

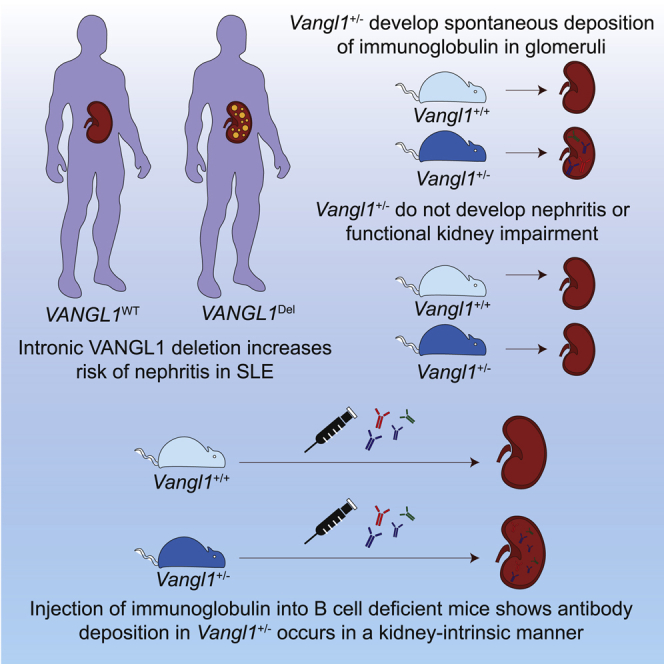
Summary
We identify an intronic deletion in VANGL1 that predisposes to renal injury in high risk populations through a kidney-intrinsic process. Half of all SLE patients develop nephritis, yet the predisposing mechanisms to kidney damage remain poorly understood. There is limited evidence of genetic contribution to specific organ involvement in SLE.1,2 We identify a large deletion in intron 7 of Van Gogh Like 1 (VANGL1), which associates with nephritis in SLE patients. The same deletion occurs at increased frequency in an indigenous population (Tiwi Islanders) with 10-fold higher rates of kidney disease compared with non-indigenous populations. Vangl1 hemizygosity in mice results in spontaneous IgA and IgG deposition within the glomerular mesangium in the absence of autoimmune nephritis. Serum transfer into B cell-deficient Vangl1+/− mice results in mesangial IgG deposition indicating that Ig deposits occur in a kidney-intrinsic fashion in the absence of Vangl1. These results suggest that Vangl1 acts in the kidney to prevent Ig deposits and its deficiency may trigger nephritis in individuals with SLE.
Keywords: glomerulonephritis, antibody, immunoglobulin, lupus nephritis, genetic, autoimmune, chronic kidney disease
Graphical abstract
Highlights
-
•
Intronic deletions in VANGL1 are associated with risks of glomerulonephritis in SLE
-
•
Vangl1+/− mice develop spontaneous deposition of immunoglobulin in glomeruli
-
•
Vangl1+/− mice do not develop glomerulonephritis despite antibody deposition
-
•
Immunoglobulin deposition in Vangl1+/− occurs in a kidney-intrinsic manner
The organ specificity risks of lupus nephritis are unclear. Jiang et al. identify a common intronic deletion in VANGL1 that increases risks of nephritis in SLE patients. Vangl1+/− mice develop spontaneous deposition of immunoglobulin in the kidney without glomerulonephritis in a kidney intrinsic manner.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease and end-organ damage is thought to result from deposition of autoantibodies. Although clinical presentation varies considerably between individuals,3,4 kidney involvement is strongly associated with adverse effects on mortality and morbidity among SLE patients.5,6 Lupus nephritis (LN) occurs in 21%–58%7,8 of SLE patients and manifests as several distinct though often overlapping histopathologic lesions.9 The mechanisms promoting different organ involvement between SLE individuals remain unclear although ethnicity,7,8 specific autoantibodies,10,11 autoantigens within affected organs12,13 and HLA14, 15, 16 have been implicated.
Genetic variation is a potent risk factor for development of SLE17 and may also influence organ-specific involvement18,19. Most reported variants predisposing to SLE are either single nucleotide variations20 (SNV) or deletions21,22 in genes with important roles in the immune system. It is increasingly recognized that relatively large structural genetic changes comprising insertions, deletions, and duplications of >1,000 base pairs called copy number variants (CNVs) represent a sizable proportion of individual genetic variability.23 Unsurprisingly, CNV in genes primarily involved in immune function have also been increasingly implicated in SLE pathogenesis.24, 25, 26
Van Gogh Like 1 (VANGL1) and Van Gogh Like 2 (VANGL2) are genes essential in the establishment of planar cell polarity (PCP). Both genes are highly conserved in vertebrates and influence PCP through interaction with several core pathway PCP genes such as Dishevelled (DVL1), Flamingo/Starry night (FMI/STAN), Prickle (PK) and Diego (DGO).27 Like most PCP genes, the VANGL are important for neural tube development.28 SNV in the murine ortholog Vangl2 results in the loop-tailed mouse model of neural tube defects (NTD),29 and SNV in VANGL1 and VANGL2 are associated with human NTD.30,31 In addition to their role in neural development, VANGL genes also regulate kidney development. Deficiency of Vangl2 in mice impairs kidney organogenesis,32 glomerular maturation, and development and repair response to glomerular injury.33 Podocyte-specific deletion of Vangl2 enhanced injury in experimental nephritis.34 Despite this known contribution of VANGL2 to kidney development, neither VANGL1 nor VANGL2 have yet been associated with human kidney disease. Here we identify a recurrent deletion in VANGL1 that predisposes to Ig deposition in the glomerulus and is associated with lupus nephritis.
Results
Copy number variation in VANGL1 associates with nephritis in SLE
To determine the role of CNV in SLE we performed a genome-wide SNV association using Affymetrix 5.0 SNP chip arrays in a cohort of SLE patients selected according to disease severity (n = 55), Sjogren’s syndrome (n = 11) and healthy controls (n = 11) (Table 1). SLE patients were classified according to the 1997 American College of Rheumatology guidelines,3 and qualitatively chosen for quantity or severity of system involvement. The Affymetrix 5.0 array has ∼500,000 SNP probes and ∼420,000 CNV probes. Array data were analyzed using R statistical language package CRNA (v2).35 All 77 arrays passed QC checks. After filtering, a total of 982 CNVs were retained for all 77 samples (Supp Data). Several CNVs were detected in more than one individual, including three SLE individuals with CNVs in VANGL1. Of the three SLE patients, two had nephritis. No Sjogren’s Sd patients or healthy controls had the CNV. The initial array analysis suggested all three SLE patients had the same double deletion of an estimated 3.17 kb of this gene spanning chr1:116,030,911–116,034,081 (NCBI36/hg18 assembly) within intron 7. This finding was supported by only two SNPs on the array but was consistent with the small size of the CNV. To confirm these findings, we performed Taqman qPCR targeting the CNV region identified in intron 7 of VANGL1 in a larger cohort of SLE patients (n = 177) (Figure 1A). In this larger SLE cohort, 18 were homozygous, 41 were heterozygous, and 119 did not have the CNV. Overall, the MAF of the CNV in the SLE cohort was similar to that of the global gnomAD frequency (MAF 0.28 versus 0.25, χ2 p = 0.7). However, we observed an association between the VANGL1 CNV and the presence of nephritis in SLE patients (χ2 = 27.06, 2 d.f., p < 0.0001). Furthermore, the correlation between 0, 1 or 2 VANGL1 copies and nephritis suggested a gene-dose increase in nephritis risk with the VANGL1 CNV. Indeed, the VANGL1 0 copies CNV was observed at twice the frequency in patients with nephritis compared to patients without nephritis (minor allele frequency (MAF) of 0.39 versus 0.17 respectively). The qPCR was also repeated in a third cohort (n = 281) and this revealed a possible, albeit non-statistically significant, trend between CNV in VANGL1 and nephritis in SLE patients (Figure S1) (χ2 = 2.1, 1 d.f., p = 0.14).
Table 1.
Demographics of patient cohorts
| Clinical | HC n = 11 | SS n = 11 | SLE 1 n = 55 | SLE 2 n = 177 | SLE 3 n = 281 |
|---|---|---|---|---|---|
| Age (years) (IQR) | 57 (56-66 years) | 48 (45-51 years) | 50 (35-60 years) | 47 (35-60 years) | |
| Gender (female) (%) | 6 (55%) | 11 (100%) | 49 (89%) | 148 (83%) | 253 (90%) |
| Oral ulcers (%) | 0 | 0 | 13 (24%) | 53 (30%) | 73 (26%) |
| Arthritis (%) | 0 | 0 | 38 (69%) | 122 (69%) | 204 (72%) |
| Raynaud’s (%) | 0 | 0 | 19 (35%) | 60 (34%) | Unknown |
| Cutaneous (%) | 0 | 0 | 30 (55%) | 99 (56%) | 153 (54%) |
| Nephritis (%) | 0 | 0 | 23 (42%) | 62 (35%) | 113 (40%) |
| Serositis (%) | 0 | 0 | 15 (27%) | 41 (23%) | 91 (32%) |
| Alopecia (%) | 0 | 0 | 7 (13%) | 39 (22%) | Unknown |
| Seizure (%) | 0 | 0 | 9 (16%) | 20 (11%) | 9 (3%) |
| Sicca (%) | 0 | 11 (100%) | 15 (27%) | 62 (35%) | Unknown |
| Myositis (%) | 0 | 0 | 0 | 7 (4%) | Unknown |
| Cytopaenia (%) | 0 | 0 | 28 (51%) | 76 (43%) | 178 (63%) |
| Antiphospholipid Ab (%) | 0 | 0 | 27 (49%) | 78 (44%) | Unknown |
| ANAs (%) | 0 | 10 (91%) | 55 (100%) | 177 (100%) | 257 (91%) |
| dsDNAs (%) | 0 | 0 | 44 (80%) | 112 (63%) | 159 (56%) |
| Anti-Sm (%) | 0 | 0 | 8 (15%) | 21 (12%) | 31 (11%) |
| Hypocomplementemia (%) | 0 | 0 | 36 (65%) | 76 (43%) | Unknown |
Figure 1.

Intronic deletions in VANGL1 associate with kidney disease
(A) Association of copy number variation in VANGL1 detected by qPCR with the presence or absence of nephritis in SLE (n = 177); χ2 2d.f., p < 0.0001 (LN = lupus nephritis).
(B) Comparison of MAF of VANGL1 CNV in esv3587290 in gnomAD global and Tiwi Islander populations (n = 120); Fisher’s exact, p < 0.0001).
(C) Representative whole genome sequencing reads of intronic deletion in VANGL1 of the maximum length reported for the CNV.
(D) Comparison of relative VANGL1 expression in PBMCs from (i) SLE patients with 0 or 2 copies of VANGL1, (ii) Healthy controls or SLE patients with or without lupus nephritis.
e) RNaseq read alignment demonstrating skipping of exon 2 in a patient homozygous for the VANGL1 CNV with read covering exon 1 and exon 3 highlighted in yellow.
Gene variants with a large difference in risk allele frequency between populations are strong candidates to explain variations in disease.38 We hypothesized that if the VANGL1 CNV predisposes to kidney disease, it may be more prevalent in populations at high risk of kidney disease. The Tiwi Islanders are an Australian Aboriginal group with rates of kidney disease 10 times greater than the non-Aboriginal population.39 In a large Tiwi Island cohort (n = 120) with an even gender distribution of 50% males and females and a median age of 40.8 years (IQR 12.7 years) who underwent whole genome sequencing (WGS), a large CNV in intron 7 of VANGL1 (esv3587290) similar to the CNVs found in patients with SLE nephritis, was identified at a frequency significantly higher compared to the global gnomAD frequency (Figure 1B, MAF 0.43 versus 0.25, χ2 p < 0.0001) and all other racial groups (Figure S2A). We explored the association between the CNV and severity of kidney disease in Tiwi. We did not observe a significant trend in later stages of CKD within patients with the CNV compared to those without (Figure S2B). When comparing Tiwi Islanders with the VANGL1 CNV compared to those without, there was no significant difference in serum creatinine (72.0 versus 65.0 μmol/L, p value 0.28) or urinary albumin-to-creatinine ratio (31.4 versus 0.9 mg/mmol, p value 0.56). Histological data are unavailable for this cohort and therefore the extent of immunoglobulin deposition or autoimmunity as a cause of chronic kidney disease could not be determined. Together, this suggests the intronic CNV in VANGL1 may predispose to the development of kidney disease, but not impact the progression of kidney damage.
To characterize the CNV in VANGL1, we first examined the Database of Genomic Variants which identifies 15 of 26 (57%) reported VANGL1 CNV located within intron 7 of VANGL1.40 These CNVs were predominantly large deletions of variable size indicating that this is a recurrent CNV region. We performed WGS in SLE patients with and without the deletion, confirming the presence and variability in size of the CNV (Figure 1c). We first tested if this CNV influenced VANGL1 expression in peripheral blood mononuclear cells (PBMCs) from SLE patients by RT-qPCR. We found no difference in exon 3 and 4 VANGL1 expression between those that had been identified with 0 CNV and 2CNV (Figure 1D) nor between healthy controls and SLE patients with or without nephritis (Figure 1D).
To detect additional splicing abnormalities potentially associated with the deletion that would not result in changes to the coding region of VANGL1, we performed RNaseq from PBMCs of patients homozygous for the VANGL1 deletion (n = 6) or lacking the deletion (n = 4). Interestingly, in two of the six patients homozygous for the deletion we observed several reads skipping or alternatively splicing exon 2, and therefore predicted to result in loss of the native VANGL1 start codon (Figure 1E). We hypothesized that this may be due to presence of a cryptic splice site within intron 7. This could either lead to translation from the next in-frame ATG start site generating a truncated protein lacking the first 98 amino acids or use an alternative out-of-frame ATG start site that would putatively encode for a different peptide and thus reduce the amount of VANGL1 protein expressed. VANGL1 protein is expressed at low levels in PBMCs, and therefore, we were not able to test for differences in the molecular weight of VANGL1 that may occur with loss of exon 1. Therefore, kidney biopsies from patients heterozygous (n = 4) or wild-type (n = 4) for the VANGL1 CNV were stained for VANGL1 using a polyclonal antibody raised against either the c-terminus (Thermofisher PA5-98739) or n-terminus (Thermofisher PA5-55231) of VANGL1 and scored by a nephropathologist for tissue expression. No obvious difference in protein expression intensity or pattern using the available polyclonal antibody was observed in heterozygous individuals (Figure S3). VANGL1 was detected primarily in tubular epithelium with some glomerular staining (Figure S3). Unfortunately, we did not have biopsy samples from patients homozygous for the CNV to measure significant impact on protein expression. Thus, recurrent intronic deletions in VANGL1 are associated with the predisposition to nephritis in SLE patients, potentially affecting RNA splicing in some patients.
Vangl1+/− mice develop spontaneous deposition of immunoglobulin in the kidney
Mice deficient in Vangl2, the other Vangl family member, have reduced glomerular numbers and size, and impaired kidney morphogenesis.32 Recent work has demonstrated a role for Vangl2 in directing podocyte repair and Vangl2 deficiency within podocytes was associated with impaired recovery after acute glomerular injury and a predisposition to focal segmental glomerulosclerosis.33 VANGL1 is highly expressed in human CD34+ and CD105+ cells, which includes endothelial cells, some T cells, and monocytes.41 We hypothesized that given the role of Vangl2 in kidney development and repair, and the functional interaction between the Vangl1 and Vangl2 in other tissues,42 the observed VANGL1 CNV may predispose to SLE nephritis in a kidney-intrinsic manner. We first examined expression of Vangl1 in murine kidney. Using immunohistochemistry, we stained kidney sections from 10-week-old adult C57BL/6 mice. Similar to the observations made of Vangl2, Vangl1 was detected largely within tubules but not within the glomeruli (Figure 2A). One possibility is that Vangl1 and Vangl2 are expressed transiently in glomeruli during development and repair.33 To determine if Vangl1 had a prominent role in glomerular development similar to Vangl2, we stained sections of D18 fetal kidneys from C57BL/6 mice but did not detect Vangl1 in these sections (data not shown).
Figure 2.

Vangl1 deficiency causes immunoglobulin deposition
(A) Immunohistochemistry demonstrating Vangl1 (brown) in kidney sections from 3-month-old wild-type C57BL/6 mice (n = 5).
(B) Representative H&E section of a D12 Vangl1−/− fetus demonstrating neural tube defects (n = 6).
(C) Immunofluorescence of immunoglobulin A or G (green), podocin (red) and DAPI in 8-week-old Vangl1+/− and Vangl1+/+ mice (n = 6/group) median, Mann-Whitney U, p < 0.05. (D) Electron microscopy demonstrating electron dense deposits consistent with immunoglobulin in 8-week-old Vangl1+/− mice (n = 6/group).
To test the consequences of Vangl1 deficiency in vivo, we obtained Vangl1 knockout mice (KOMP repository) and rederived them onto a C57BL/6 background. Vangl1 deficiency was embryonically lethal between D15 and D18 due to neural tube defects (Figure 2B). This was surprising, as published observations indicated that mice homozygous for a gene-trapped Vangl1 allele did not develop NTD, but additively worsened NTD associated with Vangl2 deficiency.42 We examined kidneys from mice lacking a single allele of Vangl1 (Vangl1+/−). Given the association of Vangl2 deficiency with impaired kidney morphogenesis we investigated if Vangl1 deficiency affects organogenesis. By indirect immunofluorescence, however, we observed spontaneous deposition of IgG and traces of IgA in glomeruli (Figure 2C), but not IgM, C3 or C4 (data not shown). Electron microscopy demonstrated significant Ig deposits within the mesangium and mild mesangial expansion (Figure 2D). We compared the IF patterns in the Vangl1 mice with biopsy reports that were available from SLE patients (who comprised part of the n = 177 SLE cohort) with biopsy-confirmed lupus nephritis and in whom we confirmed were wild-type or homozygous for the CNV but did not detect a significant difference in IF profiles (Table 2). Interestingly, by light microscopy, Vangl1+/− glomeruli appeared normal despite immunoglobulin (Ig) deposition (Figure 3A). We did not observe a significant difference in kidney size, glomeruli size, or glomeruli numbers/light field between Vangl1+/− and Vangl1+/+ littermates (Figure 3B). To ascertain the clinical consequences of Ig deposition, serum creatinine and albuminuria were measured at 6 and 9 months of age and no difference between Vangl1+/+ or Vangl1+/− mice was detected (Figure 3C). Considering the association of Vangl2 with tissue repair, we considered Vangl1 and Vangl2 may also be expressed during autoimmune glomerulonephritis. Lyn−/− mice develop proliferative glomerulonephritis by as early as 6 weeks of age (Figure S4). We stained kidney samples from 8-week-old lupus-prone Lyn−/− C57BL/6 mice with glomerulonephritis for Vangl1 and Vangl2. Interestingly, staining for Vangl1 and Vangl2 in proliferative glomeruli demonstrated scattered positivity in the glomeruli (Figure S4). Therefore, Vangl1+/− mice develop spontaneous mesangial immunoglobulin deposition with no evidence of inflammation or impairment of kidney function.
Table 2.
Immunofluorescence reports from kidney biopsies from patients wild type or homozygous for the VANGL1 CNV
| 2 CNV |
0 CNV |
||||||
|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | |
| IgG | 2+ | Trace | 3+ | 3+ | 2+ | 3+ | 2+ |
| IgM | Trace | 1+ | 1+ | Neg | 3+ | 1+ | 1+ |
| IgA | 1+ | Trace | 2+ | 1+ | 1+ | 2+ | 1+ |
| C3 | Trace | 2+ | 1+ | 3+ | 3+ | 3+ | trace |
| C1q | 1+ | Neg | 2+ | 3+ | 1+ | 1+ | neg |
Figure 3.

Vangl1+/− does not alter glomerular development
(A) Representative H&E stain of glomerular sections from 8-week-old Vangl1+/− and Vangl1+/+ (n = 6/group).
(B) Comparison of kidney volume, glomerular size and glomeruli/light field in 8-week-old mice of indicated genotype.
(C) Serum creatinine and urine albumin in 16-week-old Vangl1+/− and Vangl1+/+ mice.
(D) ANA from 16-week-old mice of indicated genotype
(E) Serum immunoglobulin of 12-month-aged mice of indicated genotype. Data are represented as median throughout.
Vangl1+/− immunoglobulin deposition occurs in a kidney-intrinsic manner
To determine the potential cause of immunoglobulin deposition, we first tested whether antibody deposition was related to increased systemic immunoglobulin production or was a manifestation of systemic autoimmunity. Flow cytometric examination of mouse splenocytes did not demonstrate significant differences in lymphocyte populations, including B cell subsets (data not shown). We considered that the Ig deposits could represent either a qualitative or quantitative abnormality of antibodies in Vangl1+/− mice, yet at 20 weeks of age there was no difference in detectable anti-nuclear antibodies (ANAs) (Figure 3D) or quantitative difference in any class of Ig (Figure 3E) produced by Vangl1+/− mice.
We next hypothesized that glomerular immunoglobulin deposition in Vangl1+/− mice is due to a kidney-intrinsic predisposition given detectable expression of Vangl1 protein in murine kidneys and the absence of systemic autoimmunity in Vangl1+/− mice. To test this, we crossed Vangl1+/− mice to C57BL/6 CD79aken/ken mice. CD79aken/ken have an N-ethyl N-nitrosourea (ENU)-induced premature stop codon in CD79a, resulting in impaired B cell signaling with complete failure of mature B cell formation, and are therefore unable to produce circulating immunoglobulin43. Murine IgG was injected intravenously daily for 5 days into both Vangl1+/−CD79aken/ken and Vangl1+/+CD79aken/ken mice. At day 6, mice were sacrificed and kidneys were examined for immunoglobulin deposition (Figure 4A). IgG was present in Vangl1+/−CD79aken/ken mouse kidneys but not in those of Vangl1+/+CD79aken/ken littermates (Figures 4B and 4C). We considered the possibility that the kidney-intrinsic predisposition to immunoglobulin deposition could increase the risk of glomerulonephritis when this is associated with autoreactive antibodies. We transferred serum from Lyn−/− mice in whom we confirmed the presence of proliferative nephritis (Figure S4) into Vangl1+/− or Vangl1+/+ littermates and injected at day 0 and day 4 with 200ul of serum as described previously with nephrotoxic serum.44 Mice were sacrificed at day 7 and kidneys preserved for histology. Three of six Vangl1+/− mice injected with Lyn−/− serum had mild segmental glomerulonephritis, whereas no Vangl1+/+ mice developed glomerulonephritis, suggesting a vulnerability to nephritis with autoreactive serum (Figure 4D). This result demonstrates that susceptibility to Ig deposition in Vangl1+/− is kidney-intrinsic and implies that one of Vangl1’s roles is to prevent immunoglobulin deposition in the kidney.
Figure 4.

Immunoglobulin occurs in a kidney intrinsic manner
(A) Schematic demonstrating experiment design testing kidney-intrinsic predisposition to kidney disease.
(B) Immunofluorescence of immunoglobulin G (red), podocin (green) and DAPI in cryosections from 8-week-old mice of B cell deficient Vangl1+/− and Vangl1+/+ mice injected with IgG.
(C) Scoring of immunofluorescent IgG deposition in mice post injection. Data are represented as median, Mann-Whitney U, p = 0.02.
Discussion
While SLE has the capacity to involve most organs, kidney involvement is common, important, and often defines disease outcomes. Here, we demonstrate a kidney-intrinsic genetic disposition to renal involvement in SLE. VANGL1 and VANGL2 are important PCP genes that have been implicated in human NTD30 and Vangl2 has been shown to play a role in kidney development and repair after kidney injury.33 Although loss of Vangl2 in mouse models results in fatal NTD, conditional loss of Vangl2 in podocytes results in impaired glomerular repair and greater injury.33 We identified a CNV in intron 7 of VANGL1, which in individuals with SLE associates with lupus nephritis in a gene-dose protective manner and is also highly prevalent in Tiwi Islanders who have a high rate of kidney disease. Interestingly, the CNV identified does not represent a single deletion but several deletions within the same intron of differing sizes in the kilobase range. In 2 of 6 patients homozygous for the deletion in intron 7 of VANGL1 CNV, RNaseq of PBMCs identified read skipping of exon 2 which encodes the start codon. The mechanism by which the VANGL1 deletion contributes to exon 2 skipping remains unclear in these individuals; however, we hypothesize this is likely to either reduce protein expression or produce a shorter or different protein. It is therefore possible that at least in a proportion of individuals with VANGL1 CNVs, there is reduced full-length VANGL1 transcript. Whether VANGL1 differential splicing varies according to cell type or organ remains unknown.
To determine the contribution of VANGL1 CNV to the development of nephritis, we examined the Vangl1+/− mouse. Although not immediately comparable with the intronic CNV in the SLE cohorts, Vangl1-deficient mice permitted examination of reduced expression of Vangl1 in the kidneys. Strikingly, Vangl1-deficient mice developed spontaneous IgA and IgG deposition in their mesangium, but IgM and complement were not present. We hypothesize that this pattern of Ig deposition may be due to disturbed endothelial expression of Vangl1 permitting passive diffusion of immunoglobulin across the endothelium of glomerular capillaries. This would explain why the monomeric immunoglobulins IgA and IgG, but not the larger pentameric IgM, are detected in the mesangium of Vangl1+/− mice. Furthermore, despite deposition of monomeric immunoglobulins in Vangl1+/− mice, there were no complement deposits nor evidence of inflammation or glomerular injury. We attribute this to the immunoglobulins deposited in Vangl1+/− mice are not autoreactive or crosslinked, and it is only in the presence of autoantibodies, such as in SLE, that antigen-antibody complexes occur, which activate complement and induce an inflammatory reaction. Indeed, the requirement for autoreactive antibody to cause pathology would permit the relatively common frequency of functional CNVs within Vangl1 from an evolutionary perspective. We hypothesize that autoreactivity of antibodies depositing in human glomeruli accounts for the difference in complement findings between Vangl1+/− mice and human SLE biopsies. In Vangl1+/− mice, autoantibodies are not autoreactive, and therefore do not form autoantibody-antigen complexes nor activate the classical complement pathway, whereas autoantibodies depositing in the kidneys of SLE patients are either precomplexed or complex with autoantigen in situ, triggering both classical and alternate complement activation with subsequent deposition. Vangl1 is known to play a role in glomerular development and is expressed in murine podocytes, consistent with glomerular and tubular staining we observed.32 We hypothesize that deficiency of Vangl1 impairs either development of glomerular or podocyte function, resulting in impaired clearance of antibody, leading to accumulation. This is consistent with accumulation of antibody in kidneys of B cell-deficient Vangl1+/− when immunoglobulin is administered directly. We hypothesize that when the antibodies that accrue are autoreactive or antibody-antigen complex, the inflammatory reaction results in glomerulonephritis.
Several important lines of further investigation remain. It is unclear how intronic CNVs in VANGL1 translate to kidney-specific injury in the absence of NTD observed in mice. Complete deficiency of VANGL1 should result in high rates of neural tube defects and therefore be under evolutionary pressure. However, the relatively high frequency of CNVs in intron 7 of VANGL1 would suggest limited selectivity against these CNVs, perhaps even balancing selection where the heterozygous deletion may confer some selective advantage for a hitherto unknown trait. Although many antibody-mediated kidney diseases can be associated with specific antibodies and epitopes, the kidney-intrinsic defect from VANGL1 deficiency may predispose to any antibody-mediated kidney disease including infection associated forms of glomerulonephritis. The precise mechanism through which VANGL1 deficiency permits this antibody deposition in such a kidney-intrinsic manner requires further elucidation.45 While in our initial cohort we observed a clear gene-dose risk of developing nephritis associated with the VANGL1 deletion, in our replication cohort we observed only a trend. One explanation may arise from the initial cohort being comprised of majority European ethnicity whereas the replication cohort were largely Spanish and therefore differences in ethnicity may account for the observed discrepancy. This raises the possibility that our findings in human SLE may be an issue predominantly for Caucasian patients compared with other ethnicities. It would be useful to test the effect of the variation in additional cohorts of different ethnicities.
The Tiwi Islanders are a population that suffer one of the highest rates of kidney disease anywhere in the world.45 The cause of kidney failure in this community may be a combination of immune mediated kidney disease, including antibody mediated postinfectious nephritis, with metabolic disease such as diabetes and hypertension.46 The exact role of VANGL1 in the different types of kidney disease and the effect on prognosis remains to be elucidated.
A heritable kidney-intrinsic predisposition to glomerulonephritis also has implications for live-related kidney donation. It may be advisable to perform paired-kidney exchange rather than related donation in circumstances where a heritable predisposition within the kidney itself is present. In summary, we have described how a common CNV in VANGL1 predisposes toward nephritis in a kidney-intrinsic manner. This offers insights into how SLE may develop predilection for specific tissues, as well as vulnerability to other forms of antibody-mediated kidney disease. The work highlights the evolving role of VANGL1 and other PCP genes in kidney injury and repair, which may have important implications for therapeutics and transplantation.
Limitations of the study
The SLE cohorts in whom the VANGL1 deletion was identified were predominantly of European ethnicity, and thus the prevalence and contribution of the deletion to nephritis in non-European groups requires exploration. Variants in VANGL1 are linked to NTD and Vangl1−/− mice are embryonically lethal from NTD, and thus it remains unclear why homozygotes for the VANGL1 deletion do not develop these defects. The precise mechanism through which Vangl1 deficiency permits antibody deposition also requires elucidation. Further, we did not correlate the deletion with specific forms of glomerulonephritis, and it would be useful to determine the effect of the VANGL1 deletion on specific histopathologic forms of glomerulonephritis.
STAR★Methods
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| IgG | Invitrogen | A28175; RRID:AB_2536161 |
| IgA | Southern Biotech | 1040-02; RRID:AB_2794370 |
| IgM | BD PharMingen | 555782; RRID:AB_396117 |
| C3 | MP Biomedicals | SKU 0855500; RRID:AB_2334931 |
| Vangl1 | Sigma | SAB4503254; RRID:AB_10748043 |
| VANGL1 | ThermoFisher | PA5-55231; RRID:AB_2649388 |
| VANGL1 | ThermoFisher | PA5-98739; RRID:AB_2813352 |
| VANGL2 | ThermoFisher | PA5-23207; RRID:AB_2540733 |
| Biological samples | ||
| Human PBMCs | This study | N/A |
| Human kidney samples | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| MiScript RT Kit | QIAGEN | 218160 |
| SYBR Green | QIAGEN | 204143 |
| Tissue-Tek O.C.T. | Sakura | 4583 |
| 2% osmium tetroxide | Electron Microscopy Sciences, USA | 19100 |
| Cacodylate buffer | This study | N/A |
| TAAB epoxy | TAAB Laboratory and Microscopy, U.K | E049 |
| Critical commercial assays | ||
| Affymetrix 5.0 SNP Chip | Affymetrix USA | #901167 |
| Taqman CN Assay | Applied Biosystem, USA | Hs05725015_cn |
| ANA ELISA | Alpha diagnostic | 5210 |
| Total serum immunoglobulin | BD Bioscience | 550487 |
| Indiko Creatinine-detect | ThermoFisher | 10015638 |
| Albumin ELISA kit | abcam | Ab108792 |
| Deposited data | ||
| RNA sequences | This study | GEO: GSE188480 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Charles River | C57BL/6 |
| Vangl1+/− | KOMP | EPD0164_3_G07 |
| CD79aKen | Australian phenomics facility | N/A |
| Lyn−/− | Australian phenomics facility | N/A |
| Oligonucleotides | ||
| Vangl1 exon three F’ | Integrated DNA Technologies | GACACAAGTCACCCCGGAATA |
| Vangl1 exon three R’ | Integrated DNA Technologies | TCCTCTGTCCGAGTAGAATCATT |
| Vangl1 exon four F’ | Integrated DNA Technologies | CCGATCCTGTGGAGGGATGA |
| Vangl1 exon four R’ | Integrated DNA Technologies | AAACACCCGTGGCATGTCA |
| Software and algorithms | ||
| Graphpad Rism 8.0 | GraphPad Software CA, USA | N/A |
| FlowJo | Tristar Inc, Stanford, US | N/A |
| RNA sequencing annotation | M Field | 10.1371/journal.pone.0143199 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Carola Vinuesa (carola.vinuesa@crick.ac.uk).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Vangl1−/− mice were created from the ES cell clone (EPD0164_3_G07) obtained from the supported KOMP Repository (www.komp.org) and generated by the Wellcome Trust Sanger Institute using CSD targeted alleles as described previously.47 Vangl+/−, Lyn−/−, and CD79a−/− mice were re-derived onto a C57BL/6 background and housed in sterile conditions. Equal numbers of male and female 12 week old mice were used unless otherwise specified and there was no gender bias in Vangl+/− mice. All mouse experiments were approved by the Institutional Ethics Committee at the Australian National University. Human subjects had written informed consent obtained as part of the Australian Point Mutation in Systemic Lupus Erythematosus study (APOSLE). The study was approved by the Australian National University and ACT Health Human Ethics Committees. Age and demographics are described in Table 1.
Method details
Study participants, saliva purification, and CNV analysis
Written informed consent was obtained as part of the Australian Point Mutation in Systemic Lupus Erythematosus study (APOSLE). The study was approved by the Australian National University and ACT Health Human Ethics Committees. Saliva was collected in Oragene DNA collection kits and purified using PrepIT DNA purification kits (Oragene) as per manufacturer’s instructions. SLE patients were classified according to 1997 revision of the American College of Rheumatology.3 Initial patients were qualitatively chosen for quantity or severity of system involvement as reported by referring clinicians but were not assessed by SLEDAI or BILAG scores. Organ involvement was reported by treating clinicians and nephritis was defined as the presence of proteinuria (> 0.5gm/day or > 3+ on dipstick)3 or biopsy confirmed lupus nephritis consistent with the 2004 revision of the WHO classification.9
Samples were analyzed for CNV using Affymetrix 5.0 SNP chip which was hybridized and scanned at the Biomolecular Research Facility (JCSMR, ANU). The Affymetrix 5.0 array has ∼500,000 SNP probes and ∼420,000 CNV probes. Array data was analyzed using R statistical programming language package CRNA (v2).35 CNVs were defined in each individual by applying the circular binary segmentation algorithm to the normalized log2(ratios of case versus pool of controls) signals. The pooled control consisted of all samples and assumed that there are no common pathogenic CNVs shared by the cases. Negative log2(ratio) values indicate copy number deletion and positive values duplication. Actual copy number was not inferred. Only CNVs supported by two or more probes were called. CNVs were filtered based on their log2(ratio) signal using an approximate empirical p value of 0.003 per individual. CNVs were further filtered by only retaining CNVs that intersected with known genes, based on the hg18 human genome assembly. Identified CNV were confirmed by qPCR using TaqMan CN Assays (Applied Biosystems, US) using the mean of 6 amplicons from CNV probe Hs05725015_cn compared with housekeeper genes. Vangl1 expression was measured by primers targeting exon 3 (F:GACACAAGTCACCCCGGAATA, R: TCCTCTGTCCGAGTAGAATCATT) and exon 4 (F:CCGATCCTGTGGAGGGATGA R: AAACACCCGTGGCATGTCA) of Vangl1. cDNA was prepared using miScript RT kit (QIAGEN) and used for qRT–PCR for miRNAs using miScript primers and SYBR Green kit from QIAGEN. Expression was normalized against HL13 and UBC housekeeper genes.
RNA sequencing
Transcriptome data were analyzed using a modified version of an existing variant detection pipeline.48
Mice and organ isolation
Vangl1−/− mice were created from the ES cell clone (EPD0164_3_G07) obtained from the supported KOMP Repository (www.komp.org) and generated by the Wellcome Trust Sanger Institute using CSD targeted alleles as described previously.47 Vangl+/−, Lyn−/−, and CD79a−/− mice were re-derived onto a C57BL/6 background and housed in sterile conditions. Equal numbers of male and female 12 week old mice were used unless otherwise specified and there was no gender bias in Vangl+/− mice. Mice were culled and kidneys set in optimum cutting temperature (OCT, Sakura) cryomolds or formalin and set in paraffin blocks.
Immunofluorescence and immunohistochemistry
Three month-old mice were sacrificed and serum and kidneys were harvested. Bound antibody was detected with anti-mouse IgG, IgA or IgM conjugated with fluorescein isothiocyanate (FITC). Immunoglobulin and C3 deposition analysis was performed on cryopreserved kidney sections. 7 μm acetone fixed sections were blocked with 3% bovine serum albumin and stained with anti-IgG Alexa Fluor 488 (Invitrogen), anti-IgA FITC (Southern Biotech), anti-IgM FITC (BD PharMingen) and anti-C3 FITC (MP Biomedicals). Fluorescence intensity was evaluated with an Olympus XI 71 microscope. Assessment of immunofluorescence, using a semiquantitative scale of 0-3+ was performed by two independent investigators. Formalin-fixed kidneys samples were sectioned and slides were stained with anti-mouse Vangl1 (SAB4503254, Sigma) and polyclonal anti-human VANGL1 (Cat PA5-55231 and Cat PA5-98739, ThermoFisher) for mouse and human samples, respectively.
Transmission electron microscopy (TEM)
Samples were primary fixed in 2% glutaraldehyde overnight, then washed 3 times in 0.1M Cacodylate buffer (pH 7.4) and postfixed with 2% osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA USA) in 0.1 M Cacodylate buffer for 2 h. En bloc staining with 2% uranyl acetate preceded dehydration through a graded series of ethanol steps. Specimens were infiltrated with TAAB low viscosity epoxy resin (TAAB Laboratory and Microscopy, England) (50:50 ethanol:TAAB resin for 2 h followed by TAAB resin for 3 h) embedded and set overnight at 70°C. Multiple levels of thin sections (90 nm) were cut from three blocks of each kidney. Thin sections mounted on copper/palladium grids were stained with Reynold’s lead citrate and viewed on a Jeol 1011 transmission electron microscope. Images were captured using a MegaView G2 digital camera and iTEM software package.
Serum injection
Three month old Vangl+/− and Vangl1+/+ mice were culled and whole blood collected. Blood was permitted to clot and centrifuged at room temperature, and serum was collected. Recipient Vangl+/−.CD79aken/ken and Vangl1+/+.CD79Aken/ken mice were injected daily with 100ul of respective serum for 7 days. At the end of the injection period, kidneys from recipient mice were cryopreserved as previously described. Lyn−/− mice in whom we confirmed the presence of proliferative nephritis were bled and allowed to clot. Serum was isolated and 200ul of serum tail-vein injected into Vangl1+/− or Vangl1+/+ recipients at day 0 and day 4. Mice were sacrificed at day 7 and kidneys isolated for histology.
Serum ANA, immunoglobulin and creatinine
Mice were bled at times indicated and serum was collected. ANAs were quantified using ELISA (Alpha Diagnostic). Total serum immunoglobulin was measured by ELISA (BD Bioscience). Serum creatinine measured using the Indiko system as per manufacturer’s instructions. Urine was collected weekly at indicated ages and urine albumin measured by ELISA as per the manufacturer’s instructions (Ebioscience).
Tiwi Islander study participants
The Tiwi Islands are located off the northern coast of Australia in the Arafura Sea, and its indigenous inhabitants, numbering approximately 2,500,49 exhibit a distinct genetic ancestry compared to other ethnicities50 and are considered most closely related to mainland Indigenous Australians (Council 2018).51 All the participants were self-identifying Tiwi Islanders and consented to collection of blood and DNA for genetic studies. A previous analysis of a separate cohort of self-identifying Tiwi Islanders (n = 73 individuals) indicated low admixture with other populations, including Europeans.50 Whole genome sequencing was performed on an Illumina HiSeq X Ten System with greater than 50x coverage. The study received the full support of the Tiwi Island Land Council and was approved by the human research ethics committees of The Northern Territory Department of Health (2012-1767),52 The Australian National University (2014-663), The University of Queensland (2012001146) and The University of Tasmania (H0012832). Chronic kidney disease was defined in the Tiwi according to the KDIGO guidelines.
Quantification and statistical analysis
All statistical comparisons were performed using Graphpad Prism. Statistical details of experiments including tests and numbers are found in the figure legends. All data expressed as medians with primary data presented to demonstrate dispersion in the figures. Significance was defined as a p value < 0.05.
Acknowledgments
RACP Jacquot NHMRC Award for Excellence, Jacquot Research Entry Scholarship, Canberra Hospital Private Practice Fund, Capital Kidney Research Fund, and NHMRC project grants to S.H.J. NHMRC program and project grants and Elizabeth Blackburn Fellowship to C.G.V. This research/project was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. Personnel of the Australian Cancer Research Foundation Biomolecular Resource Facility (JCSMR) for supporting RNA and DNA experiments. Harpreet Vohra and Mick Devoy from the MCRF facility (JCSMR) for support with flow cytometry. Stuart Read and Nikki Ross from the Australian Phenomics Facility for logistic and technical support of mouse imports.
Author contributions
S.H.J.: all aspects of study including conceptualization, investigation, and original draft writing; S.M.: conceptualization and methodology investigation; I.P.: methodology investigation and review writing and editing; M.M.: methodology investigation, software, and review writing and editing; G.D.W.: methodology and review writing and editing; M.K.: investigation and review writing and editing; M.F.: investigation and review writing and editing; M. Stanley: investigation and review writing and editing; T.L.-H.: investigation and review writing and editing; A.C.: investigation and review writing and editing; J.E.: investigation and review writing and editing; B.M.: resources, formal analysis, and review writing and editing; M. Sundaram.: resources and review writing and editing; R.T.: software and review writing and editing; P.F.C.: investigation and review writing and editing; W.H.: conceptualization, resources, and review writing and editing; H.H.: investigation and review writing and editing; M. Srivastava: investigation; K.M.: investigation and review writing and editing; I.F.: investigation and review writing and editing, R.C.: investigation and review writing and editing, R.F.: investigation and review writing and editing. S.D.: investigation and review writing and editing, M.G.: investigation and review writing and editing, V.A.: investigation and review writing and editing; M.F.: software, investigation, and review writing and editing; J.M.: conceptualization, investigation, and review writing and editing; E.C.: software, investigation, and review writing and editing; T.D.A.: software, investigation, and review writing and editing; A.R.K.: investigation and review writing and editing; M.C.C.: resources and review writing and editing; M.A.R.: investigation and review writing and editing; M.B.: software, investigation, formal analysis, and review writing and editing; C.G.V.: all aspects of study including conceptualization, methodology, resources, and review writing and editing.
Declaration of interests
The authors declare no competing interests.
Published: December 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100475.
Supplemental information
Data and code availability
RNA sequences are deposited in GEO and accession numbers are found in the STAR table.
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Chung S.A., Brown E.E., Williams A.H., Ramos P.S., Berthier C.C., Bhangale T., Alarcon-Riquelme M.E., Behrens T.W., Criswell L.A., Graham D.C., et al. International Consortium for Systemic Lupus Erythematosus Genetics Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J. Am. Soc. Nephrol. 2014;25:2859–2870. doi: 10.1681/ASN.2013050446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friebus-Kardash J., Trendelenburg M., Eisenberger U., Ribi C., Chizzolini C., Huynh-Do U., Lang K.S., Wilde B., Kribben A., Witzke O., et al. Susceptibility of BAFF-var allele carriers to severe SLE with occurrence of lupus nephritis. BMC Nephrol. 2019;20:430. doi: 10.1186/s12882-019-1623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hochberg M.C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 4.Petri M., Orbai A.M., Alarcón G.S., Gordon C., Merrill J.T., Fortin P.R., Bruce I.N., Isenberg D., Wallace D.J., Nived O., et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–2686. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yap D.Y., Tang C.S., Ma M.K., Lam M.F., Chan T.M. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol. Dial. Transplant. 2012;27:3248–3254. doi: 10.1093/ndt/gfs073. [DOI] [PubMed] [Google Scholar]
- 6.Bernatsky S., Boivin J.F., Joseph L., Manzi S., Ginzler E., Gladman D.D., Urowitz M., Fortin P.R., Petri M., Barr S., et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2550–2557. doi: 10.1002/art.21955. [DOI] [PubMed] [Google Scholar]
- 7.Feldman C.H., Hiraki L.T., Liu J., Fischer M.A., Solomon D.H., Alarcón G.S., Winkelmayer W.C., Costenbader K.H. Epidemiology and sociodemographics of systemic lupus erythematosus and lupus nephritis among US adults with Medicaid coverage, 2000-2004. Arthritis Rheum. 2013;65:753–763. doi: 10.1002/art.37795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel M., Clarke A.M., Bruce I.N., Symmons D.P. The prevalence and incidence of biopsy-proven lupus nephritis in the UK: Evidence of an ethnic gradient. Arthritis Rheum. 2006;54:2963–2969. doi: 10.1002/art.22079. [DOI] [PubMed] [Google Scholar]
- 9.Weening J.J., D’Agati V.D., Schwartz M.M., Seshan S.V., Alpers C.E., Appel G.B., Balow J.E., Bruijn J.A., Cook T., Ferrario F., et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J. Am. Soc. Nephrol. 2004;15:241–250. doi: 10.1097/01.asn.0000108969.21691.5d. [DOI] [PubMed] [Google Scholar]
- 10.Alba P., Bento L., Cuadrado M.J., Karim Y., Tungekar M.F., Abbs I., Khamashta M.A., D’Cruz D., Hughes G.R. Anti-dsDNA, anti-Sm antibodies, and the lupus anticoagulant: significant factors associated with lupus nephritis. Ann. Rheum. Dis. 2003;62:556–560. doi: 10.1136/ard.62.6.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinico R.A., Radice A., Ikehata M., Giammarresi G., Corace C., Arrigo G., Bollini B., Li Vecchi M. Anti-C1q autoantibodies in lupus nephritis: prevalence and clinical significance. Ann. N Y Acad. Sci. 2005;1050:193–200. doi: 10.1196/annals.1313.020. [DOI] [PubMed] [Google Scholar]
- 12.Mason L.J., Ravirajan C.T., Rahman A., Putterman C., Isenberg D.A. Is alpha-actinin a target for pathogenic anti-DNA antibodies in lupus nephritis? Arthritis Rheum. 2004;50:866–870. doi: 10.1002/art.20103. [DOI] [PubMed] [Google Scholar]
- 13.Amital H., Heilweil M., Ulmansky R., Szafer F., Bar-Tana R., Morel L., Foster M.H., Mostoslavsky G., Eilat D., Pizov G., Naparstek Y. Treatment with a laminin-derived peptide suppresses lupus nephritis. J. Immunol. 2005;175:5516–5523. doi: 10.4049/jimmunol.175.8.5516. [DOI] [PubMed] [Google Scholar]
- 14.Liphaus B.L., Kiss M.H., Goldberg A.C. HLA-DRB1 alleles in juvenile-onset systemic lupus erythematosus: renal histologic class correlations. Braz. J. Med. Biol. Res. 2007;40:591–597. doi: 10.1590/s0100-879x2007000400019. [DOI] [PubMed] [Google Scholar]
- 15.Vasconcelos C., Carvalho C., Leal B., Pereira C., Bettencourt A., Costa P.P., Marinho A., Barbosa P., Almeida I., Farinha F., et al. HLA in Portuguese systemic lupus erythematosus patients and their relation to clinical features. Ann. N Y Acad. Sci. 2009;1173:575–580. doi: 10.1111/j.1749-6632.2009.04873.x. [DOI] [PubMed] [Google Scholar]
- 16.Robson K.J., Ooi J.D., Holdsworth S.R., Rossjohn J., Kitching A.R. HLA and kidney disease: from associations to mechanisms. Nat. Rev. Nephrol. 2018;14:636–655. doi: 10.1038/s41581-018-0057-8. [DOI] [PubMed] [Google Scholar]
- 17.Deapen D., Escalante A., Weinrib L., Horwitz D., Bachman B., Roy-Burman P., Walker A., Mack T.M. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 18.Taylor K.E., Remmers E.F., Lee A.T., Ortmann W.A., Plenge R.M., Tian C., Chung S.A., Nititham J., Hom G., Kao A.H., et al. Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet. 2008;4:e1000084. doi: 10.1371/journal.pgen.1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez E., Nadig A., Richardson B.C., Freedman B.I., Kaufman K.M., Kelly J.A., Niewold T.B., Kamen D.L., Gilkeson G.S., Ziegler J.T., et al. BIOLUPUS and GENLES Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Ann. Rheum. Dis. 2011;70:1752–1757. doi: 10.1136/ard.2011.154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harley J.B., Alarcón-Riquelme M.E., Criswell L.A., Jacob C.O., Kimberly R.P., Moser K.L., Tsao B.P., Vyse T.J., Langefeld C.D., Nath S.K., et al. International Consortium for Systemic Lupus Erythematosus Genetics Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer O., Hauptmann G., Tappeiner G., Ochs H.D., Mascart-Lemone F. Genetic deficiency of C4, C2 or C1q and lupus syndromes. Association with anti-Ro (SS-A) antibodies. Clin. Exp. Immunol. 1985;62:678–684. [PMC free article] [PubMed] [Google Scholar]
- 22.Kemp M.E., Atkinson J.P., Skanes V.M., Levine R.P., Chaplin D.D. Deletion of C4A genes in patients with systemic lupus erythematosus. Arthritis Rheum. 1987;30:1015–1022. doi: 10.1002/art.1780300908. [DOI] [PubMed] [Google Scholar]
- 23.McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I., Maller J.B., Kirby A., et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 24.Pacheco G.V., Cruz D.C., González Herrera L.J., Pérez Mendoza G.J., Adrián Amaro G.I., Nakazawa Ueji Y.E., Angulo Ramírez A.V. Copy Number Variation of TLR-7 Gene and its Association with the Development of Systemic Lupus Erythematosus in Female Patients from Yucatan Mexico. Genet. Epigenet. 2014;6:31–36. doi: 10.4137/GEG.S16707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J.Y., Wang C.M., Chang S.W., Cheng C.H., Wu Y.J., Lin J.C., Yang B., Ho H.H., Wu J. Association of FCGR3A and FCGR3B copy number variations with systemic lupus erythematosus and rheumatoid arthritis in Taiwanese patients. Arthritis Rheumatol. 2014;66:3113–3121. doi: 10.1002/art.38813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aitman T.J., Dong R., Vyse T.J., Norsworthy P.J., Johnson M.D., Smith J., Mangion J., Roberton-Lowe C., Marshall A.J., Petretto E., et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 27.Simons M., Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu. Rev. Genet. 2008;42:517–540. doi: 10.1146/annurev.genet.42.110807.091432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kibar Z., Salem S., Bosoi C.M., Pauwels E., De Marco P., Merello E., Bassuk A.G., Capra V., Gros P. Contribution of VANGL2 mutations to isolated neural tube defects. Clin. Genet. 2011;80:76–82. doi: 10.1111/j.1399-0004.2010.01515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kibar Z., Vogan K.J., Groulx N., Justice M.J., Underhill D.A., Gros P. Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat. Genet. 2001;28:251–255. doi: 10.1038/90081. [DOI] [PubMed] [Google Scholar]
- 30.Kibar Z., Torban E., McDearmid J.R., Reynolds A., Berghout J., Mathieu M., Kirillova I., De Marco P., Merello E., Hayes J.M., et al. Mutations in VANGL1 associated with neural-tube defects. N. Engl. J. Med. 2007;356:1432–1437. doi: 10.1056/NEJMoa060651. [DOI] [PubMed] [Google Scholar]
- 31.Lei Y.P., Zhang T., Li H., Wu B.L., Jin L., Wang H.Y. VANGL2 mutations in human cranial neural-tube defects. N. Engl. J. Med. 2010;362:2232–2235. doi: 10.1056/NEJMc0910820. [DOI] [PubMed] [Google Scholar]
- 32.Yates L.L., Papakrivopoulou J., Long D.A., Goggolidou P., Connolly J.O., Woolf A.S., Dean C.H. The planar cell polarity gene Vangl2 is required for mammalian kidney-branching morphogenesis and glomerular maturation. Hum. Mol. Genet. 2010;19:4663–4676. doi: 10.1093/hmg/ddq397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocque B.L., Babayeva S., Li J., Leung V., Nezvitsky L., Cybulsky A.V., Gros P., Torban E. Deficiency of the planar cell polarity protein Vangl2 in podocytes affects glomerular morphogenesis and increases susceptibility to injury. J. Am. Soc. Nephrol. 2015;26:576–586. doi: 10.1681/ASN.2014040340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papakrivopoulou E., Vasilopoulou E., Lindenmeyer M.T., Pacheco S., Brzóska H.L., Price K.L., Kolatsi-Joannou M., White K.E., Henderson D.J., Dean C.H., et al. Vangl2, a planar cell polarity molecule, is implicated in irreversible and reversible kidney glomerular injury. J. Pathol. 2018;246:485–496. doi: 10.1002/path.5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bengtsson H., Wirapati P., Speed T.P. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics. 2009;25:2149–2156. doi: 10.1093/bioinformatics/btp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bentham J., Morris D.L., Graham D.S.C., Pinder C.L., Tombleson P., Behrens T.W., Martín J., Fairfax B.P., Knight J.C., Chen L., et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015;47:1457–1464. doi: 10.1038/ng.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Namjou B., Choi C.B., Harley I.T., Alarcón-Riquelme M.E., Kelly J.A., Glenn S.B., Ojwang J.O., Adler A., Kim K., Gallant C.J., et al. BIOLUPUS Network. Genoma en Lupus Network Evaluation of TRAF6 in a large multiancestral lupus cohort. Arthritis Rheum. 2012;64:1960–1969. doi: 10.1002/art.34361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myles S., Davison D., Barrett J., Stoneking M., Timpson N. Worldwide population differentiation at disease-associated SNPs. BMC Med. Genomics. 2008;1:22. doi: 10.1186/1755-8794-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoy W.E., Kondalsamy-Chennakesavan S., McDonald S., Wang Z. Renal disease, the metabolic syndrome, and cardiovascular disease. Ethn Dis. 2006;16:S2-46-51. [PubMed] [Google Scholar]
- 40.MacDonald J.R., Ziman R., Yuen R.K., Feuk L., Scherer S.W. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, Su A.I. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torban E., Patenaude A.M., Leclerc S., Rakowiecki S., Gauthier S., Andelfinger G., Epstein D.J., Gros P. Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc. Natl. Acad. Sci. USA. 2008;105:3449–3454. doi: 10.1073/pnas.0712126105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yabas M., Teh C.E., Frankenreiter S., Lal D., Roots C.M., Whittle B., Andrews D.T., Zhang Y., Teoh N.C., Sprent J., et al. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat. Immunol. 2011;12:441–449. doi: 10.1038/ni.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaneko Y., Nimmerjahn F., Madaio M.P., Ravetch J.V. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J. Exp. Med. 2006;203:789–797. doi: 10.1084/jem.20051900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoy W.E. Kidney disease in Aboriginal Australians: a perspective from the Northern Territory. Clin. Kidney J. 2014;7:524–530. doi: 10.1093/ckj/sfu109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoy W.E., Mathews J.D., McCredie D.A., Pugsley D.J., Hayhurst B.G., Rees M., Kile E., Walker K.A., Wang Z. The multidimensional nature of renal disease: rates and associations of albuminuria in an Australian Aboriginal community. Kidney Int. 1998;54:1296–1304. doi: 10.1046/j.1523-1755.1998.00099.x. [DOI] [PubMed] [Google Scholar]
- 47.Skarnes W.C., Rosen B., West A.P., Koutsourakis M., Bushell W., Iyer V., Mujica A.O., Thomas M., Harrow J., Cox T., et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Field M.A., Cho V., Andrews T.D., Goodnow C.C. Reliably Detecting Clinically Important Variants Requires Both Combined Variant Calls and Optimized Filtering Strategies. PLoS ONE. 2015;10:e0143199. doi: 10.1371/journal.pone.0143199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Statistics ABo . 2016. 2016 Census Quickstats.https://quickstats.censusdata.abs.gov.au/census_services/getproduct/census/2016/quickstat/036 [Google Scholar]
- 50.Thomson R.J., McMorran B., Hoy W., Jose M., Whittock L., Thornton T., Burgio G., Mathews J.D., Foote S. New Genetic Loci Associated With Chronic Kidney Disease in an Indigenous Australian Population. Front. Genet. 2019;10:330. doi: 10.3389/fgene.2019.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiwi Land Council . 2019. About the Tiwi Islands, History. Timeline: Dreamtime to 1978.https://tiwilandcouncil.com/index.cfm?fuseaction=page&p=294&l=3&id=56&smid=164&ssmid=66 [Google Scholar]
- 52.Levey A.S., Eckardt K.U., Dorman N.M., Christiansen S.L., Hoorn E.J., Ingelfinger J.R., Inker L.A., Levin A., Mehrotra R., Palevsky P.M., et al. Nomenclature for kidney function and disease: report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2020;97:1117–1129. doi: 10.1016/j.kint.2020.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequences are deposited in GEO and accession numbers are found in the STAR table.
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.