

Keywords: kidney, lymphangiogenesis, lymphatics, three-dimensional imaging, vascular endothelial growth factor receptor 3
Abstract
Expansion of renal lymphatic networks, or lymphangiogenesis (LA), is well recognized during development and is now being implicated in kidney diseases. Although LA is associated with multiple pathological conditions, very little is known about its role in acute kidney injury. The purpose of this study was to evaluate the role of LA in a model of cisplatin-induced nephrotoxicity. LA is predominately regulated by vascular endothelial growth factor (VEGF)-C and VEGF-D, ligands that exert their function through their cognate receptor VEGF receptor 3 (VEGFR3). We demonstrated that use of MAZ51, a selective VEGFR3 inhibitor, caused significantly worse structural and functional kidney damage in cisplatin nephrotoxicity. Apoptotic cell death and inflammation were also increased in MAZ51-treated animals compared with vehicle-treated animals following cisplatin administration. Notably, MAZ51 caused significant upregulation of intrarenal phospho-NF-κB, phospho-JNK, and IL-6. Cisplatin nephrotoxicity is associated with vascular congestion due to endothelial dysfunction. Using three-dimensional tissue cytometry, a novel approach to explore lymphatics in the kidney, we detected significant vascular autofluorescence attributed to erythrocytes in cisplatin alone-treated animals. Interestingly, no such congestion was detected in MAZ51-treated animals. We found increased renal vascular damage in MAZ51-treated animals, whereby MAZ51 caused a modest decrease in the endothelial markers endomucin and von Willebrand factor, with a modest increase in VEGFR2. Our findings identify a protective role for de novo LA in cisplatin nephrotoxicity and provide a rationale for the development of therapeutic approaches targeting LA. Our study also suggests off-target effects of MAZ51 on the vasculature in the setting of cisplatin nephrotoxicity.
NEW & NOTEWORTHY Little is known about injury-associated LA in the kidney and its role in the pathophysiology of acute kidney injury (AKI). Observed exacerbation of cisplatin-induced AKI after LA inhibition was accompanied by increased medullary damage and cell death in the kidney. LA inhibition also upregulated compensatory expression of LA regulatory proteins, including JNK and NF-κB. These data support the premise that LA is induced during AKI and lymphatic expansion is a protective mechanism in cisplatin nephrotoxicity.
INTRODUCTION
Acute kidney injury (AKI) is a widespread, complex condition that is a deadly complication for the critically ill. Although the burden of AKI continues to grow in prevalence, public awareness, prevention methods, and treatment strategies remain insufficient (1). AKI is associated with extended hospital stays, increased healthcare expenditure, and devastating morbidity and mortality (2). Patients who survive AKI are at risk for developing chronic kidney disease, which is associated with cardiovascular disease (3), vascular calcification (4), hypertension (5), anemia (6), and systemic inflammation (7). Current treatments for AKI remain largely supportive, thereby highlighting the need for new targets for therapies.
The lymphatic system plays an integral role in tissue fluid homeostasis, immune surveillance, and lipid absorption and is made up of complex lymphatic vessels and organs to mediate these processes. In the quiescent kidney, lymphatic vessels are primarily localized to the hilum and cortex, interwoven with the renal blood vasculature (8). De novo lymphatic vessel formation, or lymphangiogenesis (LA), is regulated in part by the vascular endothelial growth factor (VEGF) family, namely, VEGF-C and VEGF-D, which function through their cognate receptor, VEGF receptor 3 (VEGFR3; Flt4). These factors were only recently identified (for a review, see Ref. 9), which made it possible to understand LA and allowed for the discovery of other lymphatic markers including prospero-related homeodomain transcription factor (Prox-1), membrane glycoprotein podoplanin (Pdpn), and lymphatic vessel hyaluronan receptor-1 (LYVE-1).
Although the process of generating de novo lymphatics is well understood in development (10), it was only recently that LA has been identified during organ injury and disease states. LA continues to be explored in various organ systems, including cardiovascular (11) and nervous (12) systems, as well as in organ transplantation (13), among many other areas (14). Recent studies by our laboratory group (15) and others (16–21) have shown lymphatic vessel proliferation as a response to injury. Previous studies have shown that the administration of recombinant VEGF-C in a model of unilateral ureteral obstruction (UUO) attenuates inflammation and subsequent fibrosis. In a model of polycystic kidney disease, VEGF-C administration significantly reduced renal volume and cyst size (22). These studies suggested that LA induction may serve as a therapeutic avenue for kidney diseases (18). However, conflicting reports have shown that conditional lymphatic vessel-specific LYVE-1 deletion reduces inflammatory infiltrate, cytokine release, and fibrosis in both UUO and renal ischemia-reperfusion injury (20). It is important to note that different disease etiologies may elicit variable responses and consequences pertaining to the lymphatic system and remain to be fully explored.
The renal LA signaling network during injury is suggested to stem from tubular epithelial cells, which are the major sources of VEGF-C and VEGF-D in the kidney (23); in fact, VEGF-C and VEGF-D ligand expression is upregulated secondary to kidney injury and may participate in essential cellular cross talk (15). VEGF-C and VEGF-D activate VEGFR3, which subsequently activates downstream signaling pathways, such as the phosphatidylinositol 3-kinase/AKT, JNK, ERK, and NF-κB (p65) pathways. By stimulating cell migration, survival, and proliferation, these pathways are integral to LA signaling during disease (23).
MAZ51 is a reversible, ATP-competitive, VEGFR3 tyrosine kinase inhibitor (24) that has been used in a variety of organ systems and diseases (25–27). Furthermore, this inhibitor has been extensively studied in the context of cancer biology and pathogenesis, as it blocks LA signaling and suppresses tumor growth (28–30). Recently, investigators have explored the effect of VEGFR3 inhibition on cardiac remodeling after myocardial infarction and found that blockade of LA signaling inhibits lymphatic vessel proliferation and lymphatic transport while also significantly worsening inflammation, edema, and cardiac dysfunction (27).
Lymphatic vessel development and its implications in cancer have been topics of interest, as tumor lymphatic density is an indicator of poor prognosis (14, 31). It is important to explore LA in diverse disease contexts because LA is a complex and dynamic process that could have distinct implications depending on pathophysiology. This is especially true when considering LA in cancer therapeutics, as some chemotherapeutic agents, including taxanes and platinum-based agents, have recently been suggested to induce LA (32, 33). In fact, LA has been shown to be upregulated after a wide variety of renal pathologies previously explored (34). In addition to being one of the most widely used anticancer agents, cis-diamminedichloroplatinum (II) [cisplatin (Cp)] is also associated with significant nephrotoxicity, primarily targeting renal proximal tubules; however, its toxicity also affects glomeruli and the intrarenal vasculature. The effects of Cp nephrotoxicity have been extensively studied (35), but its effects on LA and its impact on AKI have yet to be explored.
Cp nephrotoxicity is a common side effect of chemotherapy for multiple solid cancers, affecting ∼30% of patients who receive Cp (36, 37). Renal tubular epithelial cells are affected by Cp, and, due to their role in LA ligand secretion and signaling, we chose to use the murine Cp nephrotoxicity model to interrogate renal LA during AKI. Additionally, signaling pathways in the kidney that contribute to and are affected by de novo LA during injury have not been described to the best of our knowledge. Here, we used MAZ51, a VEGFR3 antagonist, in Cp nephrotoxicity to determine the pathophysiology of LA on AKI outcomes.
MATERIALS AND METHODS
Reagents and general supplies were purchased from Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO), unless otherwise noted.
Study Approval
All procedures involving animals were performed in compliance with National Institutes of Health guidelines regarding the care and use of live animals. Protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham (UAB).
Cp Nephrotoxicity
Cp injury was induced in age- and weight-matched male C57BL/6J (stock no. 000664, Jackson Labs) mice (10–12 wk of age), as previously described (38). Briefly, mice were administered 20 mg/kg body wt Cp (1 mg/mL solution, BluePoint Laboratories) or vehicle (saline) by a single intraperitoneal injection. Animals were euthanized on day 3 following administration. Blood was collected by single intracardiac puncture, and serum creatinine was analyzed using liquid chromatography and tandem mass spectrometry at the UAB-University of California-San Diego (UCSD) O’Brien Center for Acute Kidney Injury Research bioanalytical core facility using previously described methods (39). All analyses were performed on day 3 following Cp administration. Urine was collected by bladder puncture at the time of euthanization, and urinary kidney injury molecule-1 (KIM-1) levels were analyzed by ELISA according to the manufacturer’s instructions (MKM100, R&D Systems).
For treatment with VEGFR3 inhibitor, mice were injected intraperitoneally with 10 mg/kg body wt MAZ51 (no. 163655-37-6, Calbiochem), as previously described (40–42), or vehicle (DMSO) once daily after Cp administration until euthanization.
Glomerular Filtration Rate Measurements
Glomerular filtration rate (GFR) was measured at day 3 post-Cp administration as previously described (43). Briefly, mice were lightly anesthetized using isoflurane (1.5% induction, 1% maintenance), and a patch on the flank of the mouse was shaved. Transdermal monitors were adhered to the skin using a double-sided adhesive patch, and FITC-sinistrin (20 mg/mL in sterile saline) was administered through a tail vein injection (MediBeacon). Mice were monitored for 2 h, after which the devices were removed and elimination kinetics of FITC-sinistrin clearance were analyzed.
Histology
Kidneys were cut transversely and fixed in 10% neutral buffered saline for 24 h, after which they were embedded in paraffin. Tissues were stained with periodic acid-Schiff hematoxylin using standard protocols. Images were acquired at ×20 magnification on a BZ-X700 All-In-One Fluorescence Microscope (Keyence, Istasca, IL). Five to eight random fields were selected from each section both in the cortex and medulla for injury blinded scoring (n = 5−6 per group). The scoring system was as follows: 0, no apparent damage; 1, up to 25% area; 2, 25–50% area; 3, 50–75% area; 4, over 75% area. Images were analyzed for infiltrating cells using ImageJ software (National Institutes of Health).
TUNEL
Transversely cut paraffin-embedded kidney sections were stained for TUNEL per the manufacturer’s instructions (Cat. No. 11684795910, Roche Applied Science, Indianapolis, IN). Briefly, tissue sections were dewaxed and rehydrated followed by incubation with proteinase K at 37°C. Sections were then stained with TUNEL reaction mixture followed by Hoechst nuclei stain. Sections were mounted and imaged using a Leica DMi8 microscope (Leica Microsystems, Buffalo Grove, IL). Entire kidney sections were imaged, and total kidney TUNEL-positive cells were analyzed using LAS X analysis software (Leica Microsystems).
Immunohistochemistry
Sections were deparaffinized and rehydrated before antigen retrieval using Trilogy, per the manufacturer’s protocol. Tissues were blocked in blocking buffer (1% BSA, 0.2% nonfat dry milk, and 0.3% Triton-X in PBS) before an overnight 4°C incubation with rat anti-mouse Ly6B.2 (CL 8993AP, clone 7/4, Cedarlane, Burlington, ON, Canada) to assess neutrophil recruitment after injury. Sections were washed and then incubated in an ABC ready-to-use kit (Vector Labs, Burlingame, CA) following the manufacturer’s instructions. Sections were exposed to cobalt-based diaminobenzidine (KPL HistoMark BLACK, SeraCare, Milford, MA), washed in distilled water, dehydrated, and then visualized by light microscopy. Images were analyzed by ImageJ software (National Institutes of Health).
Quantification of mRNA Expression
Gene expression analysis was performed as previously described (38). Briefly, total RNA was isolated from the kidneys using TRIzol (Thermo Fisher Scientific) and converted to cDNA (Qiagen). Quantitative real-time PCR was performed in triplicate using SYBR Green Master Mix (Invitrogen) and monitored using melting curve analysis. Relative mRNA expression was normalized to Gapdh as an internal control. The primers used to detect the specific genes included the following: Gapdh, forward 5′- ATCATCCCTGCATCCACT-3′ and reverse 5′- ATCCACGACGGACACATT-3′; tumor necrosis factor-α (Tnf-α), forward 5′- ACGGCATGGATCTCAAAGAC-3′ and reverse 5′- AGATAGCAAATCGGCTGACG-3′; colony stimulating factor-1 (Csf1), forward 5′- CGGGCATCATCCTAGTCTTGCTGACTGT-3′ and reverse 5′- ATAGTGGCAGTATGTGGGGGGCATCCTC-3′; chemokine (C-C motif) ligand 2 (Ccl2), forward 5′- ACTCACCTGCTGCTACTCAT-3′ and reverse 5′- CTACAGCTTCTTTGGGACA-3′; IL-6 (Il6), forward 5′- CTGCAAGAGACTTCCATCCAG-3′ and reverse 5′- AGTGGTATAGACAGGTCTGTTGG-3′; heme oxygenase (HO)-1 (Hmox1), forward 5′- GGTGATGGCTTCCTTGTACC-3′ and reverse 5′- AGTGAGGCCCATACCAGAAG-3′; Flt4, forward 5′- CCATCGAGAGTCTGGACAGC-3′ and reverse 5′- CCGGGATGGTGGTCACATAG-3′; Prox1, forward 5′- GAAGGGCTATCACCCAATCA-3′ and reverse 5′- TGAACCACTTGATGAGCTGC-3′; Lyve1, forward 5′- GGCTTTGAGACTTGCAGCTATG-3′ and reverse 5′- GCAGGAGTTAACCCAGGTGT-3′; and Pdpn, forward 5′- CTCAAGCTTCAAGATGTGGACCGTGCCAGT-3′ and reverse 5′- GAGGAATTCGGGCGAGAACCTTCCAGAAAT-3′.
Western Blot Analysis
Protein expression was performed as previously described (38). Briefly, kidneys were lysed in lysis buffer (10 mM/L Tris·HCl, 5 mM/L EDTA, 150 mM/L NaCl, 10% Nonidet P-40, and 10% Triton X-100) with protease and phosphatase (Biotool, Houston, TX) inhibitors. Total protein was quantified using a bichinchoninic acid protein assay (Thermo Fisher, Waltham, MA). Protein (75 µg) was loaded onto 10% and 12% Tris-glycine gels, resolved, and transferred onto a PVDF membrane (EMB Millipore, Billerica, MA). Membranes were blocked according to the manufacturer’s protocol for 1 h and then incubated with cleaved caspase-3 (1:1,000, Cell Signaling Technology, Danvers, MA), anti-phospho-p65 (1:1,000; Santa Cruz Biotechnology, Dallas, TX), anti-p65 (1:1,000; Cell Signaling Technology), anti-phospho-Akt (1:1,000, Cell Signaling Technology), anti-Akt (1:1,000, Cell Signaling Technology), anti-phospho-JNK (1:1,000, Cell Signaling Technology), anti-JNK (1:1,000, Cell Signaling Technology), anti-IL-6 (1:1,000, Thermo Fisher), anti-HO-1 (1:5,000, Enzo Life Sciences, Farmingdale, NY), or anti-VEGF receptor 2 (VEGFR2; 1:1,000, R&D Systems, Minneapolis, MN) followed by an incubation with peroxidase-conjugated anti-mouse (Kindle BioSciences, Greenwich, CT), anti-rabbit (Kindle BioSciences), goat anti-rat (Jackson ImmunoResearch Laboratories), or donkey anti-goat (Jackson ImmunoResearch Laboratories). Horseradish peroxidase activity was detected using the enhanced chemiluminescence KwikQuant detection system (Kindle BioSciences). Membranes were stripped and reprobed with anti-β-actin (1:10,000, Sigma-Aldrich) to confirm loading and transfer. Densitometry analysis was performed using ImageStudioLite. Protein expression was normalized to total p65, total Akt, total JNK, or β-actin, and data are presented as fold changes over controls. The specificity of antibodies was confirmed using the specsheet from the respective manufacturers to ensure that positive controls were used to identify the right molecular weight for the protein of interest. Where available, antibodies were also tested using positive and negative controls using either recombinant proteins or protein lysates from knockout mice (e.g., for HO-1).
Large-Scale Three-Dimensional Imaging and Tissue Cytometry
Kidneys were fixed in 4% paraformaldehyde and sectioned into 50-µm sections using a vibratome. Sections were stained with anti-LYVE-1 antibody (1:500, Cat. No. NB600-1008, NovusBio) or costained with endomucin (Emcn; 1:100, Cat. No. ab106100, Abcam) and von Willebrand factor (vWF; 1:200, Cat. No. NB600-586, NovusBio) antibodies. DAPI was used to stain nuclei, and Oregon green phalloidin (Cat. No. O74466, Thermo Fisher) was used for staining F-actin. Sections were mounted in Prolong Glass and cured. Confocal images were acquired with a Leica TCS SP8 confocal/2-photon system (Leica Microsystems) using a Leica HC PL APO ×20/0.75 IMM CORR CS2 objective and four-channel sequential illumination acquisition mode (in one set of slides, the blue channel was used to visualize DAPI staining, the green channel was used to collect the phalloidin signal, the red channel was used to collect the LYVE1 signal, the far red channel was used to capture endogenous fluorescence; in another set of slides, the red channel was used to collect the endomucin signal and the far red channel was used to collect the vWF signal). Volumetric tile scanning of the entire section was performed overnight, and the image volumes were digitally stitched using Leica LASX software. The resulting four-channel large-scale volumes were analyzed.
Image Processing and Analytics
Volumetric Tissue Exploration and Analysis (VTEA), an ImageJ plugin, was used to perform three-dimension (3D) tissue cytometry, as previously described (44, 45). Nuclear segmentation was completed following intensity thresholding. Nuclei were identified based on the fluorescent intensity in the respective channels. Scatterplots were generated to allow gating of the cells of interest, mapping on the image and quantitation. Endogenous fluorescence was measured in the far-red channel on maximum projections of the image volumes. An intensity threshold was applied, and the area of thresholded intensity was recorded for the entire image. The endogenous fluorescence area was reported as percentage of the total area of the kidney, which was outlined using the region of interest function in ImageJ.
Statistical Analysis
Data are presented as means ± SE. ANOVA and Tukey’s multiple comparisons post hoc test were performed for comparisons between groups. P values of <0.05 were considered significant. All analyses were performed using GraphPad Prism 8.
RESULTS
VEGFR3 Antagonism by MAZ51 Worsens Kidney Function During Cp Nephrotoxicity
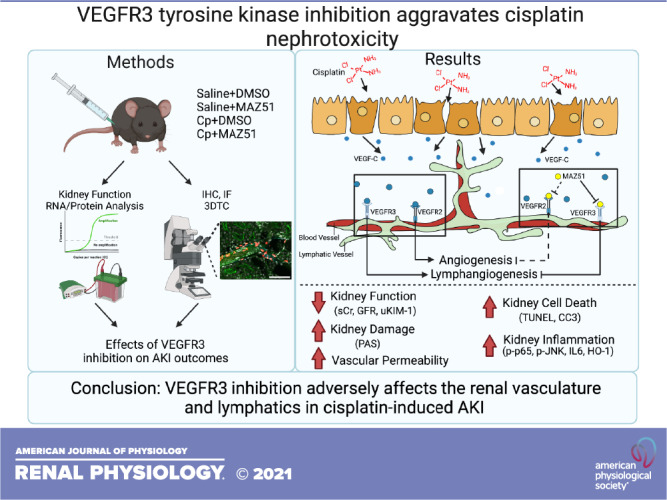
To investigate the role of LA in AKI, we used a model of kidney injury by administration of a nephrotoxic chemotherapeutic agent, Cp. We induced acute Cp nephrotoxicity using 20 mg/kg Cp (ip, dissolved in saline) and harvested at day 3 postinjury. Vegfr3-targeted deletion is lethal (46); therefore, to interrogate the effect of LA blockade in AKI, we used MAZ51 [10 mg/kg ip, dissolved in DMSO, as previously described (40–42)] as a potent VEGFR3 inhibitor at the time of Cp injection and daily until the time of harvest (Fig. 1A). We also used experimental controls: mice treated with saline and DMSO as well as mice treated with saline and MAZ51. We did not observe any effects of DMSO or MAZ51 treatment alone in control animals. However, whereas Cp and DMSO administration (Cp + DMSO) caused robust nephrotoxic injury, mice treated with Cp and MAZ51 (Cp + MAZ51) showed significantly worse function, as assessed by serum creatinine (Cp + DMSO: 0.96 ± 0.18 mg/dL vs. Cp + MAZ51: 2.1 ± 0.21 mg/dL, P = 0.0001) and GFR (Cp + DMSO: 51.2 ± 15.7 µL/min vs. Cp + MAZ51: 8.1 ± 2.8 µL/min, P = 0.0201; Fig. 1, B and C). We also investigated the degree of proximal tubule injury and found a significant rise in urinary KIM-1 in both groups, with a trend toward worse injury with Cp + MAZ51 treatment (Cp + DMSO: 999.1 ± 163.6 mg/mg urinary creatinine vs. Cp + MAZ51: 1,081 ± 88.5 ng/mg urinary creatinine, P = 0.0954; Fig. 1D). Concurrently, Cp + DMSO-treated mice had better preserved kidney architecture with significantly fewer casts and necrotic tubules in the medulla [Cp + DMSO: 3.4 ± 0.17 arbitrary units (AU) vs. Cp + MAZ51: 3.9 ± 0.11 AU, P = 0.0251], although damage in the cortex was comparable (Cp + DMSO: 3.8 ± 0.09 AU vs. Cp + MAZ51: 3.7 ± 0.13 AU, P = 0.955; Fig. 1, E–G).
Figure 1.
Vascular endothelial growth factor receptor 3 (VEGFR3) tyrosine kinase inhibition aggravates cisplatin (Cp) nephrotoxicity. A: experimental design for Cp (20 mg/kg in sterile saline) and MAZ51 (10 mg/kg in sterile DMSO) administration. Male C57Bl/6J mice were used in all experiments, and end-point measurements were made at day 3. B: serum creatinine (sCr) levels were measured and expressed as mg/dL. C: glomerular filtration rate (GFR) was measured using transcutaneous FITC-sinistrin clearance and expressed as µL/min. D: urinary kidney injury molecule-1 (KIM-1) levels were measured, normalized to urinary creatinine (uCr) levels, and expressed as ng/mg uCr. Periodic acid-Schiff-stained transverse kidney sections were imaged, analyzed for injury score in the cortex (E) and medulla (F), and expressed in arbitrary units (AU). Five to eight images from the cortex and the medulla were evaluated. The scoring system was as follows: 0, no apparent damage; 1, up to 25% area; 2, 25–50% area; 3, 50–75% area; and 4, over 75% area. G: representative periodic acid-Schiff staining from the cortex and medulla. Scale bar = 50 µm. Data are expressed as means ± SE; n = 6–24 animals per group. *P < 0.05 vs. vehicle control; #P < 0.05 vs. Cp + DMSO using one-way ANOVA followed by a Tukey’s multiple comparisons test. b.w., body weight.
Blockade of LA Exacerbates Cell Death During AKI
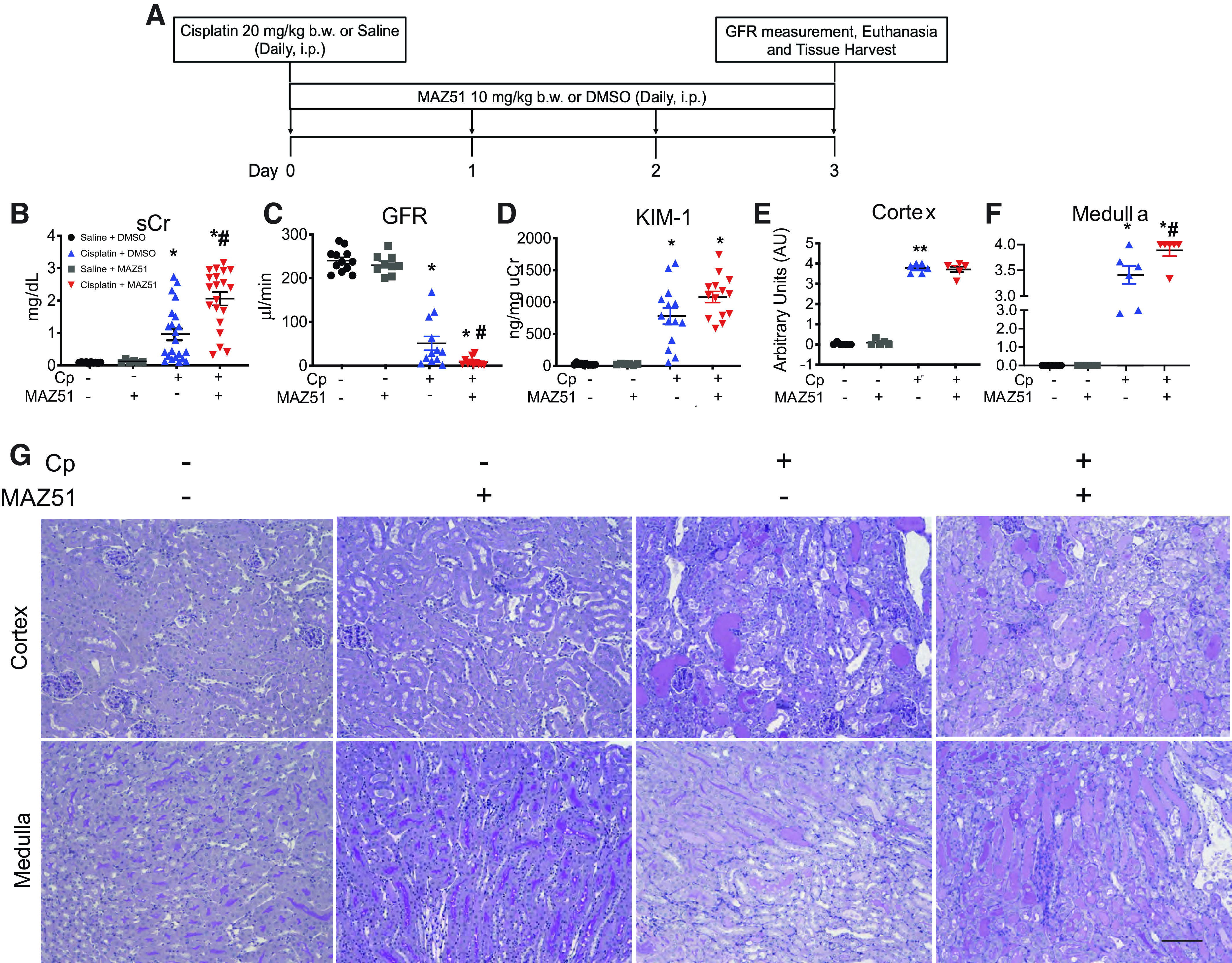
We have previously demonstrated that Cp causes kidney injury and significant apoptotic cell death (38, 47, 48). To demonstrate cell death processes during AKI and the effect of VEGFR3 antagonism on Cp-induced nephrotoxicity, we performed TUNEL on kidney sections of mice in each group (Fig. 2A). In the cortex, Cp + MAZ51-treated animals displayed a trend toward increased TUNEL-positive cells compared with Cp alone-treated controls (Cp + MAZ51: 546.8 ± 147.9 TUNEL-positive cells vs. Cp + DMSO: 258.5 ± 56.62 TUNEL-positive cells, P = 0.0910; Fig. 2B), whereas the difference between injured MAZ51-treated animals and injured controls was significant in the medulla (Cp + MAZ51: 203.3 ± 49.75 TUNEL-positive cells vs. Cp + DMSO: 56.83 ± 14.77 TUNEL-positive cells, P = 0.0052; Fig. 2C). We also assessed kidney lysates for cleaved caspase-3. Although both injury groups had an increase in apoptotic cell death, Cp + MAZ51-treated animals displayed significantly higher cleaved caspase-3 expression than Cp + DMSO-treated controls (Cp + MAZ51: 5.6 ± 0.74 AU vs. Cp + DMSO: 3.4 ± 0.54 AU, P = 0.0363; Fig. 2D).
Figure 2.
Cell death is exacerbated when lymphangiogenesis is blocked in cisplatin (Cp) nephrotoxicity. A: representative images of TUNEL staining on transverse kidney sections at day 3 after Cp administration. Whole kidney sections were imaged, and TUNEL-positive cells were quantitated in the cortex (B) and medulla (C). D: protein lysates from whole kidney tissue were analyzed for cleaved caspase-3 (CC3) expression. Anti-β-actin was used as a loading control. Densitometric values were calculated and expressed in arbitrary units (AU). Data are expressed as means ± SE; n = 6–17 animals per group. *P < 0.05 vs. vehicle control; #P < 0.05 vs. Cp + DMSO using one-way ANOVA followed by a Tukey’s multiple comparisons test. Scale bar = 100 µm.
Increased Inflammation in Cp Nephrotoxicity With VEGFR3 Inhibition
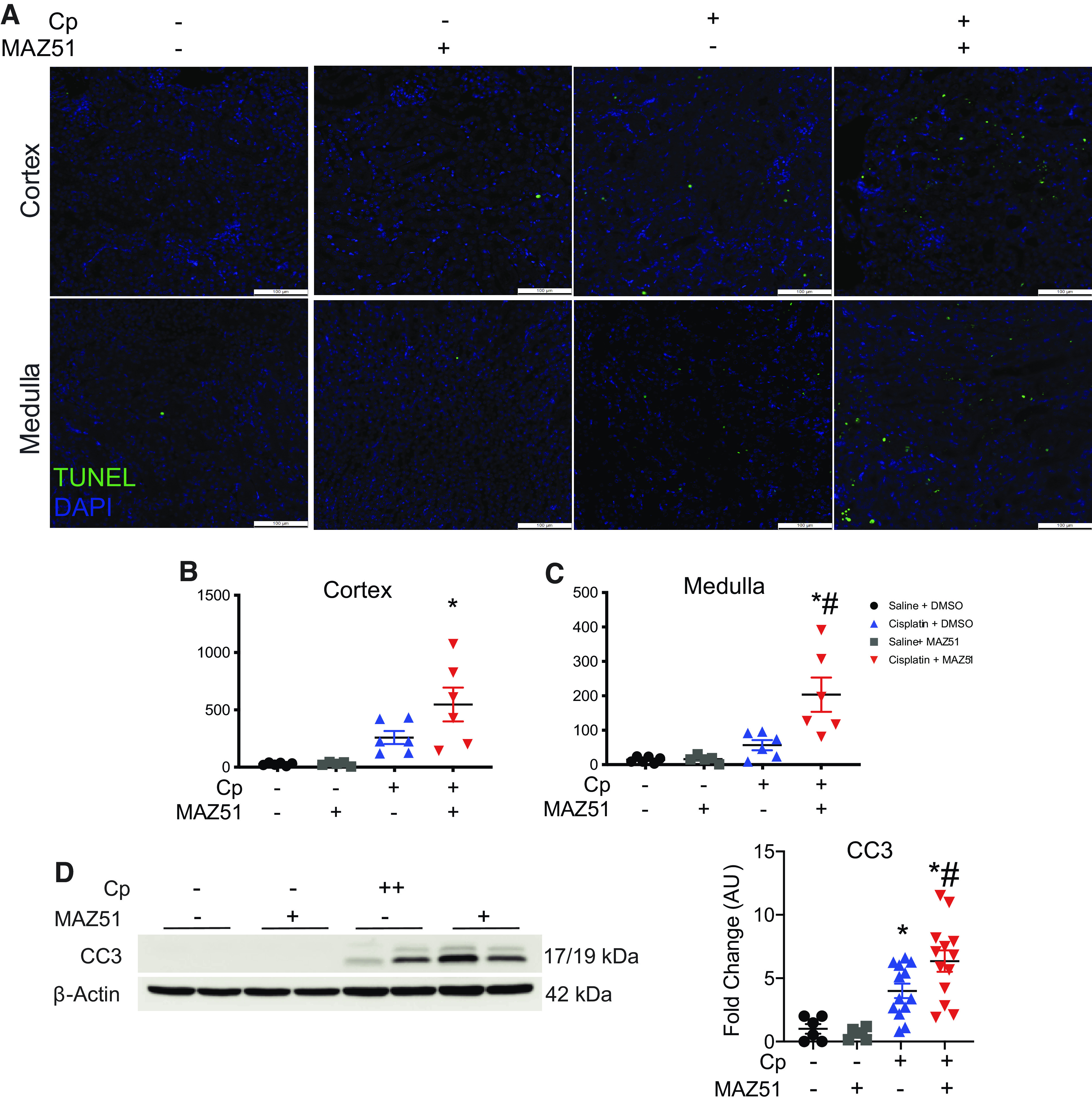
To determine the effect of VEGFR3 inhibition on intrarenal inflammation, we assessed the expression of classical inflammatory chemokines and cytokines in Cp nephrotoxicity. Although we detected a rise in mRNA levels of Csf1, Ccl2, Tnf-α, and Il6 relative to control animals, we did not detect differences between injury groups (Fig. 3, A–D). We next addressed whether VEGFR3 antagonism affects the phosphorylation of key proteins p65, Akt, and JNK as well as IL-6 expression levels, due to their role in regulating LA (49, 50). We demonstrated significantly elevated levels of phospho-p65 (Cp + MAZ51: 5.1 ± 0.83 AU vs. Cp + DMSO: 1.7 ± 0.71 AU, P = 0.005) and phospho-JNK (Cp + MAZ51: 40.9 ± 8.0 AU vs. Cp + DMSO: 13.7 ± 2.6 AU, P = 0.005) in Cp + MAZ51-treated animals compared with Cp + DMSO-treated controls. We next evaluated levels of IL-6 protein due to its role in regulating lymphangiogenic ligand synthesis (51) and detected significantly more IL-6 in Cp + MAZ51-treated kidneys compared with Cp + DMSO-treated kidneys (Cp + MAZ51: 4.6 ± 0.72 AU vs. Cp + DMSO: 2.3 ± 0.43 AU, P = 0.0094). Akt is required for the promotion of LA (52). Although we did detect a significant elevation of phospho-Akt in both injury groups compared with vehicle controls, we did not observe differences between Cp + MAZ51- and Cp + DMSO-treated groups (Cp + MAZ51: 2.07 ± 0.20 AU vs. Cp + DMSO: 2.26 ± 0.27 AU, P = 0.92; Fig. 3E). We also observed increased expression of HO-1, at both transcript (Cp + MAZ51: 9.1 ± 1.8 vs. Cp + DMSO: 3.1 ± 0.77, P = 0.0053) and protein (Cp + MAZ51: 53.0 ± 14.9 vs. Cp + DMSO: 23.6 ± 7.8, P = 0.14) levels in the Cp + MAZ51-treated group compared with Cp + DMSO-treated controls (Fig. 3, F and G), further suggesting that VEGFR3 inhibition exacerbates renal inflammation in this injury model. Although we detected upregulation of inflammation-associated proteins, we did not observe differences in inflammatory infiltrate (Supplemental Fig. S1, https://doi.org/10.6084/m9.figshare.16553616.v2); therefore, we believe that the inflammation detected is largely sourced from the proximal tubules, the maximal site of Cp-mediated injury in the kidney (53).
Figure 3.
Inhibition of lymphangiogenesis augments intrarenal inflammation in cisplatin (Cp)-induced acute kidney injury. Total RNA was isolated from whole kidney tissue at day 3 and analyzed for expression of colony stimulating factor-1 (Csf1; A), chemokine (C-C motif) ligand 2 (Ccl2; B), tumor necrosis factor-α (Tnfα; C), and interleukin-6 (Il6; D) by real-time PCR. Data were normalized to Gapdh and expressed as fold changes compared with vehicle controls. Data are expressed as means ± SE; n = 6–19 animals per group. Protein lysates from whole kidney tissue were analyzed for expression of phosphorylated (p)-p65 (NF-κB), p-Akt (RAC-α serine-threonine-protein kinase), p-JNK, and IL-6 (E) and heme oxygenase-1 (HO-1; F). Anti-β-actin was used as a loading control. Densitometric values were calculated and expressed in arbitrary units (AU). Data are expressed as means ± SE; n = 6–19 animals per group. Data were analyzed using one-way ANOVA followed by a Tukey’s multiple comparisons test. G: real-time PCR of kidneys at day 3 for expression of the HO-1 gene (Hmox1). Data were normalized to Gapdh and expressed as fold changes compared with vehicle controls. Data are expressed as means ± SE; n = 5–9 animals per group. *P < 0.05 vs. vehicle control; #P < 0.05 vs. Cp + DMSO using one-way ANOVA followed by a Tukey’s multiple comparisons test.
Dynamics of Lymphatic Vessels Are Altered With VEGFR3 Inhibition in Cp Nephrotoxicity
We have previously demonstrated a significant increase in lymphatic vessels after Cp administration (15). Using 3 D imaging and tissue cytometry (3DTC), we sought to evaluate LYVE-1-positive lymphatic vessel expansion after injury and in the context of VEGFR3 antagonism. Saline + DMSO-treated (Fig. 4A), Cp + DMSO-treated (Fig. 4B), saline + MAZ51-treated (Fig. 4C), and Cp + MAZ51-treated (Fig. 4D) kidney tissues were stained with LYVE-1 to label lymphatics, F-actin to label filamentous actin, and DAPI to label nuclei. VTEA was used to identify LYVE-1-positive nuclei. We measured serum creatinine in these mice to show the consistency and reproducibility of this experimental model and found similar results as previously noted, where Cp + MAZ51 treatment exhibited significantly worse renal injury compared with Cp + DMSO treatment (Cp + MAZ51: 2.1 ± 0.35 mg/dL vs. Cp + DMSO: 0.72 ± 0.23 mg/dL, P = 0.0146; Fig. 4E). VTEA showed no statistical difference in LYVE-1-positive lymphatic vessel abundance between Cp + DMSO and Cp + MAZ51 treatment (Cp + DMSO: 0.13 ± 0.020% total cells vs. Cp + MAZ51: 0.20 ± 0.051% total cells, P = 0.5428; Fig. 4F). We gated on LYVE-1Hi cells (Supplemental Fig. S2, https://doi.org/10.6084/m9.figshare.14569437.v2) and found that Cp + MAZ51-treated mice had a trend toward higher LYVE-1Hi lymphatic vessel density compared with Cp + DMSO-treated mice (Cp + MAZ51: 0.065 ± 0.011% total cells vs. Cp + DMSO: 0.028 ± 0.007% total cells, P = 0.0523; Fig. 4G). We also assessed LYVE-1Lo lymphatic vessel density and found no differences between Cp + MAZ51 and Cp + DMSO treatment (Cp + MAZ51: 0.13 ± 0.041% total cells vs. Cp + DMSO: 0.11 ± 0.014% total cells, P = 0.8712; Fig. 4H). Additionally, we observed no differences between saline + DMSO and saline + MAZ51 treatment (total LYVE-1-positive cells: 0.070 ± 0.014% for saline + DMSO vs. 0.085 ± 0.035% for saline + MAZ51, P = 0.99; Fig. 4F).
Figure 4.
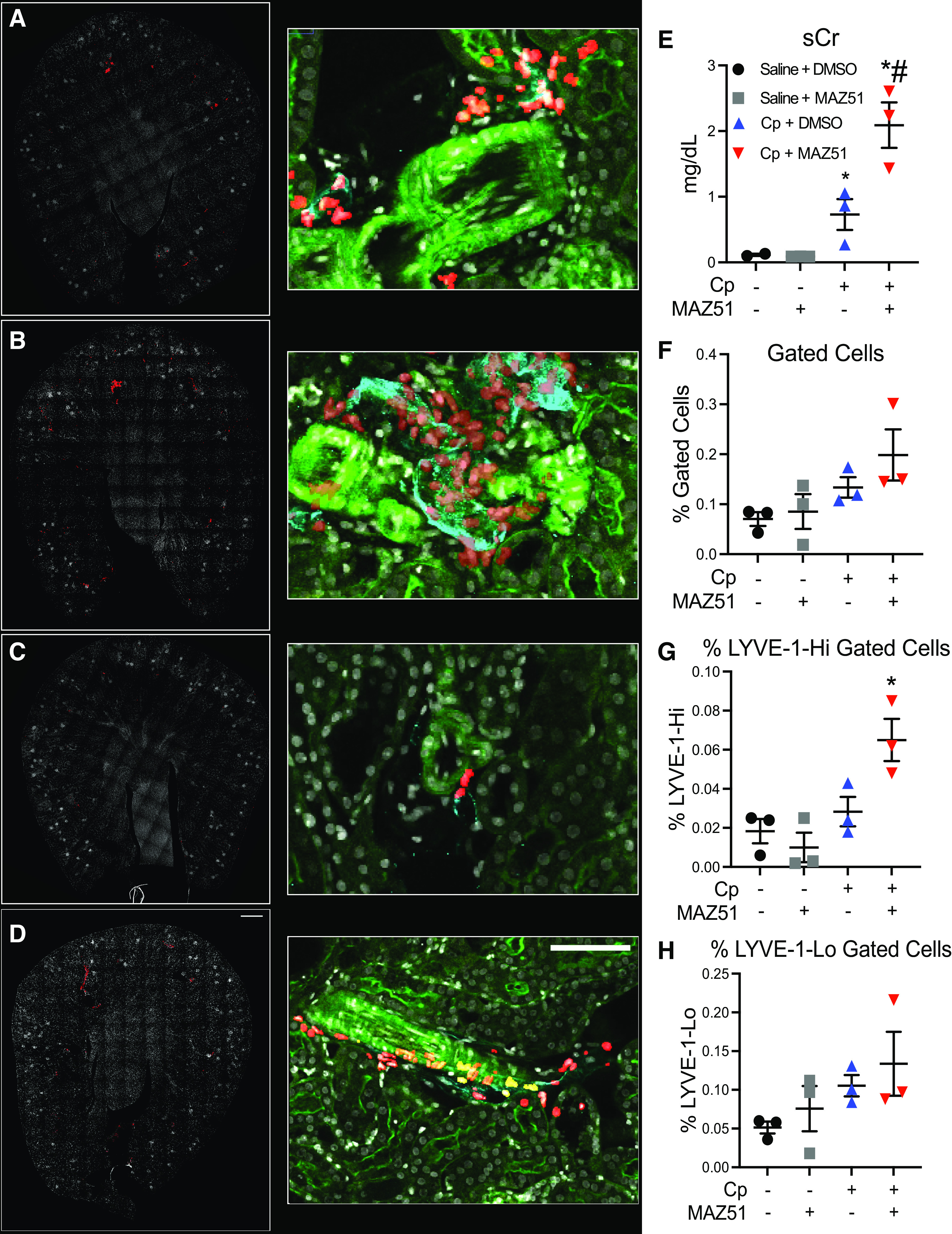
Three-dimensional confocal fluorescence microscopy and cytometry of lymphatic vessel hyaluronan receptor-1 (LYVE-1)-positive lymphatic vessels. Maximum projection of composite fluorescence images of whole kidney sections (left; scale bar = 500 µm) and high-magnification insets (middle; scale bar = 100 µm) from saline + DMSO-treated (A), cisplatin (Cp) + DMSO-treated (B), saline + MAZ51-treated (C), and Cp + MAZ51-treated (D) animals at day 3. In the left images, DAPI is shown in white and nuclei of LYVE-1-positive cells are shown in red. In the middle images, DAPI is shown in white, LYVE-1 is shown in cyan, filamentous actin is shown in green, and gated LYVE-1-positive nuclei are shown in red. E: serum creatinine (sCr) levels were measured and expressed as mg/dL at day 3. Scatterplots with percentages of total LYVE-1-positive cells (F), LYVE-1-Hi (G), and LYVE-1-Lo (H) gated cells. Quantifications obtained from Volumetric Tissue Exploration and Analysis were plotted, showing percentages out of the total number of cells. Data are expressed as means ± SE; n = 3 animals per group. *P < 0.05 vs. vehicle control; #P < 0.05 vs. Cp + DMSO using one-way ANOVA followed by a Tukey’s multiple comparisons test.
We next evaluated expression of lymphangiogenic factors. We found a trend toward suppressed expression of Lyve1 (saline + MAZ51: 0.38 ± 0.11 AU vs. saline + DMSO: 1.0 ± 0.53 AU, P = 0.95), Pdpn (saline + MAZ51: 0.83 ± 0.41 AU vs. saline + DMSO: 1.0 ± 0.32 AU, P = 0.99), Prox1 (saline + MAZ51: 0.75 ± 0.16 AU vs. saline + DMSO: 1.0 ± 0.21 AU, P = 0.46), and Flt4 (saline + MAZ51: 0.54 ± 0.13 AU vs. saline + DMSO: 1.0 ± 0.25 AU, P = 0.91) in saline + MAZ51-treated animals compared with saline + DMSO-treated controls, suggesting quiescent suppression of LA due to MAZ51. Interestingly, at day 3 post-Cp administration, we observed a trend toward increased levels of Lyve1 (Cp + MAZ51: 2.9 ± 0.75 AU vs. Cp + DMSO: 1.5 ± 0.31 AU, P = 0.21), Pdpn (Cp + MAZ51: 3.9 ± 0.77 AU vs. Cp + DMSO: 2.0 ± 0.50 AU, P = 0.093), and Flt4 (Cp + MAZ51: 2.6 ± 0.33 AU vs. Cp + DMSO: 1.9 ± 0.36 AU, P = 0.45) after Cp + MAZ51 treatment compared with Cp + DMSO treatment, with Flt4 expression significantly upregulated compared with saline + MAZ51 treatment (Cp + MAZ51: 2.6 ± 0.33 AU vs. saline + MAZ51: 0.54 ± 0.54 AU, P = 0.01). We observed similar Prox1 expression levels after Cp + DMSO and Cp + MAZ51 administration in both groups (Cp + DMSO: 0.32 ± 0.04 AU vs. Cp + MAZ51: 0.33 ± 0.04 AU, P = 0.99; Supplemental Fig. S3, A–D, https://doi.org/10.6084/m9.figshare.14569449.v2).
Cp Treatment Causes Erythrocyte-Associated Vascular Congestion That Is Absent in MAZ51-Treated Animals
We used confocal microscopy to collect the 3D volume of autofluorescence and second harmonic generation signals from kidney sections as part of the 3DTC workflow. We detected erythrocyte-associated autofluorescence after Cp + DMSO (Fig. 5, A and C), suggesting vascular congestion, which is characteristic of Cp injury (54). Interestingly, vascular congestion was absent in Cp + MAZ51-treated animals (Cp + DMSO: 5.8 ± 3.4% total cells vs. Cp + MAZ51: 0.71 ± 0.30% total cells, P = 0.0329; Fig. 5, B and C). We observed minimal autofluorescence in the saline + DMSO- and saline + MAZ51-treated groups (saline + DMSO: 1.3 ± 0.42% total cells vs. saline + MAZ51: 0.26 ± 0.30% total cells, P = 0.8807; Fig. 5C).
Figure 5.
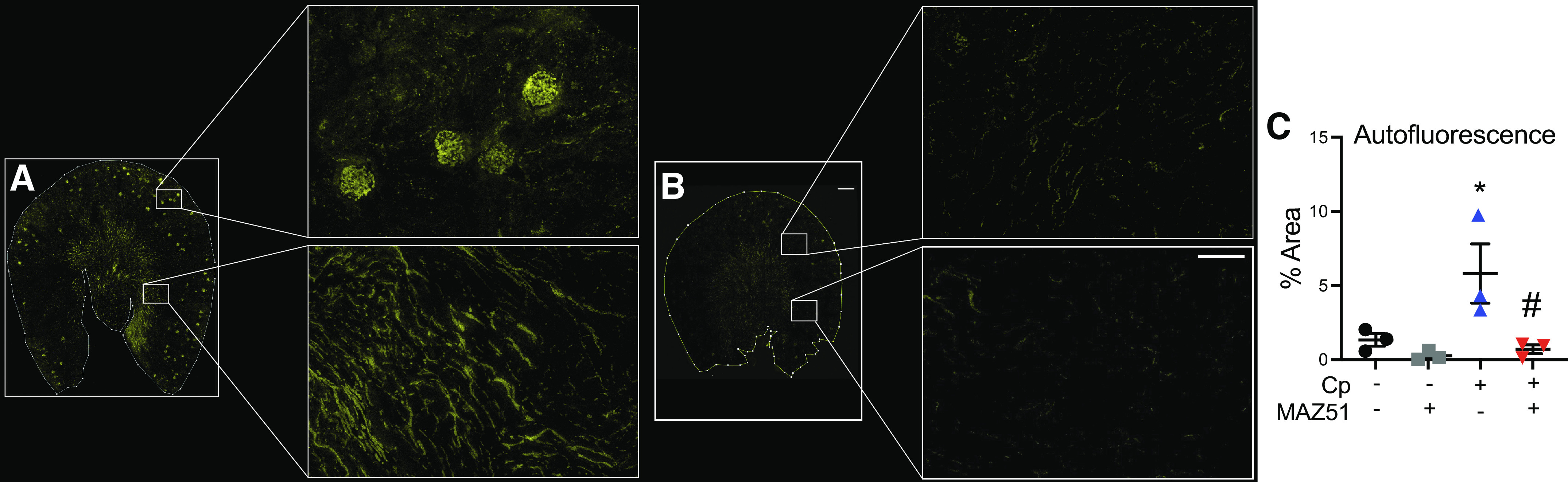
The autofluorescence imaging signature reveals erythrocyte-associated vascular congestion in cisplatin (Cp)-treated animals without MAZ51 treatment. Maximum projection images of three-dimensional volume of autofluorescence of Cp + vehicle-treated (A) and Cp + MAZ51-treated (B) whole kidney sections (left; scale bar = 500 µm) and high-magnification insets (right; scale bar = 50 µm). C: quantification obtained from volumetric tissue exploration and analysis was plotted, showing percentages of autofluorescent cells out of the total number of cells. Data are expressed as means ± SE; n = 3 animals per group. *P < 0.05 vs. vehicle control; #P < 0.05 vs. Cp + DMSO using one-way ANOVA followed by a Tukey’s multiple comparisons test.
Due to our observation that MAZ51-treated animals did not exhibit vascular congestion, we hypothesized that perhaps MAZ51 has off-target effects (29), which might affect vascular permeability. To assess the effects of MAZ51 on the blood vasculature in our injury model, we used 3DTC to assess the expression of the endothelial markers Emcn and vWF (Fig. 6, A−F). Here, we report a modest decrease in renal Emcn expression in animals treated with MAZ51 compared with their DMSO-treated controls, both at baseline (saline + MAZ51: 10.0 ± 1.2% vs. saline + DMSO: 13.8 ± 2.3%, P = 0.53) and after Cp administration (Cp + MAZ51: 9.5 ± 1.6% vs. Cp + DMSO: 11.9 ± 2.8%, P = 0.82), which was observed throughout the kidney (Fig. 6E). vWF expression also followed this trend in saline + MAZ51-treated (saline + MAZ51: 0.29 ± 0.06% vs. saline + DMSO: 0.41 ± 0.024%, P = 0.86) and Cp + MAZ51-treated (Cp + MAZ51: 0.29 ± 0.12% vs. Cp + DMSO: 0.44 ± 0.18%, P = 0.81) groups (Fig. 6F). Furthermore, it has been suggested that VEGFR3 inhibition increases vascular permeability through upregulation of VEGFR2 (55). Here, we report that MAZ51-treated animals had a trend toward increased expression of renal VEGFR2 compared with control groups both at baseline (saline + MAZ51: 1.6 ± 0.30% vs. saline + DMSO: 1.0 ± 0.31%, P = 0.57) and after Cp administration (Cp + MAZ51: 1.3 ± 0.25% vs. Cp + DMSO, 0.91 ± 0.23, P = 0.70; Fig. 6G).
Figure 6.
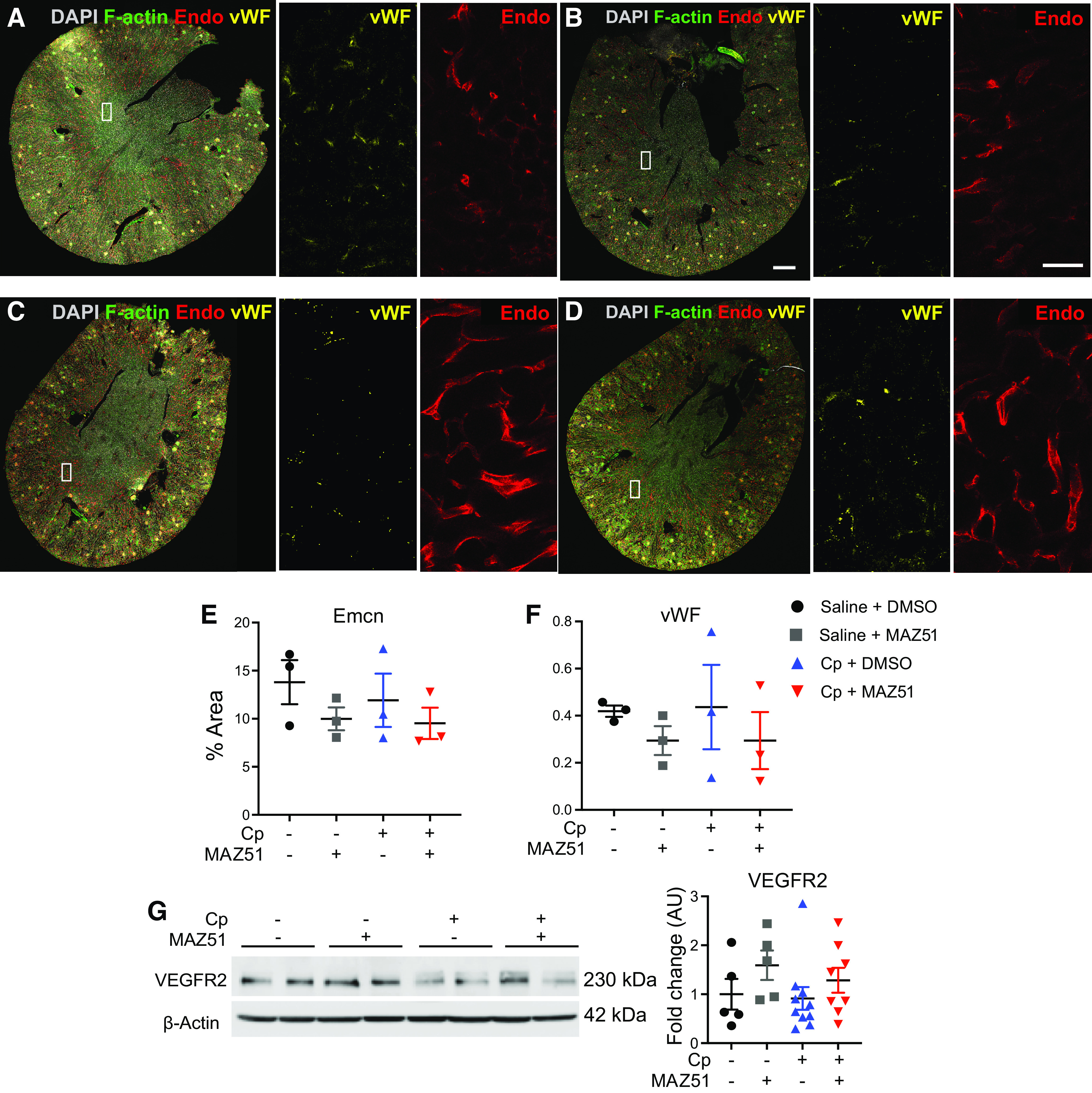
Visualization and analysis of endomucin (Emcn)-positive and von Willebrand factor (vWF)-positive cells. Large-scale three-dimensional imaging of saline + DMSO-treated (A), saline + MAZ51-treated (B), cisplatin (Cp) + DMSO-treated (C), and Cp + MAZ51-treated (D) animals at day 3. The white rectangles correspond to the two insets with Emcn (E) or wWF (F) alone. Scale bars = 500 µm in B and 50 µm in B, right inset. Quantification of the immunofluorescent area corresponding to Emcn-positive (E) and vWF (F)-positive cells. G: Western blot analysis of vascular endothelial growth factor receptor 2 (VEGFR2) expression levels. Anti-β-actin was used as a loading control. Densitometric values were calculated and expressed in arbitrary units (AU). Data are expressed as means ± SE; n = 5–10 animals per group.
DISCUSSION
The results of this work indicate that LA is a necessary process that occurs as a response to Cp-induced AKI. We used MAZ51, a highly selective VEGFR3 tyrosine kinase inhibitor. The efficacy of MAZ51 is well documented (25, 29, 40, 41, 56–58). MAZ51 minimally affects VEGFR1 and VEGFR2 at concentrations >10-fold higher than those used to inhibit VEGFR3 activity (26), highlighting its specificity to selectively block LA. Cp + MAZ51-treated mice exhibited significantly worse injury than Cp + DMSO-treated mice. Our results also indicate that blocking VEGFR3 activation significantly exacerbates renal cell death and inflammation in Cp nephrotoxicity. We used 3DTC as a novel technique to assess lymphatic expansion postinjury and found no structural differences between MAZ51-treated and vehicle-treated animals after Cp exposure; however, we interestingly found that the erythrocyte-associated vascular congestion that we observed after Cp + DMSO was absent in Cp + MAZ51 animals that displayed worse injury. Interestingly, we found evidence of vascular injury as a result of MAZ51 treatment, whereby we saw a decrease in expression of the endothelial cell markers Emcn and vWF with an increase in VEGFR2 expression.
We have previously characterized Cp nephrotoxicity as a robust inducer of renal LA (15). Other widely used kidney models, such as UUO and ischemia-reperfusion injury, have major confounding features that have the potential to augment intrarenal LA, such as urinary fluid retention and mechanical damage from clamp pressure to the renal artery, vein, and associated lymphatics, respectively. Cp administration leads to prominent VEGF-C and VEGF-D secretion into the urine and serum as well as upregulation of VEGFR3 expression in injured kidneys (15). LA has the potential to be an adaptive response to injury, as its intrinsic responsibilities include clearance of debris and excess fluid, removal of proinflammatory cytokines, and regulatory T-cell migration. This is true in the context of polycystic kidney disease (22), myocardial infarction (59), and lung transplantation (60). However, LA is a maladaptive response in the context of corneal transplantation (61) and diabetes (62), as new lymphatic vessels serve as a conduit for inflammatory cell infiltration, leading to exacerbated inflammation. Promotion of LA in cancer is also detrimental, as lymphatic vessels are associated with the dissemination of tumor cells and metastasis (63, 64). In the kidney, there have also been conflicting reports. For example, one study showed that VEGF-C administration attenuates inflammation and fibrosis post-UUO (18), whereas another study reported that de novo lymphatic vessels aggravate intrarenal inflammation and subsequently promote fibrosis (20). Furthermore, a recent study indicated that the promotion of LA via kidney-specific VEGF-D overexpression reduces blood pressure in a murine model of hypertension (65). In chronic allograft nephropathy, lymphatic expansion has been associated with renal allograft fibrosis and rejection (66, 67). Altogether, the results of these studies suggest that the beneficial or detrimental nature of LA is context and etiology specific, and further studies are needed to elucidate and confirm potential pathways that can be exploited for therapeutic use.
Cp is primarily toxic to renal proximal tubules due to their high mitochondrial density (53), although glomeruli and blood vessels are among the other compartments that are also affected (68). Most drugs enter the vascular system and are drained through the lymphatic system; however, the vast majority of drug research targets only the blood vasculature (69). Furthermore, there has been increasing interest in targeting the lymphatics for drug delivery, whereas research into the associated effects on the lymphatic vessels themselves remains unclear (70). In fact, it was recently reported that platinum-based chemotherapeutic agents directly induce VEGFR3-dependent LA in cancer, subsequently stimulating lymphatic proliferation and migration (32), highlighting the need to explore Cp and its relationship with the lymphatic system.
Our study first focused on the effects of VEGFR3 inhibition on kidney injury, function, and structural integrity after Cp administration. The S3 segment of the proximal tubule is classically regarded as the site of maximal Cp-induced injury due to its high mitochondrial content as well as its expression of organic cation transporter-2, the transporter primarily responsible for cellular Cp uptake (38, 71). We detected increased cell death by TUNEL, whereby the Cp + MAZ51-treated group had significantly more TUNEL-positive cells in the medulla compared with the Cp + DMSO-control group. This observed difference in cell death was corroborated by intrarenal cleaved caspase-3 expression. These results suggest that VEGFR3-dependent LA processes are necessary during Cp nephrotoxicity, potentially due to the indispensability of lymphatic vessels in draining inflammatory mediators and extravasated fluid (72) as well as in potentially removing chemotherapeutic drugs and other nephrotoxins from the kidney (32, 69).
Cp + MAZ51 treatment suppressed expression of major lymphatic-associated gene expression in quiescent kidneys, which supports the effects of MAZ51 in blocking LA signaling via VEGFR3 tyrosine kinase inhibition. We originally hypothesized that MAZ51-treated animals would display lesser LYVE-1-positive vessel density, as MAZ51 should block VEGFR3-mediated LA. We hypothesize that the increase in LA-associated gene expression and LYVE-1Hi vessels observed after Cp + MAZ51 treatment is due to the extent of the kidney injury and damage, where the LA response is only partially muted by MAZ51 treatment. After Cp injury, we and others have previously shown that proximal tubules express apoptotic, necrotic, and inflammatory factors in addition to LA ligands, which promote lymphatic proliferation and expansion (15, 38, 73).
We expect that AKI that would generate a significant rise in serum creatinine similar to that of our Cp + MAZ51-treated group (2.1 ± 0.21 mg/dL) would also stimulate a significant LA response. This is especially reasonable considering that LA is driven by the injury response, whereby immune cell infiltration and cytokine and growth factor expression drive lymphatic growth and regeneration. Here, we showed a significant upregulation of inflammatory proteins, including IL-6 and HO-1, after Cp + MAZ51 compared with Cp + DMSO treatment, which are largely contributed by injured proximal tubules. Due to the fact that we observed suppressed transcript levels of key LA mediators (Lyve1, Pdpn, and Flt4) in quiescence but similar levels of LA after Cp administration in DMSO- and MAZ51-treated animals, we hypothesize that the severity of injury after Cp + MAZ51 treatment causes a sustained inflammatory signal from injured proximal tubules that drives LA that supersedes MAZ51 treatment. In fact, patients with chronic interstitial nephritis and IgA nephropathy display significantly higher numbers of intrarenal Pdpn-positive lymphatic vessels compared with those with acute tubulointerstitial nephritis (74), highlighting that persistent inflammatory signaling intensifies de novo LA (75). Additionally, Cp-based chemotherapies are known to disrupt and damage the vascular endothelium (76, 77); therefore, the damage inflicted on the kidney after Cp + MAZ51 treatment in conjunction with injured lymphatic vessels would further stimulate LA. MAZ51 has also been shown to cause a significant reduction in lymphatic vessel functionality, including basal lymphatic pump function (26, 78). Future studies to understand the structural and functional integrity of renal lymphatics during Cp nephrotoxicity would be of interest.
An intriguing observation noted in this work was increased erythrocyte-associated autofluorescence in the kidneys of Cp-treated mice. This vascular complication is common in Cp-based chemotherapy (54, 79, 80). Interestingly, we did not detect this autofluorescence in kidneys from mice treated with Cp + MAZ51 or saline + MAZ51. We hypothesize that the absence of vascular congestion with MAZ51 treatment is due to the increased structural damage we observed and possible effects of MAZ51 on vascular injury and subsequent extravasation of red blood cells (26). Additionally, although VEGFR3 is restricted to the adult lymphatic endothelium, low levels of VEGF-R3 are also detectable in vascular endothelial cells, particularly in peritubular capillaries (22, 81, 82), and its expression may be upregulated in the blood vasculature during injury. In a study by Liu and colleagues (81), they showed that heterozygous mutation of VEGFR3 was required for the development of lymphatics in the kidney but did not have an adverse effect on kidney function in a model of low-dose Cp nephrotoxicity associated with mild injury. Genetic deletion of Vegfr3 (Flt4) increases vascular permeability through upregulation of VEGFR2 (55). We found that treatment with MAZ51 caused decreased expression of Emcn and vWF, with increased VEGFR2 expression, suggesting increased vascular permeability in MAZ51-treated animals and off-target effects of this inhibitor. We speculate that the effects of MAZ51 on blood vessels and lymphatics leads to worse tubular injury in Cp-induced AKI. Although it is widely accepted that MAZ51 at the dose used in this study is a robust and reliable inhibitor of VEGFR3 tyrosine kinase activity (25, 29, 40, 56–58), further studies should be conducted that address the effects of MAZ51 treatment on lymphatic vessel integrity and function.
Perspectives and Significance
De novo LA as a response to injury is only beginning to be explored in the context of the kidney. Our study describes a previously unknown role for LA in Cp nephrotoxicity and interrogated the role of VEGFR3-dependent LA signaling in this injury model. These results highlight the importance of intact LA signaling and suggest that lymphatic expansion is necessary, at least in part, for limiting Cp nephrotoxicity. Our results also suggest off-target effects of MAZ51 on the vasculature that potentially contributed to worse kidney function in this model.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16553616.v2.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14569437.v2.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.14569449.v2.
GRANTS
This work was supported by Veterans Affairs Merit Award BX004047 (to A.A.), the Indiana O’Brien Center for Advanced Microscopic Analysis (Grant P30DK079312 to T.M.E.-A.), National Institutes of Health (NIH) Grants R01DK122986 (to S.B.) and T32DK007545 (to L.M.B.), and the core resource of the UAB-UCSD O’Brien Center for AKI Research (NIH Grant P30-DK-079337, to A.A.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.M.B., T.M.E-A., and A.A. conceived and designed research; L.M.B., E.R.F., D.B., G.O., A.A.Z., A.M.T., S.K.E., S.B., G.W., M.M.K., S.W., D.R.S., S.K., A.Z., and T.M.E-A. performed experiments; L.M.B., E.R.F., D.B., G.O., A.A.Z., A.M.T., S.K.E., M.M.K., S.W., D.R.S., S.K., A.Z., T.M.E-A., and A.A. analyzed data; L.M.B., M.M.K., S.W., S.K., A.Z., T.M.E-A., and A.A. interpreted results of experiments; L.M.B., A.Z., and T.M.E-A. prepared figures; L.M.B. drafted manuscript; L.M.B., E.R.F., D.B., G.O., A.A.Z., A.M.T., S.K.E., S.B., G.W., M.M.K., S.W., D.R.S., S.K., A.Z., T.M.E-A., and A.A. edited and revised manuscript; L.M.B., E.R.F., D.B., G.O., A.A.Z., A.M.T., S.K.E., S.B., G.W., M.M.K., S.W., D.R.S., S.K., A.Z., T.M.E-A., and A.A. approved final version of manuscript.
ACKNOWLEDGMENTS
The graphical abstract was created using BioRender.
REFERENCES
- 1.Levey AS, Eckardt KU, Dorman NM, Christiansen SL, Hoorn EJ, Ingelfinger JR, Inker LA, et al. Nomenclature for kidney function and disease: report of a Kidney Disease: improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int 97: 1117–1129, 2020. doi: 10.1016/j.kint.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Neyra JA, Chawla LS. Acute kidney disease to chronic kidney disease. Crit Care Clin 37: 453–474, 2021. doi: 10.1016/j.ccc.2020.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJ, Mann JF, Matsushita K, Wen CP. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 382: 339–352, 2013. doi: 10.1016/S0140-6736(13)60595-4. [DOI] [PubMed] [Google Scholar]
- 4.Moe SM, Chen NX. Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol 19: 213–216, 2008. doi: 10.1681/ASN.2007080854. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal R, Pappas MK, Sinha AD. Masked uncontrolled hypertension in CKD. J Am Soc Nephrol 27: 924–932, 2016. doi: 10.1681/ASN.2015030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol 23: 1631–1634, 2012. doi: 10.1681/ASN.2011111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meuwese CL, Stenvinkel P, Dekker FW, Carrero JJ. Monitoring of inflammation in patients on dialysis: forewarned is forearmed. Nat Rev Nephrol 7: 166–176, 2011. doi: 10.1038/nrneph.2011.2. [DOI] [PubMed] [Google Scholar]
- 8.Russell PS, Hong J, Windsor JA, Itkin M, Phillips ARJ. Renal lymphatics: anatomy, physiology, and clinical implications. Front Physiol 10: 251, 2019. doi: 10.3389/fphys.2019.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Natale G, Bocci G, Ribatti D. Scholars and scientists in the history of the lymphatic system. J Anat 231: 417–429, 2017. doi: 10.1111/joa.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature 438: 946–953, 2005. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 11.Klaourakis K, Vieira JM, Riley PR. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat Rev Cardiol 18: 368–379, 2021. doi: 10.1038/s41569-020-00489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura R, Yoshida K, Toda M. Current understanding of lymphatic vessels in the central nervous system. Neurosurg Rev 43: 1055–1064, 2020. doi: 10.1007/s10143-019-01133-0. [DOI] [PubMed] [Google Scholar]
- 13.Wong BW. Lymphatic vessels in solid organ transplantation and immunobiology. Am J Transplant 20: 1992–2000, 2020. doi: 10.1111/ajt.15806. [DOI] [PubMed] [Google Scholar]
- 14.Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21(st) century: novel functional roles in homeostasis and disease. Cell 182: 270–296, 2020. doi: 10.1016/j.cell.2020.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zarjou A, Black LM, Bolisetty S, Traylor AM, Bowhay SA, Zhang MZ, Harris RC, Agarwal A. Dynamic signature of lymphangiogenesis during acute kidney injury and chronic kidney disease. Lab Invest 99: 1376–1388, 2019. doi: 10.1038/s41374-019-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo YC, Zhang M, Wang FX, Pei GC, Sun F, Zhang Y, He X, Wang Y, Song J, Zhu FM, Pandupuspitasari NS, Liu J, Huang K, Yang P, Xiong F, Zhang S, Yu Q, Yao Y, Wang CY. Macrophages regulate unilateral ureteral obstruction-induced renal lymphangiogenesis through C-C motif chemokine receptor 2-dependent phosphatidylinositol 3-kinase-AKT-mechanistic target of rapamycin signaling and hypoxia-inducible factor-1α/vascular endothelial growth factor-C expression. Am J Pathol 187: 1736–1749, 2017. doi: 10.1016/j.ajpath.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki Y, Ito Y, Mizuno M, Kinashi H, Sawai A, Noda Y, Mizuno T, Shimizu H, Fujita Y, Matsui K, Maruyama S, Imai E, Matsuo S, Takei Y. Transforming growth factor-β induces vascular endothelial growth factor-C expression leading to lymphangiogenesis in rat unilateral ureteral obstruction. Kidney Int 81: 865–879, 2012. doi: 10.1038/ki.2011.464. [DOI] [PubMed] [Google Scholar]
- 18.Hasegawa S, Nakano T, Torisu K, Tsuchimoto A, Eriguchi M, Haruyama N, Masutani K, Tsuruya K, Kitazono T. Vascular endothelial growth factor-C ameliorates renal interstitial fibrosis through lymphangiogenesis in mouse unilateral ureteral obstruction. Lab Invest 97: 1439–1452, 2017. doi: 10.1038/labinvest.2017.77. [DOI] [PubMed] [Google Scholar]
- 19.Lee AS, Lee JE, Jung YJ, Kim DH, Kang KP, Lee S, Park SK, Lee SY, Kang MJ, Moon WS, Kim HJ, Jeong YB, Sung MJ, Kim W. Vascular endothelial growth factor-C and -D are involved in lymphangiogenesis in mouse unilateral ureteral obstruction. Kidney Int 83: 50–62, 2013. [Erratum in Kidney Int 92: 1018, 2017]. doi: 10.1038/ki.2012.312. [DOI] [PubMed] [Google Scholar]
- 20.Pei G, Yao Y, Yang Q, Wang M, Wang Y, Wu J, Wang P, Li Y, Zhu F, Yang J, Zhang Y, Yang W, Deng X, Zhao Z, Zhu H, Ge S, Han M, Zeng R, Xu G. Lymphangiogenesis in kidney and lymph node mediates renal inflammation and fibrosis. Sci Adv 5: eaaw5075, 2019. doi: 10.1126/sciadv.aaw5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yazdani S, Navis G, Hillebrands JL, van Goor H, van den Born J. Lymphangiogenesis in renal diseases: passive bystander or active participant? Expert Rev Mol Med 16: e15, 2014. doi: 10.1017/erm.2014.18. [DOI] [PubMed] [Google Scholar]
- 22.Huang JL, Woolf AS, Kolatsi-Joannou M, Baluk P, Sandford RN, Peters DJ, McDonald DM, Price KL, Winyard PJ, Long DA. Vascular endothelial growth factor C for polycystic kidney diseases. J Am Soc Nephrol 27: 69–77, 2016. doi: 10.1681/ASN.2014090856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat Rev Nephrol 16: 289–303, 2020. doi: 10.1038/s41581-020-0260-2. [DOI] [PubMed] [Google Scholar]
- 24.Kirkin V, Mazitschek R, Krishnan J, Steffen A, Waltenberger J, Pepper MS, Giannis A, Sleeman JP. Characterization of indolinones which preferentially inhibit VEGF-C- and VEGF-D-induced activation of VEGFR-3 rather than VEGFR-2. Eur J Biochem 268: 5530–5540, 2001. doi: 10.1046/j.1432-1033.2001.02476.x. [DOI] [PubMed] [Google Scholar]
- 25.Foster RR, Satchell SC, Seckley J, Emmett MS, Joory K, Xing CY, Saleem MA, Mathieson PW, Bates DO, Harper SJ. VEGF-C promotes survival in podocytes. Am J Physiol-Renal Physiol 291: F196–F207, 2006. doi: 10.1152/ajprenal.00431.2005. [DOI] [PubMed] [Google Scholar]
- 26.Ny A, Koch M, Vandevelde W, Schneider M, Fischer C, Diez-Juan A, Neven E, Geudens I, Maity S, Moons L, Plaisance S, Lambrechts D, Carmeliet P, Dewerchin M. Role of VEGF-D and VEGFR-3 in developmental lymphangiogenesis, a chemicogenetic study in Xenopus tadpoles. Blood 112: 1740–1749, 2008. doi: 10.1182/blood-2007-08-106302. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu Y, Polavarapu R, Eskla KL, Pantner Y, Nicholson CK, Ishii M, Brunnhoelzl D, Mauria R, Husain A, Naqvi N, Murohara T, Calvert JW. Impact of lymphangiogenesis on cardiac remodeling after ischemia and reperfusion injury. J Am Heart Assoc 7: e009565, 2018. doi: 10.1161/JAHA.118.009565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JY, Hong SH, Shin M, Heo HR, Jang IH. Blockade of FLT4 suppresses metastasis of melanoma cells by impaired lymphatic vessels. Biochem Biophys Res Commun 478: 733–738, 2016. doi: 10.1016/j.bbrc.2016.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Kirkin V, Thiele W, Baumann P, Mazitschek R, Rohde K, Fellbrich G, Weich H, Waltenberger J, Giannis A, Sleeman JP. MAZ51, an indolinone that inhibits endothelial cell and tumor cell growth in vitro, suppresses tumor growth in vivo. Int J Cancer 112: 986–993, 2004. doi: 10.1002/ijc.20509. [DOI] [PubMed] [Google Scholar]
- 30.Matsuura M, Onimaru M, Yonemitsu Y, Suzuki H, Nakano T, Ishibashi H, Shirasuna K, Sueishi K. Autocrine loop between vascular endothelial growth factor (VEGF)-C and VEGF receptor-3 positively regulates tumor-associated lymphangiogenesis in oral squamoid cancer cells. Am J Pathol 175: 1709–1721, 2009. doi: 10.2353/ajpath.2009.081139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broggi MAS, Maillat L, Clement CC, Bordry N, Corthésy P, Auger A, Matter M, Hamelin R, Potin L, Demurtas D, Romano E, Harari A, Speiser DE, Santambrogio L, Swartz MA. Tumor-associated factors are enriched in lymphatic exudate compared to plasma in metastatic melanoma patients. J Exp Med 216: 1091–1107, 2019. doi: 10.1084/jem.20181618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris AR, Azimi MS, Cornelison R, Azar FN, Llaneza DC, Belanger M, Mathew A, Tkachenko S, Esparza S, Perez MJ, Rosean CB, Bostic RR, Cornelison RC, Tate KM, Peirce-Cottler SM, Paquette C, Mills A, Landen CN, Saucerman J, Dillon PM, Pompano RR, Rutkowski MA, Munson JM. Platinum chemotherapy induces lymphangiogenesis to prime tissues for tumor metastasis (Preprint). bioRxiv 781443, 2019. doi: 10.1101/781443. [DOI]
- 33.Harris AR, Perez MJ, Munson JM. Docetaxel facilitates lymphatic-tumor crosstalk to promote lymphangiogenesis and cancer progression. BMC Cancer 18: 718, 2018. doi: 10.1186/s12885-018-4619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jafree DJ, Long DA. Beyond a passive conduit: implications of lymphatic biology for kidney diseases. J Am Soc Nephrol 31: 1178–1190, 2020. doi: 10.1681/ASN.2019121320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manohar S, Leung N. Cisplatin nephrotoxicity: a review of the literature. J Nephrol 31: 15–25, 2018. doi: 10.1007/s40620-017-0392-z. [DOI] [PubMed] [Google Scholar]
- 36.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol 23: 460–464, 2003. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 37.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 38.Bolisetty S, Traylor A, Joseph R, Zarjou A, Agarwal A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol 310: F385–F394, 2016. doi: 10.1152/ajprenal.00335.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int 71: 266–271, 2007. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 40.Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484: 110–114, 2012. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 41.Hattori K, Ito Y, Honda M, Sekiguchi K, Hosono K, Shibuya M, Unno N, Majima M. Lymphangiogenesis induced by vascular endothelial growth factor receptor 1 signaling contributes to the progression of endometriosis in mice. J Pharmacol Sci 143: 255–263, 2020. doi: 10.1016/j.jphs.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Hsu M, Rayasam A, Kijak JA, Choi YH, Harding JS, Marcus SA, Karpus WJ, Sandor M, Fabry Z. Neuroinflammation-induced lymphangiogenesis near the cribriform plate contributes to drainage of CNS-derived antigens and immune cells. Nat Commun 10: 229, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Black LM, Lever JM, Traylor AM, Chen B, Yang Z, Esman SK, Jiang Y, Cutter GR, Boddu R, George JF, Agarwal A. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am J Physiol Renal Physiol 315: F1107–F1118, 2018. doi: 10.1152/ajprenal.00179.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winfree S, Ferkowicz MJ, Dagher PC, Kelly KJ, Eadon MT, Sutton TA, Markel TA, Yoder MC, Dunn KW, El-Achkar TM. Large-scale 3-dimensional quantitative imaging of tissues: state-of-the-art and translational implications. Transl Res 189: 1–12, 2017. doi: 10.1016/j.trsl.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Black LM, Winfree S, Khochare SD, Kamocka MM, Traylor AM, Esman SK, Khan S, Zarjou A, Agarwal A, El-Achkar TM. Quantitative 3-dimensional imaging and tissue cytometry reveals lymphatic expansion in acute kidney injury. Lab Invest 101: 1186–1196, 2021. doi: 10.1038/s41374-021-00609-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tammela T, Zarkada G, Wallgard E, Murtomäki A, Suchting S, Wirzenius M, Waltari M, Hellström M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Ylä-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454: 656–660, 2008. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 47.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278: F726–F736, 2000. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- 48.Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int 72: 53–62, 2007. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
- 49.Lutze G, Haarmann A, Demanou Toukam JA, Buttler K, Wilting J, Becker J. Non-canonical WNT-signaling controls differentiation of lymphatics and extension lymphangiogenesis via RAC and JNK signaling. Sci Rep 9: 4739, 2019. doi: 10.1038/s41598-019-41299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flister MJ, Wilber A, Hall KL, Iwata C, Miyazono K, Nisato RE, Pepper MS, Zawieja DC, Ran S. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-κB and Prox1. Blood 115: 418–429, 2010. doi: 10.1182/blood-2008-12-196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shinriki S, Jono H, Ueda M, Ota K, Ota T, Sueyoshi T, Oike Y, Ibusuki M, Hiraki A, Nakayama H, Shinohara M, Ando Y. Interleukin-6 signalling regulates vascular endothelial growth factor-C synthesis and lymphangiogenesis in human oral squamous cell carcinoma. J Pathol 225: 142–150, 2011. doi: 10.1002/path.2935. [DOI] [PubMed] [Google Scholar]
- 52.Zhou F, Chang Z, Zhang L, Hong YK, Shen B, Wang B, Zhang F, Lu G, Tvorogov D, Alitalo K, Hemmings BA, Yang Z, He Y. Akt/Protein kinase B is required for lymphatic network formation, remodeling, and valve development. Am J Pathol 177: 2124–2133, 2010. doi: 10.2353/ajpath.2010.091301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zsengellér ZK, Ellezian L, Brown D, Horváth B, Mukhopadhyay P, Kalyanaraman B, Parikh SM, Karumanchi SA, Stillman IE, Pacher P. Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J Histochem Cytochem 60: 521–529, 2012. doi: 10.1369/0022155412446227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luke DR, Vadiei K, Lopez-Berestein G. Role of vascular congestion in cisplatin-induced acute renal failure in the rat. Nephrol Dial Transplant 7: 1–7, 1992. doi: 10.1093/oxfordjournals.ndt.a091984. [DOI] [PubMed] [Google Scholar]
- 55.Heinolainen K, Karaman S, D'Amico G, Tammela T, Sormunen R, Eklund L, Alitalo K, Zarkada G. VEGFR3 modulates vascular permeability by controlling VEGF/VEGFR2 signaling. Circ Res 120: 1414–1425, 2017. doi: 10.1161/CIRCRESAHA.116.310477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Chen Y, Zhang L, Xing L, Xu H, Wang Y, Shi Q, Liang Q. Total saponins of panaxnotoginseng promotes lymphangiogenesis by activation VEGF-C expression of lymphatic endothelial cells. J Ethnopharmacol 193: 293–302, 2016. doi: 10.1016/j.jep.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Husseini A, Kraskauskas D, Mezzaroma E, Nordio A, Farkas D, Drake JI, Abbate A, Felty Q, Voelkel NF. Vascular endothelial growth factor receptor 3 signaling contributes to angioobliterative pulmonary hypertension. Pulm Circ 5: 101–116, 2015. doi: 10.1086/679704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breslin JW, Yuan SY, Wu MH. VEGF-C alters barrier function of cultured lymphatic endothelial cells through a VEGFR-3-dependent mechanism. Lymphat Res Biol 5: 105–113, 2007. doi: 10.1089/lrb.2007.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dubé KN, Bollini S, Matsuzaki F, Carr CA, Riley PR. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 522: 62–67, 2015. doi: 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui Y, Liu K, Monzon-Medina ME, Padera RF, Wang H, George G, Toprak D, Abdelnour E, D'Agostino E, Goldberg HJ, Perrella MA, Forteza RM, Rosas IO, Visner G, El-Chemaly S. Therapeutic lymphangiogenesis ameliorates established acute lung allograft rejection. J Clin Invest 125: 4255–4268, 2015. doi: 10.1172/JCI79693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ling S, Qi C, Li W, Xu J, Kuang W. Crucial role of corneal lymphangiogenesis for allograft rejection in alkali-burned cornea bed. Clin Exp Ophthalmol 37: 874–883, 2009. doi: 10.1111/j.1442-9071.2009.02178.x. [DOI] [PubMed] [Google Scholar]
- 62.Yin N, Zhang N, Lal G, Xu J, Yan M, Ding Y, Bromberg JS. Lymphangiogenesis is required for pancreatic islet inflammation and diabetes. PloS One 6: e28023, 2011. doi: 10.1371/journal.pone.0028023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X, Liu Z, Sun J, Song X, Bian M, Wang F, Yan F, Yu Z. Inhibition of NADPH oxidase 4 attenuates lymphangiogenesis and tumor metastasis in breast cancer. FASEB J 35: e21531, 2021. doi: 10.1096/fj.202002533R. [DOI] [PubMed] [Google Scholar]
- 64.Sleeman JP, Thiele W. Tumor metastasis and the lymphatic vasculature. Int J Cancer 125: 2747–2756, 2009. doi: 10.1002/ijc.24702. [DOI] [PubMed] [Google Scholar]
- 65.Balasubbramanian D, Baranwal G, Clark MC, Goodlett BL, Mitchell BM, Rutkowski JM. Kidney-specific lymphangiogenesis increases sodium excretion and lowers blood pressure in mice. J Hypertens 38: 874–885, 2020. doi: 10.1097/HJH.0000000000002349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, Birner P, Krieger S, Hovorka A, Silberhumer G, Laakkonen P, Petrova T, Langer B, Raab I. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol 15: 603–612, 2004. doi: 10.1097/01.asn.0000113316.52371.2e. [DOI] [PubMed] [Google Scholar]
- 67.Vass DG, Shrestha B, Haylor J, Hughes J, Marson L. Inflammatory lymphangiogenesis in a rat transplant model of interstitial fibrosis and tubular atrophy. Transpl Int 25: 792–800, 2012. doi: 10.1111/j.1432-2277.2012.01482.x. [DOI] [PubMed] [Google Scholar]
- 68.Chiruvella V, Annamaraju P, Guddati AK. Management of nephrotoxicity of chemotherapy and targeted agents: 2020. Am J Cancer Res 10: 4151–4164, 2020. [PMC free article] [PubMed] [Google Scholar]
- 69.Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer 17: 738–750, 2017. doi: 10.1038/nrc.2017.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie Y, Bagby TR, Cohen MS, Forrest ML. Drug delivery to the lymphatic system: importance in future cancer diagnosis and therapies. Expert Opin Drug Del 6: 785–792, 2009. doi: 10.1517/17425240903085128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang P, Chen J-Q, Huang W-Q, Li W, Huang Y, Zhang Z-J, Xu F-G. Renal medulla is more sensitive to cisplatin than cortex revealed by untargeted mass spectrometry-based metabolomics in rats. Sci Rep 7: 44804–44804, 2017. doi: 10.1038/srep44804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwager S, Detmar M. Inflammation and lymphatic function. Front Immunol 10: 308, 2019. doi: 10.3389/fimmu.2019.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heller F, Lindenmeyer MT, Cohen CD, Brandt U, Draganovici D, Fischereder M, Kretzler M, Anders HJ, Sitter T, Mosberger I, Kerjaschki D, Regele H, Schlöndorff D, Segerer S. The contribution of B cells to renal interstitial inflammation. Am J Pathol 170: 457–468, 2007. doi: 10.2353/ajpath.2007.060554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seeger H, Bonani M, Segerer S. The role of lymphatics in renal inflammation. Nephrol Dial Transplant 27: 2634–2641, 2012. doi: 10.1093/ndt/gfs140. [DOI] [PubMed] [Google Scholar]
- 76.Dieckmann KP, Struss WJ, Budde U. Evidence for acute vascular toxicity of cisplatin-based chemotherapy in patients with germ cell tumour. Anticancer Res 31: 4501–4505, 2011. [PubMed] [Google Scholar]
- 77.Herrmann J, Yang EH, Iliescu CA, Cilingiroglu M, Charitakis K, Hakeem A, Toutouzas K, Leesar MA, Grines CL, Marmagkiolis K. Vascular toxicities of cancer therapies. Circulation 133: 1272–1289, 2016. doi: 10.1161/CIRCULATIONAHA.115.018347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Breslin JW, Gaudreault N, Watson KD, Reynoso R, Yuan SY, Wu MH. Vascular endothelial growth factor-C stimulates the lymphatic pump by a VEGF receptor-3-dependent mechanism. Am J Physiol Heart Circ Physiol 293: H709–H718, 2007. doi: 10.1152/ajpheart.00102.2007. [DOI] [PubMed] [Google Scholar]
- 79.Oppelt P, Betbadal A, Nayak L. Approach to chemotherapy-associated thrombosis. Vasc Med 20: 153–161, 2015. doi: 10.1177/1358863X14568705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weijl NI, Rutten MF, Zwinderman AH, Keizer HJ, Nooy MA, Rosendaal FR, Cleton FJ, Osanto S. Thromboembolic events during chemotherapy for germ cell cancer: a cohort study and review of the literature. J Clin Oncol 18: 2169–2178, 2000. doi: 10.1200/JCO.2000.18.10.2169. [DOI] [PubMed] [Google Scholar]
- 81.Liu H, Hiremath C, Patterson Q, Vora S, Shang Z, Jamieson A, Fiolka R, Dean K, Dellinger M, Marciano D. Heterozygous mutation of Vegfr3 reduces renal lymphatics without renal dysfunction. J Am Soc Nephrol 32: ASN.2021010061, 2021. doi: 10.1681/ASN.2021010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kenig-Kozlovsky Y, Scott RP, Onay T, Carota IA, Thomson BR, Gil HJ, Ramirez V, Yamaguchi S, Tanna CE, Heinen S, Wu C, Stan RV, Klein JD, Sands JM, Oliver G, Quaggin SE. Ascending vasa recta are angiopoietin/Tie2-dependent lymphatic-like vessels. J Am Soc Nephrol 29: 1097–1107, 2018. doi: 10.1681/ASN.2017090962. [DOI] [PMC free article] [PubMed] [Google Scholar]