

Summary
Dendritic cells (DCs) excel at cross-presenting antigens, but their effectiveness as cancer vaccine is limited. Here, we describe a vaccination approach using mesenchymal stromal cells (MSCs) engineered to express the immunoproteasome complex (MSC-IPr). Such modification instills efficient antigen cross-presentation abilities associated with enhanced major histocompatibility complex class I and CD80 expression, de novo production of interleukin-12, and higher chemokine secretion. This cross-presentation capacity of MSC-IPr is highly dependent on their metabolic activity. Compared with DCs, MSC-IPr hold the ability to cross-present a vastly different epitope repertoire, which translates into potent re-activation of T cell immunity against EL4 and A20 lymphomas and B16 melanoma tumors. Moreover, therapeutic vaccination of mice with pre-established tumors efficiently controls cancer growth, an effect further enhanced when combined with antibodies targeting PD-1, CTLA4, LAG3, or 4-1BB under both autologous and allogeneic settings. Therefore, MSC-IPr constitute a promising subset of non-hematopoietic antigen-presenting cells suitable for designing universal cell-based cancer vaccines.
Keywords: immunoproteasome, mesenchymal stromal cells, cancer vaccine, SILACs, transcriptomics, metabolomics, immunopeptidome, immune-checkpoint blockers, universal vaccine
Graphical abstract
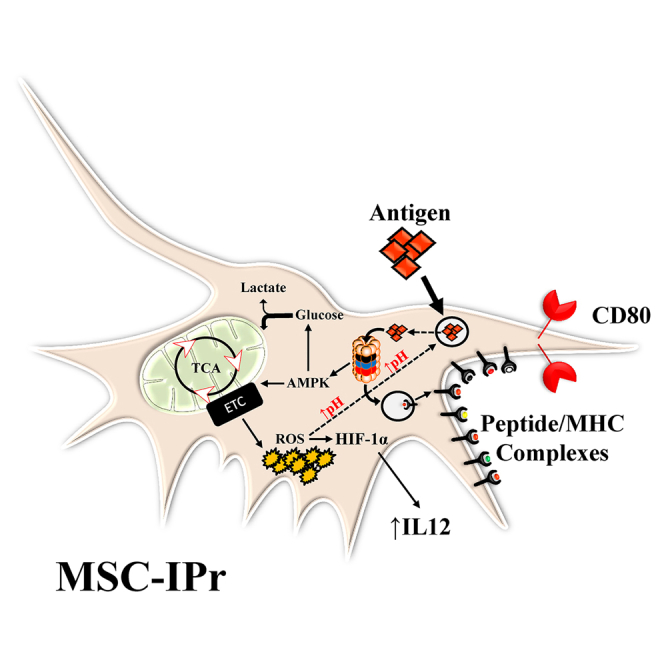
Highlights
-
•
Stable expression of the immunoproteasome in MSCs instills a pro-inflammatory phenotype
-
•
Induced metabolic alterations drive antigen cross-presentation by MSC-IPr
-
•
Engineered MSC-IPr elicit potent anti-tumoral immunity
-
•
The immunopeptidome of MSC-IPr is distinct from dendritic cells
Abusarah et al. demonstrate that mesenchymal stromal cells engineered to express the immunoproteasome (MSC-IPr) exhibit distinct antigen presentation properties, display altered metabolic signatures, and present a unique set of antigens compared with standard dendritic cells. These attributes make them ideal candidates for the future design of cell-based cancer vaccines.
Introduction
Dendritic cells (DCs) are crucial for activating cytotoxic T lymphocytes (CTLs), an essential part of the anti-tumoral immune response. Despite being clinically safe and immunogenic, the use of DCs in the development of whole-cell personalized cancer vaccines was hampered by their ineffectiveness at properly processing antigens and their limited in vivo persistence.1, 2, 3 In addition, the counts and impaired functionality of DC precursors derived from cancer-bearing patients render them inadequate for autologous cancer immunotherapy.1, 2, 3 Therefore, the generation of an antigen-presenting cell (APC) capable of bypassing the aforementioned barriers is needed.
DCs sample their environment to process and present exogenous antigen-derived peptides via their major histocompatibility complex class I (MHC class I) molecules. This mechanism, known as cross-presentation, is ideal for personalized cancer immunotherapy as it allows effective antigen presentation from a given lysate without prior knowledge of target antigens identity.4 Although cross-presentation is a known characteristic of DCs and macrophages,5,6 it can also be performed by other non-immune cell types including bone-marrow (BM)-derived mesenchymal stromal cells (MSCs) primed with interferon γ (IFNγ) (MSCγ).7 In this context, IFNγ acts as rheostat, where low levels promote a pro-inflammatory behavior and high/sustained levels are associated with immunosuppressive activity of MSCs.7 This implies that the induction of a strong inflammation in vivo can halt the APC-like function of MSCγ shortly after their administration. In addition, MSCγ express PD-L1 de novo, which impairs CTL effector functions.6,8, 9, 10, 11 To overcome these obstacles, we hypothesized that modulation of the proteasomal machinery in MSCs may permit their use as potent whole-cell-based cancer vaccines. Herein, we show that MSCs engineered to express the IPr subunits are superior to BM-derived DCs at cross-presenting antigens, resulting in potent anti-tumoral responses.
Results
IPr-expressing MSCs display an APC-like phenotype
Efficient antigen presentation relies on complex proteasomal-related machinery for the generation of MHC-bound peptides.12 The IPr is a specialized form of proteasome with peptide cleavage properties capable of generating stable and immunogenic peptide-MHC complexes.13, 14, 15, 16 In addition, its impact on functional programming of DCs is crucial.17 We therefore posited that MSCs engineered to express the IPr complex (MSC-IPr) would serve as stable and potent APCs. Transduction of MSCs using a retroviral vector encoding the IPr subunit genes (Figure S1A) led to their efficient gene expression in MSCs (Figure S1B) without altering their phenotype or differentiation plasticity (Figures S1C and S1D). However, MSC-IPr exhibited a lower proliferation potential (Figure S1E) and appeared larger in size and more granular than the control (Ctl) MSCs or MSCγ (Figures S1F–S1I). From an immunological perspective, MSC-IPr cells expressed high levels of MHC class I molecules (H2-Kb, H2-Db, and Qa2), are positive for CD80, and their PD-L1 expression remained negative (Figure S2A). Comparative analysis with mature BM-derived DCs shows higher MHCI levels on MSC-IPr than CD80, CD86, and PD-L1, which were more pronounced on the surface of DCs (Figure S2B).
Since MSCs secrete a panoply of soluble immune mediators, we next assessed whether IPr expression in MSCs affects their cytokine/chemokine profiles. Compared with DCs, both interleukin-4 (IL-4) and IL-10 were undetected in the supernatant of MSC-IPr in contrast to IL-12, which was significantly higher than DCs (Figure S2C). The chemokine profile of MSC-IPr, on the other hand, shared a closer phenotype to DCs with de novo secretion of CXCL1 and LIX in addition to enhanced MIP-1γ, RANTES, and CCL2 production (Figure S2D).
IPr expression selectively reprograms MSCs
Given that the IPr complex was introduced in resting MSCs, we first questioned its impact on the cellular degradome. Using stable isotope labeling by amino acids in cell culture (SILAC; Figure 1A), differentially expressed proteins with p values smaller than 5% were retained. Top regulated cellular processes were enriched for metabolism, protein folding, and antigen presentation and processing (Figure 1B; Figure S3). To further define these gains of function in MSC-IPr, an RNA sequencing (RNA-seq) experiment was conducted. Overall, 285 and 470 genes were significantly down- and upregulated, respectively (Figure 1C). To validate the molecular signatures differentiating MSC-IPr from Ctl MSCs, we performed a pre-ranked gene set enrichment analysis (GSEA) to assess enrichment of biological processes (Figure 1D) and pathways (Figure 1E). Consistent with the SILAC study, our findings pinpointed processes related to antigen presentation, immune responses, and metabolism that were significantly upregulated in MSC-IPr. By contrast, pathways associated with protein folding, amino acid turnover, endoplasmic reticulum (ER) stress, and pH reduction were depleted in MSC-IPr compared with Ctl MSCs (Figure 1E). Thus, both SILAC and RNA-seq studies revealed common reprogramming of specific biological processes by the IPr complex including protein folding, immunity, and metabolism.
Figure 1.

Functional characterization of MSC-IPr
(A) Representative diagram showing the design of the SILAC experiment.
(B) Bar plot showing the top enriched biological processes based on protein expression analysis from the SILAC experiment (shown in A) and corresponding adjusted p values (false discovery rate [FDR] < 0.05) from a hypergeometric test. The number of proteins contributing to the significance is shown in the x axis.
(C) Volcano plot showing the estimated fold changes (x axis) versus the minus log10 of the adjusted p values (y axis) from DESeq analysis. Significant genes with absolute value of log2 fold changes greater or equal to 2 are shown in green.
(D) Major biological processes groups based on gene expression analyses modulated in MSC-IPr in comparison with control (Ctl) MSCs (transduced with GFP-expressing retroviral particles; same backbone used for MSC-IPr). The annotations for this panel are MC (muscle contraction), IS (immune system), EO (extracellular organization), H (hemostasis), CC (cell cycle), Met (metabolism), ID (infectious diseases), GE (gene expression), and CCC (cell-cell communication).
(E) Plot showing the enriched Gene Ontology (GO) biological processes from an unbiased GSEA analysis of the differentially expressed genes between IPr and Ctl RNA-sequencing (RNA-seq) groups. The FDR threshold is set to 0.05. Features are ranked by the enrichment score from the Kolmogorov–Smirnov test (x axis).
See also Figures S1–S3.
Antigen presentation mediated by MSC-IPr is mechanistically distinct from DCs
Among the 755 differentially expressed genes detected by RNA-seq (Ctl MSCs versus MSC-IPr), 14.43% (109 genes) were related to antigen processing and presentation (Figure 2A). Upregulated genes included C-type lectins (Clec4m and CD209), proteins involved in docking and/or fusion of synaptic vesicles (Vamp3, Vamp8, and Snap23), and antigen processing (Psmb8, Psmb9, and Psmb10) and loading (Tapbp, Tap1, and Tap2). Interestingly, both Rab7a and Sec22b, which mediate endosome maturation and targeting to phagosomes, respectively, were downregulated in MSC-IPr (Figure 2A). Expression of the ER-associated proteins GPR58 (Pdai3), calnexin (Canx), and calreticulin (Calr) were also downregulated in MSC-IPr, suggesting a limited role for ER proteins in antigen processing and presentation (Figure 2A). Compared with DCs, however, the expression levels of Tapbp, Tap1, Tap2, Calr, and Nlrc5 were lower in MSC-IPr (Figure 2B). In light of these observations, we next monitored the kinetics of ovalbumin (OVA)-derived peptide-MHC complex formation at the cell surface. Although the turnover of the SIINFEKL-MHC class I signal was faster in DCs than Ctl MSCs or MSCγ, MSC-IPr dramatically sustained presentation of the peptide-MHC complex on its cell surface (Figure 2C, left panels). When the same experiment was repeated using the OVA protein, the SIINFEKL-MHC class I complex required 6 h before reaching maximal levels on the surface of DCs in contrast to 24 h for MSCγ (Figure 2C, right panels). For MSC-IPr, the peptide-MHC complex reached maximal levels 9 h post-OVA pulsing and remained stable for at least 24 h (Figure 2C, right panels). These observations correlated with the antigen presentation assay using soluble antigens (optimal at 5 mg/mL; Figure 2D; Figure S4A), demonstrating superior T cell responses when primed with MSC-IPr (Figure 2E). Given that MSCγ were ineffective at priming T cells, we suspected PD-L1 expression to be the main cause. We therefore induced PD-L1 expression on MSC-IPr by treating them with IFNγ prior to OVA pulsing and noticed no changes in T cell activation (Figures S4B and S4C). Since cytosolic delivery of captured antigen is an important step during cross-presentation, we next investigated whether Toll-like receptor (TLR) activation mediates antigen export to the cytosol.18 Interestingly, antigen cross-presentation using standard versus endotoxin-free (endograde) OVA did not lead to variations in T cell activation (Figure 2F), whereas absence of MHC class I molecules using H2-Kb/H2-Db-double deficient MSC-IPr (Figure 2G) triggered no OT-I T cell activation following SIINFEFKL or OVA pulsing (Figure 2H). Finally, DCs comprise several functionally specialized groups of which conventional type I DCs (cDC1) have the capacity to cross-present antigens and initiate highly potent anti-cancer immunity.19, 20, 21, 22, 23 We thus decided to compare the antigen presentation/cross-presentation capacity of MSC-IPr to ex-vivo-sorted cDC1 (Figure 2I). Although the T cell activation response following SIINFEKL pulsing was comparable between both cell types, antigen cross-presentation by OVA-pulsed cDC1 triggered a stronger T cell response than MSC-IPr (Figure 2J). Collectively, these results indicate that IPr expression in MSCs, and possibly other non-immune cell types (Figures S4D and S4E), instills abilities to cross-present antigens to responding T cells that are superior to standard DCs with some similarities to the cDC1 subset.
Figure 2.

Evaluating the antigen cross-presentation ability of MSC-IPr
(A) The heatmap represents the Z-scored expression level of the differentially expressed genes from the antigen presentation biological process in Ctl MSC versus MSC-IPr. Upregulated and downregulated genes are highlighted in red and blue, respectively.
(B) Transcript quantification of genes involved in antigen cross-presentation. Tested groups are (1) BM-derived mature DCs, (2) Ctl MSCs, (3) MSCγ, and (4) MSC-IPr.
(C) Representative flow cytometry analysis aimed at detecting the SIINFEKL/MHCI complex following pulsing with the SIINFELK peptide or OVA protein. Complex analysis was conducted at various time points to detect maximal activity (shown by the red arrowhead). Black histograms represent the population in question without peptide/protein pulsing (t = 0 –represents negative Ctl). The red dotted lines represent the threshold level according to unpulsed Ctls.
(D) An antigen-presentation assay conducted using MSC-IPr following pulsing with increasing concentration of OVA protein (0.01, 0.1, 1, and 5 mg/mL).
(E) Assessment of IFNγ production from OT-I CD8 T cells cultured with BM-derived mature DCs versus MSC-IPr pulsed with the SIINFEKL peptide or OVA protein for 9 h. DCs are shown in black, Ctl MSCs in gray, MSCγ in green, and MSC-IPr in red.
(F) The use of standard OVA or endotoxin-free OVA leads to similar T cell activation.
(G) Representative flow cytometry analysis of H2-Kb on MSC-IPr (red) versus MHC-class-I-deficient MSC-IPr (green). Isotype for H2-Kb (used on MSC-IPr) is shown in gray.
(H) Antigen presentation assay comparing wild type (WT) versus MHC-class-I-deficient MSC-IPr.
(I) Representative gating strategy used to sort cDC1 from the spleen of C57BL/6 mice.
(J) Antigen presentation assay comparing MSC-IPr and cDC1 following SIINFEKL or OVA pulsing.
For (B), (D) to (F), (H), and (J), n = 6/group, with ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (ANOVA test). Error bars represent standard deviation (SD). See also Figure S4.
Antigen routing in MSC-IPr is distinct from DCs
Efficient antigen cross-presentation by MSC-IPr relies on variable factors possibly involving enhanced capturing of extracellular antigens. To validate this assumption, time-dependent uptake of fluorescently labeled OVA was temporally assessed. Surprisingly, MSC-IPr exhibited a moderate increase in OVA internalization compared with BM-derived DCs (Figure 3A). This led us to speculate that intracellular routing of internalized OVA might be at play. We therefore tested the effect of various pharmacological inhibitors on cross-presentation by targeting antigen uptake, routing, or processing. The results shown in Figures 3B and 3C suggest that antigen capture by MSC-IPr is mediated by macropinocytosis as both antigen cross-presentation and uptake were impaired following dimethyl amiloride (DMA), phenylarsine oxide (PhenO), or cytochalasin (Cyto) D treatment. This observation was further confirmed using lucifer yellow CH (LY), which enters the cell by macropinocytosis (Figures 3D and 3E). Marginal inhibition in T cell activation was observed using the autophagy inhibitors 3-methyladenine (3-MA) and wortmanin (Figure 3B). However, this decrease was most likely due to diminished antigen uptake rather than autophagy inhibition (Figure 3C). Chloroquine (CQ; inhibitor of early endosome acidification) treatment, on the other hand, substantially enhanced antigen cross-presentation (Figure 3B) despite its negative impact on antigen uptake (Figure 3C), whereas brefeldin A (BFA; inhibitor of ER-to-Golgi transport), nocodazol (NocoD; inhibitor of late-stage endosomal transport), bafilomycin A (BfmA1; inhibitor of vacuolar H+ ATPase), and exotoxin A (ExoA; inhibitor of the Sec61 channel) had no effect on the cross-presentation ability of MSC-IPr (Figure 3B). In sharp contrast, addition of Eeyarestatin I (EER1), an inhibitor of the p97-associated deubiquitination process, completely abolished antigen cross-presentation (Figure 3B) without affecting antigen uptake (Figure 3C). The fact that antigen cross-presentation was unaffected by inhibitors targeting late endosomal transport, vacuolar acidification, or ER-Golgi vesicular transport indicates that MSC-IPr may not exclusively rely on the “cytosolic or vacuolar pathways,” but rather follows a route potentially involving early-recycling endosomes. In support of the latter claim, the efficiency of antigen processing is known to be negatively affected by rapid endo-lysosomal-mediated degradation of internalized antigens.24, 25, 26, 27, 28, 29 The use of the anti-malarial drug CQ confirmed this notion as blocking pH acidification in early endosomes of MSC-IPr further enhanced T cell activation as opposed to the marginal effect obtained with DCs (Figure 3F), bolstering the idea that antigen routing in MSC-IPr is distinct from DCs. When antigen processing was further evaluated over time in DCs and MSC-IPr using DQ OVA, a self-quenched OVA conjugate emitting a fluorescent signal upon proteolytic degradation, a prolonged/delayed OVA processing was detected in MSC-IPr treated with CQ (Figure 3G). In agreement with these results, a heatmap representing the pH reduction pathway revealed an overall downregulation of various V-type protein ATPases in MSC-IPr (Figure 3H) that was further confirmed by the decrease in the pHrodo Green Dextran signal, reflecting increased endosomal/vacuolar pH (Figures 3I and 3J). In sum, our data strongly indicate that antigen routing and stability in MSC-IPr are distinct from DCs and ends up preserving internalized antigens, resulting in potent T cell responses.
Figure 3.

Deciphering antigen routing in MSC-IPr
(A) Kinetic analysis of fluorescent OVA uptake by DCs (□), Ctl MSCs (▪), MSCγ (○), or MSC-IPr (●).
(B) Quantification of IFNγ production from OT-I-derived CD8 T cells co-cultured with OVA-pulsed MSC-IPr treated with various inhibitors targeting different intracellular compartments/processes. The red line represents the level of IFNγ produced by the positive Ctl (no inhibitor treatment).
(C) Representative flow cytometry assessment of fluorescent OVA uptake in the presence of selected inhibitors. The negative Ctl (no OVA) is displayed in black, whereas OVA pulsing on MSC-IPr (positive Ctl) is shown in red.
(D and E) Representative assessment of LY uptake by the different MSC populations using confocal microscopy (D) and its cognate quantification (E).
(F) A comparative antigen presentation assay using DCs or MSC-IPr in the absence or presence of CQ.
(G) Mean fluorescent intensity (MFI) quantification of DQ OVA signal in DCs or MSC-IPr in the absence or presence of CQ. The signal was obtained at different chase time points.
(H) The heatmap represents the Z-scored expression level of the differentially expressed genes from the pH reduction biological process in Ctl MSCs versus MSC-IPr. Upregulated and downregulated genes are highlighted in red and blue, respectively.
(I and J) Representative assessment of pHrodo Green by Ctl MSCs and MSC-IPr using confocal microscopy (I) and validated quantification by flow cytometry (J).
For (A), (B), (F), (G), and (J), n = 6/group, with ∗∗p < 0.01 and ∗∗∗p < 0.001. For (E), n = 816–1,982 vesicles/group, with ∗p < 0.05. Both ANOVA and Student’s t tests were used for this panel. Error bars represent SD.
The IPr complex reprograms the metabolism of MSCs
One of the IPr-affected biological processes identified by SILAC and RNA-seq is metabolism (Figures 1B and 1E). Thus, it logically follows to investigate whether the reprogrammed metabolic activities of MSC-IPr affect their ability to cross-present antigens. We first asked whether fluctuations in the expression of the glucose transporters Glut1 (Slc2a1) and Glut4 (Slc2a4) have occurred. Although the gene expression profile of Slc2a1 and Slc2a4 remained unchanged (Figure 4A), MSC-IPr exhibited higher cell surface levels of both glucose receptors, which could be due to decreased degradation/recycling from the cell surface and/or pronounced translocation from cytoplasmic vesicles to the cell surface (Figures 4B and 4C). Since the latter results suggest enhanced glucose metabolism in MSC-IPr, we next conducted a series of Seahorse experiments to assess the levels of glycolysis and mitochondrial respiration. An increase in oxygen consumption rate (OCR; reflecting oxidative phosphorylation in the mitochondria) was observed in MSC-IPr, whereas the extracellular acidification rate (ECAR; reflecting glycolysis activity) remained comparable with that of Ctl MSCs (Figure 4D). Furthermore, the percentage of activated/functional mitochondria (presented as MitoTracker Deep Redhigh MitoTracker Greenhigh) in MSC-IPr was relatively higher than in DCs, Ctl MSCs, or MSCγ (Figure 4E). To depict possible links between the observed enhanced mitochondrial function and antigen cross-presentation, MSC-IPr were treated with various pharmacological inhibitors affecting mitochondrial functions. All treatments impaired T cell responses, clearly indicating a critical function for mitochondrial activity in support of the antigen cross-presentation function of MSC-IPr (Figure 4F).
Figure 4.

Characterizing the metabolic signature of MSC-IPr
(A) Transcript quantification of Glut1 and Glut4 transporters. The white bars represent (1) BM-derived mature DCs (positive Ctl), (2) Ctl MSCs, (3) MSCγ, and (4) MSC-IPr.
(B) Flow cytometry assessment of Glut1 and Glut4 at the cell surface of Ctl MSCs (gray), MSCγ (green), and MSC-IPr (red). White histograms represent the isotype Ctl on MSC-IPr.
(C) MFI quantification for the experiment shown in (B).
(D) Assessment of metabolic activities by Seahorse. Both OCR and ECAR were assessed in Ctl MSCs (1), MSCγ (2), and MSC-IPr (3).
(E) Representative flow cytometry assessment of MitoTracker Green versus MitoTracker Red in the tested populations. The red gates represent functionally enlarged mitochondria.
(F) Assessment of MSC-IPr antigen cross-presentation in the presence of various mitochondrial/metabolic inhibitors.
(G) Representative western blot for the analysis of AMPK, FOXO1, and FOXO4.
(H) Assessment of MSC-IPr cross-presentation ability in the presence of increasing concentrations of AMPK inhibitors. The (−) group represents the condition without OVA, and the (+) group represents the positive Ctl with OVA.
(I) The cross-presentation ability of MSC-IPr derived from WT mice (1 and 2) versus MSC-IPr derived from AMPKα1−/− mice (3 and 4). OVA was added to conditions 2 and 4 only.
(J) Liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantification of AMP, ADP, ATP, and acetyl-CoA in Ctl MSCs versus MSC-IPr.
For all experiments shown in this figure, n = 6/group, with ∗∗p < 0.01 and ∗∗∗p < 0.001. Both ANOVA and Student’s t tests were used for this panel. Error bars represent SD. See also Figure S5.
The modulated mobilization of the glucose transporters (Glut1 and Glut4) combined with the increased mitochondrial activity observed in MSC-IPr might suggest an important requirement for cellular energy. Among all signaling pathways, adenosine-monophosphate (AMP)-activated protein kinase (AMPK) is considered as the energy rheostat regulating metabolism.30 As expected, AMPK and two of its target genes, forkhead family of transcription factors (FOXO)1 and FOXO4, were activated in MSC-IPr (Figure 4G). Although AMPK regulates energy homeostasis by blocking anabolic pathways, no changes in the overall translational activity of MSC-IPr were detected (Figure S5). Since AMPK triggers mitochondrial biogenesis, and mitochondrial function is central to T cell activation by MSC-IPr, we next evaluated the impact of AMPK inhibition on antigen cross-presentation. T cell activation was mitigated following pharmacological inhibition (Figure 4H) or genetic ablation (Figure 4I) of AMPK in MSC-IPr. Surprisingly, no major increase in AMP levels (the major inducer of AMPK activation) was detected in MSC-IPr, whereas the ratio of ADP to ATP and the levels of acetyl-coenzyme A (CoA) were enhanced (Figure 4J). In sum, these data indicate a link between IPr expression and cellular metabolic signatures, in particular, oxidative phosphorylation.
Oxidative phosphorylation is central to the cross-presentation function of MSC-IPr
In agreement with the enhanced metabolic activities observed in MSC-IPr, a comparative heatmap representing the ATP metabolic pathway revealed the upregulation of various metabolic genes (e.g., Hk1, Pfkl, Pgk1, Eno1, and Sdha) in MSC-IPr (Figure 5A). These observations prompted us to investigate whether such changes translate into fluctuations at the levels of glycolytic and/or tricarboxylic acid (TCA) cycle intermediates. Indeed, MSC-IPr absorbed more glucose (Figure 5B) and contained higher levels of glucose-6-phophate (G-6-P) and pyruvate (glycolysis intermediates; Figure 5C). Importantly, pyruvate was not entirely converted into lactate, but was instead used by the TCA cycle (Figure 5C). In addition, the increase in TCA cycle metabolites seems to rely mainly on glycolysis as glutamine uptake remained unchanged (Figure 5C). This enhanced mitochondrial activity translated into increased superoxide anion production as detected using the MitoSOX reagent (Figure 5D) and enhanced levels of hypoxia-inducible factor (HIF)-1α, most likely due to succinate accumulation through the TCA cycle (Figure 5E).31 To test whether mitochondrial-derived reactive oxygen species (ROS) play a role in antigen cross-presentation, MSC-IPr were treated with various antioxidants. N-Acetylcysteine (NAC) and glutathione (GSH) supplementation promoted marginal T cell activation, whereas catalase (to neutralize hydrogen peroxide) or sulforaphane (to activate the detoxifying pathway via Nrf2) treatments showed no effect (Figure 5F). T cell activation was also unaffected by apocynin (targeting the nicotinamide adenine dinucleotide phosphate oxidase NOX2) or S3QEL2 (inhibiting complex III of the electron transport chain) (Figure 5F).
Figure 5.

Comparing oxidative phosphorylation in Ctl MSCs versus MSC-IPr
(A) The heatmap represents the Z-scored expression level of the differentially expressed genes from the ATP metabolic process in Ctl MSCs versus MSC-IPr. Upregulated and downregulated genes are highlighted in red and blue, respectively.
(B) Quantification of fluorescent glucose uptake by MSCs.
(C) LC-MS/MS quantification of various glycolysis and TCA metabolites in the three MSC populations.
(D) Representative flow cytometry assessment of superoxide anion by MitoSox. DCs are shown in purple, Ctl MSCs in gray, MSCγ in green, and MSC-IPr in red.
(E) Representative western blot analysis of HIF-1α in Ctl MSCs, MSCγ, and MSC-IPr.
(F) Quantifying IFNγ levels produced by OT-I-derived CD8 T cells co-cultured with OVA-pulsed MSC-IPr treated with various inhibitors targeting ROS or mitochondrial activity.
(G) Representative western blot analysis of HIF-1α in MSC-IPr upon MMA treatment for 3 and 6 h.
(H) Transcript quantification of Sucnr1 by qPCR on DCs and MSC-IPr.
For (B) to (F) and (H), n = 6/group, with ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Both ANOVA and Student’s t tests were used for this panel. Error bars represent SD. See also Figure S6.
In sharp contrast, treatment of MSC-IPr with the vitamin E derivative α-tocopherol (lipid peroxidation blocker) or MitoTEMPO (mitochondrial inhibitor of ROS) ablated the antigen cross-presenting ability of MSC-IPr (Figure 5F), whereas succinate dehydrogenase inhibition using methymalonic acid (MMA) or 3-nitropropionic acid (3-NP) led to the opposite effect (Figure 5F). Since MMA blocks the conversion of succinate to fumarate, its use in MSC-IPr would lead to succinate buildup, which consequently resulted in HIF-1α stabilization (Figure 5G). These increases in HIF-1α levels may result from HIF prolyl-hydroxylase inhibition as MSC-IPr do not express the succinate receptor GPR91 (Figure 5H). Nevertheless, treatment with MMA enhanced the expression levels of H2-Kb/H2-Db and IL-12 production by MSC-IPr (Figure S6). Overall, these outcomes underline an important association between TCA cycle intermediates and antigen cross-presentation by MSC-IPr.
Prophylactic vaccination using MSC-IPr provides anti-tumoral responses superior to DC vaccines
To confirm the safety and tolerability of MSC-IPr vaccine in vivo, C57BL/6 female mice aged 6–8 weeks received two i.p. doses of 106 MSC-IPr at days 0 and 14. The mice were assessed by recording their weight and by taking samples from different organs upon their sacrifice at day 28. Tissue samples were sent for histological evaluation and showed no abnormalities or signs of toxicity in comparison with Ctl mice (Figure S7). Following confirmation that MSC-IPr administration to animals is well tolerated with no apparent signs of toxicity, the potency of the vaccine was next evaluated in an in vitro recall response using CD3+ T cells isolated from immunized animals. Compared with DCs, production of both IFNγ and tumor necrosis factor alpha (TNF-α) (Figure 6A) as well as the proliferation rate (Figures 6B and 6C) of T cells derived from the MSC-IPr group were superior. Thus, we evaluated the protective capacity of the vaccine using the E.G7 tumor model (expressing the OVA xenoantigen). Both DCs and MSC-IPr led to complete protection up to 7 weeks post-challenge (Figure 6D). However, the DC vaccine protective response was impaired when animals were systemically challenged with E.G7 tumor cells (Figure 6E).
Figure 6.

Characterizing the immune response generated by MSC-IPr
(A) Cytokine profiling of in-vitro-re-stimulated splenocytes derived from mice immunized at days 0 and 14. Splenocytes were collected 4 weeks following second dosing.
(B) CellTrace dilution of T cells derived from immunized mice shown in (A).
(C) Quantification of CellTrace MFI of the panel shown in (B).
(D) Prophylactic vaccination against E.G7 cells. C57BL/6 mice were vaccinated using OVA-pulsed DCs (green), Ctl MSCs (gray), MSCγ (blue), or MSC-IPr (red), followed by s.c. challenge with tumor cells at day 28. Non-immunized animals injected with the tumor cells are shown in black.
(E) Vaccination with OVA-pulsed DCs (green) or MSC-IPr (red) using similar plan as in (A), followed by challenge with tumor cells via the intravenous route on day 28. Ctl mice are shown in black, DCs in green, and MSC-IPr in red.
(F) C57BL/6 mice were vaccinated following the same plan used for (A) and then challenged with E.G7 cells (OVA-expressing EL4 cells) on one flank versus EL4 cells (non-OVA-expressing cells) on the contralateral flank. The groups are displayed as follows: non-vaccinated control mice receiving E.G7 or EL4 (white and black squares), vaccinated with DC/OVA (green and blue), and vaccinated with MSC-IPr/OVA (red and purple).
(G and H) Prophylactic vaccination against EL4 or B16 tumors following vaccination plan in (A). DCs or MSC-IPr were pulsed with tumor cell lysates and then used for vaccination. DCs are shown in green, MSC-IPr in red, and Ctl mice in black.
(I) Schematic diagram representing the vaccination schedule used with clodronate administration.
(J) Kaplan-Meier survival curve of the experiment shown in (I). Ctl mice are shown in black, MSC-IPr + liposome (in green), and MSC-IPr + clodronate in red.
(K) Schematic diagram representing the vaccination schedule used along with CD4, CD8, or NK1.1 depletion.
(L) Kaplan-Meier survival curve of the experiment shown in (K).
For (A) to (C), n = 6/group, with ∗∗p < 0.01. For (D) to (L), n = 10/group, with ∗∗p < 0.01 and ∗∗∗p < 0.001. Besides using the ANOVA test for (A) and (C), the log rank statistical test was used for the Kaplan-Meir survival curves. Error bars represent SD. See also Figure S7.
Since tumor cells produce a variety of immunosuppressive factors capable of impeding ongoing immune responses, we next questioned whether MSC-IPr can be effective at mounting an antigen-specific immune response in the presence of a secondary established tumor in the same host. For this purpose, animals were first immunized with OVA-pulsed DCs or MSC-IPr and then challenged simultaneously on both flanks with EL4 (non-OVA expressing) or E.G7 (OVA expressing). Both DCs and MSC-IPr mounted an OVA-specific immune response inhibiting E.G7 establishment, while EL4 grew on the opposite flank (Figure 6F). Notably, OVA strong immunogenicity may not reflect the standard immune response triggered against a given tumor-associated antigen. We therefore posited that the best approach to validate the potency of the MSC-IPr vaccine is to test its capacity using whole tumor cell lysates. Vaccination using MSC-IPr pulsed with the EL4 (lymphoma) or B16F0 (melanoma) lysate led to 70 and 80% protection, respectively, compared with 20 and 40% using DCs (Figures 6G and 6H). Mechanistically, these protective responses were not efferocytosis dependent as clodronate-treated mice undergoing MSC-IPr vaccination (Figure 6I) remained tumor-free despite two subsequent E.G7 re-challenges (Figure 6J). Further depletion studies (Figure 6K) revealed that both CD4 and CD8 T cells are required for MSC-IPr anti-tumoral immunity (Figure 6L).
Immune-checkpoint blockade synergizes with therapeutic vaccination to control established tumors
Given the impressive protection induced by prophylactic vaccination using MSC-IPr, we next assessed the ability of the vaccine to treat animals with pre-established tumors. Vaccination of animals with pre-established E.G7 lymphoma led to a delay in tumor growth, which was sustained for more than 6 weeks (Figure 7A). To determine the loss of therapeutic efficacy, we first questioned whether the growing tumor in MSC-IPr-vaccinated animals escaped immune rejection due to loss in OVA expression. Indeed, OVA was unapparent in the E.G7 tumors resected from vaccinated animals compared with in-vitro-grown E.G7 cells (Figure 7B). However, it is difficult to predict whether tumor escape was primarily caused by the loss of OVA expression or due to a concomitant prolongation of in vivo immune stimulation resulting in both immune-checkpoint expression and antigen loss. Indeed, cell surface analysis of CD8+-tumor-infiltrating lymphocytes (TILs) revealed the presence of PD-1+, LAG3+, and some TIM3+ CTLs (Figures 7B–7D) in tumors derived from MSC-IPr-vaccinated mice.
Figure 7.

Testing the therapeutic potency of MSC-IPr
(A) Kaplan-Meier survival curve. Tumor implantation is shown by the blue arrow, whereas vaccination is represented by black arrows. Tumor growth assessment of Ctl mice (black), DC (green), MSCγ (blue), or MSC-IPr (red). The anti-OVA western blot represents (1) OVA protein, (2) EL4, (3) cultured E.G7, and (4) E.G7 isolated from tumor masses.
(B and C) Flow cytometry assessment of immune-checkpoint expression by CD8 T cells isolated from tumor masses.
(D) A pie chart representing CD8 T cells expressing immune checkpoints.
(E) Kaplan-Meier survival curve of mice implanted with EL4 and vaccinated using lysate-pulsed MSC-IPr. MSC-IPr alone are in red, in combination with anti-41BB in dotted black line, anti-LAG3 in yellow, anti-CTLA4 in blue, or anti-PD-1 antibodies in orange. 4-1BB is represented by dotted blue line, anti-LAG3 is in gray, anti-CTL4 is in green, and anti-PD-1 is in purple.
(F) Kaplan-Meier survival curve of mice implanted as in (E) and receiving no treatment (black), anti-PD-1/4-1BB (purple), FTY720 (green), MSC-IPr + PD-1/4-1BB (orange), or MSC-IPr + PD-1/4-1BB and FTY720 (blue).
(G) Representative live in vivo imaging of mice implanted with luciferase-expressing Ctl MSCs versus MSC-IPr.
(H) Assessment of the signal decay for the experiment shown in (G).
(I and J) Experimental design and Kaplan-Meier survival curve of allogeneic vaccination using DCs (green), DCs + αPD-1 (dotted green), MSC-IPr (red), and MSC-IPr + αPD-1 (dotted red). Ctl mice are shown in black, whereas αPD-1 is in dotted black.
For (A) to (F) and (J), n = 10/group; for (G) and (H), n = 6/group, with ∗∗p < 0.01 and ∗∗∗p < 0.001. The log rank statistical test was used for the Kaplan-Meir survival curves. Error bars represent SD. See also Figures S8–S10.
Since therapeutic vaccination combined to anti-PD-1 administration failed in prolonging the therapeutic effect of the vaccine (Figure S8A), despite improved tumor infiltration by CD4+ and CD19+ cells (Figure S8B), we next tested whether enforced expression of the OVA antigen using a lentiviral vector would improve the therapeutic response. Surprisingly, treatment of mice harboring virally transduced EL4-OVA cells did not lead to the hypothesized outcome (Figure S8C). Since immunoediting against antigens can be caused by a strong selective pressure on a single antigen mediated by pro-inflammatory reactions, we repeated the vaccination protocol but used tumor lysates in combination with various immune-checkpoint blockers (αPD-1, αLAG3, or αCTLA-4) or an agonist stimulator (α4-1BB). Therapeutic vaccination of mice with pre-established tumors led to 30% survival in the group receiving the therapeutic vaccine alone (Figure 7E; Figure S9A). Although 4-1BB co-administration triggered marginal improvements (40% survival), the use of αLAG3, αCTLA-4, or αPD-1 further improved the protective responses of the vaccine, reaching 80% survival (Figure 7E). A subsequent vaccination trial combining the vaccine with αPD-1 and α4-1BB triggered a 100% mouse survival, with 8 of 10 animals achieving complete tumor elimination (Figure 7F; Figure S9B). A Similar outcome was observed using the B16F0 model, with 5 of 10 mice displaying tumor regression (Figures S10A–S10D). The loss of effectiveness observed with the use of FTY720 (inhibitor of T cell egress from secondary lymphoid organs) further bolstered the importance of active T cell recruitment in controlling tumor growth (Figure 7F; Figure S9B).
MSCs are in general known for their chemotactic and migratory abilities depending on the environmental cues surrounding them. We therefore examined whether the enhanced therapeutic response mediated by MSC-IPr corresponds to an extended in vivo survival and/or preferential migration to tumor sites and/or secondary lymphoid organs. Both Ctl MSCs and MSC-IPr engineered to express the firefly luciferase gene were administered to mice with or without pre-established tumors. Although Ctl MSCs were cleared out within 24–48 h post-administration, MSC-IPr persisted in the peritoneal cavity of injected mice up to 7 days post-administration (Figures 7G and 7H). Intriguingly, both Ctl MSCs and MSC-IPr failed to migrate to tumor sites or lymphoid organs. This suggests that although the therapeutic effectiveness of MSC-IPr may rely on both stable antigen cross-presenting ability (Figure 2C) and prolonged local in vivo survival, cells injected in the peritoneal cavity may be taken up by resident DCs that subsequently shuttle the generated peptide products to lymph nodes in order to prime responding T cells. Two in vivo studies were thus conducted to test this hypothesis. First, engineered H2-Kb/H2-Db-double deficient MSC-IPr pulsed with EL4 lysate were used along with anti-PD-1 and 4-1BB co-injection to vaccinate competent C57BL/6 mice with pre-established EL4 tumors (Figure S10E). Although no complete cure was observed, tumor growth was delayed compared with non-vaccinated mice, with all animals dying by the fifth week (Figures S10F and S10G). As these data indicate that DC cross-priming in vivo may be responsible for the observed delay in tumor growth, we next vaccinated Batf3-deficient mice (no cross-priming DCs) using EL4 lysate-pulsed MSC-IPr and obtained a similar response (Figures S10F and S10G). Altogether, our data suggest that MSC-IPr can directly present antigen to responding T cells (data from MHCI-deficient MSC-IPr), but requires endogenous DC cross-priming (data from Batf3-deficient mice) to trigger strong anti-tumoral responses. We thus vaccinated Batf3-deficient mice with EL4 lysate-pulsed H2-Kb/H2-Db-double deficient MSC-IPr to control both variables simultaneously. As shown in Figures S10H–S10J, no apparent anti-tumoral response was induced following such vaccination as all treated animals died within three weeks. In sum, these results clearly confirm that the anti-tumoral immunity mediated by MSC-IPr vaccination relies on both direct antigen presentation via MHC class I and endogenous DC cross-priming.
The anti-tumoral response of MSC-IPr is further enhanced under allogeneic settings
Personalized autologous cancer vaccines are appealing, but their manufacturing timeline and mass production limit their use. One way to overcome this would be the development of a one-size-fits-all cell-based vaccine. In fact, administration of allogeneic cells has been previously reported to elicit immune responses via three different, but not mutually exclusive, mechanisms including (1) an adjuvant-like effect triggered by allogeneic MHC-TCR interactions (priming 1∼10% of total T cells); (2) the delivery of bystander antigens to recruited endogenous DCs; and/or (3) enhanced recruitment of the short-lived KLRG1+ effector CD8 T cells.32, 33, 34 Based on this principle, we examined whether C57BL/6-derived DCs or MSC-IPr pulsed with the A20 B cell lymphoma tumor lysate would trigger beneficial anti-tumoral responses if administered to BALB/c mice pre-implanted with A20 tumor cells (Figure 7I). Although allogeneic MSC-IPr vaccination led to 60% survival (versus 0% using allogeneic DCs), its combination with αPD-1 (delivered after the second dosing) elicited complete protection versus 40% with allogeneic DCs (Figure 7J). These findings indicate that off-the-shelf allogeneic MSC-IPr formulations can be exploited as universal vaccines to trigger potent therapeutic anti-tumoral responses.
The tumor-derived antigen repertoire of MSC-IPr is distinct from DCs
The efficacy of the MSC-IPr vaccine could be considered multifactorial, involving a combination of efficient antigen cross-presentation, enhanced metabolic activities, pro-inflammatory cytokine production, and improved survival following in vivo delivery. However, the peptide repertoire generated following tumor lysate pulsing is of central importance as it enables the activation of CD8 T cells against physiologically relevant tumor antigens. Therefore, we next sought to compare the peptide repertoire of DCs and MSC-IPr following EL4 tumor lysate pulsing (Figure S11A). The set of peptides identified using MSC-IPr was not only distinct from DCs but also the number of collected peptides was over 4-fold higher (965 versus 224; Figure S11B). The peptide length in both cases was mainly eight to nine amino acids (Figure S11C) with overall similar binding affinities to various MHC class I molecules (Figure S11D). Analysis of the peptide motifs for the selected MHC class I molecules revealed a shared common hydrophobic amino acid at the C-terminal anchor position (Figure S11E). In sum, the MHC class I peptide repertoires of MSC-IPr and DCs are similar in terms of length, binding affinity, and peptide motifs, yet completely distinct from one another.
Discussion
Starting with the notion that MSCs exhibit large immune plasticity, we convey in this study a fresh introspective on their use in cancer immunotherapy. To properly behave as APCs and activate T cells, MSCs must first uptake, process, and present a given set of peptides derived from endocytosed antigens under inflammatory-promoting conditions.6 Although MSCs can effectively endocytose extracellular antigens, their ability to activate CTLs is limited, if not absent, due to their lack of antigen-processing and presentation capacity.6,7 Since the IPr complex generates peptides fitting snugly in MHC class I grooves resulting in stable peptide-MHC class I complexes, we exploited this concept and demonstrated that MSCs engineered to express the IPr complex gain an unprecedented antigen cross-presenting function.
The use of DCs in cancer immunotherapy faced various challenges. Since the lifespan of peptide-MHC complexes at the cell surface of DCs is short, the need for prolonged or sustained antigen presentation becomes essential.35, 36, 37 When assessed for their capacity to present SIINFEKL at the cell surface, MSC-IPr displayed stable peptide-MHC profiles irrespective of whether it was pulsed with a peptide or a protein. Furthermore, the ability of DCs to efficiently cross-present antigens may be limited by non-specific degradation mediated by various proteases during endosome maturation/acidification, leading to the destruction of important epitopes before they can be processed and loaded onto MHC class I.38, 39, 40 The fact that cross-presentation by MSC-IPr was unaffected by nocodazol or bafilomycin A indicates that endocytosed antigens are not sorted into mature endosomes or lysosomes. Instead, different routing most likely occurs in MSC-IPr, allowing intracellular antigen depot preservation within the endosome, from where continuous antigen processing and presentation results in the generation of sustained peptide-MHC class I complexes for efficient T cell priming.38, 39, 40 This may explain why MSC-IPr were strikingly superior to standard BM-derived DCs at triggering CD8 T cell responses. Nevertheless, the ability of MSC-IPr to process and present antigen-derived peptides (1) was qualitatively different from DCs as it occurred in an ER- and TLR-independent manner; (2) did not rely on enhanced expression of genes involved in antigen processing and peptide loading on MHC class I; and (3) was sustained despite IFNγ priming and PD-L1 expression. The sum of these immune-stimulatory effects culminated in eliciting protective and functional CTL responses in a CD4- and CD8-dependent manner.
One of the most salient observations in this study concerns the metabolic function of MSCs following IPr overexpression. Metabolic reprogramming usually results from an energy-consuming burden or fluctuation in energy levels. Initially, MSC-IPr were believed to exhibit high energy consumption due to sustained degradation of client proteins (>150 ATP molecules are required to degrade a single protein).41 As such, ATP depletion may lead to cellular starvation, which results in AMPK activation and mitochondrial biogenesis. Our data, however, do not support this hypothesis as no major fluctuations in AMP levels were detected. This begs the question: What is the link between AMPK activation and IPr overexpression in the absence of AMP depletion? Besides canonical AMPK activation pathways by the serine-threonine liver kinase B1 (LKB1) or by high intracellular calcium flux, AMPK can be activated following increased proteasomal activity.42 In these circumstances, rapid and sustained protein turnover through the IPr would trigger a “danger” proteolysis-based signal resulting in AMPK activation to upregulate starvation defenses.43 AMPK activation in this context turns on mitochondrial biogenesis, which results in increased TCA cycle intermediates and ROS production. The latter is particularly important due to its direct role in delaying endosomal acidification, while possibly disrupting endosomal membrane to allow antigen leakage into the cytoplasm.44,45 This correlates with our observations showing that blocking or neutralizing ROS mitigates antigen cross-presentation.
The currently available clinical results obtained with DC-based vaccines fell far below expectations.46 Even though a DC subset known as cDC1 can cross-present antigens slightly better than MSC-IPr, their trace numbers in peripheral blood (<0.1%) limits their therapeutic use.22,47 Engineered MSC-IPr represent therefore a robust alternative platform, devoid of DC shortcomings, and are suitable for creating distinct anti-tumor or anti-infectious immune responses superior to standard DC-based immune stimulation.
Limitations of the study
Although the use of luciferase to monitor cell migration in vivo is informative and provides important biological insights, this technique remains limited by its detection sensitivity. For instance, this approach may not provide detectable signals of smaller migrating MSC-IPr subsets to tumors or secondary lymphoid organs. Furthermore, the luciferase signal can be lost over time due to absent antibiotic selection (used in vitro to select transfected cells). Finally, a major limitation to the study is testing the potency of the human cellular ortholog (same limitation to human DCs) using an animal model. The successful completion of such study not only requires animals with fully reconstituted immune system including a wide repertoire of naive competent T cells tolerant to the host tissues but also needs a human-derived tumor compatible with the immune system initially used to reconstitute animals. Having said that, the MSC-IPr technology could be easily translated to the clinic as human MSC expansion can be initiated from healthy BM donors or by using any other permissive sources such as umbilical cords. Once a critical MSC cell number is reached, IPr engineering is conducted to generate and fully characterize a “Master Cell Bank.” As such, a vial of the “Master Cell Bank” MSC-IPr can be put in culture a week prior to vaccination to obtain the required cell dose prior to tumor lysate pulsing.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoPlus anti-mouse 4-1BB (CD137) | BioXCell | CAT# BP0239, RRID:AB_2894814 |
| InVivoPlus anti-mouse PD-1 (CD279) | BioXCell | CAT#: BP0146, RRID:AB_2894808 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | BioXCell | CAT#: BP0164, RRID:AB_2894809 |
| InVivoPlus anti-mouse LAG-3 | BioXCell | CAT#: BP0174, RRID:AB_2894812 |
| InVivoPlus anti-mouse TIM-3 (CD366) | BioXCell | CAT#: BP0115, RRID:AB_2894805 |
| InVivoPlus anti-mouse CD4 | BioXCell | CAT#: BP0003-1, RRID:AB_2891358 |
| InVivoPlus anti-mouse CD8α | BioXCell | CAT#: BP0061, RRID:AB_2891359 |
| InVivoPlus anti-mouse NK1.1 | BioXCell | CAT#: BE0036, RRID:AB_1107737 |
| Recombinant Alexa Fluor® 405 Anti-Glucose Transporter GLUT1 antibody | Abcam | CAT#: ab210438, RRID:AB_2895210 |
| Glut4 Antibody [Alexa Fluor® 647] | Novus Biologicals | Cat#: NBP1-49533AF647, RRID:AB_2895213 |
| Anti-Ovalbumin antibody | Abcam | Cat#: ab186717, RRID:AB_2895212 |
| Anti-Proteasome 20S LMP7 antibody | Abcam | Cat#: ab3329, RRID:AB_303708 |
| FoxO4 Antibody | Cell Signaling Technology | Cat#: 9472, RRID:AB_10831833 |
| HRP Anti-beta Actin antibody [AC-15] | Abcam | Cat#: ab49900, RRID:AB_867494 |
| Recombinant Anti-PSMB10/MECL1 antibody | Abcam | Cat#: ab183506, RRID:AB_2895211 |
| Recombinant Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] | Abcam | Cat#: ab140601, RRID:AB_2783797 |
| PE Rat Anti-Mouse CD105 | BD Biosciences | CAT#: 562759, RRID:AB_2737774 |
| PE-Cy7 Rat Anti-Mouse CD8a | BD Biosciences | CAT#: 552877, RRID:AB_394506 |
| APC Mouse Anti-Rat CD90/Mouse CD90.1 | BD Biosciences | CAT#: 561409, RRID:AB_10683163 |
| APC Rat Anti-Mouse CD274 | BD Biosciences | Cat#: 564715, RRID:AB_2687479 |
| APC Rat Anti-Mouse CD44 | BD Biosciences | Cat#: 559250, RRID:AB_398661 |
| APC-Cy7 Rat Anti-Mouse CD45 | BD Biosciences | Cat#: 557659, RRID:AB_396774 |
| BV421 Mouse Anti-Mouse H-2K[b] | BD Biosciences | Cat#: 562942, RRID:AB_2737908 |
| PE Hamster Anti-Mouse CD80 | BD Biosciences | Cat#: 553769, RRID:AB_395039 |
| PE Mouse Anti-Mouse H-2D[b] | BD Biosciences | Cat#: 553574, RRID:AB_394932 |
| PE Rat Anti-Mouse CD73 | BD Biosciences | Cat#: 550741, RRID:AB_393860 |
| PE Rat Anti-Mouse CD86 | BD Biosciences | Cat#: 5536922, RRID:AB_394994 |
| PE anti-mouse H-2Kb bound to SIINFEKL Antibody | BioLegend | Cat# 141604, 141603, RRID:AB_10895905, RRID:AB_10897938 |
| FoxO1 (C29H4) Rabbit mAb | Cell Signaling Technology | Cat#: 2880s, RRID:AB_2106495 |
| HIF-1α Antibody | Cell Signaling Technology | Cat #: 3716, RRID:AB_2116962 |
| K63-linkage Specific Polyubiquitin (D7A11) Rabbit mAb | Cell Signaling Technology | Cat#: 5621s, RRID:AB_10827985 |
| p38 MAPK Antibody | Cell Signaling Technology | Cat#: 9212s, RRID:AB_330713 |
| P-Akt (Ser473) Antibody | Cell signaling Technology | Cat#: #9271, RRID:AB_329825 |
| Phospho-FoxO1 (Ser256) Antibody | Cell signaling Technology | Cat#: 9461, RRID:AB_329831 |
| P-p38 MAPK (Thr180/Tyr182) Antibody | Cell signaling Technology | Cat#: 4511, RRID:AB_2139682 |
| Chemicals, peptides, and recombinant proteins | ||
| Methylmalonate | Cayman Chemical | Item No. 14885 |
| SIINFEKLE | GenScript | N/A |
| Chloroquine | InvivoGen | Cat#: tlrl-chq |
| CLODRONATE LIPOSOMES & CONTROL LIPOSOMES (PBS) | LIPOSOMA | CSKU: CP-005-005 |
| Recombinant Murine GM-CSF | PeproTech | Cat#: 315-03 |
| Recombinant Murine IFN-γ | PeproTech | Cat#: 315-05 |
| XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate | PerkinElmer | Cat#: 122799 |
| 2-Deoxy-D-glucose | Sigma-Aldrich | Cat#: D8375-1G |
| Accutase® solution | Sigma-Aldrich | Cat#: A6964 |
| Albumin from chicken egg white | Sigma-Aldrich | Cat#: A5503 |
| Alizarin Red S | Sigma-Aldrich | Cat#: A5533-25G |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma-Aldrich | Cat#: L2630-10MG |
| Oil Red O | Sigma-Aldrich | Cat#: O0625-25G |
| CellTrace Violet Cell Proliferation Kit, for flow cytometry | ThermoFisher Scientific | Cat#: C34557 |
| DQ ovalbumin | ThermoFisher Scientific | Cat#: D12053 |
| Lucifer Yellow CH | ThermoFisher Scientific | Cat#: L1177 |
| MitoSOX Red Mitochondrial Superoxide Indicator, for live-cell imaging | ThermoFisher Scientific | Cat#: M36008 |
| MitoTracker Deep Red FM | ThermoFisher Scientific | Cat#: M22426 |
| MitoTracker Green FM | ThermoFisher Scientific | Cat#: M7514 |
| Ovalbumin, Alexa Fluor 647 Conjugate | ThermoFisher Scientific | Cat#: O34784 |
| pHrodo Green Dextran, 10,000 MW, for Endocytosis | ThermoFisher Scientific | Cat# P35368 |
| pHrodo Red AM Intracellular pH Indicator | ThermoFisher Scientific | Cat#: P35372 |
| FTY720 | Tocris | N/A |
| Critical commercial assays | ||
| RNeasy Mini Kit (50) | QIAGEN | Cat#: 74104 |
| Mouse IFN-gamma DuoSet ELISA | R&D systems | Cat#: DY485-05 |
| Mouse IL-12 p70 Quantikine ELISA Kit | R&D systems | Cat#: M1270 |
| Mouse IL-6 Quantikine ELISA Kit | R&D systems | Cat#: M6000B |
| Mouse Chemokine Array C1 | RayBiotech | Cat#: AAM-CHE-1-2 |
| EasySep Mouse CD8a Positive Selection Kit | StemCell Technologies | Cat #: 18753 |
| EasySep Mouse CD8a Positive Selection Kit II | StemCell Technologies | Cat#:18953 |
| Deposited data | ||
| RNA-seq data for ctl MSCs and MSC-IPr | GEO | GEO: GSE183055 |
| Experimental models: Cell lines | ||
| Mouse: A20 | ATCC | CAT# TIB-208, RRID:CVCL1940 |
| Mouse: B16F0 | ATCC | ATCC Cat# CRL-6322, RRID:CVCL0604 |
| Mouse: E.G7-OVA [derivative of EL4] | ATCC | ATCC Cat# CRL-2113, RRID:CVCL3505 |
| Mouse: EL4 | ATCC | Cat# TIB-39, RRID:CVCL0255 |
| 293-GP2 | Clontech | RRID:CVCLWI48 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/cAnCrl | Charles River | Strain code: 028 |
| Mouse: C57BL/6NCrl | Charles River | Strain code: 027 |
| Mouse: OT1 (C57BL/6-Tg(TcraTcrb)1100Mjb/J) | The Jackson Laboratory | Strain code: 003831 |
| Mouse: Batf3-deficient mouse (B6.129S(C)-Batf3tm1Kmm/J) | The Jackson Laboratory | Strain code: 013755 |
| Mouse: MHCI-deficient mouse (B6.129P2-H2-K1tm1Bpe H2-D1tm1Bpe/DcrJ) | The Jackson Laboratory | Strain code: 019995 |
| Recombinant DNA | ||
| Murine IPr construct | This paper | n/a |
| Software and algorithms | ||
| FlowJo v10 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism-GraphPad | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Software used for statistical analysis | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Moutih Rafei (moutih.rafei.1@umontreal.ca).
Materials availability
The IPr construct and MSC-IPr generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Experimental model and subject details
Mice strains
For all experiments, C57BL/6 and BALB/c mice were purchased from Charles River (Senneville, QC, Canada) whereas OT-1 MHCI- (B6.129P2-H2-K1tm1Bpe H2-D1tm1Bpe/DcrJ) and Batf3-deficient mice (B6.129S(C)-Batf3tm1Kmm/J) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). The Vav-Cre AMPKa1-floxed mice (to isolate AMPK−/− MSCs) were a generous gift from Dr. Russel Jones (McGill University, Montreal, QC, CA). Littermate mice were interbred and housed in a pathogen-free environment at the animal facility of the Institute for Research in Immunology and Cancer (IRIC). Animal protocols were approved by the Animal Care Committee of Université de Montréal.
Cell lines
EL4, E.G7, B16F0, A20 used in this study were obtained from ATCC. 293-GP2 cells were kindly provided by Dr. Jacques Galipeau (University of Wisconsin-Madison, WI, USA). Virally transduced EL4-expressing OVA cells were a generous gift from Dr. Etienne Gagnon (Université de Montréal, Montreal, QC, Canada). EL4, A20, and EL4-OVA cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 Medium supplemented with 10% fetal bovine serum (FBS). B16F0 and 293-GP2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS. E.G7 cells were cultured RPMI 1460 supplemented with 2g/L Glucose, 10% FBS, 50 U/mL Penicillin-Streptomycin, 2 mM L-glutamine, 10mM HEPES, 1mM Sodium Pyruvate, and 0.5 mM β-Mercaptoethanol, and kept under selection using 80mg/ml of G418. All cells were maintained at 37°C in a 5% CO2 incubator. All cell culture media and reagents were purchased from Wisent Bioproducts (St-Bruno, QC, Canada).
Generation of BM-derived DCs
Mouse DCs were generated fresh for every experiment as mentioned previously with minor modifications.48 Briefly, the whole marrow from the femur of C57BL/6 mouse was flushed using RPMI 1640 supplemented with 10% FBS, 50 U/mL Penicillin-Streptomycin, 2 mM L-glutamine, 10mM HEPES, 1% MEM Non-essential Amino Acids, 1mM Sodium Pyruvate, and 0.5 mM β-Mercaptoethanol. The flushed bone marrow is mixed well to generate a single cell suspension, which is then centrifuged for 5 min at 1500 rpm to obtain cell palette. Red blood cells (RBC) lysis buffer is added to the cell pellet for 1-2 min to facilitate the removal of RBCs. The buffer is then neutralized using an equal volume of culture media and the cell suspension is centrifuged for 5 min at 1500 rpm to obtain cell palette. The obtained cell pellet is then resuspended, and cells cultured in the above-mentioned culture media supplemented with 50 ng/ml murine granulocyte macrophages-colony stimulating factor (GM-CSF). At the days 3 and 5 post collection, the cell suspension is collected, and the media removed by centrifugation for 5 min at 1500rpm. The cells are resuspended and cultured in fresh media containing GM-CSF. On day 7, the media was replaced by fresh media containing GM-CSF and LPS from Escherichia coli O111 (1 ng/ml) to stimulate DC maturation overnight. One the following day, the media is changed to remove LPS and mature DCs were assessed by flow cytometry for their expression of CD11c, CD80, CD86, MHCII and MHCI.
Generation of BM-derived MSCs
To generate MSCs, as previously described48 the femurs of 6-8 weeks old female C57BL/6 mice were flushed with Alpha Modification of Eagle’s Medium (AMEM) supplemented with 10% FBS, and 50 U/mL Penicillin-Streptomycin in 10 cm2 cell culture dish. After 48 h, non-adherent cells were removed by changing the media. On the following days, the media was changed every 3 to 4 days. When the cells reached 80% confluency, adherent cells were detached using 0.25% Trypsin, harvested, and expanded until a homogeneous population was obtained before being assessed using flow cytometry for the expression of surface markers CD44, CD45, CD73, CD90 and CD105. Generated MSCs were expanded and frozen at passage number 9 or 10. MSCs used for in vitro experiments up to passage 19. For in vivo experiments, MSCs were used up to passage 16. The pleotropic differentiation capacity of generated MSCs was evaluated by inducing their osteogenic and adipogenic differentiation as described previously.48 Briefly, for osteogenic differentiation MSCs were plated at 60% confluency and cultured for 3-4 weeks in AMEM media supplemented with 10% FBS in addition to β-glycerol phosphate (10 mM), dexamethasone (10-8 M), and ascorbic acid 2-phosphate (5 μg/mL), the media was changed every 2-3 days.49 Osteogenic differentiation was validated by staining calcium deposit using Alizarin Red S. Shortly, the cells were washed with phosphate-buffered saline (PBS), incubated for 5 min in a solution containing 2% Alizarin Red S (pH adjusted to 4.1 using ammonium hydroxide), then rinsed with distilled H2O.48 To induce adipogenic differentiation, the cells were plated at 50% confluency and for 7 days cultured in AMEM supplemented with 10% FBS, indomethacin (46 μM), 3-isobutyl-methylxanthine (0.5 mM), dexamethasone (1 μM), and insulin (10 μg/mL). The media was changed twice during 7 days.50 At the end of differentiation period, oil droplets within differentiated adipocytes were stained using Oil Red O. The cells were first fixed for 1 h, at room temperature with 4% paraformaldehyde, followed by staining for 10 min using Oil Red O solution. The solution was prepared by mixing Oil Red O (dissolved at 3.75% in isopropanol) and 2 parts distilled H2O. At the end of incubation time, the cells were rinsed with distilled H2O.48,49 The cells were visualized via transmitted light and imaged using EVOS® FL cell imaging microscope (Thermo Fisher Scientific).
Generation of Ctl MSC and MSC-IPr
A construct was designed containing the cDNA of the three inducible subunits of the murine immunoproteasome (β1i, β2i, and β5i) separated by the viral T2A sequence. The designed construct was synthesized by Life Technologies, then sub-cloned into the AP2 retroviral plasmid and sequenced at the Genomics Core Facility of IRIC. The AP2 construct contains the enhanced green fluorescence protein (eGFP) which serves as a marker for gene expression.51 The obtained construct was then co-transfected into the GP2-293 retroviral packaging cell line along with the VSV-G vector encoding the envelop protein using PolyFect® according to the manufacturer’s protocol. Supernatant containing the virus was collected at 48- and 72-h post-transfection, then centrifuged at 1500 rpm for 5 min at 4°C to remove cell debris. An ultracentrifugation at 25000 rpm for 90 min at 4°C was then conducted to concentrate the virus 10-fold. Collected virus concentrate was aliquoted and stored at −80°C. For transduction, MSCs were plated at 50%–60% confluency in a 12 well plate and transduced with the concentrated virus overnight. After their recovery and proliferation, the MSCs were re-plated at 50%–60% confluency and received two more transductions following the same steps. The transduction efficiency was confirmed by evaluating GFP expression via cell imaging and flow cytometry, in addition to immunoblotting of the IPr subunits. Successfully transduced MSCs were sorted according to GFP expression. The obtained population was assessed using flow cytometry for the expression of CD44, CD45, CD73, CD90 and CD105, expanded and frozen to be used for future experiments. Sorted MSCs were re-evaluated for their osteogenic and adipogenic differentiation potential as described above and validated by staining with Alizarin Red S and Oil Red O, respectively 4. The same steps were followed using the AP2 construct backbone to generate control (Ctrl) MSCs. The same transduction approach was used for generating Ctl MEFs and MEF-IPr.
Generation of luciferase expressing - Ctl MSCs and MSC-IPr
Plasmid # 17476 from Addgene was co-transfected into the GP2-293 retroviral packaging cell line along with the VSV-G vector encoding the envelop protein using PolyFect® according to the manufacturer’s protocol. Supernatant containing the retrovirus was collected at 48- and 72-h post-transfection, then centrifuged at 1500 rpm for 5 min at 4°C to remove cell debris. An ultracentrifugation at 25000 rpm for 90 min at 4°C was then conducted to concentrate the virus 10-fold. Collected virus concentrate was aliquoted and stored at −80°C. For transduction, Ctl MSCs or MSC-IPr were plated at 50%–60% confluency in a 12 well plate and transduced with the concentrated virus overnight. After their recovery and proliferation, the Ctl MSC and MSC-IPr were put under selection using the lowest concentrations of Puromycin to trigger cell death at 4 versus 14 mcg/ml Puromycin, respectively. The efficiency of the transduction was maintained by keeping the cell under selection using Puromycin at the pre-determined concentration and using Luciferase Assay System (Promega), according to the manufacturer’s protocol. The signal was read using Tecan Infinite M1000. Puromycin selection is stopped 24 h before every experiment.
Vaccination and tumor challenge studies
For prophylactic vaccination, 106 Ctl MSC, MSCγ, MSC-IPr or DCs, previously cultured for 9 h in the presence or absence of OVA (5 mg/ml) or tumor lysate (1 mg/ml), were used to vaccinate female C57BL/6 mice (n = 10/group) via an intraperitoneal (IP) injection. Each mouse received two doses of the same vaccine on days 0 and 14. Two weeks following the second dose of the vaccine, the mice were challenged by implanting 2 × 105 E.G7 (for OVA vaccination), EL4 or B16F0 cells (for tumor lysate vaccination) via a subcutaneous (SC) injection. Tumor growth was monitored and assessed via a caliper.
To evaluate efferocytosis, female C57BL/6 mice (n = 10/group) were injected, IP, with liposome-clodronate or control liposome 3 days (day −3), prior to the first vaccination dose using 106 MSC-IPr cells pulsed with 5mg/ml OVA, delivered IP (day 0). Two weeks later (day 14), the treatment was repeated following the same timeline. One week following the second vaccination, the mice were challenged by implanting 2 × 105 E.G7 cells via a SC injection. Tumor growth was monitored and assessed via a caliper. All tumor-free mice were then re-challenged at 4 and 8 weeks after the first challenge, with a total of three challenges.
To evaluate the role of immune cells in the immune response elicited by MSC-IPr vaccination, female C57BL/6 mice (n = 10/group) were vaccinated MSC-IPr pulsed with 5mg/ml OVA, as described above, and received additional IP injections of depleting antibodies against either αNK1.1, αCD4 or αCD8 (200 μg/mouse per dose, twice weekly for two weeks). One week following the second vaccination, the mice were challenged by implanting 2.5 × 105 E.G7 cells via a SC injection and tumor growth was monitored and assessed using a caliper.
MSC-IPr based therapeutic vaccine was evaluated as by injecting female C57BL/6 or Batf3-deficient mice (n = 10/group) SC injection with 2 × 105 tumor cells (EL4, E.G7 cells or B16F0). Three days later, mice were IP-injected with 106 DCs or MSC-IPr pulsed with either 5mg/ml OVA or 1mg/ml tumor lysate. The mice received two injections separated by 1 week. Control animals received 2 × 105 tumor cells SC alone or 2 × 105 tumor cells SC followed by administration of 106 unpulsed MSC-IPr/DCs. Treated animals were followed thereafter for tumor growth. For therapeutic vaccination in combination with the immune-checkpoint inhibitors (αPD-1, αCTLA-4, or αLAG3) or antibody agonist (α4-1BB), mice received IP-injections of the antibody or its isotype at a dose of 200 μg/mouse twice weekly for two weeks. A similar timeline and steps were conducted for allogeneic vaccination in BALB/c mice using DCs or MSC-IPr pulsed with A20 tumor cells lysate and challenged using A20 tumor cells.
MSC survival and persistence following in vivo administration.
Live imaging to assess in vivo cell persistence and migration was performed at the Small Animal Imaging Labs, RI-MUHC (Montreal, QC). C57BL/6 female mice (n = 6/group) were shaved and injected IP with 106 firefly luciferase-expressing Ctl MSC or MSC-IPr. To assess migration in the presence of a tumor load, mice were inoculated SC with 2.5 × 105 E.G7 cells 3 days before receiving 106 luciferase-expressing Ctl MSCs or MSC-IPr pulsed for 9 h with 5mg/ml OVA. Control mice received no cells only or tumor cells only. Bioluminescence signal was followed at 1, 24, 72 and 169 h post Ctl MSC or MSC-IPr injection. For each imaging session, the mice received a 0.2 mL IP injection of 15 mg/ml XenoLight D-Luciferin - K+ Salt (equivalent to 30 mg/kg). The bioluminescence signal was acquired after 10 min using Bruker In Vivo Xtreme. During acquisition, the mice were kept under 1.5 - 2.5% inhaled isoflurane anesthesia. The images were analyzed using Bruker MI 7.5 and signal plotted as sum Photons/sec.
Method details
Phenotypic analysis by flow cytometry
To assess the expression of cell surface markers, the tested cells were collected, counted, and washed by centrifuging the cells for 5 min at 1500 rpm to remove culture media followed by resuspending the cells in a buffer composed of 2% FBS in PBS. The washing step was repeated once. For staining, the cells were resuspended at the density of 106 cells/ml in cold 2% FBS in PBS and incubated with flow cytometry antibodies or their isotypes diluted according to manufacturer’s instructions for 30 min at 4°C in the dark. At the end of incubation time, the stained cells were washed twice with cold 2% FBS in PBS buffer. The cells were finally resuspended in 400 μL of cold 2% FBS in PBS and kept on ice in the dark until they were acquired by BD FACS Diva on CANTOII, then analyzed using FlowJoV10.
Immunoblotting
The cells were collected after 24 h in culture or at the end of treatment. Adherent cells were detached using 0.25% Trypsin, washed once with PBS, centrifuged for 5 min at 15000 rpm/min, counted, and 106 cell per condition were centrifuged to form a palette. The cells were lysed at room temperature using Cell Lytic lysis buffer according to the manufacturer instructions. Cell lysates were centrifuged at 4°C for 15 min at 20,000 rpm and the supernatant collected. The lysate of 106 cells was dissolved in loading buffer, boiled for 5 min, then loaded onto a 4%–12% gradient SDS-PAGE gel. Separated proteins were transferred onto activated polyvinylidene difluoride membrane, blocked for 1 h at room temperature in Tris-buffered saline and 0.1% Tween-20 buffer (TBST) containing 5% skim milk or 1% bovine serum albumin, washed three times with TBST, then incubated with primary antibodies according to manufacturer recommendations. At the end of the incubation time, the blots were washed three times with TBST followed by incubation with secondary antibodies for 1 h at room temperature. After washing three times with TBST, the proteins were revealed using enhanced chemiluminescence.
Cytokine and chemokine analysis
To assess the cytokine and chemokine profiles of DCs/MSCs, 106 cells were plated for 24 h. The following day, the media was replaced with serum-free media and left for another 24 h. The supernatant was collected, then analyzed by ELISA or chemokine arrays following manufacturer’s instructions. To evaluate the effect of the succinate dehydrogenase inhibitor MMA, the cells were treated with 10 mM MMA for 6 h followed by media replacement and incubation for 16 h. Supernatants were collected at 6 h or 24 h post-treatment to quantify IL-12 production by ELISA.
pHrodo Green Dextran stain
To evaluate cellular and endosomal pH by microscopy, 3 × 103 cells were plated in a glass bottom 96-well plate (ibidi GmbH). After 24 h, the cells were treated for 30 min; at 37°C with staining buffer (Phenol red-free DMEM containing 20mM HEPES) containing 1:2000 pHrodo Red AM (Invitrogen; P35372) and 50 μg/ml pHrodo Green Dextran (Invitrogen; P35368) prepared in AMEM. The cells were then washed, medium replaced with phenol red-free DMEM, and imaged with a confocal microscope (Zeiss; LSM 880). The fluorescence intensity was measured, and results expressed as mean fluorescence intensity. For assessment of cellular pH level by flow cytometry, the cells were plated in a 24-well glass bottom plate at the density of 30 × 103 cells/well, then treated with the staining buffer containing 40 μg/ml pHrodo Green Dextran for 30 min at 37°C. The cells were then washed once with warm staining buffer prior to collecting the cells to record the signal by BD FACS Diva on CANTOII and analyze it using FlowJoV10. In both cases, an increase in the pHrodo Green signal indicates a decrease in endosomal pH level.
Polysome analysis
To analyze polysomes, cells were plated for 24 h, washed with PBS and detached using Trypsin in the presence of 100 ug/ml of cycloheximide, spun down at 200 g for 10 min prior to lysing using a hypotonic lysis solution (50 mM Tris, 2.5 mM MgCl2, 1.5 mM KCl, 2 mM DTT, 1% Triton X-100, 0.5% sodium deoxycholate, and 100 μg/ml cycloheximide). Lysates were loaded onto 10%–50% sucrose gradients and centrifuged in an SW40 rotor (Beckman Coulter) at 110 000 g for 2 h. The absorbance at λ = 260 nm was monitored throughout the gradient using a UA-6 UV detector (ISCO).
Histological analyses
To assess the safety MSC-IPr vaccine at histological level, selected organs were harvested from C57BL/6 mice 14 days after vaccination using two IP injections of 106 MSC-IPr at days 0 and 14 versus control age and sex matched C57BL/6 mice. Histopathological assessment was conducted on organs harvested from vaccinated C57BL/6 mice, fixed in 10% formalin then mounted in paraffin. Sections were prepared then stained with hematoxylin and eosin then scanned using the NanoZoomer Digital Pathology system and NPD.scan 2.3.4 software (Hamamatsu).
Tumor cell lysate preparation
To prepare cell lysates, cultured EL4, B16F0 or A20 cells were collected by centrifugation at 1500 rpm for 5 min followed by two washing steps with PBS to remove traces of FBS. The cells were then subjected to 5 rounds of freeze and thaw in liquid nitrogen/boiling water, respectively. To remove large particles, the lysate was shredded using a G26 needle, passed through a 70 μm cell strainer, then filtered through a 0.45 μm filter. The obtained lysate was then quantified using Bradford reagent, aliquoted and stored at −80°C until use.
Analyzing CD8 T cell response in immunized mice
A group of female C57BL/6 mice (n = 10/group) received two doses of the vaccine composed of 106 Ctl MSC, MSCγ, MSC-IPr or DCs, previously cultured for 9 h in the presence or absence of OVA (5 mg/ml), on days 0 and 14. One week after the second dose, the spleens of the mice were collected used to analyze the cytokine profile of T cell reactivation in vitro following OVA re-stimulation. To assess antigen-specific CD8 T cell proliferation, splenocytes isolated from immunized mice were stained with CellTrace Violet according to the manufacturer’s instructions. Labeled cells were analyzed by flow cytometry to register basal signal. The stained cells were then co-cultured with 5mg/ml. OVA, and CellTrace Violet dilution analyzed by acquired by BD FACS Diva on CANTOII, after 48 h to assess CD8 T cell proliferation. The signal was analyzed using FlowJoV10.
Immune cell infiltration analysis
Isolated tumors were collected from mice reaching terminal point after relapse, dissected, then digested by incubation for 90 min at 37C° in a solution containing 1.6 mg/ml of type IV collagenase and 200 μg/ml DNaseI in RPMI with shaking every 15 min. Cells were separated from the rest of tumor tissue by passing the digested solution through a 70 μm cell strainer. After incubation with anti-Fcγ III/II mAb (clone 2.4G2), cells were incubated for 1 h at 4°C with the desired antibodies or proper isotypic control. Labeled cells were washed and signal acquired by BD FACS Diva on CANTOII. The signal was analyzed using FlowJoV10.
Immunoprecipitation, peptide elution for immunopeptidome analysis
Cells were cultured in serum-free media with or without tumor cell lysate (0.7 mg/ml) for 9 h, followed by washing the cells with PBS. The cells were detached using Accutase® and cell pellets were then washed 3 times with PBS prior to snap-freezing in liquid nitrogen. Thirty-five million cells from each group were pelleted prior to lysing using a 1% Triton X-100-based buffer and cleared to remove debris by centrifugation. Clarified lysates were incubated with 200 μg M1/42 linked to CNBr-activated Sepharose overnight to immunoprecipitate mouse MHC class I, then washed with lysis buffer followed by Tris-HCl with decreasing NaCl concentrations. The final elution was carried out in LoBind Eppendorf tubes using 0.1 M acetic acid and 0.1% Trifluoroacetic acid (TFA). Peptides were concentrated and desalted using solid-phase extraction (SPE) with an Empore C18 plate. Peptides were loaded directly and eluted using 80/20 acetonitrile/water (0.1% TFA). Eluted peptides were lyophilized and reconstituted in 0.1% TFA. Peptides (50% per sample) were analyzed by nano LC/MS/MS using a Waters NanoAcquity system interfaced to a ThermoFisher Fusion Lumos mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75 μm analytical column at 350 nL/min; both columns were packed with Luna C18 resin (Phenomenex). A 2-h gradient was employed. The mass spectrometer was operated using a custom data-dependent method, with MS performed in the Orbitrap at 60,000 FWHM resolution and sequential MS/MS performed using high resolution CID and EThcD in the Orbitrap at 15,000 FWHM resolution. All MS data were acquired from m/z 300-800 (Class I) and m/z 300-1500 (Class II). A 3 s cycle time was employed for all steps. Peptide analysis was conducted using the free online analysis tools GibbsCluster and NetMHCpan to stratify the peptides identified in the immunopeptidome sequencing. Binding affinity predictions are classified by the percentage rank with strong binding (SB) = < 0.5%; moderate biding (MB) = 0.5% - 2.0%; and weak binding (NB) = > 2.0%. See also Figure S11.
SILAC experiment
Cells from each group were seeded at 5 x106 cells per plate, then cultured for three passages in the presence of normal amino acids (light) versus amino acids labeled with stable heavy isotopes (heavy). At the end of the third passage, all cells were detached, collected, and lysed were combined and washed twice with ice-cold PBS, lysed in NiNTA denaturing incubation buffer (6cM Guanidinium HCl, 100cmM NaH2PO4, 20cmM 2-Chloroacetamide, 5cmM 2-Mercaptoethanol, 10cmM Tris-HCl pH 8) and sonicated. prior to protein digestion by trypsin. Analysis of the proteome was then performed by LC-MS/MS as previously described.52
Detection of peptide-MHCI complex and antigen presenting assay
To detect the formation of the SIINFEKL/MHCI complex, Ctl MSCs, MSCγ, MSC-IPr or DCs were pulsed using SIINFEKL (5 μg/ml), or OVA (5mg/ml) for 7 h. To generate MSCγ, 20 ng/ml of IFNγ was added to the MSC media overnight then media changed to remove IFNγ and used directly. At each corresponding time point, the cells were collected, washed, and stained using 25-D1.16 antibody or its isotype according to the manufacturer instructions. The signal was recorded by BD FACS Diva on CANTOII and analyzed using FlowJoV10.
To evaluate the cells’ ability to perform antigen cross-presentation to naive CD8 T cells, Ctl MSCs, MSCγ, MSC-IPr or DCs were seeded at 25 × 103 cells per well in a 24-well plate. On the following day, the cells were washed and pulsed with the antigen of interest (5 mg/ml of OVA or 5 μg/ml of the SIINFEKL peptide). At the end of the pulsing period, the cells were washed to remove excess antigen and co-cultured with 106/ml CD8 T cells purified from the spleen of OT-I male mice (6-10 weeks old) using the CD8α+ positive isolation kit according to the manufacturer’s protocol. After 72 h, supernatants were collected, centrifuged for 5 min at 1500 rpm, 4°C. Clear supernatant were used to quantify IFNγ levels by ELISA (R&D).
To generate cell-bound OVA, splenocytes were incubated in their culture media containing 5mg/ml OVA for 3 h then washed extensively before co-culture with MSC-IPr.
Monitoring antigen uptake and processing
For the evaluation of OVA uptake, 4 × 104 cells were seeded per well in 12 well plates. On the following day, Alexa Fluor® 647-conjugated OVA was added to each well at the concentration of 1 μg/ml. At the end of the incubation time, the cells were detached and washed twice with cold 2% FBS in PBS. Fluorescence was monitored by recording the signal using BD FACSCanto II. For evaluating OVA processing, the cells were incubated for 15-30 min at 37°C with 10 μg/mL DQ® ovalbumin, a self-quenched conjugate of OVA which, upon proteolytic degradation, exhibits bright green fluorescence. At the end of loading time, the cells were washed, and regular media added. The signal was chased for different time points. At the end of specified chase time, the cells were detached and washed with cold PBS containing 2% FBS. Fluorescence was monitored by acquired the signal using BD FACS Diva on CANTOII and analyzing the signal using FlowJoV10.
Assessment of macropinocytosis by confocal microscopy
For this assay, 15 × 103 cells were seeded on a sterile glass cover slide placed in the wells of a 24-well plate. On the following day, the media was changed, and LY CH added to the adherent cells at a concentration of 1 μg/ml for 24 h at 37°C. At the end of the specified incubation time, the cells were washed twice to remove excess dye, then a fixative solution containing 2% paraformaldehyde was added for 15 min at room temperature in the dark followed by two washes with PBS. The cells were then mounted on a slide in EverBrite DAPI (4′,6-diamidino-2-phenylindole)-containing mounting medium. The slides were viewed using ZEISS Axio Observer A1 confocal microscope and the results analyzed using the ImageJ software.
Quantification and statistical analysis
RNA extraction and sequencing
Total RNA was isolated from 106 cells for each group using RNeasy® mini kit (QIAGEN) according to manufacturer’s instructions. Presence of contamination with chemicals was assessed by nanodrop using 260/280 and 260/230 ratios. Quantification of total RNA was made by QuBit (ABI) and 500 ng of total RNA was used for library preparation. Quality of total RNA was assessed with the BioAnalyzer Nano (Agilent) and all samples had a RIN above 8. Library preparation was done with the KAPA mRNaseq stranded kit (KAPA, Cat no. KK8420). Ligation was made with 9 nM final concentration of Illumina index and 10 PCR cycles was required to amplify cDNA libraries. Libraries were quantified by QuBit and BioAnalyzer. All libraries were diluted to 10 nM and normalized by qPCR using the KAPA library quantification kit (KAPA; Cat no. KK4973). Libraries were pooled to equimolar concentration. Sequencing was performed with the Illumina Hiseq2000 using the Hiseq Reagent Kit v3 (200 cycles, paired-end) using 1.7 nM of the pooled library. Around 40 M paired-end PF reads was generated per sample. Library preparation and sequencing was made at the Institute for Research in Immunology and Cancer’s Genomics Platform (IRIC).
Assessment of ATP/ADP/AMP and Acetyl-CoA levels by MS
To evaluate the level of different cellular metabolites, the cells were cultured for 24 h then detached and collected to form a cell pellet by gentle centrifugation at 300 g for 5 min at 4°C. The palette is then gently resuspended with 1 mL of ice cold 150 mM NH4 formate solution (adjusted to pH 7.4 with 10% NH4OH). The sample is gently spun at 300 g for 5 min at 4°C to remove the supernatant. The cells are then lysed on ice by adding 250-400 μl of cold 65/35% (v/v) methanol /water 50mM NH4HCO3. To ensure complete lysis, the samples are mixed gently using a vortex to stir the cells followed by sonication for 2 min. The samples were transferred to −80°C for 20 min before centrifugation at 14,000 g for 5 min at 4–8°C to pellet cell debris. Finally, the metabolite-containing supernatant is transferred to a clean 1.5 mL and samples kept in - 80°C and only transfer on dry ice until analysis.
Cell extracts were analyzed using a sensitive a selective LC-MS/MS method. A calibration curve made from four to six concentrations of the reference compounds dissolved in a suitable solvent was used to estimate intracellular content in each sample. The analyte concentrations covered a range from 0.1 to 2 μM for Acetyl CoA, and from 5 to 100 μM for AMP, ADP and ATP. Calibration curves were plotted using peak area of analyte versus nominal analyte concentration, using a weighted 1/x least-squares linear regression fit. For AMP and ATP, peak area ratios of analyte versus internal standard were used. Bromo-AMP and ATP-15N5 were used as internal standards. For chromatography assessment, the analytes were separated in HILIC mode on a Thermo Biobasic-AX, 50 × 3 mm, 5 μm column at a flow rate of 0.75 ml/min. The mobile phase A consisted of 30/70 acetonitrile/water containing 2 mM ammonium acetate and 0.15% ammonium hydroxide, and mobile phase B consisted of acetonitrile. The mobile phase gradient consisted of a linear decrease in mobile phase B from 80 to 0% over a period of 2 min, and kept at 0% for 1.8 min, before being equilibrated with 80% mobile phase B for 2 min. The total run time for each injection was 6 min, and the injection volume was 3 μL. For mass spectrometry, the LC-ESI-MS/MS system was comprised of an Agilent 1100 LC system coupled to a triple quadrupole QTRAP® 4000 System (AB SCIEX) fitted with a TurboV ionization source interface, which was operated in the positive ion mode. Quantification was performed using the multiple reaction monitoring mode (MRM) with the following transitions (70 msec dwell time): m/z 610→303, collision energy of 44 (Acetyl CoA), m/z 348→136, collision energy of 29 (AMP), m/z 428→136, collision energy of 32 (ADP), and m/z 508→136, collision energy of 30 (ATP). The source dependent parameters were as follows: curtain gas (CUR), nitrogen at 25 l/min; collision gas (CAD), medium; ionspray voltage (IS), 5300 V; de-clustering potential (DP), 90 V; temperature (TEM), 650°C; ion source gas 1 (GS 1) and gas 2 (GS 2) flow rate, 40 and 60 L/min. The LC–MS/MS system was controlled by the Analyst® software version 1.6.2.
Seahorse and mitochondrial analysis
The cells were plated, and oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR) were measured using Agilent Seahorse XF Analyzer according to manufacturer’s instructions. For the assessment of mitochondria integrity and superoxide anion production, the cells were collected stained using MitoTracker DeepRed/MitoTracker Green or MitoSox Red respectively, according to the manufacturer’s protocol and assessed by BD FACS Diva on CANTOII, then analyzed using FlowJoV10.
Quantification of mitochondrial metabolic intermediates by mass spectrometry
To assess the difference in the level of different metabolic by-products produced within the mitochondria of MSC-IPr versus Ctl MSCs, cells from each group were plated for 24 h before being harvested by first aspirating the media and washing three times with ice cold normal saline (9 g NaCl per liter ddH2O). The cells were then scraped and transferred into −30°C pre-chilled 80% methanol (v/v) and stored at −80 until the day before GC/MS analysis. To ensure complete lysis, all samples were subjected to sonication using BioRuptor (UCD-200 TM, Diagenode) for 10 min (50% duty cycle of 1 min) at the “high” setting. Cellular debris were removed by centrifugation for 10 min at 14000 rpm, at 1°C. Supernatants were transferred to fresh pre-chilled tubes containing 1 μL of 80015x ng/mL D27-myristic acid internal standard. Samples were dried overnight at −4°C using a temperature-controlled vacuum centrifuge (Labconco, Kansas City, MO, USA). Just before GC/MS analysis, the samples were derivatized in two steps. First, samples were resuspended in 30 μL pyridine containing 10 mg/mL methoxyamine hydrochloride (Sigma), vortexed, and sonicated to ensure dissolution. Following 30 min of incubation at room temperature, the samples were transferred into sealed auto-injection vials containing 70 μL of N-tert-Butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) (Sigma). The vials were then incubated for 1 h at 70°C.
A volume of 1 μl was injected GC/MS analysis using an Agilent 5975C GC/MS equipped with a DB-5MS+DG (30 m x 250 μm x 0.25 μm) capillary column (Agilent J&W, Santa Clara, CA, USA). All metabolites used in this study were previously validated using authentic standards to confirm mass spectra and retention times.
Bioinformatics analysis
For the whole transcriptome experiment, raw RNA-seq counts from reads aligned to the mouse genome (mm10 assembly) were generated with Htseq-count (PMID: 25260700). Differentially expressed genes between MSC-IPr and Ctl MSC groups were calculated by DESeq2 (PMID: 25516281). Pre-ranked gene set enrichment was performed as recommended for RNA-seq data (PMID: 16199517). Custom R scripts were used to filter highly redundant biological processes. A false discovery rate of 0.05 was considered as an acceptable threshold for further investigation.
For the SILAC experiment done in biological triplicates; proteins were retained for further enrichment analysis if the t test p-value from the MSC-IPr versus control groups is smaller than 5%. Custom R scripts (https://www.R-project.org/), ggplot2 and clusterprofiler (PMID: 22455463) packages were used to create gene expression heatmaps, volcano plots, GSEA plots and enrichment bar plots.
Statistical analysis
Depending on the study, p-values were calculated using the Student’s t test, one-way analysis of variance (ANOVA) or the long rank test using GraphPad Prism. Results are represented as average mean with SD error bars, and statistical significance is represented with asterisks: ∗P˂0.05, ∗∗P˂0.01, ∗∗∗P˂0.001. Statistical tests used for bioinformatic analysis are described in their corresponding sections.
Acknowledgments
We thank Isabelle Caron and Myriam Métivier-Bélisle from the IRIC animal facility for their kind help with some of the in vivo experiments. We also thank Martin Jutras and Mihaela Friciu from the platform of Biopharmacy of Université de Montréal for their help with the metabolite study/analyses. Gas chromatography-mass spectrometry (GC-MS) analyses for TCA cycle intermediates were performed by the Metabolomics Innovation Resource funded by the Dr. John R. and Clara M. Fraser Memorial Trust, the Terry Fox Foundation, the Québec Breast Cancer Foundation, the Goodman Cancer Research Center, and McGill University. Immunopeptidome profiling was performed by Dr. Julie Rumble from Cayman Chemical Bioanalysis Services and Dr. Richard Jones from MS Bioworks.
Author contributions
J.A. and F.K. designed and performed most in vitro and in vivo experiments. N.E.H. conducted the bioinformatics analysis for the RNA-seq and SILAC experiments. N.S., M.B., K.R., S.T., and J.-P.B. designed some in vitro and in vivo experiments and flow cytometry analyses and conducted the microscopy studies. J.C. and M.J. helped in designing and conducting some metabolomic studies. L.-E.T. helped designing the Seahorse analysis. S.K. and B.A. designed and conducted the HIF-1α analysis. F.R. and J.P. designed and conducted the polysome assessment of MSCs. R.S. participated in conceiving several in vivo experiments and the immunopeptidome experiment. M.R. conceived and supervised the project, analyzed data, and wrote the paper. All authors contributed to the conception of the study, the analysis of the data, and the revision of the manuscript.
Declaration of interests
The authors declare no competing financial interests. A provisional patent has been filed to protect the MSC-IPr technology and its applications (62/835,678).
Published: December 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100455.
Contributor Information
Riam Shammaa, Email: riam.shammaa@ccrttoronto.ca.
Moutih Rafei, Email: moutih.rafei.1@umontreal.ca.
Supplemental information
Data and code availability
-
•
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code. Any analyses applied are based on previously available software and established R packages, primarily, custom R scripts (https://www.R-project.org/), ggplot2 and clusterprofiler (PMID: 22455463).
References
- 1.Figdor C.G., de Vries I.J., Lesterhuis W.J., Melief C.J. Dendritic cell immunotherapy: mapping the way. Nat. Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 2.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 3.Pinzon-Charry A., Maxwell T., López J.A. Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol. Cell Biol. 2005;83:451–461. doi: 10.1111/j.1440-1711.2005.01371.x. [DOI] [PubMed] [Google Scholar]
- 4.Wohn C., Le Guen V., Voluzan O., Fiore F., Henri S., Malissen B. Absence of MHC class II on cDC1 dendritic cells triggers fatal autoimmunity to a cross-presented self-antigen. Sci. Immunol. 2020;5:eaba1896. doi: 10.1126/sciimmunol.aba1896. [DOI] [PubMed] [Google Scholar]
- 5.François M., Romieu-Mourez R., Stock-Martineau S., Boivin M.N., Bramson J.L., Galipeau J. Mesenchymal stromal cells cross-present soluble exogenous antigens as part of their antigen-presenting cell properties. Blood. 2009;114:2632–2638. doi: 10.1182/blood-2009-02-207795. [DOI] [PubMed] [Google Scholar]
- 6.Stagg J., Pommey S., Eliopoulos N., Galipeau J. Interferon-gamma-stimulated marrow stromal cells: a new type of nonhematopoietic antigen-presenting cell. Blood. 2006;107:2570–2577. doi: 10.1182/blood-2005-07-2793. [DOI] [PubMed] [Google Scholar]
- 7.Chan J.L., Tang K.C., Patel A.P., Bonilla L.M., Pierobon N., Ponzio N.M., Rameshwar P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-γ. Blood. 2006;107:4817–4824. doi: 10.1182/blood-2006-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheng H., Wang Y., Jin Y., Zhang Q., Zhang Y., Wang L., Shen B., Yin S., Liu W., Cui L., Li N. A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008;18:846–857. doi: 10.1038/cr.2008.80. [DOI] [PubMed] [Google Scholar]
- 9.Karwacz K., Bricogne C., MacDonald D., Arce F., Bennett C.L., Collins M., Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 2011;3:581–592. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parry R.V., Chemnitz J.M., Frauwirth K.A., Lanfranco A.R., Braunstein I., Kobayashi S.V., Linsley P.S., Thompson C.B., Riley J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patsoukis N., Bardhan K., Chatterjee P., Sari D., Liu B., Bell L.N., Karoly E.D., Freeman G.J., Petkova V., Seth P., et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner C.S., Grotzke J.E., Cresswell P. Intracellular events regulating cross-presentation. Front. Immunol. 2012;3:138. doi: 10.3389/fimmu.2012.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapiro J., Claverol S., Piette F., Ma W., Stroobant V., Guillaume B., Gairin J.-E., Morel S., Burlet-Schiltz O., Monsarrat B., et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 2006;176:1053–1061. doi: 10.4049/jimmunol.176.2.1053. [DOI] [PubMed] [Google Scholar]
- 14.Heink S., Ludwig D., Kloetzel P.M., Krüger E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA. 2005;102:9241–9246. doi: 10.1073/pnas.0501711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toes R.E., Nussbaum A.K., Degermann S., Schirle M., Emmerich N.P., Kraft M., Laplace C., Zwinderman A., Dick T.P., Müller J., et al. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J. Exp. Med. 2001;194:1–12. doi: 10.1084/jem.194.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Verteuil D., Muratore-Schroeder T.L., Granados D.P., Fortier M.H., Hardy M.P., Bramoullé A., Caron E., Vincent K., Mader S., Lemieux S., et al. Deletion of immunoproteasome subunits imprints on the transcriptome and has a broad impact on peptides presented by major histocompatibility complex I molecules. Mol. Cell. Proteomics. 2010;9:2034–2047. doi: 10.1074/mcp.M900566-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Verteuil D.A., Rouette A., Hardy M.P., Lavallée S., Trofimov A., Gaucher É., Perreault C. Immunoproteasomes shape the transcriptome and regulate the function of dendritic cells. J. Immunol. 2014;193:1121–1132. doi: 10.4049/jimmunol.1400871. [DOI] [PubMed] [Google Scholar]
- 18.Zehner M., Marschall A.L., Bos E., Schloetel J.G., Kreer C., Fehrenschild D., Limmer A., Ossendorp F., Lang T., Koster A.J., et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8(+) T cells. Immunity. 2015;42:850–863. doi: 10.1016/j.immuni.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Broz M.L., Binnewies M., Boldajipour B., Nelson A.E., Pollack J.L., Erle D.J., Barczak A., Rosenblum M.D., Daud A., Barber D.L., et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell. 2014;26:938. doi: 10.1016/j.ccell.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Edelson B.T., Kc W., Juang R., Kohyama M., Benoit L.A., Klekotka P.A., Moon C., Albring J.C., Ise W., Michael D.G., et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med. 2010;207:823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildner K., Edelson B.T., Purtha W.E., Diamond M., Matsushita H., Kohyama M., Calderon B., Schraml B.U., Unanue E.R., Diamond M.S., et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silk K.M., Silk J.D., Ichiryu N., Davies T.J., Nolan K.F., Leishman A.J., Carpenter L., Watt S.M., Cerundolo V., Fairchild P.J. Cross-presentation of tumour antigens by human induced pluripotent stem cell-derived CD141(+)XCR1+ dendritic cells. Gene Ther. 2012;19:1035–1040. doi: 10.1038/gt.2011.177. [DOI] [PubMed] [Google Scholar]
- 23.Spranger S., Dai D., Horton B., Gajewski T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–723.e4. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Accapezzato D., Visco V., Francavilla V., Molette C., Donato T., Paroli M., Mondelli M.U., Doria M., Torrisi M.R., Barnaba V. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 2005;202:817–828. doi: 10.1084/jem.20051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jancic C., Savina A., Wasmeier C., Tolmachova T., El-Benna J., Dang P.M., Pascolo S., Gougerot-Pocidalo M.A., Raposo G., Seabra M.C., Amigorena S. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 2007;9:367–378. doi: 10.1038/ncb1552. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez A., Regnault A., Kleijmeer M., Ricciardi-Castagnoli P., Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999;1:362–368. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- 27.Savina A., Jancic C., Hugues S., Guermonprez P., Vargas P., Moura I.C., Lennon-Duménil A.M., Seabra M.C., Raposo G., Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 28.Savina A., Peres A., Cebrian I., Carmo N., Moita C., Hacohen N., Moita L.F., Amigorena S. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity. 2009;30:544–555. doi: 10.1016/j.immuni.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Hiltbold E.M., Vlad A.M., Ciborowski P., Watkins S.C., Finn O.J. The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J. Immunol. 2000;165:3730–3741. doi: 10.4049/jimmunol.165.7.3730. [DOI] [PubMed] [Google Scholar]
- 30.González A., Hall M.N., Lin S.C., Hardie D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020;31:472–492. doi: 10.1016/j.cmet.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 31.Koenis D.S., Medzikovic L., van Loenen P.B., van Weeghel M., Huveneers S., Vos M., Evers-van Gogh I.J., Van den Bossche J., Speijer D., Kim Y., et al. Nuclear Receptor Nur77 Limits the Macrophage Inflammatory Response through Transcriptional Reprogramming of Mitochondrial Metabolism. Cell Rep. 2018;24:2127–2140.e7. doi: 10.1016/j.celrep.2018.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C., Li Z., Zhu Z., Chai Y., Wu Y., Yuan Z., Chang Z., Wang Z., Zhang M. Allogeneic dendritic cells induce potent antitumor immunity by activating KLRG1+CD8 T cells. Sci. Rep. 2019;9:15527. doi: 10.1038/s41598-019-52151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fotaki G., Jin C., Ramachandran M., Kerzeli I.K., Karlsson-Parra A., Yu D., Essand M. Pro-inflammatory allogeneic DCs promote activation of bystander immune cells and thereby license antigen-specific T-cell responses. OncoImmunology. 2017;7:e1395126. doi: 10.1080/2162402X.2017.1395126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fotaki G., Jin C., Kerzeli I.K., Ramachandran M., Martikainen M.M., Karlsson-Parra A., Yu D., Essand M. Cancer vaccine based on a combination of an infection-enhanced adenoviral vector and pro-inflammatory allogeneic DCs leads to sustained antigen-specific immune responses in three melanoma models. OncoImmunology. 2017;7:e1397250. doi: 10.1080/2162402X.2017.1397250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harndahl M., Rasmussen M., Roder G., Dalgaard Pedersen I., Sørensen M., Nielsen M., Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 2012;42:1405–1416. doi: 10.1002/eji.201141774. [DOI] [PubMed] [Google Scholar]
- 36.Rasmussen M., Fenoy E., Harndahl M., Kristensen A.B., Nielsen I.K., Nielsen M., Buus S. Pan-Specific Prediction of Peptide-MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016;197:1517–1524. doi: 10.4049/jimmunol.1600582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Burg S.H., Visseren M.J., Brandt R.M., Kast W.M., Melief C.J. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J. Immunol. 1996;156:3308–3314. [PubMed] [Google Scholar]
- 38.Chatterjee B., Smed-Sörensen A., Cohn L., Chalouni C., Vandlen R., Lee B.C., Widger J., Keler T., Delamarre L., Mellman I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood. 2012;120:2011–2020. doi: 10.1182/blood-2012-01-402370. [DOI] [PubMed] [Google Scholar]
- 39.Cohn L., Chatterjee B., Esselborn F., Smed-Sörensen A., Nakamura N., Chalouni C., Lee B.C., Vandlen R., Keler T., Lauer P., et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 2013;210:1049–1063. doi: 10.1084/jem.20121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tacken P.J., Ginter W., Berod L., Cruz L.J., Joosten B., Sparwasser T., Figdor C.G., Cambi A. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood. 2011;118:4111–4119. doi: 10.1182/blood-2011-04-346957. [DOI] [PubMed] [Google Scholar]
- 41.Peth A., Nathan J.A., Goldberg A.L. The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J. Biol. Chem. 2013;288:29215–29222. doi: 10.1074/jbc.M113.482570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ronnebaum S.M., Patterson C., Schisler J.C. Minireview: hey U(PS): metabolic and proteolytic homeostasis linked via AMPK and the ubiquitin proteasome system. Mol. Endocrinol. 2014;28:1602–1615. doi: 10.1210/me.2014-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webster C.M., Pino E.C., Carr C.E., Wu L., Zhou B., Cedillo L., Kacergis M.C., Curran S.P., Soukas A.A. Genome-wide RNAi Screen for Fat Regulatory Genes in C. elegans Identifies a Proteostasis-AMPK Axis Critical for Starvation Survival. Cell Rep. 2017;20:627–640. doi: 10.1016/j.celrep.2017.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dingjan I., Verboogen D.R., Paardekooper L.M., Revelo N.H., Sittig S.P., Visser L.J., Mollard G.F., Henriet S.S., Figdor C.G., Ter Beest M., van den Bogaart G. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci. Rep. 2016;6:22064. doi: 10.1038/srep22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gros M., Amigorena S. Regulation of Antigen Export to the Cytosol During Cross-Presentation. Front. Immunol. 2019;10:41. doi: 10.3389/fimmu.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Constantino J., Gomes C., Falcão A., Cruz M.T., Neves B.M. Antitumor dendritic cell-based vaccines: lessons from 20 years of clinical trials and future perspectives. Transl. Res. 2016;168:74–95. doi: 10.1016/j.trsl.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 47.Sachamitr P., Hackett S., Fairchild P.J. Induced pluripotent stem cells: challenges and opportunities for cancer immunotherapy. Front. Immunol. 2014;5:176. doi: 10.3389/fimmu.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eliopoulos N., Francois M., Boivin M.N., Martineau D., Galipeau J. Neo-organoid of marrow mesenchymal stromal cells secreting interleukin-12 for breast cancer therapy. Cancer Res. 2008;68:4810–4818. doi: 10.1158/0008-5472.CAN-08-0160. [DOI] [PubMed] [Google Scholar]
- 49.Meirelles Lda.S., Nardi N.B. Murine marrow-derived mesenchymal stem cell: isolation, in vitro expansion, and characterization. Br. J. Haematol. 2003;123:702–711. doi: 10.1046/j.1365-2141.2003.04669.x. [DOI] [PubMed] [Google Scholar]
- 50.Le Blanc K., Tammik L., Sundberg B., Haynesworth S.E., Ringdén O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand. J. Immunol. 2003;57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x. [DOI] [PubMed] [Google Scholar]
- 51.Galipeau J., Li H., Paquin A., Sicilia F., Karpati G., Nalbantoglu J. Vesicular stomatitis virus G pseudotyped retrovector mediates effective in vivo suicide gene delivery in experimental brain cancer. Cancer Res. 1999;59:2384–2394. [PubMed] [Google Scholar]
- 52.Li C., McManus F.P., Plutoni C., Pascariu C.M., Nelson T., Alberici Delsin L.E., Emery G., Thibault P. Quantitative SUMO proteomics identifies PIAS1 substrates involved in cell migration and motility. Nat. Commun. 2020;11:834. doi: 10.1038/s41467-020-14581-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code. Any analyses applied are based on previously available software and established R packages, primarily, custom R scripts (https://www.R-project.org/), ggplot2 and clusterprofiler (PMID: 22455463).