

Keywords: Bowman’s capsule, glomerulus, kidney, nephron, podocytes, proximal tubules
Abstract
Kidneys, one of the vital organs in our body, are responsible for maintaining whole body homeostasis. The complexity of renal function (e.g., filtration, reabsorption, fluid and electrolyte regulation, and urine production) demands diversity not only at the level of cell types but also in their overall distribution and structural framework within the kidney. To gain an in depth molecular-level understanding of the renal system, it is imperative to discern the components of kidney and the types of cells residing in each of the subregions. Recent developments in labeling, tracing, and imaging techniques have enabled us to mark, monitor, and identify these cells in vivo with high efficiency in a minimally invasive manner. In this review, we summarize different cell types, specific markers that are uniquely associated with those cell types, and their distribution in the kidney, which altogether make kidneys so special and different. Cellular sorting based on the presence of certain proteins on the cell surface allowed for the assignment of multiple markers for each cell type. However, different studies using different techniques have found contradictions in cell type-specific markers. Thus, the term “cell marker” might be imprecise and suboptimal, leading to uncertainty when interpreting the data. Therefore, we strongly believe that there is an unmet need to define the best cell markers for a cell type. Although the compendium of renal-selective marker proteins presented in this review is a resource that may be useful to researchers, we acknowledge that the list may not be necessarily exhaustive.
INTRODUCTION
The human kidney is responsible for maintaining homeostasis in the body by regulating acid-base and electrolyte balance, controlling blood pressure, and excreting metabolic toxins and waste products. The nephron is the basic structural and functional unit of the kidney, which is composed of Bowman’s capsule, the glomerulus, tubules [proximal tubule (PT), loop of Henle, distal convoluted tubule (DCT), and collecting tubules], and the collecting duct (CD). These segments participate in different aspects of the filtration process and contain a plethora of distinct specialized cell types. For example, parietal epithelial cells (PECs) are found in Bowman’s capsule. Within the glomerulus, more than 20 different cells have been found with three major types: endothelial cells (ECs), podocytes, and mesangial cells. Further down the nephron, the PT and DCT are lined with epithelial cells. On the other hand, the connecting tubules (CNTs) and CD possess heterogeneity even within the epithelial cell types, i.e., intercalated cells (ICs) are interspersed with principal cells (PCs). Even within ICs, there is further distinction that imparts unique function to each cell type. The kidney interstitium is further composed of other cell types including ECs of peritubular capillaries (either fenestrated or nonfenestrated), vascular smooth muscle cells, pericytes (covering capillaries), fibroblasts, and resident immune cells. Discoveries made in the last two decades or more, based on advanced techniques including micropuncture, isolation of perfused tubules, and electron microscopy, indicated considerable structural and functional heterogeneity along the nephron (1). Recently, an interactive multimodal atlas encompassing both transcriptomic and epigenomic data generated from a combination of single nucleus assays for transposase-accessible chromatin using sequencing and single nuclear sequencing (snRNA-seq) has fostered, refined, and matured our understanding of unique cell types and heterogeneity in the mature human kidney (2).
Thus, the anatomy of kidney is shaped by the specific physiology of each cell type as well as their remarkable spatial relationship (3). Consequently, glomerular or tubular pathophysiology is not limited to a single cell type, i.e., it is attributed to epithelial, endothelial, mesenchymal, and/or immune cell dysfunction. For example, acidosis and alkalosis are caused by impaired IC function. Therefore, a long-term goal in renal biology is to analyze different kidney cell types in detail and to identify the gene expression profile in those cell types. Although bulk tissue RNA sequencing of different components of the kidney can provide insights into segment-specific transcriptomes, it only represents average RNA expression and not the transcriptome at the single cell level. Moreover, the presence of multiple and complex cell types in kidney tissue obfuscates data interpretation. Additionally, lack of response in a more abundant cell type might mask an apparently strong response in a minority cell type (4). Therefore, next-generation techniques such as single-cell RNA sequencing (scRNA-seq), which exploit unbiased genome-wide RNA profiling of individual cells, are used to provide gene expression data at single cell resolution (5, 6). The limitation of this approach is that the analysis of large populations of cells shadow the rare cell types and obscure miniscule differences between different individual cells. Furthermore, scRNA-seq may not be always feasible for a single cell, especially when the tissue is inflamed or fibrotic, making tissue dissection or single cell dissociation extremely challenging. Thus, snRNA-seq, which uses an easy-to-isolate high-quality nuclear preparation, has become a popular alternative to scRNA-seq. Cells like fibroblasts, which are difficult to detach from the basement membrane, are also easily captured and analyzed more thoroughly in snRNA-seq. On the flip side, immune cells are captured poorly in snRNA-seq. Moreover, to analyze the data generated from snRNA-seq, different alignment methods and parameters are required because most of the nuclear RNA is in the unspliced form (7).
With this background, it is evident that on the one hand, there is a tremendous increase in the technological development that allows for unprecedented cellular measurements and gene expression in individual renal cell types; on the other hand, each technique presents significant pros and cons, which questions the reliability, validity, and completeness of the data output. Therefore, a combined community effort from kidney experts is certainly needed to best define a marker that is specific and exclusive to a renal cell type without any discrepancy. Based on the data available, this review is aimed to provide an overview of different renal cell types, specific guide marker proteins associated with each cell type, and their association with renal pathophysiology. We anticipate that this information will provide a systematic understanding of kidney function and specific dysfunction in renal pathologies.
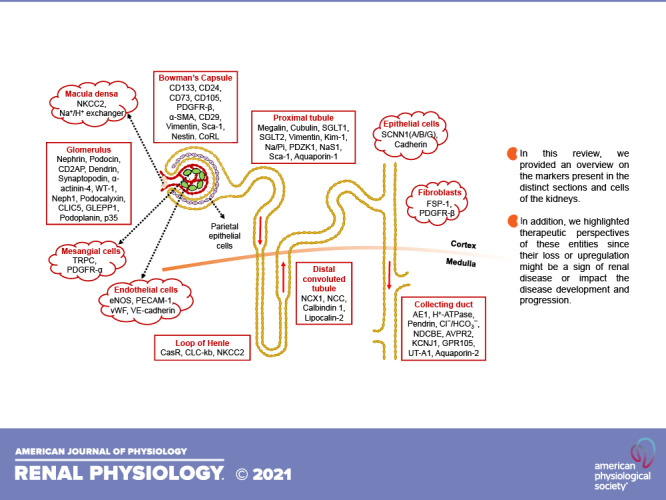
RENAL CELLS AT THE FINEST LEVEL OF SUBDIVISION
The kidney is a complex organ with various vascular compartments each including morphologically and functionally distinct cell types (Table 1). These cells possess several distinct characteristics that distinguish them from other cell types and bestow them with unique functions. In this review, we outline their localization and dynamics within the kidney. The classification is based on the available knowledge on the identification of these cells by detection of expression of their marker proteins.
Table 1.
Overview of the renal cell markers present in different segments of the nephron
| Renal Cells/Marker Proteins | Reference(s) |
|---|---|
| Bowman’s capsule | |
| CD133 | (8) |
| CD24 | (9, 10) |
| CD73 | (11, 12) |
| CD105 | (11, 12) |
| Platelet-derived growth factor receptor-β | (12) |
| α-Smooth muscle actin | (12) |
| CD29 | (11, 13, 14) |
| Vimentin | (14) |
| Stem cell antigen-1 | (14) |
| Nestin | (14) |
| Cells of renin lineage | (15, 16) |
| Glomerulus | |
| Nephrin | (17–19) |
| Podocin | (20) |
| CD2-associated protein | (21–23) |
| Dendrin | (24, 25) |
| Synaptopodin | (26) |
| α-Actinin-4 | (27) |
| Wilms’ tumor-1 | (28) |
| Kirre-like nephrin family adhesion molecule 1/Neph1 | (29) |
| Podocalyxin | (30, 31) |
| Cl− intracellular channel protein 5 | (32) |
| Glomerular epithelial protein 1 | (33) |
| Podoplanin | (34) |
| p35 | (35) |
| Proximal tubules | |
| Megalin | (36) |
| Cubilin | (37) |
| Na+-glucose cotransporter-1 | (38–42) |
| Na+-glucose cotransporter-2 | |
| Vimentin | (43, 44) |
| Kidney injury molecule-1 | |
| Na/Pi | (45) |
| PDZK1 | (46) |
| NaS1 | (47) |
| Stem cell antigen-1 | (48) |
| Aquaporin-1 | (49,50) |
| Loop of Henle | |
| Ca2+-sensing receptor | (51, 52) |
| Cl− channel-Kb | (53) |
| Na+-K+-2Cl− cotransporter-2 | (54–56) |
| Distal convoluted tubule | |
| Na+/Ca2+ exchanger isoform 1 | (57, 58) |
| Thiazide-sensitive Na+-Cl− cotransporter | (59) |
| Calbindin 1 | (58) |
| Receptor for lipocalin-2 (24p3R) | (60) |
| Collecting duct | |
| Cl−-bicarbonate transporter 1 (anion exchanger 1) | (61) |
| H+-ATPase, apical/basolateral | |
| Pendrin | |
| Cl−/ anion exchanger and SLC26A11 | |
| Na+-driven Cl−/ exchanger | |
| Arginine vasopressin receptor 2 | (62) |
| Renal inwardly rectifying K+ channel subfamily J 1 (ROMK1) | (46) |
| P2Y14 receptor (GPR105) | (63) |
| Urea transporter-A1 | (64, 65) |
| Endothelium | |
| Endothelial nitric oxide synthase | (66) |
| Platelet/endothelial cell adhesion molecule 1 | (67) |
| von Willebrand factor | (68) |
| Vascular-endothelial cadherin | (69) |
| Fibroblasts | |
| Fibroblast-specific protein 1 | (70) |
| Platelet-derived growth factor receptor-β | (71) |
| Mesangial cells | |
| Transient receptor potential canonical member 1 | (72, 73) |
| Platelet-derived growth factor receptor-α | (74) |
| Epithelial cells | |
| Epithelial Na+ channel subunits α/β/γ (SCNN1A/B/G) | (75) |
| Cadherin | (76) |
| Macula densa | |
| B isoform of Na+-K+-2Cl− cotransporter | (77) |
| Na+/H+ exchanger (isoforms 2 and 4) | (78) |
BOWMAN’S CAPSULE
The parietal epithelium of Bowman’s capsule consists of polygon-shaped epithelial cells that rest on a basement membrane. Historically, these PECs were viewed simply as cells lining the inner layer of Bowman’s capsule, but recently they have been recognized as endogenous stem cells with a potential to self-renew. In humans, a subset of PECs coexpress the common stem cell marker CD133 and renal embryonic cell marker CD24 (8–10). These CD24+CD133+ PECs are called adult parietal epithelial multipotent progenitors (APEMPs). They do not express any lineage-specific markers but demonstrate multilineage differentiation potential. APEMPs are selectively found on the urinary pole of Bowman’s capsule during nephron development (i.e., in embryonic human kidneys), but their presence decreases as development progresses, and they only represent <2% of whole cells in adult kidneys (13). Indeed, APEMPs exhibit heterogeneous potential for differentiation and regeneration into PT cells (9, 10) and podocytes (8), presumably as a compensation for their loss during renal diseases. Interestingly, gene expression profiles of glomerular and tubular APEMPs were not different from one another, indicating that those stem populations are homogenous (79). This population of cells was shown to coexpress stem cell markers CD29, CD44, CD54, and CD106, as revealed by FACS analysis of cell suspensions obtained from human embryonic kidneys (13). Of note, CD106 (vascular cell adhesion molecule 1) was used to segregate tubular-committed APEMPs from those residing in Bowman’s capsule, i.e., APEMPs lacking CD106 were localized either in the PT or in the DCT (9). Other stromal markers including CD73, CD105, platelet-derived growth factor (PDGF) receptor-β (PDGFR-β), and α-smooth muscle actin (α-SMA) were detected in the adult kidney capsule (and in the perivascular region concurrently) by immunohistochemistry (11, 12). In addition, cells derived from the mouse renal capsule were positive for mesenchymal stem cell markers CD29, vimentin, stem cell antigen-1 (Sca-1), and nestin (14). Nestin, a cytoskeleton-associated filament protein, was found to be expressed in mouse (80), rat (81), and human (82, 83) kidneys during embryogenesis. In humans, this expression is restricted to podocytes in mature glomeruli (82, 83). On the other hand, an intermediate filament protein, vimentin, is not only expressed in podocytes but also in mesangial cells of the glomerulus, the endothelium of renal capillaries, and renal stromal cells (84).
Besides APEMPs that only express stem cell proteins, Bowman’s capsule also harbors committed progenitors that coexpress podocyte markers including α-actinin-4, glomerular epithelial protein 1 (GLEPP1), nephrin, podocin, podocalyxin, synaptopodin, and the transcription factor Wilms’ tumor-1 (WT-1) (85–88). These “transitional” cells were found to be residents of Bowman’s capsule only in young and still growing glomeruli and then extend along the vascular stalk of the glomerulus at later stages of life (88–90). Lineage tracing experiments revealed that claudin-1 (87, 90, 91), p57 (87), PAX-2 (91, 92), and Ki-67-positive PECs (92) could also regenerate podocytes.
Another subset of cells lining Bowman’s capsule comprises cells of renin lineage, which extends the mapping of progenitor cells beyond the glomerular tuft since cells of renin lineage are exclusively of juxtaglomerular origin (15, 16). These landmark studies demonstrating the presence of resident multipotent progenitor cells in the adult human kidney as well as their potency for self-renewal, clonogenicity, differentiation, and migration have opened new avenues for regenerative medicine in patients with renal diseases (93, 94).
THE GLOMERULUS
Glomeruli are the key functional units of the kidney filtration apparatus. Each glomerulus consists of capillary tufts, which are structurally maintained by mesangial cells and a three-layered filtration barrier comprising ECs, the glomerular basement membrane (GBM), and highly specialized podocytes (epithelial cells). Glomerular podocytes comprise a cell body and unique actin-based foot processes (FPs) that give them an arborized morphology (95). Adjacent FPs are connected to each other via specialized intercellular junctions called as slit diaphragms (SDs). While charge selectivity is believed to be a function of the GBM, the SD functions as a size-selective sieve (96). This function of the SD is devoted to its flexible multilayered architecture, which can respond to fluctuating blood pressures (97). Under normal healthy conditions, the filtration barrier allows the passage of water, solutes, and plasma proteins smaller than albumin. However, under diseased conditions, podocytes are reorganized into a flattened shape with effacement of podocyte FPs and loss of SD substructures, culminating in massive proteinuria (>3.5 g albumin/day). Since podocytes are the key cells that are involved not only in glomerular physiology but also in the pathology underlying several nephropathies (98–101), researchers have studied and identified specific podocyte proteins that have a direct association with either the disease outcome or its prognosis and suggested new agents or therapies specific to podocytes (102–104).
Nephrin (encoded by NPHS1) is the adhesion protein that is primarily found in the kidneys (17). It is the first protein that was shown to be expressed on SDs of human podocytes (18, 19). The immunoelectron microscopy-based analysis suggested that nephrin extending from two adjacent podocyte FPs interacts with each other to form a zipper-like arrangement (18). A year later, another important SD-associated podocyte protein, podocin (encoded by NPHS2), was identified (20). The importance of podocin was underscored by the finding that localization of nephrin to the SD depended on its ability to interact with podocin via its R1160 residue at the COOH-terminal cytoplasmic tail (105, 106). This signaling complex has been shown to direct nephrin to the lipid rafts on the cell surface (107) and maintain the structural integrity of podocytes (106, 108). The most common NPHS2 disease-causing variant, the R138Q mutation in podocin, disrupts its folding and causes incomplete glycosylation (109). These events further cause podocin to relocalize from the plasma membrane and interfere with the proper trafficking of nephrin (109). As a result of this point mutation, both R138Q podocin and nephrin are retained in the endoplasmic reticulum (ER) (109), which was reported to be the subcellular destination of missense mutated nephrin molecules (110). As indicated in the later study, NPHS1 mutant proteins are also associated with improper folding and glycosylation, which is required for the plasma membrane localization of nephrin (111), leading to their retention and degradation in the ER (112). These studies demonstrated that the appropriate trafficking of podocin is a prerequisite for the successful localization of nephrin on the cell surface and stabilization of the SD. Vice versa, nephrin is also required for the appropriate localization of podocin to the SD. Recent efforts to produce kidney organoids using human induced pluripotent stem cells from patients with NPHS1 missense mutations (E725D or R460Q) have shown that the expression and overall “basal” distribution of podocin were unaffected but that nephrin was required for the recruitment of podocin to “lateral” SD domains (113, 114). In total, ∼250 distinct genetic mutations in NPHS1 (115) and >125 mutations in NPHS2 (116) have been identified. It is noteworthy that genetic defects in podocin can start at any age, whereas mutations in nephrin cause nephrotic syndrome (NS) within the first 3 mo of life (117). Therefore, while mutations in NPHS1 cause congenital NS of Finnish type in humans (17), mutations in NPHS2 are associated with autosomal recessive familial steroid-resistant NS (20). Moreover, a plethora of studies have reported downregulation of nephrin (118–125) and podocin (122–126) expression in various proteinuric kidney diseases. In fact, recent studies have demonstrated a decline in nephrin expression as an early event independent of podocyte loss, which impairs the ability of podocytes to recover after injury, thereby facilitating disease progression (127).
Another major protein, which was discovered in the late 1990s to be necessary for the SD, is CD2-associated protein (CD2AP) (22, 23). CD2AP has been detected in diverse tissues like the placenta, colon, pancreas, kidney, and thymus (128). In the kidney, it is also expressed in tubules (23); more specifically, there is robust CD2AP expression in cortical ureteric bud epithelial cells and medullary CDs at birth and PTs, DCTs, and CD epithelial cells at maturity as detected by immunofluorescent staining of the mouse kidney (21). Immunohistochemical analysis of normal mouse and human kidneys demonstrated that within the mature glomerulus, CD2AP localization was restricted to podocytes (21). Another study by Lehtonen et al. (129) described CD2AP localization in mouse embryonic and adult kidneys using in situ hybridization and immunofluorescence microscopy. In the embryonic kidney (17 days old), strong expression of CD2AP was seen in mature, medullary branches of CDs and in mature glomerular podocytes. In the adult mouse kidney, CD2AP expression was strongly retained in glomeruli and CDs, with weak but positive staining in a subset of distal tubular segments and no staining in PTs. CD2AP, an 80-kDa protein found at the cytoplasmic side of the SD, has been shown to bind to nephrin (130) and podocin (105). CD2AP functions as an adaptor protein that anchors these two SD proteins to actin filaments of podocyte cytoskeleton and is thus essential for normal podocyte function (131, 132). Furthermore, CD2AP has been shown to bind to actin-associated synaptopodin (133). Through its three adjacent SH3 domains, CD2AP can bind to different structural proteins or signaling components of the cytoskeleton simultaneously (22, 128). Besides being heavily involved in cytoskeletal rearrangements, CD2AP also plays a crucial role in cellular signaling. It interacts with the p85 regulatory subunit of phosphoinositide 3-OH kinase (PI3K) and elicits PI3K-dependent Akt signaling in podocytes (134). This CD2AP-mediated Akt activity regulates a series of biological events including actin dynamics (135) and ER stress and apoptosis (136). However, CD2AP is downregulated under diabetic conditions, which also hampers the PI3K/Akt signaling pathway (137). Furthermore, CD2AP has been shown to be required for activation of PI3K and ERK/MAPK signaling pathways by transforming growth factor-β (TGF-β), and, thus, in the absence of CD2AP, TGF-β receptors failed to engage these antiapoptotic pathways causing hyperactivation of proapoptotic p38 MAPK (138). CD2AP acquires phosphorylation by receptor tyrosine kinases in podocytes (139), which alters its affinity for nephrin. Mice devoid of CD2AP protein exhibit loss of FPs and severe proteinuria (23, 140). Heterozygosity for a defective CD2AP allele is associated with a complex renal phenotype (140), and mutations and polymorphisms in the human gene are correlated with the development of glomerulonephritis and glomerulosclerosis (141–143).
Dendrin, originally identified in the telencephalic dendrites (25), is now recognized as a member of the SD complex, where it directly interacts with nephrin and CD2AP. Immunofluorescence and Western blot analysis using antibodies against dendrin detected the expected 89-kDa and 81-kDa isoforms in the brain, but only the 81-kDa isoform was found in mouse glomeruli (24). Also, dendrin was found to colocalize with the podocyte marker synaptopodin (26, 144). Dendrin, a proline-rich protein with two putative nuclear localization signals and three PPXY motifs, also possesses proapoptotic signaling properties; i.e., in response to glomerular injury, dendrin translocates from the SD to the nucleus of podocytes (145–147). Thus, nuclear localization of dendrin serves as a useful tool or marker to assess the integrity of the SD under pathological conditions.
The SD is considered to be a modified adherens junction (148) since it contains several tight junction proteins including zonula occludens-1 (ZO-1) (149), P-cadherin, α-catenin, β-catenin, γ-catenin (148), FAT (150), p120 catenin, CASK (151), MAGI-2 (152), JAM-A, occludin, and cingulin (153). The link between these proteins and the podocyte actin cytoskeleton is formed by actin-binding proteins such as synaptopodin and α-actinin-4. Synaptopodin is a proline-rich actin-binding protein that is localized in the postsynaptic density region of podocyte FPs and neuronal dendritic spines of the rat brain (26). Functional studies have revealed that mice lacking synaptopodin demonstrated reduced synaptic plasticity due to lower dendritic spine formation in the hippocampus (154) but exhibited no structural differences within the glomeruli (155). When the podocytes were injured via protamine sulfate (PS) or lipopolysaccharide (LPS), the actin bundles (which are complexes of synaptopodin and actin filaments) had delayed reformation at the injury site (155), suggesting that synaptopodin played a role in actin filament regulation. Indeed, synaptopodin could bind to α-actinin-2 (155), α-actinin-4 (155–157), and β-catenin (157) and regulate cellular contraction, elongation, and motility. The observed differences between the cultured hippocampal cells and kidney cells were explained by the existence of different synaptopodin isoforms, e.g., neuronal Synpo-short, renal Synpo-long, and Synpo-T (155). Depending on the isoform, binding of synaptopodin could lead to the inhibition of branching of the α-actinin-induced actin filaments. Calcineurin-dependent degradation of synaptopodin by cathepsin L also causes the loss of actin stress fibers in podocytes (158). The molecular mechanisms underlying synaptopodin and actin cytoskeleton interactions involve the ability of synaptopodin to regulate various signaling molecules including small GTPases RhoA (159), Rac1 (160), and Cdc42 (161), Cdc42:IRSp53:Mena complexes (162), and adapter protein Nck1 (163). Our group has recently shown that synaptopodin could limit the expression of transient receptor potential canonical ion channel member 6 (TRPC6), another SD member protein, controlling the dynamicity of the FP and protecting podocyte function (164). Each of transient receptor potential canonical ion channel member 5 (TRPC5) and TRPC6 acted as antagonistic regulators of synaptopodin by forming molecular complexes with Rac1 and RhoA, respectively, and provided a balance between a motile phenotype and a contractile phenotype in podocytes (165). Despite these protective functions of synaptopodin reported in the past decades, a recent publication showed that any isoform of synaptopodin could be dispensable in vivo but beneficial in the protection against acute podocyte injuries (166). Interestingly, loss of synaptopodin led to lower Rac-1 and RhoA activity, which was podocyte protective (166).
α-Actinin-4 (ACTN4), one of the members of actin-bundling proteins (ACTN1–ACTN4) and known to form ∼100-kDa head-to-tail homodimers, plays an essential role in podocyte FP architecture and adhesion (27). α-Actinin monomers comprise three major distinct domains: an F-actin-binding domain (ABD) at the NH2 terminus, four spectrin-like repeats (SRs), and EF hand motifs (calmodulin-like domain) at the COOH terminus (167). While ACTN2 and ACTN3 are Ca2+ insensitive and show predominant sarcomere-specific expression (168), the other two nonmuscle isoforms, ACTN1 and ACTN4, are highly and moderately sensitive to Ca2+, respectively, and are also widely expressed throughout the body (169). However, only ACTN4 expression is detected in human kidneys, and mutations in this gene are associated with an autosomal dominant form of familial focal segmental glomerulosclerosis (FSGS) (170). Interestingly, all the major mutations, including K255E, T259I, and S262P, are located within the evolutionarily conserved ABD of ACTN4 (167). Overall, mutated ACTN4 proteins exhibit higher binding affinity to F-actin causing an alteration in the mechanical characteristics of podocytes such as cytoskeletal rigidity, aberrant localization/aggregation patterns, and significantly diminished half-life (171). Most affected individuals have only mild proteinuria in their early adulthood but a progressive decline in kidney function later. Two more mutations, albeit missense (W59R and I149del), have also been reported within the evolutionary conserved ABD. Individuals carrying these mutations exhibit proteinuria at an early age of 5 yr old that progressed to end-stage renal disease (ESRD) within 3 yr, which is much faster, earlier, and more severe compared with the other ACTN4 mutations. Besides genetic mutations, decreased expression of ACTN4 has been attributed to several glomerulopathies including IgA nephropathy, sporadic FSGS, and minimal change disease (172).
The Wilms’ tumor-1 gene (WT1), a zinc finger protein that acts both as a transcription factor and an RNA-binding protein, is expressed only in podocytes (both mouse and human), undergoes alternative splicing to generate several isoforms, and is essential for kidney development (28). Interestingly, WT-1 controls the expression of many podocyte-specific genes that localize to the SD, such as NPHS1, NPHS2, MAGI2, Kirrel, Plce1, Ptpro, Cldn5, and Nck2, by binding to their promoter regions (173). In addition to SD components, WT1 is a transcription factor and has been shown to regulate the expression of proteins that are involved in cell matrix adhesion, cytoskeletal rearrangement, and polarity maintenance in podocytes (173). Consequently, it is expected that mutations in the WT1 gene will result in a wide spectrum of renal phenotypes like disruption of podocyte development or maintenance (174). These mutations are associated with glomerulopathies such as 1) Denys-Drash syndrome, characterized by early NS with diffuse mesangial sclerosis progressing rapidly to ESRD; 2) Frasier syndrome, presenting as progressive nephropathy with proteinuria and NS accompanied with FSGS progressing to ESRD in adolescence or young adulthood, and 3) steroid-resistant NS (174). Furthermore, mice with podocyte-specific WT1 depletion display defective podocyte differentiation, anuria, kidney failure, and death within a day postbirth (175). A decrease in WT1 staining has been used as a marker to confirm podocyte injury in patients with diabetic nephropathy (DN) (176). Along with desmin, WT1 is regarded as the most sensitive marker for podocyte and glomerular damage since WT1 immunohistochemistry represents a reliable complement to a morphology-driven approach to podocyte disease (177).
Kirre-like nephrin family adhesion molecule 1, known as Kirrel1 or Neph1, shares some homology with nephrin but has a shorter extracellular part, i.e., only five IgG-like domains (29). It was first isolated by a retroviral mutagenesis gene trapping method and found to be expressed in both mouse and human kidneys, specifically in the glomerulus (29). Neph1-deficient mice develop NS at birth causing early mortality (3−8 wk postconception) due to proteinuria and FP effacement (29). In the same year, it was shown that nephrin (NPHS1) knockout mice demonstrated more severe podocyte FP effacement with gross proteinuria and edema, and death occurred much quicker (within 24 h) compared with Neph1 knockout mice (178). Both Neph1 and NPHS1 share a conserved domain for podocin binding in their COOH-termini, suggesting that both proteins are required for nephrin-dependent signaling (179). More evidence was provided in a complementary study showing that the interaction between nephrin and Neph1 was not only specific but also an important determinant of glomerular permeability (180). However, Neph1 is not only involved in maintaining the glomerular permeability barrier. As a type I transmembrane protein, it also regulates outside-in signaling in podocytes, which results in actin polymerization (181, 182). Current studies are investigating various mutations found in the Neph1 gene from human patients and their physiological consequences in NS (183). There are two more Neph1-related proteins, Neph2 (179) and Neph3 (also termed filtrin) (184), and they share a common architecture comprising a transmembrane domain, five IgG-like domains, and a cytoplasmic tail including Grb2‐ and PDZK1‐binding sites.
Podocalyxin (also known as PCLP1, MEP21, gp135, and thrombomucin) is an integral single-pass transmembrane sialoprotein that imparts a negative charge to the glomerular membrane and contributes to podocyte morphogenesis and structural integrity and is found on the apical side of rat podocyte FPs and vascular ECs (30, 31). It is a 150- to 165-kDa protein encompassing a mucin domain, a globular domain, a transmembrane domain, and a highly charged cytoplasmic tail with putative phosphorylation sites for protein kinase C and casein kinase II (185). The PSD-95/Disks-large/ZO-1 (PDZ)-binding motif (DTHL) located at the COOH terminal of podocalyxin facilitates its interactions with Na+/H+ exchanger (NHE) regulatory factors 1 and 2 (NHERF1 and NHERF2, respectively) (186), the two adaptor proteins implicated in protein trafficking, ion transport, and signaling. Furthermore, podocalyxin has been shown to interact with the actin-binding protein ezrin (187). Thus, podocalyxin performs essential cellular functions through its interaction with NHERF proteins, ezrin, and the actin cytoskeleton. Consequently, deletion of podocalyxin in mice led to defective renal functions with marked anuria and early death. The podocytes in these mice failed to develop FPs and SDs and instead formed impermeable tight junctions (188, 189). Recently, a novel heterozygous missense mutation, c.T1421G (p. L474R), was identified in the podocalyxin encoding gene in an autosomal dominant FSGS pedigree. However, the function of podocalyxin was not affected by this mutation (190). Another study has reported a heterozygous nonsense mutation in the podocalyxin gene (c.C976T; p. Arg326X) that is characterized by proteinuria and renal insufficiency (191). Interestingly, a strong association was found between poor renal outcome and reduction of podocalyxin expression with an increase in its urinary excretion (192). Thus, assessment of podocalyxin levels in renal tissues and urine was proposed as a reliable biomarker to predict the progression of DN. A recent study by Refaeli et al. (193) generated two mouse strains to study the developmental role of podocalyxin: one mouse strain had Podxl deleted from developmentally mature podocytes (Podxl Δ Pod) and in the other strain podocalyxin was heterozygous in all tissues (Podxl+/−). Histological and ultrastructural analyses were performed to gauge kidney development and function in both of these strains. While PodxlΔPod mice developed acute congenital NS characterized by FSGS and proteinuria, Podxl+/− mice had no obvious renal phenotype with a normal lifespan but were extremely susceptible to puromycine aminonucleoside-induced nephrosis. This study indicates that podocalyxin plays a critical role in the formation and maintenance of podocyte structure (193).
Another protein found to be expressed on the apical and basal membranes of mouse FPs and glomerular ECs is Cl− intracellular channel protein 5 (CLIC5) (194). CLIC5 belongs to a family of intracellular Cl− channels that are known to have a putative single transmembrane domain. CLIC5 colocalizes with podocalyxin and ezrin/radixin/moesin (ERM) complex in FPs (194). Mice lacking CLIC5 had a reduced amount of these interacting proteins, and they exhibited FP abnormalities and proteinuria (32). Ezrin, which functions as an actin-membrane linker, is also predominantly expressed in rat podocytes (194). Ezrin has been reported to be a specific marker in both undifferentiated and differentiated glomerular epithelial cells in culture, in the adult rat glomerulus, and also during glomerulogenesis (195). Furthermore, the study was extended to include glomerular disease models where podocytes were injured, and ezrin was found to be elevated in binucleated podocytes or podocytes that were partially or completely detached from the underlying GBM (195).
Glomerular epithelial protein 1 (GLEPP1), also known as protein tyrosine phosphatase receptor type O (Ptpro), is a 132-kDa receptor tyrosine phosphatase present on the apical cell surface of podocyte FPs in rabbits (33), mice (196), and humans (197). The protein contains a transmembrane domain, an intracellular phosphatase domain, and a large extracellular domain consisting of eight fibronectin type III-like repeats. GLEPP1 is believed to play an important role in maintaining podocyte integrity, structure, and function. Targeted depletion of GLEPP1 has been shown to alter podocyte structure; i.e., the typical “octopoid” podocyte structure was overtly simplified to a more “amoeboid” structure and minor FPs exhibited substantial blunting and a broader morphology along with an altered distribution of vimentin, a podocyte intermediate cytoskeletal protein (196). This structural disruption was accompanied by a loss of function, characterized by a reduction in the glomerular filtration rate. Reduction of GLEPP1 is also associated with several podocytopathies like IgA nephropathy and FSGS (198).
Podoplanin (PDPN), also called E11 antigen/GP36/aggrus, is another 43-kDa integral membrane glycoprotein that is localized on the rat podocyte surface. The level of this protein is reduced in puromycin-induced kidney injury (34). Administration of anti-PDPN antibodies to rats led to proteinuria and podocyte FP effacement (199). Furthermore, selective loss of PDPN expression was strongly correlated with enhanced proteinuria in Dahl salt-sensitive rats (200). A serendipitous yet interesting finding by Kasinath et al. (201) revealed fading of glomerular PDPN and its concomitant increase in the tubulointerstitial compartment and in the urine shortly after ischemia-reperfusion injury (IRI) in mice. The role of PDPN as an antiapoptotic factor in ANG II-induced injury in human podocytes was also underscored (202).
Glomerulosclerosis and proteinuria are primarily attributed to podocyte loss resulting from either apoptosis, detachment, or inability of podocytes to adequately proliferate. Unlike other cyclin-dependent kinases (Cdks) that are involved in cell cycle regulation, Cdk5 has been shown to modulate cell maturation, differentiation, migration, and apoptosis (35). Interestingly, in mouse kidneys, Cdk5 shows glomerular expression that is restricted to podocytes. While the levels of Cdk5 were elevated in differentiating conditionally immortalized mouse podocytes in culture and in developing rodent fetal kidneys, they declined markedly in an experimental disease model of GBM nephritis that causes podocyte dedifferentiation/proliferation (203). In contrast to other Cdks that are activated by their cognate cyclins, Cdk5 has been shown to be activated by a noncyclin protein, p35, which is also expressed constitutively in podocytes and forms a complex with Cdk5 (35). This active complex (p35/Cdk5) was found at the plasma membrane (203). Further studies showed that p35-null mice had no kidney irregularities during glomerulogenesis or renal dysfunction during adult life. However, these mice were more susceptible to external injury stimuli and their podocytes underwent apoptosis. This indicates that, although p35 does not affect glomerular development, it certainly impacts podocyte survival postinjury (35).
TUBULES
There are 43 cell types purported to be present in kidney tissue, and PTs make up a substantial portion of the entire kidney tissue (4). PT cells roughly account for 52% of the estimated 200 million tubule epithelial cells. The PT epithelial cell population that is pure, stable, and functional is characterized by CD10 (neutral endopeptidase)/CD13 (aminopeptidase M) double staining and expression of specific markers such as aquaporin-1 (AQP1) and N-cadherin (49). While these CD10+/CD13+ cells exhibit epithelial characteristics over a long time, those that are positive only for either CD10 or CD13 appear morphologically heterogeneous and do not express PT markers (204).
Some albumin molecules pass through the glomerular filtration barrier under normal physiological conditions but finally are reabsorbed by the PT epithelium (205). Megalin [an ∼600-kDa glycoprotein with a single transmembrane domain, also known as low-density lipoprotein-related protein 2 (Lrp2)], which has been identified as a target antigen in Heymann nephritis (36), and cubilin (a 460-kDa glycoprotein with no transmembrane domain), which has been identified as an albumin-binding protein in rats by affinity chromatography (37), are two large transmembrane proteins expressed on the surface of PT epithelial cells. When megalin was first identified in the proximal convoluted tubule (PCT) (36), it was also detected in glomerular epithelial cells of rats as the target antigen in a model of glomerulonephritis. However, in humans, megalin was only detected in PTs (206). Several years later, using more sensitive techniques like RT-PCR on laser microdissected glomeruli, megalin was shown to be expressed in human podocytes as well (207). Using the same approach, a year later, cubilin was also found in podocytes of rats and humans (208). Both of these glycoproteins play a pivotal role in the endocytic reabsorption of many plasma proteins that are filtered across the glomerular capillary wall (209). Consequently, albumin is absent from the urine of healthy individuals. Thus, because of a highly efficient tubular reabsorption system, the presence of minute amounts of albumin in the urine of a healthy individual is considered as a risk factor for future renal disease (210). Both inherited and acquired dysfunction of megalin and cubilin have been described in humans (211). Mutations in the lrp2 gene causes a rare autosomal recessive disorder, Donnai-Barrow facio-oculo-acousticorenal syndrome, characterized by a distinct facial appearance, developmental delays, high-grade myopia, hypertelorism, sensorineural hearing loss, and congenital diaphragmatic hernia along with proteinuria (212). Similarly, Imerslund-Gräsbeck syndrome, a rare autosomal recessive inherited defect that is characterized by proteinuria, has been ascribed to cubilin deficiency (213). In addition to the above-mentioned inherited disorders, receptor dysfunction or changes in receptor expression have been observed in several acquired disorders associated with proteinuria such as experimental models of acute and chronic renal disease including LPS-induced endotoxemia, Shiga toxin-induced nephropathy, IRI, chronic kidney disease (CKD), and diabetes (214). Furthermore, an increase in the urinary excretion of megalin and its fragments as well as cubilin has been reported in models of Alport syndrome (215), in experimental as well as human diabetes (216, 217), and in IgA nephritis (218).
In the mammalian nephron, filtered hexoses are efficiently reabsorbed, an important physiological process to retain glucose and prevent substantial energy substrate loss into the urine, primarily by a concerted action of Na+-dependent (SGLT1 and SGLT2) (38) and Na+-independent (GLUT1 and GLUT2) (219) glucose transporters. SGLT2, a low-affinity/high-capacity Na+-dependent glucose cotransporter located in the early PT, takes up the bulk of glucose (∼97%) that is filtered by the renal glomerulus (219). The presence of SGLT2 was identified predominantly on the apical side of renal PCTs (S1 and S2 segments) in the mouse and human (39–41). In contrast, using specific antibodies (immunostaining and Western blot analysis) and gene expression studies (Northern blot analysis and in situ hybridization), SGLT1 was found on the brush-border membrane of the straight renal PT (S3 segment) in the human (40), mouse (220), rat (221), rabbit (222), and pig (223). While SGLT1 is responsible for most of the dietary glucose uptake in the intestine, this high-affinity/low-capacity transporter only accounts for the absorption of residual glucose (∼3%) in the S3 segment of the late PT (42). Even though its impact on the kidney is relatively less, the presence of SGLT1 is necessary to absorb the last traces of glucose from the lumen into the blood. These two transporters differ in many aspects: 1) their affinity for glucose and Na+ (SGLT1 > SGLT2); 2) their sensitivity to phlorizin inhibitor (SGLT1 > SGLT2); 3) their predilection and selectivity for hexoses (SGLT1 can transport both glucose and galactose equally well, but SGLT2 is shown to transport glucose 10 times better than galactose); and 4) a distinct segmental distribution (224). One of the consequences of diabetes is higher renal SGLT2 expression, which eventually enhances renal glucose reabsorption and increases the glucose transport maximum (from ∼450 to 600 g/day). Drugs that directly block SGLT2 are potent therapeutics that inhibit renal glucose reabsorption, increase glucose excretion, and successively lower hyperglycemia (225, 226), as demonstrated in prospective clinical trials with patients with type 2 diabetes mellitus (227, 228) and corresponding meta-analyses (229). Since SGLT2 reabsorbs Na+ along with glucose, SGLT2 blockers promote natriuresis and glucosuria. The resultant osmotic diuresis has been shown to have an antihypertensive effect (230, 231) and cause a compensatory increase in tubuloglomerular feedback (232).
Furthermore, transcellular glucose transport is accomplished by two glucose transporters that are located on the basolateral membrane, and they allow glucose to exit from PT cells. They are low-affinity glucose transporter 2 (GLUT2) present in the S1 segment and high-affinity glucose transporter 1 (GLUT1) found in the S3 segment (233). SGLT2 has been shown to be increased in alloxan-induced diabetic rats (234), and several groups have also shown that GLUT2 in renal PTs is increased in streptozotocin-induced diabetic rats (235). Interestingly, expression of intestinal SGLT1 and GLUT2 has been reported to increase in patients with type 2 diabetes mellitus (236). Renal expression of these glucose transporters has not been investigated.
The PTs and thick ascending limbs (TALs) of the loop of Henle are susceptible to injury caused by episodes of hypoxia or exposure to nephrotoxins. Vimentin and kidney injury molecule-1 are the markers corresponding to tubular injury, and no basal expression of either of these two proteins can be observed in healthy rat kidneys (44). While these acute assaults often lead to loss of tubular cells, these injuries are only transient. This is because renal tubular cells can regenerate after such insults. However, researchers are still unable to completely discern the origin of the tubular cells that replenish the epithelial population after such damage. Whether regeneration depends on an intratubular progenitor or stem cell subpopulation or whether any enduring tubular cell survivor has the potential to divide remains controversial.
The type II Na+-coupled phosphate (Na/Pi) transporters are known to perform tubular reabsorption of inorganic phosphate (Pi), which is important for body functions, especially during periods of growth. In 1975, Baumann et al. (43) achieved the first transport assay of its kind to measure the build up of transtubular concentration differences of Pi in the renal PCT of rats and found that the rate of Pi reabsorption occurred with a higher rate in the early part of the tubule compared with the later part. Later, it has been demonstrated that Pi reabsorption is regulated by the apically localized Na/Pi cotransport system in PTs using different experimental systems including primary cultures of renal PTs, isolated tubules, and isolated brush-border membrane vesicles (45). The cloning of NaPi-2a (SLC34A1) (237) and NaPi-2c (SLC34A3) (238) phosphate transporters allowed the design of a new series of experiments to establish the physiological relevance of the isolated genes and characterize the mechanism of Pi reabsorption. While NaPi-2a reabsorbs ∼70% of Pi in the adult kidney, NaPi-2c does the remaining 30% (239). Despite a similar molecular structure, the two transporters exhibit physiological disparities, including how they adapt to dietary changes in phosphate. When Pi intake is high, NaPi-2a decreases within 1 h; however, NaPi-2c levels decrease slowly (4 h). NaPi-2a undergoes internalization in a microtubule-independent way where it is directed to the lysosomes for degradation under high phosphate conditions (240). In contrast, internalization of NaPi-2c takes place through a microtubule-dependent pathway, and instead of being targeted to lysosomes and degraded, it is accumulated in a subapical compartment (241). These differences in their regulation under dietary and hormonal stimuli and stability at the apical membrane are attributed to their differential binding to PDZ proteins, which are scaffolding proteins involved in the regulation of several receptor and transporter proteins (242). NaPi-2a has a class I PDZ-binding site at its COOH terminus (TRL) and is thus susceptible to regulation by several PDZ proteins, including NHERF family members (NHERF1, NHERF2, NHERF3, and NHERF4) (243). NaPi-2c interacts with NHERF1 and NHERF3 (PDZK1) despite the absence of a prototypical PDZ-binding motif in its COOH terminus (46).
PTs are also the site for the reabsorption of inorganic sulfate (), an indispensable and abundant anion in human plasma, which is essential for a variety of cellular processes. Stop-flow experiments in dogs revealed that circulating concentrations in plasma are mainly regulated by renal PTs and that high plasma values are likely the consequence of compromised kidney function (244). The apical membrane Na+-sulfate cotransporter NaS1 (SLC13A1) participates in sulfate reabsorption across PTs and thereby regulates blood sulfate levels (47). NaS1 prefers substrates such as sodium and sulfate, thiosulfate, and selenate. Expression of this transporter is downregulated by hypothyroidism, hypokalemia, metabolic acidosis, a high-sulfate diet, vitamin D depletion, glucocorticoids, and nonsteroidal anti-inflammatory drugs and upregulated vice versa by thyroid hormone, reduced dietary intake of sulfate, growth hormone, vitamin D supplementation, chronic renal failure, and during postnatal growth (47).
Sca-1 (also known as Ly6a) is an 18-kDa glycerophosphatidylinositol-anchored protein and a member of the Ly-6 protein family (48). It is used commonly as a marker for the detection and identification of stem cell and progenitor populations (245, 246). Sca-1 has been shown to be expressed heavily only in specific renal tubular epithelial segments of the adult mouse kidney (247), especially in the PT (but not in the CDs), a nephron segment that is extremely susceptible to IRI, and also in the loop of Henle (248). The study showed that epithelial Sca-1 can interact with TGF-β receptors I and II and inhibit TGF-β-induced canonical Smad signaling, a pathway instrumental in causing renal scarring due to fibrosis. Thus, Sca-1 has been recognized to afford a renoprotective role in IRI (248).
THE LOOP OF HENLE
The loop of Henle has a hairpin-like or a long U-shaped configuration that functions as an intermediate segment connecting the PT to the DCT. It is functionally divided into three distinct regions, e.g., the thin descending limb (DTL), the thin ascending limb (ATL), and the TAL. The glomerular filtrate is further reabsorbed in this region of nephron. Classical understanding from earlier studies is that while the DTL has high water (Pf) and low urea (Purea) permeabilities, the ATL has minimal Pf but extreme permeability to solutes like NaCl and moderate Purea. However, detailed studies on limb function, its ultrastructure, three-dimensional organization, and mathematical models have shown that the DTL has two functionally distinct segments (DTL-upper and DTL-lower). The upper 40% of the DTL has been shown to express AQP1, making it highly water permeable, but the lower 60% of the DTL lacks AQP1 channels and is thus impermeable to water (50). Furthermore, expression of the Cl− channel ClC-K1 has been shown to begin from the prebend segment at the terminal end of the DTL that continues all the way up to the ATL, accounting for high Cl− permeability. Both the DTL and ATL are now recognized as highly permeable to urea (249). The thin segments lie entirely within the renal medulla and are connected to the TAL, which is further subdivided into medullary and cortical compartments. Cells of the TAL differ from those residing in the thin limbs since they are metabolically more active and generate a steep electrochemical gradient for apical Na+ entry (due to the presence of highly active Na+-K+-ATPase). Overall, these segments are the key players in maintaining urine osmolarity.
Uromodulin (UMOD), a glycoprotein also known as Tamm-Horsfall protein (THP), is selectively expressed by epithelial cells lining the TAL of the loop of Henle and the early DCT (DCT1) in mammals (250). Mature UMOD, after its synthesis in the ER and extensive glycosylation, undergoes proteolytic cleavage by the serine protease hepsin and is released into the urine, representing the most abundant protein in human urine (250). UMOD has been implicated in concentrating the urine by exerting regulation on the Na+-K+-2Cl− cotransporter (NKCC2) (251) and renal outer medullary K+ channel (ROMK) (252) located in the TAL (253). UMOD also functions as a defense factor against ascending urinary tract infections (254) and kidney stones (255) and regulates innate immunity (256). Mutations in the UMOD gene cause defective maturation and impaired trafficking and retention of UMOD in the ER of cells located in the TAL segment (257), leading to a rare dominant progressive ER storage hereditary disease known as uromodulin-associated kidney disease (258). Hitherto, >70 uromodulin-associated kidney disease-causing UMOD mutations have been reported. Patients with uromodulin-associated kidney disease exhibit compromised urinary concentration ability, hyperuricemia associated with gout, progressive tubulointerstitial fibrosis with moderate infiltration of inflammatory cells, tubular atrophy, and renal cysts in some cases (259). Genome-wide association studies have identified UMOD as a risk factor for CKD, hypertension, and kidney stones and urinary UMOD as a useful biomarker for CKD progression (258).
The extracellular Ca2+-sensing receptor (CaSR) is a unique ∼120- to 160-kDa dimeric phospholipase C-sensitive G protein-coupled protein. CaSR, by releasing Ca2+ from the stores, functions as the master regulator of Ca2+ homeostasis (260). CaSR is expressed in high levels in the parathyroid gland, kidney, gut, and bone, where it regulates secretion of parathyroid hormone, synthesis of vitamin D (via its ability to regulate the expression of 1-α-hydroxylase enzyme), and absorption and resorption of Ca2+, respectively (261). Renal CaSR is also known to prevent hypercalcaemia, by promoting Ca2+ excretion (262) and by inhibiting 1,25-dihydroxyvitamin D [1,25(OH)2D] synthesis (263). Thus, gain- and loss-of-function mutations in the CASR gene account for severe disturbances in Ca2+ metabolism. For example, heterozygous mutations that inactivate the CASR gene cause benign (asymptomatic) familial hypocalciuric hypercalcemia, but those that are homozygous for inactivating mutations display severe hypercalcemia (264). On the other hand, CASR-activating mutations are the cause of the metabolic wasting disease known as autosomal dominant hypocalcemia or type V Bartter’s syndrome (265). In the rat kidney, abundant expression of CASR has been reported in the medullary and cortical parts of the TAL (266). Several other groups have confirmed these findings in mature mouse and human kidneys along with the developing human kidney (267–270). Besides CaSR expression in the TAL, Riccardi and colleagues (266) reported detection of CASR mRNA in glomeruli and in almost all other tubular segments, including the PCT, proximal straight tubule (PST), DCT, cortical CD (CCD), and inner medullary CD (IMCD) but not in the thin limbs and CNTs. Interestingly, Yang et al. (270) found CASR mRNA only in the DCT and CCD but not in the glomeruli or in any other tubular segments. Furthermore, using antibodies directed against CaSR, Loupy et al. (268) obtained conflicting results with no CaSR protein expression in the PCT, PST, DCT, CNT, or CCD as revealed by immunohistochemistry and immunofluorescence experiments. Thus, expression of CaSR in other parts of the kidney has remained controversial due to variable specificity and sensitivity of detection probes, antibodies, and even species (51, 52). Lately, nephron segment-specific roles of CaSR have been described (271). CaSR-mediated Ca2+ regulation is achieved by the interaction between CaSR and several variants of claudins (claudin-16 and claudin-19), which form a channel to reabsorb Ca2+ and Mg2+ (272). Apart from Ca2+ sensing, CaSR also controls ionic homeostasis by regulating the other cotransporters involved in ion reabsorption in the loop of Henle, namely, NKCC2, ATP-sensitive inward rectifier K+ channel ROMK1, and Cl− channel ClC-Kb (273). ClC-Kb is a voltage-gated Cl− channel that symports Cl− with Na+ from NKCC2 (274). Northern blot analysis revealed that the mRNA for this Cl− channel was expressed predominantly in rat kidneys, especially in the inner medulla, with a faint expression in the bladder (274). A gene expression study in microdissected nephron segments from rats revealed that the ATL of Henle’s loop, which has the highest Cl− permeability among the nephron segments, is the main site of ClC-Kb expression (274). This finding was later confirmed by immunohistochemistry and immunoelectron microscopy (53). Mutations in CLCNKB encoding kidney-specific ClC-Kb lead to Bartter’s syndrome type III, as Cl− is required to keep electroneutrality during salt resorption. Like NKCC2, ClC-Kb is regulated by CaSR (275).
Furosemide-sensitive NKCC2, with type 2 being the major subtype within the kidney, was identified as the major NaCl regulator located in the TALs in the rabbit and mouse (54–56). NKCC2 is negatively regulated by CaSR, by increased production of prostaglandin E2, which interferes with NKCC2 transport (276). Loss-of-function mutations of the SLC12A1 gene encoding NKCC2 lead to impaired renal salt retention and loss of NaCl via the tubules and TAL. The renal salt retention eventually leads to blood pressure abnormalities.
THE DISTAL CONVOLUTED TUBULE
The DCT is an extension of the TAL and starts shortly after the macula densa (MD). It is characterized by modest solute transport and very low water permeability (277). It further reabsorbs filtered NaCl and facilitates urinary dilution and generation of the osmotic gradient.
Ca2+ homeostasis is required for normal physiological functioning of all living cells. This is primarily achieved by three highly regulated systems: renal reabsorption, intestinal absorption, and bone resorption/formation. Any disturbance in these processes causes “hypercalciuria,” which is one of the leading risk factors for kidney stones. In the kidney, ∼45% of plasma Ca2+ filters through the glomerulus. However, to maintain a net Ca2+ balance, >98% of Ca2+ filtered at the glomerulus must be reabsorbed within the nephron, while in the PTs a major portion of filtered Ca2+ (∼65%) is passively reabsorbed (278), and the remaining 20% is reabsorbed in the TAL of Henle’s loop (58). Finally, regulation of Ca2+ occurs primarily in two segments of the distal nephron: the late part of the DCT (DCT2) (279) and the CNT (57). The DCT has two short segments: DCT1 (early) and DCT2 (late). Protein channels such as Na+-K+-ATPase, Na+/Ca2+ exchanger isoform 1 (NCX1), and plasma membrane ATPase type 1 b (PMCA1b) are ubiquitously expressed and found along the DCT2/CNT regions (57) in the human (280), rabbit (281), and mouse (58). However, DCT1 has been shown to express thiazide-sensitive Na+/Cl− cotransporter (NCC) and transient receptor potential melastatin subtype 6 (TRPM6) (59). The data demonstrated that the signal for NCC expression was strongest in cells of the DCT in both rats and humans, but it extended into the CNT in humans (59). Although the DCT was an acceptable site of expression for NCC in rodents, the site for the rabbit remained controversial. A combination of in situ hybridization and immunocytochemistry concluded that the DCT and not the CNT is the predominant site of NCC mRNA expression in rabbits as well (282). Furthermore, there are additional similarities between the DCT2 and CNT segments as both express transient receptor potential vanilloid type 5 (TRPV5) and the vitamin D-dependent Ca2+-binding protein calbindin-D28k (58). Calbindin-D28k has been shown to be present in the DCT and CNT of rat kidneys (283) and in the DCT and CCDs of rabbits (284) and in mice (285). Since the tight junctions in these segments are impermeable to Ca2+, Ca2+ reabsorption in the DCT2/CNT is mediated primarily by active transepithelial transport against the electrochemical gradient. Apart from these two segments, a mathematical model of PCs describing the synergistic action of the Na+/Ca2+ exchanger and Ca2+-regulated apical Na+ permeability offered important insights into Ca2+ dynamics in the CCD (286).
Most of the proteins that are filtered through the glomerulus are reabsorbed in the PT by receptor-mediated endocytosis via concerted action of the multiligand megalin-cubilin high-capacity receptor complex (287). It is believed that, under physiological conditions, the distal parts of the nephron avidly reabsorb filtered proteins, but neither the DCT nor CD involve receptor-mediated endocytosis for protein endocytosis (214). Moreover, the capacity for protein absorption in these segments is augmented when the efficiency of protein reabsorption in the PT is compromised after glomerular or PT damage. A study by Langelueddecke et al. (60) has shown that the receptor for neutrophil gelatinase-associated lipocalin (rodent 24p3/human lipocalin-2) is expressed in the DCT (not the PCT) and IMCDs of rat and mouse kidneys, where it orchestrates high-affinity receptor-mediated endocytosis of low (metallothionein)- and high (transferrin and albumin)-molecular-weight proteins, as tested in transiently transfected cultured cells. Thus, against the existing dogma, this study has cogently demonstrated that when proteins escape reabsorption in the PT due to a loss in its reabsorptive capacity following glomerular or PT damage, a surrogate high-affinity protein reabsorption mechanism in the distal nephron kicks in to compensate for the loss by carrying out exhaustive protein reabsorption, thereby limiting protein losses associated with several inherited or acquired renal diseases. In further support of this, a recent study by Weisz and colleagues (288) demonstrated the remarkable ability of PT cells to retrieve urinary albumin under normal and nephrotic conditions. The study concluded that while cubilin is responsible for the uptake of filtered albumin under normal conditions [a finding bolstered by another study where mutations in the cubilin gene were associated with albuminuria (289)], megalin had a negligible role. However, expression of megalin is essential for both active (cubilin dependent) and passive (incorporation in endocytic vesicles) uptake of albumin under normal and nephrotic states, respectively. Thus, when cubilin-mediated uptake is saturated, a second mechanism involving megalin-dependent passive uptake of albumin via its incorporation into endocytic vesicles accounts for the enormous capacity of PT cells to reclaim filtered albumin under nephrotic conditions (288).
THE COLLECTING DUCT
Originally, the CD was believed to not possess any specialized function except water reabsorption. Lately, essential and highly specific functions have been ascribed to this segment of the nephron as well. The main epithelial cell types present in the CD are PCs and ICs. ICs are specialized epithelial cells responsible for regulating acid-base homeostasis through the exchange of various ions via ion channels and pumps (290). There are three different types of ICs: type A (IC-A), type B (IC-B), and type C (non-A/non-B ICs) (291).
Due to the presence of H+-ATPase vacuolar-type proton pumps on the apical/luminal side, IC-As are the acid-secreting cells responsible for urinary acidification. While they also express anion exchanger 1 [Cl−/ exchanger (SLC4A1)], on the basolateral side, IC-As are marked by the absence of pendrin (SLC26A4), another Cl−/ exchanger. Additionally, SLC26A11, an electrogenic Cl− transporter as well as a Cl−/ anion exchanger, are also expressed at the apical pole of the IC-As (61). Loss-of-function mutations in the SLC4A1 gene cause abnormal functioning of kidney anion exchanger 1, leading to either autosomal dominant or recessive type 1 distal renal tubular acidosis syndrome. This results in defective bicarbonate reabsorption through the basolateral side of IC-As, leading to decreased urinary acidification and hyperchloremic, hypokalemic metabolic acidosis (292). On the other hand, inherited forms of distal renal tubular acidosis most often result from dysfunctional IC-As. Due to lack of acid secretion from IC-As, patients with distal renal tubular acidosis exhibit acidemia. The absence of acid secretion translates into an alkaline urine that ultimately leads to nephrocalcinosis and/or nephrolithiasis, along with hypokalemia (293).
IC-Bs, on the other hand, are basic/bicarbonate-secreting cells. Unlike IC-As, IC-Bs express H+-ATPase pumps on the basolateral pole and pendrin on the apical pole (61). IC-Bs have an electroneutral NaCl transport/reabsorption pathway that involves pendrin and the Na+-driven Cl−/ exchanger at their apical membrane (61).
Type C (non-A/non-B) ICs express both H+-ATPase pumps and pendrin on the apical side (61). All these PCs and ICs express major transporter proteins to maintain cellular osmolarity and acid-base homeostasis.
PCs express the epithelial Na+ channel (eNaC) and aquaporin 2. Other unique PC-associated markers are arginine vasopressin receptor 2 (AVPR2) and ROMK1. AVPR2 is a member of the G protein-coupled receptors. It has been postulated that in the kidney, AVPR2 is involved in electrolyte and acid-base balances and is located in various cell types within the kidney but primarily in PCs of the CD (62). Earlier studies have reported expression of AVPR2 mRNA in medullary CDs and CCDs in the developing kidneys of 16- and 19-day-old rat embryos (294). Later, evidence was provided for the localization of AVPR2 protein in the TAL, DCT, CNTs, and CDs in rat kidneys (295). While X-linked recessive congenital nephrogenic diabetes insipidus is caused by loss-of-function mutations in the AVPR2 gene, gain-of-function mutations in this gene lead to NS of inappropriate antidiuresis (296). ROMK1 is an ATP-dependent inwardly rectifying K+ channel (297) localized to the apical membranes of the distal nephron in the cortex and outer medulla, including the TAL and CNT but not in the glomerulus, PTs, and inner medulla of rats (298, 299). ROMK1 is positively regulated by CaSR. In humans, ROMK is encoded by the KCNJ1 gene, and loss-of-function mutations lead to impairment of salt retention in the kidney and are associated with a metabolic wasting disease termed “Bartter’s syndrome.” Genetically, Bartter syndrome is divided into five subtypes based on the mutations caused in five genes: type 1 (SLC12A1 encoding NKCC2), type 2 (KCNJ1 encoding ROMK1), type 3 (CLCNKB encoding ClC-Kb), type 4 (BSND encoding barttin, a subunit for ClC-Ka and ClC-Kb), and type 5 (CASR encoding CaSR). A closely related disease, termed Gitelman syndrome, is caused by dysfunctional NCC located in the tubules (300–304).
One of the leading causes of kidney failure is uncontrolled inflammation. Interestingly, proinflammatory pathways can be triggered even without an active infection. This process is termed as sterile inflammation. It has been shown that the purinergic receptor P2Y14 (GPR105) is expressed in abundance in ICs of the mouse CD and is responsible for mediating sterile inflammation (63). When cells are injured, they release UDP-glucose, which is a damage-associated molecular pattern molecule, which, in turn, activates P2Y14 (305). Although there is an established link between signaling via purinergic receptors and renal inflammation, the precise mechanism underlying their action is still unknown. Interestingly, copious purinergic receptors are expressed in the kidney, and deregulation of purinergic signaling is correlated with hypertension, CKD, acute kidney injury (AKI), DN, and glomerulonephritis (306, 307).
Urea transporters (UTs) are composed of two main types: UT-A and UT-B. While UT-As are majorly expressed in kidneys, UT-Bs are expressed in other tissues as well, including the brain. UT-A1, UT-A2, and UT-A3 are expressed in the PCs, IMCD, and TAL, respectively. The major function of UTs in the kidney is to concentrate urea. Currently, UT-A and UT-B are targets for diuretic drug treatment (64, 65, 308, 309).
CELL-BASED MARKERS
The kidney is composed of a series of specialized cell types possessing unique permeability characteristics and transport capabilities. Here, we summarize the markers present in major individual renal cells, which are necessary for the establishment of cellular interactions, polarity, filtration, and transport.
Endothelium-Associated Markers
Glomerular capillaries are lined with highly specialized (e.g., flattened and fenestrated) endothelium, which regulates the rapid flux of fluid and small solutes across the glomerular barrier (310). Glomerular ECs, along with the negatively charged luminal glycocalyx layer (consisting of a network of proteoglycans and glycoproteins), interact with both podocytes and mesangial cells and regulate intraglomerular hemodynamics, vascular reactive oxygen species production, prothrombotic and antithrombotic factors, and fibrosis (311, 312).
Nitric oxide (NO), a lipophilic gas has been implicated in many important physiological functions associated with kidneys such as regulation of renal blood flow and glomerular filtration rate, promotes natriuresis and diuresis so that kidneys are equipped to adapt to variations in dietary salt intake and thus help maintain normal blood pressure. NO is produced by a catalytic reaction that involves NO synthase (NOS). Three isoforms of NOS have been identified so far: neuronal NOS (nNOS or NOSI), inducible NOS (iNOS or NOSII), and endothelial NOS (eNOS or NOSIII) (313). Interestingly, expression of all three isoforms has been detected in kidney tissues but with different distribution profiles (66). For example, eNOS was found to be expressed in ECs of glomeruli and peritubular capillaries in the cortex and in the ECs of vascular bundles in the medulla, with a stronger immunoreactivity in cells of the renal medulla than in the cortex (314). Its expression was also found in epithelia of the TAL of the loop of Henle and CD (315). nNOS was detected predominantly in tubular epithelial cells of the MD (314) in the kidneys of rats, mice, guinea pigs, and rabbits; the intensity of the signal was much weaker in humans and pigs (316). Additionally, nNOS was also detected in the IMCD and outer medullary CD, efferent arterioles, Bowman’s capsule, and various cells at the cortical TAL in rats (317). While the mRNA transcript corresponding to iNOS has been detected in the S3 segment of the PT, cortical and medullary TAL, DCT, CCD, and IMCD in normal rat kidneys (318), expression of iNOS protein was observed in the PT, TAL, and ICs of the CD (205). Furthermore, the expression patterns of these isoforms are uniquely distinct in the developing kidneys from that observed in adults (313). Nevertheless, owing to the diffusibility of NO, it is possible that even though it is not produced locally in a nephron segment, it can still have an impact on that segment or neighboring structures. Chronic renal disease is characterized by compromised NO bioactivity. Levels of eNOS mRNA and protein as well as its activity are reduced substantially in human EC cultures exposed to erythrocytes from patients with ESRD (319). Furthermore, eNOS knockout mice develop congenital anomalies, including glomerular hypoplasia (i.e., reduction in the number of nephrons), tubular cell death, and atubular glomeruli (320, 321). eNOS deficiency has also been shown to predispose podocytes to injury in diabetes (322).
Platelet endothelial cell adhesion molecule-1 (PECAM-1 or CD31), a 130-kDa glycoprotein, consists of a single-chain molecule with an extracellular domain (composed of six Ig-like domains), a transmembrane domain, and a relatively long cytoplasmic domain. Since PECAM-1 exists in at least eight isoforms generated by alternative splicing of multiple exons encoding its cytoplasmic tail, differential combinations of these exons impact its interactions with potential intracellular signaling molecules. PECAM-1 expression is found in fenestrated renal glomerular and peritubular ECs (323), which is lost in sclerotic or fibrotic areas (67).
Circulating active levels of von Willebrand factor (vWF; factor VIII-related antigen) have shown to be increased in CKD and ESRD (68). Furthermore, vWF has been shown to contribute to the pathogenesis of acute IRI (324). Although staining for vWF has always been diffusively positive or completely negative in the fenestrated endothelium of glomeruli, its expression on ECs increases in renal glomeruli of patients with hypertension (323).
EC junctions such as tight junctions (zonulae occludens), adherent junctions (zonulae adherens), and gap junctions (desmosomes) play an important role in cellular homeostasis. Vascular-endothelial cadherin (VE-cadherin), a Ca2+-dependent transmembrane glycoprotein comprising the adherent junctions between ECs, regulates vascular integrity, endothelial barrier function, and angiogenesis (69). During inflammation, the extracellular domain of VE-cadherin undergoes proteolytic cleavage followed by shedding into the circulation as sVE-cadherin (325). Although the association of tubular epithelial injury with AKI has been exhaustively studied, the contribution of endothelial injury has not been ascertained. Fairly recently, a study by Yu et al. (326) demonstrated a direct correlation between shedding of sVE-cadherin and severe AKI and in patients with severe organ dysfunction and sepsis, suggesting that breakdown of endothelial adherent junctions contributes to renal pathogenesis. Furthermore, uremia has been reported to alter cell-to-cell junctions in ECs and impact the expression of VE-cadherin and ZO-1 (327).
Fibroblast-Associated Markers
In a seminal study on kidney fibrosis by Strutz and coworkers (70), fibroblast-specific protein 1 (FSP-1) was recognized as a marker for cells undergoing epithelial-mesenchymal transition (EMT). Authors showed that FSP-1 protein was expressed by fibroblasts and not epithelial cells and thus coined the term “fibroblast-specific protein-1.” Furthermore, the study demonstrated that while there was a low incidence of FSP-1-positive cells in healthy kidneys, a massive increase was observed in tubular epithelia in renal fibrosis, indicating that under pathological conditions, interstitial fibroblasts originate from tubular cells undergoing EMT. This work led to the widespread use of FSP-1 antibodies for the identification and detection of fibroblasts and tubular epithelium undergoing EMT (328), even though it was a persistent matter of debate in the field.
Nearly all progressive renal diseases funnel into renal fibrosis as an ultimate consequence culminating in ESRD. PDGFs essentially participate in driving processes that ultimately lead to fibrosis (329). The PDGF system consists of four isoforms: PDGF-A, PDGF-B, PDGF-C, and PDGF-D and two receptors (PDGFR-α and/or PDGFR-β chains). These receptors dimerize upon ligand binding. Whereas PDGF-A binds to the PDGFR-αα dimer only, PDGF-B has been shown to function as a ligand for all receptors. PDGF-C, on the other hand, binds to PDGFR-αα and PDGFR-αβ, whereas PDGF-D predominantly binds to PDGFR-ββ. While these two receptor chains are constitutively expressed by mesangial cells, fibroblasts, and vascular smooth muscle cells, they are not expressed by epithelial cells, such as podocytes and tubular cells, under either normal or pathological conditions (71). Upregulated PDGF and its receptor expression have been reported in nearly all rodent models of kidney injury including puromycin-induced FSGS, DN, murine lupus nephritis, IRI, mesangioproliferative anti-Thy 1.1 glomerulonephritis, nephrotoxic nephritis, ANG II-induced renal damage, murine IgA nephropathy, renal transplantation, and their corresponding human renal diseases (330).
For organs such as the kidney, where there is negligible cell turnover, cells from all renal compartments undergo decay and thus need to be replaced. In the absence of such a regeneration process, a progressive loss of the structural and functional integrity is inevitable. PDGF-B and fibroblast PDGFR-β signaling are known to assist wound healing in other organs. In contrast to other organs, little is known about the proregenerative effects of renal fibroblasts in the kidney. In a study on proximal tubular regeneration, Schiessl et al. (331) demonstrated targeted migration of interstitial PDGFR-β-positive cells to replace the cellular loss upon ablation of single tubular cells by focused laser exposure.
Mesangial Cell-Associated Markers
Renal mesangial cells are contractile irregularly shaped cells within the renal corpuscle (332), which are exclusively located in the glomerulus. They maintain the mesangial matrix and provide structural support for glomerular capillary loops. Other functions include communication with other glomerular cells by secreting growth factors and contribution to the regulation of glomerular capillary flow. Therefore, mesangial cell injury can lead to FP effacement and proteinuria (333).
Transient receptor potential canonical member 1 (TRPC1), alongside with other TRPCs (possessing seven isoforms), has been identified in cultured human mesangial cells by Western blot analysis (73) and by an immunofluorescence study in the rat kidney (72). Goel and colleagues (72) showed that TRPC1 is expressed in various parts of the rat kidney such as the tubules and DTL; however, they found that TRPC1 and transient receptor potential canonical member 4 (TRPC4) are the sole ion channels in the mouse mesangial cell line. Sours-Brothers colleagues (334) also showed that TRPC1 could physically interact with TRPC4 and TRPC6. The same group later demonstrated the possibility that TRPC1/TRPC4 interacts with stromal interaction molecule 1, which is a form of the store-operated Ca2+ channel, suggesting that TRPC1 could be part of the regulation of store-operated Ca2+ channel-mediated Ca2+ entry (334).
Renal expression of PDGFs and their receptors (PDGFRs) are well documented, as discussed above. Both PDGFR-α (74) and PDGFR-β (335, 336) are constitutively expressed by glomerular mesangial cells, interstitial fibroblasts, and vascular smooth muscle cells. PDGFR-β has been recognized as a marker for mesenchymal cells, and focal expression of PDGFR-β has also been described on parietal epithelial cells. Although PDGFRs are not expressed on glomerular ECs in vivo, glomerular ECs did express PDGFR-α and responded to PDGF-CC in vitro. Thus, PDGFR-α/PDGF-AA and PDGF-CC might have a role in glomerular capillary healing and angiogenesis (337).
Epithelial Cell-Associated Markers
The general architecture of renal epithelial cells as well as their pattern of interaction with each other and with the surrounding extracellular matrix are important for the extracellular scaffolding and maintenance of the functionally distinct segments of the kidney. Due to their ubiquity and unique characteristics, epithelial cell lines have been used widely in cell-based assays to investigate the alterations in epithelial physiology and signaling and stress pathways. Although cell culture models are widely used, it is imperative to mention that the choice of cell line and their growth conditions can significantly alter the results because they may not fully recapitulate the key aspects of a cell type or its in vivo environment. For instance, a recent study by Khundmiri et al. (338) demonstrated that transcripts expressed in none of the 14 PT cell lines fully matched the transcriptome of native PT cells. The transcripts not only matched partially but also variably with the highest percent match obtained in the opossum kidney (OK cells, 45% of proximal marker genes) followed by the pig kidney (LLC-PK1, 39%), and lower percent matches were seen in human cell lines (HK-2 cells, 26%) and lines from rodent kidneys (NRK-52E cells, 23%) (338). Park et al. (339) also substantiated this by demonstrating that OK cells cultured under continuous orbital shear stress are morphologically and functionally more similar to PT cells in vivo compared with cells maintained under static conditions, necessitating a thorough validation of conclusions derived from cell culture experiments.
ENaC [also known as Na+ channel nonneuronal 1 (SCNN1) or amiloride-sensitive Na+ channel (ASSC)] is expressed on the apical pole of epithelial cells in the distal nephron (75). The functional channel consists of an assembly of three subunits encoded by three genes: α (SCNN1A), β (SCNN1B), and γ (SCNN1G), with a stoichiometry of 1:1:1. These channels belong to the ENaC/degenerin family of proton-gated cation channels that mediate transport of Na+ through the apical membrane from the lumen into the epithelial cell (340). Since ENaCs precisely regulate Na+ balance, they play a key role in maintaining blood pressure (341). Mice devoid of any ENaC subunit succumb to kidney dysfunction and do not survive (342). Similarly, germline mutations (frameshift or missense) in an allele of SCNN1A, SCNN1B, or SCNN1G genes causes autosomal dominant Liddle syndrome (pseudoaldosteronism), characterized by an early onset of salt-sensitive hypertension with a low level of K+, metabolic alkalosis, inhibition of renin activity, and aldosterone secretion (343).
Cadherins represent one of the most important molecules that participate in renal epithelial cell-cell adhesion and are usually found at the adherens junction of epithelial cells (76). While over 50 cadherins have been described (344), the most exhaustively characterized are the classical type I cadherins: E-cadherin and N-cadherin (345). Cadherins are integral, transmembrane proteins with an extracellular domain containing Ca2+-binding sites and regions that impart them adhesive properties (346). The intracellular domain interacts with β-catenin, which is bound to α-catenin, which, in turn, connects the entire complex to the actin cytoskeleton (347). In the adult kidney, the predominant cadherins are classical N-cadherin and E-cadherin (348). The other forms, like OB-cadherin, R-cadherin, and K-cadherin, are transiently expressed during different stages of development (349). Interestingly, atypical kidney-specific cadherin (Ksp-cadherin) has been reported. Ksp-cadherin lacks the cytoplasmic catenin-binding domain, and this is how it differs from classical cadherins (350, 351). However, the functional relevance of this atypical cadherin is still elusive. Furthermore, there is a plethora of evidence that indicates differential expression of these classical cadherins in various segments of the nephron. However, these mapping studies were equivocal and difficult to interpret because of the use of different cadherin panels, different species, and poor techniques to identify the specificity of tubular segments. Therefore, a study by Prozialeck et al. (349) used dual immunofluorescence labeling technique to specifically target E-cadherin or N-cadherin, along with antibodies that serve as markers for specific nephron segments, in the adult rat kidney. The results showed that while N-cadherin is the predominant cadherin present in the PT, E-cadherin is abundant in the DCT, CD, and most medullary segments and is expressed in negligible amounts in the PT (349). In the human kidney, a complex pattern of cadherin expression has been observed (352). Epithelial cells of PTs were found to express N-cadherin and cadherin-6 (K-cadherin) (353), whereas in DCTs, E-cadherin (348) and cadherin-16 (Ksp-cadherin) (354) were present. Additionally, R-cadherin (355) and cadherin-8 (356) have been reported in the differentiating metanephric mesenchyme when kidney development is initiated. A change in E-cadherin expression or cadherin switch (357) in the kidney has been used as a surrogate marker of EMT (358).
Macula Densa Markers
The MD is a unique collection of epithelial cells located at the distal end of the TAL. Cells in MD are known to function as salt sensors that detect changes in distal tubular fluid composition. In response to these changes, MD cells transmit chemical signals to the juxtaglomerular apparatus to control renal hemodynamics, filtration rate, and renin release, a process known as tubuloglomerular feedback (359). Changes in NaCl concentration are fine tuned by a concerted action of ion transport-related intracellular events. NaCl entry into the MD cells is primarily regulated by apically located NKCC2 (BSC1). Interestingly, of the three known splice variants of NKCC2 (A, B, and F), which differ in their ionic affinities and regulation and vary in their distribution along the TAL (360–362), MD cells were shown to express the B isoform of NKCC2 (77), which has the highest affinity for Cl− relative to the other two isoforms (360, 362). With the cotransport of NaCl, the increased cellular Cl− is expelled through a basolateral channel, which results in cell depolarization along with a modest increase in cytosolic Ca2+ (78). Using patch-clamp experiments, Lapointe et al. (363) reported that upon an elevation in Ca2+ level, a nonselective cation channel, which is like transient receptor potential melastatin subtype 4, with moderate Ca2+ permeability gets activated in the basolateral membrane of MD cells. Along with NKCC2, MD cells also possess Na+/H+ exchange activity that causes cell alkalization. Functional and immunological techniques have identified that this is mediated predominantly by apical NHE2 and basolateral NHE4 isoforms (78). Traditionally, studying live MD cells has been a challenge due to their low number in each nephron. However, in a recent study, Gyarmati et al. (364) developed a method to analyze live MD cells. A chimera of a MD-specific marker, nNOS, and a tamoxifen-inducible membrane targeted green fluorescent protein was generated, and, using this, they were able to visualize the three-dimensional projections (also called the maculopodia) of MD cells (364).
SUMMARY
One of the most outstanding fundamental questions in human physiology is what types of cells form different tissues in an organ so that it can perform the requisite physiological functions. Furthermore, within those specific cell types, what are the defining marker proteins and how can an imbalance (environmental/extrinsic factors) or mutations (genetic/intrinsic factors) in those proteins lead to debilitating disease phenotypes? Historically, many cell markers have been identified through traditional and conventional cell biology experiments (e.g., immunofluorescence or immunohistochemistry). These cell markers were then expansively used to identify, isolate, or classify cell types of interest by exploiting techniques such as FACS, proteomics, confocal, deconvolution, or multiphoton microscopy. However, with the advent of scRNA-seq, more accurate characterization of cell-based markers that are unique to different cell types has become possible. Although scRNA-seq is a promising tool to investigate differential gene expression between control and impacted cells, it does not provide insights into gene regulation. Since understanding the epigenetic program that leads to a unique cell type in the kidney is essential, seminal work from Miao and colleagues (365) providing a chromatin map of the developing and adult mouse kidney based on epigenome classification at single cell resolution is extremely timely. Additionally, a recent study has exploited a FACS-based approach to first isolate and enrich specific nephron segments that have ill-defined boundaries like DCT1/DCT2 and DCT2/CNT and then couple it with scRNA-seq to resolve the cellular composition and transcriptional profiles of minority epithelial cell types in the mouse kidney distal nephron (366). However, even with the large amount of relevant and useful data that emerges from contemporary scRNA-seq and other conventional techniques, researchers find this information on cell-based markers overwhelming and strewn over thousands of publications. Furthermore, the choice of a reliable cell surface marker has always remained a controversy. This review on renal cell-based markers is a small attempt to bridge this big chasm. We first generated a nephron map that provided a cellular landscape and then compiled a list of proteins unique to a specific cell type, which is in a specific segment of nephron in the human kidney (Fig. 1). We believe that this manually curated database will serve as a comprehensive resource for renal cell markers, which can facilitate researchers worldwide to comprehensively and reliably apply the existing information on cell-based markers to their specific study. Furthermore, as we continue to advance our understanding of these proteins and multiprotein assemblies in the kidney, such information on cell surface markers will prove indispensable in the diagnosis and treatment of renal diseases. Nonetheless, considering the speed in which rare cell types and new marker proteins are identified, it is difficult to cope up and scope up with the ever-expanding arsenal of information. Therefore, we acknowledge that our inventory is only comprehensive and not exhaustive.
Figure 1.

Renal cell markers. Shown is a cartoon illustrating the spatial arrangement of a nephron with the various domains segmented and associated markers indicated in a box. The image is not drawn to scale. PDGFR, platelet-derived growth factor receptor; α-SMA, α-smooth muscle actin; TRPC, transient receptor potential canonical ion channel; eNOS, endothelial nitric oxide synthase; vWF, von Willebrand factor; PECAM-1, platelet endothelial cell adhesion molecule-1; NKCC2, Na+-K+-2Cl− cotransporter; NXC1, Na+/Ca2+ exchanger isoform 1; FSP-1, fibroblast-specific protein 1; SCNN1, Na+ channel nonneuronal 1; Sca-1, stem cell antigen-1; NCC, Na+/Cl− cotransporter; GLEPP1; glomerular epithelial protein 1; WT-1, Wilms’ tumor-1; CLIC5, Cl− intracellular channel protein 5; CaSR, Ca2+-sensing receptor; NaS1, Na+-sulfate cotransporter; NaPi, type II Na+-coupled phosphate; AE1, anion exchanger 1; UT-A1, urea transporter A1.
DISCLOSURES
J.R. has patents on novel strategies for kidney therapeutics and stands to gain royalties from their commercialization. He is the co-founder of Walden Biosciences (Cambridge, MA), a biotechnology company in which he has financial interest, including stock. Other authors have nothing to disclose and there are no competing interests.
AUTHOR CONTRIBUTIONS
J.R. and M.M.A. conceived the idea and manuscript design; S.A., Y.R.S., O.K.P., and M.M.A. drafted the manuscript; S.A., Y.R.S., and M.M.A. prepared figures and table; S.A., Y.R.S., O.K.P., and M.M.A. edited and revised the manuscript; S.A., Y.R.S., O.K.P., J.R., and M.M.A. approved the final version of the manuscript.
REFERENCES
- 1.Madsen KM, Tisher CC. Structural-functional relationships along the distal nephron. Am J Physiol Renal Physiol 250: F1–F15, 1986. doi: 10.1152/ajprenal.1986.250.1.F1. [DOI] [PubMed] [Google Scholar]
- 2.Muto Y, Wilson PC, Ledru N, Wu H, Dimke H, Waikar SS, Humphreys BD. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun 12: 2190, 2021. doi: 10.1038/s41467-021-22368-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hebert SC. Nephron heterogeneity. In: Comprehensive Physiology, edited by Terjung R, 2011. doi: 10.1002/cphy.cp080120. [DOI] [Google Scholar]
- 4.Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW, Knepper MA. Representation and relative abundance of cell-type selective markers in whole-kidney RNA-Seq data. Kidney Int 95: 787–796, 2019. doi: 10.1016/j.kint.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park J, Liu CL, Kim J, Susztak K. Understanding the kidney one cell at a time. Kidney Int 96: 862–870, 2019. doi: 10.1016/j.kint.2019.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao J, Yu Z, Chen Y, Bao M, Zou C, Zhang H, Liu D, Li T, Zhang Q, Li J, Cheng J, Mo Z. Single-cell RNA sequencing of human kidney. Sci Data 7: 4, 2020. doi: 10.1038/s41597-019-0351-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell rna sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30: 23–32, 2019. doi: 10.1681/ASN.2018090912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20: 322–332, 2009. doi: 10.1681/ASN.2008070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angelotti ML, Ronconi E, Ballerini L, Peired A, Mazzinghi B, Sagrinati C, Parente E, Gacci M, Carini M, Rotondi M, Fogo AB, Lazzeri E, Lasagni L, Romagnani P. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 30: 1714–1725, 2012. doi: 10.1002/stem.1130. [DOI] [PubMed] [Google Scholar]
- 10.Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, Serio M, Romagnani S, Romagnani P. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol 17: 2443–2456, 2006. doi: 10.1681/ASN.2006010089. [DOI] [PubMed] [Google Scholar]
- 11.Bruno S, Camussi G. Isolation and characterization of resident mesenchymal stem cells in human glomeruli. Methods Mol Biol 879: 367–380, 2012. doi: 10.1007/978-1-61779-815-3_22. [DOI] [PubMed] [Google Scholar]
- 12.Leuning DG, Engelse MA, Lievers E, Bijkerk R, Reinders ME, de Boer HC, van Kooten C, Rabelink TJ. The human kidney capsule contains a functionally distinct mesenchymal stromal cell population. PLoS One 12: e0187118, 2017. doi: 10.1371/journal.pone.0187118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lazzeri E, Crescioli C, Ronconi E, Mazzinghi B, Sagrinati C, Netti GS, Angelotti ML, Parente E, Ballerini L, Cosmi L, Maggi L, Gesualdo L, Rotondi M, Annunziato F, Maggi E, Lasagni L, Serio M, Romagnani S, Vannelli GB, Romagnani P. Regenerative potential of embryonic renal multipotent progenitors in acute renal failure. J Am Soc Nephrol 18: 3128–3138, 2007. doi: 10.1681/ASN.2007020210. [DOI] [PubMed] [Google Scholar]
- 14.Park HC, Yasuda K, Kuo MC, Ni J, Ratliff B, Chander P, Goligorsky MS. Renal capsule as a stem cell niche. Am J Physiol Renal Physiol 298: F1254–F1262, 2010. doi: 10.1152/ajprenal.00406.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pippin JW, Kaverina NV, Eng DG, Krofft RD, Glenn ST, Duffield JS, Gross KW, Shankland SJ. Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am J Physiol Renal Physiol 309: F341–F358, 2015. doi: 10.1152/ajprenal.00438.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altintas MM, Reiser J. Bridges to cross, burn, and mend: cells of renin lineage as podocyte progenitors. Am J Physiol Renal Physiol 309: F499–F500, 2015. doi: 10.1152/ajprenal.00301.2015. [DOI] [PubMed] [Google Scholar]
- 17.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998. doi: 10.1016/S1097-2765(00)80057-X. [DOI] [PubMed] [Google Scholar]
- 18.Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestila M, Jalanko H, Holmberg C, Tryggvason K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A 96: 7962–7967, 1999. doi: 10.1073/pnas.96.14.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol 10: 2440–2445, 1999. doi: 10.1681/ASN.V10112440. [DOI] [PubMed] [Google Scholar]
- 20.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000. [Erratum in Nat Genet 25: 125, 2000]. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Ruotsalainen V, Tryggvason K, Shaw AS, Miner JH. CD2AP is expressed with nephrin in developing podocytes and is found widely in mature kidney and elsewhere. Am J Physiol Renal Physiol 279: F785–F792, 2000. doi: 10.1152/ajprenal.2000.279.4.F785. [DOI] [PubMed] [Google Scholar]
- 22.Dustin ML, Olszowy MW, Holdorf AD, Li J, Bromley S, Desai N, Widder P, Rosenberger F, van der Merwe PA, Allen PM, Shaw AS. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell 94: 667–677, 1998. doi: 10.1016/S0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 23.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286: 312–315, 1999. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 24.Asanuma K, Campbell KN, Kim K, Faul C, Mundel P. Nuclear relocation of the nephrin and CD2AP-binding protein dendrin promotes apoptosis of podocytes. Proc Natl Acad Sci USA 104: 10134–10139, 2007. doi: 10.1073/pnas.0700917104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neuner-Jehle M, Denizot JP, BorbéLy AA, Mallet J. Characterization and sleep deprivation-induced expression modulation of dendrin, a novel dendritic protein in rat brain neurons. J Neurosci Res 46: 138–151, 1996. doi: 10.1002/(SICI)1097-4547(19961015)46:2<138::AID-JNR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 26.Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol 139: 193–204, 1997. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otey CA, Carpen O. Alpha-actinin revisited: a fresh look at an old player. Cell Motil Cytoskeleton 58: 104–111, 2004. doi: 10.1002/cm.20007. [DOI] [PubMed] [Google Scholar]
- 28.Kreidberg JA. Podocyte differentiation and glomerulogenesis. J Am Soc Nephrol 14: 806–814, 2003. doi: 10.1097/01.ASN.0000054887.42550.14. [DOI] [PubMed] [Google Scholar]
- 29.Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, Mathur BN, Turner CA, Geske R, Montgomery CA, Starbuck M, Brandt M, Gupta A, Ramirez-Solis R, Zambrowicz BP, Powell DR. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol 21: 4829–4836, 2001. doi: 10.1128/MCB.21.14.4829-4836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnabel E, Dekan G, Miettinen A, Farquhar MG. Biogenesis of podocalyxin–the major glomerular sialoglycoprotein–in the newborn rat kidney. Eur J Cell Biol 48: 313–326, 1989. [PubMed] [Google Scholar]
- 31.Kerjaschki D, Sharkey DJ, Farquhar MG. Identification and characterization of podocalyxin–the major sialoprotein of the renal glomerular epithelial cell. J Cell Biol 98: 1591–1596, 1984. doi: 10.1083/jcb.98.4.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierchala BA, Munoz MR, Tsui CC. Proteomic analysis of the slit diaphragm complex: CLIC5 is a protein critical for podocyte morphology and function. Kidney Int 78: 868–882, 2010. doi: 10.1038/ki.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas PE, Wharram BL, Goyal M, Wiggins JE, Holzman LB, Wiggins RC. GLEPP1, a renal glomerular epithelial cell (podocyte) membrane protein-tyrosine phosphatase. Identification, molecular cloning, and characterization in rabbit. J Biol Chem 269: 19953–19962, 1994. doi: 10.1016/S0021-9258(17)32113-0. [DOI] [PubMed] [Google Scholar]
- 34.Breiteneder-Geleff S, Matsui K, Soleiman A, Meraner P, Poczewski H, Kalt R, Schaffner G, Kerjaschki D. Podoplanin, novel 43-kD membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am J Pathol 151: 1141–1152, 1997. [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkkoetter PT, Wu JS, Ohse T, Krofft RD, Schermer B, Benzing T, Pippin JW, Shankland SJ. p35, the non-cyclin activator of Cdk5, protects podocytes against apoptosis in vitro and in vivo. Kidney Int 77: 690–699, 2010. doi: 10.1038/ki.2009.548. [DOI] [PubMed] [Google Scholar]
- 36.Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci U S A 79: 5557–5561, 1982. doi: 10.1073/pnas.79.18.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birn H, Fyfe JC, Jacobsen C, Mounier F, Verroust PJ, Orskov H, Willnow TE, Moestrup SK, Christensen EI. Cubilin is an albumin binding protein important for renal tubular albumin reabsorption. J Clin Invest 105: 1353–1361, 2000. doi: 10.1172/JCI8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poulsen SB, Fenton RA, Rieg T. Sodium-glucose cotransport. Curr Opin Nephrol Hypertens 24: 463–469, 2015. doi: 10.1097/MNH.0000000000000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, Radovic N, Jadrijevic S, Aleksic I, Walles T, Sauvant C, Sabolic I, Koepsell H. Localizations of Na+-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467: 1881–1898, 2015. doi: 10.1007/s00424-014-1619-7. [DOI] [PubMed] [Google Scholar]
- 41.Song P, Onishi A, Koepsell H, Vallon V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin Ther Targets 20: 1109–1125, 2016. doi: 10.1517/14728222.2016.1168808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 43.Baumann K, de Rouffignac C, Roinel N, Rumrich G, Ullrich KJ. Renal phosphate transport: inhomogeneity of local proximal transport rates and sodium dependence. Pflugers Arch 356: 287–298, 1975. doi: 10.1007/BF00580003. [DOI] [PubMed] [Google Scholar]
- 44.Kusaba T, Lalli M, Kramann R, Kobayashi A, Humphreys BD. Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci USA 111: 1527–1532, 2014. [Erratum in Proc Natl Acad Sci U S A 111: 5754, 2014]. doi: 10.1073/pnas.1310653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murer H, Werner A, Reshkin S, Wuarin F, Biber J. Cellular mechanisms in proximal tubular reabsorption of inorganic phosphate. Am J Physiol Cell Physiol 260: C885–C899, 1991. doi: 10.1152/ajpcell.1991.260.5.C885. [DOI] [PubMed] [Google Scholar]
- 46.Villa-Bellosta R, Barac-Nieto M, Breusegem SY, Barry NP, Levi M, Sorribas V. Interactions of the growth-related, type IIc renal sodium/phosphate cotransporter with PDZ proteins. Kidney Int 73: 456–464, 2008. doi: 10.1038/sj.ki.5002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markovich D. Na+-sulfate cotransporter SLC13A1. Pflugers Arch 466: 131–137, 2014. doi: 10.1007/s00424-013-1388-8. [DOI] [PubMed] [Google Scholar]
- 48.Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells 25: 1339–1347, 2007. doi: 10.1634/stemcells.2006-0644. [DOI] [PubMed] [Google Scholar]
- 49.Baer PC, Nockher WA, Haase W, Scherberich JE. Isolation of proximal and distal tubule cells from human kidney by immunomagnetic separation. Technical note. Kidney Int 52: 1321–1331, 1997. doi: 10.1038/ki.1997.457. [DOI] [PubMed] [Google Scholar]
- 50.Nawata CM, Evans KK, Dantzler WH, Pannabecker TL. Transepithelial water and urea permeabilities of isolated perfused Munich-Wistar rat inner medullary thin limbs of Henle’s loop. Am J Physiol Renal Physiol 306: F123–F129, 2014. doi: 10.1152/ajprenal.00491.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chattopadhyay N, Baum M, Bai M, Riccardi D, Hebert SC, Harris HW, Brown EM. Ontogeny of the extracellular calcium-sensing receptor in rat kidney. Am J Physiol Renal Physiol 271: F736–F743, 1996. doi: 10.1152/ajprenal.1996.271.3.F736. [DOI] [PubMed] [Google Scholar]
- 52.Graca JA, Schepelmann M, Brennan SC, Reens J, Chang W, Yan P, Toka H, Riccardi D, Price SA. Comparative expression of the extracellular calcium-sensing receptor in the mouse, rat, and human kidney. Am J Physiol Renal Physiol 310: F518–F533, 2016. doi: 10.1152/ajprenal.00208.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uchida S, Sasaki S, Nitta K, Uchida K, Horita S, Nihei H, Marumo F. Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J Clin Invest 95: 104–113, 1995. doi: 10.1172/JCI117626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greger R. Chloride reabsorption in the rabbit cortical thick ascending limb of the loop of Henle. A sodium dependent process. Pflugers Arch 390: 38–43, 1981. doi: 10.1007/BF00582708. [DOI] [PubMed] [Google Scholar]
- 55.Greger R, Schlatter E. Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch 392: 92–94, 1981. doi: 10.1007/BF00584588. [DOI] [PubMed] [Google Scholar]
- 56.Hebert SC, Culpepper RM, Andreoli TE. NaCl transport in mouse medullary thick ascending limbs. I. Functional nephron heterogeneity and ADH-stimulated NaCl cotransport. Am J Physiol Renal Physiol 241: F412–F431, 1981. doi: 10.1152/ajprenal.1981.241.4.F412. [DOI] [PubMed] [Google Scholar]
- 57.Boros S, Bindels RJ, Hoenderop JG. Active Ca2+ reabsorption in the connecting tubule. Pflugers Arch 458: 99–109, 2009. doi: 10.1007/s00424-008-0602-6. [DOI] [PubMed] [Google Scholar]
- 58.Loffing J, Loffing-Cueni D, Valderrabano V, Klausli L, Hebert SC, Rossier BC, Hoenderop JG, Bindels RJ, Kaissling B. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol 281: F1021–F1027, 2001. doi: 10.1152/ajprenal.0085.2001. [DOI] [PubMed] [Google Scholar]
- 59.Obermuller N, Bernstein P, Velazquez H, Reilly R, Moser D, Ellison DH, Bachmann S. Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am J Physiol Renal Physiol 269: F900–F910, 1995. doi: 10.1152/ajprenal.1995.269.6.F900. [DOI] [PubMed] [Google Scholar]
- 60.Langelueddecke C, Roussa E, Fenton RA, Wolff NA, Lee WK, Thevenod F. Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J Biol Chem 287: 159–169, 2012. doi: 10.1074/jbc.M111.308296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roy A, Al-Bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. doi: 10.2215/CJN.08880914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brown D, Weyer P, Orci L. Vasopressin stimulates endocytosis in kidney collecting duct principal cells. Eur J Cell Biol 46: 336–341, 1988. [PubMed] [Google Scholar]
- 63.Azroyan A, Cortez-Retamozo V, Bouley R, Liberman R, Ruan YC, Kiselev E, Jacobson KA, Pittet MJ, Brown D, Breton S. Renal intercalated cells sense and mediate inflammation via the P2Y14 receptor. PLoS One 10: e0121419, 2015. doi: 10.1371/journal.pone.0121419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fenton RA, Yang B. Urea transporter knockout mice and their renal phenotypes. Subcell Biochem 73: 137–152, 2014. doi: 10.1007/978-94-017-9343-8_9. [DOI] [PubMed] [Google Scholar]
- 65.Huang B, Wang H, Yang B. Water transport mediated by other membrane proteins. Adv Exp Med Biol 969: 251–261, 2017. doi: 10.1007/978-94-024-1057-0_17. [DOI] [PubMed] [Google Scholar]
- 66.Kone BC. Localization and regulation of nitric oxide synthase isoforms in the kidney. Semin Nephrol 19: 230–241, 1999. [PubMed] [Google Scholar]
- 67.Kondo S, Scheef EA, Sheibani N, Sorenson CM. PECAM-1 isoform-specific regulation of kidney endothelial cell migration and capillary morphogenesis. Am J Physiol Cell Physiol 292: C2070–C2083, 2007. doi: 10.1152/ajpcell.00489.2006. [DOI] [PubMed] [Google Scholar]
- 68.van der Vorm LN, Visser R, Huskens D, Veninga A, Adams DL, Remijn JA, Hemker HC, Rensma PL, van Horssen R, de Laat B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-stage renal disease. Clin Kidney J 13: 72–74, 2020. doi: 10.1093/ckj/sfz076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Breviario F, Caveda L, Corada M, Martin-Padura I, Navarro P, Golay J, Introna M, Gulino D, Lampugnani MG, Dejana E. Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler Thromb Vasc Biol 15: 1229–1239, 1995. doi: 10.1161/01.ATV.15.8.1229. [DOI] [PubMed] [Google Scholar]
- 70.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130: 393–405, 1995. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ostendorf T, Boor P, van Roeyen CR, Floege J. Platelet-derived growth factors (PDGFs) in glomerular and tubulointerstitial fibrosis. Kidney Int Suppl 4: 65–69, 2014. doi: 10.1038/kisup.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goel M, Sinkins WG, Zuo CD, Estacion M, Schilling WP. Identification and localization of TRPC channels in the rat kidney. Am J Physiol Renal Physiol 290: F1241–F1252, 2006. doi: 10.1152/ajprenal.00376.2005. [DOI] [PubMed] [Google Scholar]
- 73.Sours S, Du J, Chu S, Ding M, Zhou XJ, Ma R. Expression of canonical transient receptor potential (TRPC) proteins in human glomerular mesangial cells. Am J Physiol Renal Physiol 290: F1507–F1515, 2006. doi: 10.1152/ajprenal.00268.2005. [DOI] [PubMed] [Google Scholar]
- 74.Floege J, Hudkins KL, Seifert RA, Francki A, Bowen-Pope DF, Alpers CE. Localization of PDGF alpha-receptor in the developing and mature human kidney. Kidney Int 51: 1140–1150, 1997. doi: 10.1038/ki.1997.157. [DOI] [PubMed] [Google Scholar]
- 75.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 76.Gallin WJ. Evolution of the “classical” cadherin family of cell adhesion molecules in vertebrates. Mol Biol Evol 15: 1099–1107, 1998. doi: 10.1093/oxfordjournals.molbev.a026017. [DOI] [PubMed] [Google Scholar]
- 77.Yang T, Huang YG, Singh I, Schnermann J, Briggs JP. Localization of bumetanide- and thiazide-sensitive Na-K-Cl cotransporters along the rat nephron. Am J Physiol Renal Phsyiol 271: F931–F939, 1996. doi: 10.1152/ajprenal.1996.271.4.F931. [DOI] [PubMed] [Google Scholar]
- 78.Peti-Peterdi J, Chambrey R, Bebok Z, Biemesderfer D, St John PL, Abrahamson DR, Warnock DG, Bell PD. Macula densa Na+/H+ exchange activities mediated by apical NHE2 and basolateral NHE4 isoforms. Am J Physiol Renal Physiol 278: F452–F463, 2000. doi: 10.1152/ajprenal.2000.278.3.F452. [DOI] [PubMed] [Google Scholar]
- 79.Sallustio F, De Benedictis L, Castellano G, Zaza G, Loverre A, Costantino V, Grandaliano G, Schena FP. TLR2 plays a role in the activation of human resident renal stem/progenitor cells. FASEB J 24: 514–525, 2010. doi: 10.1096/fj.09-136481. [DOI] [PubMed] [Google Scholar]
- 80.Wagner N, Wagner KD, Scholz H, Kirschner KM, Schedl A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am J Physiol Regul Integr Comp Physiol 291: R779–R787, 2006. doi: 10.1152/ajpregu.00219.2006. [DOI] [PubMed] [Google Scholar]
- 81.Wang J, Lin G, Alwaal A, Zhang X, Wang G, Jia X, Banie L, Villalta J, Lin CS, Lue TF. Kinetics of label retaining cells in the developing rat kidneys. PLoS One 10: e0144734, 2015. doi: 10.1371/journal.pone.0144734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bertelli E, Regoli M, Fonzi L, Occhini R, Mannucci S, Ermini L, Toti P. Nestin expression in adult and developing human kidney. J Histochem Cytochem 55: 411–421, 2007. doi: 10.1369/jhc.6A7058.2007. [DOI] [PubMed] [Google Scholar]
- 83.Chen J, Boyle S, Zhao M, Su W, Takahashi K, Davis L, Decaestecker M, Takahashi T, Breyer MD, Hao CM. Differential expression of the intermediate filament protein nestin during renal development and its localization in adult podocytes. J Am Soc Nephrol 17: 1283–1291, 2006. doi: 10.1681/ASN.2005101032. [DOI] [PubMed] [Google Scholar]
- 84.Maretta M, Marettova E. Immunohistochemical demonstration of vimentin and S-100 protein in the kidneys. Gen Physiol Biophys 18, Suppl 1: 100–102, 1999. [PubMed] [Google Scholar]
- 85.Kabgani N, Grigoleit T, Schulte K, Sechi A, Sauer-Lehnen S, Tag C, Boor P, Kuppe C, Warsow G, Schordan S, Mostertz J, Chilukoti RK, Homuth G, Endlich N, Tacke F, Weiskirchen R, Fuellen G, Endlich K, Floege J, Smeets B, Moeller MJ. Primary cultures of glomerular parietal epithelial cells or podocytes with proven origin. PLoS One 7: e34907, 2012. doi: 10.1371/journal.pone.0034907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaverina NV, Eng DG, Freedman BS, Kutz JN, Chozinski TJ, Vaughan JC, Miner JH, Pippin JW, Shankland SJ. Dual lineage tracing shows that glomerular parietal epithelial cells can transdifferentiate toward the adult podocyte fate. Kidney Int 96: 597–611, 2019. doi: 10.1016/j.kint.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andeen NK, Nguyen TQ, Steegh F, Hudkins KL, Najafian B, Alpers CE. The phenotypes of podocytes and parietal epithelial cells may overlap in diabetic nephropathy. Kidney Int 88: 1099–1107, 2015. doi: 10.1038/ki.2015.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bariety J, Mandet C, Hill GS, Bruneval P. Parietal podocytes in normal human glomeruli. J Am Soc Nephrol 17: 2770–2780, 2006. doi: 10.1681/ASN.2006040325. [DOI] [PubMed] [Google Scholar]
- 89.Berger K, Schulte K, Boor P, Kuppe C, van Kuppevelt TH, Floege J, Smeets B, Moeller MJ. The regenerative potential of parietal epithelial cells in adult mice. J Am Soc Nephrol 25: 693–705, 2014. doi: 10.1681/ASN.2013050481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20: 333–343, 2009. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lasagni L, Angelotti ML, Ronconi E, Lombardi D, Nardi S, Peired A, Becherucci F, Mazzinghi B, Sisti A, Romoli S, Burger A, Schaefer B, Buccoliero A, Lazzeri E, Romagnani P. Podocyte regeneration driven by renal progenitors determines glomerular disease remission and can be pharmacologically enhanced. Stem Cell Reports 5: 248–263, 2015. doi: 10.1016/j.stemcr.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Muller-Deile J, Brasen JH, Pollheimer M, Ratschek M, Haller H, Pape L, Schiffer M. Graft growth and podocyte dedifferentiation in donor-recipient size mismatch kidney transplants. Transplant Direct 3: e210, 2017. doi: 10.1097/TXD.0000000000000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Romagnani P, Lasagni L, Remuzzi G. Renal progenitors: an evolutionary conserved strategy for kidney regeneration. Nat Rev Nephrol 9: 137–146, 2013. doi: 10.1038/nrneph.2012.290. [DOI] [PubMed] [Google Scholar]
- 94.Shankland SJ, Anders HJ, Romagnani P. Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr Opin Nephrol Hypertens 22: 302–309, 2013. doi: 10.1097/MNH.0b013e32835fefd4. [DOI] [PubMed] [Google Scholar]
- 95.Reiser J, Altintas MM. Podocytes. F1000Res 5: 114, 2016. doi: 10.12688/f1000research.7255.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huber TB, Benzing T. The slit diaphragm: a signaling platform to regulate podocyte function. Curr Opin Nephrol Hypertens 14: 211–216, 2005. doi: 10.1097/01.mnh.0000165885.85803.a8. [DOI] [PubMed] [Google Scholar]
- 97.Grahammer F, Wigge C, Schell C, Kretz O, Patrakka J, Schneider S, Klose M, Kind J, Arnold SJ, Habermann A, Brauniger R, Rinschen MM, Volker L, Bregenzer A, Rubbenstroth D, Boerries M, Kerjaschki D, Miner JH, Walz G, Benzing T, Fornoni A, Frangakis AS, Huber TB. A flexible, multilayered protein scaffold maintains the slit in between glomerular podocytes. JCI Insight 1: e86177, 2016. doi: 10.1172/jci.insight.86177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 54: 687–697, 1998. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 99.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 100.Nagata M. Podocyte injury and its consequences. Kidney Int 89: 1221–1230, 2016. doi: 10.1016/j.kint.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 101.Asanuma K, Mundel P. The role of podocytes in glomerular pathobiology. Clin Exp Nephrol 7: 255–259, 2003. doi: 10.1007/s10157-003-0259-6. [DOI] [PubMed] [Google Scholar]
- 102.Lee HW, Arif E, Altintas MM, Quick K, Maheshwari S, Plezia A, Mahmood A, Reiser J, Nihalani D, Gupta V. High-content screening assay-based discovery of paullones as novel podocyte-protective agents. Am J Physiol Renal Physiol 314: F280–F292, 2018. doi: 10.1152/ajprenal.00338.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pullen N, Fornoni A. Drug discovery in focal and segmental glomerulosclerosis. Kidney Int 89: 1211–1220, 2016. doi: 10.1016/j.kint.2015.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reiser J, Gupta V, Kistler AD. Toward the development of podocyte-specific drugs. Kidney Int 77: 662–668, 2010. doi: 10.1038/ki.2009.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest 108: 1621–1629, 2001. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem 276: 41543–41546, 2001. doi: 10.1074/jbc.C100452200. [DOI] [PubMed] [Google Scholar]
- 107.Simons M, Schwarz K, Kriz W, Miettinen A, Reiser J, Mundel P, Holthofer H. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol 159: 1069–1077, 2001. doi: 10.1016/S0002-9440(10)61782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martin CE, Jones N. Nephrin signaling in the podocyte: an updated view of signal regulation at the slit diaphragm and beyond. Front Endocrinol (Lausanne) 9: 302, 2018. doi: 10.3389/fendo.2018.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nishibori Y, Liu L, Hosoyamada M, Endou H, Kudo A, Takenaka H, Higashihara E, Bessho F, Takahashi S, Kershaw D, Ruotsalainen V, Tryggvason K, Khoshnoodi J, Yan K. Disease-causing missense mutations in NPHS2 gene alter normal nephrin trafficking to the plasma membrane. Kidney Int 66: 1755–1765, 2004. doi: 10.1111/j.1523-1755.2004.00898.x. [DOI] [PubMed] [Google Scholar]
- 110.Liu L, Done SC, Khoshnoodi J, Bertorello A, Wartiovaara J, Berggren PO, Tryggvason K. Defective nephrin trafficking caused by missense mutations in the NPHS1 gene: insight into the mechanisms of congenital nephrotic syndrome. Hum Mol Genet 10: 2637–2644, 2001. doi: 10.1093/hmg/10.23.2637. [DOI] [PubMed] [Google Scholar]
- 111.Yan K, Khoshnoodi J, Ruotsalainen V, Tryggvason K. N-linked glycosylation is critical for the plasma membrane localization of nephrin. J Am Soc Nephrol 13: 1385–1389, 2002. doi: 10.1097/01.asn.0000013297.11876.5b. [DOI] [PubMed] [Google Scholar]
- 112.Drozdova T, Papillon J, Cybulsky AV. Nephrin missense mutations: induction of endoplasmic reticulum stress and cell surface rescue by reduction in chaperone interactions. Physiol Rep 1: e00086, 2013. doi: 10.1002/phy2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tanigawa S, Islam M, Sharmin S, Naganuma H, Yoshimura Y, Haque F, Era T, Nakazato H, Nakanishi K, Sakuma T, Yamamoto T, Kurihara H, Taguchi A, Nishinakamura R. Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes. Stem Cell Reports 11: 727–740, 2018. doi: 10.1016/j.stemcr.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ohmori T, De S, Tanigawa S, Miike K, Islam M, Soga M, Era T, Shiona S, Nakanishi K, Nakazato H, Nishinakamura R. Impaired NEPHRIN localization in kidney organoids derived from nephrotic patient iPS cells. Sci Rep 11: 3982, 2021. doi: 10.1038/s41598-021-83501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guaragna MS, Cleto TL, Souza ML, Lutaif AC, de Castro LC, Penido MG, Maciel-Guerra AT, Belangero VM, Guerra-Junior G, De Mello MP. NPHS1 gene mutations confirm congenital nephrotic syndrome in four Brazilian cases: a novel mutation is described. Nephrology (Carlton) 21: 753–757, 2016. doi: 10.1111/nep.12667. [DOI] [PubMed] [Google Scholar]
- 116.Bouchireb K, Boyer O, Gribouval O, Nevo F, Huynh-Cong E, Moriniere V, Campait R, Ars E, Brackman D, Dantal J, Eckart P, Gigante M, Lipska BS, Liutkus A, Megarbane A, Mohsin N, Ozaltin F, Saleem MA, Schaefer F, Soulami K, Torra R, Garcelon N, Mollet G, Dahan K, Antignac C. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat 35: 178–186, 2014. doi: 10.1002/humu.22485. [DOI] [PubMed] [Google Scholar]
- 117.Hinkes B, Vlangos C, Heeringa S, Mucha B, Gbadegesin R, Liu J, Hasselbacher K, Ozaltin F, Hildebrandt F, APN Study Group. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol 19: 365–371, 2008. doi: 10.1681/ASN.2007040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huh W, Kim DJ, Kim MK, Kim YG, Oh HY, Ruotsalainen V, Tryggvason K. Expression of nephrin in acquired human glomerular disease. Nephrol Dial Transplant 17: 478–484, 2002. doi: 10.1093/ndt/17.3.478. [DOI] [PubMed] [Google Scholar]
- 119.Yuan H, Takeuchi E, Taylor GA, McLaughlin M, Brown D, Salant DJ. Nephrin dissociates from actin, and its expression is reduced in early experimental membranous nephropathy. J Am Soc Nephrol 13: 946–956, 2002. doi: 10.1681/ASN.V134946. [DOI] [PubMed] [Google Scholar]
- 120.Wernerson A, Duner F, Pettersson E, Widholm SM, Berg U, Ruotsalainen V, Tryggvason K, Hultenby K, Soderberg M. Altered ultrastructural distribution of nephrin in minimal change nephrotic syndrome. Nephrol Dial Transplant 18: 70–76, 2003. doi: 10.1093/ndt/18.1.70. [DOI] [PubMed] [Google Scholar]
- 121.Toyoda M, Suzuki D, Umezono T, Uehara G, Maruyama M, Honma M, Sakai T, Sakai H. Expression of human nephrin mRNA in diabetic nephropathy. Nephrol Dial Transplant 19: 380–385, 2004. doi: 10.1093/ndt/gfg545. [DOI] [PubMed] [Google Scholar]
- 122.Nakatsue T, Koike H, Han GD, Suzuki K, Miyauchi N, Yuan H, Salant DJ, Gejyo F, Shimizu F, Kawachi H. Nephrin and podocin dissociate at the onset of proteinuria in experimental membranous nephropathy. Kidney Int 67: 2239–2253, 2005. doi: 10.1111/j.1523-1755.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 123.Otaki Y, Miyauchi N, Higa M, Takada A, Kuroda T, Gejyo F, Shimizu F, Kawachi H. Dissociation of NEPH1 from nephrin is involved in development of a rat model of focal segmental glomerulosclerosis. Am J Physiol Renal Physiol 295: F1376–F1387, 2008. doi: 10.1152/ajprenal.00075.2008. [DOI] [PubMed] [Google Scholar]
- 124.Perysinaki GS, Moysiadis DK, Bertsias G, Giannopoulou I, Kyriacou K, Nakopoulou L, Boumpas DT, Daphnis E. Podocyte main slit diaphragm proteins, nephrin and podocin, are affected at early stages of lupus nephritis and correlate with disease histology. Lupus 20: 781–791, 2011. doi: 10.1177/0961203310397412. [DOI] [PubMed] [Google Scholar]
- 125.Koop K, Eikmans M, Baelde HJ, Kawachi H, De Heer E, Paul LC, Bruijn JA. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol 14: 2063–2071, 2003. doi: 10.1097/01.ASN.0000078803.53165.C9. [DOI] [PubMed] [Google Scholar]
- 126.Fukuda H, Hidaka T, Takagi-Akiba M, Ichimura K, Oliva Trejo JA, Sasaki Y, Wang J, Sakai T, Asanuma K, Tomino Y. Podocin is translocated to cytoplasm in puromycin aminonucleoside nephrosis rats and in poor-prognosis patients with IgA nephropathy. Cell Tissue Res 360: 391–400, 2015. doi: 10.1007/s00441-014-2100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Verma R, Venkatareddy M, Kalinowski A, Li T, Kukla J, Mollin A, Cara-Fuentes G, Patel SR, Garg P. Nephrin is necessary for podocyte recovery following injury in an adult mature glomerulus. PLoS One 13: e0198013, 2018. doi: 10.1371/journal.pone.0198013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kirsch KH, Georgescu MM, Ishimaru S, Hanafusa H. CMS: an adapter molecule involved in cytoskeletal rearrangements. Proc Natl Acad Sci USA 96: 6211–6216, 1999. doi: 10.1073/pnas.96.11.6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lehtonen S, Ora A, Olkkonen VM, Geng L, Zerial M, Somlo S, Lehtonen E. In vivo interaction of the adapter protein CD2-associated protein with the type 2 polycystic kidney disease protein, polycystin-2. J Biol Chem 275: 32888–32893, 2000. doi: 10.1074/jbc.M006624200. [DOI] [PubMed] [Google Scholar]
- 130.Shih NY, Li J, Cotran R, Mundel P, Miner JH, Shaw AS. CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am J Pathol 159: 2303–2308, 2001. doi: 10.1016/S0002-9440(10)63080-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kawachi H, Fukusumi Y. New insight into podocyte slit diaphragm, a therapeutic target of proteinuria. Clin Exp Nephrol 24: 193–204, 2020. doi: 10.1007/s10157-020-01854-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yaddanapudi S, Altintas MM, Kistler AD, Fernandez I, Moller CC, Wei C, Peev V, Flesche JB, Forst AL, Li J, Patrakka J, Xiao Z, Grahammer F, Schiffer M, Lohmuller T, Reinheckel T, Gu C, Huber TB, Ju W, Bitzer M, Rastaldi MP, Ruiz P, Tryggvason K, Shaw AS, Faul C, Sever S, Reiser J. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J Clin Invest 121: 3965–3980, 2011. [Erratum in J Clin Invest 122: 780, 2012]. doi: 10.1172/JCI58552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huber TB, Kwoh C, Wu H, Asanuma K, Godel M, Hartleben B, Blumer KJ, Miner JH, Mundel P, Shaw AS. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest 116: 1337–1345, 2006. doi: 10.1172/JCI27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, Egger L, Lecha RL, Borner C, Pavenstadt H, Shaw AS, Walz G, Benzing T. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol 23: 4917–4928, 2003. doi: 10.1128/MCB.23.14.4917-4928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17: 428–437, 2007. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 136.Xavier S, Niranjan T, Krick S, Zhang T, Ju W, Shaw AS, Schiffer M, Bottinger EP. TbetaRI independently activates Smad- and CD2AP-dependent pathways in podocytes. J Am Soc Nephrol 20: 2127–2137, 2009. doi: 10.1681/ASN.2008070806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ha TS, Hong EJ, Han GD. Diabetic conditions downregulate the expression of CD2AP in podocytes via PI3-K/Akt signalling. Diabetes Metab Res Rev 31: 50–60, 2015. doi: 10.1002/dmrr.2562. [DOI] [PubMed] [Google Scholar]
- 138.Schiffer M, Mundel P, Shaw AS, Bottinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem 279: 37004–37012, 2004. doi: 10.1074/jbc.M403534200. [DOI] [PubMed] [Google Scholar]
- 139.Tossidou I, Teng B, Worthmann K, Muller-Deile J, Jobst-Schwan T, Kardinal C, Schroder P, Bolanos-Palmieri P, Haller H, Willerding J, Drost DM, de Jonge L, Reubold T, Eschenburg S, Johnson RI, Schiffer M. Tyrosine phosphorylation of CD2AP affects stability of the slit diaphragm complex. J Am Soc Nephrol 30: 1220–1237, 2019. doi: 10.1681/ASN.2018080860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsvetkov D, Hohmann M, Anistan YM, Mannaa M, Harteneck C, Rudolph B, Gollasch M. A CD2AP mutation associated with focal segmental glomerulosclerosis in young adulthood. Clin Med Insights Case Rep 9: 15–19, 2016. doi: 10.4137/ccrep.s30867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lowik MM, Groenen PJ, Pronk I, Lilien MR, Goldschmeding R, Dijkman HB, Levtchenko EN, Monnens LA, van den Heuvel LP. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int 72: 1198–1203, 2007. doi: 10.1038/sj.ki.5002469. [DOI] [PubMed] [Google Scholar]
- 142.Hyvonen ME, Ihalmo P, Sandholm N, Stavarachi M, Forsblom C, McKnight AJ, Lajer M, Maestroni A, Lewis G, Tarnow L, Maestroni S, Zerbini G, Parving HH, Maxwell AP, Groop PH, Lehtonen S. CD2AP is associated with end-stage renal disease in patients with type 1 diabetes. Acta Diabetol 50: 887–897, 2013. doi: 10.1007/s00592-013-0475-9. [DOI] [PubMed] [Google Scholar]
- 143.Takano T, Bareke E, Takeda N, Aoudjit L, Baldwin C, Pisano P, Matsuda J, El Andalousi J, Muhtadie L, Bernard C, Majewski J, Miyazaki T, Yamamura KI, Gupta IR. Recessive mutation in CD2AP causes focal segmental glomerulosclerosis in humans and mice. Kidney Int 95: 57–61, 2019. doi: 10.1016/j.kint.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 144.Patrakka J, Xiao Z, Nukui M, Takemoto M, He L, Oddsson A, Perisic L, Kaukinen A, Szigyarto CA, Uhlen M, Jalanko H, Betsholtz C, Tryggvason K. Expression and subcellular distribution of novel glomerulus-associated proteins dendrin, ehd3, sh2d4a, plekhh2, and 2310066E14Rik. J Am Soc Nephrol 18: 689–697, 2007. doi: 10.1681/ASN.2006060675. [DOI] [PubMed] [Google Scholar]
- 145.Asanuma K, Akiba-Takagi M, Kodama F, Asao R, Nagai Y, Lydia A, Fukuda H, Tanaka E, Shibata T, Takahara H, Hidaka T, Asanuma E, Kominami E, Ueno T, Tomino Y. Dendrin location in podocytes is associated with disease progression in animal and human glomerulopathy. Am J Nephrol 33: 537–549, 2011. doi: 10.1159/000327995. [DOI] [PubMed] [Google Scholar]
- 146.Duner F, Patrakka J, Xiao Z, Larsson J, Vlamis-Gardikas A, Pettersson E, Tryggvason K, Hultenby K, Wernerson A. Dendrin expression in glomerulogenesis and in human minimal change nephrotic syndrome. Nephrol Dial Transplant 23: 2504–2511, 2008. doi: 10.1093/ndt/gfn100. [DOI] [PubMed] [Google Scholar]
- 147.Kodama F, Asanuma K, Takagi M, Hidaka T, Asanuma E, Fukuda H, Seki T, Takeda Y, Hosoe-Nagai Y, Asao R, Horikoshi S, Tomino Y. Translocation of dendrin to the podocyte nucleus in acute glomerular injury in patients with IgA nephropathy. Nephrol Dial Transplant 28: 1762–1772, 2013. doi: 10.1093/ndt/gfs500. [DOI] [PubMed] [Google Scholar]
- 148.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol 11: 1–8, 2000. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 149.Schnabel E, Anderson JM, Farquhar MG. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol 111: 1255–1263, 1990. doi: 10.1083/jcb.111.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Inoue T, Yaoita E, Kurihara H, Shimizu F, Sakai T, Kobayashi T, Ohshiro K, Kawachi H, Okada H, Suzuki H, Kihara I, Yamamoto T. FAT is a component of glomerular slit diaphragms. Kidney Int 59: 1003–1012, 2001. doi: 10.1046/j.1523-1755.2001.0590031003.x, 10.1046/j.1523-1755.2001.00583.x. [DOI] [PubMed] [Google Scholar]
- 151.Lehtonen S, Lehtonen E, Kudlicka K, Holthofer H, Farquhar MG. Nephrin forms a complex with adherens junction proteins and CASK in podocytes and in Madin-Darby canine kidney cells expressing nephrin. Am J Pathol 165: 923–936, 2004. doi: 10.1016/S0002-9440(10)63354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lehtonen S, Ryan JJ, Kudlicka K, Iino N, Zhou H, Farquhar MG. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc Natl Acad Sci U S A 102: 9814–9819, 2005. doi: 10.1073/pnas.0504166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. Slit diaphragms contain tight junction proteins. J Am Soc Nephrol 20: 1491–1503, 2009. doi: 10.1681/ASN.2008101117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Deller T, Korte M, Chabanis S, Drakew A, Schwegler H, Stefani GG, Zuniga A, Schwarz K, Bonhoeffer T, Zeller R, Frotscher M, Mundel P. Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc Natl Acad Sci USA 100: 10494–10499, 2003. doi: 10.1073/pnas.1832384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J, Mundel P. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest 115: 1188–1198, 2005. doi: 10.1172/JCI200523371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Patrie KM, Drescher AJ, Welihinda A, Mundel P, Margolis B. Interaction of two actin-binding proteins, synaptopodin and alpha-actinin-4, with the tight junction protein MAGI-1. J Biol Chem 277: 30183–30190, 2002. doi: 10.1074/jbc.M203072200. [DOI] [PubMed] [Google Scholar]
- 157.Kannan N, Tang VW. Synaptopodin couples epithelial contractility to alpha-actinin-4-dependent junction maturation. J Cell Biol 211: 407–434, 2015. [Erratum in J Cell Biol 211: 933, 2015]. doi: 10.1083/jcb.201412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 8: 485–491, 2006. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 160.Buvall L, Wallentin H, Sieber J, Andreeva S, Choi HY, Mundel P, Greka A. Synaptopodin is a coincidence detector of tyrosine versus serine/threonine phosphorylation for the modulation of rho protein crosstalk in podocytes. J Am Soc Nephrol 28: 837–851, 2017. doi: 10.1681/ASN.2016040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Klambt V, Mao Y, Schneider R, Buerger F, Shamseldin H, Onuchic-Whitford AC, Deutsch K, Kitzler TM, Nakayama M, Majmundar AJ, Mann N, Hugo H, Widmeier E, Tan W, Rehm HL, Mane S, Lifton RP, Alkuraya FS, Shril S, Hildebrandt F. Generation of monogenic candidate genes for human nephrotic syndrome using 3 independent approaches. Kidney Int Rep 6: 460–471, 2021. doi: 10.1016/j.ekir.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yanagida-Asanuma E, Asanuma K, Kim K, Donnelly M, Young Choi H, Hyung Chang J, Suetsugu S, Tomino Y, Takenawa T, Faul C, Mundel P. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol 171: 415–427, 2007. doi: 10.2353/ajpath.2007.070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Buvall L, Rashmi P, Lopez-Rivera E, Andreeva S, Weins A, Wallentin H, Greka A, Mundel P. Proteasomal degradation of Nck1 but not Nck2 regulates RhoA activation and actin dynamics. Nat Commun 4: 2863, 2013. doi: 10.1038/ncomms3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yu H, Kistler A, Faridi MH, Meyer JO, Tryniszewska B, Mehta D, Yue L, Dryer S, Reiser J. Synaptopodin limits TRPC6 podocyte surface expression and attenuates proteinuria. J Am Soc Nephrol 27: 3308–3319, 2016. doi: 10.1681/ASN.2015080896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, Pavenstädt H, Pavenstaedt H, Hsu HH, Schlondorff J, Ramos A, Greka A. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal 3: ra77, 2010. [Erratum in Sci Signal 3: er11, 2010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ning L, Suleiman HY, Miner JH. Synaptopodin is dispensable for normal podocyte homeostasis but is protective in the context of acute podocyte injury. J Am Soc Nephrol 31: 2815–2832, 2020. doi: 10.1681/ASN.2020050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Feng D, DuMontier C, Pollak MR. The role of alpha-actinin-4 in human kidney disease. Cell Biosci 5: 44, 2015. doi: 10.1186/s13578-015-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Beggs AH, Byers TJ, Knoll JH, Boyce FM, Bruns GA, Kunkel LM. Cloning and characterization of two human skeletal muscle alpha-actinin genes located on chromosomes 1 and 11. J Biol Chem 267: 9281–9288, 1992. doi: 10.1016/S0021-9258(19)50420-3. [DOI] [PubMed] [Google Scholar]
- 169.Burridge K, Feramisco JR. Non-muscle alpha actinins are calcium-sensitive actin-binding proteins. Nature 294: 565–567, 1981. doi: 10.1038/294565a0. [DOI] [PubMed] [Google Scholar]
- 170.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24: 251–256, 2000. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 171.Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, Pollak MR. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2: e167, 2004. doi: 10.1371/journal.pbio.0020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liu Z, Blattner SM, Tu Y, Tisherman R, Wang JH, Rastaldi MP, Kretzler M, Wu C. Alpha-actinin-4 and CLP36 protein deficiencies contribute to podocyte defects in multiple human glomerulopathies. J Biol Chem 286: 30795–30805, 2011. doi: 10.1074/jbc.M111.255984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kann M, Ettou S, Jung YL, Lenz MO, Taglienti ME, Park PJ, Schermer B, Benzing T, Kreidberg JA. Genome-wide analysis of Wilms’ tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J Am Soc Nephrol 26: 2097–2104, 2015. doi: 10.1681/ASN.2014090940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Niaudet P, Gubler MC. WT1 and glomerular diseases. Pediatr Nephrol 21: 1653–1660, 2006. doi: 10.1007/s00467-006-0208-1. [DOI] [PubMed] [Google Scholar]
- 175.Dong L, Pietsch S, Tan Z, Perner B, Sierig R, Kruspe D, Groth M, Witzgall R, Grone HJ, Platzer M, Englert C. Integration of cistromic and transcriptomic analyses identifies Nphs2, Mafb, and Magi2 as Wilms’ tumor 1 target genes in podocyte differentiation and maintenance. J Am Soc Nephrol 26: 2118–2128, 2015. doi: 10.1681/ASN.2014080819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Su J, Li SJ, Chen ZH, Zeng CH, Zhou H, Li LS, Liu ZH. Evaluation of podocyte lesion in patients with diabetic nephropathy: Wilms’ tumor-1 protein used as a podocyte marker. Diabetes Res Clin Pract 87: 167–175, 2010. doi: 10.1016/j.diabres.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 177.Funk J, Ott V, Herrmann A, Rapp W, Raab S, Riboulet W, Vandjour A, Hainaut E, Benardeau A, Singer T, Jacobsen B. Semiautomated quantitative image analysis of glomerular immunohistochemistry markers desmin, vimentin, podocin, synaptopodin and WT-1 in acute and chronic rat kidney disease models. Histochem Cell Biol 145: 315–326, 2016. doi: 10.1007/s00418-015-1391-6. [DOI] [PubMed] [Google Scholar]
- 178.Putaala H, Soininen R, Kilpelainen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet 10: 1–8, 2001. doi: 10.1093/hmg/10.1.1. [DOI] [PubMed] [Google Scholar]
- 179.Sellin L, Huber TB, Gerke P, Quack I, Pavenstadt H, Walz G. NEPH1 defines a novel family of podocin interacting proteins. FASEB J 17: 115–117, 2003. doi: 10.1096/fj.02-0242fje. [DOI] [PubMed] [Google Scholar]
- 180.Liu G, Kaw B, Kurfis J, Rahmanuddin S, Kanwar YS, Chugh SS. Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J Clin Invest 112: 209–221, 2003. doi: 10.1172/JCI200318242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, Hattori S. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J Biol Chem 283: 9177–9186, 2008. doi: 10.1074/jbc.M707247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol 27: 8698–8712, 2007. doi: 10.1128/MCB.00948-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Solanki AK, Widmeier E, Arif E, Sharma S, Daga A, Srivastava P, Kwon SH, Hugo H, Nakayama M, Mann N, Majmundar AJ, Tan W, Gee HY, Sadowski CE, Rinat C, Becker-Cohen R, Bergmann C, Rosen S, Somers M, Shril S, Huber TB, Mane S, Hildebrandt F, Nihalani D. Mutations in KIRREL1, a slit diaphragm component, cause steroid-resistant nephrotic syndrome. Kidney Int 96: 883–889, 2019. doi: 10.1016/j.kint.2019.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ihalmo P, Palmen T, Ahola H, Valtonen E, Holthofer H. Filtrin is a novel member of nephrin-like proteins. Biochem Biophys Res Commun 300: 364–370, 2003. doi: 10.1016/S0006-291X(02)02854-1. [DOI] [PubMed] [Google Scholar]
- 185.Vitureira N, Andres R, Perez-Martinez E, Martinez A, Bribian A, Blasi J, Chelliah S, Lopez-Domenech G, De Castro F, Burgaya F, McNagny K, Soriano E. Podocalyxin is a novel polysialylated neural adhesion protein with multiple roles in neural development and synapse formation. PLoS One 5: e12003, 2010. doi: 10.1371/journal.pone.0012003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Li Y, Li J, Straight SW, Kershaw DB. PDZ domain-mediated interaction of rabbit podocalyxin and Na+/H+ exchange regulatory factor-2. Am J Physiol Renal Physiol 282: F1129–F1139, 2002. doi: 10.1152/ajprenal.00131.2001. [DOI] [PubMed] [Google Scholar]
- 187.Orlando RA, Takeda T, Zak B, Schmieder S, Benoit VM, McQuistan T, Furthmayr H, Farquhar MG. The glomerular epithelial cell anti-adhesin podocalyxin associates with the actin cytoskeleton through interactions with ezrin. J Am Soc Nephrol 12: 1589–1598, 2001. doi: 10.1681/ASN.V1281589. [DOI] [PubMed] [Google Scholar]
- 188.Doyonnas R, Kershaw DB, Duhme C, Merkens H, Chelliah S, Graf T, McNagny KM. Anuria, omphalocele, and perinatal lethality in mice lacking the CD34-related protein podocalyxin. J Exp Med 194: 13–27, 2001. doi: 10.1084/jem.194.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kang HG, Lee M, Lee KB, Hughes M, Kwon BS, Lee S, McNagny KM, Ahn YH, Ko JM, Ha IS, Choi M, Cheong HI. Loss of podocalyxin causes a novel syndromic type of congenital nephrotic syndrome. Exp Mol Med 49: e414, 2017. doi: 10.1038/emm.2017.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nielsen JS, McNagny KM. The role of podocalyxin in health and disease. J Am Soc Nephrol 20: 1669–1676, 2009. doi: 10.1681/ASN.2008070782. [DOI] [PubMed] [Google Scholar]
- 191.Lin FJ, Yao L, Hu XQ, Bian F, Ji G, Jiang GR, Gale DP, Ren HQ. First identification of PODXL nonsense mutations in autosomal dominant focal segmental glomerulosclerosis. Clin Sci (Lond) 133: 9–21, 2019. doi: 10.1042/CS20180676. [DOI] [PubMed] [Google Scholar]
- 192.Wang R, Yao C, Liu F. Association between renal podocalyxin expression and renal dysfunction in patients with diabetic nephropathy: a single-center, retrospective case-control study. Biomed Res Int 2020: 7350781, 2020. doi: 10.1155/2020/7350781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Refaeli I, Hughes MR, Wong AK, Bissonnette ML, Roskelley CD, Wayne Vogl A, Barbour SJ, Freedman BS, McNagny KM. Distinct functional requirements for podocalyxin in immature and mature podocytes reveal mechanisms of human kidney disease. Sci Rep 10: 9419, 2020. doi: 10.1038/s41598-020-64907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hatano R, Takeda A, Abe Y, Kawaguchi K, Kazama I, Matsubara M, Asano S. Loss of ezrin expression reduced the susceptibility to the glomerular injury in mice. Sci Rep 8: 4512, 2018. doi: 10.1038/s41598-018-22846-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hugo C, Nangaku M, Shankland SJ, Pichler R, Gordon K, Amieva MR, Couser WG, Furthmayr H, Johnson RJ. The plasma membrane-actin linking protein, ezrin, is a glomerular epithelial cell marker in glomerulogenesis, in the adult kidney and in glomerular injury. Kidney Int 54: 1934–1944, 1998. doi: 10.1046/j.1523-1755.1998.00195.x. [DOI] [PubMed] [Google Scholar]
- 196.Wharram BL, Goyal M, Gillespie PJ, Wiggins JE, Kershaw DB, Holzman LB, Dysko RC, Saunders TL, Samuelson LC, Wiggins RC. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J Clin Invest 106: 1281–1290, 2000. doi: 10.1172/JCI7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wiggins RC, Wiggins JE, Goyal M, Wharram BL, Thomas PE. Molecular cloning of cDNAs encoding human GLEPP1, a membrane protein tyrosine phosphatase: characterization of the GLEPP1 protein distribution in human kidney and assignment of the GLEPP1 gene to human chromosome 12p12-p13. Genomics 27: 174–181, 1995. doi: 10.1006/geno.1995.1021. [DOI] [PubMed] [Google Scholar]
- 198.Hill GS, Karoui KE, Karras A, Mandet C, Van Huyen JD, Nochy D, Bruneval P. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. I. Immunohistochemical studies. Kidney Int 79: 635–642, 2011. doi: 10.1038/ki.2010.466. [DOI] [PubMed] [Google Scholar]
- 199.Matsui K, Breiteneder-Geleff S, Kerjaschki D. Epitope-specific antibodies to the 43-kD glomerular membrane protein podoplanin cause proteinuria and rapid flattening of podocytes. J Am Soc Nephrol 9: 2013–2026, 1998. doi: 10.1681/ASN.V9112013. [DOI] [PubMed] [Google Scholar]
- 200.Koop K, Eikmans M, Wehland M, Baelde H, Ijpelaar D, Kreutz R, Kawachi H, Kerjaschki D, de Heer E, Bruijn JA. Selective loss of podoplanin protein expression accompanies proteinuria and precedes alterations in podocyte morphology in a spontaneous proteinuric rat model. Am J Pathol 173: 315–326, 2008. doi: 10.2353/ajpath.2008.080063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kasinath V, Yilmam OA, Uehara M, Yonar M, Jiang L, Li X, Qiu W, Eskandari S, Ichimura T, Abdi R. Urine podoplanin heralds the onset of ischemia-reperfusion injury of the kidney. Am J Physiol Renal Physiol 316: F957–F965, 2019. doi: 10.1152/ajprenal.00538.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Eisenreich A, Langer S, Herlan L, Kreutz R. Regulation of podoplanin expression by microRNA-29b associates with its antiapoptotic effect in angiotensin II-induced injury of human podocytes. J Hypertens 34: 323–331, 2016. doi: 10.1097/HJH.0000000000000799. [DOI] [PubMed] [Google Scholar]
- 203.Griffin SV, Hiromura K, Pippin J, Petermann AT, Blonski MJ, Krofft R, Takahashi S, Kulkarni AB, Shankland SJ. Cyclin-dependent kinase 5 is a regulator of podocyte differentiation, proliferation, and morphology. Am J Pathol 165: 1175–1185, 2004. doi: 10.1016/S0002-9440(10)63378-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Van der Hauwaert C, Savary G, Gnemmi V, Glowacki F, Pottier N, Bouillez A, Maboudou P, Zini L, Leroy X, Cauffiez C, Perrais M, Aubert S. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling. PLoS One 8: e66750, 2013. doi: 10.1371/journal.pone.0066750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tojo A, Kinugasa S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int J Nephrol 2012: 481520, 2012. doi: 10.1155/2012/481520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kerjaschki D, Horvat R, Binder S, Susani M, Dekan G, Ojha PP, Hillemanns P, Ulrich W, Donini U. Identification of a 400-kD protein in the brush borders of human kidney tubules that is similar to gp330, the nephritogenic antigen of rat Heymann nephritis. Am J Pathol 129: 183–191, 1987. [PMC free article] [PubMed] [Google Scholar]
- 207.Prabakaran T, Nielsen R, Larsen JV, Sorensen SS, Feldt-Rasmussen U, Saleem MA, Petersen CM, Verroust PJ, Christensen EI. Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS One 6: e25065, 2011. doi: 10.1371/journal.pone.0025065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Prabakaran T, Christensen EI, Nielsen R, Verroust PJ. Cubilin is expressed in rat and human glomerular podocytes. Nephrol Dial Transplant 27: 3156–3159, 2012. doi: 10.1093/ndt/gfr794. [DOI] [PubMed] [Google Scholar]
- 209.Gburek J, Verroust PJ, Willnow TE, Fyfe JC, Nowacki W, Jacobsen C, Moestrup SK, Christensen EI. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J Am Soc Nephrol 13: 423–430, 2002. doi: 10.1681/ASN.V132423. [DOI] [PubMed] [Google Scholar]
- 210.Cravedi P, Remuzzi G. Pathophysiology of proteinuria and its value as an outcome measure in chronic kidney disease. Br J Clin Pharmacol 76: 516–523, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sun J, Hultenby K, Axelsson J, Nordstrom J, He B, Wernerson A, Lindstrom K. Proximal tubular expression patterns of megalin and cubilin in proteinuric nephropathies. Kidney Int Rep 2: 721–732, 2017. doi: 10.1016/j.ekir.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Storm T, Tranebjærg L, Frykholm C, Birn H, Verroust PJ, Nevéus T, Sundelin B, Hertz JM, Holmström G, Ericson K, Christensen EI, Nielsen R. Renal phenotypic investigations of megalin-deficient patients: novel insights into tubular proteinuria and albumin filtration. Nephrol Dial Transplant 28: 585–591, 2013. doi: 10.1093/ndt/gfs462. [DOI] [PubMed] [Google Scholar]
- 213.Namour F, Dobrovoljski G, Chery C, Audonnet S, Feillet F, Sperl W, Gueant JL. Luminal expression of cubilin is impaired in Imerslund-Grasbeck syndrome with compound AMN mutations in intron 3 and exon 7. Haematologica 96: 1715–1719, 2011. doi: 10.3324/haematol.2011.043984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nielsen R, Christensen EI, Birn H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int 89: 58–67, 2016. doi: 10.1016/j.kint.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 215.Vinge L, Lees GE, Nielsen R, Kashtan CE, Bahr A, Christensen EI. The effect of progressive glomerular disease on megalin-mediated endocytosis in the kidney. Nephrol Dial Transplant 25: 2458–2467, 2010. doi: 10.1093/ndt/gfq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ogasawara S, Hosojima M, Kaseda R, Kabasawa H, Yamamoto-Kabasawa K, Kurosawa H, Sato H, Iino N, Takeda T, Suzuki Y, Narita I, Yamagata K, Tomino Y, Gejyo F, Hirayama Y, Sekine S, Saito A. Significance of urinary full-length and ectodomain forms of megalin in patients with type 2 diabetes. Diabetes Care 35: 1112–1118, 2012. doi: 10.2337/dc11-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Thrailkill KM, Nimmo T, Bunn RC, Cockrell GE, Moreau CS, Mackintosh S, Edmondson RD, Fowlkes JL. Microalbuminuria in type 1 diabetes is associated with enhanced excretion of the endocytic multiligand receptors megalin and cubilin. Diabetes Care 32: 1266–1268, 2009. doi: 10.2337/dc09-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Seki T, Asanuma K, Asao R, Nonaka K, Sasaki Y, Oliva Trejo JA, Kurosawa H, Hirayama Y, Horikoshi S, Tomino Y, Saito A. Significance of urinary full-length megalin in patients with IgA nephropathy. PLoS One 9: e114400, 2014. doi: 10.1371/journal.pone.0114400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ghezzi C, Loo DD, Wright EM. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61: 2087–2097, 2018. doi: 10.1007/s00125-018-4656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, Friedrich A, Scherneck S, Rieg T, Cunard R, Veyhl-Wichmann M, Srinivasan A, Balen D, Breljak D, Rexhepaj R, Parker HE, Gribble FM, Reimann F, Lang F, Wiese S, Sabolic I, Sendtner M, Koepsell H. Na+-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61: 187–196, 2012. doi: 10.2337/db11-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Balen D, Ljubojević M, Breljak D, Brzica H, Z˘Lender V, Koepsell H, Sabolić I. Revised immunolocalization of the Na+-d-glucose cotransporter SGLT1 in rat organs with an improved antibody. Am J Physiol Cell Physiol 295: C475–C489, 2008. doi: 10.1152/ajpcell.00180.2008. [DOI] [PubMed] [Google Scholar]
- 222.Morrison AI, Panayotova-Heiermann M, Feigl G, Scholermann B, Kinne RK. Sequence comparison of the sodium-D-glucose cotransport systems in rabbit renal and intestinal epithelia. Biochim Biophys Acta 1089: 121–123, 1991. doi: 10.1016/0167-4781(91)90093-2. [DOI] [PubMed] [Google Scholar]
- 223.Koepsell H, Korn K, Raszeja-Specht A, Bernotat-Danielowski S, Ollig D. Monoclonal antibodies against the renal Na+-D-glucose cotransporter. Identification of antigenic polypeptides and demonstration of functional coupling of different Na+-cotransport systems. J Biol Chem 263: 18419–18429, 1988. doi: 10.1016/S0021-9258(19)81375-3. [DOI] [PubMed] [Google Scholar]
- 224.Sabolic I, Skarica M, Gorboulev V, Ljubojevic M, Balen D, Herak-Kramberger CM, Koepsell H. Rat renal glucose transporter SGLT1 exhibits zonal distribution and androgen-dependent gender differences. Am J Physiol Renal Physiol 290: F913–F926, 2006. doi: 10.1152/ajprenal.00270.2005. [DOI] [PubMed] [Google Scholar]
- 225.Chao EC. SGLT-2 inhibitors: a new mechanism for glycemic control. Clin Diabetes 32: 4–11, 2014. doi: 10.2337/diaclin.32.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Nespoux J, Vallon V. Renal effects of SGLT2 inhibitors: an update. Curr Opin Nephrol Hypertens 29: 190–198, 2020. doi: 10.1097/MNH.0000000000000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJ, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW, Investigators CT. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380: 2295–2306, 2019. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 228.Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Furtado RH, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Bhatt DL, Leiter LA, McGuire DK, Wilding JP, Sabatine MS. Comparison of the effects of glucagon-like peptide receptor agonists and sodium-glucose cotransporter 2 inhibitors for prevention of major adverse cardiovascular and renal outcomes in type 2 diabetes mellitus. Circulation 139: 2022–2031, 2019. doi: 10.1161/CIRCULATIONAHA.118.038868. [DOI] [PubMed] [Google Scholar]
- 229.Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RH, Bhatt DL, Leiter LA, McGuire DK, Wilding JP, Sabatine MS. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 393: 31–39, 2019. doi: 10.1016/S0140-6736(18)32590-X. [DOI] [PubMed] [Google Scholar]
- 230.Cherney DZ, Cooper ME, Tikkanen I, Pfarr E, Johansen OE, Woerle HJ, Broedl UC, Lund SS. Pooled analysis of Phase III trials indicate contrasting influences of renal function on blood pressure, body weight, and HbA1c reductions with empagliflozin. Kidney Int 93: 231–244, 2018. doi: 10.1016/j.kint.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 231.Kohan DE, Fioretto P, Tang W, List JF. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int 85: 962–971, 2014. doi: 10.1038/ki.2013.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 302: R75–R83, 2012. doi: 10.1152/ajpregu.00357.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr 89: 3–9, 2003. doi: 10.1079/BJN2002763. [DOI] [PubMed] [Google Scholar]
- 234.Vestri S, Okamoto MM, de Freitas HS, Aparecida Dos Santos R, Nunes MT, Morimatsu M, Heimann JC, Machado UF. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol 182: 105–112, 2001. doi: 10.1007/s00232-001-0036-y. [DOI] [PubMed] [Google Scholar]
- 235.Marks J, Carvou NJ, Debnam ES, Srai SK, Unwin RJ. Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J Physiol 553: 137–145, 2003. doi: 10.1113/jphysiol.2003.046268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dyer J, Wood IS, Palejwala A, Ellis A, Shirazi-Beechey SP. Expression of monosaccharide transporters in intestine of diabetic humans. Am J Physiol Gastrointest Liver Physiol 282: G241–G248, 2002. doi: 10.1152/ajpgi.00310.2001. [DOI] [PubMed] [Google Scholar]
- 237.Magagnin S, Werner A, Markovich D, Sorribas V, Stange G, Biber J, Murer H. Expression cloning of human and rat renal cortex Na/Pi cotransport. Proc Natl Acad Sci USA 90: 5979–5983, 1993. doi: 10.1073/pnas.90.13.5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K. Growth-related renal type II Na/Pi cotransporter. J Biol Chem 277: 19665–19672, 2002. doi: 10.1074/jbc.M200943200. [DOI] [PubMed] [Google Scholar]
- 239.Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch 447: 763–767, 2004. doi: 10.1007/s00424-003-1072-5. [DOI] [PubMed] [Google Scholar]
- 240.Levi M, Lotscher M, Sorribas V, Custer M, Arar M, Kaissling B, Murer H, Biber J. Cellular mechanisms of acute and chronic adaptation of rat renal Pi transporter to alterations in dietary Pi. Am J Physiol Renal Physiol 267: F900–F908, 1994. doi: 10.1152/ajprenal.1994.267.5.f900. [DOI] [PubMed] [Google Scholar]
- 241.Segawa H, Yamanaka S, Ito M, Kuwahata M, Shono M, Yamamoto T, Miyamoto K. Internalization of renal type IIc Na-Pi cotransporter in response to a high-phosphate diet. Am J Physiol Renal Physiol 288: F587–F596, 2005. doi: 10.1152/ajprenal.00097.2004. [DOI] [PubMed] [Google Scholar]
- 242.Gisler SM, Stagljar I, Traebert M, Bacic D, Biber J, Murer H. Interaction of the type IIa Na/Pi cotransporter with PDZ proteins. J Biol Chem 276: 9206–9213, 2001. doi: 10.1074/jbc.M008745200. [DOI] [PubMed] [Google Scholar]
- 243.Giral H, Lanzano L, Caldas Y, Blaine J, Verlander JW, Lei T, Gratton E, Levi M. Role of PDZK1 protein in apical membrane expression of renal sodium-coupled phosphate transporters. J Biol Chem 286: 15032–15042, 2011. doi: 10.1074/jbc.M110.199752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hierholzer K, Cade R, Gurd R, Kessler R, Pitts R. Stop-flow analysis of renal reabsorption and excretion of sulfate in the dog. Am J Physiol 198: 833–837, 1960. doi: 10.1152/ajplegacy.1960.198.4.833. [DOI] [PubMed] [Google Scholar]
- 245.Dekel B, Zangi L, Shezen E, Reich-Zeliger S, Eventov-Friedman S, Katchman H, Jacob-Hirsch J, Amariglio N, Rechavi G, Margalit R, Reisner Y. Isolation and characterization of nontubular Sca+Lin− multipotent stem/progenitor cells from adult mouse kidney. J Am Soc Nephrol 17: 3300–3314, 2006. doi: 10.1681/ASN.2005020195. [DOI] [PubMed] [Google Scholar]
- 246.Ito CY, Li CY, Bernstein A, Dick JE, Stanford WL. Hematopoietic stem cell and progenitor defects in Sca-1/Ly-6A-null mice. Blood 101: 517–523, 2003. doi: 10.1182/blood-2002-06-1918. [DOI] [PubMed] [Google Scholar]
- 247.Blake PG, Madrenas J, Halloran PF. Ly-6 in kidney is widely expressed on tubular epithelium and vascular endothelium and is up-regulated by interferon gamma. J Am Soc Nephrol 4: 1140–1150, 1993. doi: 10.1681/ASN.V451140. [DOI] [PubMed] [Google Scholar]
- 248.Camarata TD, Weaver GC, Vasilyev A, Arnaout MA. Negative regulation of TGFbeta signaling by stem cell antigen-1 protects against ischemic acute kidney injury. PLoS One 10: e0129561, 2015. doi: 10.1371/journal.pone.0129561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Dantzler WH, Layton AT, Layton HE, Pannabecker TL. Urine-concentrating mechanism in the inner medulla: function of the thin limbs of the loops of Henle. Clin J Am Soc Nephrol 9: 1781–1789, 2014. doi: 10.2215/CJN.08750812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Devuyst O, Olinger E, Rampoldi L. Uromodulin: from physiology to rare and complex kidney disorders. Nat Rev Nephrol 13: 525–544, 2017. doi: 10.1038/nrneph.2017.101, 10.1038/nrrheum.2017.107. [DOI] [PubMed] [Google Scholar]
- 251.Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castaneda-Bueno M, Gamba G, Bachmann S. Activation of the bumetanide-sensitive Na+,K+,2Cl− cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem 286: 30200–30210, 2011. doi: 10.1074/jbc.M111.222968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J Biol Chem 286: 2224–2235, 2011. doi: 10.1074/jbc.M110.149880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tokonami N, Takata T, Beyeler J, Ehrbar I, Yoshifuji A, Christensen EI, Loffing J, Devuyst O, Olinger EG. Uromodulin is expressed in the distal convoluted tubule, where it is critical for regulation of the sodium chloride cotransporter NCC. Kidney Int 94: 701–715, 2018. doi: 10.1016/j.kint.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 254.Bates JM, Raffi HM, Prasadan K, Mascarenhas R, Laszik Z, Maeda N, Hultgren SJ, Kumar S. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection: rapid communication. Kidney Int 65: 791–797, 2004. doi: 10.1111/j.1523-1755.2004.00452.x. [DOI] [PubMed] [Google Scholar]
- 255.Mo L, Huang HY, Zhu XH, Shapiro E, Hasty DL, Wu XR. Tamm-Horsfall protein is a critical renal defense factor protecting against calcium oxalate crystal formation. Kidney Int 66: 1159–1166, 2004. doi: 10.1111/j.1523-1755.2004.00867.x. [DOI] [PubMed] [Google Scholar]
- 256.Olinger AK. What does uromodulin do? Clin J Am Soc Nephrol 16: 150–153, 2021. doi: 10.2215/CJN.06390420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Bernascone I, Vavassori S, Di Pentima A, Santambrogio S, Lamorte G, Amoroso A, Scolari F, Ghiggeri GM, Casari G, Polishchuk R, Rampoldi L. Defective intracellular trafficking of uromodulin mutant isoforms. Traffic 7: 1567–1579, 2006. doi: 10.1111/j.1600-0854.2006.00481.x. [DOI] [PubMed] [Google Scholar]
- 258.Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int 80: 338–347, 2011. doi: 10.1038/ki.2011.134. [DOI] [PubMed] [Google Scholar]
- 259.Nasr SH, Lucia JP, Galgano SJ, Markowitz GS, D’Agati VD. Uromodulin storage disease. Kidney Int 73: 971–976, 2008. doi: 10.1038/sj.ki.5002679. [DOI] [PubMed] [Google Scholar]
- 260.Hebert SC, Brown EM, Harris HW. Role of the Ca2+-sensing receptor in divalent mineral ion homeostasis. J Exp Biol 200: 295–302, 1997. doi: 10.1242/jeb.200.2.295. [DOI] [PubMed] [Google Scholar]
- 261.Diaz-Soto G, Rocher A, Garcia-Rodriguez C, Nunez L, Villalobos C. The calcium-sensing receptor in health and disease. Int Rev Cell Mol Biol 327: 321–369, 2016. doi: 10.1016/bs.ircmb.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 262.Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL, Pollak MR, Goltzman D, Brown EM. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab 297: E915–E923, 2009. doi: 10.1152/ajpendo.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Bland R, Walker EA, Hughes SV, Stewart PM, Hewison M. Constitutive expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in a transformed human proximal tubule cell line: evidence for direct regulation of vitamin D metabolism by calcium. Endocrinology 140: 2027–2034, 1999. doi: 10.1210/endo.140.5.6683. [DOI] [PubMed] [Google Scholar]
- 264.Egbuna OI, Brown EM. Hypercalcaemic and hypocalcaemic conditions due to calcium-sensing receptor mutations. Best Pract Res Clin Rheumatol 22: 129–148, 2008. doi: 10.1016/j.berh.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Mancilla EE, De Luca F, Baron J. Activating mutations of the Ca2+-sensing receptor. Mol Genet Metab 64: 198–204, 1998. doi: 10.1006/mgme.1998.2716. [DOI] [PubMed] [Google Scholar]
- 266.Riccardi D, Lee WS, Lee K, Segre GV, Brown EM, Hebert SC. Localization of the extracellular Ca2+-sensing receptor and PTH/PTHrP receptor in rat kidney. Am J Physiol Renal Physiol 271: F951–F956, 1996. doi: 10.1152/ajprenal.1996.271.4.f951. [DOI] [PubMed] [Google Scholar]
- 267.Crisi GM, Rockwell GF, Braden GL, Campfield TJ. Immunolocalization of the calcium-sensing receptor in developing human kidney. Pediatr Res 74: 133–140, 2013. doi: 10.1038/pr.2013.72. [DOI] [PubMed] [Google Scholar]
- 268.Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, de la Faille R, Bourgeois S, Bruneval P, Mandet C, Christensen EI, Faure H, Cheval L, Laghmani K, Collet C, Eladari D, Dodd RH, Ruat M, Houillier P. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest 122: 3355–3367, 2012. doi: 10.1172/JCI57407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Toka HR, Pollak MR, Houillier P. Calcium sensing in the renal tubule. Physiology (Bethesda) 30: 317–326, 2015. doi: 10.1152/physiol.00042.2014. [DOI] [PubMed] [Google Scholar]
- 270.Yang T, Hassan S, Huang YG, Smart AM, Briggs JP, Schnermann JB. Expression of PTHrP, PTH/PTHrP receptor, and Ca2+-sensing receptor mRNAs along the rat nephron. Am J Physiol Renal Physiol 272: F751–F758, 1997. doi: 10.1152/ajprenal.1997.272.6.F751. [DOI] [PubMed] [Google Scholar]
- 271.Hannan FM, Kallay E, Chang W, Brandi ML, Thakker RV. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat Rev Endocrinol 15: 33–51, 2018. doi: 10.1038/s41574-018-0115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Gunzel D, Yu AS. Function and regulation of claudins in the thick ascending limb of Henle. Pflugers Arch 458: 77–88, 2009. doi: 10.1007/s00424-008-0589-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Gamba G, Friedman PA. Thick ascending limb: the Na+:K+:2Cl− co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. Pflugers Arch 458: 61–76, 2009. doi: 10.1007/s00424-008-0607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Uchida S, Sasaki S, Furukawa T, Hiraoka M, Imai T, Hirata Y, Marumo F. Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J Biol Chem 268: 3821–3824, 1993. [Erratum in J Biol Chem 269: 19192, 1994]. doi: 10.1016/S0021-9258(18)53545-6. [DOI] [PubMed] [Google Scholar]
- 275.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 17: 171–178, 1997. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 276.Abdullah HI, Pedraza PL, McGiff JC, Ferreri NR. Calcium-sensing receptor signaling pathways in medullary thick ascending limb cells mediate COX-2-derived PGE2 production: functional significance. Am J Physiol Renal Physiol 295: F1082–F1089, 2008. doi: 10.1152/ajprenal.90316.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol 289: F699–F720, 2005. doi: 10.1152/ajprenal.00043.2005. [DOI] [PubMed] [Google Scholar]
- 278.Sutton RA, Dirks JH. The renal excretion of calcium: a review of micropuncture data. Can J Physiol Pharmacol 53: 979–988, 1975. doi: 10.1139/y75-136. [DOI] [PubMed] [Google Scholar]
- 279.Costanzo LS, Windhager EE, Ellison DH. Calcium and sodium transport by the distal convoluted tubule of the rat. 1978. J Am Soc Nephrol 11: 1562–1580, 2000. [PubMed] [Google Scholar]
- 280.Biner HL, Arpin-Bott MP, Loffing J, Wang X, Knepper M, Hebert SC, Kaissling B. Human cortical distal nephron: distribution of electrolyte and water transport pathways. J Am Soc Nephrol 13: 836–847, 2002. doi: 10.1681/ASN.V134836. [DOI] [PubMed] [Google Scholar]
- 281.Hoenderop JG, Hartog A, Stuiver M, Doucet A, Willems P, Bindels RJ. Localization of the epithelial Ca2+ channel in rabbit kidney and intestine. J Am Soc Nephrol 11: 1171–1178, 2000. doi: 10.1681/ASN.V1171171. [DOI] [PubMed] [Google Scholar]
- 282.Bachmann S, Velazquez H, Obermuller N, Reilly RF, Moser D, Ellison DH. Expression of the thiazide-sensitive Na-Cl cotransporter by rabbit distal convoluted tubule cells. J Clin Invest 96: 2510–2514, 1995. doi: 10.1172/JCI118311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bindels RJ, Hartog A, Timmermans JA, van Os CH. Immunocytochemical localization of calbindin-D28k, calbindin-D9k and parvalbumin in rat kidney. Contrib Nephrol 91: 7–13, 1991. doi: 10.1159/000420150. [DOI] [PubMed] [Google Scholar]
- 284.Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, Bindels RJ. Molecular identification of the apical Ca2+ channel in 1,25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem 274: 8375–8378, 1999. doi: 10.1074/jbc.274.13.8375. [DOI] [PubMed] [Google Scholar]
- 285.Rhoten WB, Bruns ME, Christakos S. Presence and localization of two vitamin D-dependent calcium binding proteins in kidneys of higher vertebrates. Endocrinology 117: 674–683, 1985. doi: 10.1210/endo-117-2-674. [DOI] [PubMed] [Google Scholar]
- 286.Tang Y, Stephenson JL. Calcium dynamics and homeostasis in a mathematical model of the principal cell of the cortical collecting tubule. J Gen Physiol 107: 207–230, 1996. doi: 10.1085/jgp.107.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3: 256–266, 2002. doi:10.1038/nrn797, 10.1038/nrn799. [DOI] [PubMed] [Google Scholar]
- 288.Ren Q, Weyer K, Rbaibi Y, Long KR, Tan RJ, Nielsen R, Christensen EI, Baty CJ, Kashlan OB, Weisz OA. Distinct functions of megalin and cubilin receptors in recovery of normal and nephrotic levels of filtered albumin. Am J Physiol Renal Physiol 318: F1284–F1294, 2020. doi: 10.1152/ajprenal.00030.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Bedin M, Boyer O, Servais A, Li Y, Villoing-Gaude L, Tete MJ, et al. Human C-terminal CUBN variants associate with chronic proteinuria and normal renal function. J Clin Invest 130: 335–344, 2020. doi: 10.1172/jci129937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wagner CA, Devuyst O, Bourgeois S, Mohebbi N. Regulated acid-base transport in the collecting duct. Pflugers Arch 458: 137–156, 2009. doi: 10.1007/s00424-009-0657-z. [DOI] [PubMed] [Google Scholar]
- 291.Rao R, Bhalla V, Pastor-Soler NM. Intercalated cells of the kidney collecting duct in kidney physiology. Semin Nephrol 39: 353–367, 2019. doi: 10.1016/j.semnephrol.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.DuBose TJ, Goode AK. Isolated renal tubular disorder: mechanisms and clinical expression. In: Schrier’s Diseases of the Kidney, edited by Coffman TM, Falk RJ, Molitoris B, Neilson EG, Schrier RW.. Philadelphia, PA: Wolters Kluwer Lippincott, Williams & Wilkins, 2007, 587–614. [Google Scholar]
- 293.Sebastian A, McSherry E, Morris RC Jr.. Renal potassium wasting in renal tubular acidosis (RTA): its occurrence in types 1 and 2 RTA despite sustained correction of systemic acidosis. J Clin Invest 50: 667–678, 1971. doi: 10.1172/JCI106537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ostrowski NL, Young WS, 3rd, Knepper MA, Lolait SJ. Expression of vasopressin V1a and V2 receptor messenger ribonucleic acid in the liver and kidney of embryonic, developing, and adult rats. Endocrinology 133: 1849–1859, 1993. doi: 10.1210/endo.133.4.8404628. [DOI] [PubMed] [Google Scholar]
- 295.Sarmiento JM, Ehrenfeld P, Anazco CC, Reyes CE, Troncoso S, Figueroa CD, Muller-Esterl W, Gonzalez CB. Differential distribution of the vasopressin V receptor along the rat nephron during renal ontogeny and maturation. Kidney Int 68: 487–496, 2005. doi: 10.1111/j.1523-1755.2005.00426.x. [DOI] [PubMed] [Google Scholar]
- 296.Pan Y, Metzenberg A, Das S, Jing B, Gitschier J. Mutations in the V2 vasopressin receptor gene are associated with X-linked nephrogenic diabetes insipidus. Nat Genet 2: 103–106, 1992. doi: 10.1038/ng1092-103. [DOI] [PubMed] [Google Scholar]
- 297.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362: 31–38, 1993. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 298.Kohda Y, Ding W, Phan E, Housini I, Wang J, Star RA, Huang CL. Localization of the ROMK potassium channel to the apical membrane of distal nephron in rat kidney. Kidney Int 54: 1214–1223, 1998. doi: 10.1046/j.1523-1755.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- 299.Wang WH, Hebert SC. The molecular biology of renal K channels. In: The Kidney: Physiology and Pathophysiology (3rd ed.), edited by Seldin D, Giebisch G. Philadelphia, PA: Lippincott-Raven, 2000, p. 235–250. [Google Scholar]
- 300.Walsh PR, Tse Y, Ashton E, Iancu D, Jenkins L, Bienias M, Kleta R, Van’t Hoff W, Bockenhauer D. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J 11: 302–309, 2018. doi: 10.1093/ckj/sfx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183–188, 1996. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 302.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14: 152–156, 1996. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 303.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 304.Kleta R, Bockenhauer D. Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol 104: p73–p80, 2006. doi: 10.1159/000094001. [DOI] [PubMed] [Google Scholar]
- 305.Lazarowski ER, Shea DA, Boucher RC, Harden TK. Release of cellular UDP-glucose as a potential extracellular signaling molecule. Mol Pharmacol 63: 1190–1197, 2003. doi: 10.1124/mol.63.5.1190. [DOI] [PubMed] [Google Scholar]
- 306.Arulkumaran N, Turner CM, Sixma ML, Singer M, Unwin R, Tam FW. Purinergic signaling in inflammatory renal disease. Front Physiol 4: 194, 2013. doi: 10.3389/fphys.2013.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Burnstock G, Evans LC, Bailey MA. Purinergic signalling in the kidney in health and disease. Purinergic Signal 10: 71–101, 2014. doi: 10.1007/s11302-013-9400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yang B, Bankir L, Gillespie A, Epstein CJ, Verkman AS. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J Biol Chem 277: 10633–10637, 2002. doi: 10.1074/jbc.M200207200. [DOI] [PubMed] [Google Scholar]
- 309.Zhang S, Zhao Y, Wang S, Li M, Xu Y, Ran J, Geng X, He J, Meng J, Shao G, Zhou H, Ge Z, Chen G, Li R, Yang B. Discovery of novel diarylamides as orally active diuretics targeting urea transporters. Acta Pharm Sin B 11: 181–202, 2021. doi: 10.1016/j.apsb.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Deen WM, Lazzara MJ, Myers BD. Structural determinants of glomerular permeability. Am J Physiol Renal Physiol 281: F579–F596, 2001. doi: 10.1152/ajprenal.2001.281.4.F579. [DOI] [PubMed] [Google Scholar]
- 311.Fogo AB, Kon V. The glomerulus–a view from the inside–the endothelial cell. Int J Biochem Cell Biol 42: 1388–1397, 2010. doi: 10.1016/j.biocel.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 312.Haraldsson B, Nystrom J. The glomerular endothelium: new insights on function and structure. Curr Opin Nephrol Hypertens 21: 258–263, 2012. doi: 10.1097/MNH.0b013e3283522e7a. [DOI] [PubMed] [Google Scholar]
- 313.Han KH, Lim JM, Kim WY, Kim H, Madsen KM, Kim J. Expression of endothelial nitric oxide synthase in developing rat kidney. Am J Physiol Renal Physiol 288: F694–F702, 2005. doi: 10.1152/ajprenal.00085.2004. [DOI] [PubMed] [Google Scholar]
- 314.Han KH, Jung JY, Chung KY, Kim H, Kim J. Nitric oxide synthesis in the adult and developing kidney. Electrolyte Blood Press 4: 1–7, 2006. doi: 10.5049/EBP.2006.4.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Lee J. Nitric oxide in the kidney: its physiological role and pathophysiological implications. Electrolyte Blood Press 6: 27–34, 2008. doi: 10.5049/EBP.2008.6.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Bachmann S, Bosse HM, Mundel P. Topography of nitric oxide synthesis by localizing constitutive NO synthases in mammalian kidney. Am J Physiol Renal Physiol 268: F885–F898, 1995. doi: 10.1152/ajprenal.1995.268.5.f885. [DOI] [PubMed] [Google Scholar]
- 317.Terada Y, Tomita K, Nonoguchi H, Marumo F. Polymerase chain reaction localization of constitutive nitric oxide synthase and soluble guanylate cyclase messenger RNAs in microdissected rat nephron segments. J Clin Invest 90: 659–665, 1992. doi: 10.1172/JCI115908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Ahn KY, Mohaupt MG, Madsen KM, Kone BC. In situ hybridization localization of mRNA encoding inducible nitric oxide synthase in rat kidney. Am J Physiol Renal Physiol 267: F748–F757, 1994. doi: 10.1152/ajprenal.1994.267.5.F748. [DOI] [PubMed] [Google Scholar]
- 319.Bonomini M, Pandolfi A, Di Pietro N, Sirolli V, Giardinelli A, Consoli A, Amoroso L, Gizzi F, De Lutiis MA, Felaco M. Adherence of uremic erythrocytes to vascular endothelium decreases endothelial nitric oxide synthase expression. Kidney Int 67: 1899–1906, 2005. doi: 10.1111/j.1523-1755.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- 320.Heeringa P, van Goor H, Itoh-Lindstrom Y, Maeda N, Falk RJ, Assmann KJ, Kallenberg CG, Jennette JC. Lack of endothelial nitric oxide synthase aggravates murine accelerated anti-glomerular basement membrane glomerulonephritis. Am J Pathol 156: 879–888, 2000. doi: 10.1016/S0002-9440(10)64957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Forbes MS, Thornhill BA, Park MH, Chevalier RL. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am J Pathol 170: 87–99, 2007. doi: 10.2353/ajpath.2007.060610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Yuen DA, Stead BE, Zhang Y, White KE, Kabir MG, Thai K, Advani SL, Connelly KA, Takano T, Zhu L, Cox AJ, Kelly DJ, Gibson IW, Takahashi T, Harris RC, Advani A. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol 23: 1810–1823, 2012. doi: 10.1681/ASN.2011121170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Pusztaszeri MP, Seelentag W, Bosman FT. Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem 54: 385–395, 2006. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
- 324.Ono S, Matsui H, Noda M, Kasuda S, Yada N, Yoshimoto K, Akiyama M, Miyata T, Sugimoto M, Nishio K. Functional regulation of von Willebrand factor ameliorates acute ischemia-reperfusion kidney injury in mice. Sci Rep 9: 14453, 2019. doi: 10.1038/s41598-019-51013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Blaise S, Polena H, Vilgrain I. Soluble vascular endothelial-cadherin and auto-antibodies to human vascular endothelial-cadherin in human diseases: two new biomarkers of endothelial dysfunction. Vasc Med 20: 557–565, 2015. doi: 10.1177/1358863X15591201. [DOI] [PubMed] [Google Scholar]
- 326.Yu WK, McNeil JB, Wickersham NE, Shaver CM, Bastarache JA, Ware LB. Vascular endothelial cadherin shedding is more severe in sepsis patients with severe acute kidney injury. Crit Care 23: 18, 2019. doi: 10.1186/s13054-019-2315-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Maciel RA, Cunha RS, Busato V, Franco CR, Gregorio PC, Dolenga CJ, Nakao LS, Massy ZA, Boullier A, Pecoits-Filho R, Stinghen AE. Uremia impacts VE-cadherin and ZO-1 expression in human endothelial cell-to-cell junctions. Toxins (Basel) 10: 404, 2018. doi: 10.3390/toxins10100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Nishitani Y, Iwano M, Yamaguchi Y, Harada K, Nakatani K, Akai Y, Nishino T, Shiiki H, Kanauchi M, Saito Y, Neilson EG. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 68: 1078–1085, 2005. doi: 10.1111/j.1523-1755.2005.00500.x. [DOI] [PubMed] [Google Scholar]
- 329.Boor P, Ostendorf T, Floege J. PDGF and the progression of renal disease. Nephrol Dial Transplant 29: i45–i54, 2014. doi: 10.1093/ndt/gft273. [DOI] [PubMed] [Google Scholar]
- 330.Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol 19: 12–23, 2008. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- 331.Schiessl IM, Grill A, Fremter K, Steppan D, Hellmuth MK, Castrop H. Renal Interstitial platelet-derived growth factor receptor-beta cells support proximal tubular regeneration. J Am Soc Nephrol 29: 1383–1396, 2018. doi: 10.1681/ASN.2017101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Schlondorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol 20: 1179–1187, 2009. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]
- 333.Kitching AR, Hutton HL. The players: cells involved in glomerular disease. Clin J Am Soc Nephrol 11: 1664–1674, 2016. doi: 10.2215/CJN.13791215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Sours-Brothers S, Ding M, Graham S, Ma R. Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med (Maywood) 234: 673–682, 2009. doi: 10.3181/0809-RM-279. [DOI] [PubMed] [Google Scholar]
- 335.Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF. Developmental patterns of PDGF B-chain, PDGF-receptor, and alpha-actin expression in human glomerulogenesis. Kidney Int 42: 390–399, 1992. doi: 10.1038/ki.1992.300. [DOI] [PubMed] [Google Scholar]
- 336.Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF. PDGF-receptor localizes to mesangial, parietal epithelial, and interstitial cells in human and primate kidneys. Kidney Int 43: 286–294, 1993. doi: 10.1038/ki.1993.45. [DOI] [PubMed] [Google Scholar]
- 337.Boor P, van Roeyen CR, Kunter U, Villa L, Bucher E, Hohenstein B, Hugo CP, Eriksson U, Satchell SC, Mathieson PW, Eitner F, Floege J, Ostendorf T. PDGF-C mediates glomerular capillary repair. Am J Pathol 177: 58–69, 2010. doi: 10.2353/ajpath.2010.091008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Khundmiri SJ, Chen L, Lederer ED, Yang CR, Knepper MA. Transcriptomes of major proximal tubule cell culture models. J Am Soc Nephrol 32: 86–97, 2021. doi: 10.1681/ASN.2020010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Park HJ, Fan Z, Bai Y, Ren Q, Rbaibi Y, Long KR, Gliozzi ML, Rittenhouse N, Locker JD, Poholek AC, Weisz OA. Transcriptional programs driving shear stress-induced differentiation of kidney proximal tubule cells in culture. Front Physiol 11: 587358, 2020. doi: 10.3389/fphys.2020.587358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 579: 95–132, 2016. doi: 10.1016/j.gene.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Busst CJ. Blood pressure regulation via the epithelial sodium channel: from gene to kidney and beyond. Clin Exp Pharmacol Physiol 40: 495–503, 2013. doi: 10.1111/1440-1681.12124. [DOI] [PubMed] [Google Scholar]
- 342.Bonny O, Hummler E. Dysfunction of epithelial sodium transport: from human to mouse. Kidney Int 57: 1313–1318, 2000. doi: 10.1046/j.1523-1755.2000.00968.x. [DOI] [PubMed] [Google Scholar]
- 343.Tetti M, Monticone S, Burrello J, Matarazzo P, Veglio F, Pasini B, Jeunemaitre X, Mulatero P. Liddle syndrome: review of the literature and description of a new case. Int J Mol Sci 19: 812, 2018. doi: 10.3390/ijms19030812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci 114: 629–641, 2001. doi: 10.1242/jcs.114.4.629. [DOI] [PubMed] [Google Scholar]
- 345.Wheelock MJ, Johnson KR. Cadherin-mediated cellular signaling. Curr Opin Cell Biol 15: 509–514, 2003. doi: 10.1016/S0955-0674(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 346.Petruzzelli L, Takami M, Humes HD. Structure and function of cell adhesion molecules. Am J Med 106: 467–476, 1999. doi: 10.1016/S0002-9343(99)00058-3. [DOI] [PubMed] [Google Scholar]
- 347.Adams CL, Nelson WJ. Cytomechanics of cadherin-mediated cell-cell adhesion. Curr Opin Cell Biol 10: 572–577, 1998. doi: 10.1016/S0955-0674(98)80031-8. [DOI] [PubMed] [Google Scholar]
- 348.Nouwen EJ, Dauwe S, van der Biest I, De Broe ME. Stage- and segment-specific expression of cell-adhesion molecules N-CAM, A-CAM, and L-CAM in the kidney. Kidney Int 44: 147–158, 1993. doi: 10.1038/ki.1993.225. [DOI] [PubMed] [Google Scholar]
- 349.Prozialeck WC, Lamar PC, Appelt DM. Differential expression of E-cadherin, N-cadherin and beta-catenin in proximal and distal segments of the rat nephron. BMC Physiol 4: 10, 2004. doi: 10.1186/1472-6793-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Thomson RB, Aronson PS. Immunolocalization of Ksp-cadherin in the adult and developing rabbit kidney. Am J Physiol Renal Physioil 277: F146–F156, 1999. doi: 10.1152/ajprenal.1999.277.1.F146. [DOI] [PubMed] [Google Scholar]
- 351.Thomson RB, Igarashi P, Biemesderfer D, Kim R, Abu-Alfa A, Soleimani M, Aronson PS. Isolation and cDNA cloning of Ksp-cadherin, a novel kidney-specific member of the cadherin multigene family. J Biol Chem 270: 17594–17601, 1995. doi: 10.1074/jbc.270.29.17594. [DOI] [PubMed] [Google Scholar]
- 352.Igarashi P. Following the expression of a kidney-specific gene from early development to adulthood. Nephron Exp Nephrol 94: e1–6, 2003. doi: 10.1159/000070812. [DOI] [PubMed] [Google Scholar]
- 353.Paul R, Ewing CM, Robinson JC, Marshall FF, Johnson KR, Wheelock MJ, Isaacs WB. Cadherin-6, a cell adhesion molecule specifically expressed in the proximal renal tubule and renal cell carcinoma. Cancer Res 57: 2741–2748, 1997. [PubMed] [Google Scholar]
- 354.Thedieck C, Kuczyk M, Klingel K, Steiert I, Muller CA, Klein G. Expression of Ksp-cadherin during kidney development and in renal cell carcinoma. Br J Cancer 92: 2010–2017, 2005. doi: 10.1038/sj.bjc.6602597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Goto S, Yaoita E, Matsunami H, Kondo D, Yamamoto T, Kawasaki K, Arakawa M, Kihara I. Involvement of R-cadherin in the early stage of glomerulogenesis. J Am Soc Nephrol 9: 1234–1241, 1998. doi: 10.1681/ASN.V971234. [DOI] [PubMed] [Google Scholar]
- 356.Blaschke S, Mueller CA, Markovic-Lipkovski J, Puch S, Miosge N, Becker V, Mueller GA, Klein G. Expression of cadherin-8 in renal cell carcinoma and fetal kidney. Int J Cancer 101: 327–334, 2002. doi: 10.1002/ijc.10623. [DOI] [PubMed] [Google Scholar]
- 357.Loh CY, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP, Looi CY. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: signaling, therapeutic implications, and challenges. Cells 8: 1118, 2019. doi: 10.3390/cells8101118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 119: 1429–1437, 2009. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Bell PD, Lapointe JY, Peti-Peterdi J. Macula densa cell signaling. Annu Rev Physiol 65: 481–500, 2003. doi: 10.1146/annurev.physiol.65.050102.085730. [DOI] [PubMed] [Google Scholar]
- 360.Gimenez I, Isenring P, Forbush B. Spatially distributed alternative splice variants of the renal Na-K-Cl cotransporter exhibit dramatically different affinities for the transported ions. J Biol Chem 277: 8767–8770, 2002. doi: 10.1074/jbc.C200021200. [DOI] [PubMed] [Google Scholar]
- 361.Plata C, Meade P, Hall A, Welch RC, Vazquez N, Hebert SC, Gamba G. Alternatively spliced isoform of apical Na+-K+-Cl− cotransporter gene encodes a furosemide-sensitive Na+-Cl− cotransporter. Am J Physiol Renal Physiol 280: F574–F582, 2001. doi: 10.1152/ajprenal.2001.280.4.F574. [DOI] [PubMed] [Google Scholar]
- 362.Plata C, Meade P, Vazquez N, Hebert SC, Gamba G. Functional properties of the apical Na+-K+-2Cl− cotransporter isoforms. J Biol Chem 277: 11004–11012, 2002. doi: 10.1074/jbc.M110442200. [DOI] [PubMed] [Google Scholar]
- 363.Lapointe JY, Bell PD, Sabirov RZ, Okada Y. Calcium-activated nonselective cationic channel in macula densa cells. Am J Physiol Renal Physiol 285: F275–F280, 2003. doi: 10.1152/ajprenal.00313.2002. [DOI] [PubMed] [Google Scholar]
- 364.Gyarmati G, Shroff UN, Riquier-Brison A, Kriz W, Kaissling B, Neal CR, Arkill KP, Ahmadi N, Gill IS, Moon JY, Desposito D, Peti-Peterdi J. A new view of macula densa cell microanatomy. Am J Physiol Renal Physiol 320: F492–F504, 2021. doi: 10.1152/ajprenal.00546.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Miao Z, Balzer MS, Ma Z, Liu H, Wu J, Shrestha R, Aranyi T, Kwan A, Kondo A, Pontoglio M, Kim J, Li M, Kaestner KH, Susztak K. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat Commun 12: 2277, 2021. doi: 10.1038/s41467-021-22266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Chen L, Chou CL, Knepper MA. Targeted single-cell RNA-seq identifies minority cell types of kidney distal nephron. J Am Soc Nephrol 32: 886–896, 2021. doi: 10.1681/ASN.2020101407. [DOI] [PMC free article] [PubMed] [Google Scholar]