexecutive MCI, and cluster-derived normal group.
Quantification and statistical analysis
All analyses were performed using the R statistical software, version 4.0.2 (https://www.r-project.org). Group differences in continuous variables were analyzed with an ANOVA or with independent-sample t tests. ANOVAs were followed, when relevant, by Tukey post hoc comparisons. Chi-square tests were applied to evaluate differences in categorical variables. Longitudinal data were analyzed using the R WRS2 package,91 and were based on a repeated-measures analysis of variance (RM-ANOVA), with group (MCI-AD, MCI-CN) serving as the between-subjects factor and time (baseline, follow-up) as the within-subject factor. Mauchly’s test was applied to test for violations in the assumption of sphericity, followed by Greenhouse-Geisser corrections, if necessary. A Kaplan-Meier survival curve was generated to estimate the time to AD diagnosis based on information from follow-up visits. A log rank test was used to compare survival curves between the subgroups. The plots for comparing cognitive scores, PET, and CSF between subgroups were based on the R RainCloudPlots package.92 Regional imaging results were displayed on a surface using the R ggseg (https://ggseg.github.io/ggseg/) package.
Acknowledgments
Research reported in this publication was supported by the National Institute On Aging of the National Institutes of Health under Award Number R01AG062590. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of HealthNational Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association, Alzheimer's Drug Discovery Foundation, Araclon Biotech, BioClinica, Inc., Biogen, Bristol-Myers Squibb Company, CereSpir, Inc., Cogstate, Eisai, Elan Pharmaceuticals, Inc., Eli Lilly and Company, EuroImmun, F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc., Fujirebio, GE Healthcare, IXICO Ltd.,Janssen Alzheimer Immunotherapy Research & Development, LLC., Johnson & Johnson Pharmaceutical Research & Development LLC., Lumosity, Lundbeck, Merck & Co., Inc.,Meso Scale Diagnostics, LLC., NeuroRx Research, Neurotrack Technologies, Novartis Pharmaceuticals Corporation, Pfizer Inc., Piramal Imaging, Servier, Takeda Pharmaceutical Company, and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (https://www.fnih.org/). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Validation data used in the preparation of this study were provided by the OASIS-3 database (https://www.oasis-brains.org/). OASIS-3 (principal investigators: T. Benzinger, D. Marcus, and J. Morris; NIH P50 AG00561, P30 NS09857781, P01 AG026276, P01 AG003991, R01 AG043434, UL1 TR000448, R01 EB009352).
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of the ADNI and/or provided data but did not participate in analysis or writing of this article. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Author contributions
Conceptualization, K.K. and E.D.; methodology, K.K.; software, K.K.; validation, K.K.; formal analysis, K.K.; investigation, K.K., K.S.G., A.B., M.S., and E.D.; resources, K.K. and E.D.; data curation, K.K.; writing – original draft, K.K. and E.D.; writing – review & editing, K.K., K.S.G., A.B., M.S., and E.D.; visualization, K.K. and E.D.; supervision, E.D.
Declaration of interests
The authors declare no competing interests.
Published: December 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100467.
Supplemental information
Data and code availability
All raw data including MRI, CSF, and cognitive scores are available through the ADNI data archive (http://adni.loni.ucs.edu/). Data used for validation is available from the OASIS dataset (https://www.oasis-brains.org).
The custom code for training the deep learning model was written in Python with TensorFlow 2.0. The code is available publicly: https://github.com/rlckd/MCI-subtype. Any additional information required to reanalyze the data reported in this work is available from the Lead Contact upon request.
References
- 1.Petersen R.C., Roberts R.O., Knopman D.S., Boeve B.F., Geda Y.E., Ivnik R.J., Smith G.E., Jack C.R., Jr. Mild cognitive impairment: ten years later. Arch. Neurol. 2009;66:1447–1455. doi: 10.1001/archneurol.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manly J.J., Tang M.X., Schupf N., Stern Y., Vonsattel J.P.G., Mayeux R. Frequency and course of mild cognitive impairment in a multiethnic community. Ann. Neurol. 2008;63:494–506. doi: 10.1002/ana.21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeCarli C. Mild cognitive impairment: prevalence, prognosis, aetiology, and treatment. Lancet Neurol. 2003;2:15–21. doi: 10.1016/s1474-4422(03)00262-x. [DOI] [PubMed] [Google Scholar]
- 4.Bowen J., Teri L., Kukull W., McCormick W., McCurry S.M., Larson E.B. Progression to dementia in patients with isolated memory loss. Lancet. 1997;349:763–765. doi: 10.1016/S0140-6736(96)08256-6. [DOI] [PubMed] [Google Scholar]
- 5.Petersen R.C., Smith G.E., Waring S.C., Ivnik R.J., Tangalos E.G., Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch. Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 6.Petersen R.C., Stevens J.C., Ganguli M., Tangalos E.G., Cummings J.L., DeKosky S.T. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1133–1142. doi: 10.1212/wnl.56.9.1133. [DOI] [PubMed] [Google Scholar]
- 7.Meyer J.S., Xu G., Thornby J., Chowdhury M.H., Quach M. Is mild cognitive impairment prodromal for vascular dementia like Alzheimer’s disease? Stroke. 2002;33:1981–1985. doi: 10.1161/01.str.0000024432.34557.10. [DOI] [PubMed] [Google Scholar]
- 8.Ferman T.J., Smith G.E., Kantarci K., Boeve B.F., Pankratz V.S., Dickson D.W., Graff-Radford N.R., Wszolek Z., Van Gerpen J., Uitti R., et al. Nonamnestic mild cognitive impairment progresses to dementia with Lewy bodies. Neurology. 2013;81:2032–2038. doi: 10.1212/01.wnl.0000436942.55281.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koepsell T.D., Monsell S.E. Reversion from mild cognitive impairment to normal or near-normal cognition: risk factors and prognosis. Neurology. 2012;79:1591–1598. doi: 10.1212/WNL.0b013e31826e26b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grundman M., Petersen R.C., Ferris S.H., Thomas R.G., Aisen P.S., Bennett D.A., Foster N.L., Jack C.R., Jr., Galasko D.R., Doody R., et al. Alzheimer’s Disease Cooperative Study Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch. Neurol. 2004;61:59–66. doi: 10.1001/archneur.61.1.59. [DOI] [PubMed] [Google Scholar]
- 11.Winblad B., Palmer K., Kivipelto M., Jelic V., Fratiglioni L., Wahlund L.O., Nordberg A., Bäckman L., Albert M., Almkvist O., et al. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 2004;256:240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- 12.Tabert M.H., Manly J.J., Liu X., Pelton G.H., Rosenblum S., Jacobs M., Zamora D., Goodkind M., Bell K., Stern Y., Devanand D.P. Neuropsychological prediction of conversion to Alzheimer disease in patients with mild cognitive impairment. Arch. Gen. Psychiatry. 2006;63:916–924. doi: 10.1001/archpsyc.63.8.916. [DOI] [PubMed] [Google Scholar]
- 13.Reisberg B., Ferris S.H., de Leon M.J., Franssen E.S.E., Kluger A., Mir P., Borenstein J., George A.E., Shulman E., Steinberg G., et al. Stage-specific behavioral, cognitive, and in vivo changes in community residing subjects with age-associated memory impairment and primary degenerative dementia of the Alzheimer type. Drug Dev. Res. 1988;15:101–114. [Google Scholar]
- 14.Petersen R.C., Doody R., Kurz A., Mohs R.C., Morris J.C., Rabins P.V., Ritchie K., Rossor M., Thal L., Winblad B. Current concepts in mild cognitive impairment. Arch. Neurol. 2001;58:1985–1992. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 15.Edmonds E.C., Delano-Wood L., Clark L.R., Jak A.J., Nation D.A., McDonald C.R., Libon D.J., Au R., Galasko D., Salmon D.P., Bondi M.W., Alzheimer’s Disease Neuroimaging Initiative Susceptibility of the conventional criteria for mild cognitive impairment to false-positive diagnostic errors. Alzheimers Dement. 2015;11:415–424. doi: 10.1016/j.jalz.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Machulda M.M., Lundt E.S., Albertson S.M., Kremers W.K., Mielke M.M., Knopman D.S., Bondi M.W., Petersen R.C. Neuropsychological subtypes of incident mild cognitive impairment in the Mayo Clinic Study of Aging. Alzheimers Dement. 2019;15:878–887. doi: 10.1016/j.jalz.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han J.W., Kim T.H., Lee S.B., Park J.H., Lee J.J., Huh Y., Park J.E., Jhoo J.H., Lee D.Y., Kim K.W. Predictive validity and diagnostic stability of mild cognitive impairment subtypes. Alzheimers Dement. 2012;8:553–559. doi: 10.1016/j.jalz.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Hughes T.F., Snitz B.E., Ganguli M. Should mild cognitive impairment be subtyped? Curr. Opin. Psychiatry. 2011;24:237–242. doi: 10.1097/YCO.0b013e328344696b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack C.R., Jr., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., Holtzman D.M., Jagust W., Jessen F., Karlawish J., et al. Contributors NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubois B., Feldman H.H., Jacova C., Hampel H., Molinuevo J.L., Blennow K., DeKosky S.T., Gauthier S., Selkoe D., Bateman R., et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 21.Albert M.S., DeKosky S.T., Dickson D., Dubois B., Feldman H.H., Fox N.C., Gamst A., Holtzman D.M., Jagust W.J., Petersen R.C., et al. The Diagnosis of Mild Cognitive Impairment due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Focus (Madison) 2013;11:96–106. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noh Y., Jeon S., Lee J.M., Seo S.W., Kim G.H., Cho H., Ye B.S., Yoon C.W., Kim H.J., Chin J., et al. Anatomical heterogeneity of Alzheimer disease: based on cortical thickness on MRIs. Neurology. 2014;83:1936–1944. doi: 10.1212/WNL.0000000000001003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young A.L., Marinescu R.V., Oxtoby N.P., Bocchetta M., Yong K., Firth N.C., Cash D.M., Thomas D.L., Dick K.M., Cardoso J., et al. Genetic FTD Initiative (GENFI) Alzheimer’s Disease Neuroimaging Initiative (ADNI) Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with Subtype and Stage Inference. Nat. Commun. 2018;9:4273. doi: 10.1038/s41467-018-05892-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ten Kate M., Dicks E., Visser P.J., van der Flier W.M., Teunissen C.E., Barkhof F., Scheltens P., Tijms B.M., Alzheimer’s Disease Neuroimaging Initiative Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain. 2018;141:3443–3456. doi: 10.1093/brain/awy264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong A., Toledo J.B., Honnorat N., Doshi J., Varol E., Sotiras A., Wolk D., Trojanowski J.Q., Davatzikos C., Alzheimer’s Disease Neuroimaging Initiative Heterogeneity of neuroanatomical patterns in prodromal Alzheimer’s disease: links to cognition, progression and biomarkers. Brain. 2017;140:735–747. doi: 10.1093/brain/aww319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davatzikos C., Xu F., An Y., Fan Y., Resnick S.M. Longitudinal progression of Alzheimer’s-like patterns of atrophy in normal older adults: the SPARE-AD index. Brain. 2009;132:2026–2035. doi: 10.1093/brain/awp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwak K., Niethammer M., Giovanello K.S., Styner M., Dayan E., Alzheimer’s Disease Neuroimaging Initiative Differential Role for Hippocampal Subfields in Alzheimer’s Disease Progression Revealed with Deep Learning. Cereb. Cortex. 2021:bhab223. doi: 10.1093/cercor/bhab223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaMontagne P.J., Benzinger T.L.S., Morris J.C., Keefe S., Hornbeck R., Xiong C., Grant E., Hassenstab J., Moulder K., Vlassenko A.G., et al. OASIS-3: Longitudinal neuroimaging, clinical, and cognitive dataset for normal aging and Alzheimer disease. medRxiv. 2019 doi: 10.1101/2019.12.13.19014902. [DOI] [Google Scholar]
- 29.Huang G., Liu Z., Van Der Maaten L., Weinberger K.Q. CVPR; 2017. Densely connected convolutional networks. In Proceedings - 30th IEEE Conference on Computer Vision and Pattern Recognition; pp. 2261–2269. [Google Scholar]
- 30.Landau S.M., Lu M., Joshi A.D., Pontecorvo M., Mintun M.A., Trojanowski J.Q., Shaw L.M., Jagust W.J., Alzheimer’s Disease Neuroimaging Initiative Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Ann. Neurol. 2013;74:826–836. doi: 10.1002/ana.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Wilde A., Reimand J., Teunissen C.E., Zwan M., Windhorst A.D., Boellaard R., van der Flier W.M., Scheltens P., van Berckel B.N.M., Bouwman F., Ossenkoppele R. Discordant amyloid-β PET and CSF biomarkers and its clinical consequences. Alzheimers Res. Ther. 2019;11:78. doi: 10.1186/s13195-019-0532-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosconi L., McHugh P.F. FDG- and amyloid-PET in Alzheimer’s disease: is the whole greater than the sum of the parts? Q. J. Nucl. Med. Mol. Imaging. 2011;55:250–264. [PMC free article] [PubMed] [Google Scholar]
- 33.Landau S.M., Mintun M.A., Joshi A.D., Koeppe R.A., Petersen R.C., Aisen P.S., Weiner M.W., Jagust W.J., Alzheimer’s Disease Neuroimaging Initiative Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann. Neurol. 2012;72:578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landau S.M., Harvey D., Madison C.M., Reiman E.M., Foster N.L., Aisen P.S., Petersen R.C., Shaw L.M., Trojanowski J.Q., Jack C.R., Jr., et al. Alzheimer’s Disease Neuroimaging Initiative Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan E.L., Meier P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958;53:457–481. [Google Scholar]
- 36.Barnes J., Bartlett J.W., van de Pol L.A., Loy C.T., Scahill R.I., Frost C., Thompson P., Fox N.C. A meta-analysis of hippocampal atrophy rates in Alzheimer’s disease. Neurobiol. Aging. 2009;30:1711–1723. doi: 10.1016/j.neurobiolaging.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takao H., Hayashi N., Ohtomo K. A longitudinal study of brain volume changes in normal aging. Eur. J. Radiol. 2012;81:2801–2804. doi: 10.1016/j.ejrad.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Wang J., Knol M.J., Tiulpin A., Dubost F., de Bruijne M., Vernooij M.W., Adams H.H.H., Ikram M.A., Niessen W.J., Roshchupkin G.V. Gray matter age prediction as a biomarker for risk of dementia. Proc. Natl. Acad. Sci. USA. 2019;116:21213–21218. doi: 10.1073/pnas.1902376116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang G., Yu S., Dong H., Slabaugh G., Dragotti P.L., Ye X., Liu F., Arridge S., Keegan J., Guo Y., et al. DAGAN: Deep De-Aliasing Generative Adversarial Networks for Fast Compressed Sensing MRI Reconstruction. IEEE Trans. Med. Imaging. 2018;37:1310–1321. doi: 10.1109/TMI.2017.2785879. [DOI] [PubMed] [Google Scholar]
- 40.Nogovitsyn N., Souza R., Muller M., Srajer A., Hassel S., Arnott S.R., Davis A.D., Hall G.B., Harris J.K., Zamyadi M., et al. Testing a deep convolutional neural network for automated hippocampus segmentation in a longitudinal sample of healthy participants. Neuroimage. 2019;197:589–597. doi: 10.1016/j.neuroimage.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 41.Hosseini-Asl E., Keynton R., El-Baz A. Alzheimer’s disease diagnostics by adaptation of 3D convolutional network. In Proceedings - International Conference on Image Processing. ICIP. 2016:126–130. [Google Scholar]
- 42.Greenspan H., Van Ginneken B., Summers R.M. Guest Editorial Deep Learning in Medical Imaging: Overview and Future Promise of an Exciting New Technique. IEEE Trans. Med. Imaging. 2016;35:1153–1159. [Google Scholar]
- 43.Cheng B., Liu M., Zhang D., Munsell B.C., Shen D. Domain Transfer Learning for MCI Conversion Prediction. IEEE Trans. Biomed. Eng. 2015;62:1805–1817. doi: 10.1109/TBME.2015.2404809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H., Habes M., Wolk D.A., Fan Y., Alzheimer’s Disease Neuroimaging Initiative and the Australian Imaging Biomarkers and Lifestyle Study of Aging A deep learning model for early prediction of Alzheimer’s disease dementia based on hippocampal magnetic resonance imaging data. Alzheimers Dement. 2019;15:1059–1070. doi: 10.1016/j.jalz.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wee C.Y., Liu C., Lee A., Poh J.S., Ji H., Qiu A., Alzheimers Disease Neuroimage Initiative Cortical graph neural network for AD and MCI diagnosis and transfer learning across populations. Neuroimage Clin. 2019;23:101929. doi: 10.1016/j.nicl.2019.101929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansson O., Zetterberg H., Buchhave P., Londos E., Blennow K., Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 47.Mattsson N., Zetterberg H., Hansson O., Andreasen N., Parnetti L., Jonsson M., Herukka S.K., van der Flier W.M., Blankenstein M.A., Ewers M., et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 48.Pascoal T.A., Therriault J., Mathotaarachchi S., Kang M.S., Shin M., Benedet A.L., Chamoun M., Tissot C., Lussier F., Mohaddes S., et al. Alzheimer’s Disease Neuroimaging Initiative Topographical distribution of Aβ predicts progression to dementia in Aβ positive mild cognitive impairment. Alzheimers Dement. (Amst.) 2020;12:e12037. doi: 10.1002/dad2.12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jansen W.J., Ossenkoppele R., Knol D.L., Tijms B.M., Scheltens P., Verhey F.R.J., Visser P.J., Aalten P., Aarsland D., Alcolea D., et al. Amyloid Biomarker Study Group Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313:1924–1938. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iaccarino L., Sala A., Perani D., Alzheimer’s Disease Neuroimaging Initiative Predicting long-term clinical stability in amyloid-positive subjects by FDG-PET. Ann. Clin. Transl. Neurol. 2019;6:1113–1120. doi: 10.1002/acn3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tam A., Dansereau C., Iturria-Medina Y., Urchs S., Orban P., Sharmarke H., Breitner J., Bellec P., Alzheimer’s Disease Neuroimaging Initiative A highly predictive signature of cognition and brain atrophy for progression to Alzheimer’s dementia. Gigascience. 2019;8:giz055. doi: 10.1093/gigascience/giz055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Habes M., Grothe M.J., Tunc B., McMillan C., Wolk D.A., Davatzikos C. Disentangling Heterogeneity in Alzheimer’s Disease and Related Dementias Using Data-Driven Methods. Biol. Psychiatry. 2020;88:70–82. doi: 10.1016/j.biopsych.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Callen D.J.A., Black S.E., Gao F., Caldwell C.B., Szalai J.P. Beyond the hippocampus: MRI volumetry confirms widespread limbic atrophy in AD. Neurology. 2001;57:1669–1674. doi: 10.1212/wnl.57.9.1669. [DOI] [PubMed] [Google Scholar]
- 54.Jack C.R., Jr., Shiung M.M., Gunter J.L., O’Brien P.C., Weigand S.D., Knopman D.S., Boeve B.F., Ivnik R.J., Smith G.E., Cha R.H., et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology. 2004;62:591–600. doi: 10.1212/01.wnl.0000110315.26026.ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langella S., Sadiq M.U., Mucha P.J., Giovanello K.S., Dayan E., Alzheimer’s Disease Neuroimaging Initiative Lower functional hippocampal redundancy in mild cognitive impairment. Transl. Psychiatry. 2021;11:61. doi: 10.1038/s41398-020-01166-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadiq M.U., Langella S., Giovanello K.S., Mucha P.J., Dayan E. Accrual of functional redundancy along the lifespan and its effects on cognition. Neuroimage. 2021;229:117737. doi: 10.1016/j.neuroimage.2021.117737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 58.Braak H., Alafuzoff I., Arzberger T., Kretzschmar H., Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jack C.R., Jr., Petersen R.C., Xu Y., O’Brien P.C., Smith G.E., Ivnik R.J., Boeve B.F., Tangalos E.G., Kokmen E. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55:484–489. doi: 10.1212/wnl.55.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badhwar A., McFall G.P., Sapkota S., Black S.E., Chertkow H., Duchesne S., Masellis M., Li L., Dixon R.A., Bellec P. A multiomics approach to heterogeneity in Alzheimer’s disease: focused review and roadmap. Brain. 2020;143:1315–1331. doi: 10.1093/brain/awz384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clark L.R., Delano-Wood L., Libon D.J., McDonald C.R., Nation D.A., Bangen K.J., Jak A.J., Au R., Salmon D.P., Bondi M.W. Are empirically-derived subtypes of mild cognitive impairment consistent with conventional subtypes? J. Int. Neuropsychol. Soc. 2013;19:635–645. doi: 10.1017/S1355617713000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nettiksimmons J., DeCarli C., Landau S., Beckett L., Alzheimer’s Disease Neuroimaging Initiative Biological heterogeneity in ADNI amnestic mild cognitive impairment. Alzheimers Dement. 2014;10:511–521.e1. doi: 10.1016/j.jalz.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ewers M., Franzmeier N., Suárez-Calvet M., Morenas-Rodriguez E., Caballero M.A.A., Kleinberger G., Piccio L., Cruchaga C., Deming Y., Dichgans M., et al. Alzheimer’s Disease Neuroimaging Initiative Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019;11:eaav6221. doi: 10.1126/scitranslmed.aav6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Digma L.A., Madsen J.R., Reas E.T., Dale A.M., Brewer J.B., Banks S.J., Alzheimer’s Disease Neuroimaging Initiative Tau and atrophy: domain-specific relationships with cognition. Alzheimers Res. Ther. 2019;11:65. doi: 10.1186/s13195-019-0518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wirth M., Villeneuve S., Haase C.M., Madison C.M., Oh H., Landau S.M., Rabinovici G.D., Jagust W.J. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurol. 2013;70:1512–1519. doi: 10.1001/jamaneurol.2013.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jack C.R., Jr., Bernstein M.A., Borowski B.J., Gunter J.L., Fox N.C., Thompson P.M., Schuff N., Krueger G., Killiany R.J., Decarli C.S., et al. Alzheimer’s Disease Neuroimaging Initiative Update on the magnetic resonance imaging core of the Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 2010;6:212–220. doi: 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shaw L.M., Vanderstichele H., Knapik-Czajka M., Clark C.M., Aisen P.S., Petersen R.C., Blennow K., Soares H., Simon A., Lewczuk P., et al. Alzheimer’s Disease Neuroimaging Initiative Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trojanowski J.Q., Vandeerstichele H., Korecka M., Clark C.M., Aisen P.S., Petersen R.C., Blennow K., Soares H., Simon A., Lewczuk P., et al. Alzheimer’s Disease Neuroimaging Initiative Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010;6:230–238. doi: 10.1016/j.jalz.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anderson V.M., Schott J.M., Bartlett J.W., Leung K.K., Miller D.H., Fox N.C. Gray matter atrophy rate as a marker of disease progression in AD. Neurobiol. Aging. 2012;33:1194–1202. doi: 10.1016/j.neurobiolaging.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang X., Mormino E.C., Sun N., Sperling R.A., Sabuncu M.R., Yeo B.T.T. Bayesian model reveals latent atrophy factors with dissociable cognitive trajectories in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2016;113:E6535–E6544. doi: 10.1073/pnas.1611073113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson P.M., Hayashi K.M., de Zubicaray G., Janke A.L., Rose S.E., Semple J., Herman D., Hong M.S., Dittmer S.S., Doddrell D.M., Toga A.W. Dynamics of gray matter loss in Alzheimer’s disease. J. Neurosci. 2003;23:994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frisoni G.B., Testa C., Zorzan A., Sabattoli F., Beltramello A., Soininen H., Laakso M.P. Detection of grey matter loss in mild Alzheimer’s disease with voxel based morphometry. J. Neurol. Neurosurg. Psychiatry. 2002;73:657–664. doi: 10.1136/jnnp.73.6.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frisoni G.B., Pievani M., Testa C., Sabattoli F., Bresciani L., Bonetti M., Beltramello A., Hayashi K.M., Toga A.W., Thompson P.M. The topography of grey matter involvement in early and late onset Alzheimer’s disease. Brain. 2007;130:720–730. doi: 10.1093/brain/awl377. [DOI] [PubMed] [Google Scholar]
- 74.Scahill R.I., Schott J.M., Stevens J.M., Rossor M.N., Fox N.C. Mapping the evolution of regional atrophy in Alzheimer’s disease: Unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. USA. 2002;99:4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ashburner J., Friston K.J. Unified segmentation. Neuroimage. 2005;26:839–851. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 76.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38:95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 77.Landau S.M., Harvey D., Madison C.M., Koeppe R.A., Reiman E.M., Foster N.L., Weiner M.W., Jagust W.J., Alzheimer’s Disease Neuroimaging Initiative Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging. 2011;32:1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kingma D.P., Ba J.L. Adam: A method for stochastic optimization. In 3rd International Conference on Learning Representations. ICLR 2015 - Conference Track Proceedings. 2015 [Google Scholar]
- 79.Platt J. Fast Training of Support Vector Machines using Sequential Minimal Optimization. In Advances in Kernel Methods—Support Vector Learning. 1998:185–208. [Google Scholar]
- 80.Cox D.R. The Regression Analysis of Binary Sequences. J. R. Stat. Soc. B. 1958;20:215–242. [Google Scholar]
- 81.Liaw A., Wiener M. Classification and Regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 82.Sadr J., Jarudi I., Sinha P. The role of eyebrows in face recognition. Perception. 2003;32:285–293. doi: 10.1068/p5027. [DOI] [PubMed] [Google Scholar]
- 83.Fischl B., van der Kouwe A., Destrieux C., Halgren E., Ségonne F., Salat D.H., Busa E., Seidman L.J., Goldstein J., Kennedy D., et al. Automatically parcellating the human cerebral cortex. Cereb. Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- 84.Desikan R.S., Ségonne F., Fischl B., Quinn B.T., Dickerson B.C., Blacker D., Buckner R.L., Dale A.M., Maguire R.P., Hyman B.T., et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 85.Klein A., Tourville J. 101 labeled brain images and a consistent human cortical labeling protocol. Front. Neurosci. 2012;6:171. doi: 10.3389/fnins.2012.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rey A. Presses Universitaries De France; 1958. L’examen clinique en psychologie. [The clinical examination in psychology.] [Google Scholar]
- 87.Rosen W.G. Verbal Fluency in Aging and Dementia. J. Clin. Neuropsychol. 1980;2:135–146. [Google Scholar]
- 88.Kaplan E., Goodglass H.W.S. Lea Febiger; 1983. The Boston Naming Test. [Google Scholar]
- 89.Reitan R.M. Validity of the trail making test as an indicator of organic brain damage. Percept. Mot. Skills. 1958;8:271–276. [Google Scholar]
- 90.Ward J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963;58:236–244. [Google Scholar]
- 91.Mair P., Wilcox R. Robust statistical methods in R using the WRS2 package. Behav. Res. Methods. 2020;52:464–488. doi: 10.3758/s13428-019-01246-w. [DOI] [PubMed] [Google Scholar]
- 92.Allen M., Poggiali D., Whitaker K., Marshall T.R., Kievit R.A. Raincloud plots: a multi-platform tool for robust data visualization. Wellcome Open Res. 2019;4:63. doi: 10.12688/wellcomeopenres.15191.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data including MRI, CSF, and cognitive scores are available through the ADNI data archive (http://adni.loni.ucs.edu/). Data used for validation is available from the OASIS dataset (https://www.oasis-brains.org).
The custom code for training the deep learning model was written in Python with TensorFlow 2.0. The code is available publicly: https://github.com/rlckd/MCI-subtype. Any additional information required to reanalyze the data reported in this work is available from the Lead Contact upon request.