

Abstract
Existing studies have revealed that microRNAs (miRNAs) have a role in cognitive deficits in Alzheimer’s disease (AD). However, the function and pathophysiological mechanism of deregulated miRNAs underlying AD pathology remain to be investigated. The present study aimed to clarify the role and mechanism of miR-148a-3p in AD. RNA sequencing, qRT-PCR, and western blot analysis were used to identify the aberrant expression and signaling of miR-148a-3p within cells, mice, and patients with AD. Molecular biology techniques involving luciferase reporter assays, gene overexpression and silencing, chromatin immunoprecipitation, and adeno-associated virus-based miRNA overexpression were used to explore the biological function and mechanisms of miR-148a-3p. Downregulation of miR-148a-3p was identified in AD. Upregulation of miR-148a-3p was found to protect neuronal cells against Aβ-associated tau hyperphosphorylation by directly targeting p35/CDK5 and PTEN/p38 mitogen-activated protein kinase (MAPK) pathways. A mutual regulatory link between miR-148a-3p and PTEN using a feedforward arrangement was confirmed via promotion of transcription and expression of miR-148a-3p by way of the PTEN/Akt/CREB pathway. Significantly, in vivo targeting of miR-148a-3p signaling ameliorated cognitive deficits by decreasing p35/PTEN-elicited tau hyperphosphorylation, accompanied by feedforward transduction of the PTEN/Akt/CREB pathway. In conclusion, the present study implicated the miR-148a-3p/p35/PTEN pathway as an essential contributor to tau hyperphosphorylation and feedforward regulation in AD.
Keywords: Alzheimer’s disease, cAMP-response element-binding protein, cognition, microRNA, p35, phosphatase and tensin homolog deleted on chromosome 10, tau protein
Graphical abstract
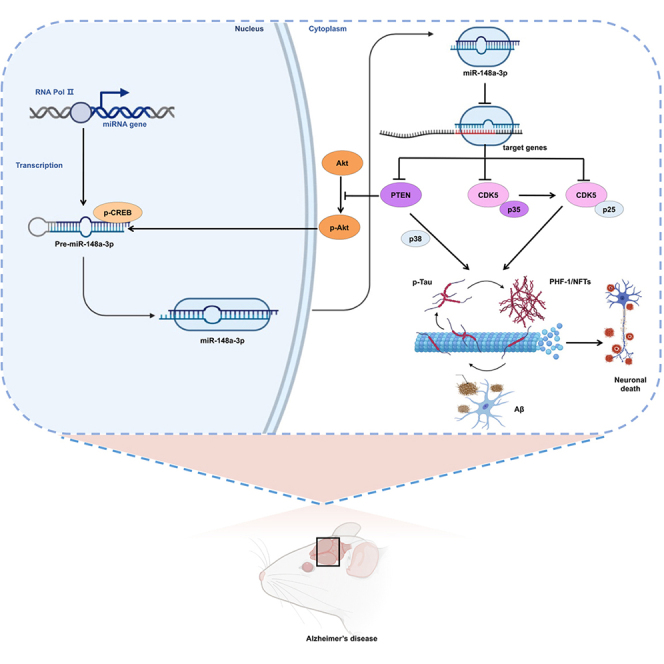
The present findings identified a novel function for miR-148a-3p in the pathogenesis of AD and revealed the underlying mechanisms involved in miR-148a-3p/p35/PTEN signaling, allowing the rescue of AD-like symptoms. The miR-148a-3p mimic-based therapy and unique mechanism promise to be therapeutic targets and prognostic biomarkers for the treatment of AD.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, manifested histopathologically as extracellular senile plaques formed by amyloid-β peptides (Aβ) and intracellular neurofibrillary tangles (NFTs) consisting of aberrantly hyperphosphorylated tau proteins.1 As the exact etiology of their formation and their pathogenesis remain primarily unelucidated, therapies effective in preventing the development of AD, or halting its progression, have yet to be found. NFTs are considered a key event in AD, and evidence suggests that they are closely associated with cognitive deficit in such patients.2 Intracellular hyperphosphorylated tau proteins are critical components of NFTs, resulting in structural abnormalities in neuronal microtubules and dysfunction in axonal transport.3 More importantly, insoluble aggregations of NFTs inside neurons and glial cells cause neurotoxic transformation, inducing dendritic aberrance, synaptic loss, and ultimately a decline in recollection.2,4 Therefore, inhibition of tau hyperphosphorylation through early modification therapy of upstream target signaling may be most effective in preventing AD.
Emerging evidence indicates that microRNAs (miRNAs) are implicated in multiple pathological processes in AD.5, 6, 7, 8 As key factors in the regulation of gene expression, miRNAs operate by inhibition of protein expression through post-transcriptional binding to the 3′ untranslated regions (UTRs) of target messenger RNAs (mRNAs).9,10 It is hypothesized that abnormal regulation of miRNA-targeted networks plays a key role in AD pathological processes via regulation of genes involving Aβ overproduction,11, 12, 13 tau hyperphosphorylation,14,15 glial-activation-induced neuroinflammation,16,17 synaptic dysfunction,18,19 and neuronal apoptosis.20,21 Despite reported involvement of novel miRNAs in AD pathogenesis, the identity of sensitive mRNAs involved in the diagnosis, treatment, and pathophysiological implications of AD remains unclear.
In the present study, we report novel functions and molecular mechanisms of miR-148a-3p, a member of a highly conservative miR-148a cluster, in the decline in cognition in AD pathology, also characterized as a potential noninvasive biomarker in the diagnosis and treatment of AD. Here, we demonstrated that downregulation of miR-148a-3p contributed to tau hyperphosphorylation, and thus the progression of AD, having substantial diagnostic and therapeutic significance in a clinical setting. We then revealed that miR-148a-3p targeted cyclin-dependent kinase 5 regulatory subunit 1 (CDK5R1; p35) and phosphatase and tensin homolog deleted on chromosome 10 (PTEN), which were involved in tau hyperphosphorylation. In addition, the PTEN/protein kinase B/cAMP-response element-binding protein (Akt/CREB) signaling pathway exhibited feedforward autoregulation of miR-148a-3p. Notably, the experimental strategy targeting miR-148a-3p signaling rescued cognitive-behavioral abnormalities, neurodegeneration, and tau hyperphosphorylation in AD mice. The current findings implicate the miR-148a-3p/p35/PTEN pathway as a critical contributor to tau hyperphosphorylation and feedforward autoregulation in AD.
Results
miR-148a-3p is downregulated during AD progression
High-throughput sequencing was used to identify deregulated miRNAs in amyloid precursor protein (APP)/presenilin-1 (PS1) mice of different ages compared with age-matched wild-type (WT) controls. A literature search was conducted for 12 miRNAs that were significantly downregulated as the APP/PS1 mice aged to narrow candidate molecules (Figure 1A, Table S1). Nine miRNAs were excluded owing to the abnormal expression that occurs in a variety of tumors and that is less likely to act in AD. Of the remaining miRNAs, miR-148a-3p exhibited continuous and significant downregulation from an early (3 months) to an established (9 months) stage of progressive AD, accompanied by a high area under the curve value (AUC; 0.799; specificity, 16.7%; sensitivity, 95.8%), which discriminated APP/PS1 from WT control mice (Figure 1B, p < 0.05), suggesting that it represents a novel miRNA associated with the AD pathological process.
Figure 1.

miR-148a-3p is downregulated during AD progression
(A) miR-148a-3p expression in the cortex of APP/PS1 mice using RNA-sequencing analysis at different stages of AD (n = 3). Two-way hierarchical clustering of miRNAs in sequencing experiments was performed to differentiate miRNAs with differential expression levels at different stages of AD. The color scale indicates the relative expression of an miRNA in a particular age group: red represents high relative expression, and green represents low relative expression. (B) Receiver operating characteristic (ROC) curves discriminate APP/PS1 mice from WT control mice in terms of miR-148a-3p levels from miRNA profiling. (C and D) Decreased expression of miR-148a-3p in the hippocampus (C) and cortex (D) of APP/PS1 mice and SAMP8 mice (n = 4). (E) Decreased expression of miR-148a-3p in APPswe cell lines from 24 to 48 h (n = 5). (F) Reduced levels of miR-148a-3p in the serum of AD patients compared with healthy age-matched volunteers (HAVs) (n = 15–21). (G) Correlation analysis of serum miR-148a-3p levels and MMSE score using Pearson correlation. (H) ROC curves discriminate AD patients from HAVs through serum miR-148a-3p levels (n = 15–21). Results represent means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding control.
To further verify this association as AD progressed, the brain tissues of APP/PS1 and senescence-accelerated mouse prone 8 (SAMP8) mice at different ages, presenting AD-symptom-like cognitive decline, Aβ accumulation, and tau hyperphosphorylation, as frequently reported,22,23 were analyzed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). The results demonstrated that, consistent with miRNA profiling, miR-148a-3p expression decreased in the hippocampus and cortex of APP/PS1 and SAMP8 mice at 6 and 9 months of age (Figures 1C and 1D, p < 0.05–0.01). miR-148a-3p expression decreased in a time-dependent fashion (Figure 1E, all p < 0.001) in an in vitro model that used copper to trigger the toxicity of Aβ in SH-SY5Y cells transfected with the Swedish mutant form of human APP (referred to as “APPswe cells”), combined with decreased cell viability in response to copper (Figure S1, all p < 0.001).
The clinical significance of miR-148a-3p expression was evaluated in AD patients compared with healthy age-matched volunteers (HAVs) using qRT-PCR. The results demonstrated that miR-148a-3p expression levels in serum were lower in AD patients than in HAVs (Figure 1F, p < 0.05). A strong positive correlation between Mini-Mental State Examination (MMSE) score and serum miR-148a-3p level in AD patients was identified (Figure 1G, R2 = 0.20, p < 0.05), indicating that miR-148a-3p represented a blood biomarker with good diagnostic value (Figure 1H, AUC, 0.777; specificity, 90%; sensitivity, 45.5%; p < 0.05). Collectively, these results suggest that miR-148a-3p is downregulated during AD progression and may be considered a prognostic biomarker for the diagnosis of AD.
miR-148a-3p has neuroprotective effects and attenuates tau hyperphosphorylation in AD cells
To explore the functional role of miR-148a-3p in AD, miR-148a-3p mimics, anti-miR-148a-3p, or scrambled controls were transfected into APPswe cells, which were then treated with copper. miR-148a-3p mimics increased the viability of APPswe cells, while anti-miR-148a-3p decreased viability following copper exposure compared with the corresponding scrambled control (Figure 2A, both p < 0.001). Flow cytometry revealed that the total degree of apoptosis, representing both early and late apoptosis, in APPswe cells transfected with miR-148a-3p mimics was reduced, but exacerbated when transfected with anti-miR-148a-3p (Figures 2B and 2C, p < 0.01–0.001). These results indicate that upregulation of miR-148a-3p exerted a neuroprotective effect in AD.
Figure 2.

miR-148a-3p provides neuroprotection to APPswe cells and inhibits tau hyperphosphorylation
(A) Cell viability of copper-injured APPswe cells transfected with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled control (Scr/Anti-scr) as measured by CCK-8 assay. (B) Representative images of copper-injured APPswe cells transfected with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled control (Scr/Anti-scr) assessed by flow cytometry. (C) Quantification of the percentage of cell apoptosis. (D and E) Representative western blot images (D) and quantification (E) of tau phosphorylation at AT8, Ser199, Ser396, and Ser404 sites in copper-injured APPswe cells transfected with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled control (Scr/Anti-scr). Results represent means ± SEM, n = 6. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus Scr/Anti-scr. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Additional western blot analysis indicated that miR-148a-3p mimics inhibited tau phosphorylation at a number of epitopes, including AT8, Ser199, Ser396, and Ser404 (Figures 2D and 2E, p < 0.05–0.001), and, conversely, tau phosphorylation was exacerbated at these epitopes when transfection was conducted with anti-miR-148a-3p (p < 0.05–0.001). In addition, miR-148a-3p mimics and anti-miR-148a-3p did not influence APP expression (Figure S2). Thus, these findings suggest that inhibition of tau hyperphosphorylation contributes to miR-148a-3p-mediated neuroprotection in AD.
miR-148a-3p attenuates tau hyperphosphorylation by targeting p35
To establish how miR-148a-3p exerts neuroprotection in AD, bioinformatics analysis was conducted using online databases and correlation analysis of RNA sequencing. Among the predicted miR-148a-3p targets in three databases (Figure 3A, Table S2), the majority of predicted mRNAs were excluded because they are reported to be primarily expressed in tumors and other unrelated diseases. Of predicted targets reported to be associated with AD, including CDK5R1 (p35), PTEN, low-density lipoprotein receptor (LDLR), and quaking (QKI), p35 and PTEN mRNAs were found to display a strong negative correlation with miR-148a-3p expression in APP/PS1 mice as the disease progressed (Figures 3B and 3C, both p < 0.05). The subsequent dual-luciferase reporter assay indicated that the predicted LDLR and QKI genes were not specific targets of miR-148a-3p (Figure S3).
Figure 3.

miR-148a-3p attenuates tau hyperphosphorylation by the direct targeting of p35
(A) Venn diagram of predicted targets of miR-148a-3p as defined using three databases: TargetScan, miRDB, and Tarbase. (B and C) Correlation analysis of miR-148a-3p with p35 (CDK5R1) (B) and PTEN (C) mRNA in APP/PS1 mouse cortex at different stages of AD. (D) Potential binding site for miR-148a-3p in the 3′ UTR of p35 mRNA as suggested by bioinformatics analysis. (E) Construction of wild-type (Wt) and mutant (Mut) 3′ UTR of p35 within a pGL3 vector. (F) Relative luciferase activity after co-transfection with a plasmid constructed with a Wt or Mut p35 3′ UTR with scrambled control (Scr) or miR-148a-3p mimic (148a) in HEK293 cells. (G) Representative western blot images of p35 expression in APPswe cells after transfection with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled controls (Scr/Anti-scr). (H) Quantification of p35 protein expression. (I) p35 mRNA gene expression levels quantified by qRT-PCR after transfection of APPswe cells with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled controls (Scr/Anti-scr). (J and K) Representative western blot images of p35 and CDK5 expression in APPswe cells (J) and quantification (K) after overexpression of p35 (exclusion of nonspecific band of p35). (L) Binding of p35 and CDK5 using an immunoprecipitation assay in APPswe cell homogenates immunoprecipitated with anti-p35 antibody. (M and N) Representative western blot images of p35, p25, and CDK5 expression (M) and quantification (N) in APPswe cells transfected with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled controls (Scr/Anti-scr). (O and P) Representative western blot images of the phosphorylation of tau protein at AT8, S199, S396, and S404 sites (O) and quantification (P) after transfection of APPswe cells with scrambled control (Scr), miR-148a-3p mimic (148a), and p35. Results represent means ± SEM, n = 5. ∗∗p < 0.01, ∗∗∗p < 0.001 versus Scr or Anti-scr; $p < 0.05, $$p < 0.01, $$$p < 0.001 versus 148a. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
miR-148a-3p was found to have a conserved target site in the 3′ UTR of the p35 gene and had a mirSVR score of −1.1160 as a predicted target of miR-148a-3p (Figures 3D and 3E, Table S2). The dual-luciferase reporter assay indicated that the luciferase activity of the WT p35 mRNA 3′ UTR was attenuated by co-transfection of miR-148a-3p mimics, which was undetected when mutant 3′ UTR was expressed (Figure 3F, p < 0.001), suggesting that miR-148a-3p directly targets the predicted p35 mRNA 3′ UTR sequence. qRT-PCR and western blot analysis were then performed to determine the influence of miR-148a-3p on p35 gene and protein expression. miR-148a-3p mimics were found to downregulate p35 protein expression (Figures 3G and 3H, p < 0.001), while anti-miR-148a-3p upregulated p35 protein expression (p < 0.001). Owing to miR-148a-3p mimics or anti-miR-148a-3p not causing a change in p35 mRNA expression (Figure 3I), miR-148a-3p was extrapolated to target the 3′ UTR of p35, thereby suppressing its translation.
p35 is a neuron-specific regulatory protein of cyclin-dependent protein kinase 5 (CDK5), an important kinase involved in abnormal hyperphosphorylation. Typically, CDK5 upregulation was demonstrated by p35 overexpression (Figures 3J and K, p < 0.01), while the direct binding of p35 to CDK5 was determined using an immunoprecipitation assay (Figure 3L). Notably, miR-148a-3p mimics suppressed the expression of p35, p25, and CDK5 (Figures 3M and 3N, p < 0.01–0.001), while anti-miR-148a-3p caused the reverse (p < 0.01–0.001). Consistently, the inhibitory effect of miR-148a-3p on tau phosphorylation was reversed by p35 overexpression (Figures 3O and 3P, p < 0.05–0.001; Figure S4). Therefore, these findings suggest that miR-148a-3p acted upstream of the inhibition of tau hyperphosphorylation in a cascade that regulated p35/CDK5 signaling.
miR-148a-3p regulates the PTEN/Akt/CREB signaling pathway through a negative feedforward loop
Consistent with the RNA-sequencing analysis described above, a mirSVR score of −1.1250 (Table S2) and a conserved target site in the 3′ UTR of PTEN (Figure 4A) indicated that PTEN represented an additional mRNA to which miR-148a-3p was able to bind. Co-transfection of miR-148a-3p mimics with WT PTEN mRNA 3′ UTR led to inhibition of luciferase activity (Figures 4B and 4C, p < 0.01), while mutation of the predicted miR-148a-3p binding sites in the 3′ UTR of PTEN abolished luciferase activity, identifying PTEN as a direct target of miR-148a-3p in vitro. Furthermore, PTEN protein expression levels were downregulated by miR-148a-3p mimics and upregulated by anti-miR-148a-3p (Figures 4D and 4E, p < 0.001 and 0.05), while no variation in PTEN mRNA expression was observed when miR-148a-3p mimics or anti-miR-148a-3p were used (Figure 4F). Jointly, these results suggest that PTEN is under the regulatory control of miR-148a-3p via the 3′ UTR binding of PTEN, thereby reducing protein production.
Figure 4.

miR-148a-3p directly targets PTEN and regulates the PTEN/Akt/CREB signaling pathway through a negative feedforward loop
(A) Binding site for miR-148a-3p in the 3′ UTR of PTEN mRNA as found by bioinformatics analysis. (B) Construction of wild-type (Wt) and mutant (Mut) 3′ UTR of PTEN in the pGL3 vector. (C) Relative luciferase activity after co-transfection with a plasmid constructed with a Wt PTEN 3′ UTR or Mut PTEN 3′ UTR with scrambled control (Scr) or miR-148a-3p mimic (148a) in HEK293 cells (n = 4). (D and E) Representative western blot images of PTEN protein expression (D) and quantification of PTEN protein expression (E) in APPswe cells after transfection with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled controls (Scr/Anti-scr) (n = 4). (F) PTEN mRNA expression as measured by qRT-PCR after transfection of APPswe cells with miR-148a-3p mimic (148a), anti-miR-148a-3p (Anti-148a), and scrambled controls (Scr/Anti-scr) (n = 4). (G) Top 10 most enriched pathways in KEGG pathway analysis of the predicted mRNA targets. (H–J) Representative western blot images of p-PTEN, PTEN, p-Akt, and Akt expression (H) in APPswe cells transfected with PTEN-overexpressing plasmid (PTEN), basic vector (Vector), PTEN siRNA, and scrambled siRNA (Scr siRNA) and quantification of p-PTEN/PTEN (I) and p-Akt/Akt (J) (n = 6). (K) miR-148a-3p expression quantified in APPswe cells by qRT-PCR after transfection with PTEN-overexpressing plasmid (PTEN), basic vector (Vector), PTEN siRNA, and scrambled siRNA (Scr siRNA) (n = 4). (L) miR-148a-3p expression in APPswe cells measured by qRT-PCR after transfection with Akt-overexpressing plasmid (Akt), basic vector (Vector), Akt siRNA, and scrambled siRNA (Scr siRNA) (n = 4). (M) miR-148a-3p expression measured by qRT-PCR in APPswe cells treated with trehalose, IGF-1, PBS, and LY294002 (n = 4). (N) Luciferase activity in a luciferase reporter assay constructed by cloning the putative promoter of miR-148a-3p into a pGL4 basic vector when PTEN was overexpressed (PTEN) or silenced (PTEN siRNA) and Akt was overexpressed (Akt) or silenced (Akt siRNA) in HEK293 cells (n = 4). (O) Luciferase activity in the luciferase reporter assay constructed by cloning the putative promoter of miR-148a-3p into a pGL4 basic vector when treated with IGF-1 and LY294002 in HEK293 cells (n = 4). Results represent means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus corresponding controls. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To further explore the signaling pathways of miR-148a-3p, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of miR-148a-3p predicted targets indicated that PTEN/Akt signaling was the most relevant enriched pathway associated with AD (Figure 4G). As confirmed by western blot analysis, PTEN overexpression inhibited Akt phosphorylation, while PTEN silencing caused the opposite effect (Figures 4H–4J, p < 0.01–0.001), indicating that PTEN acted as an Akt phosphorylation inhibitor. Furthermore, miR-148a-3p expression was negatively regulated by PTEN overexpression or silencing (Figure 4K, both p < 0.001), but positively regulated by Akt overexpression or silencing (Figure 4L, p < 0.001 and 0.01). In addition, the stimulation of Akt by insulin-like growth factor 1 (IGF-1) and the inhibition of phosphatidylinositol 3-kinase (PI3K) by LY294002 both increased miR-148a-3p expression (Figure 4M, both p < 0.01; Figure S5, both p < 0.001). These findings indicate an involvement of PTEN/Akt signaling in miR-148a-3p-mediated neuroprotection.
To validate the regulatory interplay of PTEN/Akt with miR-148a-3p, a luciferase reporter assay to detect miR-148a-3p translation was performed by cloning the 3-kDa putative promoter of miR-148a-3p into a pGL4 basic vector. The results demonstrated that PTEN overexpression, Akt silencing, and PI3K inhibition significantly suppressed luciferase activity of the miR-148a-3p promoter (Figures 4N and 4O, p < 0.05–0.01). Conversely, the opposite stimulus involving PTEN silencing, Akt overexpression, or induction by IGF-1 increased luciferase activity (all p < 0.001). Together, these results imply that miR-148a-3p is not only a proteogenic suppressor of PTEN, but also a feedforward effector of the PTEN/Akt pathway at a transcriptional level.
The PTEN/Akt pathway regulates miR-148a-3p expression in a CREB-dependent manner
Because the transcription of miR-148a-3p was found to be a feedforward regulator through the signaling pathway of its target PTEN, the manner in which PTEN signaling influenced the expression of miR-148a-3p was investigated in AD pathology. As revealed by western blot analysis, CREB was found to be an important downstream effector of the PTEN/Akt pathway, demonstrated by PTEN overexpression, Akt silencing, and PI3K inactivation decreasing the ratio of phosphorylated CREB to total CREB, whereas it was increased when the converse PTEN and Akt intervention was performed (Figures 5A–5E, p < 0.05–0.001). Using the bioinformatics application rVista 2.0, an evolutionarily conserved putative CREB-binding site was identified 1,217 bp upstream of the miR-148a-3p cluster (Figures 5F and 5G). The results of the chromatin immunoprecipitation (ChIP) assay confirmed that DNA fragments containing the putative binding site were precipitated by the CREB antibody (Figures 5H and 5I).
Figure 5.

PTEN/Akt pathway regulates miR-148a-3p expression in a CREB-dependent manner
(A and B) Representative western blot images of p-CREB and CREB (A) and quantification of p-CREB/CREB ratio (B) after transfection of APPswe cells with PTEN-expressing plasmid (PTEN), vector control (Vector), PTEN siRNA, and scrambled siRNA (Scr siRNA) (n = 6). (C–E) Representative western blot images of p-Akt, Akt, p-CREB, and CREB (C) and quantification of ratio of p-Akt/Akt (D) and of p-CREB/CREB (E) after transfection of APPswe cells with Akt-expressing plasmid (Akt), basic vector control (Vector), Akt siRNA, and scrambled siRNA (Scr siRNA) (n = 6). (F) Potential binding sites of CREB with the miR-148a-3p promoter. (G) Binding motif of CREB transcription factor with the miR-148a-3p promoter. (H) Quantitation of ChIP assay results by qRT-PCR (n = 4). (I) ChIP assay with CREB antibody and PCR amplification of the region from the miR-148a-3p promoter. (J) Luciferase activity of wild-type miR-148a-3p promoter construct (BS Wt) or CREB-binding-site mutant construct (BS Mut) in HEK293 cells transfected with CREB-expressing plasmid (CREB) and its vector control (Vector) (n = 4). (K) Expression of miR-148a-3p using qRT-PCR after transfection of APPswe cells with CREB-overexpressing plasmid (CREB), vector control (Vector), CREB siRNA, and scrambled siRNA (Scr siRNA) (n = 4). Results represent means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus vector or Scr siRNA. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant.
To confirm whether CREB regulated the transcription of miR-148a-3p via this binding site, the luciferase reporter containing the putative or mutant binding site of miR-148a-3p was co-expressed with a plasmid overexpressing CREB in HEK293 cells. A significant increase in luciferase activity was observed in the putative reporter compared with that in the mutant (Figure 5J, p < 0.001). Consistent with the findings described above, CREB overexpression resulted in a substantial 6-fold increase in miR-148a-3p expression, while CREB silencing led to a 2-fold decrease in its expression (Figure 5K, p < 0.001 and 0.01). The above results indicate that CREB regulated miR-148a-3p transcription by its direct binding to the promoter of miR-148a-3p. Collectively, these findings suggest that feedforward action occurred between miR-148a-3p and PTEN/Akt via CREB-dependent signaling.
PTEN/p38 MAPK is an additional pathway contributing to the inhibitory effects of miR-148a-3p on tau phosphorylation
Previous studies have suggested a significant role for aberrant PTEN signaling in tau phosphorylation.24,25 Coincidentally, the phosphorylation sites of tau proteins Ser396, Ser404, Ser199, and AT8 were upregulated by PTEN overexpression and downregulated by PTEN silencing (Figures 6A and 6B, p < 0.05–0.01). The laser confocal microscopy images demonstrated that AD-relevant tau phosphorylation epitopes, paired helical filament (PHF)-1 (Ser396/404), and total tau were stained in the cytoplasm of APPswe cells. It was observed that PTEN was distributed in the cytoplasm and nucleus. A further relationship between PTEN and these tau proteins was analyzed by Pearson’s coefficient, which displayed that PTEN had strong co-localization with PHF-1, from the high value of 0.53 ± 0.025, and moderate co-localization with total tau, from the value of 0.27 ± 0.097, suggesting that PTEN preferred co-localization in the cytoplasm with phosphorylated tau to total tau (Figures 6C and 6D, p < 0.05). Of the most prominent kinases involved in tau phosphorylation, p38 mitogen-activated protein kinase (MAPK) was upregulated by PTEN overexpression and downregulated by PTEN silencing (Figures 6E and 6F, both p < 0.05), accompanied by a negative change in phosphorylated extracellular signal-regulated kinase-1/2 (p-ERK1/2) (both p < 0.05) and a limited change in the phosphorylated glycogen synthase kinase-3β (p-GSK3β)/GSK3β ratio in response to a change in PTEN in APPswe cells. Furthermore, PTEN-induced phosphorylation was inhibited by miR-148a-3p (Figures 6G and 6H, p < 0.05–0.001). Therefore, miR-148a-3p may have a dual regulatory role concerning tau phosphorylation via the p35/CDK5 and PTEN/p38 MAPK pathways.
Figure 6.

PTEN/p38 MAPK signaling contributes to the inhibitory effects of miR-148a-3p on tau phosphorylation
(A and B) Representative western blot images of phosphorylation of tau protein at AT8, S199, S396, and S404 sites and Tau (A) and quantification of the ratios of AT8/Tau, S199/Tau, S396/Tau, and S404/Tau (B) after transfection of APPswe cells with PTEN-expressing plasmid (PTEN), vector control (Vector), PTEN siRNA, and scrambled siRNA (Scr siRNA). (C) Co-localization of PTEN with PHF-1, the Ser396/404 sites of tau phosphorylation, and total tau (Tau) in APPswe cells using an immunofluorescence assay. Scale bar, 50 μm. (D) Quantitative analysis of co-localization of PTEN with PHF-1 and Tau using Pearson coefficient in Fiji ImageJ software. (E and F) Representative western blot images of p-PTEN, PTEN, pTS396, pTS404, Tau, p-p38 MAPK, p38 MAPK, p-ERK1/2, ERK1/2, p-GSK3β, and GSK3β (E) and quantification of PTEN expression and ratios of p-PTEN/PTEN, S396/Tau, S404/Tau, p-GSK3β/GSK3β, p-ERK1/2/ERK1/2, p-JNK/JNK, and p-p38 MAPK/p38 MAPK (F) after transfection of APPswe cells with PTEN-expressing plasmid (PTEN), vector control (Vector), PTEN siRNA, and scrambled siRNA (Scr siRNA). (G and H) Representative western blot images of phosphorylation of tau protein at AT8, S199, S396, and S404 sites; total tau; p-PTEN; and PTEN (G) and quantification (H) after transfection of APPswe cells with PTEN-overexpressing plasmid (PTEN) and PTEN-expressing plasmid plus miR-148a-3p mimic (PTEN+148a). Results represent means ± SEM, n = 5. ∗n < 0.05, ∗∗n < 0.01 versus vector or Scr siRNA or PTEN + Tau. $p < 0.05, $$p < 0.01, $$$p < 0.001 versus PTEN. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
miR-148a-3p overexpression or PTEN silencing rescues cognitive deficit in AD mice
As the results demonstrated that miR-148a-3p/p35/PTEN was involved in AD pathology, the therapeutic potential of the miR-148a-3p-mediated pathway toward AD was examined in 6-month-old APP/PS1 mice, which were infused with adeno-associated viruses (AAVs) transfected with miR-148a-3p mimic, PTEN small interfering RNAs (siRNAs), or scrambled sequences. Initially, miR-148a-3p and PTEN levels in the cortex and hippocampus of 6-month-old APP/PS1 mice changed (Figures S6A–S6F, all p < 0.05). The administration of miR-148a-3p mimics or PTEN siRNAs in APP/PS1 mice enhanced spatial learning capability (Figure 7A) and hippocampus-dependent memory (Figures 7C and 7D, p < 0.05–0.001), with the mice displaying a relatively precise and definite travel path in the Morris water maze (MWM) test (Figure 7E). As demonstrated by immunoreactive staining, miR-148a-3p mimics or PTEN siRNA treatment ameliorated tau pathology in the hippocampus and cortex, involving AT8 (the residues at Ser202/Thr205 hyperphosphorylated initially), Tau 1 (tau with Ser198/199/202 sites nonphosphorylated), and PHF-1 (mature hyperphosphorylated forms of tau) (Figures 8A–8F, p < 0.05–0.001), aside from Tau 5 (total tau) (Figures 8G and 8H). Similarly, miR-148a-3p mimic or PTEN siRNA treatment ameliorated neuronal degeneration as illustrated by neuronal-specific nuclear protein (NeuN) and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining in the cortex and hippocampus of APP/PS1 mice (Figures 8I–8L, p < 0.05–0.001).
Figure 7.

miR-148a-3p overexpression or PTEN silencing rescues spatial cognitive behavior in AD mice
(A) Comparison of the latency of finding the platform during 5 training days in the Morris water maze test in APP/PS1 mice infused with AAVs containing miR-148a-3p mimic, PTEN siRNA, or scrambled control. (B) Swimming speed in the navigation test. (C) Increased number of platform crossings by APP/PS1 mice treated with miR-148a-3p mimic or PTEN siRNA in the probe test. (D) Increased duration within the target quadrant of miR-148a-3p mimic- or PTEN siRNA-treated APP/PS1 mice in the probe test. (E) Representative images of tracings representing the route of mice finding the platform in the probe test on days 6 and 7. Results represent means ± SEM, n = 10. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus WT mice. #p < 0.05, ###p < 0.001 versus APP/PS1 scrambled controls.
Figure 8.

miR-148a-3p overexpression or PTEN silencing ameliorated tau pathology and neurodegeneration in AD mice
(A–H) Representative images and quantitative analysis, respectively, of immunoreactivity of AT8 (A and B), Tau 1 (C and D), PHF-1 (E and F), and Tau 5 (G and H) using immunohistochemistry of the hippocampus and cerebral cortex in APP/PS1 mice treated with miR-148a-3p mimics, PTEN siRNAs, and scrambled controls. (I and J) Representative images (I) and quantitative analysis of immunoreactivity (J) of NeuN staining in the hippocampus and cerebral cortex of APP/PS1 mice treated with miR-148a-3p mimics, PTEN siRNAs, and scrambled controls. (K and L) Representative images (K) and quantitative analysis of immunoreactivity (L) of TUNEL staining of the hippocampus and cerebral cortex tissue of APP/PS1 mice treated with miR-148a-3p mimics, PTEN siRNAs, and scrambled controls. Scale bar, 50 μm. Results represent means ± SEM, n = 3. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus WT mice. #p < 0.05, ##p < 0.01, ###p < 0.001 versus APP/PS1 scrambled controls.
Measurement of the signaling pathway first elucidated co-localization of PTEN and p35 with phosphorylated tau PHF-1 in the hippocampus and cortex of differentially treated APP/PS1 mice (Figures 9A–9D). Subsequently, p35, p25, and CDK5 expression decreased in the hippocampus of miR-148a-3p-treated APP/PS1 mice, in parallel with downregulated tau phosphorylation at Ser199, Ser396, and Ser404 epitopes compared with APP/PS1 scrambled control mice (Figures 9E and 9F, p < 0.05–0.001). The critical molecular factors within the feedforward loop of miR-148a-3p were created in PTEN siRNA-treated APP/PS1 mice, indicative of increased ratios of p-PTEN/PTEN, p-Akt/Akt, and p-CREB/CREB and the upregulation of miR-148a-3p in the hippocampus compared with the scrambled APP/PS1 control (Figures 9G–9I, p < 0.05–0.001). These findings demonstrate that a target-specific strategy to rebuild the miR-148a-3p/p35/PTEN pathway ameliorated AD-like symptoms and attenuated tau phosphorylation in vivo.
Figure 9.

miR-148a-3p overexpression or PTEN silencing protects miR-148a-3p/p35/PTEN signaling and PTEN/Akt/CREB feedforward in AD mice
(A–D) Co-localization of p35 (A and B) and PTEN (C and D) with phosphorylated tau PHF-1 in the hippocampus (A and C) and cortex (B and D) of APP/PS1 mice treated with miR-148a-3p mimics, PTEN siRNAs, and scrambled controls. Scale bar, 50 μm. (E and F) Representative western blot images of pTS396, pTS404, pTS199, Tau, p35, p25, CDK5, p-PTEN, and PTEN (E) and quantification of ratios of S396/Tau, S404/Tau, S199/Tau, and p-PTEN/PTEN and expression of p25, p35, and CDK5 (F) in the hippocampus of APP/PS1 mice treated with miR-148a-3p mimic and scrambled control. (G and H) Representative western blot images of pTS396, pTS404, pTS199, Tau, p-PTEN, PTEN, p-Akt, Akt, p-CREB, and CREB (G) and quantification of ratios of S396/Tau, S404/Tau, S199/Tau, p-PTEN/PTEN, p-Akt/Akt, and p-CREB/CREB (H) in the hippocampus of APP/PS1 mice treated with PTEN siRNA and scrambled control. (I) miR-148a-3p expression in the hippocampus of APP/PS1 mice treated with PTEN siRNA and scrambled control. Results represent means ± SEM, n = 5. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus WT mice. #p < 0.05, ##p < 0.01, ###p < 0.001 versus APP/PS1 scrambled controls. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
The present study uncovered novel signaling pathways of miR-148a-3p/p35/PTEN in AD, suggesting that miR-148a-3p has potential clinical diagnostic value with biochemical cascades in AD pathology. Based on these findings, miR-148a-3p was downregulated in AD progression, and upregulation of miR-148a-3p reduced tau hyperphosphorylation and eventually protected neuronal cells against Aβ-induced injury by targeting p35 and PTEN. A regulatory feedforward link between miR-148a-3p and PTEN/Akt/CREB signaling was found via the promotion of miR-148a-3p expression by CREB as a downstream effector of the PTEN/Akt pathway. Significantly, the in vivo targeting of miR-148a-3p/PTEN signaling ameliorated cognitive deficit by increasing the transduction of the Akt/CREB feedforward pathway and reduced tau phosphorylation by decreasing p35/PTEN-elicited tau hyperphosphorylation (Figure 10).
Figure 10.

Neuroprotective mechanism of miR-148a-3p/p35/PTEN signaling as established in the present study
Aβ, amyloid-β peptides; Akt, protein kinase B; CDK5, cyclin-dependent protein kinase 5; CREB, cAMP-response element binding protein; NFTs, neurofibrillary tangles; p25, a proteolytic fragment of p35; p35, cyclin-dependent kinase 5 regulatory subunit 1; p38, p38 mitogen-activated protein kinase; PHF, paired helical filament; PTEN, phosphatase and tensin homolog deleted on chromosome 10; RNA Pol II, RNA polymerase II.
miR-148a-3p dysfunction in AD
As is well known, research has focused on the identification of miR-148a-3p as a tumor suppressor in a number of cancers.26, 27, 28, 29, 30 Little discussion has involved its function in AD. The novelty of the present study stems from the insight that miR-148a-3p is involved in driving the pathogenesis of AD. A substantial reduction in miR-148a-3p occurs as the disease progresses, revealed by RNA sequencing and verified in a variety of AD models, indicating that miR-148a-3p is a causative factor in AD. A reduction of miR-148a-3p was found in the hippocampus and cortex, two regions susceptible to AD, highlighting tissue-specific abnormalities of miR-148a-3p as cognitive decline occurs and progresses.
Tau proteins become abnormally hyperphosphorylated in the early stages of AD, proceeding progressively to pre-neurofibrillary tangles and intraneuronal and extraneuronal NFTs,31 resulting in axonal dysfunction, neuritic atrophy, and neuronal death. Although the etiology of AD is multifactorial and undefined, tau hyperphosphorylation has been suggested to be the pathology that is most correlated with cognitive decline.32 There are approximately 40 serine-threonine phosphorylation sites in tau proteins related to AD, including Ser199, Ser396, Ser404, and Thr205, epitopes that lead to PHF aggregates in pre-tangles to neurofibrillary changes,33 eventually inducing region-specific neuronal apoptosis and degeneration.34 A range of kinases has been shown to phosphorylate tau protein, such as CDK5, GSK3β, and MAPKs.35 As tau hyperphosphorylation involves the abnormal post-transcriptional modification of kinases directly involved in the process of tau protein phosphorylation,36 the misregulation of miRNAs contributes greatly to the pathogenic risks of AD.
In the present study, miR-148a-3p was found to have a neuroprotective effect by inhibition of abnormal tau phosphorylation at multiple sites involved in the different stages of tau phosphorylation for the complicated neuropathological events in AD. More importantly, the results of AD pathology and spatial cognitive decline demonstrated that miR-148a-3p improved learning and memory capability and prevented the onset of histopathological changes by reducing tau hyperphosphorylation and reducing neuronal degeneration. Consistent with in vivo findings, miR-148a-3p levels in the blood of AD patients were closely positively correlated with the cognitive scores, providing desirable rates of specificity and sensitivity for the disease. The findings are possibly a function of the favorable effects of miR-148a in attenuating the loss of nigral neurons in Parkinson’s disease.37 Therefore, miR-148a-3p may represent a good prognostic factor and a promising neuroprotective target for inhibition of hyperphosphorylation of tau that could contribute to the treatment of AD.
p35 and PTEN are novel targets of miR-148a-3p in the brain
Several studies showed a role for miR-148a-3p in some neurologic disorders via action on different target genes. Rho-associated coiled-coil kinase 1 (ROCK1) is a potential target of miR-148a-3p, which, by specifically binding to ROCK, attenuated Aβ25-35-induced neurotoxicity in SH-SY5Y cells.38 Other reported target genes of miR-148a-3p, such as synaptojanin-1 (SYNJ1),39 DNA methyltransferase 1 (DNMT1),40 and programmed cell death 1/programmed cell death ligand 1 (CD274),41 may be involved in neuroimmune function and cell apoptosis; among these, overexpression of miR-148a-3p is shown to inhibit neuronal apoptosis via activation of the PI3K/AKT signaling pathway by suppression of SYNJI expression in the hippocampal neurons of rats with febrile seizures.39 These findings imply that miR-148a-3p may exert neuroprotective effects in certain neurologic disorders via specific targets at the epigenetic level. However, the underlying mechanism of action of miR-148a-3p on AD still needs to be elucidated.
In the present study, the first inhibitory effect of miR-148a-3p upregulation of tau phosphorylation was mediated by targeting p35, with the signature of decreased p35/CDK5 signaling. p35 is known to be a chief neural-specific activator of CDK5, among the most critical kinases for the regulation of tau hyperphosphorylation in AD.35 The manifestation of the CDK5-p35 complex differs depending on whether the conditions are physiological or pathological. CDK5 activity is physiologically autoinhibited and limited to membrane targeting owing to the ubiquitination pathway undertaken by the p10 fragment of p35.42 Under pathological conditions, neurotoxic stimulation increases intracellular calcium levels, which leads to activation of calpain, a protease responsible for the cleavage of the p10 fragment from p35 to produce p25. As a result of loss of the regulation of ubiquitination by the p10 fragment, the truncated p35 by-product p25 causes hyperactivation and mislocalization of CDK5, leading to severe neuropathological aberrations, including hyperphosphorylated tau-mediated NFT formation and neuronal death, resulting in AD.43 Here, we found that increased p35 levels preceded those of CDK5 in response to copper-induced Aβ stimulation in vitro, consistent with the feedback activation of CDK5 by upregulation of p35 in a glutamate-induced excitotoxic neurodegeneration model.44 In addition, p35 expression was found to have continually increased in mouse cortical samples during AD progression, negatively correlating with miR-148a-3p levels. Notably, miR-148a-3p was confirmed to repress the expression of p35 and subsequently inhibit the expression of p25 and CDK5 in vitro. Supplementation in vivo with miR-148a-3p coincidentally caused negative regulation of the expression of p35, p25, and CDK5. Further specific upregulation of p35 blocked the beneficial effects of miR-148a-3p on tau phosphorylation at multiple epitopes. Therefore, novel mechanistic control exerted by miR-148a-3p, but dependent on p35/CDK5 signaling, regarding tau phosphorylation in AD was elucidated.
The second inhibitory effect of miR-148a-3p on tau phosphorylation was attributed to a different target, PTEN, deregulation of which was closely linked to results suggesting a link to abnormal tau phosphorylation in AD. A number of studies have reported that the targeting of PTEN by miR-148a-3p is responsible for the promotion of differentiation, proliferation, and macrophage activation in tumor and inflammation-related diseases.45, 46, 47, 48 The present study indicated that PTEN is a direct target of miR-148a-3p and is responsible for attenuation of tau hyperphosphorylation, through co-localization with phosphorylated tau proteins in neuronal cells. In combination with the well-documented functions of miR-148a-3p, we propose that miR-148a-3p/PTEN signaling may contribute to neuroprotection in AD, at least partially due to the reduction in tau phosphorylation.
Of the downstream signaling of PTEN due to regulation by miR-148a-3p, PTEN/Akt signaling represented the principally enriched pathway associated with AD. An extensive literature search has demonstrated that PTEN causes deregulation of the PI3K/Akt pathway in conjunction with increased GSK3β and MAPK activity,49, 50, 51 playing a crucial role in the onset and progression of AD. Correspondingly, we have shown that PTEN negatively regulates PI3K/Akt signaling and subsequently induces the activation of p38 MAPK, which is associated with the effective kinase/phosphatase system leading to tau phosphorylation. These results are consistent with the observation that downregulation of PTEN-mediated Akt signaling is correlated with the elevation of kinases and phosphatases in tau phosphorylation.
Novel regulatory mechanism between miR-148a-3p and PTEN/Akt/CREB is established
It is worth noting that a novel reciprocal regulatory mechanism between miR-148a-3p and PTEN was established. As revealed above, miR-148a-3p suppressed PTEN expression by targeting its 3′ UTR, thereby inactivating the Akt signaling pathway. Conversely, overexpression of PTEN or silencing of Akt led to the suppression of miR-148a-3p expression, and the silencing of PTEN or activation of Akt increased miR-148a-3p expression. Thus, a novel correlation between miR-148a-3p and PTEN emerged based on the manifestation that miR-148a-3p/PTEN signaling functioned as a negative regulatory feedback loop.
Further investigation demonstrated the crucial role of CREB in the regulation of PTEN/Akt signaling transduction presented by its interference with PTEN or Akt in vitro and shown by levels of activated CREB phosphorylation in the hippocampus of APP/PS1 mice by PTEN silencing in vivo. CREB is a critical effector involved in Akt signaling that contributes to neuronal survival, neurogenesis, and neuroplasticity.52 As a well-studied transcription factor, CREB modulates the transcription of a number of miRNAs by binding to their promoter regions, thereby promoting signaling transduction.53 Mechanistic studies further found that CREB bound to the promoter region of miR-148a-3p, thereby inducing its transcription. Correspondingly, the existence of a negative feedforward loop involving miR-148a-3p/PTEN/Akt/CREB in APP/PS1 mice is well established in the recovery of spatial cognitive deficits and relief of neuronal degeneration.
In conclusion, our findings identified a novel function for miR-148a-3p in the progression of AD and revealed the mechanisms involved in miR-148a-3p/p35/PTEN signaling, allowing the rescue of AD-like cognitive impairment and tau pathology. These findings provide a potential therapeutic target, namely, miR-148a-3p/p35/PTEN signaling, for the treatment of AD.
Materials and methods
Animals and treatment
Zhishan Healthcare Research Institute (Beijing, China) provided APP/PS1 mice and nontransgenic littermates, SAMP8 mice, and senescence-accelerated mouse resistance 1 (SAMR1) mice. Consent for the experiments was provided by the Animal Care and Use Committee of the Institute of Medicinal Biotechnology (approval no. IMB-D8-2018071102).
RNA-sequencing assay
The APP/PS1 and WT mice were divided by age into 1-, 3-, 6-, and 9-month-old groups. Total RNA from the cerebral cortex of the mice was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Samples were strictly qualified for RNA integrity and concentration. High-throughput sequencing was performed by Sangon Biotech (Shanghai) Co. Ltd. (Shanghai, China) for analysis of miRNA and mRNA profiles, as previously reported.54
Adenovirus vectors and intracerebroventricular injections
Recombinant AAVs containing miR-148a-3p and PTEN siRNA that expressed green fluorescent protein (GFP) from a cytomegalovirus enhancer plus chicken β-actin (CAG) promoter were acquired from SyngenTech Co. (Beijing, China). Titers of the vectors ranged from 4.2 × 1011 to 3 × 1012. An AAV with a scrambled sequence was used as a control.
The 6-month-old APP/PS1 and WT control mice were randomly divided into the following groups, consisting of 10 mice in each group, 5 males and 5 females: WT control mice or APP/PS1 mice injected with AAV scrambled sequences, AVV-miR-148a-3p, or AVV-PTEN siRNA. Intracerebroventricular injections were performed using the following stereotactic coordinates: +1.0 mm anteroposterior, −0.5 mm mediolateral, and −3 mm dorsoventral to the bregma. A total volume of 1 μL was injected using a microinjector at a rate of 0.2 μL/min.
Human blood collection and preparation
Blood samples from 21 AD patients at Xuanwu Hospital of Capital Medical University and 15 HAVs were obtained. Biomarker measurement experiments were approved by the ethics committee of Xuanwu Hospital of Capital Medical University (approval no. linyanshen2014033; Beijing, China). Serum was separated by centrifugation at 1,000 × g for 15 min at 4°C. Total RNA was extracted using an miRNeasy serum/plasma kit (Qiagen, Duesseldorf, Germany) and stored at −80°C until required.
Plasmid transfection
SH-SY5Y cells (ATCC, Manassas, VA, USA) transfected with the Swedish mutant form of human APP, named APPswe cells, are an established cell model of AD in which copper triggers Aβ overproduction.55, 56, 57 They were cultured in Dulbecco's modified Eagle medium/nutrient mixture F-12 (DMEM/F12) supplemented with 2 mM L-glutamine, 10% fetal bovine serum (FBS; Gibco/Invitrogen, Grand Island, NY, USA), and 400 μM G418 (Sigma Chemical Co., St. Louis, MO, USA). miR-148a-3p mimics, antisense miR-148a-3p oligonucleotides (anti-miR-148a-3p), and scrambled controls were synthesized by GenePharma (Shanghai, China). pCMV-PTEN, pCMV-myr-Akt1, and pCMV-CREB were purchased from OriGene (Beijing, China) and transfected at a final concentration of 2 μg/mL. siRNAs for PTEN, Akt1, and CREB1 were obtained from Cell Signaling Technology (Danvers, MA, USA) and transfected at a final concentration of 100 nM. Cells were plated in culture plates 24 h before transfection. All plasmids were transfected transiently using Lipofectamine 3000 (Invitrogen).
Cell viability and apoptosis assay
Cell survival was assessed using a CCK-8 assay (Vazyme Biotech, Nanjing, China) and measured with a Spark 20M multimode microplate reader (Tecan Group Ltd., Mannedorf, Switzerland). APPswe cell apoptosis following transfection with various plasmids was measured by flow cytometry after staining with fluorescein isothiocyanate (FITC)-labeled Annexin-V and propidium iodide (PI) (BD Biosciences, San Jose, CA, USA). Following staining, cells were analyzed in a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA).
Immunofluorescence and confocal microscopy
APPswe cells were seeded into 20-mm-diameter confocal glass-bottom cell culture dishes at a density of 1.0 × 105 cells and transfected with PTEN-overexpressing plasmid. 48h after transfection, the cells were processed as described previously.57 Fluorescence images and data were acquired using a laser confocal microscope (LSM780; Carl Zeiss; Jena, Germany) with 20×/numeric aperture (NA) = 0.5 and 40×/NA = 0.75 objectives, using primary antibodies against PHF-1 (Abcam, Cambridge, MA, USA), Tau (Proteintech, Chicago, IL, USA), and PTEN (Abcam). Fiji ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to provide a quantitative measurement of the co-localization of PTEN with PHF-1 and Tau by Pearson’s correlation coefficient, which estimated the degree of overlap between fluorescence signals obtained in two channels. The degree of co-localization from the Pearson’s coefficient values was categorized as very strong (0.85 to 1.0), strong (0.49 to 0.84), moderate (0.1 to 0.48), weak (−0.26 to 0.09), and very weak (−1 to −0.27) based on a previously published description.58
qRT-PCR analysis
Total cellular RNA and tissue RNA were extracted using TRIzol reagent (Invitrogen). miRNA was reverse transcribed using a TaqMan miRNA reverse-transcription kit (Thermo Fisher Scientific, Waltham, MA, USA). PCR was performed using TaqMan Universal PCR Master Mix (Thermo Fisher Scientific). Relative miR-148a-3p expression was calculated using the 2−ΔΔCT method. Results were normalized from the geometric mean of two internal housekeeping miRNAs, miR-106a-5p and miR-17-5p. mRNA was reverse transcribed to complementary DNA (cDNA) using a HiScript III 1st Strand cDNA synthesis kit (Vazyme Biotech), after which qPCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech). Primers are listed in Table 1.
Table 1.
Primers used for PCR and miRNA reverse transcription
| Primer name | Primer sequence |
|---|---|
| PTEN-F | 5′-TTGTGGTCTGCCAGCTAAAGGT-3′ |
| PTEN-R | 5′-GAACTTGTCTTCCCGTCGTGTG-3′ |
| CDK5R1-F | 5′-GCAGATCCAAGGGGGCAG-3′ |
| CDK5R1-R | 5′-GGGATAAAACCGCTCACCGA-3′ |
| GAPDH-F | 5′-CAAATTCCATGGCACCGTCA-3′ |
| GAPDH-R | 5′-AGCATCGCCCCACTTGATTT-3′ |
| Pten-F | 5′-ATTGGCTGCTGTCCTGCTGTT-3′ |
| Pten-R | 5′-GGTTAAGTCATTGCTGCTGTGTCT-3′ |
| Cdk5r1-F | 5′-GCTGTCCTGCTGACCTGTCTGTA-3′ |
| Cdk5r1-R | 5′-TTCTTGTCCTCCTGACCGCTCTC-3′ |
| Actb-F | 5′-GAGATTACTGCTCTGGCTCCTA-3′ |
| Actb-R | 5′-GGACTCATCGTACTCCTGCTTG-3′ |
| miR-148a-3p-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAGGTA TTCGCACTGGATACGACACAAAG-3′ |
| miR-148a-3p-F | 5′-GCGCGTCAGTGCACTACAGAA-3′ |
| miR-148a-3p-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| miR-17-5p-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTC GCACTGGATACGACCTACCT-3′ |
| miR-17-5p-F | 5′-GCGCAAAGTGCTTACAGTGC-3′ |
| miR-17-5p-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| miR-106a-5p-RT | 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCA CTGGATACGACCTACCT-3′ |
| miR-106a-5p-F | 5′-CGCGAAAAGTGCTTACAGTGC-3′ |
| miR-106a-5p-R | 5′-AGTGCAGGGTCCGAGGTATT-3′ |
| U6-RT | 5′-CGCTTCACGAATTTGCGT-3′ |
| U6-F | 5′-CTCGCTTCGGCAGCACA-3′ |
| U6-R | 5′-CGCTTCACGAATTTGCGT-3′ |
F, forward primer; R, reverse primer; RT, reverse-transcription primer.
Western blot analysis
Protein samples were obtained from APPswe cells following plasmid transfection and from the cortex and hippocampus of APP/PS1, SAMP8, and control mice using M-PER mammalian protein extraction reagent (Invitrogen) and an ultrasonic homogenizer (Ningbo Scientz Biotechnology Co., Ltd., Ningbo, China). Proteins were separated on 10% SDS-polyacrylamide electrophoresis gels and transferred to a polyvinylidene fluoride membrane (Merck Millipore, Billerica, MA, USA). Membranes were blocked using 5% skimmed milk in Tris-buffered saline/Tween 20 (TBST) for 1 h and then incubated overnight with the appropriate primary antibody at 4°C. The membrane was washed three times with TBST and incubated with horseradish peroxidase (HRP)-conjugated goat anti-(rabbit or mouse) antibody (1:8,000) for 1 h at room temperature. After incubation with primary and secondary antibodies, protein bands on the membranes were acquired using a Fusion-FX6 imaging system (Vilber Lourmat, Paris, France), and relative protein concentration was quantified. The antibodies used for western blot analysis are listed in Table 2.
Table 2.
Primary and secondary antibodies used in western blot analysis
| Primary antibody | Dilution | Source | Secondary antibody |
|---|---|---|---|
| Anti- tau (phospho S202 + T205) rabbit mAb | 1:1,000 | Abcam | HRP-conjugated goat anti-rabbit or anti-mouse IgG (H+L) (1:10,000, EarthOx) |
| Anti-tau (phospho S404) rabbit mAb | 1:1,000 | Abcam | |
| Anti-tau (phospho S396) rabbit mAb | 1:1,000 | Abcam | |
| Anti-tau (phospho S199) rabbit mAb | 1:5,000 | Abcam | |
| Anti-Tau rabbit mAb | 1:2,000 | Abcam | |
| Anti-p35 rabbit pAb | 1:500 | Abcam | |
| Anti-CDK5 rabbit mAb | 1:2,000 | Abcam | |
| Anti-PTEN (phospho S380) rabbit mAb | 1:1,000 | Abcam | |
| Anti-PTEN rabbit mAb | 1:1,000 | Abcam | |
| Anti-Akt (phospho S473) rabbit mAb | 1:5,000 | Abcam | |
| Anti-Akt rabbit mAb | 1:1,000 | CST | |
| Anti-CREB (phospho S133) rabbit mAb | 1:5,000 | Abcam | |
| Anti-CREB rabbit mAb | 1:500 | Abcam | |
| Anti-p38 MAPK (phospho T180/Y182) rabbit mAb | 1:1,000 | CST | |
| Anti-p38 MAPK rabbit mAb | 1:1,000 | Abcam | |
| Anti-p44/42 MAPK (ERK1/2) (phospho T202/Y204) mouse mAb | 1:2,000 | CST | |
| Anti-p44/42 MAPK (ERK1/2) rabbit mAb | 1:1,000 | CST | |
| Anti-GSK-3β (phospho S9) rabbit mAb | 1:1,000 | Abcam | |
| Anti-GSK-3β rabbit mAb | 1:1,000 | CST | |
| Anti-APP rabbit mAb | 1:1,000 | CST | |
| Anti-GAPDH rabbit mAb | 1:5,000 | Proteintech | |
| Anti-α-tubulin rabbit pAb | 1:5,000 | Proteintech | |
| Anti-β-actin mouse mAb | 1:5,000 | Proteintech |
Abcam, Cambridge, MA, USA; CST, Cell Signaling Technology, Danvers, MA, USA; Proteintech, Rosemont, IL, USA; EarthOx Life Sciences, Millbrae, CA, USA.
ChIP assay
ChIP assay was performed using a Pierce magnetic ChIP kit (Thermo Fisher Scientific) in accordance with the manufacturer's protocol. Briefly, SH-SY5Y cells were cross-linked in 1% formaldehyde for 10 min at room temperature, and the reaction was terminated using glycine solution from the ChIP kit. The cells were lysed using extraction buffer and then sonicated to obtain DNA fragments. After centrifugation, 10 μL of the supernatant was used as input, and the remaining supernatant was used to perform the ChIP assay using a specific antibody for CREB (Merck Millipore). The miR-148a-3p promoter region was analyzed using real-time PCR, primers for which were as follows: forward, ACCTCTGCTGATGACACGAG; reverse, TTGGGTTTGGAGACGACCTG.
Luciferase activity assay
HEK293 cells were cultured in 24-well plates 24 h before transfection. WT and mutant (Mut) 3′ UTR luciferase constructs of p35 and PTEN mRNA containing the binding sites of miR-148a-3p were synthesized by Sangon Biotech and were transfected with miR-148a-3p or scrambled mimics. The 2,000 bp upstream of the miR-148a-3p promoter in the WT-luciferase and Mut-luciferase constructs were synthesized by Sangon Biotech and transfected into HEK293 cells with CREB or the vector in addition to Renilla. The luminescence of luciferase was measured after 48 h using a dual-luciferase reporter assay system (Promega, Madison, WI, USA) in a Spark 20M multimode microplate reader (Tecan Group Ltd.). The assay was normalized using Renilla luminescence.
Immunoprecipitation assay
An immunoprecipitation (IP) assay was performed using a Pierce Classic Magnetic IP/Co-IP kit (Thermo Fisher Scientific) in accordance with the manufacturer's protocol. After the cells were lysed, an anti-p35 antibody (Cell Signaling Technology) was added to cell lysate, and the quantity of purified protein mixture was analyzed by Western blotting.
Cognitive behavior testing
The cognitive capability of the mice was assessed using an MWM test, in which a water navigation task and exploration of an area were evaluated as a measure of spatial learning and memory capability, respectively.59 Briefly, mice were first subjected to a water navigation task performed in a 1.2-m-diameter white bucket with a hidden platform, with four trials per day consisting of 60 s of swimming during each trial on 5 consecutive days. The path taken during swimming and the time to reach the hidden platform (known as escape latency) were recorded. Exploration of the space was conducted on the 6th and 7th days, whereon the platform was removed to allow the mice to swim freely for 60 s. The time each mouse spent in the location of the platform and the frequency of crossing over the location were recorded.
Histochemical analysis
WT control and APP/PS1 mice, after injection with scrambled sequences, miR-148a-3p, or PTEN siRNA, were anesthetized using isoflurane and perfused through the heart, first with normal saline and then with 4% paraformaldehyde (PFA). The brains were removed, dissociated, and then cut into sections (4–10 μm) for histochemical and immunohistochemical analysis. The primary and secondary antibodies used in the histochemical analysis are listed in Table 3. The pre-frontal cortical and hippocampal regions were imaged using a Pannoramic DESK slide scanner and Caseviewer 2.3 software (3DHISTECH Ltd., Budapest, Hungary).
Table 3.
Primary and secondary antibodies used in histochemical analysis
| Primary antibody | Dilution | Source | Secondary antibody |
|---|---|---|---|
| Anti-CDK5R1 mouse mAb | 1:200 | Proteintech | Cy3-conjugated goat anti-mouse IgG (H+L) (1:200, Servicebio) |
| Anti-PHF-1 rabbit mAb | 1:100 | Abcam | Alexa Fluor 488-conjugated goat anti-rabbit IgG (H+L) (1:200, Servicebio) |
| Anti-PTEN mouse mAb | 1:200 | Proteintech | Cy3-conjugated goat anti-mouse IgG (H+L) (1:200, Servicebio) |
| Anti-Tau-1 mouse mAb | 1:200 | Millipore | HRP-conjugated goat anti-mouse IgG (H+L) (1:200, Servicebio) |
| Anti-Tau-5 mouse mAb | 1:200 | Millipore | HRP-conjugated goat anti-mouse IgG (H+L) (1:200, Servicebio) |
| Anti-tau (phospho S202 + T205) rabbit mAb | 1:200 | Abcam | HRP-conjugated goat anti-rabbit IgG (H+L) (1:200, Servicebio) |
| Anti-NeuN rabbit pAb | 1:500 | Servicebio | HRP-conjugated goat anti-rabbit IgG (H+L) (1:200, Servicebio) |
Proteintech, Rosemont, IL, USA; Abcam, Cambridge, MA, USA; Merck Millipore, Billerica, MA, USA; CST, Cell Signaling Technology, Danvers, MA, USA; Servicebio, Wuhan, China.
Brain cell apoptosis was measured using TUNEL (Roche, Basel, Switzerland) staining. The pre-frontal cortex and hippocampus were harvested from PFA-perfused mice. The brain was sliced into 4-μm-thick sections, and routine histological protocols were performed for histological staining.60
Bioinformatics analysis
The potential target genes of miR-148a-3p were predicted by computer-aided algorithms using miRNA target prediction databases, including TargetScan (http://www.targetscan.org/), TarBase (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex), and miRDB (http://www.mirdb.org). The miRanda database (http://www.microrna.org/microrna/home.do) was further used to evaluate the reliability of the binding sites through mir-SVR score and PhastCons score. The lower the mirSVR score, the stronger the binding stability of miRNA-mRNA. The higher the PhastCons score, the better the evolutionary conservation of untranslated regions of genes in various species.
Statistical analysis
The data represent means ± standard error of the mean (SEM). Data were analyzed using a one-way ANOVA, followed by a Tukey multiple comparison test, or a Student t test. p < 0.05 was considered statistically significant. Analysis was performed using GraphPad Prism version 7.0 software (GraphPad Software, La Jolla, CA, USA). Navigation tasks and speed of swimming in the MWM test were analyzed using SPSS version 25.0 software (IBM, Armonk, NY, USA), using repeated-measures ANOVA.
Acknowledgments
This study was supported by National Natural Science Foundation of China, China (U1803281 and 8167344), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences, China (2018RC350013), and CAMS Initiative for Innovative Medicine, China (2021-I2M-1-030).
Author contributions
R.L. and Z.L. conceived and directed the projects. R.L. obtained the funding. L.Z., H.J., J.L., L.W., K.Z., M.L., and G.M.A. performed the experiments and analyzed the data. R.L., L.Z., H.J., and G.M.A. wrote the paper. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.11.019.
Contributor Information
Zhuorong Li, Email: lizhuorong@imb.pumc.edu.cn.
Rui Liu, Email: liurui@imb.pumc.edu.cn.
Supplemental information
References
- 1.Alexiou A., Kamal M.A., Ashraf G.M. Editorial: the Alzheimer's disease challenge. Front. Neurosci. 2019;13:768. doi: 10.3389/fnins.2019.00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mamun A.A., Uddin M.S., Mathew B., Ashraf G.M. Toxic tau: structural origins of tau aggregation in Alzheimer's disease. Neural Regen. Res. 2020;15:1417–1420. doi: 10.4103/1673-5374.274329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holtzman D.M., Carrillo M.C., Hendrix J.A., Bain L.J., Catafau A.M., Gault L.M., Goedert M., Mandelkow E., Mandelkow E.M., Miller D.S., et al. Tau: from research to clinical development. Alzheimers Dement. 2016;12:1033–1039. doi: 10.1016/j.jalz.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 4.Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.L., et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottero V., Potashkin J.A. Meta-analysis of gene expression changes in the blood of patients with mild cognitive impairment and alzheimer's disease dementia. Int. J. Mol. Sci. 2019;20:5403. doi: 10.3390/ijms20215403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takousis P., Sadlon A., Schulz J., Wohlers I., Dobricic V., Middleton L., Lill C.M., Perneczky R., Bertram L. Differential expression of microRNAs in Alzheimer's disease brain, blood, and cerebrospinal fluid. Alzheimers Dement. 2019;15:1468–1477. doi: 10.1016/j.jalz.2019.06.4952. [DOI] [PubMed] [Google Scholar]
- 7.Denk J., Oberhauser F., Kornhuber J., Wiltfang J., Fassbender K., Schroeter M.L., Volk A.E., Diehl-Schmid J., Prudlo J., Danek A., et al. Specific serum and CSF microRNA profiles distinguish sporadic behavioural variant of frontotemporal dementia compared with Alzheimer patients and cognitively healthy controls. PLoS One. 2018;13:e0197329. doi: 10.1371/journal.pone.0197329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dehghani R., Rahmani F., Rezaei N. MicroRNA in Alzheimer's disease revisited: implications for major neuropathological mechanisms. Rev. Neurosci. 2018;29:161–182. doi: 10.1515/revneuro-2017-0042. [DOI] [PubMed] [Google Scholar]
- 9.Santovito D., Egea V., Weber C. Small but smart: MicroRNAs orchestrate atherosclerosis development and progression. Biochim. Biophys. Acta. 2016;1861:2075–2086. doi: 10.1016/j.bbalip.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Hata A., Kashima R. Dysregulation of microRNA biogenesis machinery in cancer. Crit. Rev. Biochem. Mol. Biol. 2016;51:121–134. doi: 10.3109/10409238.2015.1117054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P., Xu Y., Wang B., Huang J., Li Q. miR-34a-5p and miR-125b-5p attenuate Aβ-induced neurotoxicity through targeting BACE1. J. Neurol. Sci. 2020;413:116793. doi: 10.1016/j.jns.2020.116793. [DOI] [PubMed] [Google Scholar]
- 12.An F., Gong G., Wang Y., Bian M., Yu L., Wei C. Correction: MiR-124 acts as a target for Alzheimer's disease by regulating BACE1. Oncotarget. 2018;9:24871. doi: 10.18632/oncotarget.25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hebert S.S., Horre K., Nicolai L., Papadopoulou A.S., Mandemakers W., Silahtaroglu A.N., Kauppinen S., Delacourte A., Strooper B.D. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D., Tang H., Li X.Y., Deng M.F., Wei N., Wang X., Zhou Y.F., Wang D.Q., Fu P., Wang J.Z., et al. Targeting the HDAC2/HNF-4A/miR-101b/AMPK pathway rescues tauopathy and dendritic abnormalities in alzheimer's disease. Mol. Ther. 2017;25:752–764. doi: 10.1016/j.ymthe.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L., Liu J., Wang Q., Jiang H., Zeng L., Li Z., Liu R. MicroRNA-200a-3p mediates neuroprotection in Alzheimer-related deficits and attenuates amyloid-beta overproduction and tau hyperphosphorylation via coregulating BACE1 and PRKACB. Front. Pharmacol. 2019;10:806. doi: 10.3389/fphar.2019.00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing H., Guo S., Zhang Y., Zheng Z., Wang H. Upregulation of microRNA-206 enhances lipopolysaccharide-induced inflammation and release of amyloid-β by targeting insulin-like growth factor 1 in microglia. Mol. Med. Rep. 2016;14:1357–1364. doi: 10.3892/mmr.2016.5369. [DOI] [PubMed] [Google Scholar]
- 17.Han C., Guo L., Yang Y., Guan Q., Shen H., Sheng Y., Jiao Q. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer's disease. Brain Behav. 2020;10:e01627. doi: 10.1002/brb3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y., Hu M., Zhang J., Teng Z.Q., Chen C. A novel mechanism of synaptic and cognitive impairments mediated via microRNA-30b in Alzheimer's disease. EBioMedicine. 2019;39:409–421. doi: 10.1016/j.ebiom.2018.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Liu D., Huang H.Z., Wang Z.H., Hou T.Y., Yang X., Pang P., Wei N., Zhou Y.F., Dupras M.J., et al. A novel MicroRNA-124/PTPN1 signal pathway mediates synaptic and memory deficits in alzheimer's disease. Biol. Psychiatry. 2018;83:395–405. doi: 10.1016/j.biopsych.2017.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Yang K., Feng S., Ren J., Zhou W. Upregulation of microRNA-196a improves cognitive impairment and alleviates neuronal damage in hippocampus tissues of Alzheimer's disease through downregulating LRIG3 expression. J. Cell Biochem. 2019;120:17811–17821. doi: 10.1002/jcb.29047. [DOI] [PubMed] [Google Scholar]
- 21.Gasiorowski K., Brokos B., Leszek J., Tarasov V.V., Ashraf G.M., Aliev G. Insulin resistance in alzheimer disease: p53 and MicroRNAs as important players. Curr. Top Med. Chem. 2017;17:1429–1437. doi: 10.2174/1568026617666170103161233. [DOI] [PubMed] [Google Scholar]
- 22.Chen L., Xu S., Wu T., Shao Y., Luo L., Zhou L., Ou S., Tang H., Huang W., Guo K., et al. Studies on APP metabolism related to age-associated mitochondrial dysfunction in APP/PS1 transgenic mice. Aging (Albany NY) 2019;11:10242–10251. doi: 10.18632/aging.102451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farr S.A., Roesler E., Niehoff M.L., Roby D.A., McKee A., Morley J.E. Metformin improves learning and memory in the SAMP8 mouse model of alzheimer's disease. J. Alzheimers Dis. 2019;68:1699–1710. doi: 10.3233/JAD-181240. [DOI] [PubMed] [Google Scholar]
- 24.Zhao J., Chen Y., Xu Y., Pi G. Effects of PTEN inhibition on the regulation of tau phosphorylation in rat cortical neuronal injury after oxygen and glucose deprivation. Brain Inj. 2016;30:1150–1159. doi: 10.3109/02699052.2016.1161828. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X., Li F., Bulloj A., Zhang Y.W., Tong G., Zhang Z., Liao F.F., Xu H. Tumor-suppressor PTEN affects tau phosphorylation, aggregation, and binding to microtubules. FASEB J. 2006;20:1272–1274. doi: 10.1096/fj.06-5721fje. [DOI] [PubMed] [Google Scholar]
- 26.Wang W., Dong J., Wang M., Yao S., Tian X., Cui X., Fu S., Zhang S. miR-148a-3p suppresses epithelial ovarian cancer progression primarily by targeting c-Met. Oncol. Lett. 2018;15:6131–6136. doi: 10.3892/ol.2018.8110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Lacerda J.Z., Ferreira L.C., Lopes B.C., Aristizábal-Pachón A.F., Bajgelman M.C., Borin T.F., Zuccari D.A.P.C. Therapeutic potential of melatonin in the regulation of MiR-148a-3p and angiogenic factors in breast cancer. Microrna. 2019;8:237–247. doi: 10.2174/2211536608666190219095426. [DOI] [PubMed] [Google Scholar]
- 28.Wang X., Liang Z., Xu X., Li J., Zhu Y., Meng S., Li S., Wang S., Xie B., Ji A., et al. miR-148a-3p represses proliferation and EMT by establishing regulatory circuits between ERBB3/AKT2/c-myc and DNMT1 in bladder cancer. Cell Death Dis. 2016;7:e2503. doi: 10.1038/cddis.2016.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B., Wang W., Li Z., Chen Z., Zhi X., Xu J., Li Q., Wang L., Huang X., Wang L., et al. MicroRNA-148a-3p enhances cisplatin cytotoxicity in gastric cancer through mitochondrial fission induction and cyto-protective autophagy suppression. Cancer Lett. 2017;410:212–227. doi: 10.1016/j.canlet.2017.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie Q., Yu Z., Lu Y., Fan J., Ni Y., Ma L. microRNA-148a-3p inhibited the proliferation and epithelial-mesenchymal transition progression of non-small-cell lung cancer via modulating Ras/MAPK/Erk signaling. J. Cell Physiol. 2019;234:12786–12799. doi: 10.1002/jcp.27899. [DOI] [PubMed] [Google Scholar]
- 31.Zempel H., Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37:721–732. doi: 10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 33.Paudel H.K., Lew J., Ali Z., Wang J.H. Brain proline directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer’s paired helical filaments. J. Biol. Chem. 1993;268:23512–23518. [PubMed] [Google Scholar]
- 34.Lovestone S., Reynolds C.H. The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience. 1997;78:309–324. doi: 10.1016/s0306-4522(96)00577-5. [DOI] [PubMed] [Google Scholar]
- 35.Ferrer I., Gomez-Isla T., Puig B., Freixes M., Ribé E., Dalfóo E., Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 36.Bazrgar M., Khodabakhsh P., Mohagheghi F., Prudencio M., Ahmadiani A. Brain microRNAs dysregulation: implication for missplicing and abnormal post-translational modifications of tau protein in Alzheimer's disease and related tauopathies. Pharmacol. Res. 2020;155:104729. doi: 10.1016/j.phrs.2020.104729. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Q., Liu H., Cheng J., Zhu Y., Xiao Q., Bai Y., Tao J. Neuroprotective effects of lithium on a chronic MPTP mouse model of Parkinson's disease via regulation of alpha-synuclein methylation. Mol. Med. Rep. 2019;19:4989–4997. doi: 10.3892/mmr.2019.10152. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H., Liu W., Ge H., Li K. Aberrant expression of miR-148a-3p in Alzheimer's disease and its protective role against amyloid-β induced neurotoxicity. Neurosci. Lett. 2021;756:135953. doi: 10.1016/j.neulet.2021.135953. [DOI] [PubMed] [Google Scholar]
- 39.Xu J., Sun M., Li X., Huang L., Gao Z., Gao J., Xie A. MicroRNA expression profiling after recurrent febrile seizures in rat and emerging role of miR-148a-3p/SYNJ1 axis. Sci. Rep. 2021;11:1262. doi: 10.1038/s41598-020-79543-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai Y., Lang L., Zhao W., Niu R. Long non-coding RNA HOXA11-AS promotes non-small cell lung cancer tumorigenesis through microRNA-148a-3p/DNMT1 regulatory Axis. Onco Targets Ther. 2019;12:11195–11206. doi: 10.2147/OTT.S198367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashizawa M., Okayama H., Ishigame T., Thar Min A.K., Saito K., Ujiie D., Murakami Y., Kikuchi T., Nakayama Y., Noda M., et al. miRNA-148a-3p regulates immunosuppression in DNA mismatch repair-deficient colorectal cancer by targeting PD-L1. Mol. Cancer Res. 2019;17:1403–1413. doi: 10.1158/1541-7786.MCR-18-0831. [DOI] [PubMed] [Google Scholar]
- 42.Mapelli M., Massimiliano L., Crovace C., Seeliger M.A., Tsai L.H., Meijer L., Musacchio A. Mechanism of CDK5/p25 binding by CDK inhibitors. J. Med. Chem. 2005;48:671–679. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- 43.Kimura T., Ishiguro K., Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao L., Xiao H., Ai L.Q., Chen C., Lin S., Zhou Y., Ye J., Liu W. Vps35 deficiency impairs cdk5/p35 degradation and promotes the hyperphosphorylation of tau protein in retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2020;61:1. doi: 10.1167/iovs.61.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai S.W., Han Y., Wang G.P. miR-148a-3p exhaustion inhibits necrosis by regulating PTEN in acute pancreatitis. Int. J. Clin. Exp. Pathol. 2018;11:5647–5657. [PMC free article] [PubMed] [Google Scholar]
- 46.He H., Cai M., Zhu J., Xiao W., Liu B., Shi Y., Yang X., Liang X., Zheng T., Hu S., et al. miR-148a-3p promotes rabbit preadipocyte differentiation by targeting PTEN. In Vitro Cell. Dev. Biol. Anim. 2018;54:241–249. doi: 10.1007/s11626-018-0232-z. [DOI] [PubMed] [Google Scholar]
- 47.Qingjuan L., Xiaojuan F., Wei Z., Chao W., Pengpeng K., Hongbo L., Sanbing Z., Jun H., Min Y., Shuxia L. miR-148a-3p overexpression contributes to glomerular cell proliferation by targeting PTEN in lupus nephritis. Am. J. Physiol. Cell Physiol. 2016;310:C470–C478. doi: 10.1152/ajpcell.00129.2015. [DOI] [PubMed] [Google Scholar]
- 48.Huang F., Zhao J.L., Wang L., Gao C.C., Liang S.Q., An D.J., Bai J., Chen Y., Han H., Qin H.Y. miR-148a-3p mediates Notch signaling to promote the differentiation and M1 activation of macrophages. Front. Immunol. 2017;8:1327. doi: 10.3389/fimmu.2017.01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rai S.N., Dilnashin H., Birla H., Singh S.S., Zahra W., Rathore A.S., Singh B.K., Singh S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019;35:775–795. doi: 10.1007/s12640-019-0003-y. [DOI] [PubMed] [Google Scholar]
- 50.Robbins H.L., Hague A. The PI3K/Akt pathway in tumors of endocrine tissues. Front Endocrinol. (Lausanne) 2016;6:188. doi: 10.3389/fendo.2015.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng R., Zhang Z.H., Chen C., Chen Y., Jia S.Z., Liu Q., Ni J.Z., Song G.L. Selenomethionine promoted hippocampal neurogenesis via the PI3K-Akt-GSK3beta-Wnt pathway in a mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017;485:6–15. doi: 10.1016/j.bbrc.2017.01.069. [DOI] [PubMed] [Google Scholar]
- 52.Moosavi F., Hosseini R., Saso L., Firuzi O. Modulation of neurotrophic signaling pathways by polyphenols. Drug Des. Devel. Ther. 2015;10:23–42. doi: 10.2147/DDDT.S96936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi J. Regulatory networks between neurotrophins and miRNAs in brain diseases and cancers. Acta Pharmacol. Sin. 2015;36:149–157. doi: 10.1038/aps.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeng L., Jiang H.L., Ashraf G.M., Li Z.R., Liu R. MicroRNA and messenger RNA profiling of cerebral cortex in a transgenic mouse model of Alzheimer’s disease by RNA sequencing. Neural Regen. Res. 2021;16:2099–2108. doi: 10.4103/1673-5374.308104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Velagapudi R., Ajileye O.O., Okorji U., Jain P., Aderogba M.A., Olajide O.A. Agathisflavone isolated from Anacardium occidentale suppresses SIRT1-mediated neuroinflammation in BV2 microglia and neurotoxicity in APPSwe-transfected SH-SY5Y cells. Phytother. Res. 2018;32:1957–1966. doi: 10.1002/ptr.6122. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J., Liu Q., Chen Q., Liu N.Q., Li F.L., Lu Z.B., Qin C., Zhu H., Huang Y.Y., He W., et al. Nicotine attenuates beta-amyloid-induced neurotoxicity by regulating metal homeostasis. FASEB J. 2006;20:1212–1214. doi: 10.1096/fj.05-5214fje. [DOI] [PubMed] [Google Scholar]
- 57.Liu R., Meng F., Zhang L., Liu A., Qin H., Lan X., Li L., Du G. Luteolin isolated from the medicinal plant Elsholtzia rugulosa (Labiatae) prevents copper-mediated toxicity in β-amyloid precursor protein Swedish mutation overexpressing SH-SY5Y cells. Molecules. 2011;16:2084–2096. doi: 10.3390/molecules16032084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinchuk V., Wu Y., Grossenbacher-Zinchuk O. Bridging the gap between qualitative and quantitative colocalization results in fluorescence microscopy studies. Sci. Rep. 2013;3:1365. doi: 10.1038/srep01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McClean P.L., Parthsarathy V., Faivre E., Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J. Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamaguchi H., Shen J. Histological analysis of neurodegeneration in the mouse brain. Methods Mol. Biol. 2013;1004:91–113. doi: 10.1007/978-1-62703-383-1_8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.