

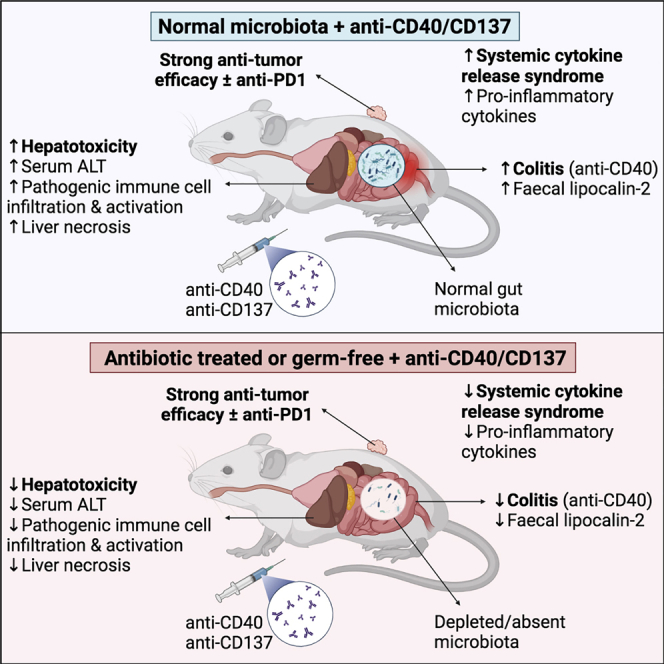
Summary
Immune agonist antibodies (IAAs) are promising immunotherapies that target co-stimulatory receptors to induce potent anti-tumor immune responses, particularly when combined with checkpoint inhibitors. Unfortunately, their clinical translation is hampered by serious dose-limiting, immune-mediated toxicities, including high-grade and sometimes fatal liver damage, cytokine release syndrome (CRS), and colitis. We show that the immunotoxicity, induced by the IAAs anti-CD40 and anti-CD137, is dependent on the gut microbiota. Germ-free or antibiotic-treated mice have significantly reduced colitis, CRS, and liver damage following IAA treatment compared with conventional mice or germ-free mice recolonized via fecal microbiota transplant. MyD88 signaling is required for IAA-induced CRS and for anti-CD137-induced, but not anti-CD40-induced, liver damage. Importantly, antibiotic treatment does not impair IAA anti-tumor efficacy, alone or in combination with anti-PD1. Our results suggest that microbiota-targeted therapies could overcome the toxicity induced by IAAs without impairing their anti-tumor activity.
Keywords: immune agonist antibody, immunotherapy, liver damage, cytokine release syndrome, gut microbiota, anti-CD40, anti-CD137
Graphical abstract
Highlights
-
•
Antibiotic-treated and germ-free mice are protected from IAA-induced toxicity
-
•
Antibiotic treatment does not impair IAA + anti-PD1 anti-tumor efficacy
-
•
Gut microbiota modulates IAA-induced immune responses in liver
-
•
MyD88 mediates IAA-induced CRS and anti-CD137-induced liver damage
Blake et al. investigate the impact of the gut microbiota on responses to the immune agonist antibody (IAA) immunotherapies, anti-CD40 and anti-CD137. The study reveals that, in mice, the hepatotoxicity, colitis, and cytokine release induced by these immunotherapies is dependent on the gut microbiota, while their anti-tumor efficacy is not.
Introduction
The development of immune checkpoint inhibitors (ICIs) targeting PD1/PDL1 and CTLA41 has revolutionized the treatment of cancer, inducing long-term, potentially curative responses in some patients, particularly in those with advanced melanoma.2 Unfortunately, ICIs are only efficacious in a relatively small portion of patients, and their efficacy is limited to tumor types with a significant immune infiltrate. To improve response rates, approaches that combine ICIs with other immunotherapies, such as immune agonist antibodies (IAAs), are currently being evaluated. IAAs target immune co-activating/co-stimulatory receptors such as CD40 and CD137, which are expressed on a range of myeloid and lymphoid cells. IAAs provide activating signals via these receptors to induce downstream processes, including enhanced immune cell proliferation, survival, antigen presentation, and cytotoxicity.3 IAAs are therefore attractive cancer immunotherapies as they induce both direct anti-tumor immunity and increase immune cell infiltration into tumors, sensitizing them to ICI therapies.4 As of September 2021, there were 104 active, recruiting, or upcoming phase I to III clinical trials evaluating IAAs, predominantly in combination with other immunotherapies such as ICIs (https://clinicaltrials.gov).
The clinical translation of IAAs has been hampered by serious dose-limiting toxicities, including grade 3 or higher immune-mediated side effects, such as cytokine release syndrome (CRS), liver damage, and death. For example, clinical trials of several CD40 agonists have identified dose-limiting toxicity due to CRS and grade 3 to 4 hematological and liver toxicities.5,6 Trials of another IAA, targeting CD137 (urelumab), were halted due to severe dose-dependent liver toxicity,7 while an anti-CD28 IAA (TGN1412) induced a rapid CRS in patients in a phase I trial that required long-term hospitalization of participants in intensive care.8 Several strategies are now being assessed to better manage IAA-induced immunotoxicity, including limiting the dose, co-administering immunosuppressive corticosteroids,5 or administering IAAs locally to tumors.9 These strategies could, however, come at the expense of anti-tumor efficacy by dampening anti-tumor immune responses.10
The composition and activity of the microbiota (particularly the gut microbiota) has been shown to strongly influence immune responses in a broad range of contexts11 and has been linked to numerous diseases in the liver, such as alcoholic and non-alcoholic fatty liver disease,12,13 primary sclerosing cholangitis,14 and cirrhosis.15 In mice, specific bacterial species have also been shown to modulate immune-mediated liver damage induced by concanavalin A.16,17 Recent studies have also found that the gut microbiota plays a critical role in the efficacy of the ICIs, anti-PD1,18, 19, 20 and anti-CTLA4.21 Here, we report that antibiotic-treated or germ-free (GF) mice were protected against the liver damage, CRS, and colitis induced by treatment with the IAAs anti-CD40 or anti-CD137. Importantly, antibiotic treatment did not significantly impair the anti-tumor efficacy of anti-CD40 or anti-CD137 alone or in combination with the ICI anti-PD1. Our data provide a strong rationale to investigate if the composition of the gut microbiota can predict patients at risk of high-grade IAA-induced immunotoxicity and suggest that microbiota-targeted interventions may reduce the immunotoxicity associated with IAAs, overcoming a critical roadblock to their clinical application.
Results
Antibiotic treatment reduces the immunotoxicity of anti-CD40 without reducing its anti-tumor efficacy
Due to the close interaction between the liver and gut microbiota via the hepatic portal vein,22 we hypothesized that the hepatotoxicity and systemic CRS associated with anti-CD40 was mediated by the gut microbiota. To assess this, we first investigated the impact of antibiotic treatment on responses to anti-CD40 in tumor-bearing C57BL/6 mice (Figure 1A). Antibiotic-exposed (ABX) mice had an ∼4 log-fold reduction in fecal bacterial load compared to unexposed mice (no ABX) (Figure S1A). Mice have been previously shown to experience similar anti-CD40-induced immunotoxicity23 as observed in patients,6,7 and concordantly, we observed that in normally colonized specific and opportunistic pathogen-free (SOPF) mice, treatment with anti-CD40 induced rapid onset liver damage and CRS that peaked 24 h following therapy initiation. Liver immunotoxicity was potently reduced in ABX mice (Figures 1B–1D). Serum alanine aminotransferase (ALT), a marker of hepatocellular damage, was significantly elevated following anti-CD40 treatment in SOPF mice but was almost completely inhibited in ABX mice (Figure 1B). Furthermore, histological analysis of livers collected 24 h after anti-CD40 treatment revealed large areas of hepatocellular necrosis, which were almost completely absent in ABX mice (Figure 1C). Some necrotic areas were associated with thrombi in portal blood vessels, and these were also dramatically reduced in ABX mice (Figure S1B). Histopathology confirmed that ABX mice had significantly reduced liver damage and necrosis following anti-CD40 treatment (Figures 1D and S1C). ABX mice also had significantly reduced levels of the proinflammatory cytokines TNFα and IL6 in serum following anti-CD40 treatment (Figures 1E and 1F), indicating that antibiotic treatment also significantly abrogated the systemic CRS induced by anti-CD40. Serum levels of interferon (IFN)γ, a key lymphocyte effector cytokine, were elevated following anti-CD40 treatment in both untreated (no ABX) and ABX mice (Figure 1G).
Figure 1.

Antibiotic treatment significantly reduces anti-CD40 immunotoxicity without impacting its anti-tumor efficacy
(A) Overview of the experimental design.
(B) Levels of ALT in serum collected 24 h after control (PBS) or anti-CD40 treatment (100 μg intraperitoneally [i.p.]) in antibiotic-treated (ABX) and untreated (no ABX) mice.
(C) Representative H&E-stained liver lateral cross-sections collected 24 h after PBS or anti-CD40 treatment. Large areas of hepatocellular necrosis are highlighted by dotted green lines.
(D) Liver histological score at 24 h after control or anti-CD40 treatment.
(E–G) Levels of (E) TNFα, (F) IL6, and (G) IFNγ in serum collected 24 h after control or anti-CD40 treatment.
(H) Lipocalin-2 levels in feces collected 24 h after control or anti-CD40 treatment.
(I) MC38 tumor growth in ABX and no ABX mice injected i.p. every 4 days with 3 doses of PBS (control) or anti-CD40 (100 μg) once tumors reached a size of ∼40 to 50 mm2 (day 9).
(J) AT3 tumor growth in ABX and no ABX mice. Mice were treated with either PBS (control) or anti-CD40 (100 μg i.p.) once tumors reached ∼40 to 50 mm2 (Day 17). 4 days after the first control/anti-CD40 treatment, mice were injected i.p. every 4 days with 3 doses of PBS (control) or anti-PD1 (200 μg i.p.). n = 6–15 mice per group.
Statistical significance was determined using a Mann-Whitney test (B–H) or one-way ANOVA with Tukey’s post-test analysis (final tumor sizes in each group analyzed) (I and J). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001. N.S., not significant. Data are represented as mean ± SEM. Results shown are pooled from 2 independent experiments (B–G and I), or from independent single experiments (H and J).
We observed that anti-CD40-treated mice were reluctant to produce stool, suggesting that anti-CD40 could induce gastrointestinal inflammation, as has been reported clinically.6,7 Histological analysis of colons identified numerous enterocyte-derived apoptotic bodies and increased lymphocyte infiltration in the lamina propria following anti-CD40 treatment (Figure S1D). While similar lesions were observed in anti-CD40-treated ABX mice, they were much less severe and were reduced in distribution. To quantitate gut inflammation, we assessed fecal lipocalin-2 levels in stool samples.24 Anti-CD40-treated mice had significantly elevated levels of lipocalin-2 in their stool samples, which was potently inhibited in ABX mice (Figure 1H). Histological analysis of lung or skin sections indicated that anti-CD40 did not induce immune cell infiltration or tissue damage in these organs (Figures S1E and S1F).
Next, we investigated whether antibiotic treatment altered the anti-tumor efficacy of anti-CD40 against established subcutaneous tumors. Antibiotic treatment had no impact on the efficacy of anti-CD40 in suppressing MC38 tumor growth (Figure 1I). We also observed that anti-CD40 induced immune alterations in MC38 tumors, consistent with previous reports,25 which were not significantly altered in ABX mice (Figures S2A–S2D). Due to their potential to act synergistically with ICIs,4,26 most clinical trials are assessing the efficacy of IAAs in combination with ICIs, such as anti-PD1.3 Concerningly, the efficacy of anti-PD1 as a monotherapy has been shown to be strongly reduced by antibiotic treatment.27 We evaluated the impact of antibiotic treatment on the efficacy of anti-CD40 + anti-PD1 combination therapy against anti-PD1-insensitive AT3 tumors.25 As expected, anti-PD1 was ineffective at suppressing AT3 tumor growth alone (Figure 1J). Treating mice with a single dose of anti-CD40 sensitized AT3 tumors to anti-PD1, with the combination treatment resulting in potent suppression of tumor growth which, importantly, was not reduced in ABX mice (Figure 1J).
Finally, we also investigated whether a shorter course of antibiotics was sufficient to reduce anti-CD40-induced immunotoxicity. Mice treated with antibiotics starting either 3 or 7 days before anti-CD40 treatment had significantly reduced serum ALT levels following treatment (Figure S2E). The CRS induced by anti-CD40 was also significantly reduced in mice treated with antibiotics for at least 7 days prior to anti-CD40 treatment (Figures S2F and S2G). Consistent with our previous observations, IFNγ levels in serum following anti-CD40 treatment were not significantly altered by antibiotic treatment (Figure S2H). Shorter durations of antibiotic treatment also had no significant impact on the anti-tumor efficacy of anti-CD40 against MC38 tumors (Figure S2I). Taken together, these data suggest that antibiotic treatment could be a feasible strategy to reduce anti-CD40-induced immunotoxicity without compromising anti-tumor efficacy either alone or in combination with anti-PD1.
Antibiotic treatment also reduces the immunotoxicity induced by anti-CD137 treatment
Urelumab, an anti-CD137 IAA, induces severe liver damage in patients, particularly when administered at higher doses.7 Anti-CD137 similarly induces liver damage in mice,28 which is increased in severity when combined with anti-PD1.29 We next investigated whether antibiotic treatment also reduced the immunotoxicity induced by anti-CD137 (Figure 2A). Consistent with previous reports,28,30 anti-CD137-induced liver damage peaked at 11 days following treatment initiation (Figures 2B–2D). As we observed for anti-CD40, anti-CD137-induced serum ALT levels were significantly reduced in ABX mice (Figure 2B). Histological analysis of the livers confirmed that antibiotic treatment significantly reduced anti-CD137-induced liver damage (Figures 2C and 2D). While CRS is not a typical side effect of anti-CD137 treatment,7 we found that anti-CD137-induced IFNγ levels in serum were significantly reduced by antibiotic treatment (Figure 2E). Tumor necrosis factor (TNF)α or interleukin (IL)6 levels in serum were not significantly altered by anti-CD137 treatment (Figures S3A and S3B).
Figure 2.

Antibiotic treatment significantly reduces the immunotoxicity induced by anti-CD137
(A) Overview of the experimental design.
(B–E) Serum (B) ALT levels, (C) liver histological score, (D) representative H&E-stained liver cross-sections (areas of immune infiltration are indicated with arrows), and (E) IFNγ levels in serum assessed at 11 days after initiation of control (PBS) or anti-CD137 treatment (100 μg i.p., 3 doses 4 days apart) in ABX or no ABX mice.
(F and G) Frequency of (F) MC38 tumor rejection and (G) MC38 tumor growth in ABX and no ABX mice treated with PBS (control), anti-CD137 (100 μg i.p.), anti-PD1 (200 μg i.p), or anti-CD137 (100 μg i.p.) + anti-PD1 (200 μg i.p). Treatment was repeated 3 times 4 days apart and initiated when tumors were ∼40 to 50 mm2 (day 10).
(H) Levels of ALT in serum collected 11 days after treatment initiation. n = 6–20 mice per group.
Statistical significance was determined by a Mann-Whitney test (B–F and H) or one-way ANOVA with Tukey’s post-test analysis (final tumor sizes in each group analyzed) (G). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗∗p ≤ 0.0001. N.S., not significant. Data are represented as mean ± SEM. Results shown are pooled from 2 independent experiments (B–E) or from a single experiment (F–H).
We next investigated whether antibiotic treatment affected the anti-tumor activity of anti-CD137 against the PD1-sensitive MC38 cell line,31 alone or in combination with anti-PD1. Anti-CD137 or anti-PD1 alone induced a modest reduction to MC38 tumor growth, which was dramatically increased when the therapies were combined (Figures 2F and 2G). Importantly, the anti-tumor efficacy of anti-CD137 alone, anti-PD1 alone, or the anti-CD137 + anti-PD1 combination was not significantly different in ABX mice (Figures 2F and 2G). The infiltration and activation (% Ki67+) of CD8+ T cells into tumors induced by anti-CD137 was also not significantly altered in ABX mice (Figures S3C and S3D). Furthermore, anti-CD137 treatment reduced the proportion of immunosuppressive regulatory T cells (Tregs) in the tumor, which was not significantly altered in ABX mice (Figure S3E). As previously reported,29 we found that anti-CD137 + anti-PD1 combination therapy induced significantly greater liver damage than anti-CD137 treatment alone. Critically, this was also significantly abrogated in ABX mice (Figure 2H). Taken together, these data indicate that antibiotic treatment potently reduces the immunotoxicity of anti-CD137 alone or combined with anti-PD1 without significantly impacting anti-tumor efficacy.
GF mice have significantly reduced anti-CD40- and anti-CD137-induced immunotoxicity, which is restored by fecal microbiota transplant (FMT)
We next investigated responses to anti-CD40 and anti-CD137 in GF mice and in GF mice recolonized by FMT (GF+FMT) (Figure 3A). Successful colonization of GF mice by FMT was confirmed by 16S rRNA gene sequencing (Figure 3B). Similar to ABX mice, GF mice were protected against liver damage following anti-CD40 treatment, and FMT was sufficient to restore anti-CD40-induced liver damage (Figures 3C and 3D). GF mice also had significantly reduced levels of TNFα (Figure 3E), but not IL6 (data not shown), in serum following anti-CD40 treatment. As we observed in ABX mice, anti-CD40-induced IFNγ levels in serum were not significantly different between GF and GF+FMT mice (Figure 3F). Anti-CD137-induced liver damage was significantly reduced in GF mice (Figure 3G). As we observed in ABX mice, IL6 levels in serum were not significantly elevated in anti-CD137-treated GF+FMT mice (data not shown). Interestingly, however, anti-CD137 induced significantly increased levels of TNFα in serum of GF+FMT mice (Figure 3H), which was not observed in SOPF mice (Figure S3A). Anti-CD137-induced TNFα release was significantly inhibited in GF mice (Figure 3H). IFNγ levels in serum following anti-CD137 treatment were also significantly reduced in GF mice compared to GF+FMT mice (Figure 3I). Taken together, these data demonstrate that the gut microbiota plays a causal role in mediating the immunotoxicity induced by the IAAs anti-CD40 and anti-CD137.
Figure 3.

Fecal microbiota transplant (FMT) restores anti-CD40 and anti-CD137 induced immunotoxicity in germ-free (GF) mice
(A) Overview of the experimental design. Material for FMT sourced from age-matched specified and opportunistic pathogen-free (SOPF) mice.
(B) 16S rRNA gene sequencing was used to profile the composition of the fecal microbiota in SOPF and GF+FMT mice and revealed the microbiota of GF+FMT mice was similar to that of SOPF mice. Each boxplot represents a fecal sample from an individual mouse. Taxa with a mean relative abundance of <1% summed and visualized as a single feature.
(C–F) Serum (C) ALT levels, (D) liver histological score, (E) serum TNFα, and (F) IFNγ levels 24 h after control (PBS i.p.) or anti-CD40 treatment (100 μg i.p.) in GF or GF+FMT mice.
(G–I) Liver histological score (G), serum TNFα (H), and serum IFNγ (I) levels in samples collected from GF and GF+FMT mice 11 days after treatment initiation with anti-CD137 (100 μg i.p., 3 doses 4 days apart) or PBS.
(J) Linear regression analysis between serum ALT 24 h post-anti-CD40 treatment and fecal bacterial load (qPCR quantification of 16S rRNA gene copies).
(K–M) Levels of (K) ALT, (L) TNFα, and (M) IL6 in serum collected 24 h after control/anti-CD40 treatment of GF mice colonized with monocultures of Enterobacter cloacae, Clostridium scindens, or Akkermansia muciniphila. n = 5–17 mice per group.
Statistical significance was determined using a Mann-Whitney (C–I), Kruskal-Wallis test with Dunn’s post-test analysis (K–M), or linear regression analysis (E). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001. N.S., not significant. Data are represented as mean ± SEM. Results shown are from single independent experiments (B, D, G–I, and K–M) or pooled from 3 independent experiments (C, E, F, and J).
Interestingly, we found that there was a strong correlation between bacterial load in feces and levels of ALT following anti-CD40 treatment (Figure 3J). These data suggest that overall bacterial load in the gut microbiota may be an important factor mediating hepatotoxicity following IAA treatment. Consistent with this, colonizing GF mice (Table S1) with Enterobacter cloacae, a Gram-negative lipopolysaccharide (LPS)-producing commensal,32 Clostridium scindens, which is known to metabolize secondary bile acids,33 or Akkermansia muciniphila, which has been shown to modulate the liver damage induced by concanavalin A16 was not sufficient to restore anti-CD40-induced ALT, TNFα, or IL6 (Figures 3K–3M), which were comparable to levels observed in GF mice.
Antibiotic treatment suppresses anti-CD40-induced inflammatory responses in the liver
We next used RNA sequencing (RNA-seq) to profile gene expression in the livers of untreated and antibiotic-treated mice 24 h following control or anti-CD40 treatment. Multi-dimensional scaling analysis revealed that the primary sources of variation in the gene expression data were driven by anti-CD40 treatment followed by antibiotic treatment (Figure 4A). Treatment with anti-CD40 profoundly altered gene expression in the liver. >1,500 genes were significantly differentially expressed (false discovery rate [FDR] < 0.05) at least 2-fold in comparison to control mice (Table S2). Pathway analysis revealed that anti-CD40 treatment induced dramatic changes in the expression of a broad array of immune and metabolic pathways including the upregulation of genes involved in inflammatory responses, such as neutrophil degranulation, neutrophil and leukocyte chemotaxis, cellular response to LPS, cellular response to IFNγ and IFNβ, TNF signaling, and TLR signaling (Figures 4B and 4C; Table S3). Consistent with the well-established links between dysregulated lipid metabolism and liver inflammation and damage,34 anti-CD40 treatment led to a significant downregulation of genes involved in metabolism of lipids, amino acids, and bile acids (Figures 4B and 4C).
Figure 4.

Antibiotic treatment potently modulates anti-CD40-induced changes in gene expression and lipid metabolism in the liver
RNA sequencing was used to profile gene expression in liver samples collected from no ABX or ABX mice 24 h after treatment with control (PBS) or anti-CD40 (100 μg i.p.).
(A) Multidimensional scaling (MDS) analysis of the RNA sequencing data.
(B) Heatmap showing differentially expressed genes in selected statistically enriched Gene Ontology (GO) terms. Intensity represents Z score of log2 library size normalized counts.
(C and D) Top GO terms enriched (FDR < 0.05) among genes that were differentially expressed between (C) control and anti-CD40 treated mice and between (D) no ABX and ABX mice following anti-CD40 treatment. A complete list of enriched pathways and GO terms is available in Table S3.
(E and F) Ccl3 (E) and Ccl4 (F) normalized gene expression (log2 count per million). Data represents median and interquartile range. Statistical significance was assessed using EdgeR.
(G and H) Expression of (G) inflammatory response genes or (H) lipid and bile acid metabolism genes, which were significantly down- (orange) or up- (blue) regulated (FDR < 0.05) in ABX mice following anti-CD40 treatment. n = 5 mice per group from a single experiment.
(I–L) Negatively charged masses were quantitated in liver sections collected from no ABX, ABX, and GF mice by MALDI-mass spectrometry imaging. Four masses of interest identified as (I) cholesterol sulfate, (J) taurallocholic acid, (K) arachidonic acid, and (L) C17 sphingosine are shown as they showed the most substantive differences between no ABX and ABX or GF mice. All mice were treated with anti-CD40.
Anti-CD40-treated ABX mice had significantly altered expression of >500 genes (at least 2-fold) in comparison to control mice treated with anti-CD40 (Table S2). Genes involved in inflammation, chemotaxis, neutrophil degranulation, and TNF and TLR signaling had significantly reduced expression in anti-CD40-treated ABX mice (Figures 4D and S4A; Table S3). Genes involved in chemotaxis of monocytes and neutrophils that were downregulated in ABX + anti-CD40 mice included Ccl3 (Mip1α) and Ccl4 (Mip1β) (Figures 4E and 4F), and numerous inflammatory response genes were also downregulated (Figure 4G). These data indicate that the gut microbiota enhances the hepatotoxicity induced by anti-CD40 via the induction of inflammatory response pathways, which are dampened by antibiotic treatment.
Antibiotic treatment modulates anti-CD40-induced changes to lipid and bile acid metabolism in the liver
Our liver transcriptomics data also indicated that antibiotic treatment reduced the impact of anti-CD40 on pathways involved in metabolism, in particular, lipid and bile acid metabolism (Figures 4B and 4D; Table S3). Many genes involved in these processes were upregulated in comparison to anti-CD40-treated mice that were not treated with antibiotics (Figure 4H). Interestingly, genes involved in metabolism-related pathways, including PPAR signaling, lipid metabolism, and oxidative processes, were downregulated in ABX mice in the absence of anti-CD40 treatment (Figure S4B; Table S3). To confirm the microbiota-mediated alterations to lipid metabolism in the context of anti-CD40 treatment, we performed mass spectrometry imaging (MSI) to generate a spatially resolved map of the liver lipidome in untreated, ABX, and GF mice following anti-CD40 treatment. Consistent with previous reports,35 GF and ABX mice had an accumulation of cholesterol in their livers (Figure 4I). Additionally, we observed that ABX and GF mice had significantly reduced levels of bioactive lipids in the liver, including the bile acid taurallocholic acid, arachidonic acid, and C17 sphingosine (Figures 4J–4L). Targeted liquid chromatography-mass spectrometry (LC-MS) analysis of livers from GF and GF+FMT mice following anti-CD40 or control treatment also revealed that the levels of a number of primary and secondary bile acids in the liver were altered by the microbiota and, interestingly, by anti-CD40 treatment itself (Figure S4C). Antibiotic treatment also had a similar effect on liver bile acid levels (data not shown). Bioactive lipids such as sphingosines36 and microbiota-generated secondary bile acids37,38 are known to modulate metabolism and immune responses in the liver in other disease contexts. We therefore examined if inhibition of sphingosine kinase 1 (SPHK1) or bile acid sequestration via a 2% cholestyramine diet39 altered anti-CD40-induced immunotoxicity. Treating mice with the SPHK1 inhibitor PF-54340 did not reduce anti-CD40-induced liver damage or CRS compared to vehicle-treated controls (Figures S4D–S4F). As anticipated, many primary and secondary bile acids were potently reduced in mice fed a cholestyramine diet, without significantly altering the composition of the gut microbiota (Figures S4G and S4H), but these mice did not have significantly altered anti-CD40-induced liver damage (Figure S4I). Interestingly, we found that the CRS induced by anti-CD40 was partially dependent on bile acid metabolism, as mice fed the cholestyramine diet had significantly reduced levels of IL6 (Figure S4J), but not TNFα (Figure S4K), following anti-CD40 treatment. These data suggest that microbiota-mediated modulation of bile acid metabolism alters the systemic IL6 release, but not the liver damage, induced by anti-CD40.
The gut microbiota mediate the pathogenic immune cell infiltration into the liver induced by anti-CD40 and anti-CD137
Our liver RNA-seq data indicated that antibiotic-driven depletion of the gut microbiota potently modulated the inflammatory response to anti-CD40 in the liver. To investigate this further, we profiled liver immune cell infiltration and activation 24 h after anti-CD40 treatment, when anti-CD40-induced serum ALT was at its peak (Figure S5A). While antibiotic treatment did not alter overall leukocyte infiltration into the liver following anti-CD40 treatment (Figure S5B), the infiltration of CD11b+Ly6G– cells (macrophages and monocytes) was almost completely blocked in ABX mice (Figure 5A). Furthermore, macrophages and monocytes were less activated in ABX mice following anti-CD40 treatment, as indicated by significantly reduced cell surface expression of CD80 (Figure 5B) and CD86 (Figure S5C). Consistent with the effects on monocyte and macrophage activation being mediated via the microbiota, monocyte and macrophage activation was also potently reduced in GF compared to GF+FMT mice (Figures 5C and S5D). Interestingly, anti-CD40-induced infiltration of macrophages/monocytes was not significantly different between GF and GF+FMT mice (Figure S5E), suggesting that the effects of the gut microbiota are primarily at the level of activation. Anti-CD40-induced infiltration/activation of CD11c+MHCII+ conventional dendritic cells (cDCs) was also strongly abrogated in ABX mice (Figures S5F and S5G), while anti-CD40-induced alterations to CD8+ T cells, natural killer (NK)-cells, natural killer T cell (NKT)-like cells, and CD4+ T cells (CD4+TCRβ+) were not significantly different (Figures S5H–S5K).
Figure 5.

The gut microbiota modulate anti-CD40- and anti-CD137-induced inflammatory immune cell infiltration and activation in the liver
Flow cytometry analysis was performed on liver cells collected from no ABX and ABX mice or GF and GF mice recolonized by a fecal microbiota transplant (GF+FMT) 24 h after treatment with control (PBS) or anti-CD40 (100 μg i.p.).
(A–C) The number of macrophages/monocytes (CD11b+Ly6G–) per gram of liver (A) and (B and C) the frequency of liver macrophages/monocytes expressing CD80 in (B) no ABX and ABX mice or (C) GF and GF+FMT mice.
(D–F) Levels of (D) ALT, (E) TNFα, and (F) IL6 in serum collected 24 h after control or anti-CD40 treatment of mice that were either treated with control PBS-loaded liposomes (Lip-PBS) or clodronate-loaded liposomes (Lip-Clod) 24 h prior to anti-CD40 treatment.
(G and H) The number of neutrophils per gram of liver in (G) ABX and no ABX or (H) GF and GF+FMT mice.
(I) Heatmap showing the normalized expression of differentially expressed genes in the Reactome neutrophil degranulation pathway. Intensity represents the Z score of log2 library size normalized counts.
(J–L) Levels of (J) ALT, (K) TNFα, and (L) IL6 in serum collected 24 h after control or anti-CD40 treatment of mice that were either treated with PBS or anti-Ly6G (500 μg i.p.) 16 h prior to anti-CD40 treatment.
(M and N) The number of CD8+ T cells in livers collected from (M) ABX and no ABX mice or (N) GF and GF+FMT mice 11 days after treatment initiation with anti-CD137 (100 μg i.p., 3 doses 4 days apart) or PBS. n = 5–10 mice per group.
Statistical significance was determined using a Mann-Whitney test. ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001. N.S., not significant. Data are represented as mean ± SEM. Results shown are pooled from two independent experiments (A–H and J–M) or a single experiment (N).
Myeloid cells have previously been implicated in anti-CD40-induced liver damage.41,42 We evaluated the toxicity of anti-CD40 in mice depleted of macrophages via clodronate liposome injection. Mice injected with clodronate loaded liposomes had ∼70% fewer liver macrophages (CD11b+Ly6G−F4/80+) compared to control mice (Figure S6A), a level of depletion comparable with other studies.43,44 Mice depleted of their macrophages had potently reduced anti-CD40-induced liver damage (Figure 5D) and serum TNFα (Figure 5E) but, interestingly, did not have altered serum IL6 levels (Figure 5F). These data indicate that macrophages mediate anti-CD40-induced liver damage and systemic TNFα secretion. Macrophage depletion also modulated the liver immune response to anti-CD40, significantly reducing cDCs and NK infiltration, but not inflammatory monocyte or neutrophil infiltration, following treatment (Figures S6B–S6E).
We observed that neutrophils (CD11b+Ly6G+) were significantly increased in livers of mice following anti-CD40 treatment. While infiltration of neutrophils into the liver was not significantly reduced by antibiotic treatment (Figure 5G), neutrophil infiltration was significantly reduced in GF compared to GF+FMT mice following anti-CD40 treatment (Figure 5H). Furthermore, our RNA-seq data identified the neutrophil degranulation pathway as one of the most potently modulated pathways in ABX + anti-CD40-treated mice (Figure 5I). Taken together these data suggested that microbiota-mediated changes to neutrophil recruitment or activation could mediate anti-CD40-induced immunotoxicity in addition to macrophages. As anticipated,45 injection with an anti-Ly6G antibody almost completely eliminated liver neutrophils (Figure S6F). Consistent with our hypothesis, neutrophil depletion potently protected mice against anti-CD40-induced liver damage (Figure 5J) but, interestingly, did not significantly alter anti-CD40-induced TNFα (Figure 5K) or IL6 (Figure 5L) levels in serum. Neutrophil depletion had little impact on the frequency of other liver immune cell populations (Figures S6G–S6I) and, in fact, increased the number of macrophages in the liver following anti-CD40 treatment (Figure S6J). Given that depletion of neutrophils protected against anti-CD40-induced hepatotoxicity without reducing macrophage infiltration, these data suggest that neutrophils mediate the liver damage induced by anti-CD40 downstream of macrophages. Depletion of macrophages or neutrophils did not prevent anti-CD40-induced colitis, as assessed by fecal lipocalin-2 levels (Figures S6K and S6L).
We also assessed immune cell infiltration in the livers of control and ABX mice 11 days following anti-CD137 treatment initiation (i.e., when anti-CD137 induced toxicity peaks). As we observed for anti-CD40-treated mice, ABX mice had significantly reduced immune cell infiltration into the liver following anti-CD137 treatment including, notably, CD8+ T cells (Figure 5M). GF mice similarly had significantly reduced anti-CD137-induced CD8+ T cell liver infiltration compared to GF+FMT mice (Figure 5N). Given that anti-CD137-induced liver damage is known to be driven by pathogenic CD8+ T cells,28,30 these data indicate that signals from the microbiota are required for the pathogenic infiltration of CD8+ T cells into the liver induced by anti-CD137. As we observed in anti-CD40-treated mice, ABX and GF mice also had significantly reduced monocyte/macrophage infiltration into the liver of anti-CD137-treated mice (Figures S6M and S6N); however, macrophage depletion did not significantly alter anti-CD137-induced liver damage or CD8+ T cell liver infiltration (Figures S6O and S6P), although it did reduce anti-CD137-induced IFNγ in serum (Figure S6Q). Anti-CD137-induced infiltration of cDCs and CD4+ T cells into the liver was also significantly reduced by antibiotic treatment (Figures S6R and S6S). Anti-CD137-driven NK cell activation-induced cell death46 was not altered in ABX mice (Figure S6T). These data indicate that the gut microbiota mediates the liver toxicity induced by anti-CD40 and anti-CD137 by influencing the immune cell infiltration and activation of different immune cell populations in the liver.
The liver damage and CRS induced by anti-CD40 is dependent on TNFα and type I IFN (IFN-I) signaling, respectively
Next, we assessed whether specific cytokine signaling pathways mediate the influence of the microbiota on the immunotoxicity induced by anti-CD40. Given that anti-CD40-induced TNFα levels in serum were reduced in ABX and GF mice (Figures 1E and 3E) and the TNFα signaling pathway was potently modulated in the livers of anti-CD40 treated ABX mice (Figure 6A), we hypothesized that the gut microbiota modulates the immunotoxicity induced by anti-CD40 by amplifying the macrophage response to anti-CD40 ligation, leading them to produce pathogenic levels of TNFα. Consistent with our hypothesis, treating mice with an anti-TNFα-blocking antibody concurrently with anti-CD40 treatment completely abrogated anti-CD40-induced liver damage (Figure 6B). Furthermore, the systemic release of both TNFα (Figure 6C) and IL6 (Figure 6D) was also completely inhibited in mice treated with an anti-TNFα-blocking antibody. Blocking TNFα signaling also potently modulated liver immune responses to anti-CD40. While anti-CD40-induced infiltration of macrophages or monocytes was not significantly altered (Figure 6E), their activation, as assessed by the upregulation of CD80 and CD86 cell surface expression, was completely inhibited by TNFα blockade (Figures 6F and 6G). A similar effect on cDCs was also observed (Figures S7A–S7C). Blocking TNFα signaling also prevented the infiltration of neutrophils, NK cells, and CD8+ T cells into the liver following anti-CD40 treatment (Figure 6H, S7D, and S7E). Anti-CD40-induced colitis was also dependent on TNFα signaling, as mice treated with anti-TNFα-blocking antibody had significantly reduced fecal lipocalin-2 levels (Figure S7F). In contrast, anti-CD137-induced serum ALT levels and the infiltration of pathogenic CD8+ T cells were not altered in anti-TNFα-treated mice (Figures S7G and S7H). However, we did observe that the anti-TNFα blockade significantly increased serum levels of IFNγ following anti-CD137 treatment (Figure S7I), suggesting that IFNγ signaling might compensate for the inhibition of TNF signaling. We also observed that anti-CD40-induced expression of IL1β was significantly reduced in livers of ABX mice (Figure S7J); however, administration of an anti-IL1β-blocking antibody had no impact on anti-CD40-induced liver damage or CRS (Figures S7K and S7M).
Figure 6.

TNFα and type I interferon signaling mediate anti-CD40-induced liver damage and cytokine release syndrome, respectively
(A) Heatmap showing differentially expressed genes in the KEGG TNF signaling pathway in livers from no ABX and ABX mice 24 h after treatment with anti-CD40 (100 μg i.p.). Intensity represents the Z score of log2 library size normalized counts.
(B–H) Levels of (B) ALT, (C) TNFα, (D) IL6 in serum, (E) number of monocytes/macrophages (CD11b+Ly6G–) per gram of liver, frequency of liver monocytes/macrophages expressing (F) CD80 or (G) CD86, and (H) number of neutrophils (CD11b+Ly6G+) per gram of liver 24 h after control (PBS) or anti-CD40 treatment. Indicated groups were also treated concurrently with PBS or anti-TNF (200 μg i.p.).
(I) Heatmap showing selected downregulated interferon stimulated genes in livers from no ABX and ABX mice 24 h after treatment with anti-CD40.
(J–L) Levels of (J) ALT, (K) TNFα, and (L) IL6 in serum in co-housed Ifnar−/− or wild-type C57BL/6 (Ifnar+/+) mice after control (PBS) or anti-CD40 treatment. n = 7–10 mice per group.
Statistical significance was determined using a Mann-Whitney test. ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001. N.S., not significant. Data are represented as mean ± SEM. Results shown are from single independent experiments.
Our RNA-seq data also suggested that anti-CD40-induced immunotoxicity might be mediated via IFN-I signaling (Figure 6I), a pathway which is known to be influenced by the gut microbiota in other contexts.47 Interestingly, there was no significant difference in anti-CD40-induced liver damage between interferon alpha receptor (Ifnar)−/− and wild-type mice (Figure 6J); however, we did observe a potent decrease in anti-CD40-induced CRS in Ifnar−/− mice (Figures 6K and 6L). Anti-CD40, anti-tumor efficacy against MC38 tumors was not significantly altered in Ifnar−/− mice (Figure S7N). In conclusion, we found that TNF signaling plays a critical role in the liver damage and CRS induced by anti-CD40, while IFN-I signaling regulates anti-CD40-induced CRS only.
The immunotoxicity of anti-CD40 and anti-CD137 is dependent on MyD88 signaling
Pattern recognition receptors such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs) recognize specific microbial products from the gut microbiota such as LPS (TLR4)48 and peptidoglycan (NOD2).49 Our RNA-seq data suggested that TLR signaling was altered in antibiotic-treated mice compared to control mice following anti-CD40 treatment (Figure 7A), suggesting a potential pathway through which the microbiota could mediate effects on anti-CD40-induced immunotoxicity. We evaluated anti-CD40-induced immunotoxicity in Myd88−/− (an essential adaptor downstream of many TLRs48) mice and found that anti-CD40-induced serum ALT levels in Myd88−/− mice were not significantly different compared to wild-type control mice (Figure 7B). Consistent with our data showing that anti-CD40-induced liver damage is mediated by a pathogenic infiltration of immune cells, infiltration of monocytes/macrophages, cDCs, NK cells, and neutrophils following anti-CD40 treatment was not significantly altered in Myd88−/− mice (Figures S8A–S8D). Interestingly, Myd88−/− mice did have significantly reduced TNFα and IL6 levels in serum following anti-CD40 treatment (Figures 7C and 7D), indicating that a MyD88-dependent pathway mediates the CRS associated with anti-CD40 treatment. We also examined anti-CD40-induced CRS and liver damage in Tlr2−/−, Tlr4−/−, and Tlr5−/− mice; however, apart from a modest increase in serum IL6 in Tlr2−/− mice, CRS and liver damage induced by anti-CD40 was comparable to wild-type controls (Figures S8E–S8M). Nod2−/− mice also had similar levels of anti-CD40-induced ALT, TNFα, and IL6 compared to wild-type controls (Figures 7N–7P). The anti-tumor efficacy of anti-CD40 against MC38 tumors was also not significantly altered in Myd88−/−, Tlr4−/−, or Nod2−/− mice (Figures S8Q–S8S). These data suggest that while a MyD88-dependent pathway mediates the CRS induced by anti-CD40, this pathway is not required for anti-CD40-induced liver damage nor its anti-tumor efficacy. Taking all our data together indicates that anti-CD40 induces CRS in a manner that is dependent on the gut microbiota, MyD88 and IFN-Isignaling, and, in the case of IL6 release, bile acid metabolism, while, in contrast, anti-CD40-induced liver damage is mediated by the gut microbiota via TNFα signaling and a pathogenic activation of macrophages and neutrophils in the liver (Figure 7E).
Figure 7.

MyD88 signaling is required for anti-CD40-induced CRS and anti-CD137-induced CRS and liver damage
(A) Heatmap showing the normalized expression of differentially expressed genes in the Reactome TLR cascades pathway in livers from no ABX or ABX mice 24 h after treatment with anti-CD40 (100 μg i.p.). Intensity represents the Z score of log2 library size normalized counts.
(B–D) Levels of (B) ALT, (C) TNFα, and (D) IL6 in serum of littermate Myd88−/− and wild-type (Myd88+/+) mice collected 24 h after control (PBS) or anti-CD40 treatment.
(E) Overview of pathways, immune cells, and cytokines that mediate the influence of the gut microbiota on anti-CD40-induced CRS and liver damage.
(F–I) ALT levels in serum (F), number of CD8+ T cells (CD8+CD3+) per gram of liver (G), frequency of liver CD8+ T cells expressing granzyme β (H), and IFNγ levels in serum (I) of co-housed Myd88−/− and wild-type C57BL/6 (Myd88+/+) mice 11 days after initiation of control (PBS) or anti-CD137 treatment (100 μg i.p., 3 doses 4 days apart).
(J) Overview of pathways and immune cells that mediate the influence of the gut microbiota on anti-CD137-induced liver damage and systemic IFNγ release. n = 8–20 mice per group.
Statistical significance was determined using a Mann-Whitney. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. N.S., not significant. Data are represented as mean ± SEM. Results shown are pooled from 2 independent experiments (B–D) or from single independent experiments (A and F–I).
Given the potent effect of MyD88 on anti-CD40-induced CRS, we also examined whether the immunotoxicity induced by anti-CD137 was altered in Myd88−/− mice. Interestingly, we found that Myd88−/− mice had significantly reduced serum ALT levels (Figure 7F), suggesting that the gut microbiota modulates anti-CD137-induced liver damage via a MyD88-dependent pathway. Consistent with these data, the anti-CD137-induced infiltration of pathogenic CD8+ T cells was significantly reduced in Myd88−/− mice (Figures 7G and 7H). Anti-CD137-driven infiltration of monocytes/macrophages and neutrophils was also reduced in Myd88−/− mice, as were the proportion of activated CD8+ T cells expressing PD1 and Ki67 (Figures S9A–S9D). Serum IFNγ levels induced by anti-CD137 were also significantly reduced in Myd88−/− mice (Figure 7I). In conclusion, our study demonstrates that anti-CD137-induced liver damage is dependent on the gut microbiota, MyD88 signaling, and CD8+ T cells, while anti-CD137-induced systemic IFNγ release is dependent on the gut microbiota, MyD88 signaling, and macrophages (Figure 7J).
Discussion
The development of novel cancer immunotherapies, such as ICIs, holds substantial promise to significantly improve treatment outcomes for a number of cancer types; however, their efficacy is limited to tumors with significant immune infiltration. IAAs can sensitize immunologically cold tumors to ICIs by driving lymphocyte infiltration into these tumors and upregulating the expression of checkpoint receptors.50 Because of the potential of IAAs to improve responses to ICIs, research into IAAs has experienced a resurgence of interest over the last few years. Promisingly, one IAA, anti-ICOS, has recently progressed to phase III clinical trials,51 and 104 current phase I/II trials (https://clinicaltrials.gov) are assessing other IAAs, predominantly combined with other immunotherapies. Clinical trials testing anti-CD40 + anti-PD1 in pancreatic cancers insensitive to almost all treatments are also showing promising early results.52 Despite these advances, a significant obstacle to the clinical translation of IAAs is serious dose-limiting toxicities that include CRS, serious liver damage, and death.3 Here, we report that the gut microbiota is a critical mediator of the immunotoxicity induced by the IAAs anti-CD40 and anti-CD137. GF or antibiotic-treated mice were protected from the liver damage, CRS, and colitis induced by these IAAs. Importantly, given previous reports that the efficacy of ICIs is dependent on signals from the gut microbiota and can be abrogated by antibiotic treatment,18, 19, 20,27 we found that antibiotic treatment did not impair the anti-tumor efficacy of anti-CD40 or anti-CD137 in combination with the ICI anti-PD1. Interestingly, antibiotic treatment did not reduce the efficacy of anti-PD1 against MC38 tumors, indicating that anti-PD1 efficacy in this model is less dependent on an intact microbiota than the B16, MCA-205, and RET tumor models used in previous studies.18, 19, 20 Taken together, our data suggest that microbiota-targeted interventions may be a highly tractable approach to reduce the immunotoxicity associated with IAA immunotherapies, while also preserving anti-tumor efficacy.
We found that the gut microbiota mediated IAA-induced immunotoxicity in the liver by regulating the pathogenic infiltration of immune cells following treatment. GF and antibiotic-treated mice had significantly reduced infiltration and/or activation of monocytes, macrophages, and cDCs following anti-CD40 treatment. Depleting macrophages or neutrophils or blocking TNFα production was sufficient to prevent anti-CD40 induced liver damage, which is consistent with previous reports.41,53, 54, 55, 56 Our study adds to these previous reports by demonstrating that the gut microbiota is critically required for anti-CD40-induced pathogenic immune cell infiltration/activation in the liver and for anti-CD40-induced cytokine release. While both macrophages and neutrophils were required for anti-CD40-induced liver damage, only macrophage depletion reduced anti-CD40-induced systemic TNFα release. These data suggest that the gut microbiota enhance anti-CD40-induced macrophage activation in liver, which, in turn, promote increased liver neutrophil activation and degranulation, thereby significantly enhancing anti-CD40-induced liver damage. We also have identified roles for MyD88 and IFN-I signaling in mediating anti-CD40-induced CRS (IL6 and TNFα) but not liver damage. Interestingly, changes to bile acid metabolism also significantly influenced anti-CD40-induced IL6, but not TNFα, release. These data suggest that the gut microbiota mediates the liver damage and CRS induced by anti-CD40 via multiple, distinct pathways. Given that both IL6 and TNFα mediate the organ damage observed in a number of other contexts including sepsis,57 graft versus host disease (GVHD),58 and CRS induced by other immunotherapies such as blinatumomab or CAR-T cells,59 our study provides a clear rationale to investigate these pathways and the role of the gut microbiota in similar disease settings.
We also found that the gut microbiota potently modulate the liver damage induced by anti-CD137 in a MyD88-and CD8+ T cell-dependent manner. Previous studies have shown that CD8+ T cells are responsible for anti-CD137-induced liver damage.28,30 Consistent with these data, GF and ABX mice had significantly reduced anti-CD137-driven CD8+ T cell liver infiltration, indicating that the gut microbiota mediates anti-CD137-induced liver damage by enhancing the infiltration of pathogenic CD8+ T cells into the liver. In contrast to anti-CD40, we found a critical role for MyD88 signaling in anti-CD137-induced CD8+ T cell liver infiltration and liver damage, as well as systemic IFNγ secretion. MyD88-mediated sensing of the microbiota has been shown to influence T cell responses in other pathologies, such as type 1 diabetes and GVHD, directly60 or indirectly by influencing innate immune cell populations that subsequently influence T cell responses.61 Finally, we found that antibiotic treatment significantly reduced the liver damage induced by anti-CD137 + anti-PD1 combination therapy without impairing anti-tumor efficacy, which is of particular importance given this combination therapy is being actively investigated in clinical trials.62
Given our findings that combining antibiotic treatment with IAA immunotherapy significantly reduces IAA-induced immunotoxicity, our study has important implications for the development and use of IAAs clinically. There are approximately 26 different IAAs targeting 8 different co-activating receptors that are currently in phase I to III clinical trials.3 Serious grade 3 or above immune-related adverse events have been reported in ∼15% to 30% of patients treated with anti-CD405,6 and in ∼20% of patients treated with anti-CD137.7 Our data suggest that profiling the composition of the gut microbiota in patients could be a useful prognostic tool to identify individuals at greater risk of serious side effects. We encourage the collection of fecal samples from ongoing clinical trials so that this can be further assessed. Our data also suggest that microbiota-targeted interventions, including antibiotic therapy, FMT, probiotics, or small molecule inhibitors of microbial processes,63 may be useful approaches to abrogate the immunotoxicity induced by IAAs, without compromising their efficacy. For example, FMT has been shown to effectively treat the colitis induced by the ICI anti-CTLA4 in a small number of patients.64 Any interventions that can reduce immunotherapy-associated immunotoxicity will reduce cancer patient morbidity due to adverse immune toxicity and reduce the significant health care system costs associated with managing these adverse events. Simultaneously, such strategies can also enhance the efficacy of cancer immunotherapies by enabling higher doses of therapies to be administered due to a reduced risk of serious side effects.
Limitations of the study
While our study has demonstrated a critical role for the gut microbiota in mediating the immunotoxicity induced by IAAs in pre-clinical mouse models, clinical studies are now required to demonstrate that similar mechanisms are at play in patients receiving these therapies. Our study used murine antibodies targeting CD40 and CD137; therefore, further studies investigating the effects of the gut microbiota on the immunotoxicity induced by anti-CD40 and anti-CD137 human antibodies in humanized mice would be beneficial. Additionally, while we have identified a link between bacterial load and anti-CD40-induced immunotoxicity, our study does not rule out the possibility that specific taxa or bacterial products in the gut microbiota drive IAA-induced immunotoxicity. Finally, our study assessed the effect of antibiotic treatment on the efficacy of anti-CD40/CD137 alone or in combination with anti-PD1 against two subcutaneous tumor models. Further research should confirm these findings in additional orthotopic and spontaneous tumor models prior to assessment in the clinic.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD137, clone 3H3 (Isotype: Rat IgG2a) | Bio X Cell | Cat#BE0239; RRID:AB_2687721 |
| Anti-CD40: clone FGK4.5 (Isotype, Rat IgG2a) | Bio X Cell | Cat#BE0016; RRID:AB_1107647 |
| Anti-PD1: clone RMP1.14 (Isotype Rat 1gG2a) | Bio X Cell | Cat#BE0146; RRID:AB_10949053 |
| Rat IgG2a Isotype control: Clone 2A3 | Bio X Cell | Cat#BE0089; RRID:AB_1107769 |
| Anti-TNF: clone XT3.11 (Isotype Rat IgG1) | Bio X Cell | Cat#BE0058; RRID:AB_1107764 |
| Anti-IL1β: clone B122 (Isotype Armenian Hamster IgG) | Bio X Cell | Cat#BE0246;RRID:AB_2687727 |
| Anti-Ly6G: Clone 1A8 (Isotype Rat IgG2a) | Leinco Technologies | Cat#L280; RRID:AB_2737551 |
| FC blocking antibody Clone 2.4G2 (anti-CD16/32) | BD Biosciences | Cat#553141; RRID:AB_394656 |
| B220 PE-CF594 | BD Biosciences | Cat#562313; RRID:AB_11154399 |
| MHCII BV711 | BD Biosciences | Cat#563414; RRID:AB_2738191 |
| MHCII Vioblue | Miltenyi | Cat#130-123-278; RRID:AB_2802021 |
| NK1.1 APC | Miltenyi | Cat#130-117-528; RRID:AB_2727975 |
| Ly6C FITC | BD Biosciences | Cat#553104; RRID:AB_394628 |
| LY6G PE-Cy7 | BD Biosciences | Cat#560601; RRID:AB_1727562 |
| Ly6G PerCP-Vio700 | Miltenyi | Cat#130-103-861; RRID:AB_2660050 |
| TCRB FITC | BD Biosciences | Cat#553171, RRID:AB_394683 |
| FoxP3 Alexa647 | BD Biosciences | Cat#560401; RRID:AB_1645201 |
| F4/80 BV421 | BD Biosciences | Cat#565411; RRID:AB_2734779 |
| Ki67 BV395 | BD Biosciences | Cat#564071;RRID:AB_2738577 |
| PD1 PE | Miltenyi | Cat#130-102-299; RRID:AB_2661364 |
| CD3 biotin | eBioscience | Cat#11-0032-82; RRID:AB_2572431 |
| CD3-BV421 | BD Biosciences | Cat#562600; RRID:AB_11153670 |
| CD3 PE-Vio770 | Miltenyi | Cat#130-113-702; RRID:AB_2726243 |
| CD4 BV510 | BD Biosciences | Cat#563106; RRID:AB_2687550 |
| CD4 PE-Cy7 | Biolegend | Cat#100528; RRID:AB_312729 |
| CD8 APC-Cy7 | BD Biosciences | Cat#557654; RRID:AB_396769 |
| CD8 BUV395 | BD Biosciences | Cat#563786; RRID:AB_2732919 |
| CD11b BV711 | BD Biosciences | Cat#563168; RRID:AB_2716860 |
| CD11b PE | BD Biosciences | Cat#557397; RRID:AB_396680 |
| CD11c APC-Cy7 | BD Biosciences | Cat#561241; RRID:AB_10611727 |
| CD19 PerCP-Cy5.5 | BD Biosciences | Cat#551001; RRID:AB_394004 |
| CD19 PE-Vio770 | Miltenyi | Cat#130-113-732; RRID:AB_2733208 |
| CD80 FITC | Miltenyi | Cat#130-102-532; RRID:AB_2659265 |
| CD86 PE | Miltenyi | Cat#130-102-604; RRID:AB_2660746 |
| Granzyme B PE-CF594 | BD Biosciences | Cat#562462; RRID:AB_2737618 |
| Bacterial and virus strains | ||
| Enterobacter cloacae | PMID: 2974683 | N/A |
| Akkermansia muciniphila | DSMZ | Cat#DSM 22959 |
| Clostridium scindens | DSMZ | Cat#DSM 5676 |
| Biological samples | ||
| Mouse cecal samples for gavaging | C57BL/6J | Isolated from mice housed at SOPF facility, mice aged 6-8 weeks |
| Chemicals, peptides, and recombinant proteins | ||
| Streptavidin PE-dazzle | BioLegend | Cat#405207 |
| DAPI | BD Biosciences | Cat#564907 |
| Zombie Aqua | BioLegend | Cat#423101 |
| Phosphate buffered solution (PBS) | Sigma-Aldrich | Cat#D8537-500ML |
| Neomycin trisulfate salt hydrate | Sigma-Aldrich | Cat#N1876 |
| Ampicillin sodium salt | Sigma-Aldrich | Cat#A0166 |
| Dulbecco’s modified eagle medium | GIBCO | Cat#11960-044 |
| Fetal bovine serum | Assay Matrix | Cat#ASFBS-U |
| 2mM Glutamine | GIBCO | Cat#35050061 |
| 1mM Sodium pyruvate | GIBCO | Cat#11360070 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat#P4333-100ML |
| Trypsin-EDTA | Sigma-Aldrich | Cat#T4049-500ML |
| SYBR Green PCR Master Mix | Invitrogen | Cat#4309155 |
| TRIzol | Ambion | Cat#10296-028 |
| chloroform | Sigma-Aldrich | Cat#C2432 |
| 100% isopropanol | Sigma-Aldrich | Cat#I9516 |
| RNase-free water | Adelab | Cat#FISBIOUPW |
| 3M sodium acetate | Sigma-Aldrich | Cat#S2889 |
| Collagenase type IV | Thermofisher Scientific | Cat#17104019 |
| DNase I | Roche | Cat#4716728001 |
| RPMI 1640 | Sigma-Aldrich | Cat#R8758-500ML |
| Percoll | GE healthcare | Cat#P1644 |
| Pharm-Lyse Buffer | BD Biosciences | Cat#555899 |
| Bovine Serum Albumin | AusGeneX | Cat#PBSA |
| 0.5M EDTA | Invitrogen | CAT#15575038 |
| Clodronate Liposomes & PBS Liposomes | clodronate liposomes.com | N/A |
| PF-543 Hydrochloride | Sigma-Aldrich | Cat#PZ0234 |
| Mucin from porcine stomach | Sigma-Aldrich | Cat#M1778-10G |
| Oxoid Brain heart infusion agar | Thermofisher Scientific | Cat#CM1136 |
| Acetonitrile Optima® LC/MS | Fisher Scientific | Cat#A955-4 |
| Zirconia/silica beads 1 mm diameter | BioSpec Products | Cat#11079110z |
| Sodium Hydroxyde | Sigma-Aldrich | Cat#S5881 |
| n-Hexane hypergrade for LCMS LiChrosolv® | Sigma-Aldrich | Cat#1037011000 |
| Methanol hypergrade for LC-MS LiChrosolv® | Merck | Cat#1060351000 |
| Pierce Formic acid 99% 10x 1mL ampules | ThermoFisher Scientific | Cat#28905 |
| 5β-CHOLANIC ACID-3α, 7α, 12α-TRIOL-2,2,4,4-d4 (Cholic acid-D4) | Steraloids | Cat#C1900-015 |
| 23-NOR-5β-CHOLANIC ACID-3α, 7α, 12α-TRIOL (norcholic acid) | Steraloids | Cat#N2450-000 |
| 5β-CHOLANIC ACID-3α, 7α-DIOL-2,2,4,4-d4 (chenodeoxycholic acid-D4) | Steraloids | Cat#C0940-015 |
| 5β-CHOLANIC ACID-3α, 7α, 12α-TRIOL N-(CARBOXYMETHYL)-AMIDE-2,2,4,4 -d4 (glycocholic acid-D4) | Steraloids | Cat#C1925-015 |
| 5β-CHOLANIC ACID-3α, 12α-DIOL-2,2,4,4-d4 (deoxycholic acid-D4) | Steraloids | Cat#C1070-015 |
| 5β-CHOLANIC ACID-3α, 7α-DIOL N-(CARBOXYMETHYL)-AMIDE-2,2,4,4 -d4 (glycochenodeoxycholic acid-D4) | Steraloids | Cat#C0960-015 |
| β-Muricholic Acid | Santa Cruz | Cat#sc477731 |
| 5β-CHOLANIC ACID-3α, 6β, 7α-TRIOL (alpha muricholic acid) | Steraloids | Cat#C1890-000 |
| 5β-CHOLANIC ACID-3α, 6α, 7β-TRIOL (omega muricholic acid) | Steraloids | Cat#C1888-000 |
| 5β-CHOLANIC ACID-3α, 6β, 7β, -TRIOL N-(2-SULPHOETHYL)-AMIDE SODIUM SALT (TAURO β-MURICHOLIC ACID SODIUM SALT) | Steraloids | Cat#C1899-000 |
| Taurocholic acid | Santa Cruz | Cat#sc220189 |
| Taurolithocholic acid | Cayman Chemicals | Cat#17275 |
| 5β-CHOLANIC ACID-3α, 6α, 7α-TRIOL (hyocholic acid) | Steraloids | Cat#C1850-000 |
| 5β-CHOLANIC ACID-3α, 7α-DIOL N-(2-SULPHOETHYL)-AMIDE (taurochenodeoxycholic acid) | Steraloids | Cat#C0990-000 |
| 5β-CHOLANIC ACID-3α, 12α-DIOL N-(2-SULPHOETHYL)-AMIDE SODIUM SALT (taurodeoxycholic acid) | Steraloids | Cat#C1162-000 |
| 5β-CHOLANIC ACID-3α, 7β-DIOL N-(2-SULPHOETHYL)-AMIDE SODIUM SALT (tauroursodeoxycholic acid) | Steraloids | Cat#C1052-000 |
| 5β-CHOLANIC ACID-3α-OL N-(CARBOXYMETHYL)-AMIDE (glycolithocholic acid) | Steraloids | Cat#C1435-000 |
| 5β-CHOLANIC ACID-3α, 7α-DIOL N-(CARBOXYMETHYL)-AMIDE SODIUM SALT (glycochenodeoxycholic acid) | Steraloids | Cat#C0962-000 |
| 5β-CHOLANIC ACID-3α, 7β-DIOL N-(CARBOXYMETHYL)-AMIDE (glycoursodeoxycholic acid) | Steraloids | Cat#C1025-000 |
| 5β-CHOLANIC ACID-3α, 7α, 12α-TRIOL N-(CARBOXYMETHYL)-AMIDE SODIUM SALT (glycocholic acid) | Steraloids | Cat#C1927-000 |
| 5β-CHOLANIC ACID-3α, 12α-DIOL N-(CARBOXYMETHYL)-AMIDE SODIUM SALT (glycodeoxycholic acid) | Steraloids | Cat#C1087-000 |
| Cholic Acid 7-sulfate | Cayman Chemical | Cat#9002532 |
| Cholic acid | Sigma-Aldrich | Cat#C1129 |
| Deoxycholic acid | Sigma-Aldrich | Cat#D2510 |
| Lithocholic acid | Sigma-Aldrich | Cat#L6250 |
| Chenodeoxycholic acid | Sigma-Aldrich | Cat#C1050000 |
| 5β-CHOLANIC ACID-3α, 7β-DIOL (ursodeoxycholic acid) | Steraloids | Cat#C1020-000 |
| Critical commercial assays | ||
| Liquid ALT (SGPT) Reagent Set | Pointe Scientific | Cat#A7526 |
| MycoAlert™ assay | Lonza | Cat#9002-93-1 |
| Mouse IFN-γ enzyme-linked immunosorbent assay (ELISA) kit | BD OptEIA, BD Biosciences | Cat#555138 |
| Mouse TNF-α Uncoated ELISA Kit | Invitrogen | Cat#88-7324-88 |
| Mouse IL-6 Uncoated ELISA Kit | Invitrogen | Cat#88-7064-88 |
| DuoSet® Lipocalin-2/NGAL ELISA | R&D Systems | Cat#DY1857-05 |
| DNeasy PowerLyzer PowerSoil Kit | QIAGEN | Cat#12855-100 |
| DNA-free DNA Removal Kit | Thermofisher Scientific | Cat#AM1906 |
| Qubit dsDNA BR Assay Kit | Thermofisher Scientific | Cat#Q32853 |
| NEBNext High-Fidelity 2X PCR Master Mix | New England Biolabs, Inc. | Cat#50-591-079 |
| Sera-Mag Select™ | Cytiva | Cat#29343045 |
| LC Column 150x21 mm Kinetex® 2.6μm C18 100Å | Phenomenex | Cat#00F-4462-AN |
| OASIS HLB 1cc Vac RC Cartridge, 60 mg sorbent per cartridge, 30 μm particle size | Waters | Cat#186000381 |
| Deposited data | ||
| 16 s rRNA gene sequence data | This paper | SRA Bioproject: PRJNA668656 |
| RNA-Seq data | This paper | GEO: GSE159761 |
| Experimental models: Cell lines | ||
| MC38 | Donated by Dr. Susan Woods (SAHMRI) | N/A |
| AT3 | Donated by Fernando Souza-Fonseca-Guimaraes (WEHI) | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#000664, |
| Mouse: C57BL/6 (Myd88−/−). 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#009088 |
| Mouse: C57BL/6 (Tlr2−/−). 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#004650 |
| Mouse: C57BL/6 (Tlr4−/−). 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#007227 |
| Mouse: C57BL/6 (Tlr5−/−). 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#008377 |
| Mouse: C57BL/6 (Ifnar−/−). 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#028288 |
| Mouse: C57BL/6 (Nod2−/−). 6-16 weeks old, raised under specific opportunistic pathogen free conditions | The Jackson Laboratory (raised in SAHMRI bioresources) | Cat#005763 |
| Mouse: C57BL/6, 6-16 weeks old, raised under germ free conditions. | Translational Research Institute | N/A |
| Oligonucleotides | ||
| 16S Fwd: 5′-TCCTACGGGAGGCAGCAGT-3′ | Nadkarni et al. 65 | N/A |
| 16S Rv: 5′-GGACTACCAGGGTATCTAATCCTGTT-3′ | Nadkarni et al. 65 | N/A |
| SANGER 27-Fwd: 5′-AGAGTTTGATCMTGGCTCAG-3′ | Yang et al. 66 | N/A |
| SANGER 1492-Rv: 5′-CGGTTACCTTGTTACGACTT-3′ | Yang et al. 66 | N/A |
| SANGER 515-Fwd:5′-GTGCCAGCMGCCGCGGTA A-3′ | Yang et al. 66 | N/A |
| Software and algorithms | ||
| FlowJo™ | FlowJo, LLC | Version 10 |
| GraphPad Prism | GraphPad Software Inc. | Version 8 |
| Caseviewer | 3DHistech | Version 2.4 |
| Biorender | Biorender.com | N/A |
| Seq Scanner | Thermofisher Scientific | Version 2 |
| Profinder | Agilent Technologies | Version B.06.00 Build 6.0.625.0 |
| MasHunter PCDL Manager | Agilent Technologies | Version B.04.00 Build 92.1 Service Pack 1 |
| MassHunter Workstation Software LC/MS Data Acquisition for 6200 series TOF/6500 series Q-TOF | Agilent Technologies | Version B.05.01 Build 5.01.5125.1 |
| QIIME2 | Bolyen et al.67 | Version 2019.10 |
| FastQC | Babraham Bioinformatics | Version 0.11.3 |
| MultiQC | Ewels et al.68 | Version 1.8 |
| Trimmomatic | Bolger et al.69 | Version 0.38 |
| HiSAT2 | Kim et al.70 | Version 2.1.0 |
| FeatureCounts | Liao et al.71 | Version 1.5.0-p2 |
| R | R Core Team | Version 3.6.3 |
| SVASeq | Leek et al.72 | Version 3.3 |
| EdgeR | Robinson et al.73 | Version 3.26 |
| R code for analysis and plots | This paper | https://bitbucket.org/lynnlab/anti_cd40 |
| Matplotlib | Hunter74 | Version 3.3.2 |
| Custom Python scripts | This paper | https://bitbucket.org/lynnlab/anti_cd40 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Lynn (david.lynn@sahmri.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
C57BL/6 mice were used as experimental models to generate all data included in this study. C57BL/6 mice had a normal health/immune status except where specific genetically deficient or gnotobiotic (germ-free) mice were used. Mice were not subject to any procedures or drug treatment prior to use in experiments. Mice were maintained under standardized conditions with a 12 hr/day light cycle and controlled temperature (18-24°C) and humidity (45%–75%). Mice were co-housed in sex-specific groups of up to 5 mice per cage under Specific and Opportunistic Pathogen Free (SOPF) or gnotobiotic (GF) conditions, with details of each specific condition detailed below. Cages were provided with autoclaved play tunnels, paper and cotton nesting materials. Mice had access ad libitum to autoclaved water and commercial food pellets either irradiated diet (TD2918 Teklad 18% protein rodent diet, Envigo) for SOPF conditions or autoclaved diet (2018SX Teklad global 18% protein, Envigo) for GF conditions. The MC38 murine colon adenocarcinoma cell line75 or AT-3 breast cancer line76 were used to model cancer in mice. In vitro cell line culture conditions are outlined in the Method Details section. Cells were routinely tested for mycoplasma contamination using the MycoAlert™ assay (Lonza, USA) and were also confirmed to be free of 13 common mouse pathogens by commercial PCR testing (Compath). Cell line authentication was not undertaken.
SOPF C57BL/6 mice were bred and maintained in individually ventilated cages (TECNIPLAST) at the South Australian Health and Medical Research Institute (SAHMRI). Colony founders were sourced from the Jackson Laboratory. C57BL/6 Tlr4−/− and Myd88−/− mice were generated by backcrossing to C57BL/6 mice for littermate controls. C57BL/6 Tlr2−/−, Tlr5−/−, Myd88−/− (anti-CD137 experiment, Figures 7F–7I) and Nod2−/− mice were co-housed with wild-type SOPF C57BL/6 mice at 3-5 weeks of age for at least 4 weeks prior to use in experiments. The genotype of the mice was confirmed by a commercial provider (Garvan Institute) using PCR protocols provided by Jackson Laboratories.
GF C57BL/6 mice were purchased from the Translational Research Institute, Brisbane and housed at SAHMRI in positively pressurized, high-efficiency particulate air-filtered isolators (Park Biosciences) or HEPA filtered ISO-P cages (TECNIPLAST). GF mice were tested for sterility regularly, with water, feces and bedding tested commercially (ComPath) or by assessing bacterial load in feces by 16S rRNA gene RT-qPCR. Both male and female mice were used except for mice inoculated with AT-3 breast cancer cells, where only female mice were used.76 All experiments and procedures were executed in accordance with protocols approved by the SAHMRI Animal Ethics Committee.
Method details
Key experimental findings were replicated in independent experiments and the data from individual experiments pooled for analysis as stated in the figure legends. Stratification of mice to treatment groups was not undertaken except where tumor growth assessed, then mice were allocated to treatment groups to ensure similar tumor sizes at time of treatment initiation. Bile acid quantification and histological liver scoring were undertaken in a blinded fashion. Researchers were not blinded to treatment group allocation in other experiments. Sample size estimation was based on power calculations performed using data from a pilot experiment assessing the impact of antibiotic treatment on anti-CD40 induced ALT, IL6 and TNFα. A power calculator provided by the University of British Columbia https://www.stat.ubc.ca/∼rollin/stats/ssize/n2.html was used to estimate the appropriate sample size using parameters from this pilot experiment. A sample size of 8-10 was estimated to be required to have statistical power = 0.85 to detect significant differences between groups with 95% confidence. Data was not subject to any inclusion/exclusion criteria.
Antibiotic treatment
Mice were treated with neomycin (0.5mg/ml) and ampicillin (1mg/ml) dissolved in sterile drinking water. Mice had free access to treated drinking water throughout the experiments and the water was replaced three times weekly. Control mice had access to untreated drinking water ad libitum.
Fecal microbiota transplant into GF mice
To establish an intestinal microbiota, gnotobiotic mice received fecal microbiota transplants from SOPF mice. The cecal contents of aged-matched healthy, untreated control mice were extracted under anaerobic conditions. Cecal contents were pooled and diluted 3-fold in anaerobic (PBS). Gnotobiotic mice were administered the cecal material suspension via oral gavage, with a sterile flexible canicular. Successful microbiota colonization was confirmed by 16S rRNA gene qPCR or 16S rRNA gene sequencing.
Bacterial culture and transplant into GF mice
Enterobacter cloacae was isolated from the cecal contents of a SOPF C57BL/6 mouse at SAHMRI Bioresources that was colonized with high levels of Enterobacter (as shown by 16S rRNA gene sequencing) as described in Lynn et al.65
Akkermansia muciniphila (DSM 22959) and Clostridium scindens (DSM 5676) were purchased commercially (DSMZ, Germany). Monocultures were prepared from a pure culture plate under aerobic conditions using brain and heart infusion broth at 37 ᵒC. Briefly, 200 mL BHI broth or BHI with mucin (5%) was inoculated with 20 mL of the overnight culture and grown to an OD600 of 0.9. Pellets were produced by spinning down bacteria and washed in 1X PBS and resuspended in 1X PBS at 1x 109 CFU/mL. 100 μl of the suspension was administered to GF mice via oral gavage. Oral gavage was performed two times, 2-6 days apart and the monocultures were allowed to establish for 3 weeks prior to tumor inoculation.
Macrophage depletion
To deplete macrophages, mice were injected intravenously with 10 μL/g of bodyweight with either PBS, or clodronate loaded liposomes (5mg/ml) as per manufacturer’s instructions (Clodronateliposomes.org, the Netherlands). Depletion was performed 2-24 hours prior to administration of IAAs.
Cell line culture
The MC38 cell line, derived from C57BL/6 murine colon adenocarcinoma cells,75 was kindly donated by Dr. Susan Woods (SAHMRI). The AT-3 breast cancer line, established from cells of the primary mammary gland carcinoma of a MTAG transgenic mouse,76 was kindly donated by Fernando Souza-Fonseca-Guimaraes, (Walter and Elisa Hall Institute). Cell lines were cultured in culture medium (Dulbecco’s modified eagle medium, 10% Fetal bovine serum, 2 mM glutamine, 1mM Sodium Pyruvate and Penicillin-Streptomycin), at 37°C 10% CO2. Cell lines were passaged 3 times weekly in log phase by detaching adherent cells using trypsin-EDTA, centrifuging at 350 g and reseeded in flasks at a 1:5-1:10 dilution.
Tumor inoculation and monitoring
Cell lines in log phase at 60%–85% confluency were detached by trypsin-EDTA, washed once and resuspended to 1x107 cells/ml in DMEM without any additives. 1x106 cells (100 μL) were injected subcutaneously into the flank of mice. Female and male mice were injected with MC38 tumors while the AT-3 breast cancer cell line was injected into female mice only. Tumor growth was measured with Vernier Calipers weekly until the tumor area reached > 50mm2 after which tumor growth was measured 3 times weekly. Area was calculated as width x breadth. Mice were humanely euthanized through CO2 exposure when they reached a maximum tumor size of 1000 mm3.
Antibody administration
Mice were injected intraperitoneally (i.p.) with 100 μg of anti-CD40: clone FGK4.5 (Isotype, Rat IgG2a); anti-CD137: clone 3H3 (Isotype: Rat IgG2a), Rat IgG2a isotype control, clone 2A3; 200 μg of anti-PD1: clone RMP1.14 (Isotype Rat 1gG2a); anti-TNF: clone XT3.11 (Isotype Rat 1gG1); anti-IL1 β: clone B122 (Isotype Armenian hamster IgG) or 500 μg anti-Ly6G (Isotype Rat IgG2a) that were all purchased commercially (Bio X Cell or Leinco Technologies) and diluted in a solution of PBS for injection. Control mice were injected with either PBS vehicle control or Rat IgG2a isotype control. Neither PBS nor isotype control IgG2a induced serum ALT, cytokines or impacted tumor growth. Mice were treated as indicated in figure legends.
DNA extraction from fecal samples
Fecal samples were collected from mice at various time-points. Fecal samples were aseptically collected from mice and immediately snap frozen on dry ice and stored at −80°C until processing. Fecal samples were individually weighed and resuspended in 1mL of PBS, vortexed into a uniform suspension then pelleted by centrifugation at 16,200 g for 10 minutes. DNA was extracted from the fecal pellets using the DNeasy PowerLyzer PowerSoil Kit (QIAGEN). The manufacturer’s protocol was followed with the following modifications: fecal pellets were resuspended in 750 μL of Powersoil bead solution and 60 μL of C1 solution. The samples were incubated at 65°C for 10 minutes prior to bead beating.
Quantitative real-time PCR analysis to determine bacterial load
Quantitative real-time PCR (RT-qPCR) was undertaken to assess bacterial load in collected fecal samples. RT-qPCR was performed on a Quant Studio 7 Flex Real-time PCR system using primers outlined in Key resources table that target the conserved region of the 16S rRNA gene.67 Each reaction consisted of 5 μL SYBR green, 0.2 μM of each forward and reverse primer and 3 μL DNA template diluted in sterile water. Reactions were performed in triplicate and run against a no template control containing sterile water, substituted for the DNA template. The amplification program was 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 s and 60°C for 60 s. The total bacterial load was quantified against a standard curve of serial dilutions of E. coli genomic DNA using the calculation described in Nadkarni et al.67
16S rRNA gene sequencing
DNA extracted from fecal pellets were used to generate amplicons of the V4 hypervariable region of the 16S rRNA gene as described previously.65 Sequencing of the amplicon library was performed using an Illumina Miseq system (2 × 300bp run). 16S rRNA library preparation and sequencing was performed by the SAHMRI Genomics core. Paired-end 16S rRNA gene sequences were demultiplexed and imported into QIIME2 (release 2019.9) for processing.77 Sequences were error corrected, and counts of error-corrected reads per sample, which we refer to herein as exact sequence variants (ESVs), were generated with DADA2 version 1.8.78 A phylogenetic tree of error-corrected sequences was constructed with FastTree.79 Taxonomy was assigned to sequences with the sklearn plugin for QIIME2 with an 80% confidence threshold, using the GreenGenes 13.8 database.66 Further statistical analysis was carried out in R version 3.6.3, with graphing performed using ggplot2.
16S rRNA SANGER sequencing for identification of novel bacterial strains
Sanger sequencing to identify bacterial monocultures was undertaken using a protocol described by the University of Pennsylvania School of Veterinary Medicine, Center for Host-Microbial Interactions. Briefly, PCR amplification of DNA extracted from fecal samples using a reaction consisting of NEBNext High-Fidelity PCR Master Mix, 0.2 μM 27F primer, 0.2 μM 1492R primer and DNA template between 150-250ng/μL. The amplification program was set at 95°C for 2 minutes, 30 cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 1 minute 40 s, and 1 cycle of 72°C for 5 minutes. Samples were then incubated for 5 minutes with Sera-Mag™ Select DNA beads and placed on a magnetic stand until beads aggregated, and the solution was clear. Beads were then washed twice with 80% ethanol by incubating on a magnetic stand for 30 s and supernatant was discarded. Samples were then removed from the magnetic stand, resuspended in PCR grade water and incubated for 2 minutes, then placed back on a magnetic stand until the liquid was clear. Supernatant was then collected and Qubit quantification was performed on samples using the Qubit™ dsDNA BR Assay Kit following manufactures instructions. DNA concentrations was then adjusted to 25ng/μL with PCR grade water. Samples were sequenced by Australian Genome Research Facility, with each reaction consisted of sample at 25ng/μL, 3 μM 515F primer and PCR grade water. Sequences were analyzed using Seq Scanner 2 to confirm monoculture purity. Chromatograms were visually inspected to confirm that a single unique product was sequenced. The species identify was assigned via a BLASTn search against the NCBI nr nucleotide database as previously described.68
Liver RNA extraction
Liver sections (50-100 mg) were collected, snap frozen and stored at −80°C. Frozen liver samples were ground into powder and resuspended in TRIzol and incubated at room temperature for 15 minutes. The liver suspension was centrifuged for 2 minutes at 12,000 g and supernatant collected to eliminate particulate matter. RNA was extracted following the manufacturer’s protocol. Genomic DNA was removed from the RNA elution with the DNA-free DNA Removal Kit (Thermofisher) following the manufacturer’s instructions. Following DNase treatment, purity of RNA was improved by performing a sodium acetate re-precipitation by adding 1/10 dilution of 3M sodium acetate and 100% ethanol to reach a 70% ethanol concentration. Samples were then incubated overnight at 20°C. Samples were then spun for 30 min at 12000 g, washed twice in ethanol, allowed to air dry, and resuspended in RNase free water. RNA was quantitated using the QuBit RNA Broad Range Assay Kit (QIAGEN) and RNA integrity was confirmed using a Bioanalyser (ThermoFisher).
RNA Sequencing
Library preparation and RNA sequencing of liver RNA was undertaken by the SAHMRI Genomics core facility. Briefly, total RNA was converted to strand specific Illumina compatible sequencing libraries using the Nugen Universal Plus mRNA mRNA-Seq library kit from Tecan (Mannedorf, Switzerland) as per the manufacturer’s instructions (MO1442 v2). Briefly, 500 ng of total RNA was polyA selected and the mRNA fragmented prior to reverse transcription and second strand cDNA synthesis using dUTP. The resultant cDNA was end repaired before the ligation of Illumina-compatible barcoded sequencing adapters. The cDNA libraries were strand selected and PCR amplified for 12 cycles prior to assessment using an Agilent Tapestation to assess quality and a Qubit fluorescence assay for quantification. Sequencing pools were generated by mixing equimolar amounts of compatible sample libraries based on the Qubit measurements. Sequencing of the library pool was done using an Illumina Novaseq 6000 using a S1 flowcell with 2x100 bp paired-end reads.
Fastq read quality was visualized with FastQC version v0.11.3 and summarized with MultiQC.80 Adaptor sequences were removed with Cutadapt v2.8,69 and remaining sequences were quality filtered with Trimmomatic v0.3881 where a sliding window with a minimum PHRED score of 20 with a window size of 4, together with average quality of 30. Ribosomal RNA levels were estimated with SortMeRNA v2.1.70 Reads were then aligned to GRCm38 mouse genome (Ensembl release 99 annotation) with HiSAT2 v2.1.0 on default alignment parameters.71 Feature Counts v1.5.0-p2 was used to count aligned reads.72 Per sample counts were then imported into R v3.6.3 for further statistical analysis. Gene sets were filtered for at least 1 count per million in 3 samples prior to analysis and unknown sources of variation were removed using SVAseq.73 EdgeR was used to normalize the data (using trimmed mean of M-values method) and perform differential expression analyses (with the glmLRT function).82 Pathways and Gene Ontology (GO) enrichment analysis was performed using a hypergeometric test implemented in R version 3.6.
Serum alanine aminotransferase (ALT) assay
Serum ALT levels were measured using the Liquid ALT Reagent Set (Pointe Scientific) following the manufacturer’s instructions with the following modification. The reaction was scaled down from a 1 mL volume to 200 μL, to allow multiple samples to be run on a 96-well clear bottom black sided plate. ALT levels were measured on an EnSpire® Multimode Plate Reader (PerkinElmer) spectrophotometer at a temperature of 37°C.
Cytokine ELISA
Serum IFNγ levels were measured using the Mouse IFNγ enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences). Serum TNFα levels were measured using the Mouse TNFα Uncoated ELISA Kit (Invitrogen). Serum IL6 were measured using the Mouse IL6 Uncoated ELISA Kit (Invitrogen). All ELISAs were run in accordance with the manufacturer’s specifications.
Fecal Lipcalin-2 ELISA
Lipocalin-2 was measured in fecal samples using a method described previously.24 Briefly, fecal samples were weighed and then resuspended in PBS with 0.1% Tween® 20 and vortexed for 20 minutes to generate homogeneous suspensions. Samples were then spun at 13,800 g for 10 minutes at 4°C and then supernatant removed and diluted 1/100 in ELISA buffer and Lipocalin-2 detected using the DuoSet® Mouse Lipocalin-1/NGAL Kit. ELISA was run in accordance with the manufacturer’s specifications.
Flow cytometry
Tumors and livers were collected for immunophenotyping by flow cytometry. Single-cell suspensions were generated by mincing tumor tissue with scissors and incubating with 1mg/ml Collagenase type IV, 500 ng/ml DNase I and 2% FBS in RPMI 1640 for 45 minutes at 37°C. Tumor samples were then pushed through a 40 μm cell strainer and washed with PBS. Livers were pushed through a 70 μm cell strainer and leukocytes were selected by resuspending cells in a single layer of 37.5% Percoll and centrifuging samples for 12 minutes at 690 g, supernatant was discarded. Pharm-lyse buffer (BD Bioscience) was added to the pellet to lyse erythrocytes. Liver/tumor samples were resuspended in FC blocking antibody Clone 2.4G2 (anti-CD16/32) in FACS buffer (PBS, 0.1% BSA 2mM EDTA) in order to block Fc receptors and stained with the antibodies outlined in the Key resources table (all from BD Biosciences, eBioscience, Miltenyi or Biolegend) and incubated for 30 minutes on ice. In some experiments, intracellular staining was performed to detect transcription factors in cells. Samples were washed in FACS buffer and fixed in FoxP3 staining buffer (eBioscience) for 30 minutes on ice. Samples were then washed with perm buffer and intracellular antibodies; FoxP3, Granzymeβ or Ki67 (BD Bioscience) were added to samples and incubated for 30 minutes on ice; samples were washed with perm buffer followed by FACS buffer. Cells were then resuspended in FACS buffer and run on the BD LSRFortessa™ X-20 (BD bioscience). Dead cells were stained with DNA binding dyes DAPI (BD Bioscience) or ZombieAqua (Biolegend) prior to running on a flow cytometer and were excluded from analysis. Liquid counting beads (BD Biosciences) were added to samples to determine absolute counts. Data was analyzed using FlowJo™ v10 (FlowJo).
Isolation and profiling of bile acids from mouse liver
Snap frozen liver segments (∼50-100 mg) were spiked with a known concentration of each internal standard (D4-chenodeoxycholic acid, D4-cholic acid, D4-deoxycholic acid, D4-glycodeoxycholic acid, D4-glycochenodeoxycholic acid and norcholic acid) and then 500 μL of 0.2 M NaOH were added. After homogenization in a bead beater using zirconia/silica beads (1mm diameter, Daintree Scientific), the liver homogenates were incubated for 20 minutes at room temperature. Fats were removed through liquid:liquid extraction using hexane and bile acids isolated from the aqueous phase through solid phase extraction using OASIS-HLB columns (Waters) as previously described.35 Bile acids were eluted in 90% acetonitrile: water, dried using a nitrogen stream and reconstitute in 1:1 methanol:water. Chromatographic separation and bile acid identification were achieved through liquid chromatography-mass spectrometry as previously described.83
MALDI mass spectrometry imaging (MSI)
Snap frozen liver tissue was placed inside a Shandon Cryotome E (Thermo Scientific, MA, USA) at −20°C for 30 minutes prior to sectioning. Tissues were mounted onto the chuck with enough O.C.T. to securely hold the tissue while ensuring no O.C.T. contacted with the region to be sectioned, 10 μm tissue sections were cut and thaw mounted directly onto ITO slides (Bruker, Bremen, Germany). Tissue sections were washed by briefly dipping into cold 150 mM ammonium formate solution repeatedly for 20 s (method adapted from Angel et al.74). Slides were blotted dry around the tissue and placed in a vacuum desiccator for 60 minutes. After drying the slide was mounted onto the underside of the glass cold finger in a sublimation chamber (Ace Glass Incorporated, NJ, USA). Norharmane matrix (5 mg) was placed on a foil disk in the bottom of the sublimation chamber (prepared by air drying 200 μL of 25 mg/mL Norharmane in acetone on the foil disk). Sublimation was achieved by placing the system under vacuum (0.1 mBar) filling the cold finger with ice water and heating the matrix to 140°C for 10 minutes.
Norharmane coated liver sections were imaged using a timsTOF FleX mass spectrometer (Bruker, Bremen, Germany). The instrument was operated in negative ion mode and calibrated with Agilent ESI-L Low concentration tunning mix (Agilent technologies, CA, USA). Data was acquired with the following settings: m/z range 50-1250; laser energy 60% (a.u.); 250 shots per pixel with the 10kHz Smartbeam 3D laser, using a 20 μm pixel and step size. Imaging data was imported into SCiLS Lab (2020) software, spectra were aligned by moving peaks to local maxima before exporting the data in .csv format containing m/z, x-, y-coordinates and intensity information for each tissue section.
Mass identification from MALDI MSI
Custom Python scripts were written to process and clean mass spectra csv files and generate 2D visualizations (available from https://bitbucket.org/lynnlab/anti_cd40). Peaks with extremely low intensity (intensity < 100 AU) in all samples were excluded. Mean intensity fold change from No ABX to ABX and GF groups was calculated for each m/z peak. Peaks exhibiting an absolute log2 fold change of > 0.5 in either comparison were selected for further analysis. 2D visualization of selected mass peaks in tissue sections was performed using Matplotlib 3.3.2.84 Peaks were annotated with potential lipid matches found by searching the Lipid Maps Structure Database.85
Organ histological analysis
Portions of the liver, ear (skin), lung and colon were fixed in 10% neutral buffered formalin for 1-7 days and transferred to an 80% ethanol solution for storage. Organs were then embedded in a paraffin block and lateral cross-sections were cut and stained with hematoxylin and eosin (H&E) (Histology Services, University of Adelaide). Slides were scanned using a SCN400 F Brightfield and Fluorescence Slide Scanner (Leica Microsystems) at 20X magnification and the CaseViewer software (3DHISTECH Ltd) was used to visualize samples. Liver, skin and lung sections were histologically scored in accordance with the protocol described in Mayer et al.86 Liver sections (3-5 separate lobes/mouse) were scored by trained pathologists in a blinded manner.
Quantification and statistical analysis
Data was analyzed using a Mann-Whitney test, Kruskal-Wallis test with Dunn’s post-test analysis or one-way ANOVA with Tukey’s post-test analysis or as otherwise stated in the figure legends using GraphPad Prism 8 (GraphPad Software Inc.). Data are presented as means ± standard error of the mean (SEM) with P values ≤ 0.05 considered statistically significant.
Acknowledgments
We thank Anna Acuna, Chris Christou, the late Tim Kuchel, and the SAHMRI Bioresources/PIRL facility staff for assistance with mouse husbandry and breeding. We would also like to thank Randall Grose for assistance with flow cytometry, Mark Van der Hoek for assistance with RNA-seq, and Marten Snel, Paul Trim, and Jacob Truong for assistance with MSI. Several figures were created using Biorender.com. This work was supported by an Ideas grant (APP1180799) awarded to D.J.L. and S.J.B. and a Synergy grant (APP1183640) awarded to F.O.G. from the Australian National Health and Medical Research Council. This work was also supported by an EMBL Australia Group Leader award to D.J.L., a Tour De Cure Pioneering Research Grant (RSP-264-18/19), and a Flinders Foundation Health Seed Research Grant and by funding awarded to F.O.G. from The Health Research Board Ireland (ILP-POR-2019-004 and MRCG-2018-16).
Author contributions
Conceptualization, S.J.B. and D.J.L.; methodology, S.J.B., F.J.R., J.C.-M., F.O.G., and M.A.L.; investigation, S.J.B., J.J., J.C.-M., G.L.E., Y.C.T., S.C.B., D.J.T., A.S., N.E.S., J.W.F., H.K., F.O.G., M.A.L., and D.J.L.; writing – original draft, S.J.B. and D.J.L.; funding acquisition, S.J.B. and D.J.L.; writing – review & editing, J.J., F.J.R., D.L.W., S.L.W., and M.A.L.; data curation, F.J.R. and J.R.S.; visualization, F.J.R. and J.R.S.; project administration, G.L.E. and D.J.L.; formal analysis, J.W.F. and H.K.
Declaration of interests
S.J.B. and D.J.L. are co-inventors on International Patent Application no. PCT/AU2020/051278 related to this project. D.J.L. also receives funding from GSK for research not related to this project. All other authors declare no competing interests.
Published: December 8, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100464.
Supplemental information
Differentially expressed genes (FDR < 0.05) as determined by RNA sequencing of liver samples collected from control (No ABX) or antibiotic treated (ABX) mice 24 h after treatment with PBS (control) or anti-CD40.
Data and code availability
The 16S rRNA gene sequencing data have been deposited at NCBI Sequence Read Archive under BioProject: PRJNA668656 and the RNA-Seq data have been deposited in the Gene Expression Omnibus (GEO): GSE159761 and are publicly available as of the date of publication.
All data analysis code has been deposited in the Lynn Laboratory Bitbucket repository (https://bitbucket.org/lynnlab/anti_cd40).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Topalian S.L., Drake C.G., Pardoll D.M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok J.D., Chiarion-Sileni V., Gonzalez R., Rutkowski P., Grob J.J., Cowey C.L., Lao C.D., Wagstaff J., Schadendorf D., Ferrucci P.F., et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayes P.A., Hance K.W., Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018;17:509–527. doi: 10.1038/nrd.2018.75. [DOI] [PubMed] [Google Scholar]
- 4.Lesokhin A.M., Callahan M.K., Postow M.A., Wolchok J.D. On being less tolerant: enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci. Transl. Med. 2015;7:280sr1. doi: 10.1126/scitranslmed.3010274. [DOI] [PubMed] [Google Scholar]
- 5.Johnson P., Challis R., Chowdhury F., Gao Y., Harvey M., Geldart T., Kerr P., Chan C., Smith A., Steven N., et al. Clinical and biological effects of an agonist anti-CD40 antibody: a Cancer Research UK phase I study. Clin. Cancer Res. 2015;21:1321–1328. doi: 10.1158/1078-0432.CCR-14-2355. [DOI] [PubMed] [Google Scholar]
- 6.Vonderheide R.H., Flaherty K.T., Khalil M., Stumacher M.S., Bajor D.L., Hutnick N.A., Sullivan P., Mahany J.J., Gallagher M., Kramer A., et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J. Clin. Oncol. 2007;25:876–883. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 7.Segal N.H., Logan T.F., Hodi F.S., McDermott D., Melero I., Hamid O., Schmidt H., Robert C., Chiarion-Sileni V., Ascierto P.A., et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2017;23:1929–1936. doi: 10.1158/1078-0432.CCR-16-1272. [DOI] [PubMed] [Google Scholar]
- 8.Suntharalingam G., Perry M.R., Ward S., Brett S.J., Castello-Cortes A., Brunner M.D., Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 9.Aznar M.A., Tinari N., Rullán A.J., Sánchez-Paulete A.R., Rodriguez-Ruiz M.E., Melero I. Intratumoral Delivery of Immunotherapy-Act Locally, Think Globally. J. Immunol. 2017;198:31–39. doi: 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- 10.Riudavets M., Mosquera J., Garcia-Campelo R., Serra J., Anguera G., Gallardo P., Sullivan I., Barba A., Del Carpio L., Barnadas A., et al. Immune-Related Adverse Events and Corticosteroid Use for Cancer-Related Symptoms Are Associated With Efficacy in Patients With Non-small Cell Lung Cancer Receiving Anti-PD-(L)1 Blockade Agents. Front. Oncol. 2020;10:1677. doi: 10.3389/fonc.2020.01677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spencer S.P., Fragiadakis G.K., Sonnenburg J.L. Pursuing Human-Relevant Gut Microbiota-Immune Interactions. Immunity. 2019;51:225–239. doi: 10.1016/j.immuni.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan Y., Llorente C., Lang S., Brandl K., Chu H., Jiang L., White R.C., Clarke T.H., Nguyen K., Torralba M., et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–511. doi: 10.1038/s41586-019-1742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moschen A.R., Kaser S., Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol. Metab. 2013;24:537–545. doi: 10.1016/j.tem.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Sabino J., Vieira-Silva S., Machiels K., Joossens M., Falony G., Ballet V., Ferrante M., Van Assche G., Van der Merwe S., Vermeire S., Raes J. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut. 2016;65:1681–1689. doi: 10.1136/gutjnl-2015-311004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acharya C., Sahingur S.E., Bajaj J.S. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight. 2017;2:94416. doi: 10.1172/jci.insight.94416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W., Lv L., Shi D., Ye J., Fang D., Guo F., Li Y., He X., Li L. Protective Effect of Akkermansia muciniphila against Immune-Mediated Liver Injury in a Mouse Model. Front. Microbiol. 2017;8:1804. doi: 10.3389/fmicb.2017.01804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J., Wei Y., He J., Cui G., Zhu Y., Lu C., Ding Y., Xue R., Bai L., Uede T., et al. Natural killer T cells play a necessary role in modulating of immune-mediated liver injury by gut microbiota. Sci. Rep. 2014;4:7259. doi: 10.1038/srep07259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Routy B., Le Chatelier E., Derosa L., Duong C.P.M., Alou M.T., Daillère R., Fluckiger A., Messaoudene M., Rauber C., Roberti M.P., et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 19.Matson V., Fessler J., Bao R., Chongsuwat T., Zha Y., Alegre M.L., Luke J.J., Gajewski T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopalakrishnan V., Spencer C.N., Nezi L., Reuben A., Andrews M.C., Karpinets T.V., Prieto P.A., Vicente D., Hoffman K., Wei S.C., et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vétizou M., Pitt J.M., Daillère R., Lepage P., Waldschmitt N., Flament C., Rusakiewicz S., Routy B., Roberti M.P., Duong C.P., et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Racanelli V., Rehermann B. The liver as an immunological organ. Hepatology. 2006;43(2, Suppl 1):S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 23.Kimura K., Kakimi K., Wieland S., Guidotti L.G., Chisari F.V. Activated intrahepatic antigen-presenting cells inhibit hepatitis B virus replication in the liver of transgenic mice. J. Immunol. 2002;169:5188–5195. doi: 10.4049/jimmunol.169.9.5188. [DOI] [PubMed] [Google Scholar]
- 24.Chassaing B., Srinivasan G., Delgado M.A., Young A.N., Gewirtz A.T., Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS ONE. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ngiow S.F., Young A., Blake S.J., Hill G.R., Yagita H., Teng M.W., Korman A.J., Smyth M.J. Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res. 2016;76:6266–6277. doi: 10.1158/0008-5472.CAN-16-2141. [DOI] [PubMed] [Google Scholar]
- 26.Bajor D.L., Mick R., Riese M.J., Huang A.C., Sullivan B., Richman L.P., Torigian D.A., George S.M., Stelekati E., Chen F., et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. OncoImmunology. 2018;7:e1468956. doi: 10.1080/2162402X.2018.1468956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinato D.J., Howlett S., Ottaviani D., Urus H., Patel A., Mineo T., Brock C., Power D., Hatcher O., Falconer A., et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019;5:1774–1778. doi: 10.1001/jamaoncol.2019.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu L., Strahotin S., Hewes B., Zhang B., Zhang Y., Archer D., Spencer T., Dillehay D., Kwon B., Chen L., et al. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. J. Immunol. 2007;178:4194–4213. doi: 10.4049/jimmunol.178.7.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartkowiak T., Jaiswal A.R., Ager C.R., Chin R., Chen C.H., Budhani P., Ai M., Reilley M.J., Sebastian M.M., Hong D.S., Curran M.A. Activation of 4-1BB on Liver Myeloid Cells Triggers Hepatitis via an Interleukin-27-Dependent Pathway. Clin. Cancer Res. 2018;24:1138–1151. doi: 10.1158/1078-0432.CCR-17-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J., Blake S.J., Harjunpää H., Fairfax K.A., Yong M.C., Allen S., Kohrt H.E., Takeda K., Smyth M.J., Teng M.W. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016;76:5288–5301. doi: 10.1158/0008-5472.CAN-16-0194. [DOI] [PubMed] [Google Scholar]
- 31.Ngiow S.F., Young A., Jacquelot N., Yamazaki T., Enot D., Zitvogel L., Smyth M.J. A Threshold Level of Intratumor CD8+ T-cell PD1 Expression Dictates Therapeutic Response to Anti-PD1. Cancer Res. 2015;75:3800–3811. doi: 10.1158/0008-5472.CAN-15-1082. [DOI] [PubMed] [Google Scholar]
- 32.White P.J., Gaston M.A., Wilkinson S.G. Composition of O-antigenic lipopolysaccharides from Enterobacter cloacae. Microbiol. Immunol. 1984;28:1169–1179. doi: 10.1111/j.1348-0421.1984.tb00775.x. [DOI] [PubMed] [Google Scholar]
- 33.Marion S., Studer N., Desharnais L., Menin L., Escrig S., Meibom A., Hapfelmeier S., Bernier-Latmani R. In vitro and in vivo characterization of Clostridium scindens bile acid transformations. Gut Microbes. 2019;10:481–503. doi: 10.1080/19490976.2018.1549420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bechmann L.P., Hannivoort R.A., Gerken G., Hotamisligil G.S., Trauner M., Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012;56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 35.Sayin S.I., Wahlström A., Felin J., Jäntti S., Marschall H.U., Bamberg K., Angelin B., Hyötyläinen T., Orešič M., Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Spiegel S., Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma C., Han M., Heinrich B., Fu Q., Zhang Q., Sandhu M., Agdashian D., Terabe M., Berzofsky J.A., Fako V., et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiorucci S., Biagioli M., Zampella A., Distrutti E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018;9:1853. doi: 10.3389/fimmu.2018.01853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scaldaferri F., Pizzoferrato M., Ponziani F.R., Gasbarrini G., Gasbarrini A. Use and indications of cholestyramine and bile acid sequestrants. Intern. Emerg. Med. 2013;8:205–210. doi: 10.1007/s11739-011-0653-0. [DOI] [PubMed] [Google Scholar]
- 40.Pitman M.R., Costabile M., Pitson S.M. Recent advances in the development of sphingosine kinase inhibitors. Cell. Signal. 2016;28:1349–1363. doi: 10.1016/j.cellsig.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Byrne K.T., Leisenring N.H., Bajor D.L., Vonderheide R.H. CSF-1R-Dependent Lethal Hepatotoxicity When Agonistic CD40 Antibody Is Given before but Not after Chemotherapy. J. Immunol. 2016;197:179–187. doi: 10.4049/jimmunol.1600146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medina-Echeverz J., Ma C., Duffy A.G., Eggert T., Hawk N., Kleiner D.E., Korangy F., Greten T.F. Systemic Agonistic Anti-CD40 Treatment of Tumor-Bearing Mice Modulates Hepatic Myeloid-Suppressive Cells and Causes Immune-Mediated Liver Damage. Cancer Immunol. Res. 2015;3:557–566. doi: 10.1158/2326-6066.CIR-14-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tavares A.J., Poon W., Zhang Y.N., Dai Q., Besla R., Ding D., Ouyang B., Li A., Chen J., Zheng G., et al. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc. Natl. Acad. Sci. USA. 2017;114:E10871–E10880. doi: 10.1073/pnas.1713390114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiedner G., Hertel S., Johnston M., Dries V., van Rooijen N., Kochanek S. Selective depletion or blockade of Kupffer cells leads to enhanced and prolonged hepatic transgene expression using high-capacity adenoviral vectors. Mol. Ther. 2003;7:35–43. doi: 10.1016/s1525-0016(02)00017-5. [DOI] [PubMed] [Google Scholar]
- 45.Daley J.M., Thomay A.A., Connolly M.D., Reichner J.S., Albina J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 46.Vinay D.S., Kwon B.S. Therapeutic potential of anti-CD137 (4-1BB) monoclonal antibodies. Expert Opin. Ther. Targets. 2016;20:361–373. doi: 10.1517/14728222.2016.1091448. [DOI] [PubMed] [Google Scholar]
- 47.Zheng D., Liwinski T., Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30:492–506. doi: 10.1038/s41422-020-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ignacio A., Morales C.I., Câmara N.O., Almeida R.R. Innate Sensing of the Gut Microbiota: Modulation of Inflammatory and Autoimmune Diseases. Front. Immunol. 2016;7:54. doi: 10.3389/fimmu.2016.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al Nabhani Z., Dietrich G., Hugot J.P., Barreau F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017;13:e1006177. doi: 10.1371/journal.ppat.1006177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vonderheide R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell. 2018;33:563–569. doi: 10.1016/j.ccell.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garber K. Immune agonist antibodies face critical test. Nat. Rev. Drug Discov. 2020;19:3–5. doi: 10.1038/d41573-019-00214-5. [DOI] [PubMed] [Google Scholar]
- 52.Bear A.S., Vonderheide R.H., O’Hara M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell. 2020;38:788–802. doi: 10.1016/j.ccell.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stone M.L., Lee J., Herrera V.M., Graham K., Lee J.W., Huffman A., Coho H., Tooker E., Myers M.I., Giannone M., et al. TNF blockade uncouples toxicity from antitumor efficacy induced with CD40 chemoimmunotherapy. JCI Insight. 2021;6:146314. doi: 10.1172/jci.insight.146314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siwicki M., Gort-Freitas N.A., Messemaker M., Bill R., Gungabeesoon J., Engblom C., Zilionis R., Garris C., Gerhard G.M., Kohl A., et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci. Immunol. 2021;6:eabi7083. doi: 10.1126/sciimmunol.abi7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacoberger-Foissac C., Blake S.J., Liu J., McDonald E., Triscott H., Nakamura K., Smyth M.J., Teng M.W. Concomitant or delayed anti-TNF differentially impact on immune-related adverse events and antitumor efficacy after anti-CD40 therapy. J. Immunother. Cancer. 2020;8:e001687. doi: 10.1136/jitc-2020-001687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonnans C., Thomas G., He W., Jung B., Chen W., Liao M., Heyen J., Buetow B., Pillai S., Matsumoto D., et al. CD40 agonist-induced IL-12p40 potentiates hepatotoxicity. J. Immunother. Cancer. 2020;8:e000624. doi: 10.1136/jitc-2020-000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bozza F.A., Salluh J.I., Japiassu A.M., Soares M., Assis E.F., Gomes R.N., Bozza M.T., Castro-Faria-Neto H.C., Bozza P.T. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit. Care. 2007;11:R49. doi: 10.1186/cc5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tvedt T.H.A., Ersvaer E., Tveita A.A., Bruserud Ø. Interleukin-6 in Allogeneic Stem Cell Transplantation: Its Possible Importance for Immunoregulation and As a Therapeutic Target. Front. Immunol. 2017;8:667. doi: 10.3389/fimmu.2017.00667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maude S.L., Barrett D., Teachey D.T., Grupp S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsuoka S., Hashimoto D., Kadowaki M., Ohigashi H., Hayase E., Yokoyama E., Hasegawa Y., Tateno T., Chen X., Aoyama K., et al. Myeloid differentiation factor 88 signaling in donor T cells accelerates graft-versus-host disease. Haematologica. 2020;105:226–234. doi: 10.3324/haematol.2018.203380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wen L., Ley R.E., Volchkov P.Y., Stranges P.B., Avanesyan L., Stonebraker A.C., Hu C., Wong F.S., Szot G.L., Bluestone J.A., et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chester C., Sanmamed M.F., Wang J., Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. 2018;131:49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 63.Cully M. Microbiome therapeutics go small molecule. Nat. Rev. Drug Discov. 2019;18:569–572. doi: 10.1038/d41573-019-00122-8. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y., Wiesnoski D.H., Helmink B.A., Gopalakrishnan V., Choi K., DuPont H.L., Jiang Z.D., Abu-Sbeih H., Sanchez C.A., Chang C.C., et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat. Med. 2018;24:1804–1808. doi: 10.1038/s41591-018-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lynn M.A., Tumes D.J., Choo J.M., Sribnaia A., Blake S.J., Leong L.E.X., Young G.P., Marshall H.S., Wesselingh S.L., Rogers G.B., Lynn D.J. Early-Life Antibiotic-Driven Dysbiosis Leads to Dysregulated Vaccine Immune Responses in Mice. Cell Host Microbe. 2018;23:653–660.e5. doi: 10.1016/j.chom.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 66.DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K., Huber T., Dalevi D., Hu P., Andersen G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nadkarni M.A., Martin F.E., Jacques N.A., Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology (Reading) 2002;148:257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 68.Geva-Zatorsky N., Sefik E., Kua L., Pasman L., Tan T.G., Ortiz-Lopez A., Yanortsang T.B., Yang L., Jupp R., Mathis D., et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell. 2017;168:928–943.e11. doi: 10.1016/j.cell.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
- 70.Kopylova E., Noé L., Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 71.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 73.Leek J.T. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014;42:e161. doi: 10.1093/nar/gku864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Angel P.M., Spraggins J.M., Baldwin H.S., Caprioli R. Enhanced sensitivity for high spatial resolution lipid analysis by negative ion mode matrix assisted laser desorption ionization imaging mass spectrometry. Anal. Chem. 2012;84:1557–1564. doi: 10.1021/ac202383m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corbett T.H., Griswold D.P., Jr., Roberts B.J., Peckham J.C., Schabel F.M., Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35:2434–2439. [PubMed] [Google Scholar]
- 76.Stewart T.J., Abrams S.I. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J. Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 77.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J., Holmes S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Price M.N., Dehal P.S., Arkin A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ewels P., Magnusson M., Lundin S., Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caparrós-Martín J.A., Lareu R.R., Ramsay J.P., Peplies J., Reen F.J., Headlam H.A., Ward N.C., Croft K.D., Newsholme P., Hughes J.D., O’Gara F. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome. 2017;5:95. doi: 10.1186/s40168-017-0312-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hunter J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- 85.Sud M., Fahy E., Cotter D., Brown A., Dennis E.A., Glass C.K., Merrill A.H., Jr., Murphy R.C., Raetz C.R., Russell D.W., Subramaniam S. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007;35:D527–D532. doi: 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mayer C.T., Tian L., Hesse C., Kühl A.A., Swallow M., Kruse F., Thiele M., Gershwin M.E., Liston A., Sparwasser T. Anti-CD4 treatment inhibits autoimmunity in scurfy mice through the attenuation of co-stimulatory signals. J. Autoimmun. 2014;50:23–32. doi: 10.1016/j.jaut.2013.08.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed genes (FDR < 0.05) as determined by RNA sequencing of liver samples collected from control (No ABX) or antibiotic treated (ABX) mice 24 h after treatment with PBS (control) or anti-CD40.
Data Availability Statement
The 16S rRNA gene sequencing data have been deposited at NCBI Sequence Read Archive under BioProject: PRJNA668656 and the RNA-Seq data have been deposited in the Gene Expression Omnibus (GEO): GSE159761 and are publicly available as of the date of publication.
All data analysis code has been deposited in the Lynn Laboratory Bitbucket repository (https://bitbucket.org/lynnlab/anti_cd40).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.