

Abstract
Aims:
In order to better predict the pharmacokinetics (PK) of antibodies in children, and to facilitate dose optimization of antibodies in paediatric patients, there is a need to develop systems PK models that integrate ontogeny-related changes in human physiological parameters.
Methods:
A population-based physiological-based PK (PBPK) model to characterize antibody PK in paediatrics has been developed, by incorporating age-related changes in body weight, organ weight, organ blood flow rate and interstitial volumes in a previously published platform model. The model was further used to perform Monte Carlo simulations to investigate clearance vs. age and dose–exposure relationships for infliximab.
Results:
By estimating only one parameter and associated interindividual variability, the model was able to characterize clinical PK of infliximab from two paediatric cohorts (n = 141, 4–19 years) reasonably well. Model simulations demonstrated that only 50% of children reached desired trough concentrations when receiving FDA-labelled dosing regimen for infliximab, suggesting that higher doses and/or more frequent dosing are needed to achieve target trough concentrations of this antibody.
Conclusion:
The paediatric PBPK model presented here can serve as a framework to characterize the PK of antibodies in paediatric patients. The model can also be applied to other protein therapeutics to advance precision medicine paradigm and optimize antibody dosing regimens in children.
Keywords: infliximab (Remicade), monoclonal antibodies, paediatrics, physiologically-based pharmacokinetics, population pharmacokinetics
1 ∣. INTRODUCTION
Determination of an optimal dosing regimen for monoclonal antibodies (mAbs) in paediatrics is challenging, due to limited clinical experience with these molecules. Often, the adult dosing regimen is extrapolated to paediatrics based on body weight (BW) or body surface area (BSA).1 However, the validity of this practice remains in question since there is a lack of consensus regarding whether the pharmacokinetics (PK) of mAbs differ significantly between adults and children.2 It is reported that infants and young children achieve a lower plasma exposure of mAbs compared to adults when the same BW-based doses are given, while BSA-based dosing may result in higher drug exposure in infants compared to adults.2-4 The higher fraction of extracellular fluid volume and faster rate of extravasation in young children compared to adults may contribute to differences in mAb disposition between these two populations.5 In addition, reported low expression levels of FcRn and relatively higher concentrations of endogenous IgG in infants6,7 may contribute to higher elimination of mAbs in children. Moreover, the lymph flow,8-11 hematopoietic cell concentrations11,12 and endogenous IgG,9 which play roles in antibody disposition, have been reported to be age-dependent. Additional differences in organ composition between adults and children may also affect tissue PK of mAbs in these populations, despite the plasma PK being similar across different age groups.1,13,14 As such, there is a need to develop systems PK models that can mathematically integrate physiological changes reported between adults and children, and help with a priori prediction of mAb PK in the plasma and site-of-action of paediatric population.
Physiologically-based pharmacokinetic (PBPK) models are widely used systems PK models to establish exposure–response relationships for drugs, and to facilitate the selection of a safer and more effective dose in special populations like paediatrics. We have developed a platform PBPK model for mAbs in the past, which can characterize the PK of mAb in various preclinical species and humans reasonably well.15 In this study, we have extended our platform PBPK model towards paediatrics, and evaluated the ability of this model to predict the PK of mAb in different age groups. In order to accurately capture the dynamic changes in physiological properties that happen throughout the childhood, we have used a series of recently published comprehensive equations that describe the relationships between organ weight, blood flow and age.16 We have also included a continuous relationship between age and interstitial volume fractions of adipose17,18 and muscle tissues,19 which has been previously reported to change between infants and adults.
The ability of the paediatric PBPK model to predict the PK of mAbs was evaluated using clinical PK data of infliximab (IFX). In order to capture the inter-individual variability (IIV) observed in the clinical PK of mAbs, the PBPK model was further evolved to account for the variability in the key PK parameters.15 In fact, such population PBPK modelling approach20 has been applied to adults,21 but no such application yet exists for the paediatric population. After establishing the population PBPK model, Monte Carlo simulations were used to critically evaluate how well different IFX dosing regimens commonly used in clinical care achieve target IFX trough concentrations in children. Our simulations suggest that a more intense dosing regimen may be needed to achieve the targeted trough concentrations of IFX in the majority of paediatric patients, which is consistent with recently published recommendations for IFX dosing in children with inflammatory bowel disease (IBD).22,23
2 ∣. METHODS
2.1 ∣. Clinical dataset for model validation
Clinical PK of IFX was obtained from two cross-sectional cohorts of children receiving intravenous IFX as part of routine clinical care at either the Children's Mercy Kansas City Infusion Center (cohort 1, IRB#14100454)24 or Lucile Packard Children's Hospital Stanford University (cohort 2, protocol# 44562).25 Both studies were approved by the local Institutional Review Board with waiver of consent. Cohort 1 collected the trough concentrations from patients who received stable IFX dosing (i.e., no changes in dose level or frequency for more than two dosings), and one sample was collected from each patient. Cohort 2 obtained the trough concentrations as part of standard clinical practice from patients who received IFX induction and maintenance or just maintenance therapy. One to three samples were collected from each patient. In cohort 1, the NF-κβ luciferase gene-reporter assay (GRA)26 was used to analyse IFX concentrations, with the lower limit of quantitation (LLOQ) of 0.65 μg/mL and the upper limit of quantitation (ULOQ) of 40 μg/mL. In cohort 2, the PROMETHEUS® Anser® IFX27 was used with LLOQ and ULOQ of 0.56 μg/mL and 27 μg/mL. Patient demographics (Table 1) were comparable between the two cohorts.
TABLE 1.
Patient demographics
| Characteristic | Cohort 1 (n = 93) | Cohort 2 (n = 48) | All (n = 141) |
|---|---|---|---|
| Age | |||
| Mean (SD) | 14.7 (3.84) | 14.4 (3.50) | 14.6 (3.72) |
| Median (range) | 16.0 (4.0–19.0) | 15.0 (6.2–19.0) | 15.0 (4.0–19.0) |
| Female sex, n (%) | 41 (44.1) | 22 (45.8) | 63 (44.7) |
| Weight, kg | |||
| Mean (SD) | 58.7 (23.2) | 50.2 (17.1) | 55.8 (21.6) |
| Median (range) | 54.5 (14.2–138) | 52.8 (21.0–87.7) | 54.0 (14.2–138) |
| Diagnosis, n (%) | |||
| IBD | 69 (74.2) | 48 (100) | 117 (83.0) |
| JIA | 16 (17.2) | 0 | 16 (11.3) |
| Uveitis | 8 (8.60) | 0 | 8 (5.67) |
| Treatment with induction, n (%) | 0 | 19 (39.6) | 19 (13.5) |
| Use of immunomodulator, n (%) | 39 (41.9) | 43 (89.6) | 82 (58.2) |
| Dosing regimen | |||
| < 5 mg/kg, n/ total (%) | 2/93 (2.06) | 5/48 (10.2) | 7/141 (4.79) |
| Q4W, n | 1 | 2 | 3 |
| Q5–7W, n | 1 | 0 | 1 |
| Q8W, n | 0 | 2 | 2 |
| > Q8W, n | 0 | 1 | 1 |
| 5–7.5 mg/kg, n/ total (%) | 33/93 (34.0) | 26/48 (53.1) | 59/141 (40.4) |
| Q4W, n | 4 | 4 | 8 |
| Q5–7W, n | 13 | 9 | 22 |
| Q8W, n | 16 | 11 | 27 |
| > Q8W, n | 0 | 2 | 2 |
| 7.5–10 mg/kg, n/ total (%) | 38/93 (39.2) | 11/48 (22.4) | 49/141 (33.6) |
| Q4W, n | 17 | 1 | 18 |
| Q5–7W, n | 17 | 7 | 24 |
| Q8W, n | 4 | 3 | 7 |
| 10–12.5 mg/kg, n/ total (%) | 20/93 (20.6) | 6/48 (12.2) | 26/141 (17.8) |
| Q4W, n | 12 | 1 | 13 |
| Q5–7W, n | 4 | 4 | 8 |
| > 12.5 mg/kg, n/ total (%) | 4/93 (4.12) | 1/48 (2.04) | 5/141 (3.42) |
| Q4W, n | 3 | 1 | 4 |
| Q5–7W, n | 1 | 0 | 1 |
| Albumin (g/dL) | |||
| Mean (SD) | 4.31 (0.512) | 3.51 (0.561) | 4.03 (0.645) |
| Median (range) | 4.30 (3.1–7.7) | 3.60 (2.3–4.7) | 4.10 (2.3–7.7) |
| CRP > 10 mg/dL, n (%) | 1 (1.08)a | 15 (31.3) | 16 (11.4) |
| ADA detectable, n (%) | 6 (6.45) | 4 (8.33) | 10 (7.09) |
Data were not available for 13 patients. IBD, inflammatory bowel disease; JIA, juvenile idiopathic arthritis; CRP, C-creatinine protein; ADA, anti-drug antibodies.
2.2 ∣. PBPK model structure and physiological parameters
A detailed structure (Figure 1) for the mAb PBPK model has been described in our previous publication.15 Mathematical equations describing the continuous relationships between age and BW, organ weight and blood flow rate were obtained from our recent publication16 and incorporated into the PBPK model (Tables S1-S4). Utilizing these equations allowed us to derive the body weight-normalized organ weight and organ flow rate as a function of age.16 Then, each patient's actual body weight and height data recorded at the time of blood sampling visit were used as an input, allowing the model to calculate patient-specific physiological parameters based on individual's age, body weight, height and sex.
FIGURE 1.

(A) Structure of the whole-body platform PBPK model of mAb in paediatrics. All organs are represented by a rectangular compartment and connected in an anatomical manner with blood flow (solid arrows) and lymphatic flow (dashed arrows). Organ weights and organ blood flow rates in each organ are a function of age, body weight, and sex. (B) Structure of the organ level PBPK model of mAb in paediatrics. Each organ within the model, except blood and lymph node, is divided into plasma, blood cell, endosomal, interstitial and cellular sub-compartments. Drug-specific parameters were fixed and taken from a previous publication (Table S1) and only degradation rate of FcRn unbound IgG (Kdeg) was estimated accounting for interindividual variability
As physiological properties are continuously changing within the paediatric age range, the time-varying nature of physiology for each patient was accounted for using Equation 1.
| (1) |
where Agei is the actual age of patient i and is the initial age when this patient entered the study. Thus, we were able to derive the physiological parameters based on an individual's age, BW and sex, which were changed for each patient in real time. The mathematical equation (Equation 2) that describes the change in fractional ratio of interstitial volume over total tissue volume (fIS) as a function of age was derived from published data.13
| (2) |
where a is 0.358 and 0.466 and b is −0.0459 and −0.0542 for muscle and fat, respectively. For all tissues across all ages, lymph flow was assumed to be 0.2% of plasma flow for a given tissue,28 and endosomal volume to be 0.5% of total tissue volume.29
2.3 ∣. PBPK model fitting and parameter estimation
IFX PK data from two cohorts were simultaneously fitted with the PBPK model. Data were censored if observed concentrations were below the LLOQ or above the ULOQ. The censoring interval for LLOQ and ULOQ were 0–0.5 μg/mL and 40–150 μg/mL, respectively. Most of the drug-specific parameters were taken from the previous publication, where they have been optimized for typical antibodies across different species and in humans,30 and assumed to be the same across the age range (Table S5). Only degradation rate of IgG that is unbound to FcRn (Kdeg) was estimated with the Stochastic Approximation Expectation Maximization (SAEM) algorithm in Monolix (2019R2). Kdeg was assumed to be lognormally distributed, and was characterized using the following equation:
| (3) |
where Kdegi and Kdegpop are individual and typical values of population parameters, and ηi denotes the IIV random effects, which are normally distributed with a mean of 0 and a variance of . The tested residual error models included additive, proportional and combined error models. Model qualification was guided by the precision of parameter estimates, goodness-of-fit (GOF) plots, % shrinkage and visual predictive check (VPC) plots. Albumin levels, immunomodulator use (azathioprine, 6-mercaptopurine or methotrexate), C-reactive protein (CRP) levels, type of disease (IBD, JIA, uveitis) and study type (cohort 1 vs. 2) were tested as covariates on the Kdeg, as these parameters have been reported previously to influence the PK of IFX.31-37 Statistical tests were made using Pearson's correlation or analysis of variance (ANOVA) and Wald test, where a P-value of <.05 was considered statistically significant.
2.4 ∣. Monte Carlo simulation
The paediatric PBPK model with the estimated value for Kdeg was used in the mlxR package (version 4.0) in R software to simulate plasma and tissue PK profiles of IFX in 0–20-year-old male and female children. The simulations accounted for IIV of Kdeg without residual variability. For each dosing regimen in each age and sex group, we simulated the concentrations every 1 hour for 1000 patients for at least eight doses to ensure reaching the steady state. BW for the simulated patients in each age group were randomly sampled, assuming they were lognormally distributed, using the mean values and standard deviation reported from the Centers for Disease Control and Prevention (CDC).38 We simulated five doses (5, 7.5, 10, 12 and 15 mg/kg) with different frequencies [every 4 weeks (Q4W), every 6 weeks (Q6W), and every 8 weeks (Q8W)]. The median, 5th and 95th percentiles were determined based on 1000 individual simulations, and 90% confidence intervals (CI) were derived using R software. Based on our clinical experience and published literature,22 we examined IFX target trough concentrations of 3, 5, 7 and 16 μg/mL. The percentage of patients who achieved the target trough concentration for each of the different dosing regimens was calculated.
We also investigated the age vs. area under the concentration–time curve (AUC) and age vs. clearance relationships for IFX. We calculated the AUC during one dosing interval at steady state (AUCss) for each 1000 simulated patient, and calculated the median, 5th and 95th percentiles, and derived 90% CI for each age and sex group. Clearance and BW-normalized clearance were calculated by dividing dose (mg) or BW-based dose (mg/kg) by AUCss. The predicted AUCss, clearance and BW-normalized clearance in different age groups were compared with clinically observed data reported in the literature.31,39-43
2.5 ∣. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.
3 ∣. RESULTS
The PBPK model was able to characterize the PK of IFX from the two cohorts, with ages ranging from 4 to 19 years and BW ranging from 14.2 to 138 kg, reasonably well (Figure 2). The Kdeg was estimated with good precision [relative standard error (RSE) = 3.62%] and the optimized typical value of Kdeg for IFX was 44.6 h−1, which was slightly higher than previously published typical values for mAb (15.3 h−1).30 The estimated IIV of Kdeg was 33.6% (RSE = 8.62%) and shrinkage was 4.19%. A proportional error model best described the residual variability with an estimate of 32.3% (RSE = 10.4%). Covariate analysis demonstrated no statistically significant predictors of Kdeg in the PBPK model using Pearson's correlation tests/ANOVA and Wald tests (Table S6). In addition, no significant patterns were observed in the plots of predicted vs. observed concentration when stratified by weight, sex, dosing regimens and all covariates (Figures S1-S4). No patterns were observed in the delta plot for each covariate (Figure S5). As such, no covariates were included in the final population PBPK model.
FIGURE 2.
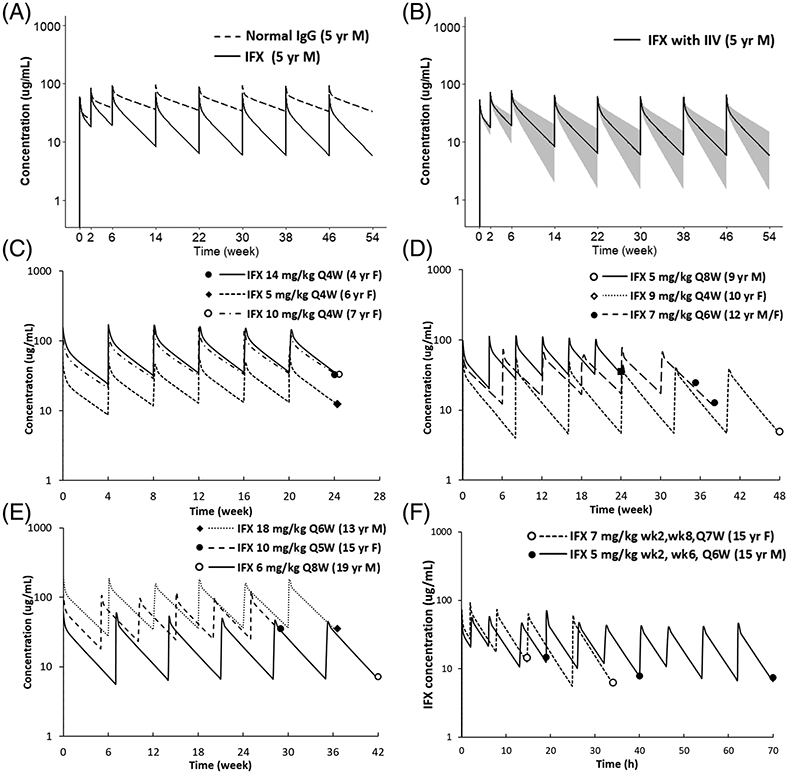
(A) Comparison of plasma PK profiles of a typical antibody and infliximab. Figure displays simulated plasma PK profiles of a typical antibody and infliximab in a 5-year-old male (weight 18.3 kg) after receiving infliximab 5 mg/kg at weeks 0, 2 and 6 and then every 8 weeks. (B) Population simulation of infliximab plasma PK profiles. Simulated infliximab plasma PK profiles in the 5-year-old male paediatric population (weight 18.3 kg, CV 14.5%) accounting for inter-individual variability of Kdeg. The black line represents the median, and the shaded area represents the 90% CI. (C–F) represent predicted and observed infliximab plasma PK profiles in paediatrics for different ages, sex and treatment indications (i.e., induction vs. maintenance). Figure displays individual prediction (solid lines) and observed infliximab concentration (solid dots) for (C) 4–7 years every 4 weeks maintenance therapy; (D) 8–12 years every 4–8 weeks maintenance therapy; (E) 13–20 years every 5–8 weeks maintenance therapy; (F) 15 years induction therapy
Figure 2(A) shows the simulated plasma PK profiles of IFX in comparison with the PK of a nonspecific mAb in a 5-year-old child, at an FDA-approved dosing regimen of IFX in children (i.e., 5 mg/kg Q8W). Figure 2(B) shows the simulated plasma PK profile of IFX from Figure 2(A) with 90% CI, which accounts for the IIV. The model predictions for several representative patients, across different age groups, disease types and dosing regimens, along with their observed data, are shown in Figure 2(C)-(F). These representative patients were chosen to cover the entire age ranges of the two cohorts and represent different indications, dose levels and dose frequencies. The model was able to predict the PK of IFX across age, sex, dosing regimens, treatment indication (i.e., induction versus maintenance) and disease type, reasonably well.
Figure 3(A) shows the predicted vs. observed concentrations of IFX. Before accounting for IIV stemming from Kdeg, 70% of predictions were within twofold of the observed data, whereas, after accounting for IIV stemming from Kdeg, more than 99% of predictions were within twofold of the observed concentrations, as shown in Figure 3(B). Figure 3(C)-(F) shows the plots of population weighted residuals (WRES) and individual WRES with respect to the time and prediction, demonstrating that the points were scattered evenly around the horizontal zero-line without any pattern.
FIGURE 3.

Diagnostic plots. The plots of population (A) and individual (B) prediction vs. observed concentration along with the identity line (solid line) and two-fold boundary lines (dashed lines) stratified by different age groups. The plots of population (C) and individual (D) weighted residuals vs. time since first dose, and population (E) and individual (F) weighted residuals vs. observations, along with spline interpolation (blue lines)
Figure 4 shows the median and 90% CI for clearance, BW-normalized clearance and AUCss in children from 0 to 20 years of age, based on population simulation results. The PBPK model-predicted PK parameters were similar to the parameter values reported in different clinical studies (Figure 4). Although total clearance (mL/day) increased with age, BW-normalized clearance (mL/day/kg) and AUCss remained steady from 0 to 20 years of age, suggesting no significant change in systemic clearance or drug exposure during childhood growth and development.
FIGURE 4.

Relationships between age and PK parameters. The plots display PBPK model simulated values of (A) total clearance (mL/day), (B) body weight normalized clearance (mL/day/kg), and (C) AUC0-tau at steady state for infliximab in 0–20-year-old subjects, superimposed over the published data collected from Refs. 31, 39-41 (red circles). The black line and the shaded area represent the median and the 90% CI, respectively
The PBPK model was also used to predict the percentage of children that achieve the target trough concentration at different, commonly used, dosing regimens. The results from this analysis are summarized in Table 2. Of note, here we present results based on simulations conducted for 10-year-old males. However, the percentages were similar for children between 0–20 years and for both males and females (data not shown), and the information summarized in Table 2 is applicable to children in general. We found that when receiving FDA-labelled 5 mg/kg Q8W regimen, only 30% and 50% of children achieved a trough concentration >5 μg/mL and >3 μg/mL, respectively. More intense dosing strategies (e.g., doses ≥10 mg/kg and/or drug frequency ≤Q6W) were required to achieve concentrations >5 μg/mL in >80% of patients. For higher target trough concentrations (e.g., >7 μg/mL), more frequent dosing was more efficient in achieving target trough concentrations than dose escalation (i.e., mg/kg). For example, 10 mg/kg Q6W and 10 mg/kg Q4W dosing regimens resulted in >90% of children achieving trough concentrations >7 μg/mL and >16 μg/mL, respectively.
TABLE 2.
Percentage of paediatrics predicted to achieve target infliximab trough concentrations by dosing regimen
| Percentage (%) achieved target Ctrough |
||||
|---|---|---|---|---|
| > 3 ug/mL | > 5 ug/mL | > 7 ug/mL | > 16 ug/mL | |
| 5 mg/kg | ||||
| Q8W | 73.7 | 50.8 | 30.1 | 2.80 |
| Q6W | 94.1 | 82.8 | 66.3 | 16.7 |
| Q4W | 99.8 | 98.7 | 95.9 | 60.1 |
| 7.5 mg/kg | ||||
| Q8W | 87.2 | 68.7 | 53.9 | 13.5 |
| Q6W | 97.9 | 92.6 | 85.0 | 38.9 |
| Q4W | 100 | 99.6 | 98.7 | 85.0 |
| 10 mg/kg | ||||
| Q8W | 91.9 | 80.8 | 66.7 | 23.0 |
| Q6W | 98.8 | 96.7 | 91.3 | 58.1 |
| Q4W | 100 | 99.8 | 99.6 | 93.3 |
| 12.5 mg/kg | ||||
| Q8W | 94.4 | 87.0 | 76.0 | 34.6 |
| Q6W | 99.4 | 97.9 | 95.4 | 70.2 |
| Q4W | 100 | 100 | 99.8 | 96.7 |
| 15 mg/kg | ||||
| Q8W | 96.6 | 89.4 | 82.9 | 46.4 |
| Q6W | 99.6 | 98.7 | 97.0 | 79.5 |
| Q4W | 100 | 100 | 99.8 | 98.0 |
Percentages were calculated based on the results of 1000 simulations in 10-year-old male populations accounting for inter-individual variability and without residual error.
4 ∣. DISCUSSION
Whether the PK of mAbs in children is different from those in adults remains unclear. However, it is known that children have marked variability in the PK of mAb such as IFX, and target trough concentration achievement in children frequently requires higher IFX dosing for IBD, JIA and uveitis.44,45 In addition, studies that use the population PK modelling approach indicate there are relationships between body weight and IFX PK parameters.46 To investigate if there are any physiological bases for observing the differences in the PK of mAb between adults and paediatrics, and to develop a mathematical model that can characterize population PK of mAbs in paediatrics, we have extended our previously established platform PBPK model for mAbs to pediatrics.15 We have also evaluated the ability of this model to characterize the clinical PK of IFX (a prototype drug) in different age groups, across different BW, sex, disease types and dosing regimen. In the future, we hope to apply our model to other mAbs commonly prescribed to children, in order to advance the field of paediatric precision therapeutics and optimize mAb dosing strategies for children.
While several population PK models of IFX have been published, PBPK models offer the potential opportunity to provide more mechanistic insight into age-dependent changes in the PK of mAb. In addition, two paediatric PBPK models of mAbs have been published recently.9,47 However, these models do not estimate IIV in the PK mAbs based on observed individual data. To our knowledge, this is the first study that combines a paediatric PBPK model with population approaches, which allow the model to account for the dynamic changes in the physiology of children along with the IIV observed in the physiological parameters. This is an important feature of the model considering the variability in the PK data from paediatric populations and availability of only sparse data.20 Recently, Malik and Edginton have investigated the PK of IFX in paediatrics using a PBPK model established based on adult data.48 The model included age-dependent physiological information based on a PK-Sim database,3 and parameters related to target-mediated drug disposition (TMDD) and anti-drug antibody (ADA) were considered. However, their model failed to capture the observed paediatric trough concentration, suggesting the accuracy of characterizing age-dependent changes in physiological parameters is an important feature of the paediatric PBPK model, and additional age-dependent factors may need to be considered when extrapolating the adult PBPK model to paediatrics. On the other hand, our approach allowed us to characterize the IIV in the clinical PK of model mAb IFX, following estimation of just one physiological parameter and associated variability. The PBPK model was able to well-capture the IFX PK data across different ages, weights and sex, and the estimated CV% of Kdeg indicated high variability in the elimination parameter between paediatric patients. Of significance to our PBPK model is that by using a consistent population Kdeg value and solely accounting for the paediatric physiology and anatomy, we are able to fit the trough concentrations across 4–19 year olds, which also validates paediatric physiology and anatomy parameterization of the model.16
We have incorporated a series of published equations that describe physiological parameters and age relationships into the PBPK model,16 allowing us to account for real-time growth and maturation of the individuals throughout the time course of drug exposure. This time-variant PBPK model is especially relevant for chronic disorders that start in childhood and continue through adulthood (e.g., IBD, JIA and uveitis).49 Age-dependent changes in interstitial volumes of adipose17,18 and muscle19 were also incorporated into the PBPK model. However, such information is not available for other tissues. It is important to note that this is a key parameter, since tissue interstitial volume directly determines the interstitial concentrations at the site-of-action. To evaluate the effect of this parameter, we further simulated tissue PK profiles of the mAb in 1 month-old, 1, 2, 5, 10 and 15-year-old children, as shown in Figure S6. We observed that the PK profiles of mAb in fat and muscle were significantly different among different age groups, and the PK was not always parallel to the plasma PK profiles. However, for other tissues that did not have age-dependent changes in interstitial volume, the PK profiles were similar across different age groups and parallel to the plasma PK profiles. As such, more information about age-dependent changes in tissue composition is needed to better understand tissue PK of mAbs in paediatrics.5 It should be noticed that we aim to demonstrate the ability of the PBPK model to simulate tissue PK profiles, and thus whenever actual tissue data is available in the future, they can be superimposed with our simulations to further validate the model.
While this model serves as a foundation for the development of a platform paediatric PBPK model for mAbs, there are many other physiological parameters that could change with age that are not accounted for in this model, since there is no robust quantitative information that can facilitate mathematical characterization of the continuous changes in these parameters with age. Malik et al.11 have reported that the extravasation rate of mAb is approximately three times higher in neonates than in adults, and organ capillary density follows a U-shaped relationship as a function of age. Both of these age-dependent changes in physiology can affect mAb convection from plasma to interstitial space, and may be relevant to a paediatric PBPK model. Lymphatic tissue has also been reported to change with age,8 but specific lymph flow rate data in paediatrics is not yet available. Therefore, here we have assumed that paediatric lymph flow is proportional to the regional blood flow, and set it at 0.2% of the plasma flow28 (i.e., similar to adults). Other studies have set the lymph flow at 2–2.6-fold higher in neonates than in adults based on allometry scaling from adult data9 or animal studies,10,11 highlighting the differences in PBPK approaches due to gaps in the availability of paediatric physiological data. Of note, while the lymph flow is not a highly sensitive parameter when it comes to plasma PK of mAbs, changes in this parameter can lead to significant changes in tissue PK of mAbs, which is hard to measure in the clinic.
There are studies that also support that haematopoietic cells are involved in the homeostasis of mAb due to the expression and function of FcRn in these cells.50,51 In the current PBPK model, however, only endothelial cells are assumed to express the FcRn. Since it is reported that hematopoietic cell concentrations at birth are about twofold higher than in adults and decline with age,11,12 incorporating age-dependent mAb homeostasis via haematopoietic cells may be included in the paediatric PBPK model to refine it further.11 Endogenous mAb can also compete with exogenous mAb for FcRn in the endosomal compartment.52 The ontogeny of endogenous mAb has been reported previously, with the overall endogenous mAb concentration reaching 50% of the adult level by the end of the first year of life.9 In addition, increased mAb clearance has been observed in patients with higher endogenous mAb burden associated with disease status.53 While the current PBPK model did not account for the contribution of endogenous mAb, age-dependent and/or disease-related changes in endogenous mAb in the endosomal compartment can be easily included in the model. It is important to note that since all the PK data used in this investigation came from children that were 4 years of age or older, any model misspecification issues related to this assumption may not have been noticeable during our analysis. Similarly, due to the paucity of data and lack of consistency in reported ontogeny-related changes in FcRn expression6,7 in animals and humans,3 here we have not incorporated age-related changes in FcRn expression in the PBPK model. Hardiansyah and Ng54 used a minimal PBPK model to analyse the relationship between age, weight and FcRn concentration, suggesting that FcRn expression was inversely proportional to age. However, other studies have conversely reported FcRn expression to increase with age until the end of puberty.6,7 Ultimately, more studies are needed for quantitative measurement of FcRn in paediatric organs before incorporation of this data into the PBPK model. As such, several assumptions had to be made during the development of our PBPK model, due to the limited physiologic information available for paediatrics. However, since the model presented here is a platform model, it can be easily updated to account for any new information about age-dependent factors as it becomes available.
In our PBPK model, drug-specific and system-specific parameters including Kdeg, pinocytosis rate, association and dissociation rate constant between antibodies and FcRn, and FcRn concentration, altogether describe the dynamics of intracellular processing and antibody trafficking at the cellular level.15 We have optimized the Kdeg value to represent the PK property specific to IFX. The optimized Kdeg value is able to capture the PK data of IFX in both adult (Figure S7) and paediatric populations (Figure S8) reported in the literature.31,39,55-60 Our model is able to predict the reported peak concentrations (Cmax), trough concentrations and distribution phase of IFX, for both single and multiple dosages, demonstrating the usability of the current PBPK model to predict the PK profiles of IFX. However, other parameters such as pinocytosis rate or the parameters related to antibody-FcRn binding might be different for IFX compared to other antibodies.61 Therefore, once the evidence in this field is more concrete, one can easily change the Kdeg values or replace/add this parameter with other parameters to further optimize the PBPK model.
As prior clinical studies reported IFX to display linear PK when given at doses that are commonly used in clinical practice,62 we did not include target-mediated drug disposition (TMDD) in the current PBPK model. Additionally, it has been previously reported that a wide range of TNF-α levels (0.1–100 pM) had minimal effect on IFX clearance according to sensitivity analysis.9 For mAbs other than IFX, where targets play important roles in drug disposition, one would need to incorporate soluble or membrane target into the platform paediatric PBPK model presented here.
Anti-drug antibodies (ADA) may affect the PK and pharmacodynamics (PD) of mAbs, but several factors such as limitations in ADA quantification, relative rarity of immunogenicity in clinical trials, and yet to be characterized tissue distribution of ADA-mAb complexes, make it challenging to incorporate ADA into physiologically-based PK/PD models.63 Corresponding to the clinical observations that approximately 7–10% of patients develop ADA during maintenance therapy,64 11% of patients in our dataset developed ADA. Only half of these patients had detectable IFX concentrations and, as such, it was felt that the current dataset provided limited information about the ADA and IFX PK relationship. However, data below the LLOQ (primarily from patients with ADA) or above the ULOQ were treated as interval censored data in an attempt to be comprehensive and include this relevant information into the PBPK model.
Markers that detect acute systemic inflammation in paediatric IBD like CRP and erythrocyte sedimentation rate (ESR) have been identified as covariates of the IFX clearance.23,65 Our analysis indicated that CRP was not a significant predictor of Kdeg, while information on ESR was limited in our patient cohorts; hence, they are not included in the PBPK model. Of note, in the future, if one has established the relationship between these disease-related biomarkers and physiological parameters, they can be incorporated into the PBPK model.
The estimated residual variability (32.3%) in our study was within the range of the reported values for IFX PK models in paediatrics.31,65 The between-subject variability and assay error may contribute to this uncertainty. The assays used to analyse IFX concentrations in our study have reported variability of 20% for cohort 166 and cohort 2,27 which can contribute to the observed residual variability. While we did not include between-subject variability in our model, since most patients in the two cohorts had two or fewer measurements, it has been reported that incorporating the time-varying covariates in the model may improve between-subject variability.65 In addition, since we did not have rich data and a long enough follow-up period for each patient to build a reliable relationship between age and covariates, only the covariate values measured at the time of dosing and PK sampling were incorporated into the covariate analysis.
Monte Carlo simulations allowed us to account for IIV in Kdeg and predict a range of concentrations for different paediatric ages and for various dosing regimens. The predicted systemic exposure (AUCss) and clearance, along with 90% CI, for patients of 0–20 years provided insight into whether linear adult-to-paediatric extrapolation is appropriate and whether the same BW-based dosing strategies can be applied to the entire paediatric age range. Although total clearance (mL/day) of IFX increased with age, when normalized by BW, clearance per kg remained unchanged across different age groups. This corresponded to the clinical observations that weight-normalized IFX clearance in young patients (including infants with Kawasaki disease) was similar to the clearance in adult patients with RA,39,67-69 and agreed with statements that clearance of most mAbs were comparable between adults and paediatrics after adjusting for body size.70,71 However, our finding contradicted some review articles which concluded that most mAbs show a higher BW-normalized clearance in paediatrics than in adults.1,72 One reason for this discrepancy may be that the youngest patient in our dataset was only 4 years of age and data from younger children are needed to validate our simulated observations. In addition, our PBPK model predicts that the weight-normalized clearance is relative consistent across different paediatric ages, which may seem to contradict some studies that use a population PK model approach and conclude a nonlinear relationship between weight and IFX clearance.23,31,34,65 However, one should be cautious when directly comparing the estimated parameters (i.e., clearance, volume of distribution) between PBPK modelling and population PK modelling approaches, since both systems have their unique features and emphases. For example, the PBPK models consider the BW-related changes in organ weights and flow rates, where the physiology and autonomy parameterization have been incorporated. On the other hand, the population PK models usually account for BW-related changes in clearance or volume of distribution, which are empirical parameters.39 Moreover, body weight and other covariates such as albumin level and immunomodulator usage altogether may contribute to the estimations of these empirical PK parameters.31-37 Therefore, in a population PK model, one may not use just the body weight–clearance relationship to assess the effect of weight on clearance. Of note, we are concluding that in the age range we have studied, there is no clear trend between age and weight-normalized clearance. Once more data for children younger than 4 years are available, further investigation on the effect of age on antibody clearance is needed before making any solid conclusions regarding this subject matter.
Of paramount importance to clinicians are the results from our population PBPK simulation. Using the standard FDA-labelled 5 mg/kg Q8W dosing of IFX, the majority of patients fall far below trough concentration of 5–16 μg/mL, which are considered to be adequate for IBD treatment,73 associated with improved clinical outcomes based on clinical trial results,22,74,75 recommended by clinical guideline,76 and desired based on our own clinical experience. Our simulations also showed that higher doses and/or shorter dosing intervals are needed to achieve target troughs for paediatrics (Table 2).73,77 This finding corresponds well with previous publications that have used a population PK approach for dose optimization of IFX in children.22,74
In summary, here we have developed a population PBPK model to characterize paediatric PK of mAbs. Age-related changes in BW, organ weight and organ blood flow rate were incorporated into the PBPK model, along with the changes in interstitial volumes of adipose and muscle. The model was used to characterize clinical PK of IFX by estimating just one model parameter and associated IIV. The PBPK model was further used to simulate age vs. clearance and the dose–exposure relationship for IFX. Our results suggest that a more intense dosing regimen may be needed to achieve the targeted trough concentrations of IFX in the majority of the paediatric patients.
Supplementary Material
What is already known about this subject
There is no consensus on whether the PK of antibodies differ significantly between adults and children.
PBPK models can provide mechanistic insight into age-dependent changes in the PK of drug molecules.
What this study adds
A paediatric PBPK model combined with population approaches has been developed to enable a priori prediction of antibody PK across different age groups, and characterization of interindividual variability in the clinical PK of antibodies in paediatrics.
The PBPK model suggests that higher doses and/or more frequent dosing of infliximab than the FDA-labelled dosing regimen may be needed to achieve desired trough concentrations of the antibody in the majority of paediatric patients.
ACKNOWLEDGEMENTS
At the time of this work R.S.F. was supported by a University of Kansas General Research Fund grant, a CTSA grant from NCATS awarded to the University of Kansas for Frontiers: University of Kansas Clinical and Translational Science Institute (# KL2TR002367) and a Kansas Institute of Precision Medicine Centers of Biomedical Research Excellence grant from NIGMS awarded to the University of Kansas Medical Center (# P20GM130423). V.S. was supported by a career development grant from NIDDK (5K23DK115827).
M.L.B. receives support from the National Institutes of Health (5R01HD089928–03, 5U24TR001608-4, HHSN275201800003I, 3U24TR001608-04S1), FDA Arthritis Advisory Committee, and the Childhood Arthritis and Rheumatology Research Alliance.
A.F. is a scientific consultant for Takeda Pharmaceuticals unrelated to the submitted work.
This work was supported by the following grants to D.K.S.: National Institute of General Medical Sciences grant [GM114179], National Institute of Allergy and Infectious Diseases grant [AI138195], and National Cancer Institute grant [R01CA246785]; supported by the following grant to M.L.B.: National Institute of Child Health and Human Development [5R01HD089928-03], and supported by the following grant to V.S.: National Institute of Diabetes and Digestive and Kidney Diseases [DK115827].
Funding information
Center for Strategic Scientific Initiatives, National Cancer Institute, Grant/Award Number: R01CA246785; Kansas Institute of Precision Medicine Centers of Biomedical Research Excellence grant from NIGMS awarded to the University of Kansas Medical Center, Grant/Award Number: P20GM130423; National Institute of Allergy and Infectious Diseases, Grant/Award Number: AI138195; National Institute of Child Health and Human Development, Grant/Award Number: 5R01HD089928-03; National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: DK115827; National Institute of General Medical Sciences, Grant/Award Numbers: GM114179, GM130423; National Institutes of Health, Grant/Award Numbers: 3U24TR001608-04S1, 5U24TR001608-4, HHSN275201800003I; NIDDK, Grant/Award Number: 5K23DK115827; University of Kansas Clinical and Translational Science Institute, Grant/Award Number: KL2TR002367
Footnotes
COMPETING INTERESTS
With the exception of any potential conflicts mentioned in the Acknowledgments section, all authors report no conflicts of interest related to this work.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Temrikar ZH, Suryawanshi S, Meibohm B. Pharmacokinetics and clinical pharmacology of monoclonal antibodies in pediatric patients. Paediatr Drugs. 2020;22(2):199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edlund H, Melin J, Parra-Guillen ZP, Kloft C. Pharmacokinetics and pharmacokinetic-pharmacodynamic relationships of monoclonal antibodies in children. Clin Pharmacokinet. 2015;54(1):35–80. [DOI] [PubMed] [Google Scholar]
- 3.Malik P, Edginton A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin Drug Metab Toxicol. 2018;14(6):585–599. [DOI] [PubMed] [Google Scholar]
- 4.Mahmood I. Pharmacokinetic considerations in designing pediatric studies of proteins, antibodies, and plasma-derived products. Am J Ther. 2016;23(4):e1043–e1056. [DOI] [PubMed] [Google Scholar]
- 5.Friis-Hansen B. Body water compartments in children: changes during growth and related changes in body composition. Pediatrics. 1961;28: 169–181. [PubMed] [Google Scholar]
- 6.Tian Z, Sutton BJ, Zhang X. Distribution of rat neonatal Fc receptor in the principal organs of neonatal and pubertal rats. J Recept Signal Transduct Res. 2014;34(2):137–142. [DOI] [PubMed] [Google Scholar]
- 7.Cianga C, Cianga P, Plamadeala P, Amalinei C. Nonclassical major histocompatibility complex I-like Fc neonatal receptor (FcRn) expression in neonatal human tissues. Hum Immunol. 2011;72(12):1176–1187. [DOI] [PubMed] [Google Scholar]
- 8.Snyder WS, Cook MJ, Nasset ES, Karhausen LR, Howells GP, Tipton IH. Report of the Task Group on Reference Man. Oxford: Pergamon Press; 1975. [Google Scholar]
- 9.Pan X, Stader F, Abduljalil K, et al. Development and application of a physiologically-based pharmacokinetic model to predict the pharmacokinetics of therapeutic proteins from full-term neonates to adolescents. AAPS J. 2020;22(4):76. [DOI] [PubMed] [Google Scholar]
- 10.Johnson SA, Vander Straten MC, Parellada JA, Schnakenberg W, Gest AL. Thoracic duct function in fetal, newborn, and adult sheep. Lymphology. 1996;29(2):50–56. [PubMed] [Google Scholar]
- 11.Malik PRV, Edginton AN. Integration of ontogeny into a physiologically based pharmacokinetic model for monoclonal antibodies in premature infants. J Clin Pharmacol. 2020;60(4):466–476. [DOI] [PubMed] [Google Scholar]
- 12.Shearer WT, Rosenblatt HM, Gelman RS, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. 2003; 112(5):973–980. [DOI] [PubMed] [Google Scholar]
- 13.Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45(10):1013–1034. [DOI] [PubMed] [Google Scholar]
- 14.Ginsberg G, Hattis D, Miller R, Sonawane B. Pediatric pharmacokinetic data: implications for environmental risk assessment for children. Pediatrics. 2004;113(4 Suppl):973–983. [PubMed] [Google Scholar]
- 15.Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1): 67–86. [DOI] [PubMed] [Google Scholar]
- 16.Chang HP, Kim SJ, Shah DK. Age-related changes in pediatric physiology: quantitative analysis of organ weights and blood flows. AAPS J. 2021;23(3):50. [DOI] [PubMed] [Google Scholar]
- 17.Baker GL. Human adipose tissue composition and age. Am J Clin Nutr. 1969;22(7):829–835. [DOI] [PubMed] [Google Scholar]
- 18.Boulton TJ, Dunlop M, Court JM. The growth and development of fat cells in infancy. Pediatr Res. 1978;12(9):908–911. [DOI] [PubMed] [Google Scholar]
- 19.Dickerson JW, Widdowson EM. Chemical changes in skeletal muscle during development. Biochem J. 1960;74(2):247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krauss M, Tappe K, Schuppert A, Kuepfer L, Goerlitz L. Bayesian population physiologically-based pharmacokinetic (PBPK) approach for a physiologically realistic characterization of interindividual variability in clinically relevant populations. PLoS ONE. 2015;10(10): e0139423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malik PRV, Hamadeh A, Phipps C, Edginton AN. Population PBPK modelling of trastuzumab: a framework for quantifying and predicting inter-individual variability. J Pharmacokinet Pharmacodyn. 2017;44(3): 277–290. [DOI] [PubMed] [Google Scholar]
- 22.Winter DA, Joosse ME, de Wildt SN, Taminiau J, de Ridder L, Escher JC. Pharmacokinetics, pharmacodynamics, and immunogenicity of infliximab in pediatric inflammatory bowel disease: a systematic review and revised dosing considerations. J Pediatr Gastroenterol Nutr. 2020;70(6):763–776. [DOI] [PubMed] [Google Scholar]
- 23.Bauman LE, Xiong Y, Mizuno T, et al. Improved population pharmacokinetic model for predicting optimized infliximab exposure in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2020;26(3):429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Funk RS, Shakhnovich V, Cho YK, et al. Factors associated with reduced infliximab exposure in the treatment of pediatric autoimmune disorders: a cross-sectional prospective convenience sampling study. Pediatr Rheumatol Online J. 2021;19(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piester T, Frymoyer A, Christofferson M, Yu H, Bass D, Park KT. A mobile infliximab dosing calculator for therapy optimization in inflammatory bowel disease. Inflamm Bowel Dis. 2018;24(2):227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlov IY, Carper J, Lazar-Molnar E, Delgado JC. Clinical laboratory application of a reporter-gene assay for measurement of functional activity and neutralizing antibody response to infliximab. Clin Chim Acta. 2016;453:147–153. [DOI] [PubMed] [Google Scholar]
- 27.Wang SL, Ohrmund L, Hauenstein S, et al. Development and validation of a homogeneous mobility shift assay for the measurement of infliximab and antibodies-to-infliximab levels in patient serum. J Immunol Methods. 2012;382(1–2):177–188. [DOI] [PubMed] [Google Scholar]
- 28.Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50(1):3–20. [DOI] [PubMed] [Google Scholar]
- 29.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709. [DOI] [PubMed] [Google Scholar]
- 30.Chang HP, Kim SJ, Shah DK. Whole-body pharmacokinetics of antibody in mice determined using enzyme-linked immunosorbent assay and derivation of tissue interstitial concentrations. J Pharm Sci. 2021; 110(1):446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fasanmade AA, Adedokun OJ, Blank M, Zhou H, Davis HM. Pharmacokinetic properties of infliximab in children and adults with Crohn's disease: a retrospective analysis of data from 2 phase III clinical trials. Clin Ther. 2011;33(7):946–964. [DOI] [PubMed] [Google Scholar]
- 32.Petitcollin A, Leuret O, Tron C, et al. Modeling immunization to infliximab in children with Crohn's disease using population pharmacokinetics: a pilot study. Inflamm Bowel Dis. 2018;24(8):1745–1754. [DOI] [PubMed] [Google Scholar]
- 33.Ternant D, Ducourau E, Perdriger A, et al. Relationship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br J Clin Pharmacol. 2014;78(1):118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dotan I, Ron Y, Yanai H, et al. Patient factors that increase infliximab clearance and shorten half-life in inflammatory bowel disease: a population pharmacokinetic study. Inflamm Bowel Dis. 2014;20(12): 2247–2259. [DOI] [PubMed] [Google Scholar]
- 35.Kevans D, Murthy S, Mould DR, Silverberg MS. Accelerated clearance of infliximab is associated with treatment failure in patients with corticosteroid-refractory acute ulcerative colitis. J Crohns Colitis. 2018;12(6):662–669. [DOI] [PubMed] [Google Scholar]
- 36.Matsuoka K, Hamada S, Shimizu M, et al. Factors contributing to the systemic clearance of infliximab with long-term administration in Japanese patients with Crohn's disease: analysis using population pharmacokinetics. Int J Clin Pharmacol Ther. 2020;58(2):89–102. [DOI] [PubMed] [Google Scholar]
- 37.Dreesen E, Faelens R, Van Assche G, et al. Optimising infliximab induction dosing for patients with ulcerative colitis. Br J Clin Pharmacol. 2019;85(4):782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuczmarski RJ, Ogden CL, Guo SS, et al. 2000 CDC Growth Charts for the United States: Methods and Development. Vital and Health Statistics Series 11, Data from the National Health Survey. Hyattsville, MD: Department of Health and Human Services; 2002. [PubMed] [Google Scholar]
- 39.Adedokun OJ, Xu Z, Padgett L, et al. Pharmacokinetics of infliximab in children with moderate-to-severe ulcerative colitis: results from a randomized, multicenter, open-label, phase 3 study. Inflamm Bowel Dis. 2013;19(13):2753–2762. [DOI] [PubMed] [Google Scholar]
- 40.Hadigan C, Baldassano R, Braegger CP, et al. Pharmacokinetics of infliximab (anti-TNF∞) in children with Crohn's disease: a multicenter trial. J Pediatr Gastroenterol Nutr. 1999;29(4):525. [Google Scholar]
- 41.Klotz U, Teml A, Schwab M. Clinical pharmacokinetics and use of infliximab. Clin Pharmacokinet. 2007;46(8):645–660. [DOI] [PubMed] [Google Scholar]
- 42.Cech P, Maly T, Mala L, Zahalka F. Body composition of elite youth pentathletes and its gender differences. Sport Sci. 2013;6(2): 29–35. [Google Scholar]
- 43.Bindler RC, Howry LB, Wilson BA, Shannon MT, Stang CL. Prentice Hall Pediatric Drug Guide. Englewood Cliffs, NJ: Pearson; 2005. [Google Scholar]
- 44.Sukumaran S, Marzan K, Shaham B, Reiff A. High dose infliximab in the treatment of refractory uveitis: does dose matter? ISRN Rheumatol. 2012;2012:765380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tambralli A, Beukelman T, Weiser P, Atkinson TP, Cron RQ, Stoll ML. High doses of infliximab in the management of juvenile idiopathic arthritis. J Rheumatol. 2013;40(10):1749–1755. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Langevin BA, Zhou H, Xu Z. Model-aided adults-to-children pharmacokinetic extrapolation and empirical body size-based dosing exploration for therapeutic monoclonal antibodies—is allometry a reasonable choice? J Clin Pharmacol. 2020;60(12): 1573–1584. [DOI] [PubMed] [Google Scholar]
- 47.Basu S, Lien YTK, Vozmediano V, et al. Physiologically based pharmacokinetic modeling of monoclonal antibodies in pediatric populations using PK-Sim. Front Pharmacol. 2020;11:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malik PRV, Edginton AN. Physiologically-based pharmacokinetic modeling vs. allometric scaling for the prediction of infliximab pharmacokinetics in pediatric patients. CPT Pharmacometrics Syst Pharmacol. 2019;8(11):835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abduljalil K, Jamei M, Rostami-Hodjegan A, Johnson TN. Changes in individual drug-independent system parameters during virtual paediatric pharmacokinetic trials: introducing time-varying physiology into a paediatric PBPK model. AAPS J. 2014;16(3):568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu X, Meng G, Dickinson BL, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol. 2001;166(5): 3266–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179(7):4580–4588. [DOI] [PubMed] [Google Scholar]
- 52.Li L, Gardner I, Dostalek M, Jamei M. Simulation of monoclonal antibody pharmacokinetics in humans using a minimal physiologically based model. AAPS J. 2014;16(5):1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waldmann TA, Strober W. Metabolism of immunoglobulins. In: Bach FH, Good RA, eds. Clinical Immunobiology. Vol.3 New York: Elsevier; 1976:71–95. [Google Scholar]
- 54.Hardiansyah D, Ng CM. Effects of the FcRn developmental pharmacology on the pharmacokinetics of therapeutic monoclonal IgG antibody in pediatric subjects using minimal physiologically-based pharmacokinetic modelling. MAbs. 2018;10(7):1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fasanmade AA, Adedokun OJ, Ford J, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65(12):1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baldassano R, Braegger CP, Escher JC, et al. Infliximab (REMICADE) therapy in the treatment of pediatric Crohn's disease. Am J Gastroenterol. 2003;98(4):833–838. [DOI] [PubMed] [Google Scholar]
- 57.Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum. 2007;56(9):3096–3106. [DOI] [PubMed] [Google Scholar]
- 58.Singh N, Rosenthal CJ, Melmed GY, et al. Early infliximab trough levels are associated with persistent remission in pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2014;20(10): 1708–1713. [DOI] [PubMed] [Google Scholar]
- 59.Candon S, Mosca A, Ruemmele F, Goulet O, Chatenoud L, Cézard JP. Clinical and biological consequences of immunization to infliximab in pediatric Crohn's disease. Clin Immunol. 2006;118(1):11–19. [DOI] [PubMed] [Google Scholar]
- 60.Hämäläinen A, Sipponen T, Kolho KL. Serum infliximab concentrations in pediatric inflammatory bowel disease. Scand J Gastroenterol. 2013;48(1):35–41. [DOI] [PubMed] [Google Scholar]
- 61.Chung S, Nguyen V, Lin YL, et al. An in vitro FcRn-dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. MAbs. 2019; 11(5):942–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sorkin MR, Walker JA, Kabaria SR, Torosian NP, Alabi CA. Responsive antibody conjugates enable quantitative determination of intracellular bond degradation rate. Cell Chem Biol. 2019;26(12): 1643–1651. [DOI] [PubMed] [Google Scholar]
- 63.Gómez-Mantilla JD, Trocóniz IF, Parra-Guillén Z, Garrido MJ. Review on modeling anti-antibody responses to monoclonal antibodies. J Pharmacokinet Pharmacodyn. 2014;41(5):523–536. [DOI] [PubMed] [Google Scholar]
- 64.Denmark VK, Mayer L. Current status of monoclonal antibody therapy for the treatment of inflammatory bowel disease: an update. Expert Rev Clin Immunol. 2013;9(1):77–92. [DOI] [PubMed] [Google Scholar]
- 65.Xiong Y, Mizuno T, Colman R, et al. Real-world infliximab pharmacokinetic study informs an electronic health record-embedded dashboard to guide precision dosing in children with Crohn's disease. Clin Pharmacol Ther. 2021;109(6):1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pavlov IY, Carper J, Lázár-Molnár E, Delgado JC. Clinical laboratory application of a reporter-gene assay for measurement of functional activity and neutralizing antibody response to infliximab. Clin Chim Acta. 2016;453:147–153. [DOI] [PubMed] [Google Scholar]
- 67.Hyams J, Crandall W, Kugathasan S, et al. Induction and maintenance infliximab therapy for the treatment of moderate-to-severe Crohn's disease in children. Gastroenterology. 2007;132(3):863–873. quiz 1165–1166 [DOI] [PubMed] [Google Scholar]
- 68.Burns JC, Best BM, Mejias A, et al. Infliximab treatment of intravenous immunoglobulin-resistant Kawasaki disease. J Pediatr. 2008; 153(6):833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hyams J, Damaraju L, Blank M, et al. Induction and maintenance therapy with infliximab for children with moderate to severe ulcerative colitis. Clin Gastroenterol Hepatol. 2012;10(4):391–399. [DOI] [PubMed] [Google Scholar]
- 70.Xu Z, Davis HM, Zhou H. Rational development and utilization of antibody-based therapeutic proteins in pediatrics. Pharmacol Ther. 2013;137(2):225–247. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Wei X, Bajaj G, et al. Challenges and considerations for development of therapeutic proteins in pediatric patients. J Clin Pharmacol. 2015;55(S3):S103–S115. [DOI] [PubMed] [Google Scholar]
- 72.Shi R, Derendorf H. Pediatric dosing and body size in biotherapeutics. Pharmaceutics. 2010;2(4):389–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kang B, Choi SY, Choi YO, et al. Infliximab trough levels are associated with mucosal healing during maintenance treatment with infliximab in paediatric Crohn's disease. J Crohns Colitis. 2019;13(2): 189–197. [DOI] [PubMed] [Google Scholar]
- 74.Frymoyer A, Hoekman DR, Piester TL, et al. Application of population pharmacokinetic modeling for individualized infliximab dosing strategies in Crohn disease. J Pediatr Gastroenterol Nutr. 2017;65(6): 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoekman DR, Brandse JF, de Meij TG, et al. The association of infliximab trough levels with disease activity in pediatric inflammatory bowel disease. Scand J Gastroenterol. 2015;50(9):1110–1117. [DOI] [PubMed] [Google Scholar]
- 76.Turner D, Ruemmele FM, Orlanski-Meyer E, et al. Management of paediatric ulcerative colitis, part 2: acute severe colitis—an evidence-based consensus guideline from the European Crohn's and Colitis Organization and the European Society of Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018; 67(2):292–310. [DOI] [PubMed] [Google Scholar]
- 77.Vande Casteele N, Ferrante M, Van Assche G, et al. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology. 2015;148(7):1320–1329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.