

Summary
Acute lymphoblastic leukemia (ALL) dissemination to the central nervous system (CNS) is a challenging clinical problem whose underlying mechanisms are poorly understood. Here, we show that primary human ALL samples injected into the femora of immunodeficient mice migrate to the skull and vertebral bone marrow and provoke bone lesions that enable passage into the subarachnoid space. Treatment of leukemia xenografted mice with a biologic antagonist of receptor activator of nuclear factor κB ligand (RANKL) blocks this entry route. In addition to erosion of cranial and vertebral bone, samples from individuals with B-ALL also penetrate the blood-cerebrospinal fluid barrier of recipient mice. Co-administration of C-X-C chemokine receptor 4 (CXCR4) and RANKL antagonists attenuate both identified routes of entry. Our findings suggest that targeted RANKL and CXCR4 pathway inhibitors could attenuate routes of leukemia blast CNS invasion and provide benefit for B-ALL-affected individuals.
Keywords: B cell acute lymphoblastic leukemia, B-ALL, CNS invasion, RANKL, CXCR4, xenograft model, PDX, Osteoprotegerin, OPG
Graphical abstract
Highlights
-
•
Primary human B-ALL cells use multiple independent transit mechanisms to invade the CNS
-
•
B-ALL migrate to skull or vertebral bone marrow in patient-derived xenograft (PDX) mice
-
•
B-ALL blasts provoke bone lesions through which they enter the subarachnoid space
-
•
RANKL and CXCR4 antagonists attenuate B-ALL entry into CNS in PDX mice
Rajakumar et al. show that inhibitors targeting immune receptors attenuate primary human B cell leukemia (B-ALL) cells transplanted into mice from invading the central nervous system (CNS). The findings in this study support clinical efforts to block these specific mechanisms of CNS invasion in B-ALL-affected individuals with targeted therapies.
Introduction
Acute lymphoblastic leukemia (ALL) involving the central nervous system (CNS) at initial diagnosis or relapse poses a major clinical problem. Conventional CNS-directed ALL therapy, including intrathecal chemotherapy and cranial irradiation, have greatly improved survival rates,1,2 but are associated with irreversible neurocognitive dysfunction, endocrine disorders, and an increased risk of secondary malignant brain tumors.3, 4, 5, 6 Thus, there is an unmet need to define the mechanisms of CNS invasion and develop targeted therapies of similar or better efficacy, but with lower toxicity to prevent and treat CNS leukemia, particularly in children with ALL.
B-ALL blasts can disseminate to the subarachnoid space of the brain and spinal cord (CNS), creating leptomeningeal metastasis, but the routes of invasion are not well established. Direct breach of the blood-brain barrier (BBB) by ALL blasts into the brain parenchyma is rarely observed.7, 8, 9 Leukemic blasts are thought to invade the subarachnoid space of the CNS through multiple routes.8 ALL blasts may transit into the cerebrospinal fluid (CSF) or subarachnoid space through the blood-CSF barrier (BCSFB) formed by epithelial cells of the choroid plexus, within the brain ventricles.8,10,11 Circulating ALL blasts may also access the subarachnoid space through tight junction-interconnected endothelial cells of the meningeal microvessels that form the blood-leptomeningeal (BLM) barrier.8,11 In one recent study, B-ALL blasts entered the CNS through vessels connecting vertebral or calvarial bone marrow with the subarachnoid space.12
Identification of the mechanisms that mediate leukemic blast dissemination to the CNS is needed to design effective targeted therapies. The C-X-C chemokine receptor 4 (CXCR4) and its cognate ligand stromal cell-derived factor 1 (SDF-1) are involved in the migration of ALL cells to the CNS.10,12,13 Human B-ALL blasts and cell lines can express messenger RNA (mRNA) for the chemokine receptor, CXCR4,10,13,14 a phenotype associated with extramedullary organ infiltration in pediatric ALL.10,15 SDF-1 is abundant in CNS blood vessels13 and BCSFB tissues.10 A CXCR4+ ALL cell line injected into mice migrated to the vasculature of the skull bone marrow, and a CXCR4-SDF-1 antagonist inhibited vascular homing of these cells.13 Engagement of the receptor activator of nuclear factor κB (RANK) with its ligand RANKL controls bone-specific metastatic behavior of breast cancer and melanoma cells.16 We recently reported that RANK-RANKL interaction critically regulates B-ALL-mediated long bone destruction;17 however, its role in CNS invasion is unknown. Given these observations, we examined CXCR4-SDF-1 and RANK-RANKL interactions as potential routes of CNS invasion by primary human B-ALL blasts.
Here, patient-derived xenograft (PDX) models of two high-risk primary human B-ALL subtypes were used to identify the mechanisms of leukemic blast migration into the CNS. We found that human leukemic blasts migrated from the femur to cranial and vertebral bone marrow, eroded the surrounding bone, and transited into the subarachnoid space or breached the BCSFB. We identify routes of B-ALL blast entry into the CNS and provide evidence that antagonists of RANKL and CXCR4 pathways may protect against leukemic blast invasion of the CNS. These results suggest that treatments targeting these interactions may reduce the risk of CNS relapse and improve outcomes for B-ALL-affected individuals with CNS disease.
Results
Human BCR-ABL1 B-ALL blasts invade cranial and vertebral bone in PDX mice
We reported that primary human B-ALL blasts cause RANK-RANKL-dependent trabecular bone loss in PDX models.17 Treatment of these PDX mice with osteoprotegerin (OPG), a soluble decoy receptor for RANKL,18,19 protected the bone against this B-ALL-mediated effect.17 Therefore, we asked whether primary human B-ALL blasts isolated at diagnosis from an individual with the high-risk BCR-ABL1 subtype can similarly invade cranial and vertebral bones and whether this novel route of entry was mediated by RANK-RANKL interaction. We implanted diagnostic B-ALL blasts (ID: 090233; BCR-ABL1; Table S1) by intrafemoral (i.f.) injection into the right femora of NOD.Prkdcscid/scidIl2rgtm1Wjl/SzJ (NSG) mice. The control group was treated with immunoglobulin G (IgG)1-Fc protein and the experimental group with the RANKL antagonist recombinant OPG-Fc (rOPG-Fc). Treatments were initiated at the time of leukemic blast injection, and the mice were examined 6 weeks later (Figure S1). This protocol was designed to determine whether rOPG-Fc could attenuate the CNS dissemination of leukemic blasts. The frequency of human leukemic blasts recovered from the brain and spinal cord was insufficient for reliable flow cytometric quantitation. However, flow cytometry analysis of human CD45+CD19+ cells revealed no difference in leukemic blast engraftment in the long bones or spleen between the IgG1-Fc control and rOPG-Fc-treatment groups, reflecting that RANKL-targeted therapy does not compromise leukemic blast survival (Figures 1A, 1B, and S2A).
Figure 1.

Effects of rOPG-Fc in early CNS leukemic blast invasion in a BCR-ABL1 PDX model
Human BCR-ABL1 leukemic blasts were injected orthotopically into the right femur of NSG mice and treated with IgG1-Fc control or rOPG-Fc. Mice were euthanized after 6 weeks of leukemic blast injection and treatment (n = 3 mice per group).
(A) Leukemic blast engraftment was assessed by flow cytometric analysis of cells from the injected right femur, non-injected right tibia, and spleen using antibodies specific for human CD45 and CD19.
(B) The percentage of CD45+CD19+ leukemic blasts gated on live singlets is shown.
(C and D) Representative sections of (C) vertebra or (D) calvaria of the IgG1-Fc- or rOPG-Fc-treated mice stained with TRAP or anti-human CD19 antibody. The dotted box represents a magnified view. The dotted outline and brown color on CD19-stained images indicate leukemic blasts.
(E and F) CD19 protein (y axis) was quantified in multiple regions of interest (ROIs; 25 μm) that include the entire bone marrow from the (E) vertebral and (F) calvarial bones of the IgG1-Fc- and rOPG-Fc-treated mice (x axis). The graph shows the mean staining intensity (2 μm per pixel) of CD19 protein (arbitrary units [au]). Each dot represents ROIs from 3 biological replicates and 4 technical replicates (n = 3 mice per group).
In (B), (E), and (F), data are means ± SDs. A 2-tailed unpaired t test with Welch’s correction was performed between the 2 groups. p value with a 95% confidence interval is indicated for each comparison. ∗∗∗∗p < 0.0001. BM, bone marrow; SA, subarachnoid space; SC, spinal cord. Scale bars, 100 μm. See also Figures S1, S2, and S3.
We next considered whether leukemic blasts may invade the skull and vertebral bone marrow before invasion of the subarachnoid space. We, therefore, performed histological analyses of the roof of the skull or calvaria and vertebrae of BCR-ABL1 B-ALL PDX mice 6 weeks after leukemic blast engraftment and treatment with either IgG1-Fc or rOPG-Fc (Figures S1A and S1B). In contrast to non-transplanted NSG mice (Figure S3), IgG1-Fc-treated PDX mice displayed multinucleated tartrate-resistant acid phosphatase (TRAP)+ osteoclasts and CD19+ B-ALL cells in the vertebral and calvarial bone marrow (Figures 1C and 1D). In contrast, rOPG-Fc-treated PDX mice had fewer CD19+ leukemic blasts and greater structural preservation of vertebral and calvarial bones (Figures 1C and 1D, ∗∗∗∗p < 0.0001; Figures 1E, 1F, and S1B). Thus, although we did not detect leukemic blasts in the subarachnoid space by flow cytometry, primary B-ALL blasts conferred destructive invasion of cranial and vertebral bone marrow. Overall, in this setting, the rOPG-Fc treatment appeared to restrain B-ALL blast-mediated invasion of cranial and vertebral bones.
We have previously reported that p53−/−; Rag2−/−; Prkdcscid/scid triple mutant (TM) mice display spontaneous B-ALL with infiltration of the subarachnoid space and clinical signs of CNS leukemia.20 TM leukemic blasts expressed cell-surface RANKL and conferred long bone and vertebral bone destruction when engrafted into immunodeficient recipient mice.17 Here, we asked whether these TM leukemic blasts generate bone channels that may provide access to the subarachnoid space. Compared to nonleukemic SCID mice, RANKL+ CD19+ TM leukemic blasts infiltrated the bone marrow in the calvaria, which is evident near the frontal olfactory lobe and at the caudal end near the cerebellum (Figure 2A). TM leukemic cells were evident inside bone channels that appeared contiguous with the subarachnoid space. Similar evidence of bone channels from the bone marrow into the subarachnoid space and the skeletal muscles were seen in the vertebrae of these mice (Figure 2B). Here, we demonstrate that TM mouse and human BCR-ABL1 leukemic blasts invade the calvarial and vertebral bone marrow, which may provide an access route to the subarachnoid space. Moreover, RANKL antagonism in the BCR-ABL1 PDX model protect the cranial and vertebral bones from leukemic blast invasion.
Figure 2.

Leukemic blast invasion into the CNS of TM mice
Various CNS anatomical locations were assessed in leukemic TM mice versus age- and sex-matched nonleukemic SCID mice (n = 3 mice per group). TM mice between 8 and 12 weeks of age showing symptoms of lymphadenopathy, domed head, or hind-limb paralysis were euthanized. The whole brain with an intact skull and vertebrae with a spinal cord were fixed in 10% phosphate-buffered formalin, decalcified in PBS + 14% EDTA, paraffin embedded, sectioned, and stained with TRAP, CD19, or RANKL.
(A) Representative sagittal sections of brain and skull showing calvarial bone marrow and subarachnoid space in the (1) frontal and (2) caudal regions.
(B) (1–3) Representative cross-sections of a vertebra showing the bone marrow, subarachnoid space, spinal cord, and skeletal muscles. Purple staining on TRAP images indicates multinucleated osteoclasts. Brown staining indicates CD19+ or RANKL+ cells. The box represents a magnified view. The dotted outline shows leukemic blasts. The red arrows indicate bone channels that connect the calvarial or vertebral bone marrow with the subarachnoid space or skeletal muscles. SM, skeletal muscle. Scale bars, 500, 100, or 50 μm.
Impact of RANKL antagonist on B-ALL invasion of the CNS
Next, we investigated the degree of CNS invasion by primary human B-ALL blasts at 10 weeks after transplantation, when PDX mice were visibly moribund (Figure S1). Primary B-ALL blasts from the same individual (BCR-ABL1; Table S1) were i.f. injected into NSG mice that were randomized to rOPG-Fc or IgG1-Fc treatment for 10 weeks. By this point, CD45+CD19+ leukemic blasts had infiltrated the subarachnoid space of the CNS (Figure S4). Consistent with the targeted action of rOPG-Fc, the frequency of human CD45+CD19+ leukemic blasts quantified by flow cytometry in the injected femur, tibia, and spleen did not differ between the rOPG-Fc and IgG1-Fc control-treated groups (Figures 3A, 3B, and S2B). We observed a trend in the reduction of the frequency of CD45+CD19+ leukemic blasts in the brain and spinal cord including the subarachnoid space (CNS) of the rOPG-Fc-treated mice compared to control mice that did not reach significance (Figure S4).
Figure 3.

Effects of rOPG-Fc in CNS leukemic blast invasion at a clinical endpoint in a BCR-ABL1 PDX model
Human BCR-ABL1 leukemic blasts were injected orthotopically into the right femur of NSG mice and treated with IgG1-Fc control or rOPG-Fc. Ten weeks after leukemic blast injection and treatment, mice with visible symptoms of leukemia were euthanized (n = 5 mice per group).
(A and B) Ex vivo cells from the injected right femur, non-injected right tibia, and spleen were assessed for leukemic blast engraftment using antibodies specific for human CD45 and CD19 by flow cytometry.
(B) The percentage of CD45+CD19+ leukemic blasts gated on live singlets is shown.
(C–F) Representative sections of the (C) vertebra or (E) calvaria of the IgG1-Fc- or rOPG-Fc-treated mice stained with TRAP or anti-human CD19 antibody. CD19 protein (y axis) was quantified in multiple ROIs (25 μm) that include the entire bone marrow from the (D) vertebral and (F) calvarial bones of the IgG1-Fc- and rOPG-Fc-treated mice (x axis). The graph shows the mean staining intensity (2 μm per pixel) of CD19 protein (au). Each dot represents ROIs from 3 biological replicates and 4 technical replicates (n = 3 mice per group).
In (B), (D), and (F), data are means ± SDs. A 2-tailed unpaired t test with Welch’s correction was performed between the 2 groups. p value with a 95% confidence interval is indicated for each comparison. ∗∗∗∗p < 0.0001. The dotted box represents a magnified view. The dotted outline and brown color on the CD19-stained images indicate leukemic blasts. The red arrows indicate bone channels that connect the bone marrow and subarachnoid space. The purple staining on TRAP images (purple arrows) indicates multinucleated osteoclasts. Scale bars, 100 μm. See also Figures S1, S2, and S4.
In this heavy disease burden setting, we asked whether rOPG-Fc treatment affected leukemic blast invasion of the vertebral and cranial bones. IgG1-Fc control-treated mice displayed massive human CD19+ leukemic blast infiltration into the vertebral bone marrow accompanied by prominent TRAP staining reflecting multinucleated osteoclast activity at the boundaries between leukemic blasts and residual bone (Figure 3C, top). Although the vertebrae of rOPG-Fc-treated mice also displayed leukemic blast invasion, they displayed greater bone retention compared to control-treated mice (Figures 3C [bottom], 3D [∗∗∗∗p < 0.0001], and S1B). The vertebral bones of IgG1-Fc-treated animals manifested bone erosions in the form of channels that were continuous with the subarachnoid space and packed with leukemic blasts (Figure 3C, top). These bone channels were not observed in rOPG-Fc-treated mice (Figure 3C, bottom). However, in both groups, leukemic blast invasion of the subarachnoid space was detected both by immunohistology (Figure 3C) and by flow cytometric analysis (Figure S4). The bone-sparing effects of rOPG-Fc were even more apparent in the skull, where the treatment protected the calvarium from leukemic cell infiltration (Figures 3E, 3F [∗∗∗∗p < 0.0001], and S1B). Thus, calvarial and vertebral bone invasion displayed RANKL dependence at this late disease stage. However, RANKL antagonism alone did not prevent leukemic blast invasion of the subarachnoid space, although a trend in reduction was observed (Figure S4). These results collectively suggested that later in disease progression, leukemic blasts used additional mechanisms to breach the blood-leptomeningeal and blood-cerebrospinal barriers.
CXCR4-mediated CNS leukemia dissemination
ALL blasts can express the chemokine receptor CXCR4, suggesting that the presence of cognate ligand SDF-1 in the CSF may promote CNS dissemination.10,12,21 We tested the contribution of this mechanism by treating B-ALL PDX mice with AMD3100 octahydrochloride,22,23 an inhibitor of CXCR4 binding to SDF-1. Primary B-ALL blasts (BCR-ABL1; Table S1) were i.f. injected, and the PDX recipients were treated with AMD3100 (Figure S1A). Eight weeks after leukemic blast injection and continuous treatment, the long bones, the cranium, and the brain were examined. As expected, there was heavy leukemic blast engraftment in the long bones (Figures S5A and S5B), and CXCR4 was readily detected on the cell surface of the CD45+CD19+ leukemic blasts (Figure S5B). TRAP and anti-human CD19 antibody staining of brain and skull sagittal sections of control-treated PDX mice identified leukemic blast infiltration in the subarachnoid space and calvarial bone marrow (Figure 4A). The cortical skull bone displayed channels packed with leukemic blasts consistent with the passage of leukemic blasts between calvarial bone marrow and the brain subarachnoid space (Figure 4A [1–3]). Leukemic blasts also invaded the growth plate of the occipital and sphenoid bones at the base of the skull (Figure S5C). In comparison to these control-treated PDX mice (Figure 4A [1–3]), AMD3100 treatment reduced leukemic blast invasion of the calvarial bone marrow and brain subarachnoid space (Figures 4B [1–3], 4D and 4E [∗∗∗∗p < 0.0001], and S1B). Of interest, we did not detect human CD19+ leukemic blasts in either the lateral or fourth brain ventricles in control-treated mice (Figure S5C) or the AMD3100-treated mice (Figure S5D), suggesting that these leukemic blasts did not breach the BCSFB. Similar to observations in the calvarial bone marrow and subarachnoid space of the brain, the vertebral bone marrow and subarachnoid space of the spinal cord of control-treated PDX mice showed massive leukemic blast invasion (Figure 4A) compared to control animals that did not receive leukemic grafts (Figure S3). In control-treated mice, vertebral cortical bone lesions were replete with B-ALL blasts, illustrating their transit between the bone marrow and subarachnoid space of the spinal cord (Figure 4A). As we observed in the calvaria, AMD3100 treatment reduced leukemia invasion of the vertebral bone marrow (Figures 4F [∗∗∗∗p < 0.0001] and S1B) and impeded their transit from the vertebral bone marrow into the spinal subarachnoid space through bone channels (Figures 4B, 4G [∗∗∗∗p < 0.0001], and S1B). Overall, CXCR4 inhibition abrogated B-ALL invasion of the bone marrow and the transit from the bone marrow into the subarachnoid space in the skull and vertebrae.
Figure 4.

Transit of BCR-ABL1 B-ALL blasts to the subarachnoid space through the skull or vertebral colonization in a PDX setting
Human BCR-ABL1 leukemic blasts were injected orthotopically into the right femur of NSG mice and treated with IgG1-Fc control or AMD3100 or rOPG-Fc at the time of leukemic blast injections (n = 3 mice per group). Mice were euthanized 8 weeks later. The whole brain with an intact skull and vertebral bones with an entire spinal cord were fixed, sectioned, and stained with TRAP or anti-human CD19 antibody.
(A–C), Representative sagittal sections of the brain and skull showing calvarial bone marrow and brain subarachnoid space in the (1) frontal and (2–3) caudal region, and vertebra showing the bone marrow, subarachnoid space, and spinal cord of IgG1-Fc- or AMD3100- or rOPG-Fc-treated mice. The dotted outline and brown color in the CD19-stained images indicates leukemic blasts. The dotted box and # represent a magnified view. The red arrows indicate bone channels that connect the bone marrow and subarachnoid space. The purple staining on TRAP images (purple arrows) indicates multinucleated osteoclasts.
(D–G) CD19 protein (y axis) was quantified in multiple ROIs (25 μm) from the calvarial bone marrow, brain subarachnoid space, vertebral bone marrow, and spinal cord subarachnoid space of the IgG1-Fc-, AMD3100-, and rOPG-Fc-treated mice (x axis). The graph shows the mean staining intensity (2 μm per pixel) of CD19 protein (au). Each dot represents ROIs from 3 biological replicates and 3 or 4 technical replicates (n = 3 mice per group). Data are means ± SDs. Data were analyzed using 1-way ANOVA (95% confidence interval) among the 3 groups; p values represent Bonferroni’s multiple comparison test, ∗∗∗∗p < 0.0001.
(H) A time-lapse illustration of CD19+ leukemic blasts in the calvarial and vertebral bone marrow and subarachnoid space of the treatment groups of BCR-ABL1 PDX mice.
Scale bars, 100 μm. See also Figures S1, S3, and S5.
RANKL-mediated B-ALL transit into the subarachnoid space
We asked whether RANKL antagonist rOPG-Fc could hinder the direct migration of leukemic blasts from the skull or vertebral bone marrow into the subarachnoid space. Human BCR-ABL1 B-ALL blasts (Table S1) were xenografted into cohorts of NSG mice, randomized for treatment with IgG1-Fc control or rOPG-Fc that was initiated concurrently with leukemic blast injection. The PDX cohorts were aged for 8 weeks to model the condition of substantial leukemia burden (Figure S1A). RANKL+ leukemic blast invasion in the calvarial bone marrow and the brain subarachnoid space was identified by TRAP, RANKL, and human anti-CD19 antibody staining of samples from IgG1-Fc-treated mice (Figures 4A and S5C). In contrast, rOPG-Fc treatment resulted in lower levels of leukemic blast invasion into the calvarial bones (Figure 4C), associated with a reduced invasion of the sphenoid and occipital bones at the base of the cranium (Figure S5E) and with a reduction in leukemic blast transit from the calvarial bone marrow into the brain subarachnoid space (Figures 4C [1–3], 4D [∗∗∗∗p < 0.0001], and 4E [∗∗∗∗p < 0.0001]). We observed similar rOPG-Fc effects in the vertebral bones where CD19+ RANKL+ leukemic blasts were reduced in the bone marrow (Figures 4C, 4F [∗∗∗∗p < 0.0001], and S5E) and absent from the spinal cord subarachnoid space at this disease stage compared to control-treated mice (Figures 4C and 4G [∗∗∗∗p < 0.0001]). These results demonstrated that RANKL antagonism attenuated leukemic blast transit from the calvarial and vertebral bones into the brain or spinal cord subarachnoid space. Therefore, in addition to the effects of CXCR4 inhibition, RANKL antagonism in this BCR-ABL1 PDX model blocked retention and expansion of B-ALL blasts in the calvarial and vertebral bone marrow at early (6-week), mid- (8-week), and late (10-week) disease stages, and prevented leukemic cell transit into the subarachnoid space at a mid- (8-week) disease stage. The progression of disease and CNS entry routes is illustrated in Figure 4H.
CXCR4- and RANKL-mediated cell invasion in MLL/KMT2A rearranged ALL
To determine whether the mechanisms of CNS invasion displayed by primary human BCR-ABL1 samples were observed in another high-risk leukemia subtype, we evaluated a primary sample of MLL/KMT2A-rearranged (MLLr) infant ALL, a leukemia that is associated with a high rate of CNS involvement and poor outcomes.24 The sample had been isolated from an infant that displayed CNS leukemia at diagnosis (ID: 9037; Table S1). The B-ALL blasts were injected into the right femurs of cohorts of NSG mice, and treatment was initiated with either IgG1-Fc control or CXCR4 antagonist AMD3100, and continued for either 4 or 6 weeks (Figure S1A). The long bones had a heavy CD45+CD19+ leukemic blast engraftment both 4 and 6 weeks after treatment (Figures S6A and S6B). The CD19+ CXCR4+ and RANKL+ B-ALL blasts (Figures 5, 6, S7A, and S7F) invaded the calvarial and vertebral bones and were associated with the development of bone channels that appeared to permit leukemic cell transit between the bone marrow and subarachnoid space (Figures 5A and 6A). In control-treated mice, these cells invaded the occipital and sphenoid bones at the skull base as well as the skeletal muscles that surround the vertebra (Figures 5A, 6A, S6C, and S7A), concordant with observations in TM mice (Figure 2B). AMD3100 treatment did not reduce leukemic blast infiltration of the calvarial and vertebral bone marrow (Figures 5B, 5E, 5F, 6B, 6E, and 6F) or the generation of bone lesions, which allowed leukemic cell transit from bone marrow to the subarachnoid space (Figures 5B, 5G, 5H, 6B, 6G, and 6H). We did not observe any effects on the burden of leukemic blast engraftment of the bones of the cranial base after 4 or 6 weeks of AMD3100 treatment (Figures S6D, S6G, S7B, and S7E) compared to the control group. In contrast to these tissue sites, 4 weeks of AMD3100 treatment had a potent effect on leukemic cell invasion of the skeletal muscles that surround the vertebra (Figures 5B and 5I [∗∗p < 0.01]), and we observed a similar trend in leukemic blast reduction after 6 weeks of treatment compared to control-treated mice (Figures 6B and 6I). As observed in the BCR-ABL1 PDX mice 4 weeks after leukemic blast injection (Figure S5C), the MLLr leukemic blasts did not appear to breach the BCSFB in the control-treated group (Figure S6C). Overall, CXCR4 blockade reduced the migration of BCR-ABL1 ALL blasts from the skull and vertebral bone marrow into the subarachnoid space (Figures 4B and 4D–4G), but this treatment did not produce the same effect on the MLLr B-ALL sample blasts, suggesting that the latter cells may use other routes of entry.
Figure 5.

B-ALL transit through the skull and vertebral bones in an MLL-rearranged B-ALL
Human MLLr NSG-passaged leukemic blasts were injected into the right femurs of NSG mice and treated with IgG1-Fc control or AMD3100 or rOPG-Fc or combined AMD3100 and rOPG-Fc (n = 3 mice per group). After 4 weeks of leukemic blast injection and treatment, mice were euthanized. The whole brain with an intact skull and vertebral bones with a spinal cord were fixed, sectioned, and stained with anti-human CD19 or CXCR4 antibodies.
(A–D) Representative sagittal sections of the brain and skull showing calvarial bone marrow and brain subarachnoid space in the (1) caudal region, and vertebra showing the bone marrow, subarachnoid space, spinal cord, and skeletal muscles of IgG1-Fc- or AMD3100- or rOPG-Fc- or combined AMD3100- and rOPG-Fc-treated mice. Brown staining indicates CD19 or CXCR4 abundance. The box represents a magnified view. The dotted outline indicates leukemic blasts. The red arrows indicate bone channels that connect the bone marrow and subarachnoid space.
(E–I) CD19 protein (y axis) was quantified in multiple ROIs (25 μm) from the (E) calvarial and (F) vertebral bone marrow, (G) and brain and (H) spinal cord subarachnoid space, and (I) skeletal muscles of the IgG1-Fc-, AMD3100-, rOPG-Fc-, or AMD3100 + rOPG-Fc-treated mice (x axis). The graph shows the mean staining intensity (2 μm per pixel) of CD19 protein (au). Each dot represents multiple ROIs from 3 biological replicates and 2 or 3 technical replicates (n = 3 mice per group).
Data are means ± SDs. All of the data were analyzed using non-parametric Kruskal-Wallis 1-way analysis of variance (ANOVA) (95% confidence interval) among the 3 groups; p values represent post hoc Dunn’s test, in which ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Scale bars, 100 μm. See also Figures S1, S6, and S7.
Figure 6.

B-ALL blast transit by breaching the blood-cerebrospinal fluid barrier in MLL-rearranged PDX mice
Human MLLr NSG-passaged leukemic blasts were injected into the right femurs of NSG mice and treated with IgG1-Fc control or AMD3100 or rOPG-Fc or combined AMD3100 and rOPG-Fc (n = 4 mice per group). After 6 weeks of leukemic blast injection and treatment, the mice were euthanized. The whole brain with an intact skull and vertebral bones spinal cord was fixed, sectioned, and stained with anti-human CD19 or CXCR4 antibodies.
(A–D) Representative sagittal sections of the brain and skull showing calvarial bone marrow and brain subarachnoid space in the (1) caudal region, lateral ventricle (LV) and fourth ventricles (FVs), and vertebra showing the bone marrow, subarachnoid space, spinal cord, and skeletal muscles of IgG1-Fc- or AMD3100- or rOPG-Fc- or combined AMD3100- and rOPG-Fc-treated mice. Brown staining indicates CD19 or CXCR4 abundance. The box represents a magnified view. The dotted outline indicates leukemic blasts. The red arrows indicate bone channels that connect the bone marrow and subarachnoid space.
(E–J) CD19 protein (y axis) was quantified in multiple ROIs (25 μm) from the (E) calvarial and (F) vertebral bone marrow, (G) brain and (H) spinal cord subarachnoid space, (I) skeletal muscles, and (J) lateral and fourth brain ventricles of the IgG1-Fc-treated, AMD3100-treated, rOPG-Fc-treated, or AMD3100 + rOPG-Fc-treated mice (x axis). The graph shows the mean staining intensity (2 μm per pixel) of CD19 protein (au). Each dot represents multiple ROIs from 4 biological replicates and 2 or 3 technical replicates (n = 4 mice per group).
Data are means ± SDs. All of the data were analyzed using non-parametric Kruskal-Wallis 1-way ANOVA (95% confidence interval) among the 3 groups; p values represent post hoc Dunn’s test, in which ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Scale bars, 100 μm. See also Figures S1, S6, and S7.
At both 4 and 6 weeks after leukemic blast injection and coincident treatment, rOPG-Fc either as a single agent or given in combination with AMD3100 reduced CD19+ CXCR4+ RANKL+ MLLr leukemic blast invasion of the calvarial and vertebral bones (Figures 5C, 5D, 5E and 5F [∗∗∗p < 0.001 and ∗∗p < 0.01], 6C, 6D, and 6E and 6F [∗∗∗∗p < 0.0001]), brain and spinal cord subarachnoid space (Figures 5C, 5D, 5G and 5H [∗∗∗p < 0.001], 6C, 6D, and 6G [∗∗p < 0.01] and 6H [∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001]), sphenoid and occipital bones (Figures S6E, S6F, and S6G [∗∗p < 0.01], S7C, S7D, and S7E [∗p < 0.05 and ∗∗p < 0.01]), and skeletal muscles (Figures 5C, 5D, 5I [∗p < 0.05 and ∗∗∗∗p < 0.0001], 6C, 6D, and 6I [∗∗∗∗p < 0.0001]). These data demonstrated that these MLLr B-ALL blasts transit through the skull or vertebral bone lesions into the subarachnoid space and skeletal muscles was RANKL mediated at all disease stages modeled in these PDX studies.
B-ALL blast transit to the CNS through the BCSFB
The MLLr infant B-ALL example studied here was taken from an individual who displayed CNS involvement at diagnosis. Therefore, we hypothesized that these leukemic cells may also breach the BCSFB. Similar to our observations in the BCR-ABL1 PDX mice (Figure S5C), control-treated MLLr PDX mice at 4 weeks after leukemia cell injection did not display evidence of leukemic blasts in the ventricles (Figure S6C). We identified CD19+ CXCR4+ leukemic blasts in the lateral and fourth ventricles of MLLr PDX mice at 6 weeks after leukemic blast injections (Figures 6A and S7A), coincident with gross signs of CNS leukemia in these animals, including domed head (Figure 6A). AMD3100 and rOPG-Fc either as single agents or in combination attenuated B-ALL blast transit through the BCSFB (Figures 6B–6D and 6J [∗∗p < 0.01]). In contrast to control-treated mice, rOPG-Fc or the rOPG-Fc and AMD3100 combination-treated animals did not exhibit CNS symptoms (e.g., domed head [Figure 6A]) and also showed reduced leukemic blast invasion of the calvarial and vertebral bone marrow and the leptomeninges of the CNS. Overall, these data demonstrate that B-ALL blast transit through the BCSFB at the advanced disease stage modeled here is mediated independently by both RANKL and CXCR4 mechanisms.
Discussion
Prophylaxis and treatment of CNS involvement in children with ALL poses a major clinical challenge. Cranial irradiation, either as treatment or prophylaxis, has reduced the risk of CNS leukemia relapse, but it is associated with irreversible neurocognitive late effects and is used only in rare circumstances.25, 26, 27 Prophylactic CNS-directed chemotherapy therapy is the standard of care; however, relapse still occurs in 3%–8% of pediatric ALL cases, either confined to the CNS or combined with bone marrow disease.2,28,29 In addition, CNS-directed chemotherapy in children is associated with neurocognitive dysfunction. Therefore, there is an unmet need to develop efficacious therapeutic approaches to prevent and treat CNS leukemia that has limited neurotoxicity. A central rationale for the present study is that these approaches could include targeting mechanisms of leukemic blast entry into the CNS.
We have identified mechanisms by which B-ALL blasts enter the CNS using both spontaneous mouse models of B-ALL and primary human B-ALL sample xenograft (PDX) models. We previously reported that TM mice spontaneously develop B-ALL with neurological impairment and leukemic blast infiltration into the CNS subarachnoid space.20 We later showed that these RANKL+ TM leukemic blasts cause bone destruction in Rag2−/− recipient mice.17 RANKL is a critical osteoclast differentiation factor.30, 31, 32 RANK-RANKL interaction between tumor cells and the bone microenvironment drives a cycle of bone destruction and tumor growth.33 RANK-RANKL interaction is also involved in breast and prostate cancer cell invasion of the bone.16,34,35 In B-ALL-affected pediatric and adult subjects, periosteal reactions and osteolytic lesions in the skull have been reported.36, 37, 38 A recent study39 showed invasion of the skull bone marrow by ALL blasts in newly diagnosed pediatric subjects before the initiation of therapy. Here, we report that RANKL+ TM leukemic blasts invade the skull and vertebral bone marrow cavities, create cortical bone lesions, and transit to the subarachnoid space of the brain and spinal cord and the skeletal muscles.
Given these observations in the TM mouse model, we interrogated mechanisms of human B-ALL blast dissemination from the femur to the CNS by xenotransplantation into NSG mice. NSG mice is an immunodeficient strain that lacks mature T, B, and functional natural killer (NK) cells, and expresses a variant of the inhibitory checkpoint protein SIRP alpha capable of binding to the human cognate receptor CD47, collectively resulting in a permissive environment for the engraftment of primary human leukemic cells.40, 41, 42 Here, we investigated the mechanisms of leukemic blast transit into the CNS using examples of both the MLLr and BCR-ABL1 B-ALL subtypes, as they are associated with CNS involvement and poor outcomes. The calvarium houses hematopoietically active marrow43 and most B cells found in the CNS of normal mice are derived from the calvaria and reach the meninges through specialized vasculature.44 We reasoned that the calvarium may provide a reservoir for leukemic blast dissemination into the CNS. Here, we report that for the patient sample examined, BCR-ABL1 B-ALL blasts injected into the femur of mouse recipients invaded calvarial and vertebral bone marrow well before B-ALL blasts were evident in the subarachnoid space by flow cytometry. Compared to control-treated PDX mice, rOPG-Fc treatment protected the calvarial and vertebral bones from leukemic blast invasion. In BCR-ABL1 B-ALL PDX mice analyzed at a later time point, administration of rOPG-Fc continued to protect the skull and vertebral bones from leukemic blast invasion. However, by this point, leukemic blasts had also infiltrated the subarachnoid space of the rOPG-Fc-treated mice, although leukemic invasion was reduced compared to the control-treated group. These data suggested that in the setting of heavy disease burden, leukemic blasts can use alternate entry routes of CNS entry, including the blood-leptomeningeal barrier and the BCSFB.
Previous studies have identified associations between CNS leukemia and leukemic cell expression of molecules that may aid CNS entry. These observations include interleukin-15 (IL-15) expression on leukemic blasts observed together with activated NK cells,45 vascular endothelial growth factor (VEGF) expression on leukemic blasts,46 IL-7 receptor expression on B-ALL blasts,47 and association of B-ALL CD79a expression with CNS infiltration and relapse.48 Moreover, chemokine pathways may also contribute to ALL migration into the CNS.21,49 B-ALL cells can express CXCR4, and the cognate ligand SDF-1 is present at high concentrations in the BCSFB tissues.10,12 We hypothesized that CXCR4-expressing B-ALL blasts would migrate toward the SDF-1-rich CSF-containing subarachnoid space. Using the CXCR4 small-molecule inhibitor AMD3100,22,23 we asked whether blocking B-ALL blast interaction with SDF-1 could alter their dissemination into the CSF-rich subarachnoid space, and if so, by what routes. AMD3100-treated BCR-ABL1 PDX mice displayed a reduced leukemic cell invasion into the skull and vertebral bone marrow, and they were not observed to further penetrate the subarachnoid space. In contrast, CD19+CXCR4+ MLLr B-ALL blasts created skull and vertebral cortical bone channels and invaded the subarachnoid space of these PDX mice. Our results demonstrate that CXCR4 antagonism restrained blasts from a BCR-ABL1 but not from an MLLr B-ALL leukemic patient sample from advancing through skull and vertebral bone marrow into the subarachnoid space. The peripheral leukemia burden became extensive in later time points examined in the MLLr PDX mice, enabling the leukemic blasts to access other transit routes such as the blood-leptomeningeal barrier to enter the leptomeninges. Since we studied one example of each leukemia subtype, we cannot determine whether their observed differential CXCR4 dependence is characteristic of each subtype or instead reflects the differential aggressiveness of these two tumor exemplars in the PDX setting.
We asked whether B-ALL blasts can transit directly from the skull or vertebral bone marrow into the subarachnoid space, as we previously observed in the TM mouse model. Previous work showed that the NALM-6 B-ALL cell line spreads within the bone microenvironment by migrating into the perivascular domains of the calvarial bone marrow.13 B-ALL cells can use bridging laminin-rich vessels to migrate from the calvarial or vertebral bone marrow toward the CSF in the subarachnoid space.12 We reasoned that human primary B-ALL blasts could resorb the skull and vertebral cortical bone, creating RANK-RANKL-mediated bone channels, and thereby transit directly into the subarachnoid space. To test this idea, we treated PDX mice engrafted with either BCR-ABL1 or MLLr primary ALL blasts with rOPG-Fc and examined leukemic blast transit routes in recipient mice at multiple times after leukemic cell i.f. injection. In control-treated mice, B-ALL blasts created bone channels in the skull and vertebral cortical bones to access the subarachnoid space. In contrast, rOPG-Fc treatment protected the skull and vertebral bone marrow from leukemic blast entry into the subarachnoid space. At times chosen to model a mid-disease stage, neither BCR-ABL1 nor MLLr B-ALL blasts in control-treated mice breached the BCSFB, further emphasizing the importance of the skull and vertebral bone-mediated transit route. Our results demonstrate that B-ALL blast penetrates the subarachnoid space by establishing RANKL-mediated skull and vertebral bone channels.
Our PDX study design enabled analyses of leukemia blast CNS transit routes over time. Even at a later disease stage in MLLr PDX mice, rOPG-Fc treatment either alone or together with a CXCR4 antagonist, protected the calvarial, vertebral, sphenoid, and occipital bones and the skeletal muscle from blast infiltration. While rOPG-Fc treatment alone could not entirely block leukemic blast entry, it significantly reduced leukemic blast invasion into the subarachnoid space. These observations suggest that these MLLr leukemic blasts use additional transit routes, likely through the blood-leptomeningeal or BCSFBs. Cytokines and chemokines can regulate the migration of both normal leukocytes and leukemia cells across the BCSFB.50, 51, 52 A recent study10 posited that CNS invasion is a generic property of B-ALL blasts, suggesting that they transited through the BCSFB in PDX mice even when the patient from whom the cells were isolated was not diagnosed with CNS disease based upon CSF findings. However, this study did not identify a functional association between a specific chemokine receptor and propensity for CNS invasion.10 Here, we demonstrate that MLLr B-ALL blasts can breach the BCSFB releasing CD19+ CXCR4+ cells into the brain ventricles. In this PDX model of late-stage disease, therapeutic RANKL and CXCR4 antagonism independently restrained leukemic blast transit through the BCSFB route. Thus, our results indicate that RANKL and CXCR4 antagonism can inhibit the migration of primary B-ALL blasts through the BCSFB into the subarachnoid space.
Limitations of the study
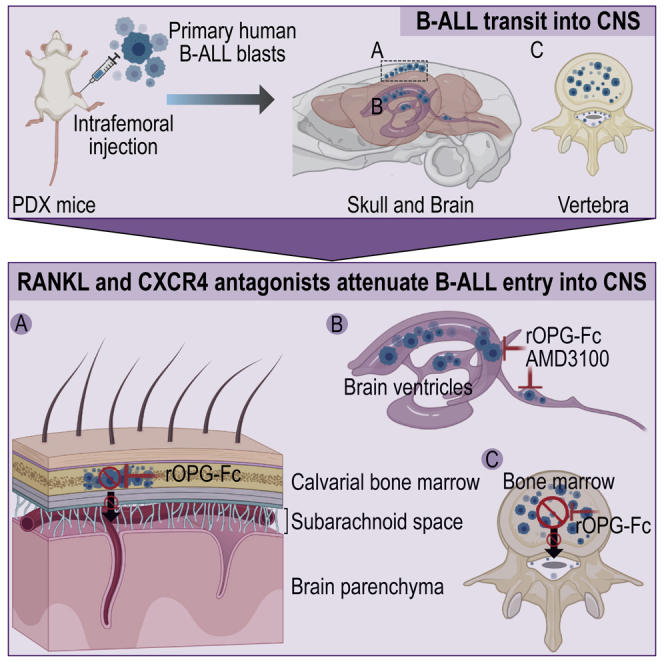
BCR-ABL1 and MLLr B-ALL affected individuals have a high rate of CNS involvement and poor outcomes. Our in-depth study of examples of these two high-risk tumor subtypes identifies multiple independent transit mechanisms of leukemic blast into the CNS through treatment of PDX mice with RANK-RANKL- and CXCR4-SDF1-targeted therapies (Figure 7). Primary B-ALL blasts migrated to the skull and vertebra bone marrow cavities, where they provoked bone lesions enabling passage into the subarachnoid space by a RANKL-mediated mechanism and that targeted rOPG therapy effectively opposed. Our results do not address whether these patterns of CNS invasion are generalized to BCR-ABL1 and/or MLLr leukemia subtypes. Evaluation of this question will require a sufficient sample size for each B-ALL subtype, reliance on higher throughput approaches than those used in this study, and will benefit from a longitudinal study design to examine CNS invasion at diagnosis, in response to therapy and at relapse.
Figure 7.

Schematic representation of B-ALL blast transit routes to the CNS
Sagittal sections of the mouse brain showing RANKL- and CXCR4-mediated B-ALL transit routes.
(A) In the PDX model, we show that B-ALL blasts fail to breach the blood-brain barrier, as we did not observe leukemic blasts in the brain parenchyma. However, at a late disease stage, BCR-ABL1 and MLLr B-ALL blasts may have reached the subarachnoid space by breaching the blood-leptomeningeal barrier.
(B) We identified a calvarial or vertebral bone marrow-mediated leukemic blast transit into the subarachnoid space in PDX mice. RANKL antagonist rOPG-Fc treatment protected the skull and vertebral bone marrow from B-ALL blast invasion and subsequent skull or vertebral bone marrow-mediated leukemic blast transit into the subarachnoid space.
(C) BCR-ABL1 B-ALL blasts did not breach the blood-cerebrospinal fluid barrier in PDX mice. In contrast, CD19+ CXCR4+MLLr B-ALL blasts breached the blood-cerebrospinal fluid barrier found in the LV, third ventricle, and FV of the brain at a late-disease stage, and CXCR4 and rOPG-Fc antagonism using AMD3100 and rOPG-Fc prevented B-ALL blast transit through the blood-cerebrospinal fluid barrier.
In PDX models, we show that human BCR-ABL1 and MLLr B-ALL blasts in the femur migrate and invade the skull and vertebral bone marrow at an early disease stage and enter the subarachnoid space by a RANK-RANKL-mediated pathway. We identify a second CXCR4-SDF1-dependent pathway through which a BCR-ABL1 leukemia migrated from cranial and vertebral bone marrow into the subarachnoid space. In addition, an MLLr leukemia breached the BCSFB through both RANKL- and CXCR4-mediated pathways. These findings provide a rationale to evaluate clinically available RANKL and CXCR4 pathway inhibitors to reduce CNS involvement of ALL in combination with current multi-agent chemotherapy. Appropriately designed studies are needed to investigate whether these transit routes into the CNS are specific and/or generalizable to B-ALL genetic subtypes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 (UCHT1) | BioLegend | Cat# 300430; RRID:AB_893301 |
| CD45 (2D1) | BD Biosciences | Cat# 347463; RRID:AB_400306 |
| CD19 (4G7) | BD Biosciences | Cat# 349209; RRID:AB_400407 |
| RANKL (12A668) | Abcam | Cat# ab45039; RRID:AB_2205935 |
| CD19 (EPR5906) | Abcam | Cat# ab134114; RRID:AB_2801636 |
| CD19 (EP169) | BioGenex | Cat# AN729-5M |
| CD19 (EPR23174-145) | Abcam | Cat# ab245235; RRID:AB_2895109 |
| CXCR4 (UMB2) | Abcam | Cat# ab124824; RRID:AB_10975635 |
| Biotinylated secondary antibody, Mouse and Rabbit SS Link-label IHC detection system | BioGenex | Cat# QP900-LE (HK340-9KT-multi link) |
| Biotinylated secondary antibody, Mouse and Rabbit HRP/DAB (ABC) detection kit | Abcam | Cat# ab64264 |
| CXCR4 (12G5) | BD Biosciences | Cat# 560669; RRID:AB_1727435 |
| Biotinylated rOPG-Fc (in-house); Biotinylated in-house using EZ-link sulpho-NHS biotin kit | rOPG-Fc: Amgen and NHS-Biotin kit: Thermoscientific | Cat# 21326 (NHS Biotin Kit) |
| Streptavidin-PerCP-Cy5.5 | e-Bioscience (Thermo-Fisher Scientific) | Cat# 45-4317-82; RRID:AB_10311495 |
| Biological samples | ||
| Patient-Derived B-ALL Samples | Table S1 | Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human OPG-Fc protein (RANKL antagonist) | Amgen | Amgen |
| IgG1-Fc protein (produced in-house using pFUSE-Fc plasmid) | Invivogen | Catalog# pfuse-hg1fc2; plasmid construct |
| AMD3100 octahydrochloride (CXCR4 antagonist) | Sigma-Aldrich | A5602-5MG |
| Experimental models: organisms/strains | ||
| Mouse: p53−/−; Rag2−/−; Prkdcscid/scid triple-mutant (TM) | The center for Phenogenomics (TCP) (Toronto, Canada) housed and bred the TM mice. | N/A |
| Mouse: p53+/−; Rag2−/−; Prkdcscid/scid nonleukemic control | The center for Phenogenomics (TCP) (Toronto, Canada) housed and bred the nonleukemic SCID control mice. | N/A |
| Mouse: NOD.Prkdcscid/scidIl2rg tm1Wjl/SzJ (NSG) | The University Health Network (UHN), Max bell-animal resource center (Toronto, Canada), housed and bred the NSG mice in pathogen-free (SPF) conditions. | N/A |
| Software and algorithms | ||
| GraphPad Prism | https://www.graphpad.com/ | Version 8.4.1, for Mac OS X |
| FlowJo v.10 (Tree Star, Ashland, OR) | https://www.flowjo.com/ | Version 10 (Tree Star, Ashland, OR) |
| NDP.view2 | https://www.hamamatsu.com/us/en/product/type/U12388-01/index.html | Version-U12388-01 |
| QuPath (Quantitative Pathology and Bioimage Analysis) | https://qupath.github.io | Version 0.2.3 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Jayne S. Danska (jayne.danska@sickkids.ca)
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Study design
PDX experiments were performed using two B-ALL patient samples taken during diagnosis (ID 090233, 9037) (Table S1). Due to the limited availability, B-ALL blasts from the primary MLLr patient sample (ID 9037) were expanded in NOD.Prkdcscid/scidIl2rg tm1Wjl/SzJ (NSG) mice and the NSG-passaged MLLr B-ALL blasts were used in experiments where mice were treated with RANKL and CXCR4 antagonists. Samples were selected for in vivo experiments based on previous experiments and engraftment testing, where > 80% of human leukemic blasts engraft in the NSG BM and the CNS within 8 to 10 weeks after leukemia blast injection. For PDX experiments, mice were randomly assigned to the IgG1-Fc control or rOPG-Fc or AMD3100 treatment groups.
Mice
The center for Phenogenomics (TCP) (Toronto, Canada) housed the p53−/−; Rag2−/−; Prkdcscid/scid triple mutant (TM), and p53+/−; Rag2−/−; Prkdcscid/scid nonleukemic SCID control mice. The TCP animal care committee approved all experimental procedures. TM and nonleukemic SCID control mice were generated and genotyped as previously described20 and maintained on acidified water. Mice were monitored daily for labored breathing and peripheral lymphadenopathy indicative of systemic leukemia as well as CNS signs, including domed head, ataxic gait, and hind limb paralysis or paresis. TM and nonleukemic SCID male or female mice (8 to 12 weeks old) were used in the experiments. The University Health Network (UHN), Max bell-animal resource center (Toronto, Canada), housed the NSG mice in pathogen-free (SPF) conditions. NSG female mice (8-weeks old) were used in the PDX experiments. The UHN animal care committee approved all experimental procedures with the NSG mice.
Human B-ALL samples
The Research Ethics Boards at the Hospital for Sick Children (Toronto, Canada) and UHN (Toronto, Canada) approved this study. Bone marrow or peripheral blood samples from newly diagnosed patients with ALL were obtained with informed consent. Mononuclear cells were isolated using Ficoll-Paque density gradient separation (GE Healthcare) according to the manufacturer’s instructions. Isolated mononuclear cells were frozen in 90% (v/v) fetal calf serum (FCS) with 10% (v/v) dimethylsulphoxide (DMSO), stored in liquid nitrogen, and used for PDX studies. The age, gender, cytogenetics, and risk-group of the B-ALL patients are indicated in Table S1.
Method details
B-ALL PDX
Primary patient B-ALL samples were depleted of CD3+ cells by immunomagnetic bead separation using CD3 microbeads (Miltenyi Biotech). Cells were processed through an AutoMACS Pro Separator (Miltenyi Biotech), as per the manufacturer’s instruction. CD3-labeled cells were separated using the “depletes” program on AutoMACS Pro Separator. The purity of the depletion was assessed by flow cytometry using an antibody specific for human CD3-PerCP-Cy5.5 (1:30 dilution, clone: UCHT1, BioLegend), which is always greater than 99%. NSG female mice (8-weeks old) were sub- lethally irradiated (200 cGy) using a 137Cs γ-irradiator, 24 h before injection of human leukemic blasts as previously described.17,53 Human leukemic blasts were injected orthotopically into the BM niche of the right femur as previously described.17,53 Briefly, mice were anesthetized by isoflurane inhalation, and the area of the injection site just below the patella or femur kneecap was cleaned using a sterile alcohol wipe. A 27.5-gauge needle was used to drill through the knee, and CD3-depleted B-ALL blasts (5 × 105 cells in 30 μL volume) was directly injected into the right femur bone using a 28.5-gauge insulin syringe.
Treatment with IgG1-Fc control protein (in-house) or recombinant human OPG-Fc fusion protein (Amgen) or AMD3100 octahydrochloride,23 a CXCR4 antagonist (Sigma-Aldrich), was initiated at the time of leukemic blast injections for up to ten weeks. rOPG-Fc was administered three times per week (3 mg/kg) by subcutaneous injections. The dose, frequency, and route of administering rOPG-Fc injections were based on the pharmacodynamics and pharmacokinetics analyses in healthy young rats.54 AMD3100 octahydrochloride was administered to PDX mice five times per week (5 mg/kg) up to eight weeks by subcutaneous injections based on established studies.55,56 Mice were monitored daily for visible signs of morbidity, including weight loss, ruffled coat, difficulty breathing, abnormal posture, and inactivity. Mice were sacrificed at periods between six to ten weeks after leukemic blast injections and treatment.
Lymphoblast extraction
PDX mice were euthanized by CO2 inhalation. Staining media (SM) was prepared by adding 10mM HEPES (pH 7.2) and 2% calf serum (Wisent Inc.) to Hank’s balanced salt solution (GIBCO). Bone marrow cells were isolated from human leukemic blast injected right femur and non-injected right tibia by flushing bones with staining media using a 27.5-gauge needle. Once flushed, cells were further disrupted using a 25.5-gauge needle and filtered through a 70 μm nitex nylon mesh and centrifuged for 5 min at 400 x g, 4°C. Red blood cells were lysed and removed by re-suspending the BM cell pellets in 2 × 107 cells per ml of 1X Gey’s lysis solution (in-house). Cells were incubated on ice for 3 min, washed in SM, and pelleted again by centrifugation for 5 min at 400 x g, 4°C. Cells were re-suspended in SM, and the number of viable cells was determined by trypan blue (Sigma Aldrich, Oakville, Canada) exclusion method.
To isolate lymphoblasts from spleen and CNS, PDX mice were sacrificed by CO2 inhalation. The spleen, brain, and spinal cord were dissected from mice. The skull bones were opened, and an incision was made at the caudal end of the spine. The brain along with the leptomeningeal layer was dissected out of the skull, and an 18.5-gauge needle filled with 1X PBS was used for flushing the spinal cord out of the vertebral column. The isolated entire brain and spinal cord along with the leptomeninges were placed in a 50 mL falcon tube with SM. Single-cell suspensions were made by dissociating the organs in SM with a 3cc syringe plunger, and the tissues in SM were filtered through a 70 μm mesh cell strainer (BD Biosciences, San Diego, USA). Cell suspensions from the spleen were centrifuged (400 x g, 5 min at 4°C), and red blood cells were further lysed using 1X Gey’s lysis solution. Cells were washed and re-suspended in SM for flow cytometric analysis. Lymphoblast from the brain and spinal cord (CNS) tissues were isolated using layered percoll gradient (70% followed by 37% and 30% percoll (GE healthcare)). After the addition of percoll gradient, tubes were centrifuged at 400 x g, 20 min at room temperature. Lymphoblast cells were collected from the 37% and 70% interface, washed with SM and re-suspended in RPMI 1640 with 10% calf serum.
Flow cytometry
Human BCR-ABL1 B-ALL injected NSG mice were treated with either IgG1-Fc control (in-house protein) or rOPG-Fc (Amgen) at the time of leukemic blast injections and were sacrificed at six weeks and ten weeks after leukemic blast injection and treatment. Single cell suspensions (1 × 106 cells) isolated from the right femur, non-injected right tibia, spleen, and CNS of PDX mice were aliquoted into staining tubes. SM was added and centrifuged at 300 x g for 5 min at 4°C. Cells were stained with human-specific cell-surface antibodies for CD45-FITC (1:30 dilution, clone: 2D1, BD Biosciences), CD19-PE (1:30 dilution, clone: 4G7, BD Biosciences), along with Human TruStain Fc-receptor blocking solution [BioLegend] and incubated for 30 min at RT. Cells were washed with SM and centrifuged at 300 x g for 5 min at 4°C. Amine-reactive fixable Zombie UV dye [BioLegend] in PBS was added and incubated for 30 min at RT. Cells were washed in SM and centrifuged at 300 x g for 5 min at 4°C. Cells were immediately fixed with BD Cytofix-Cytoperm buffer [BD PharMingen], which uses a single step cell fixation and permeabilization. Cells were incubated on ice for 30 min, washed in 1X BD Perm-Wash buffer and centrifuged at 800 x g for 5 min at 4°C. Cells were re-suspended in SM and filtered through a 70 μm nitex mesh. The stained cells were acquired using an LSRFortessa cell analyzer (BD Biosciences).
Moreover, single-cell suspensions isolated from the bone marrow of BCR-ABL1 PDX mice that were treated with IgG1-Fc control were stained with human-specific cell-surface antibodies for CXCR4-PECy7 (1:150 dilution, clone: 12G5, BD Biosciences), CD45-FITC (1:30 dilution, clone: 2D1, BD Biosciences), CD19-PE (1:30 dilution, clone: 4G7, BD Biosciences). In addition, isolated lymphoblasts from the CNS of MLLr PDX mice after treatment with IgG1-Fc control were stained with antibodies specific for cell-surface CD45-FITC (1:30 dilution, clone: 2D1, BD Biosciences), CD19-PE (1:30 dilution, clone: 4G7, BD Biosciences), CXCR4-PECy7 (1:150 dilution, clone: 12G5, BD Biosciences), and biotinylated rOPG-Fc+Streptavidin-PerCP-Cy5.5 (to detect RANKL) as previously described.17 The stained cells were acquired using an LSRFortessa cell analyzer (BD Biosciences) as described above.
Bone histology
TM mice between 8-12 weeks of age, showing symptoms of lymphadenopathy, domed head, or hind-limb paralysis, and nonleukemic SCID control mice were euthanized. For PDX experiments, primary BCR-ABL1 B-ALL or NSG-passaged MLLr B-ALL blasts-injected NSG mice were treated with IgG1-Fc control (in-house protein) or rOPG-Fc (Amgen) or AMD3100 octahydrochloride (Sigma) or a combination of rOPG-Fc and AMD3100 at the time of leukemic blast injections. Mice were sacrificed at eight weeks or four and six weeks after leukemic blast injection and treatment for BCR-ABL1 and MLLr PDX mice, respectively. The whole brain with an intact skull and skull only or vertebral bones with an intact spinal cord were dissected and were fixed in 10% neutral buffered formalin (Sigma-Aldrich) for 120 h and 48 h respectively. Tissues were rinsed in PBS for 15 min (3 times) and decalcified in 14% (w/v) EDTA dissolved in PBS (pH 7.5). A fresh solution was added each day until decalcification was achieved (10 days). Tissues were then processed and embedded in paraffin and 4-μm-thick sections were cut and used for histological and immunohistochemistry analyses. Tartrate-resistant acid phosphatase (TRAP) stain was done by standard techniques at the TCP pathology core facility (Toronto, ON).
Immunohistochemistry (IHC)
For CD19 and CXCR4 staining, slides of paraffin-embedded bone as described above in the bone histology section were dewaxed and rehydrated using the EZ-Retriever microwave and the EZ-AR common solution (Biogenex, USA). Antigen retrieval was performed in the EZ-Retriever microwave using EZ-AR2 solution, according to the manufacturer’s instructions. Slides were cooled for 20 min at RT, washed in distilled water for 5 min and transferred to wash buffer (1X DPBS, Dulbecco’s Phosphate-Buffered Saline) (GIBCO, Life technologies). For RANKL staining, slides of paraffin-embedded bone as described above in the bone histology section were dewaxed, rehydrated, and digested with a ready-to-use Trypsin kit (Carezyme I, Bio-care Medical). Slides with trypsin were incubated at 37°C (10 min) for antigen retrieval.
Dewaxed and rehydrated paraffin-embedded bone sections from BCR-ABL1 PDX mice were stained for CD19 as described below. Slides were incubated with peroxide block solution (Biogenex, USA) followed by incubations in serum-free protein block (Dako, USA), avidin block and biotin block (Biogenex, USA) at RT. Slides were incubated in pre-diluted ready-to-use anti-human CD19 (clone: EP169) rabbit monoclonal antibody (Biogenex, USA) overnight at 4°C according to the manufacturer’s instruction. Next, a highly specific Super Sensitive (SS) Link-Label IHC Detection System (Biogenex, USA) was used to detect bound primary antibodies (QP900-LE). After washing, slides were incubated with pre- diluted ready-to-use biotinylated anti-rabbit secondary antibody optimized for Biogenex primary antibodies (SS anti-mouse and rabbit, HK340-9KT-multi-link, Biogenex, USA) for 30 min at RT. As an experimental control to detect non-specific secondary antibody-binding, tissue sections were incubated with pre-diluted ready-to-use biotinylated anti-rabbit secondary antibody without the addition of the primary antibody for 30 min at RT. All slides were washed in DPBS three times for 10 min each and incubated with HRP (horse-radish peroxidase) labeled streptavidin for 30 min at RT (HK330-9KT-label, Biogenex, USA). Slides were washed and DAB (3,3′- diaminobenzidine) chromogen in substrate buffer (HK124, Biogenex, USA) was added to the tissue sections and incubated for 10 min at RT. Slides were rinsed in deionized water and counterstained in Mayer’s hematoxylin (1.0 g/l, MHS32, Sigma-Aldrich) for one minute, then rinsed in tap water and left to air-dry overnight. Slides were dipped in Histo-Clear (National diagnostics, Fisher Scientific) and were coverslipped using Eukitt quick hardening mounting media (Sigma-Aldrich). In addition, bone sections from BCR-ABL1 PDX mice were stained with anti-mouse or human RANKL (clone: 12A668; 1:25 dilution; Abcam, ab45039) primary antibody and processed as previously described for human CD19 staining.17
Dewaxed and rehydrated paraffin-embedded bone sections (as described above in the bone histology section) from TM and nonleukemic SCID mice were stained with anti-mouse CD19 (clone: EPR23174-145; 1:750 dilution; Abcam, ab245235) or anti-mouse or human RANKL (clone: 12A668; 1:25 dilution; Abcam, ab45039) primary antibodies and processed as previously described for human CD19 staining.17 Bone sections from MLLr PDX mice were stained with anti-human CD19 (clone: EPR5906; 1:250 dilution; Abcam, ab134114) or anti-mouse or human RANKL (clone: 12A668; 1:25 dilution; Abcam, ab45039) primary antibodies and processed as previously described for human CD19 staining.17 Bone sections from MLLr PDX mice were stained with an anti-human CXCR4 (clone: UMB2) primary antibody (1:250 dilution; Abcam, ab124824), using the same method as previously described for human CD19 staining.17 The anti-human CXCR4 antibody recognizes the non-phosphorylated C terminus of CXCR4. Therefore, lambda protein phosphatase treatment (2000U; New England Biolabs, P0753S) was performed on each section for 1 h at RT before primary antibody incubation to dephosphorylate the samples, as per the manufacturer’s instructions.
Imaging
TRAP, CD19, RANKL, and CXCR4-stained slides were imaged using Hamamatsu NanoZoomer 2.0-R digital slide scanner (40X magnification) at the TCP pathology core facility. Slides were viewed using NDP.view2 (version-U12388-01).
Image analysis
CD19-stained calvaria, brain with calvaria and vertebral bone with spinal cord FFPE sections were imaged as described above. Using QuPath (v.0.2.3),57 an open source bioimage software, we evaluated the invasion of CD19+ leukemic blasts in the various CNS anatomical regions of PDX mice (Figure S1B). Multiple regions of interest (ROI; 25 μm) were drawn around the entire calvarial bone marrow, subarachnoid space, ventricles, skeletal muscles, and vertebral bone sections including all CD19+ stained areas (Figure S1B). In QuPath, we used the intensity feature mode with a resolution of 2 μm pixel size to calculate the mean DAB staining intensity for each ROI which indicates CD19+ leukemic blasts in each normalized area of interest. To reduce bias, the field of view used to calculate the mean staining intensity was a larger area that included all CD19+ stained regions including any adjacent bone space. For example, in the vertebral sections, we chose the entire bone area to calculate the DAB staining intensity (Figure S1B). Thus, the mean staining intensity value variation based on the leukemic blasts load considers the bone space. Each assessment was also normalized to the area.
Quantification and statistical analysis
All data are reported as means ± SD, as detailed in the figure legends. The two-group analysis was performed using a two-tailed unpaired Student’s t test with or without Welch’s correction for heteroscedasticity. One-way analysis of variance with Bonferroni’s or Dunn’s correction was used to evaluate differences among multiple comparisons. Statistical differences with two-tailed probability values of p < 0.05 were considered significant. All data were analyzed using GraphPad Prism software, version 8.4.1, for Mac OS X.
Acknowledgments
We thank William Dougall at Amgen for providing the rOPG-Fc fusion protein. We thank M. Ganguly and the team at the Centre for Phenogenomics for performing tissue sectioning and TRAP staining. We thank S. Mortin-Toth for advice with immunohistochemistry (IHC) staining procedures. This work was supported by grants to J.S.D. from the Leukemia and Lymphoma Society of Canada (524579) and to J.S.D. and C.J.G. from the Ontario Institute of Cancer Research (AL-TRI-FR-HSK). J.S.D. and C.J.G. receive support from the SickKids Foundation. J.S.D. is supported by the Anne and Max Tanenbaum Chair in Molecular Medicine, University of Toronto, Canada.
Author contributions
S.A.R., C.J.G., and J.S.D. conceived and designed the study and S.A.R. and J.S.D. oversaw the study and wrote the manuscript. S.A.R. performed all of the experiments and analyzed the data. I.G. assisted with the tissue processing and maintenance of TM mice. M.D.M. and J.K.H. provided samples from affected individuals. All of the authors commented on and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure that the study questionnaires were prepared in an inclusive way. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: December 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100470.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. There are no codes generated in this paper. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
References
- 1.Hunger S.P., Lu X., Devidas M., Camitta B.M., Gaynon P.S., Winick N.J., Reaman G.H., Carroll W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J. Clin. Oncol. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui C.H., Howard S.C. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol. 2008;9:257–268. doi: 10.1016/S1470-2045(08)70070-6. [DOI] [PubMed] [Google Scholar]
- 3.Demopoulos A., DeAngelis L.M. Neurologic complications of leukemia. Curr. Opin. Neurol. 2002;15:691–699. doi: 10.1097/01.wco.0000044765.39452.2d. [DOI] [PubMed] [Google Scholar]
- 4.Pui C.H., Cheng C., Leung W., Rai S.N., Rivera G.K., Sandlund J.T., Ribeiro R.C., Relling M.V., Kun L.E., Evans W.E., Hudson M.M. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N. Engl. J. Med. 2003;349:640–649. doi: 10.1056/NEJMoa035091. [DOI] [PubMed] [Google Scholar]
- 5.Hijiya N., Hudson M.M., Lensing S., Zacher M., Onciu M., Behm F.G., Razzouk B.I., Ribeiro R.C., Rubnitz J.E., Sandlund J.T., et al. Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA. 2007;297:1207–1215. doi: 10.1001/jama.297.11.1207. [DOI] [PubMed] [Google Scholar]
- 6.Sanders K.E., Ha C.S., Cortes-Franco J.E., Koller C.A., Kantarjian H.M., Cox J.D. The role of craniospinal irradiation in adults with a central nervous system recurrence of leukemia. Cancer. 2004;100:2176–2180. doi: 10.1002/cncr.20280. [DOI] [PubMed] [Google Scholar]
- 7.Ginsberg L.E., Leeds N.E. Neuroradiology of leukemia. AJR Am. J. Roentgenol. 1995;165:525–534. doi: 10.2214/ajr.165.3.7645463. [DOI] [PubMed] [Google Scholar]
- 8.Frishman-Levy L., Izraeli S. Advances in understanding the pathogenesis of CNS acute lymphoblastic leukaemia and potential for therapy. Br. J. Haematol. 2017;176:157–167. doi: 10.1111/bjh.14411. [DOI] [PubMed] [Google Scholar]
- 9.Price R.A. Histopathology of CNS leukemia and complications of therapy. Am. J. Pediatr. Hematol. Oncol. 1979;1:21–30. [PubMed] [Google Scholar]
- 10.Williams M.T., Yousafzai Y.M., Elder A., Rehe K., Bomken S., Frishman-Levy L., Tavor S., Sinclair P., Dormon K., Masic D., et al. The ability to cross the blood-cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukemia blasts. Blood. 2016;127:1998–2006. doi: 10.1182/blood-2015-08-665034. [DOI] [PubMed] [Google Scholar]
- 11.Shechter R., London A., Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat. Rev. Immunol. 2013;13:206–218. doi: 10.1038/nri3391. [DOI] [PubMed] [Google Scholar]
- 12.Yao H., Price T.T., Cantelli G., Ngo B., Warner M.J., Olivere L., Ridge S.M., Jablonski E.M., Therrien J., Tannheimer S., et al. Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature. 2018;560:55–60. doi: 10.1038/s41586-018-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sipkins D.A., Wei X., Wu J.W., Runnels J.M., Côté D., Means T.K., Luster A.D., Scadden D.T., Lin C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu S., Gessner R., Taube T., Korte A., von Stackelberg A., Kirchner R., Henze G., Seeger K. Chemokine IL-8 and chemokine receptor CXCR3 and CXCR4 gene expression in childhood acute lymphoblastic leukemia at first relapse. J. Pediatr. Hematol. Oncol. 2006;28:216–220. doi: 10.1097/01.mph.0000212908.14642.a5. [DOI] [PubMed] [Google Scholar]
- 15.Crazzolara R., Kreczy A., Mann G., Heitger A., Eibl G., Fink F.M., Möhle R., Meister B. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2001;115:545–553. doi: 10.1046/j.1365-2141.2001.03164.x. [DOI] [PubMed] [Google Scholar]
- 16.Jones D.H., Nakashima T., Sanchez O.H., Kozieradzki I., Komarova S.V., Sarosi I., Morony S., Rubin E., Sarao R., Hojilla C.V., et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–696. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- 17.Rajakumar S.A., Papp E., Lee K.K., Grandal I., Merico D., Liu C.C., Allo B., Zhang L., Grynpas M.D., Minden M.D., et al. B cell acute lymphoblastic leukemia cells mediate RANK-RANKL-dependent bone destruction. Sci. Transl. Med. 2020;12:eaba5942. doi: 10.1126/scitranslmed.aba5942. [DOI] [PubMed] [Google Scholar]
- 18.Teitelbaum S.L. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 19.Simonet W.S., Lacey D.L., Dunstan C.R., Kelley M., Chang M.S., Lüthy R., Nguyen H.Q., Wooden S., Bennett L., Boone T., et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 20.Gladdy R.A., Taylor M.D., Williams C.J., Grandal I., Karaskova J., Squire J.A., Rutka J.T., Guidos C.J., Danska J.S. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell. 2003;3:37–50. doi: 10.1016/s1535-6108(02)00236-2. [DOI] [PubMed] [Google Scholar]
- 21.Gómez A.M., Martínez C., González M., Luque A., Melen G.J., Martínez J., Hortelano S., Lassaletta Á., Madero L., Ramírez M. Chemokines and relapses in childhood acute lymphoblastic leukemia: a role in migration and in resistance to antileukemic drugs. Blood Cells Mol. Dis. 2015;55:220–227. doi: 10.1016/j.bcmd.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 22.De Clercq E. The bicyclam AMD3100 story. Nat. Rev. Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 23.Hatse S., Princen K., Bridger G., De Clercq E., Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–262. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- 24.Pieters R., De Lorenzo P., Ancliffe P., Aversa L.A., Brethon B., Biondi A., Campbell M., Escherich G., Ferster A., Gardner R.A., et al. Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated With the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J. Clin. Oncol. 2019;37:2246–2256. doi: 10.1200/JCO.19.00261. [DOI] [PubMed] [Google Scholar]
- 25.Vagace J.M., de la Maya M.D., Caceres-Marzal C., Gonzalez de Murillo S., Gervasini G. Central nervous system chemotoxicity during treatment of pediatric acute lymphoblastic leukemia/lymphoma. Crit. Rev. Oncol. Hematol. 2012;84:274–286. doi: 10.1016/j.critrevonc.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Genschaft M., Huebner T., Plessow F., Ikonomidou V.N., Abolmaali N., Krone F., Hoffmann A., Holfeld E., Vorwerk P., Kramm C., et al. Impact of chemotherapy for childhood leukemia on brain morphology and function. PLoS ONE. 2013;8:e78599. doi: 10.1371/journal.pone.0078599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacola L.M., Krull K.R., Pui C.H., Pei D., Cheng C., Reddick W.E., Conklin H.M. Longitudinal Assessment of Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated on a Contemporary Chemotherapy Protocol. J. Clin. Oncol. 2016;34:1239–1247. doi: 10.1200/JCO.2015.64.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagedorn N., Acquaviva C., Fronkova E., von Stackelberg A., Barth A., zur Stadt U., Schrauder A., Trka J., Gaspar N., Seeger K., et al. Resistant Disease Committee of the International BFM Study Group Submicroscopic bone marrow involvement in isolated extramedullary relapses in childhood acute lymphoblastic leukemia: a more precise definition of “isolated” and its possible clinical implications, a collaborative study of the Resistant Disease Committee of the International BFM Study Group. Blood. 2007;110:4022–4029. doi: 10.1182/blood-2007-04-082040. [DOI] [PubMed] [Google Scholar]
- 29.Pui C.H. Central nervous system disease in acute lymphoblastic leukemia: prophylaxis and treatment. Hematology Am. Soc. Hematol. Educ. Program. 2006:142–146. doi: 10.1182/asheducation-2006.1.142. [DOI] [PubMed] [Google Scholar]
- 30.Kong Y.Y., Yoshida H., Sarosi I., Tan H.L., Timms E., Capparelli C., Morony S., Oliveira-dos-Santos A.J., Van G., Itie A., et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 31.Lacey D.L., Timms E., Tan H.L., Kelley M.J., Dunstan C.R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 32.Anderson D.M., Maraskovsky E., Billingsley W.L., Dougall W.C., Tometsko M.E., Roux E.R., Teepe M.C., DuBose R.F., Cosman D., Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 33.Dougall W.C. Molecular pathways: osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin. Cancer Res. 2012;18:326–335. doi: 10.1158/1078-0432.CCR-10-2507. [DOI] [PubMed] [Google Scholar]
- 34.Santini D., Perrone G., Roato I., Godio L., Pantano F., Grasso D., Russo A., Vincenzi B., Fratto M.E., Sabbatini R., et al. Expression pattern of receptor activator of NFκB (RANK) in a series of primary solid tumors and related bone metastases. J. Cell. Physiol. 2011;226:780–784. doi: 10.1002/jcp.22402. [DOI] [PubMed] [Google Scholar]
- 35.Sisay M., Mengistu G., Edessa D. The RANK/RANKL/OPG system in tumorigenesis and metastasis of cancer stem cell: potential targets for anticancer therapy. OncoTargets Ther. 2017;10:3801–3810. doi: 10.2147/OTT.S135867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogalsky R.J., Black G.B., Reed M.H. Orthopaedic manifestations of leukemia in children. J. Bone Joint Surg. Am. 1986;68:494–501. [PubMed] [Google Scholar]
- 37.Müller H.L., Horwitz A.E., Kühl J. Acute lymphoblastic leukemia with severe skeletal involvement: a subset of childhood leukemia with a good prognosis. Pediatr. Hematol. Oncol. 1998;15:121–133. doi: 10.3109/08880019809167227. [DOI] [PubMed] [Google Scholar]
- 38.Shahnazi M., Khatami A., Shamsian B., Haerizadeh B., Mehrafarin M. Bony lesions in pediatric acute leukemia: pictorial essay. Iran. J. Radiol. 2012;9:50–56. doi: 10.5812/iranjradiol.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao W., Liang C., Gen Y., Wang C., Zhao C., Sun L. Role of diffusion-weighted imaging for detecting bone marrow infiltration in skull in children with acute lymphoblastic leukemia. Diagn. Interv. Radiol. 2016;22:580–586. doi: 10.5152/dir.2016.15167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarry J.E., Murphy K., Perry R., Sanchez P.V., Secreto A., Keefer C., Swider C.R., Strzelecki A.C., Cavelier C., Récher C., et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J. Clin. Invest. 2011;121:384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takenaka K., Prasolava T.K., Wang J.C., Mortin-Toth S.M., Khalouei S., Gan O.I., Dick J.E., Danska J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- 42.Theocharides A.P., Jin L., Cheng P.Y., Prasolava T.K., Malko A.V., Ho J.M., Poeppl A.G., van Rooijen N., Minden M.D., Danska J.S., et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 2012;209:1883–1899. doi: 10.1084/jem.20120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butcher E.C. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 44.Brioschi S., Wang W.L., Peng V., Wang M., Shchukina I., Greenberg Z.J., Bando J.K., Jaeger N., Czepielewski R.S., Swain A., et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science. 2021;373:eabf9277. doi: 10.1126/science.abf9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frishman-Levy L., Shemesh A., Bar-Sinai A., Ma C., Ni Z., Frenkel S., Muench V., Bruckmueller H., Vokuhl C., Debatin K.M., et al. Central nervous system acute lymphoblastic leukemia: role of natural killer cells. Blood. 2015;125:3420–3431. doi: 10.1182/blood-2014-08-595108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Münch V., Trentin L., Herzig J., Demir S., Seyfried F., Kraus J.M., Kestler H.A., Köhler R., Barth T.F.E., Te Kronnie G., et al. Central nervous system involvement in acute lymphoblastic leukemia is mediated by vascular endothelial growth factor. Blood. 2017;130:643–654. doi: 10.1182/blood-2017-03-769315. [DOI] [PubMed] [Google Scholar]
- 47.Alsadeq A., Lenk L., Vadakumchery A., Cousins A., Vokuhl C., Khadour A., Vogiatzi F., Seyfried F., Meyer L.H., Cario G., et al. IL7R is associated with CNS infiltration and relapse in pediatric B-cell precursor acute lymphoblastic leukemia. Blood. 2018;132:1614–1617. doi: 10.1182/blood-2018-04-844209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lenk L., Carlet M., Vogiatzi F., Spory L., Winterberg D., Cousins A., Vossen-Gajcy M., Ibruli O., Vokuhl C., Cario G., et al. CD79a promotes CNS-infiltration and leukemia engraftment in pediatric B-cell precursor acute lymphoblastic leukemia. Commun. Biol. 2021;4:73. doi: 10.1038/s42003-020-01591-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buonamici S., Trimarchi T., Ruocco M.G., Reavie L., Cathelin S., Mar B.G., Klinakis A., Lukyanov Y., Tseng J.C., Sen F., et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature. 2009;459:1000–1004. doi: 10.1038/nature08020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams M.T., Yousafzai Y., Cox C., Blair A., Carmody R., Sai S., Chapman K.E., McAndrew R., Thomas A., Spence A., et al. Interleukin-15 enhances cellular proliferation and upregulates CNS homing molecules in pre-B acute lymphoblastic leukemia. Blood. 2014;123:3116–3127. doi: 10.1182/blood-2013-05-499970. [DOI] [PubMed] [Google Scholar]
- 51.Sørensen T.L., Tani M., Jensen J., Pierce V., Lucchinetti C., Folcik V.A., Qin S., Rottman J., Sellebjerg F., Strieter R.M., et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Invest. 1999;103:807–815. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hühmer A.F., Biringer R.G., Amato H., Fonteh A.N., Harrington M.G. Protein analysis in human cerebrospinal fluid: physiological aspects, current progress and future challenges. Dis. Markers. 2006;22:3–26. doi: 10.1155/2006/158797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazurier F., Doedens M., Gan O.I., Dick J.E. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat. Med. 2003;9:959–963. doi: 10.1038/nm886. [DOI] [PubMed] [Google Scholar]
- 54.Capparelli C., Morony S., Warmington K., Adamu S., Lacey D., Dunstan C.R., Stouch B., Martin S., Kostenuik P.J. Sustained antiresorptive effects after a single treatment with human recombinant osteoprotegerin (OPG): a pharmacodynamic and pharmacokinetic analysis in rats. J. Bone Miner. Res. 2003;18:852–858. doi: 10.1359/jbmr.2003.18.5.852. [DOI] [PubMed] [Google Scholar]
- 55.Azab A.K., Runnels J.M., Pitsillides C., Moreau A.S., Azab F., Leleu X., Jia X., Wright R., Ospina B., Carlson A.L., et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang Y., Chen B.J., Deoliveira D., Mito J., Chao N.J. Selective enhancement of donor hematopoietic cell engraftment by the CXCR4 antagonist AMD3100 in a mouse transplantation model. PLoS ONE. 2010;5:e11316. doi: 10.1371/journal.pone.0011316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. There are no codes generated in this paper. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.