

Abstract
Idiopathic pulmonary arterial hypertension (PAH) is a fatal and progressive disease. Sustained vasoconstriction due to pulmonary arterial smooth muscle cell (PASMC) contraction and concentric arterial remodeling due partially to PASMC proliferation are the major causes for increased pulmonary vascular resistance and increased pulmonary arterial pressure in patients with precapillary pulmonary hypertension (PH) including PAH and PH due to respiratory diseases or hypoxemia. We and others observed upregulation of TRPC6 channels in PASMCs from patients with PAH. A rise in cytosolic Ca2+ concentration ([Ca2+]cyt) in PASMC triggers PASMC contraction and vasoconstriction, while Ca2+-dependent activation of PI3K/AKT/mTOR pathway is a pivotal signaling cascade for cell proliferation and gene expression. Despite evidence supporting a pathological role of TRPC6, no selective and orally bioavailable TRPC6 antagonist has yet been developed and tested for treatment of PAH or PH. In this study, we sought to investigate whether block of receptor-operated Ca2+ channels using a nonselective blocker of cation channels, 2-aminoethyl diphenylborinate (2-APB, administered intraperitoneally) and a selective blocker of TRPC6, BI-749327 (administered orally) can reverse established PH in mice. The results from the study show that intrapulmonary application of 2-APB (40 µM) or BI-749327 (3–10 µM) significantly and reversibly inhibited acute alveolar hypoxia-induced pulmonary vasoconstriction. Intraperitoneal injection of 2-APB (1 mg/kg per day) significantly attenuated the development of PH and partially reversed established PH in mice. Oral gavage of BI-749327 (30 mg/kg, every day, for 2 wk) reversed established PH by ∼50% via regression of pulmonary vascular remodeling. Furthermore, 2-APB and BI-749327 both significantly inhibited PDGF- and serum-mediated phosphorylation of AKT and mTOR in PASMC. In summary, the receptor-operated and mechanosensitive TRPC6 channel is a good target for developing novel treatment for PAH/PH. BI-749327, a selective TRPC6 blocker, is potentially a novel and effective drug for treating PAH and PH due to respiratory diseases or hypoxemia.
Keywords: BI-749327, calcium signaling, hypoxic pulmonary vasoconstriction, pulmonary hypertension, transient receptor potential channel
INTRODUCTION
Idiopathic pulmonary arterial hypertension (PAH) is a fatal and progressive disease in which elevated pulmonary vascular resistance (PVR) results in elevation of pulmonary arterial pressure (PAP) and right ventricular (RV) afterload, causing right heart failure and death, if untreated (1). Despite the recent progress and advancement of new therapeutic approaches, the 5-yr survival of patients with advanced PAH remains poor. NYHA functional classification is an important predictor of future survival based on the REVEAL Registry (2–4). Sustained pulmonary vasoconstriction due to pulmonary arterial smooth muscle cell (PASMC) contraction and concentric pulmonary arterial medial hypertrophy due to PASMC proliferation and migration are two of the major contributors to elevated PVR/PAP in patients with PAH (5–7). Clinical observations reinforce the importance of continuous monitoring of RV function in patients with PAH (8) and the continuous identification of new targets and development of novel therapies that improve pulmonary hemodynamics and RV function in patients with PAH (3, 9–17).
An increase in cytosolic Ca2+ concentration ([Ca2+]cyt) in PASMCs triggers PASMC contraction and pulmonary vasoconstriction (18, 19), whereas Ca2+-dependent activation of signaling cascades associated with the cell cycle and gene expression stimulates PASMC proliferation and induces pulmonary vascular remodeling or pulmonary arterial medial hypertrophy (20–23). Platelet-derived growth factor (PDGF)-mediated PASMC migration and proliferation also contribute to muscularization of pulmonary arterioles and precapillary arterioles (24–26), which is also an important contributor to elevated PVR/PAP in patients with PAH. In human and animal PASMCs, at least three classes of Ca2+-permeable cation channels are responsible for increasing [Ca2+]cyt via Ca2+ influx: voltage-dependent Ca2+ channels (VDCC) that are opened by membrane depolarization; receptor-operated Ca2+ channels (ROCC) that are opened by ligand binding to membrane receptors and intracellular second messengers like diacylglycerol (DAG); and store-operated Ca2+ channels (SOCC) that are opened by depletion of Ca2+ from intracellular Ca2+ stores such as sarcoplasmic (SR) or endoplasmic (ER) reticulum (27, 28). All of these Ca2+-permeable channels are involved in the regulation of PASMC excitation-contraction coupling, cell mobility and migration, and cell proliferation.
TRPC6 is a transient receptor potential (TRP) channel of the classical TRPC subfamily (29–31). TRPC6, encoded by the TRPC6 gene (Ch. 11q22.1), is expressed in the lungs and heart, and is highly distributed in vascular smooth muscle cells (SMC) and cardiomyocytes (32, 33). Upregulated expression and increased activity of TRPC6 channels play an important role in the development of hypertension and cardiac hypertrophy (33–38). The homotetrameric TRPC6 channels is highly permeable to Ca2+ in comparison with other cations (e.g., PCa/PNa = 5/1). Vasoconstrictors like angiotensin II (Ang-II), serotonin (5-HT), and endothelin-1 (ET-1) activate TRPC6, whereas vasodilators like nitric oxide (NO) and cGMP (39) inhibit TRPC6 channels. TRPC6 is not only a receptor-operated Ca2+ (or cation) channel (40–42), but also a mechanosensitive cation channel (32, 33); mechanical stretch synergistically regulates TRPC6 channels activated by receptor-mediated signaling. In addition to being an important subunit to form receptor-operated Ca2+ channels, TRPC6 is also involved in forming store-operated Ca2+ channels or regulating store-operated Ca2+ entry (33, 43, 44).
We and others previously reported that TRPC6 was upregulated in PASMCs from patients with idiopathic PAH (45, 46) and animals with experimental PH (21, 47). In addition, genetic deletion of trpc6 in mice significantly inhibited alveolar hypoxia-induced pulmonary vasoconstriction (48, 49) and ameliorated the development and progression of experimental pulmonary hypertension (PH) (48). It is well established that TRPC6, along with other nonselective cation channels, plays an important physiological role in regulating PASMC contraction and proliferation and has a pathogenic role in PAH and PH due to respiratory diseases or hypoxemia.
Many divalent or trivalent cations and small molecule inhibitors are available to block TRPC6 channels or receptor-operated and store-operated Ca2+ entry like Gd3+ and 2-aminoethyl diphenylborinate (2-APB) (50–53); few of them, however, can selectively block TRPC6 channels (54). Recently, a selective TRPC6 antagonist, BI-749327 (with IC50 of 13–19 nM for human and animal TRPC6), has been demonstrated to improve renal and cardiac fibrosis (55). BI-749327 is much more selective for TRPC6 than other TRPC channels based on IC50 for different TRPC channels and other types of cation channels (e.g., 85-fold more selective for TRPC6 than for TRPC3 and 42-fold more selective for TRPC6 than for TRPC7) (55).
In this study, we sought to investigate whether nonselective inhibition of TRP channels with 2-APB and selective inhibition of TRPC6 channels with BI-749327 inhibit acute hypoxic pulmonary vasoconstriction in ex vivo isolated perfused/ventilated lung experiments and reverse established PH in in vivo animal experiments. Furthermore, we examined whether inhibition of TRPC6 channels was sufficient to inhibit growth factor-mediated phosphorylation of AKT/mTOR in PASMCs. The data from this study provide compelling evidence that TRPC6, and other TRP cation channels, is a promising target to develop novel therapies for PAH and PH due to lung diseases and/or hypoxemia.
METHODS AND MATERIALS
Experimental Animals
Eight-week-old male mice weighing ∼25 g were used for measuring pulmonary hemodynamics in in vivo experiments using intact mice, determining lung vascular reactivity in ex vivo experiments using isolated perfused/ventilated lungs, and detecting pulmonary vascular remodeling in ex vivo angiography experiments. All animal care and experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego (La Jolla, CA) and executed according to the IACUC guidelines complying with national and international regulations. All the mice were housed in standard cages with 12-h light and dark cycles. Mice food and tap water were provided ad libitum.
To induce experimental PH, mice were placed in normobaric hypoxic chambers (Cat. No. A30271-P, BioSpherix, Lacona, NY) equilibrated with hypoxic gas mixture (10% O2 in N2) for 4 wk. The hypoxic chamber (30-in. wide, 20-in. deep, and 20-in. high) contained an O2 sensor (Cat. No. ProOx P110-E702) that continuously monitored O2 concentration and PO2 inside of the chamber. The normoxic control mice were placed in the same animal room in which animals were exposed to room air (21% O2). The feed, bedding, and water for mice were changed once a week.
In Vivo Pharmacological Experiments
In this study, two types of experimental protocol (prevention and reversal) were used to examine the effects of 2-aminoethyl diphenylborinate (2-APB, D9754, Sigma-Aldrich, St. Louis, MO), a nonselective blocker of TRP cation channels, and BI-749327 (BI, HY-111925, MedChemExpress, NJ), a selective blocker of TRPC6 channels (55).
In the prevention experiments, 2-APB was intraperitoneally (ip) administered once a day (every day) at the beginning of hypoxic exposure and for the entire duration of hypoxic exposure (4 wk). The prevention experimental protocol was designed to test whether the drug prevents (or attenuates) the animals from developing experimental PH. For the prevention experiments using 2-APB, mice were randomly divided into three groups (n = 5 mice per group): 1) Normoxia + vehicle (DMSO) group (Nor + Veh), 2) Hypoxia + vehicle group (Hyp + Veh), and 3) Hypoxia + 2-APB group (Hyp + 2-APB). In the Hyp + Veh and Hyp + 2-APB groups, DMSO and 2-APB (1 mg/kg body wt) were intraperitoneally (ip) injected, respectively, into mice at the same time each day for 4 wk.
In the reversal experiments, BI-749327 was orally administered (by mouth), or 2-APB was intraperitoneally (ip) administered, once a day (every day) for 2 wk after mice were exposed to normobaric hypoxia for 4 wk and had developed PH. The reversal experimental protocol was designed to test whether the drug reverses or regresses (at least partially) established experimental PH. For the reversal experiments using BI-749327, mice were first randomly divided into three groups (n = 5–10 for each group): 1) Normoxia + vehicle (oil) group (Nor + Veh), 2) Hypoxia + vehicle group (Hyp + Veh), and 3) Hypoxia + BI group (Hyp + BI). To examine the effect of different doses of BI, the Hyp + BI group was further divided into several subgroups (n = 5–10 for each subgroup) for, for example, 3, 10, 20, and 30 mg/kg of BI (orally, every day). Hyp + Veh and Hyp + BI groups were first placed in a normobaric hypoxic chamber (10% O2) for 4 wk, and then oil and BI-749327 were orally administered daily via oral gavage, respectively, for two more weeks during hypoxia. The entire experimental (or hypoxic exposure) duration was 6 wk.
Following hypoxic exposure and drug treatment, mice were anesthetized by continuous inhalation of isoflurane (1.5%) and then right heart catheterization (RHC) was conducted to measure right ventricle pressure (RVP) and right ventricle (RV) contractility (RV-±dP/dt) using a pressure transducer catheter (Millar Instruments, PVR1030, Houston, TX) inserted into the RV via the external right jugular vein. Baseline calibration was performed for the catheter before each measurement to ensure that the basal pressure was zero. RVP and RV-±dP/dt were recorded and analyzed using the AD Instruments Lab Chart software. The mean pulmonary arterial pressure (mPAP) was estimated based on the right ventricular systolic pressure (RVSP) using the equation: mPAP (in mmHg) = 0.61 × RVSP + 2 (72, 73).
In addition, we measured systemic arterial pressure (SAP) in control mice and mice treated with BI-749327. After hypoxic exposure and treatment with BI-749327 at dose 30 mg/kg per day for 2 wk, mice were anesthetized by inhalation of 1.5% isoflurane. The left carotid artery was carefully isolated and the Millar pressure catheter PV 1030 (ADInstruments, CO) was inserted to carotid artery to continuously measure systolic (sSAP) and diastolic (dSAP) SAP using the Millar data acquisition system (ADInstruments, CO). The mean SAP was calculated by the following equation: mean SAP = (sSAP + 2dSAP)/3.
Determination of Fulton Index
To determine RV hypertrophy, or the ratio of the weight of the RV to the weight of the left ventricle (LV) and septum (S) [RV/(LV + S)], also referred to as Fulton Index, the heart was carefully removed. After removal of the right and left atria, the RV wall was isolated from the remaining heart. The dry weight of the RV as well as the LV and septum (S) were carefully measured using a fine balance to calculate Fulton Index. The index was measured for each experimental mouse to show RV hypertrophy in mice with experimental PH.
Ex Vivo Experiments Using Isolated, Perfused/Ventilated Lungs
Pulmonary arterial pressure (PAP) was measured in isolated perfused/ventilated mouse lungs as described previously (56, 57). Briefly, male mice were anesthetized using pentobarbital sodium (120 mg/kg ip). The trachea was then carefully isolated and intubated immediately after anesthetization. Mice were ventilated with normoxic gas (21% O2) using a rodent mini ventilator (Minivent 845, Harvard Apparatus, Holliston, MA). To prevent coagulation, heparin was injected into the lung vasculature via the RV. A pressure transducer catheter (P75 type 379; Hugo Sachs Elektronik-Harvard Apparatus) was then inserted into the main pulmonary artery (PA) via the RV and used to continuously measure PAP. The left side of the heart was cannulated with a polyethylene tube to drain the perfusate.
Physiological salt solution (PSS) contained 120 mM NaCl, 4.3 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 19 mM NaHCO3, 1.1 mM KH2PO4, 10 mM glucose, and 20% fetal bovine serum (pH 7.4). 40 mM K+ (40 K)-containing PSS was prepared by replacing equimolar NaCl with 35.7 mM KCl to adjust osmolarity. A peristaltic pump (ISM 834; Ismatec, Glattbrugg, Switzerland) was used to superfuse PSS continuously through the isolated and ventilated lung at a flow rate of 1 mL/min. Initially, the mouse lung was challenged by superfusing 40 K-PSS three times (3 min/challenge) to obtain a stable basal PAP when the mouse was ventilated with normoxic gas (21% O2). After a stable baseline was achieved, the lung was ventilated with hypoxic gas mixture (1% O2; PO2 was ∼7.5 Torr) for 4 min to obtain a stable or sustained increase in PAP (due to alveolar hypoxic pulmonary vasoconstriction). The effect of 2-APB or BI-749327 on alveolar hypoxia-induced rise in PAP or alveolar hypoxic pulmonary vasoconstriction was examined by intrapulmonary superfusion of 40 µM 2-APB or various doses (1–10 µM) of BI-749327. Data were acquired and analyzed using PowerLab 8/30 software from Lab Chart ADInstruments.
Ex Vivo Lung Angiogram
Lung angiogram was performed according to the previously published methods (56, 58–60). Briefly, C57Bl/6 male mice were anesthetized using sodium pentobarbital (120 mg/kg ip). After removal of the anterior chest wall, heparin (20 IU) was immediately injected into the RV to prevent blood clotting. A PE-20 tube was then inserted into the main PA through the RV and warm PBS (pH = 7.4 at 36–38°C) was superfused through the tube at a speed of 0.05 mL/min for 3 min to flush out residual blood in the pulmonary vasculature. Freshly prepared Microfil polymer mixture (MV-122, Flow-Tech Inc., Carver, MA) was then gently instilled into the PA perfusate via a syringe pump (Farmingdale, NY) at the same speed for 1–2 min until the polymer mixture reached the peripheral branches of pulmonary arteries. At the end of preparation, the Microfil-filled lungs and heart were dissected and wetted in PBS overnight at 4°C. The next day, the Microfil-filled lungs were dehydrated in ethanol by immersing the lungs (1 h for each concentration) into 50%, 70%, 80%, 95%, and 100% ethanol solutions, respectively, for two times. The dehydrated lungs were finally put into a methyl salicylate (Sigma-Aldrich) solution and then placed on a shaker overnight. When the lungs became translucent and the Microfil polymer was clearly visible, the lungs were photographed (pixel size 3,584 width × 2,748 height) with a digital camera (MU1000, FMA050, Amscope, CA) through a dissecting microscope (WILD M651, Leica, Switzerland). The peripheral branch images of the pulmonary vasculature (1 mm from the edge of the lung) were selected using Adobe Photoshop software and quantified by Image J to calculate the total length of vascular branches, the number of branches, and the number of branch junctions in the pulmonary vascular networks.
Cell Culture
Normal human PASMCs were purchased from Lonza (Cat. No. CC2581, Basel, Switzerland) and cultured in 100-mm petri dishes in an incubator under a humidified atmosphere of 5% CO2 and 95% air at 37°C. VascuLife SMC basal medium without antimicrobials and phenol red (Lifeline Cell Technologies, Cat. No. LM-0002) supplemented with LifeFactors kit (Lifeline Cell Technologies, Cat. No. LS-1040) including 5% fetal bovine serum, 5 ng/mL recombinant human fibroblast growth factor-β, 5 μg/mL recombinant human insulin, 50 μg/mL ascorbic acid, 10 mM l-glutamine, 5 ng/mL recombinant human epidermal growth factor, 30 mg/mL gentamicin, and 15 μg/mL amphotericin B was used as a growth medium for human PASMCs. After reaching 70%–90% confluence, the cells were subcultured by trypsinization with 0.05% trypsin-EDTA (Thermo Fisher Scientific) and plated in 6-mm petri dishes or 6-well petri dishes. The medium was changed during the next 24 h after passaging and then every other day.
HEK-293 cells, purchased from Invitrogen, were cultured in high-glucose DMEM (Invitrogen) supplemented with growth factors and antibiotics such as 10% fetal bovine serum (FBS; Invitrogen), 100 IU/mL penicillin, and 100 µg/mL streptomycin (Sigma-Aldrich). Cells were cultured in a humidified environment at 37°C and 5% CO2 in an incubator. When cells attained 70%–90% confluence, cells were trypsinized and replated on 25-mm cover slips for electrophysiological (patch clamp) experiments and in 60-mm petri dishes for Western blot experiments.
The membrane impermeant BAPTA (2 mM) and membrane permeable BAPTA-AM (25 µM) were used to chelate extracellular Ca2+ in the culture medium and intracellular Ca2+ in the cytosol and intracellular organelles, respectively (61–63). Dissolving 1.8 mM BAPTA in culture medium reduced extracellular free [Ca2+] from 1.8 mM to ∼482 nM. BAPTA-AM (at micromolar range) can be accumulated in the cytosol and intracellular stores to chelate Ca2+ in millimolar range (61, 64).
Cell Proliferation Assay
PASMC from normal subjects were plated in 96-well plates at the density of 5 × 103 cells/well and cultured in the growth media for 24 h. Then, the cells were incubated in the basal medium with 0.3% FBS to be synchronized into G0/G1 phase (65). The cells were then cultured in the media with 5% FBS + vehicle or 5% FBS + BI-749327 (250 nM) for 48 h. To measure the number of viable cells, the cells were incubated with CCK8 solution (Cell Counting Kit-8, Cat. No. K1018, ApexBio, Houston, TX) for 2 h. The absorbance was measured at 450 nm using a microplate reader (iMark, Bio-Rad Laboratories, Hercules, CA) to assess cell viability.
TRPC6 Transfection and Patch Clamp Experiment
HEK293 cells were transiently transfected with 1 µg of human TRPC6 construct (Addgene, Watertown, MA) using X-tremeGENE 360 DNA Transfection Reagent (Roche, Basel, Switzerland) for 4–6 h in Opti-MEM Reduced Serum Medium (Gibco). Then, the Opti-MEM medium was replaced with 10% FBS DMEM. Twenty-four to 48 h later, cells were collected for protein isolation or were used for electrophysiological experiments. The transfected cells were identified by green fluorescence.
Whole cell cation currents were recorded with the patch clamp technique using an Axopatch-200B amplifier and a DigiData 1550B low-noise data acquisition (Molecular Devices). For recording whole cell nonselective cation currents through TRPC6 channels (ITRPC6), we used HEK-293 cells transiently transfected with the human TRPC6 gene and then recorded the currents 24–48 h after transfection.
Briefly, a glass coverslip plated with cells was mounted onto a Plexiglas perfusion chamber on the stage of a Nikon inverted microscope. For recording whole cell cation currents, cells on the coverslip were bathed in physiological salt solution (PSS) containing 141 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, and 10 glucose (pH was adjusted to 7.4 with NaOH). The pipette (intracellular) solution contained 120 mM CsCl, 120 mM aspartic acid, 0.6475 mM CaCl2, 4 mM MgCl2, 10 mM HEPES, 10 mM EGTA, and 5 mM Na2ATP (pH was adjusted to 7.2 with CsOH). We used patch pipettes (2–3 MΩ) that were fabricated on a Sutter Instrument (Novato, CA) puller from borosilicate glass tubes and were fire polished by a Narishige microforge (Narishige Scientific Instruments, Tokyo, Japan). Command voltage protocols and data acquisition were performed using pCLAMP-10 software (Axon Instruments). The series resistance was at a range of 4–9 MΩ when the whole cell configuration was formed. Series resistance compensation was performed for most of cells. Currents were filtered at 1–2 kHz and digitized at 2–5 kHz. To obtain a full-scale of current-voltage (I-V) relationship curve for whole cell cation currents, or ITRPC6, in TRPC6-transfected HEK-293 cells, we used voltage step protocol: the cells were depolarized to a series of test potentials (for 500 ms) ranging from −100 to +100 mV (in 20 mV increments) from a holding potential of 0 mV. We also used voltage ramp protocol to elicit inward and outward currents, the voltage ramp was given to cells from −100 mV to +100 mV for 1,000 ms. All experiments were performed at room temperature (22–24°C).
Western Blot Analysis
Human PASMCs were washed with ice-cold PBS and suspended into 1× RIPA buffer (Millipore) supplemented with a protease inhibitor cocktail tablet (Roche). The cell lysates were then centrifuged at 12,000 g for 15 min, and the supernatant was collected. Protein concentrations were measured by a Nano-Drop spectrophotometer (Thermo Fisher Scientific). Samples were applied on SDS-PAGE (8%) and proteins were transferred onto nitrocellulose membranes by electroblot. Membranes were blocked in 5% bovine serum albumen [in 1× Tris-buffered saline (TBS) containing 0.1% Tween 20] for 1 h at room temperature and incubated overnight at 4°C with primary antibodies for phosphor AKT (pAKT) (Cell Signaling Technology, Cat. No. 4060S, 1:1,000), phosphor mTOR (pmTOR) (Cell Signaling Technology, Cat. No. 2971S, 1:1,000), mTOR (Cell Signaling Technology, Cat. No. 2983S, 1:1,000), pan AKT (Cell Signaling Technology, Cat. No. 4691S, 1:1,000), and β-actin (Santa Cruz Biotechnology, Cat. No. sc-47778; 1:2,000; as a loading control). The membranes were incubated with anti-mouse (Cell Signaling Technology, Cat. No. 7076S) or anti-rabbit (Cell Signaling, Cat. no. 7074S) secondary antibodies for 1 h at room temperature the next day. Blots were developed using ECL Prime Western Blotting Detection Regents (GE Healthcare, UK). Bands were visualized using ImageJ software. The band intensity of pAKT, AKT, pmTOR, and mTOR from cells treated with growth medium (including 5% FBS and multiple growth factors) or PDGF-BB in the presence or absence of BAPTA-AM, BAPTA, 2-APB, or BI 749327 was normalized to the level of control cells (incubated in 0.3% FBS media) and expressed in arbitrary units.
Drugs and Special Reagents
All drugs and chemicals were obtained from Sigma Aldrich (St. Louis, MO) unless otherwise stated. For ex vivo experiments using isolated perfused/ventilated mouse lung and in vitro experiments using cultured cells, BI-749327 or 4-((6-aminopyridazin-3-yl) piperidin-1-yl)(4-(4-(trifluoromethyl)phenoxy)phenyl)methanone (HY-111925; MedChemExpress, NJ) was prepared as a 11 mM stock solution in DMSO. For in vivo experiments using intact mice, BI-749327-containing solution was freshly prepared daily using 90% corn oil and 10% DMSO at a concentration of 5 mg/mL. For in vitro, ex vivo, and in vivo experiments, 2-aminoethoxydiphenyl borate (2-APB, Sigma) was prepared as an 88 mM stock solution in DMSO. BAPTA (HY-100168; 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, MedChemExpress, NJ) and BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester] (HY-100545, MedChemExpress, NJ) were prepared as concentrated stock solution in DMSO. Recombinant human PDGF-BB (SRP3138) was prepared as a stock solution in DMSO. All drug and chemical stock solutions were aliquoted in DMSO or distilled water and kept frozen at −20°C until experimentation. Aliquots of the stock solutions were then diluted into PSS (BI-749327 and 4-APB for ex vivo experiments) and cell culture media (BAPTA and PDGF-BB for in vitro experiments) to make final concentrations designated for each experiment. Similar dilution of DMSO alone was used as a vehicle control in perfusate and culture media. The pH values of all solutions were checked after the addition of drugs and were readjusted to 7.4.
Statistics
The summarized data are expressed as means ± standard error (SE). Statistical analysis was performed using paired or unpaired Student’s t test (for two groups) or one-way ANOVA and post hoc test (Student-Newman-Keuls) (for multiple groups). Differences were considered statistically significant at P < 0.05. A significant difference is expressed in the figures or figure legends as *P < 0.05, **P < 0.01, or ***P < 0.001.
RESULTS
Sustained pulmonary vasoconstriction due to PASMC contraction and concentric pulmonary vascular remodeling due, at least in part, to PASMC proliferation and migration are important causes for the elevated PVR and PAP in patients with PAH (7, 28, 66). A rise in [Ca2+]cyt due to Ca2+ influx through various Ca2+-permeable cation channels in the plasma membrane and Ca2+ mobilization from intracellular Ca2+ stores is a major trigger for PASMC contraction (18, 19, 67) and a critical stimulus for PASMC proliferation and migration (20, 23, 68–70). TRPC6 is a receptor-operated and mechanosensitive cation channel that plays an important role in the regulation of smooth muscle excitation-contraction coupling and cell proliferation and migration (33, 34). The experiments were designed to examine whether block of nonselective cation channels and store-operated cation channels with 2-APB (52, 53, 71) and block of TRPC6 channels with a specific TRPC6 inhibitor BI-749327 (55) inhibit pulmonary vasoconstriction and reverses established experimental PH.
To confirm the selective inhibitory effect of BI-749327 on TRPC6 channels, we measured whole cell cation currents in HEK-293 cells transiently transfected with the human TRPC6 gene. In comparison to mock-transfected HEK cells, transient transfection of TRPC6 enabled HEK cells to generate huge whole cell cation currents when a series of test potentials (−100 to +100 in 20 mV increment) were applied to the cells (Fig. 1, A–D). Extracellular application of 10 nM BI-749327 significantly and reversibly reduced the whole cell cation currents through TRPC6 channels in TRPC6-transfected HEK cells [Fig. 1A (bottom), B and C (right), and D]. Superfusion of BI-749327 started decreasing the amplitude of TRPC6 currents almost immediately; the maximal inhibition took place at ∼2 min (Fig. 1E). These data indicate that BI-749327 is a potent TRPC6 blocker that rapidly inhibits cation currents through TRPC6 channels. We previously reported, with a similar experimental approach, that extracellular application of 100 µM 2-APB significantly decreased whole cell cation currents in HEK-293 cells transiently transfected with TRPC6 (48).
Figure 1.

BI-749327 (BI) reversibly inhibits cation currents in HEK-293 cells transiently transfected with the human TRPC6 gene. A: representative whole cell cation currents, elicited by depolarization from a holding potential of 0 mV to a series of test potentials ranging from −100 to +100 mV (in 20 mV increment) in mock- (−TRPC6, top) and TRPC6-transfected (+TRPC6, bottom) HEK cells before (Control), during (BI-749327), and after (Washout) extracellular application of 10 nM BI-749327. B: amplitudes of cation currents at −100 mV and +100 mV (means ± SE) in mock (−TRPC6, left) and TRPC6-transfected (+TRPC6, right) HEK cells before (Control), during (BI-749327), and after (Washout) extracellular application of BI-749327. *P < 0.05, ***P < 0.001 vs. Control (blue) and Washout (green); ##P < 0.01 vs. BI-749327 (red) with one-way ANOVA. C: the current-voltage (I-V) relationship curves in mock- (−TRPC6, left) TRPC6-transfected (+TRPC6, right) HEK cells before (Control, blue), during (BI-749327, red) and after (Washout, green) extracellular application of 10 nM BI-749327. Data are expressed as means ± SE (n = 6 cells). The I-V curves obtained from the cells before (Control) and during (BI-749327) treatment with BI-749327 are significantly different (P < 0.05 with one-way ANOVA). D: representative cation currents elicited by a ramp protocol from −100 mV to + 100 mV (for 1,100 ms) in TRPC6-transfected cell before (Cont, blue), during (BI, red), and after (Wash, green) extracellular application of BI-749327. E: representative time-course of BI-749327-mediated decrease in the amplitude of whole cell cation currents elicited by a test potential of +100 mV (from a holding potential of 0 mV). The horizontal bar indicates the time when BI-749327 was superfused to the cell (gray area).
Block of Cation Channels with 2-APB and TRPC6 Channels with BI-749327 Inhibits Hypoxic Pulmonary Vasoconstriction
In the isolated perfused/ventilated mouse lung, alveolar ventilation of hypoxic gas mixture (1% O2 and 5% CO2 in N2; the alveolar PO2 was ∼10 Torr) via a tracheal intubation tube caused a rapid and reversible increase in pulmonary arterial pressure (PAP) due to hypoxic pulmonary vasoconstriction (HPV). Removal of extracellular Ca2+ (Ca2+-free) from PSS superfused into pulmonary artery abolished alveolar HPV. Upon restoration of Ca2+ in the perfusate (to 1.8 mM), the hypoxia-induced increase in PAP was fully recovered (Fig. 2A). These data indicate that HPV is fully dependent of Ca2+ influx in the isolated perfused/ventilated lung preparation.
Figure 2.

Inhibition of TRPC6 channels with BI-749327 (BI) reversibly attenuates acute hypoxia-induced pulmonary vasoconstriction (HPV) in isolated perfused/ventilated mouse lungs. A: representative record showing changes in pulmonary artery pressure (PAP) before, during, and after ventilation of hypoxic gas (H, 1% O2 in N2, for 4 min) when physiological salt solution (PSS) with or without (Ca2+-free) 1.8 mM Ca2+ was perfused into pulmonary artery. B: representative records (left) depicting alveolar hypoxia (H)-induced changes in PAP before, during, and after intrapulmonary perfusion of 2-aminoethyl diphenylborinate (2-APB, 40 µM), a nonselective blocker of cation channels. Summarized data (means ± SE, n = 5 lungs or animals, right) showing the hypoxia-induced increases in PAP before (Cont), during (2-APB), and after (Rec) intrapulmonary superfusion of 2-APB. *P < 0.05 vs. Cont and Rec (ANOVA). C: representative records (left) showing hypoxia (H)-induced PAP changes before, during, and after intrapulmonary perfusion of the selective TRPC6 blocker, BI-749327, at concentrations of 1 (a), 3 (b), or 10 (c) µM, respectively. Summarized data (means ± SE, n = 5 lungs or animals, right) showing the hypoxia-induced increases in PAP before (Cont), during (BI), and after (Rec) intrapulmonary superfusion of 1 (a, n = 7), 3 (b, n = 7), or 10 (c, n = 6 lungs or animals) µM of BI-749327 (BI). **P < 0.01, ***P < 0.001 vs. Cont and Rec (ANOVA). D: dose-response curve depicting the percentage inhibition of hypoxia-induced increases in PAP during intrapulmonary application of 0 (Control), 1, 3, and 10 µM of BI. Data are shown as means ± SE (n = 6–7 lungs or animals). **P < 0.01, ***P < 0.001 vs. 0 µM BI (control); §§P < 0.01 vs. 1 µM BI; ##P < 0.01 vs. 3 µM BI (ANOVA).
Intrapulmonary perfusion of 2-APB (40 µM), a nonselective blocker of cation channels (48), significantly and reversibly inhibited HPV (Fig. 2B), suggesting that Ca2+ influx through 2-APB-sensitive nonselective cation channels is also required for HPV in mouse lungs. Furthermore, intrapulmonary perfusion of BI-749327, a selective TRPC6 blocker (55), at 1–10 µM resulted in a dose-dependent inhibition of HPV in isolated perfused/ventilated mouse lungs (Fig. 2C). The IC50 of BI-749327-mediated inhibition of acute alveolar HPV was estimated to be ∼1.1 µM based on the dose-response curve (Fig. 2D). These data indicate that inhibition of TRPC6 with the nonselective blocker 2-APB or the selective blocker BI-749327 significantly and reversibly inhibits alveolar hypoxia-mediated pulmonary vasoconstriction.
2-APB Inhibits the Development of Experimental PH in Mice
We conducted a set of experiments to determine and compare pulmonary hemodynamics in normoxic control mice and mice exposed to 4 wk of hypoxia and 6 wk of hypoxia. Right ventricular (RV) systolic pressure (RVSP), a surrogate measure for systolic pulmonary arterial pressure (PAP), in normoxic control mice was 21.98 ± 1.83 mmHg (n = 20; means ± SD). Four weeks of hypoxic exposure significantly increased RVSP to 35.89 ± 2.16 mmHg (n = 10, P < 0.001 vs. normoxic control, ANOVA), whereas 6 wk of hypoxia also significantly increased RVSP to 37.10 ± 3.60 mmHg (n = 23, P < 0.001 vs. normoxic control, ANOVA) in mice. There was no significant difference, however, of RVSP in mice exposed to 4 wk of hypoxia and 6 wk of hypoxia (P = 0.26, ANOVA).
To examine whether 2-APB-sensitive nonselective cation channels including TRPC1/3/5/6, TRPV6, TRPM3/7/8, and TRPP2 (74–80) are involved in the development of experimental PH (i.e., chronic hypoxia-induced PH), we first conducted a prevention experiment in which 2-APB was intraperitoneally injected (ip) at the beginning of hypoxic exposure and continuously injected once a day (every day) for 4 wk during hypoxia (Fig. 3, A and B). Chronically exposing mice to normobaric hypoxia (10% O2) for 4 wk significantly increased RVSP, mean PAP (mPAP, estimated by the value of RVSP using the formula: mPAP = 0.61 × RVSP + 2) (72, 73), and RV contractility (RV-±dP/dt) (Fig. 3C, a and b). Chronic hypoxia slightly increased RV contractile index [(RV-+(dP/dt)max)/RVSP]; but did not change heart rate (Fig. 3C, a and b). The increased RVSP and mPAP in mice exposed to normobaric hypoxia for 4 wk was also associated with a significant increase in RV hypertrophy determined by the Fulton Index [RV/(LV + S)] (Fig. 3D), while the heart rate was not changed in chronically hypoxic mice in comparison to normoxic mice (Fig. 3Cb). In the prevention experiment (Fig. 3A), intraperitoneal injection of 2-APB (1 mg/kg body wt, every day) significantly inhibited the development of PH (Fig. 3C) and RV hypertrophy (Fig. 3D). 2-APB (1 mg/kg, ip, every day for 4 wk) resulted in significant inhibition of hypoxia-induced increases in RVSP (by 47.3 ± 6.0%, n = 10; P < 0.001), RV-+(dP/dt)max (by 50.2 ± 7.6%, n = 10; P < 0.001) and Fulton Index (by 44.7 ± 7.1%, n = 5; P < 0.001) (Fig. 3, Cb and D).
Figure 3.
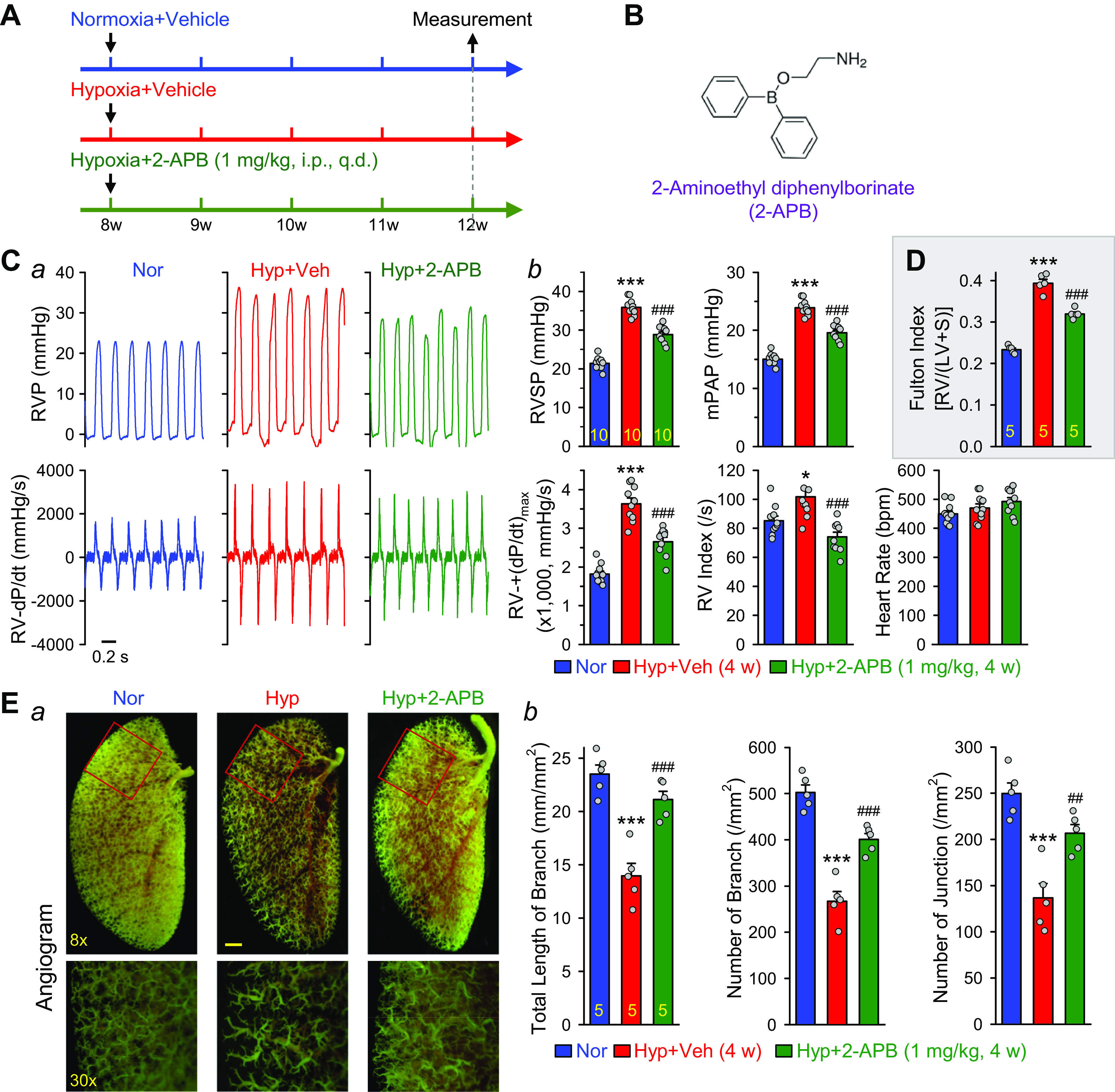
Block of cation channels with 2-aminoethyl diphenylborinate (2-APB) inhibits the development and progression of hypoxia-induced pulmonary hypertension (HPH) in mice. A: experimental protocol for the prevention experiment using 2-APB. Eight-week-old C57Bl/6 mice are subjected to normoxia (Nor) or hypoxia (10% O2) for 4 wk. 2-APB (1 mg/kg/day) was intraperitoneally injected (ip) at the beginning of hypoxia exposure and continuously injected for 4 wk during hypoxia. B: chemical structure of 2-APB. C, a: representative records of right ventricular pressure (RVP) (top) and right ventricle (RV) contractility (RV-±dP/dt) (bottom) in normoxic (Nor) control mice, hypoxic (Hyp) mice, and Hyp mice receiving 2-APB (1 mg/kg ip, every day). Summarized data (means ± SE, b) showing right ventricular systolic pressure (RVSP), estimated mean pulmonary arterial pressure (mPAP), RV-+(dP/dt)max, RV contractility index [RV-+(dP/dt)max/RVSP], and heart rate in Nor (n = 10), Hyp (n = 10), and Hyp + 2-APB (n = 10) mice. *P < 0.05, ***P < 0.001 vs. Nor; ###P < 0.001 vs. Hyp (ANOVA). D: summarized data (means ± SE) showing Fulton Index, the ratio of the RV weight to the left ventricle (LV) and septum (S) weight [RV/(LV+S)] in Nor (n = 5), Hyp (n = 5), and Hyp + 2-APB (n = 5) mice. ***P < 0.001 vs. Nor; ###P < 0.001 vs. Hyp (ANOVA). E, a: representative images of angiograph of the left lung at ×8 (top) and ×30 (bottom) magnification from Nor, Hyp, and Hyp + 2-APB mice. Summarized data (means ± SE, b) showing the total length of lung vascular branches (left), the number of lung vascular branches (middle), and the number of lung vascular branch junctions (right) per square millimeter of the selected lung area from Nor (n = 5), Hyp (n = 5), and Hyp + 2-APB (n = 5) mice. ***P < 0.001 vs. Nor; ##P < 0.01, ###P < 0.001 vs. Hyp (ANOVA).
In addition to the hemodynamic and RV hypertrophy measurements, we also conducted angiography experiments in Nor, Hyp + Veh (DMSO) and Hyp + 2-APB mice. As shown in Fig. 3E, a and b, chronic hypoxia (10% O2 for 4 wk) significantly decreased the total length of lung vascular branches (from 23.5 ± 1.2 to 13.9 ± 1.2 mm/mm2, n = 5; P < 0.001), the number of lung vascular branches (from 502.2 ± 16.5 to 267.3 ± 20.8 per mm2, n = 5; P < 0.001), and the number of lung vascular branch junctions (from 249.6 ± 11.5 to 136.5 ± 15.8 per mm2, n = 5; P < 0.001). Intraperitoneal injection of 2-APB (1 mg/kg, every day for 4 wk) partially recovered the total length of branches (from 13.9 ± 1.2 to 21.1 ± 0.8 mm/mm2, n = 5; P < 0.001), the number of branches (from 267.3 ± 20.8 to 400.6 ± 13.0 per mm2, n = 5; P < 0.001), and the number of junction (from 136.5 ± 15.8 to 206.7 ± 9.3 per mm2, n = 5; P < 0.01) (Fig. 3Eb). The angiography data are consistent with the hemodynamic data showing that 2-APB, by blocking nonselective cation channels, significantly inhibits hypoxia-mediated increases in PAP and hypoxia-induced pulmonary vascular remodeling.
2-APB Partially Reverses the Established Experimental Pulmonary Hypertension in Mice
To establish a potential therapeutic effect of 2-APB, it is important to examine whether 2-APB can reverse established PH. We conducted a reversal experiment using mice with chronic hypoxia-induced PH. As shown in the experimental protocol (Fig. 4A), 8-wk-old mice were randomly divided into three groups: 1) Nor mice, 2) Hyp + Veh mice, and 3) Hyp + 2-APB mice. The Hyp + Veh and Hyp + 2-APB mice were first exposed to normobaric hypoxia (10% O2) for 4 wk without injection of vehicle or 2-APB. When PH was established at the end of the 4-wk exposure to hypoxia, the mice were then intraperitoneally injected, once a day, with vehicle (DMSO) or 1 mg/kg of 2-APB (dissolved into DMSO) (Fig. 4B) for two more weeks under hypoxic conditions before the hemodynamic and angiography measurements were conducted.
Figure 4.
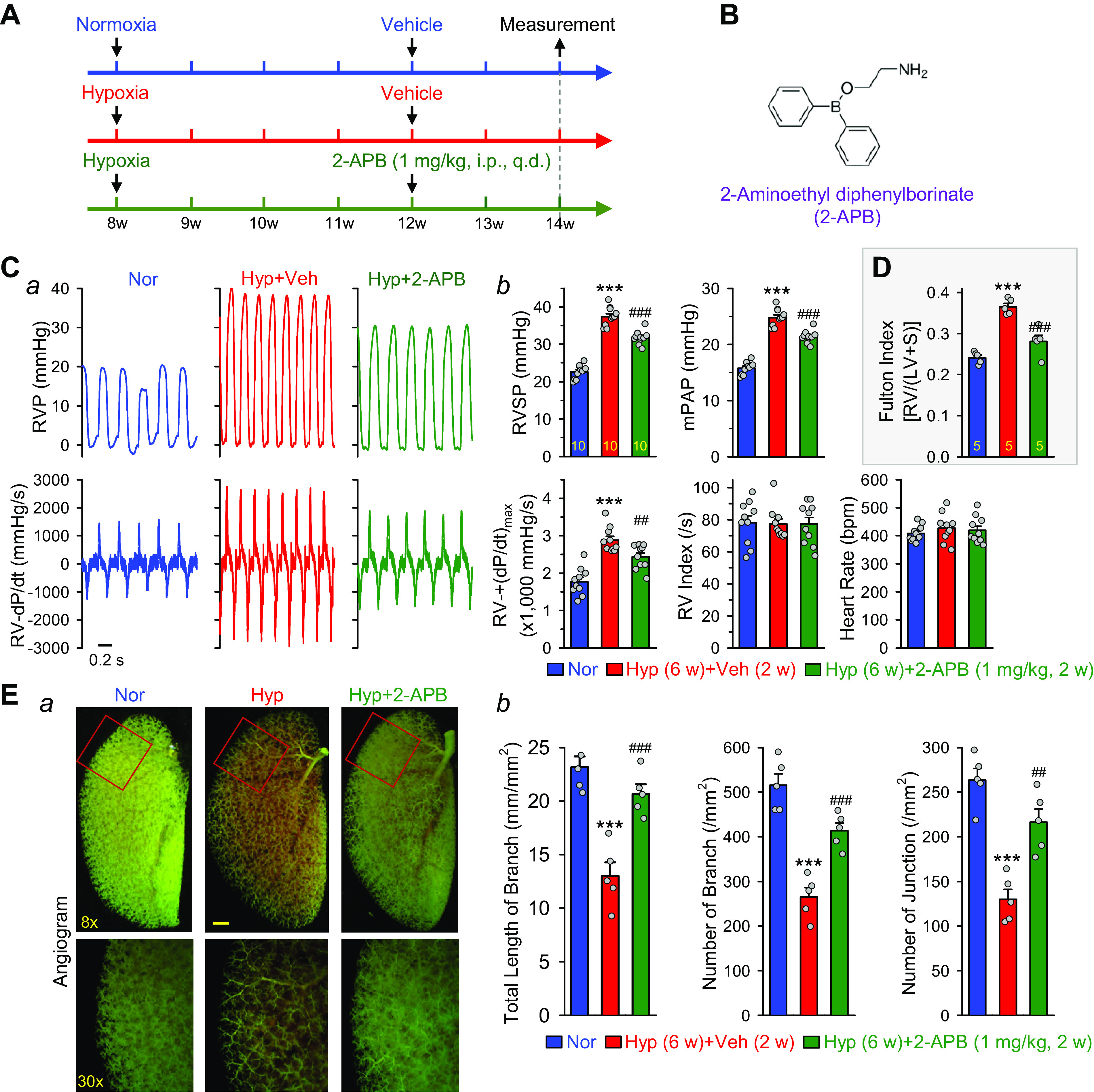
Inhibition of cation channels with 2-aminoethyl diphenylborinate (2-APB) partially reverses hypoxia-induced pulmonary hypertension (HPH) in mice. A: experimental protocol for the reversal experiment using 2-APB. Eight-week-old C57Bl/6 mice are subjected to normoxia (Nor) or hypoxia (10% O2) for 4 wk at first. 2-APB (1 mg/kg/day) was then intraperitoneally injected (ip) for two more weeks during hypoxia. B: chemical structure of 2-APB. C, a: representative records of right ventricular pressure (RVP) (top) and right ventricle (RV) contractility (RV-±dP/dt) (bottom) in normoxic (Nor) control mice, hypoxic (Hyp), mice and Hyp mice receiving 2-APB (1 mg/kg ip, every day). Summarized data (means ± SE, b) showing right ventricular systolic pressure (RVSP), estimated mean pulmonary arterial pressure (mPAP), RV-+(dP/dt)max, RV contractility index [RV-+(dP/dt)max/RVSP] and heart rate in Nor (n = 10), Hyp (n = 10), and Hyp + 2-APB (n = 10) mice. ***P < 0.001 vs. Nor; ##P < 0.01, ###P < 0.001 vs. Hyp (ANOVA). D: summarized data (means ± SE) showing Fulton Index, the ratio of the RV weight to the left ventricle (LV) and septum (S) weight [RV/(LV+S)] in Nor (n = 5), Hyp (n = 5), and Hyp + 2-APB (n = 5) mice. ***P < 0.001 vs. Nor; ###P < 0.001 vs. Hyp (ANOVA). E, a: representative images of angiograph of the left lung at × (top) and ×30 (bottom) magnification from Nor, Hyp, and Hyp + 2-APB (1 mg/kg/day for 2 wk after 4-wk of hypoxia exposure) mice. Summarized data (means ± SE, b) showing the total length of lung vascular branches (left), the number of lung vascular branches (middle), and the number of lung vascular branch junctions (right) per square millimeter of the selected lung area from Nor (n = 5), Hyp (n = 5), and Hyp + 2-APB (n = 5) mice. ***P < 0.001 vs. Nor; ##P < 0.01, ###P < 0.001 vs. Hyp (ANOVA).
Four weeks of exposure to hypoxia increased RVSP by 14.8 ± 0.8 mmHg (from 22.6 ± 0.6 to 37.4 ± 0.8 mmHg, n = 10; P < 0.001), mPAP by 9.0 ± 0.5 mmHg (from 15.8 ± 0.4 to 24.8 ± 0.5 mmHg, n = 10; P < 0.001), RV-+(dP/dt)max by 1,110.6 ± 105.0 mmHg/s (from 1,769.9 ± 116.3 to 2,880.4 ± 105.4 mmHg/s, n = 10; P < 0.001), and Fulton Index by 0.124 ± 0.008 (from 0.241 ± 0.006 to 0.365 ± 0.008, n = 10; P < 0.001) (Fig. 4, C and D). Intraperitoneal injection of 2-APB (1 mg/kg, every day) for 2 wk attenuated the hypoxia-induced increases in RVSP by 38.1 ± 3.8% (from 14.8 ± 0.8 to 9.1 ± 0.6 mmHg; P < 0.001), mPAP by 38.1 ± 3.8% (from 9.0 ± 0.5 to 5.6 ± 0.3 mmHg; P < 0.001), RV-+(dP/dt)max by 40.0 ± 9.2% (from 1,110.6 ± 105.0 to 667.0 ± 102.0 mmHg/s; P < 0.01), and Fulton Index by 68.0 ± 12.1% (from 0.124 ± 0.008 to 0.040 ± 0.015; P < 0.001) (Fig. 4, C and D). Although 2-APB significantly reduced RVSP and RV-±dP/dt, it did not reduce RV contractile index (RV-+(dP/dt)max/RVSP). These results indicate that intraperitoneal injection of 2-APB (1 mg/kg, every day for 2 wk) to mice with hypoxia-induced PH partially reverses established PH (by ∼40% on RVSP and mPAP) and RV hypertrophy (by 68% on Fulton Index).
In addition to hemodynamics, we also tested the effect of 2-APB on pulmonary vascular remodeling by angiography. Four-week exposure of mice to normobaric hypoxia (10% O2) resulted in significant pulmonary vascular remodeling as indicated by the decreases in the total number of lung vascular branches (from 23.2 ± 1.0 to 13.0 ± 1.3 mm/mm2, n = 5; P < 0.001), the number of vascular branches (from 515.2 ± 25.6 to 264.8 ± 21.1 branches per mm2, n = 5; P < 0.001), and the number of vascular branch junctions (from 263.4 ± 13.0 to 129.8 ± 11.4 junctions per mm2, n = 5; P < 0.001) (Fig. 4E, a and b). The decreased total number of branches shown in the angiogram in chronically hypoxic mice was mainly due to sustained pulmonary vasoconstriction, concentric pulmonary vascular wall thickening, and in situ thrombosis that preclude the contrast from filling into small pulmonary arteries and arterioles. Intraperitoneal injection of 2-APB (1 mg/kg, every day, for two weeks) after 4-wk hypoxic exposure partially reversed the total length of branches (from 13.0 ± 1.3 to 20.7 ± 0.9 mm/mm2, P < 0.001), the number of branches (from 264.8 ± 21.1 to 243.4 ± 17.3 branches/mm2, P < 0.001), and the number of junctions (from 129.8 ± 11.4 to 216.2 ± 15.0 junctions/mm2, P < 0.01) (Fig. 4Eb). 2-APB (1 mg/kg ip, every day for 2 wk) partially reversed the hypoxia-induced decreases in the total length of branches by 75.4 ± 8.9% (from −10.2 ± 1.3 to −2.5 ± 0.9 mm/mm2; P < 0.001), the number of branches by 59.3 ± 6.9% (from −250.4 ± 21.1 to −101.8 ± 17.3 branches/mm2; P < 0.001), and the number of junctions by 64.7 ± 11.2% (from −133.6 ± 11.4 to −47.2 ± 15.0 junctions/mm2; P < 0.01) (Fig. 4Eb). These results indicate that 2-APB (1 mg/kg ip, every day for 2 wk) efficiently results in regression of pulmonary vascular remodeling in chronically hypoxic mice, which is one of the major contributors to reducing PAP (RVSP and mPAP), decreasing RV afterload, and inhibiting RV hypertrophy (Fulton Index).
Inhibition of TRPC6 Channels by Oral Administration of BI-749327 Partially Reverses Established PH in Mice
2-APB (at 10–100 µM) is known to block multiple TRP channels and IP3 receptors (74–80). Therefore, the inhibitory or reversal effects of 2-APB on experimental PH (shown in Figs. 3 and 4) might be due to the inhibition of multiple targets. To examine whether specific inhibition of TRPC6 channels is therapeutically beneficial for PH, we conducted a reversal experiment using an orally bioavailable TRPC6 blocker, BI-749327 (IC50 = 13 nM for mouse TRPC6 and 19 nM for human TRPC6 determined by patch clamp experiments) (55).
The experimental protocol for the reversal experiments (Fig. 5A) using BI-749327 (Fig. 5B) was the same as that for the 2-APB experiment (Fig. 4A). Eight-week-old mice were randomly divided into the following groups: 1) Nor mice, 2) Hyp + Veh (90% corn oil) mice, and 3) Hyp + BI-749327 (dissolved in 90% corn oil) with oral administration (orally), via oral gavage, of BI-749327 (BI, every day, for 2 wk). The Hyp + Veh and Hyp + BI mice were first exposed to normobaric hypoxia (10% O2 for 4 wk) to develop PH and RV hypertrophy. Then the mice were orally administered, once a day, with vehicle in Hyp + Veh mice and various doses of BI-749327 (3, 10, 20, and 30 mg/kg) in Hyp + BI mice for two more weeks under hypoxic conditions before the hemodynamic and angiography measurements were conducted.
Figure 5.
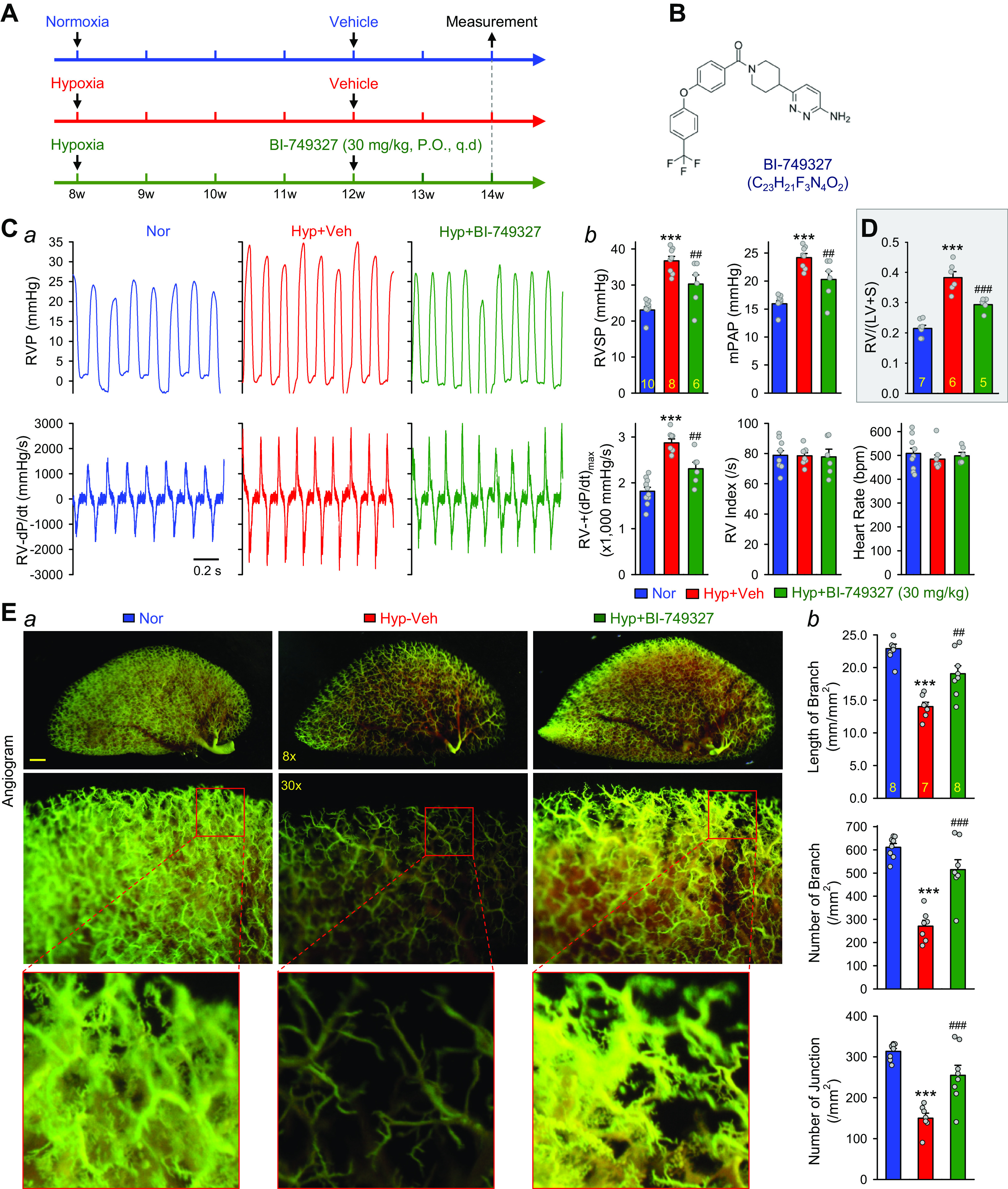
Inhibition of TRPC6 channels with BI-749327 (BI) partially reverses hypoxia-induced pulmonary hypertension in mice. A: experimental protocol for the reversal experiment using BI-749327 (BI). Eight-week-old C57Bl/6 mice were subjected to normoxia (Nor) or hypoxia (10% O2) for 4 wk at first. BI-749327 (30 mg/kg/day) was then orally administered (orally) via oral gavage for two more weeks during hypoxia. B: chemical structure of BI-749327. C, a: representative records of right ventricular pressure (RVP) (top) and right ventricle (RV) contractility (RV-±dP/dt) (bottom) in normoxic (Nor) control mice, hypoxic (Hyp) mice, and Hyp mice receiving BI (30 mg/kg, orally, every day for 2 wk). Summarized data (means ± SE, b) showing right ventricular systolic pressure (RVSP), estimated mean pulmonary arterial pressure (mPAP), RV-+(dP/dt)max, RV contractility index [RV-+(dP/dt)max/RVSP], and heart rate in Nor (n = 10), Hyp (n = 8), and Hyp+BI (n = 6) mice. ***P < 0.001 vs. Nor; ##P < 0.05 vs. Hyp (ANOVA). D: summarized data (means ± SE) showing Fulton Index, the ratio of the RV weight to the left ventricle (LV) and septum (S) weight [RV/(LV+S)] in Nor (n = 7), Hyp (n = 6), and Hyp + BI-749327 (n = 5) mice. ***P < 0.001 vs. Nor; ###P < 0.001 vs. Hyp (ANOVA). E, a: representative images of angiograph of the left lung at ×8 (top) and ×30 (middle) magnification from Nor, Hyp, and Hyp + BI (30 mg/kg, orally, every day for 2 wk) mice. Images in the bottom panels are enlarged images from the selected areas of the ×30 images in the middle panels. Summarized data (means ± SE, b) showing the total length of lung vascular branches (top), the number of lung vascular branches (middle), and the number of vascular branch junctions (lower) per square millimeter of the selected lung area from Nor (n = 8), Hyp (n = 7), and Hyp + BI (n = 8) mice. ***P < 0.001 vs. Nor; ##P < 0.01, ###P < 0.001 vs. Hyp (ANOVA).
Like the reversal experiments for 2-APB, in the reversal experiments for BI-749327 (Fig. 5), hypoxic exposure increased RVSP by 13.6 ± 3.5 mmHg (from 23.1 ± 2.8 to 36.7 ± 3.5 mmHg, P < 0.001), mPAP by 8.2 ± 0.7 mmHg (from 16.0 ± 1.6 to 24.2 ± 2.0 mmHg, P < 0.001), and RV-±(dP/dt)max by 1,057.2 ± 87.9 mmHg/s (from 1,814.9 ± 89.3 to 2,872.1 ± 87.9 mmHg/s, P < 0.001) (Fig. 5C). Oral administration of 3, 10, and 20 mg/kg BI (every day for 2 wk) after 4 wk of normobaric hypoxic exposure had a negligible effect on hypoxia-mediated changes in pulmonary hemodynamics and on lung vascular remodeling. Oral administration of 30 mg/kg BI (every day for 2 wk) in mice with established PH, however, inhibited hypoxia-induced increases in RVSP by 47.2 ± 18.4% (from 13.6 ± 1.2 to 7.2 ± 2.5 mmHg; P < 0.01), mPAP by 47.4 ± 17.7% (from 8.2 ± 0.7 to 4.3 ± 1.5 mmHg; P < 0.01), and RV-+(dP/dt)max by 53.3 ± 13.3% (from 1,057.2 ± 87.9 to 493.4 ± 141.1 mmHg/s; P < 0.01) (Fig. 5C). Furthermore, the BI-74932-induced inhibition of hypoxia-induced increases in RVSP and mPAP was associated with significant inhibition of Fulton Index (Fig. 5D). These results indicate that oral administration of BI-749327 (30 mg/kg, every day), a selective TRPC6 blocker, to mice efficiently reverses established PH (by ∼65% on RVSP and mPAP) and RV hypertrophy with little effect on RV contractile index.
To examine whether the inhibitory or therapeutic effect of BI-749327 is selective to the pulmonary circulation in mice with experimental PH, we also tested the effect of BI-749327 on systemic arterial pressure (SAP). As shown in Fig. 6, the same treatment with BI-749327 (30 mg/kg, orally, for 2 wk) in mice pre-exposed to 4-wk hypoxia had little effect on SAP and heart rate (Fig. 6). These results indicate that oral administration of BI-749327 at the dose of 30 mg/kg selectively and significantly ameliorate established PH with little effect on systemic hemodynamics.
Figure 6.

Inhibition of TRPC6 channels with BI-749327 (BI) negligibly affects systemic arterial pressure and heart rate in mice. Summarized data (means ± SE) showing systolic, diastolic, and calculated mean systemic arterial systolic (SAP) (A) and heart rate (B), determined by carotid artery catheterization in normoxic (Nor, n = 5) control mice, hypoxic (Hyp for 6 wk) mice (n = 5), and Hyp (for 6 wk) mice receiving BI (30 mg/kg, orally, every day for 2 wk, n = 5).
In addition to hemodynamics, we also examined the effect of different concentrations of BI-749327 on pulmonary vascular remodeling determined by angiogram (Fig. 5E). Exposure to normobaric hypoxia (10% O2) decreased the total number of lung vascular branches by 8.3 ± 1.0 mm/mm2 (from 22.2 ± 0.7 to 13.9 ± 1.0 mm/mm2; P < 0.001), the number of vascular branches by 344.3 ± 40.1 (from 636.1 ± 13.4 to 291.9 ± 40.1 branches/mm2; P < 0.01), and the number of vascular branch junctions by 180.3 ± 20.1 junctions/mm2 (from 321.9 ± 7.6 to 141.6 ± 20.1 junctions/mm2; P < 0.01) (Fig. 5E, a and b). It must be noted that the decreased number of lung vascular branches and junctions does not necessarily indicate that the branches were lost during the 6 wk of normobaric hypoxic exposure. The decreased total length and reduced number of branches and junctions in chronically hypoxic mice were mainly due to sustained vasoconstriction, concentric pulmonary vascular wall thickening (or intraluminal narrowing), and occlusive lesions that prevented the injected contrast from filling into small pulmonary arteries and arterioles.
Oral administration of BI-749327 (30 mg/kg, every day, for 2 wk) after 4-wk hypoxic exposure in mice significantly reversed pulmonary vascular remodeling (Fig. 5E, a and b). BI-749327 restored or reversed the hypoxia-mediated decreases in the total length of branches by 74.7 ± 21.5% (from −8.3 ± 1.0 to −2.1 ± 1.8 mm/mm2, P < 0.05), the number of branches by 69.3 ± 20.2% (from −344.3 ± 40.1 to −105.6 ± 69.5 branches/mm2, P < 0.01), and the number of junctions by 70.7 ± 21.1% (from −180.3 ± 20.1 to −52.8 ± 38.0 junctions/mm2, P < 0.01) (Fig. 5Eb). These data indicate that the selective TRPC6 blocker BI-749327 (30 mg/kg, orally, every day, for 2 wk) efficiently reverses established PH in mice by inhibiting HPV and regression of remodeled pulmonary vasculature during hypoxia.
We previously reported that TRPC6 was upregulated in PASMCs from patients with idiopathic PAH (45–47). The observations from the current study imply that the upregulated TRPC6 in PASMCs from patients with PAH is a good therapeutic target for developing novel and efficient treatment for PAH and PH due to lung diseases and/or hypoxemia. The orally available drug, BI-749327, is potentially a novel and promising drug for treatment of PAH and PH due to respiratory diseases or hypoxemia or precapillary PH resulting from pulmonary arterial contraction, pulmonary arteriole muscularization, and pulmonary arterial and arteriole occlusive lesions.
Ca2+ Is Involved in Growth Factor-Mediated Phosphorylation of AKT and mTOR in PASMC
An increase in [Ca2+]cyt, which can rapidly increase nuclear [Ca2+] (81) and stored [Ca2+] in the sarcoplasmic and endoplasmic reticulum (82–84), activates cytoplasmic mitogen-activated protein kinases and nuclear Ca2+-sensitive transcriptional factors, thereby promoting cell proliferation (20, 65, 85). The PI3K/AKT/mTOR signaling cascade is one of the major pathways involved in cell proliferation and protein expression (86–91) and is implicated in the development of PAH/PH (69, 92). Genetic deletion of AKT1 or mTOR ameliorates experimental PH in mice (93, 94). The next set of experiments was designed to examine whether the BI-749327-induced inhibitory effect on Ca2+ influx was associated with inhibition of the AKT/mTOR signaling cascade that is required for PASMC proliferation.
We first used BAPTA (a strong Ca2+ chelator) to chelate extracellular Ca2+ in the culture media and BAPTA-AM (a cell-permeant Ca2+ chelator that can be accumulated in intracellular organelles) to chelate intracellular Ca2+ (61–63) to examine the effect of chelation of extracellular and/or intracellular free Ca2+ on the phosphorylation (p) of AKT (pAKT) and mTOR (pmTOR) in PASMCs induced by growth medium (GM, including 5% FBS and multiple growth factors) (Fig. 7A, a and b) and PDGF-BB (Fig. 7B). Chelation of extracellular Ca2+ by BAPTA or chelation of intracellular Ca2+ by BAPTA-AM significantly inhibited GM-mediated (Fig. 7A) and PDGF-BB-mediated (Fig. 7B, a and b) increases in AKT and mTOR phosphorylation. Although BAPTA + BAPTA-AM almost abolished GM- and PDGF-mediated increases in pAKT, BAPTA-AM or BAPTA + BPATA-AM actually decreased basal pmTOR level in PASMCs (Fig. 7Ab). Furthermore, the in vitro cell proliferation experiments showed that incubation of human PASMCs in 5% FBS media-containing BI-749327 (250 nM) significantly attenuated PASMC proliferation (Fig. 7C). Forty-eight hours incubation of synchronized or growth-arrested PASMCs in 5% FBS media significantly increased the number of viable cells (from 0.4779 ± 0.0133 to 0.8766 ± 0.0203, P < 0.001, n = 18), while 250 nM BI-749327 resulted in a 50.26 ± 3.36% (n = 18) inhibition of 5% FBS-mediated PASMC proliferation (Fig. 7C). These results provide strong evidence that PDGF (and other growth factors)-mediated activation of AKT/mTOR signaling, indicative of increased pAKT/AKT and pmTOR/mTOR, and growth medium-mediated cell proliferation, are dependent of Ca2+ influx in PASMCs.
Figure 7.
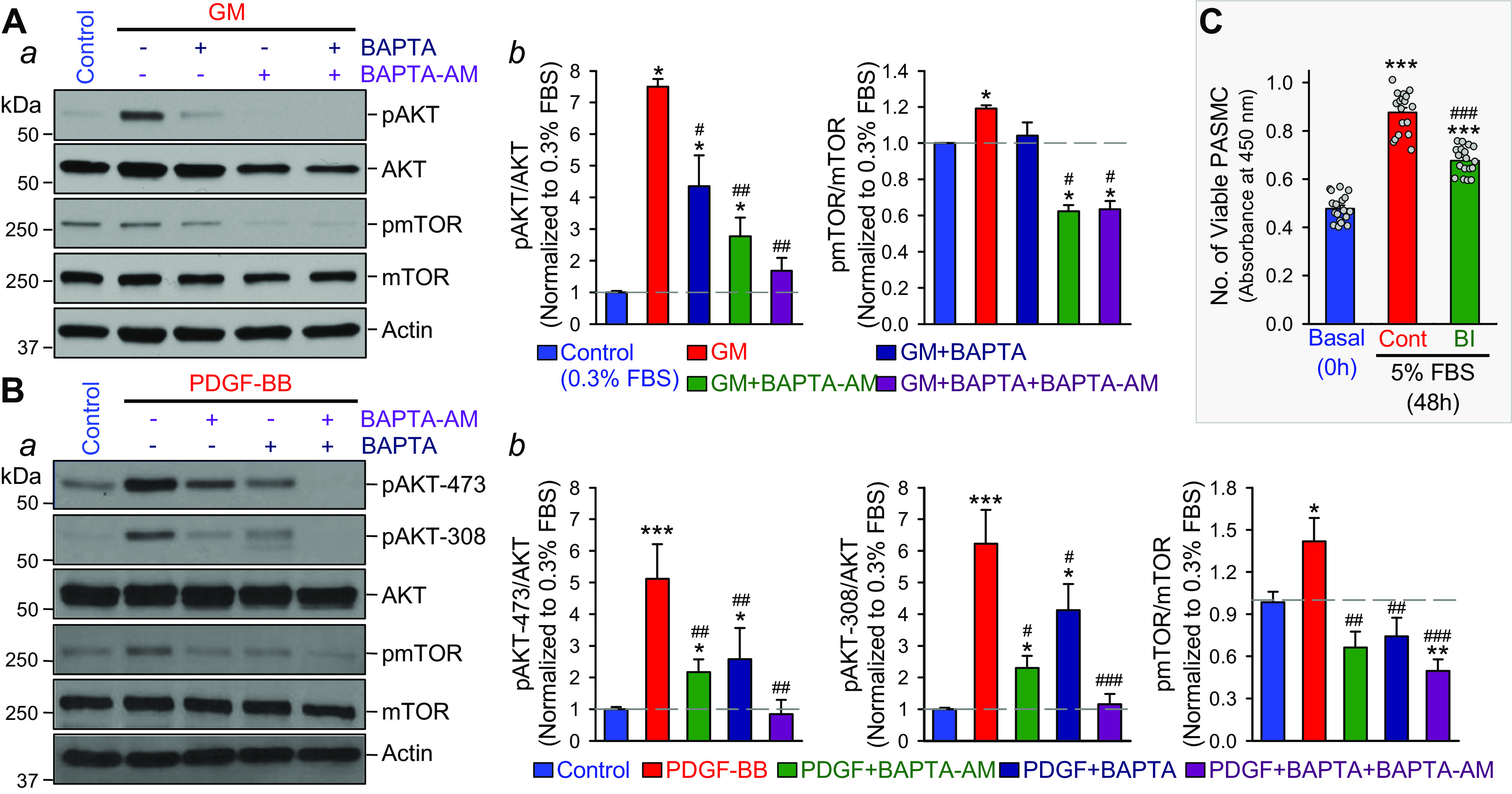
Growth factor-mediated phosphorylation of AKT and mTOR is dependent of extracellular and intracellular Ca2+ in human pulmonary arterial smooth muscle cells (PASMCs) and inhibition of TRPC6 channels with BI-749327 (BI) attenuates growth factor-mediated PASMC proliferation. A, a: Western blot analysis on phosphorylated AKT (pAKT), total AKT (AKT), phosphorylated mTOR (pmTOR), and total mTOR (mTOR) in control PASMCs and PASMCs treated with growth medium (GM) in the absence (−) and presence (+) of BAPTA (2 mM), a potent Ca2+ chelator that decreases extracellular free Ca2+ concentration to 400–500 nM, and BAPTA-AM (25 µM), a membrane-permeable BAPTA that significantly decreases intracellular free Ca2+ concentration. Summarized data (means ± SE, b) showing the ratio of pAKT/AKT and pmTOR/mTOR in control PASMCs (Control) and GM-treated PASMCs in the absence and presence of BAPTA and/or BAPTA-AM. *P < 0.05 vs. Control; #P < 0.05, ##P < 0.01 vs. GM (ANOVA). B, a: Western blot analysis on pAKT at Ser473 (pAKT-473), pAKT at Ser308 (pAKT-308), AKT, pmTOR, and mTOR in control PASMCs and PASMCs treated with platelet-derived growth factor (PDGF-BB, 10 ng/mL) in the absence (−) and presence (+) of BAPTA and/or BAPTA-AM. Summarized data (means ± SE, b) showing the ratio of pAKT-473/AKT (left), pAKT-308/AKT (middle), and pmTOR/mTOR (right) in control PASMCs (Control) and PDGF-treated PASMCs in the absence and presence of BAPTA and/or BAPTA-AM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. PDGF (ANOVA). C: summarized data (means ± SE) showing the changes of number of viable cells in normal human PASMC cultured under basal condition (Basal, 0 h) and in 5% FBS-containing growth media in the absence (Cont) or presence (BI) of BI-749327 (250 nM) for 48 h. ***P < 0.001 vs. Basal; ###P < 0.001 vs. 5% FBS without (Cont) BI.
Block of TRPC6 Channels with BI-749327 Inhibits PDGF-Mediated Phosphorylation of AKT and mTOR in PASMC
To examine whether Ca2+ influx through nonselective cation channels including TRPC6 channels affects the AKT/mTOR signaling, we conducted two sets of experiments using 2-APB (a nonselective blocker of cation channels) (75, 79, 80) and BI-749327 (a specific blocker of TRPC6 channels) (55) in PASMCs incubated in serum-free basal medium containing only PDGF-BB (10 ng/mL) and in 5% FBS-containing growth media (GM) supplemented with multiple growth factors (5 ng/mL hEGF, 5 ng/mL hFGF-β, and 5 μg/mL human insulin).
Nonselective inhibition of cation channels with 50 µM 2-APB (for 24 h) significantly inhibited PDGF-BB-mediated phosphorylation of AKT and mTOR (Fig. 8A, a and b). 2-APB inhibited the PDGF-mediated increases in pAKT/AKT by 47.7 ± 1.6% (n = 6; P < 0.001) and pmTOR/mTOR by 145.8 ± 21.9% (n = 6; P < 0.01) (Fig. 8Ac). Although low doses of BI-749327 (0.15 and 0.25 µM) for short-time treatment (6 and 24 h) had a negligible effect on PDGF-mediated increases in pmTOR and pAKT (Fig. 7B), the high dose of BI-749327 (0.25 µM, for 48-h incubation) significantly inhibited PDGF-mediated increases in pAKT and pmTOR (Fig. 8C, a and b). In PASMCs incubated in 10 ng/mL PDGF-containing medium, BI-749327 inhibited the PDGF-mediated increases in pAKT/AKT by 24.0 ± 1.5.6% (n = 6; P < 0.001) and pmTOR/mTOR by 104.9 ± 18.0% (n = 6; P < 0.01) (Fig. 8Ac). These results indicate that inhibition of TRP channels (79, 80) and IP3 receptors (75) with 2-APB or selective inhibition of TRPC6 channels with BI-749327 significantly inhibit PDGF-BB-mediated phosphorylation of AKT and mTOR in human PASMCs. PDGF is a critical growth factor implicated in the development and progression of PAH and experimental PH by its stimulation of PASMC proliferation (24, 65, 95–98). The 48% inhibition by 2-APB (a nonselective TRP blocker) and 24% inhibition of BI-749327 (a specific TRPC6 blocker) (55) of PDGF-BB-mediated increases in pAKT/AKT and pmTOR/mTOR ratio is likely one of the critical mechanisms involved in the regression of pulmonary vascular remodeling in PH.
Figure 8.
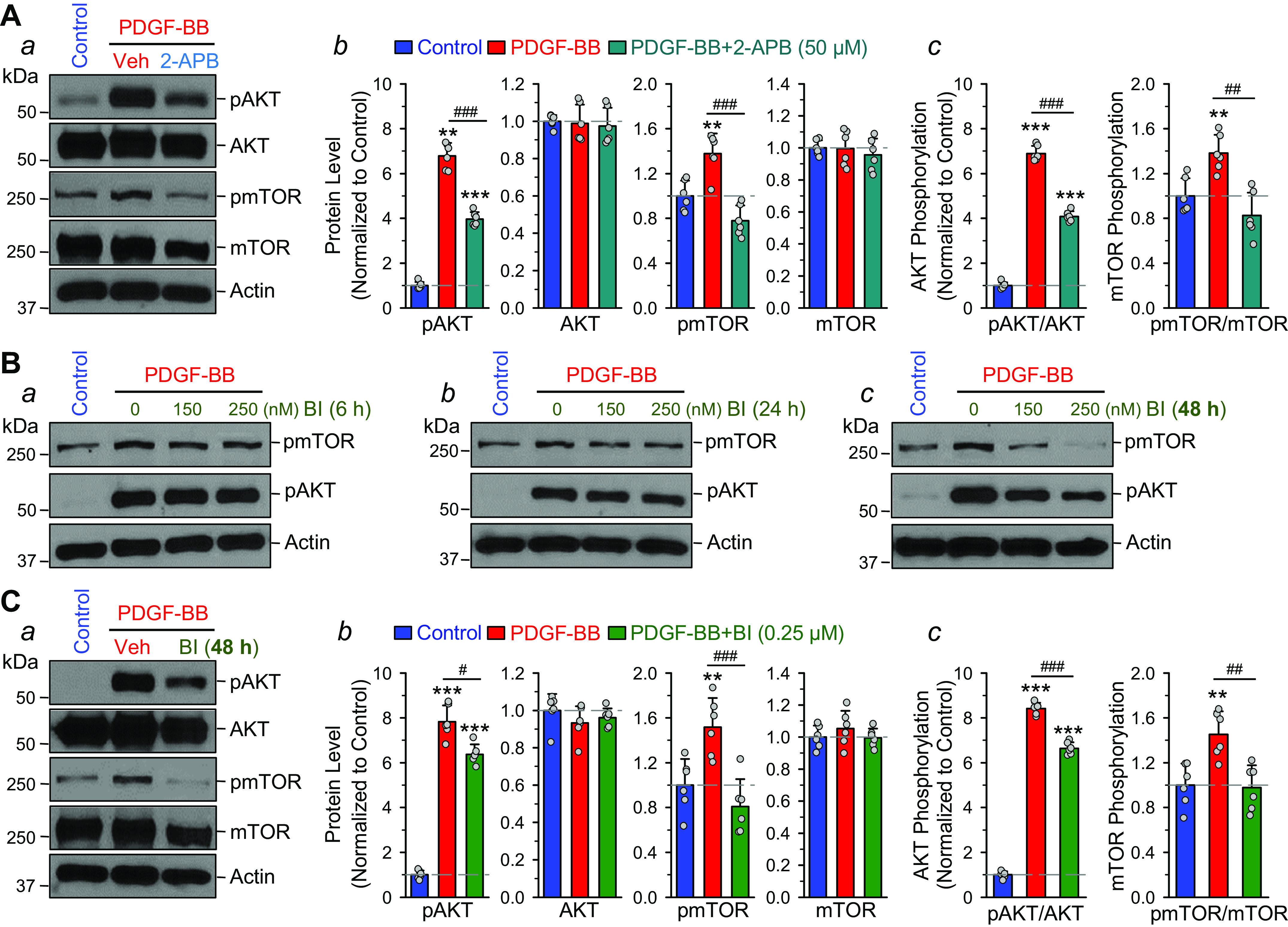
Block of cation channels with 2-aminoethyl diphenylborinate (2-APB) and block of TRPC6 channels with BI-749327 (BI) inhibit PDGF-induced AKT/mTOR phosphorylation in human pulmonary arterial smooth muscle cells (PASMCs). A, a: Western blot analysis on pAKT, AKT, pmTOR, and mTOR in control PASMCs and PASMCs treated with PDGF-BB (10 ng/mL) in the absence (vehicle control, Veh) and presence of 2-APB (50 µM, for 24 h), a nonselective blocker of cation channels. Summarized data (means ± SE) showing the relative protein levels of pAKT, AKT, pmTOR, and mTOR (b, normalized to Control) as well as the ratio of pAKT/AKT and pmTOR/mTOR (c, normalized to Control) in control PASMCs (Control) and PDGF-treated PASMCs in the absence (PDGF-BB) and presence (PDGF-BB + 2-APB) of 50 µM 2-APB. **P < 0.01, ***P < 0.001 vs. Control; ##P < 0.01, ###P < 0.001 vs. PDGF-BB. B: Western blot analysis on pAKT and pmTOR in control PASMCs and PASMCs treated with PDGF-BB (10 ng/mL) in the absence (Veh) and presence of 150 and 250 nM of BI-749327, a selective blocker of TRPC6 channels, for 6 (a), 24 (b), and 48 (c) h. C, a: Western blot analysis on pAKT, AKT, pmTOR, and mTOR in control PASMCs and PASMCs treated with PDGF-BB (10 ng/mL) in the absence (Veh) and presence of BI-749327 (BI, 250 nM for 48 h), a selective blocker of TRPC6 channels. Summarized data (means ± SE) showing the relative protein levels of pAKT, AKT, pmTOR, and mTOR (b, normalized to Control) as well as the ratio of pAKT/AKT and pmTOR/mTOR (c, normalized to Control) in control PASMCs (Control) and PDGF-treated PASMCs in the absence (PDGF-BB) and presence (PDGF-BB + BI-749327) of 250 nM BI-949327. **P < 0.01, ***P < 0.001 vs. Control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. PDGF-BB.
In addition to PDGF-BB-mediated activation of AKT/mTOR, we also examined the effects of 2-APB and BI-749327 on the phosphorylation of AKT and mTOR in PASMC stimulated by 5% FBS-containing GM. Nonselective blockade of TRP channels with 50 µM 2-APB (for 24 h) abolished 30-min GM-mediated increases in pAKT/AKT and pmTOR/mTOR ratio (Fig. 9A, a and b); 2-APB inhibited GM-mediated increases in pAKT/AKT by 97.7 ± 1.2% (n = 6; P < 0.001) and pmTOR/mTOR by 88.1 ± 12.1% (n = 6; P < 0.001) (Fig. 9Ac). Although low doses of BI-749327 (0.25, 0.75, 2.25, 7.5, and 22.5 µM; for 24 h) had a negligible effect on GM-mediated increases in pmTOR and pAKT (Fig. 9, B and C), the high dose of BI-749327 (75 µM) for 48-h treatment significantly inhibited PDGF-mediated increases in pAKT and pmTOR (Fig. 9D, a and b). Neither GM nor 2-APB significantly affected the total expression of AKT and mTOR in PASMC. The inability of BI-749327 to inhibit GM-mediated phosphorylation of AKT/mTOR at the dose of 0.25 µM indicates that 5% serum and multiple growth factors activate multiple cation channels and other Ca2+ channels and transporters to induce Ca2+-dependent phosphorylation of AKT/mTOR and cell proliferation. In PASMCs incubated in GM, BI-749327 at a high dose (75 µM for 48 h) inhibited PDGF-mediated increases in pAKT/AKT by 35.2 ± 3.4% (n = 6; P < 0.001) and in pmTOR/mTOR by 68.2 ± 7.4% (n = 6; P < 0.01) (Fig. 9Dc).
Figure 9.
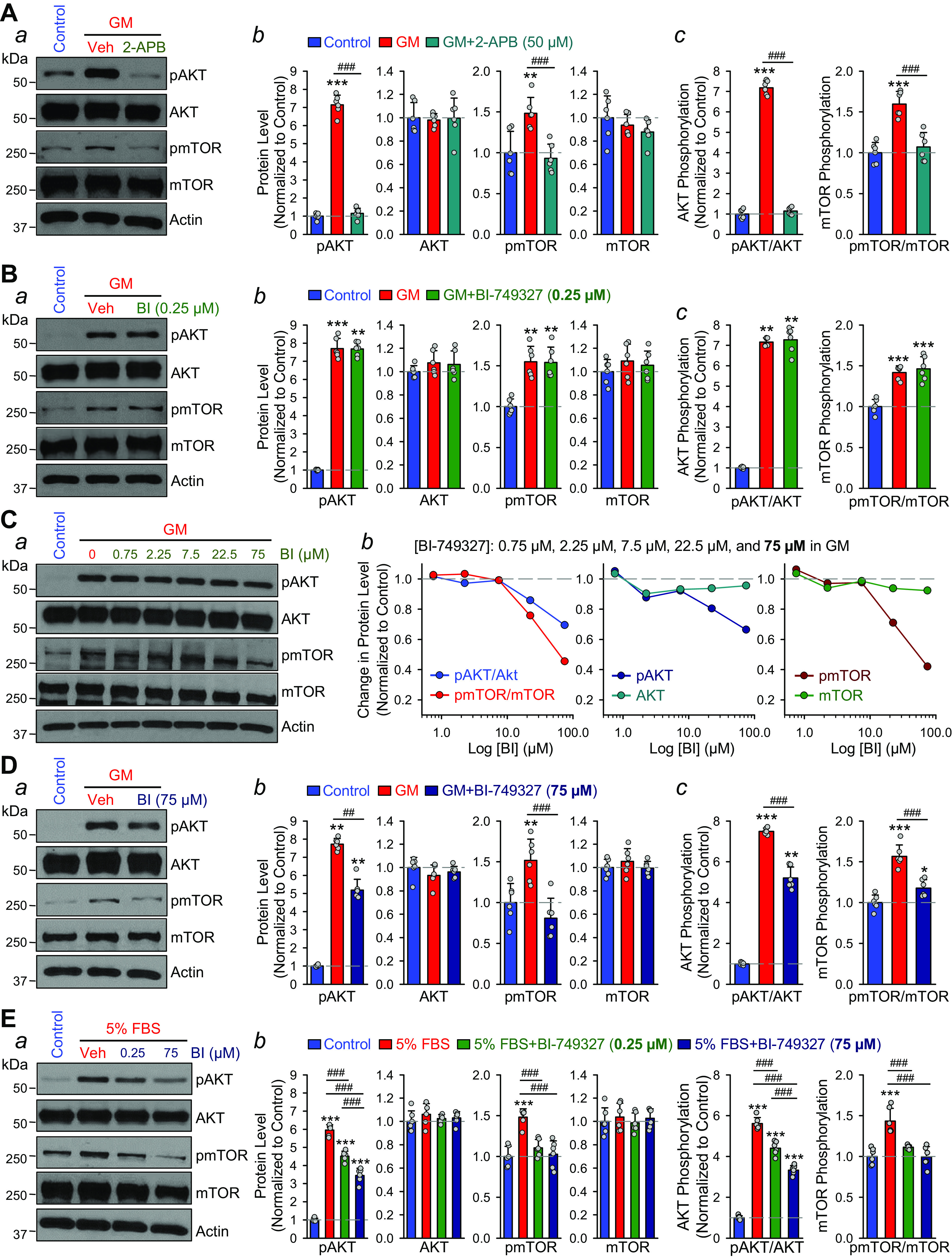
Block of cation channels with 2-aminoethyl diphenylborinate (2-APB) and block of TRPC6 with BI-749327 (BI) inhibit growth media (GM)- and serum (5% FBS)-induced AKT/mTOR phosphorylation in human pulmonary arterial smooth muscle cells (PASMCs). A, a: Western blot analysis on pAKT, AKT, pmTOR, and mTOR in control PASMCs and PASMCs treated with GM (including 5% FBS, SMC basal medium, 5 ng/mL hEGF, 5 ng/mL hFGF-β, 5 μg/mL human insulin, 50 μg/mL ascorbic acid, 10 mM l-glutamine, 30 mg/mL gentamicin, and 15 μg/mL amphotericin B) for 30 min in the absence (vehicle control, Veh) and presence (2-APB) of 2-APB (50 µM for 24 h), a nonselective blocker of cation channels. Summarized data (means ± SE) showing the relative protein levels of pAKT, AKT, pmTOR, and mTOR (b, normalized to Control) as well as the ratio of pAKT/AKT and pmTOR/mTOR (c, normalized to Control) in control PASMCs (Control) and GM-treated PASMCs in the absence (GM) and presence (GM + 2-APB) of 50 µM 2-APB. **P < 0.01, ***P < 0.001 vs. Control; ###P < 0.001 vs. GM (ANOVA). B, a: Western blot analysis on pAKT, AKT, pmTOR, and mTOR in control PASMCs and PASMCs treated with GM (for 30 min) in the absence (Veh) and presence (BI) of BI-749327 (0.25 µM for 48 h), a selective blocker of TRPC6 channels. Summarized data (means ± SE) showing the relative protein levels of pAKT, AKT, pmTOR, and mTOR (b, normalized to Control) as well as the ratio of pAKT/AKT and pmTOR/mTOR (c) in control PASMCs (Control) and GM-treated PASMCs in the absence (GM) and presence (GM + BI 749327) of 0.25 µM BI-749327. **P < 0.01, ***P < 0.001 vs. Control (ANOVA). C, a: Western blot analysis on pAKT, AKT, pmTOR and mTOR in control PASMCs and PASMCs treated with GM (for 30 min) in the absence (Veh) and presence of 0.75, 2.25, 7.5, 22.5, 75 µM BI-749327 (for 48 h). Dose-response curves (b) showing the ratio of pAKT/AKT and pmTOR/mTOR as well as the relative protein levels of pAKT, AKT, pmTOR, and mTOR (normalized to Control) in control PASMCs (Control) and GM-treated PASMCs in the absence (GM) and presence (GM + BI 749327) of 0.75, 2.25, 7.5, 22.5, 75 µM of BI-749327. D, a: Western blot analysis on pAKT, AKT, pmTOR, and mTOR in control PASMCs and PASMCs treated with GM in the absence (Veh) and presence (BI) of 75 µM BI-749327 (for 48 h). Summarized data (means ± SE) showing the relative protein levels of pAKT, AKT, pmTOR, and mTOR (b, normalized to Control) as well as the ratio of pAKT/AKT and pmTOR/mTOR (c) in control PASMCs (Control) and GM-treated PASMCs in the absence (GM) and presence (GM + BI 749327) of 75 µM BI-749327. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Control; ##P < 0.01, ###P < 0.001 vs. GM. E, a: Western blot analysis on pAKT, AKT, pmTOR and mTOR in control PASMCs and PASMCs treated with 5% fetal bovine serum (FBS)-containing medium without growth factors (5% FBS) in the absence (Veh) and presence (BI) of 0.25 and 75 µM BI-749327 (for 48 h). Summarized data (means ± SE, b) showing the relative protein levels of pAKT, AKT, pmTOR, and mTOR (normalized to Control) as well as the ratio of pAKT/AKT and pmTOR/mTOR in control PASMCs (Control) and 5% FBS medium-treated PASMCs in the absence (5% FBS) and presence (5% FBS + BI-749327) of 0.25 and 75 µM BI-749327. ***P < 0.001 vs. Control; ###P < 0.001 vs. 5% FBS (ANOVA).
Since a high dose (75 µM) of BI-749327 had to be used to significantly inhibit GM-mediated increases in pAKT/AKT and pmTOR/mTOR, we conducted another set of experiments using only 5% serum (FBS) as the stimulus for activating the AKT/mTOR signaling cascade in PASMCs. As shown in Fig. 9E, the low dose (0.25 µM) of BI-749327, which was insufficient to inhibit GM-mediated phosphorylation of AKT and mTOR (Fig. 9, B and C), resulted in a significant inhibition of 5% FBS-mediated increases in the pAKT/AKT and pmTOR/mTOR ratios (Fig. 9E, a and b). BI-749327 (0.25 µM) attenuated 5% FBS-mediated increases in pAKT/AKT by 26.3 ± 3.0% (n = 6; P < 0.001) and pmTOR/mTOR by 74.0 ± 2.3% (n = 6; P < 0.001) (Fig. 9Ec). In this set of experiments, 75 µM BI-749327 further inhibited 5% FBS-induced increases in pAKT/AKT by 49.5 ± 2.0% (n = 6; P < 0.001) and pmTOR/mTOR by 101.2 ± 11.8% (n = 6; P < 0.001) in PASMCs (Fig. 9Ec).
The inhibitory effects of 2-APB and BI-749327 on growth factor-mediated phosphorylation of AKT/mTOR were apparently due to attenuation of Ca2+ influx through TRPC6 (and other cation channels) in PASMCs. Given that the AKT/mTOR signaling is a major pathway for cell proliferation and gene expression, the 2-APB- or BI-749327-induced inhibitory effect on the AKT/mTOR signaling cascade would contribute to the regression of pulmonary vascular remodeling and partial reversal of experimental PH (Figs. 4 and 5). These results suggest that inhibition of TRPC6 attenuates PASMC proliferation in a dose-dependent manner. This inhibitory effect also depends on the mitogenic factors and growth factors. These data also indicate that specific growth factors, such as PDGF, stimulate PASMC proliferation via activating the AKT/mTOR signaling cascade due, at least partially, to Ca2+ influx through BI-749327-sensitive TRPC6 channels, whereas multiple growth factors and 5% serum may activate AKT/mTOR signaling by inducing Ca2+ influx through different cation channels and ion transporters in the plasma membrane and Ca2+ release from IP3-sensitive intracellular stores.
DISCUSSION
In this study, we report that: 1) acutely (10 min) blocking nonselective cation channels with 2-APB (40 µM) or the TRPC6 channel with BI-749327 (3–10 µM) significantly and reversibly inhibited alveolar hypoxia-induced pulmonary vasoconstriction in isolated perfused/ventilated mouse lungs; 2) chronic treatment of mice with intraperitoneal injection of 2-APB (1 mg/kg, every day for 4 wk) significantly inhibited the development of hypoxia-induced PH (by 45%–50%), pulmonary vascular remodeling (by 40%–75%), and RV hypertrophy (by 45%); 3) treatment of mice with 2-APB (1 mg/kg ip, every day for 2 wk) or BI-749327 (30 mg/kg, orally, every day for 2 wk) (55) partially reversed established experimental PH as indicated by significantly improved pulmonary hemodynamics (by 40% with 2-APB and 65% with BI, respectively), regressed lung vascular remodeling (by 60%–75% with 2-APB and 70%–75% with BI, respectively) and inhibited RV hypertrophy (by 68% with 2-APB); and 4) blocking cation channels with 2-APB or TRPC6 with BI-749327 significantly inhibited growth factor-mediated activation of the AKT/mTOR signaling cascade (determined by the increased ratio of pAKT/AKT and pmTOR/mTOR), a major signaling pathway associated with PASMC proliferation. These data indicate that nonselective inhibition of TRPC channels with 2-APB and selective inhibition of TRPC6 channels with BI-749327 are both sufficient to ameliorate established PH via inhibition of pulmonary vasoconstriction and regression of pulmonary vascular remodeling.
An increase in [Ca2+]cyt in PASMCs is a major trigger for PASMC contraction and pulmonary vasoconstriction (18, 19, 99–101) and is an important stimulus for PASMC proliferation/migration and concentric pulmonary vascular wall thickening or medial hypertrophy (28, 85). Receptor-operated Ca2+ channels and store-operated Ca2+ channels formed in part by TRP channels (including TRPC6) and Orai/STIM channels are important Ca2+-permeable cation channels responsible for regulating [Ca2+]cyt in PASMCs and thus pulmonary vasoconstriction and vascular remodeling (20, 33, 34, 62, 65, 70, 102, 103). We previously reported that upregulation of TRPC6 channels and STIM2 proteins are, at least in part, responsible for the enhanced Ca2+ influx and augmented resting [Ca2+]cyt in PASMCs from patients with idiopathic PAH (22, 45, 46, 65). In addition, upregulated TRPC6 channels and increased receptor- and store-operated Ca2+ entry are implicated in highly proliferative PASMCs from animals with experimental PH (21, 47). TRPC6 is not only a receptor-operated cation channel activated by diacylglycerol (a critical intracellular messenger upon receptor activation), but also a mechanosensitive channel responsible for mechanical stretch- and shear stress-mediated Ca2+ influx in smooth muscle and endothelial cells (104). Membrane stretch synergistically activates TRPC6 channels with diacylglycerol (32, 51, 105). Upregulated TRPC6 in PASMCs from patients with PAH would significantly enhance mechanical stretch-mediated Ca2+ influx and receptor-operated Ca2+ entry. Inhibition of ligand-mediated and mechanosensitive Ca2+ influx through upregulated TRPC6 in PASMCs from patients with idiopathic PAH and animals with experimental PH is thus a promising therapeutic strategy for PAH and PH due to lung diseases and/or hypoxemia.
2-APB (2-aminoethyl diphenylborinate), at 10–100 µM, is known to block multiple TRP channels including TRPC1/3/5/6, TRPV6, TRPM3/7/8, and TRPP2 (74–77, 80). In HEK cells transfected with TRPC6, 75 µM 2-APB resulted in 90% inhibition of cation currents through TRPC6 channels (80). The IC50 for 2-APB-mediated inhibition of TRPC6 channels was reported to be ∼10.4 µM (106). 2-APB also induces functional uncoupling between STIM1 and Orai1 to inhibit store-operated Ca2+ entry (53, 107). In addition, 2-APB inhibits sarcoplasmic or endoplasmic reticulum Ca2+-Mg2+ ATPase (SERCA, IC50 ≈ 325 µM) (108), volume-regulated anion channels (IC50 ≈ 122.8 µM) (109), GABA receptors (IC50 ≈ 23 µM) (110), and IP3 receptors (IC50 ≈ 42 µM) (71, 74, 75, 80). As shown in this study, 45 µM 2-APB resulted in 50% inhibition of alveolar hypoxia-induced pulmonary vasoconstriction in isolated perfused/ventilated mouse lungs (Fig. 1B), indicating that Ca2+ influx through 2-APB-sensitive nonselective cation channels is involved in induction of HPV (48).
In the in vivo reversal experiments, we intraperitoneally injected 2-APB at a dose of 1 mg/kg body wt, once a day for 2 wk at the end of the 4 wk of hypoxic exposure, when RVSP (and mPAP) was significantly increased; two more weeks of 2-APB injection or treatment decreased RVSP by 5.7 mmHg (or 38%, P < 0.01) and mPAP by 3.4 mmHg (or 38%, P < 0.001) (Fig. 4C). The body weight of the mice used in this study was around 25 g, and the total amount of 2-APB administered to each mouse daily was ∼0.025 mg. On average, a mouse has ∼58 mL of blood per kg of body weight, so a mouse weighing 25 g would have a total blood volume of ∼1.5 (or 6% of the mouse’s body weight). Therefore, each intraperitoneal injection of 2-APB at the dose of 1 mg per kg body weight would result in a concentration of 74 µM in the blood. In in vitro experiments using HEK cells transfected with TRPC6, 75 µM 2-APB resulted in 90% inhibition of TRPC6 currents (80). In PASMCs transfected with TRPC6, 100 µM 2-APB resulted in 79% inhibition of outward cation currents at +90 mV and 37% inhibition of inward cation currents at −90 mV (48); the IC50 for 2-APB-mediated inhibition of TRPC6 was 10.4 µM (106). Given that 10–100 µM 2-APB can block multiple channels (including TRPC, TRPV, and TRPM) and receptors (e.g., IP3 receptor), our in vivo pharmacological experiments indicate that nonselective inhibition of various cation channels with 2-APB is efficient to prevent (or attenuate) the development of PH (Fig. 3) and reverse established PH (Fig. 4).
BI-749327 is an orally bioavailable and selective TRPC6 blocker with IC50 of 13–19 nM for human and animal TRPC6. The selectivity of BI-749327 for TRPC6 is 42- to 85-fold higher than the selectivity for other TRPC channels (e.g., TRPC3 and TRPC7), while BI-749327 has a long terminal half-life (t1/2 is 8.5–13.5 hr) for mice at doses of 3–30 mg/kg (orally). The efficacy of BI-749327 was tested in in vivo mouse models of renal and cardiac fibrosis. The study clearly demonstrated that orally administered BI-749327 was a selective TRPC6 inhibitor with favorable bio-availability and pharmacokinetics to ameliorate disease conditions associated with TRPC6 (55). In vivo, BI-749327 (30 mg/kg/day) has been reported to improve left heart function and inhibit fibrosis in mice (55). In this study, we first conducted a dose-response experiment and found that oral administration of 30 mg/kg BI-749327 via oral gavage (every day) for 2 wk, after 4-wk hypoxic exposure, decreased RVSP by 6.4 mmHg (or 47%, P < 0.01) and mPAP by 3.9 mmHg (or 47%, P < 0.01). BI-749327 significantly decreased RV-+(dP/dt)max due to its inhibitory effect on RVSP, but did not alter the RV contractile index or the ratio of RV-+(dP/dt)max to RVSP (Fig. 5C), indicating that 2-wk oral administration of BI-749327 does not cause a negative inotropic effect on RV. The inhibitory effect of BI-749327 on pulmonary hemodynamics was mainly due to the regression of pulmonary vascular remodeling and inhibition of pulmonary vasoconstriction. Two-week oral administration of BI-749327 resulted in 74%, 69.3%, and 70.7% recovery of hypoxia-induced decreases in the total length of lung vascular branches, the number of branches, and the number of vascular junctions, respectively (Fig. 5E). Furthermore, intrapulmonary perfusion of 10 µM BI-749327 resulted in 84% inhibition (P < 0.001) of alveolar hypoxia-induced pulmonary vasoconstriction. The low concentrations of BI-749327 we tested in this study (3, 10, and 20 mg/kg) did not have significant inhibitory effect on pulmonary hemodynamics and lung vascular remodeling, which implies that the low concentration (3–10 mg/kg body wt) of BI-749327 was insufficient to have a favorable pharmacokinetics and thus can be rapidly eliminated from the blood (55).
If all orally administered BI-749327 (at 30 mg/kg body wt) is absorbed into blood, the total amount of BI-749327 would be ∼0.75 mg per day (given the average body weight of a mouse is 25 g and the mouse blood volume is 6% of its body weight), and the maximal concentration of BI-749327 (MW = 442.43) in the blood would be estimated to be 1.13 mM. However, the actual concentration of BI-749327 in the blood would be much lower than 1.13 mM because of its oral administration, incomplete intestinal absorption, and potential acid and enzyme-mediated degradation and breakdown in the gastrointestinal system. More pharmacokinetic and pharmacodynamic studies are needed to estimate actual concentration of BI-749327 in the blood and pulmonary vasculature when given orally. It is also possible that regression of remodeled pulmonary arteries and arterioles requires a high concentration of BI-794327 to block TRPC6 channels in different cells and tissues.
In the in vitro experiments, 0.25 µM BI-749327 (for 48 h) significantly inhibited PDGF-mediated phosphorylation of AKT and mTOR by 24% (P < 0.001) and 104.9% (P < 0.001), respectively (Fig. 7Cc), whereas 75 µM BI-749327 significantly inhibited multiple growth factors-mediated phosphorylation of AKT and mTOR by 35% (P < 0.001) and 68% (P < 0.001), respectively (Fig. 8Dc). BI-749327 (0.25 µM) inhibited serum-mediated AKT and mTOR phosphorylation by 26.3% (P < 0.001) and 75.0% (P < 0.001), respectively. In the ex vivo experiments using isolated perfused/ventilated lungs, intrapulmonary perfusion of 10 µM BI-749327 significantly inhibited acute alveolar hypoxia-induced pulmonary vasoconstriction (Fig. 2, C and D). These data indicate that BI-749327 is an efficient inhibitor of the AKT/mTOR signaling pathway in PASMCs by blocking Ca2+ influx through TRPC6 or receptor-operated and mechanosensitive cation channels and inhibiting PASMC proliferation.
In PAH and severe PH due to lung diseases and/or hypoxemia, sustained pulmonary vasoconstriction, concentric pulmonary arterial wall thickening, arteriole muscularization, obliterative intimal lesions, and in situ microvascular thrombosis all contribute to the elevated PVR/PAP, which reinforces burden or increases afterload for the RV. At the late stage of the disease, uncompensated RV hypertrophy and failure due to increased afterload are the major risk factors for RV failure and death in patients with PAH and severe PH associated with cardiopulmonary and vascular diseases. Prevention or inhibition of the development and progression of pulmonary vasculopathy, induction of pulmonary vasodilation, regression of remodeled and fibrotic pulmonary arteries and arterioles, inhibition of pulmonary vascular inflammatory response, and inhibition of RV hypertrophy and cardiac fibrosis would all contribute to reversing PAH and PH-associated RV hypertrophy and failure. Inhibition of TRPC6 channels in various types of cells would thus exert therapeutic effect on PAH and PH via multiple pathways: 1) inhibition of PASMC contraction and pulmonary vasoconstriction (47–49, 111, 112) by blocking ligand or agonist-mediated activation of receptors and synthesis of diacylglycerol (105, 113), 2) inhibition of myogenic tone in the pulmonary artery (PA) by attenuating mechanosensitive Ca2+ influx (32, 104, 114) and inducing vasodilation (36, 39), 3) inhibition of PASMC proliferation (65, 115) and concentric pulmonary vascular remodeling (21, 45–47, 116–118), 4) inhibition of RV hypertrophy and fibrosis (34, 35, 37, 38, 55, 119–123), 5) inhibition of platelet activation (124–126) and thrombosis; 6) inhibition of lung fibrosis (125) and edema (127), and 7) inhibition of inflammatory response by attenuating leukocyte transendothelial migration (128) and neutrophil chemotaxis (129, 130).
There are technical limitations using lung angiography to accurately determine pulmonary vasculature remodeling. The angiogram method, by determining the total length of lung vascular branches and the numbers of vascular branches and branch junctions, should be used with standard histological measurement of lung vascular wall thickness to quantitatively define the changes of pulmonary vascular structure in animal models with experimental PH. We will explore the possibility whether we can directly measure and compare the histological changes (e.g., wall thickness and muscularization) and lung angiography alterations in the same mouse using left (for angiogram via injection of Microfil through the left PA) and right (for histology) lungs. Nevertheless, we recently show that lung angiography parameters are consistent with the lung histological data (e.g., lung vascular wall thickness) in demonstrating pulmonary vascular remodeling in mice with experimental PH (59). We believe that the ability to exhibit or examine the entire pulmonary vasculature via lung angiography is an important method to indicate distal pulmonary arterial remodeling in comparison with the standard histological method in small animal models of experimental PH.
In conclusion, inhibition of TRPC6, a receptor-operated and mechanosensitive nonselective cation channel that is expressed in various types of cells, is a good strategy to develop novel therapeutic approach for PAH and PH in addition to its potential therapeutic effect on cancer (131) and Alzheimer’s disease (132). Oral administration of BI-794327, a newly developed specific blocker of TRPC6 channels, can effectively reverse, at least in part, established experimental PH in mice. Oral administration of BI-794327 not only improves pulmonary hemodynamics (e.g., RVSP, a surrogate measure of pulmonary arterial systolic pressure), but also alleviates pulmonary vascular remodeling in mice with established experimental PH. More clinical studies in patients with PH are needed to define the therapeutic potential of inhibition of TRPC6 channels or BI-749327 (a selective TRPC6 antagonist) on different subgroups of PH.
GRANTS
This study is supported in part by grants from the National Lung, Heart, and Blood Institute of the National Institutes of Health (HL135807 and HL142214) and A.B. was supported by the American Heart Association Postdoctoral Fellowship (20POST35210959).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.P.J., M.X., A.M., and J.X.-J.Y. and conceived and designed research; P.P.J., N.L., M.X., J.C., A.B., T.Z., S.P., M.Z., C.P., M.M., R.P., and A.B. performed experiments; P.P.J., N.L., M.X., J.C., A.B., T.Z., S.P., C.P., M.M., R.P., A.B., N.H.K., D.V.-J., P.A.T., J.Y-J.S., J.W., J.G.N.G., A.M., and J.X.-J.Y. analyzed data; P.P.J., N.L., M.X., J.C., A.B., T.Z., M.Z., C.P., M.M., R.P., A.B., N.H.K., D.V.-J., P.A.T., J.Y.-J.S., J.W., J.G.N.G., A.M., and J.X.-J.Y. interpreted results of experiments; P.P.J., M.X., J.C., A.B., T.Z., A.B., J.W., A.M., and J.X.-J.Y., prepared figures; P.P.J., N.L., M.X., and J.X.-J.Y. drafted manuscript; P.P.J., N.L., J.C., A.B., T.Z., S.P., M.Z., C.P., M.M., R.P., A.B., N.H.K., D.V.-J., P.A.T., J.Y.-J.S., J.W., J.G.N.G., A.M., and J.X.-J.Y. edited and revised manuscript; P.P.J., N.L., M.X., J.C., A.B., T.Z., S.P., M.Z., C.P., M.M., R.P., A.B., N.H.K., D.V.-J., P.A.T., J.Y.-J.S., J.W., J.G.N.G., A.M., and J.X.-J.Y. approved final version of manuscript.
REFERENCES
- 1.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 351: 1425–1436, 2004. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 2.Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142: 448–456, 2012. doi: 10.1378/chest.11-1460. [DOI] [PubMed] [Google Scholar]
- 3.Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, Benza RL. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest 148: 1043–1054, 2015. doi: 10.1378/chest.15-0300. [DOI] [PubMed] [Google Scholar]
- 4.Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, Krichman A, Liou TG, Raskob GE, Wason P, Feldkircher K, Turner M, McGoon MD. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US Contemporary Registries. Chest 139: 128–137, 2011. doi: 10.1378/chest.10-0075. [DOI] [PubMed] [Google Scholar]
- 5.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 54: S10–S19, 2009. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53: 1801887, 2019. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren X, Johns RA, Gao WD. Right heart in pulmonary hypertension: from adaptation to failure. Pulm Circ 9: 204589401984561, 2019. doi: 10.1177/2045894019845611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 109: 1239–1244, 2012. doi: 10.1073/pnas.1120385109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 304: H1708–H1718, 2013. doi: 10.1152/ajpheart.00746.2012. [DOI] [PubMed] [Google Scholar]
- 11.Corris PA, Nikkho S, Fernandes P. The innovative drug development initiative (IDDI) of the Pulmonary Vascular Research Institute (PVRI): a series of four guidance documents for academics, drug regulators and industry partners. Pulm Circ 10: 2045894020963031, 2020. doi: 10.1177/2045894020963031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 301: H1798–H1809, 2011. doi: 10.1152/ajpheart.00654.2011. [DOI] [PubMed] [Google Scholar]
- 13.Shen H, Zhang J, Wang C, Jain PP, Xiong M, Shi X, Lei Y, Chen S, Yin Q, Thistlethwaite PA, Wang J, Gong K, Yuan ZY, Yuan JX, Shyy JY. MDM2-mediated ubiquitination of angiotensin-converting enzyme 2 contributes to the development of pulmonary arterial hypertension. Circulation 142: 1190–1204, 2020. doi: 10.1161/CIRCULATIONAHA.120.048191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sodimu A, Bartolome S, Igenoza OP, Chin KM. Hemodynamic effects of fluoxetine in pulmonary arterial hypertension: an open label pilot study. Pulm Circ 10: 2045894020971954, 2020. doi: 10.1177/2045894020971954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toshner M, Spiekerkoetter E, Bogaard H, Hansmann G, Nikkho S, Prins KW. Repurposing of medications for pulmonary arterial hypertension. Pulm Circ 10: 2045894020941494, 2020. doi: 10.1177/2045894020941494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voelkel NF, Peters-Golden M. A new treatment for severe pulmonary arterial hypertension based on an old idea: inhibition of 5-lipoxygenase. Pulm Circ 10: 2045894019882635, 2020. doi: 10.1177/2045894019882635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Dong J, Martin M, He M, Gongol B, Marin TL, Chen L, Shi X, Yin Y, Shang F, Wu Y, Huang HY, Zhang J, Zhang Y, Kang J, Moya EA, Huang HD, Powell FL, Chen Z, Thistlethwaite PA, Yuan ZY, Shyy JY. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am J Respir Crit Care Med 198: 509–520, 2018. doi: 10.1164/rccm.201712-2570OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 19.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature 372: 231–236, 1994. [Erratum in Nature 372: 812, 1994]. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 20.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280: H746–H755, 2001. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 21.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 22.Song MY, Makino A, Yuan JX. STIM2 contributes to enhanced store-operated Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 1: 84–94, 2011. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283: L144–L155, 2002. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- 24.Ntokou A, Dave JM, Kauffman AC, Sauler M, Ryu C, Hwa J, Herzog EL, Singh I, Saltzman WM, Greif DM. Macrophage-derived PDGF-B induces muscularization in murine and human pulmonary hypertension. JCI Insight 6: e139067, 2021. doi: 10.1172/jci.insight.139067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 7: 308ra159, 2015. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep 23: 1152–1165, 2018. doi: 10.1016/j.celrep.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev 96: 1261–1296, 2016. doi: 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- 28.Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012. doi: 10.1152/ajpheart.00944.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai Y, Yu X, Chen H, Horne D, White R, Wu X, Lee P, Gu Y, Ghimire-Rijal S, Lin DC, Huang X. Structural basis for pharmacological modulation of the TRPC6 channel. eLife 9: e53311, 2020. doi: 10.7554/eLife.53311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clapham DE, Runnels LW, Strubing C. The TRP ion channel family. Nat Rev Neurosci 2: 387–396, 2001. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 31.Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 95: 645–690, 2015. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res 104: 1399–1409, 2009. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 33.Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circ Res 99: 119–131, 2006. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- 34.Kinoshita H, Kuwahara K, Nishida M, Jian Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, Li Y, Nakagawa Y, Usami S, Fujiwara M, Yamada Y, Minami T, Ueshima K, Nakao K. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res 106: 1849–1860, 2010. doi: 10.1161/CIRCRESAHA.109.208314. [DOI] [PubMed] [Google Scholar]
- 35.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 116: 3114–3126, 2006. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M, Nakaya M, Sato Y, Kita S, Iwamoto T, Hirano K, Inoue R, Kurose H. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler Thromb Vasc Biol 31: 2278–2286, 2011. doi: 10.1161/ATVBAHA.110.221010. [DOI] [PubMed] [Google Scholar]
- 37.Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 25: 5305–5316, 2006. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, Marino JP Jr, Birnbaumer L, Schnackenberg CG, Kass DA. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci USA 111: 1551–1556, 2014. [Erratum in Proc Natl Acad Sci USA 111: 6115, 2014]. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol 586: 4209–4223, 2008. doi: 10.1113/jphysiol.2008.156083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez-Miguel I, Cidad P, Perez-Garcia MT, Lopez-Lopez JR. Differences in TRPC3 and TRPC6 channels assembly in mesenteric vascular smooth muscle cells in essential hypertension. J Physiol 595: 1497–1513, 2017. doi: 10.1113/JP273327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279: 18887–18894, 2004. doi: 10.1074/jbc.M311274200. [DOI] [PubMed] [Google Scholar]
- 42.Itsuki K, Imai Y, Hase H, Okamura Y, Inoue R, Mori MX. PLC-mediated PI(4,5)P2 hydrolysis regulates activation and inactivation of TRPC6/7 channels. J Gen Physiol 143: 183–201, 2014. doi: 10.1085/jgp.201311033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG, Muallem S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 42: 205–211, 2007. doi: 10.1016/j.ceca.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9: 636–645, 2007. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. doi: 10.1161/CIRCULATIONAHA.108.782458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 48.Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. doi: 10.1165/rcmb.2014-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F, Gudermann T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci USA 103: 19093–19098, 2006. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bon RS, Beech DJ. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels—mirage or pot of gold? Br J Pharmacol 170: 459–474, 2013. doi: 10.1111/bph.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harteneck C, Gollasch M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr Pharm Biotechnol 12: 35–41, 2011. doi: 10.2174/138920111793937943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hofer A, Kovacs G, Zappatini A, Leuenberger M, Hediger MA, Lochner M. Design, synthesis and pharmacological characterization of analogs of 2-aminoethyl diphenylborinate (2-APB), a known store-operated calcium channel blocker, for inhibition of TRPV6-mediated calcium transport. Bioorg Med Chem 21: 3202–3213, 2013. doi: 10.1016/j.bmc.2013.03.037. [DOI] [PubMed] [Google Scholar]
- 53.Schild A, Bhardwaj R, Wenger N, Tscherrig D, Kandasamy P, Dernic J, Baur R, Peinelt C, Hediger MA, Lochner M. Synthesis and pharmacological characterization of 2-aminoethyl diphenylborinate (2-APB) derivatives for inhibition of store-operated calcium entry (SOCE) in MDA-MB-231 breast cancer cells. Int J Mol Sci 21: 5604, 2020. doi: 10.3390/ijms21165604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maier T, Follmann M, Hessler G, Kleemann HW, Hachtel S, Fuchs B, Weissmann N, Linz W, Schmidt T, Lohn M, Schroeter K, Wang L, Rutten H, Strubing C. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br J Pharmacol 172: 3650–3660, 2015. doi: 10.1111/bph.13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S, Bian H, Zeveleva S, Zhen X, Blair NT, Chong JA, Hessler DP, Bedja D, Zhu G, Muller GK, Ranek MJ, Pantages L, McFarland M, Netherton MR, Berry A, Wong D, Rast G, Qian HS, Weldon SM, Kuo JJ, Sauer A, Sarko C, Moran MM, Kass DA, Pullen SS. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci USA 116: 10156–10161, 2019. doi: 10.1073/pnas.1815354116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jain PP, Hosokawa S, Xiong M, Babicheva A, Zhao T, Rodriguez M, Rahimi S, Pourhashemi K, Balistrieri F, Lai N, Malhotra A, Shyy JY, Valdez-Jasso D, Thistlethwaite PA, Makino A, Yuan JX. Revisiting the mechanism of hypoxic pulmonary vasoconstriction using isolated perfused/ventilated mouse lung. Pulm Circ 10: 2045894020956592, 2020. doi: 10.1177/2045894020956592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoo HY, Zeifman A, Ko EA, Smith KA, Chen J, Machado RF, Zhao YY, Minshall RD, Yuan JX. Optimization of isolated perfused/ventilated mouse lung to study hypoxic pulmonary vasoconstriction. Pulm Circ 3: 396–405, 2013. doi: 10.4103/2045-8932.114776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jain PP, Zhao T, Xiong M, Song S, Lai N, Zheng Q, Chen J, Carr SG, Babicheva A, Izadi A, Rodriguez M, Rahimi S, Balistrieri F, Rahimi S, Simonson T, Valdez-Jasso D, Thistlethwaite PA, Shyy JY, Wang J, Makino A, Yuan JX. Halofuginone, a promising drug for treatment of pulmonary hypertension. Br J Pharmacol 178: 3373–3394, 2021. doi: 10.1111/bph.15442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiong M, Jain PP, Chen J, Babicheva A, Zhao T, Alotaibi M, Kim NH, Lai N, Izadi A, Rodriguez M, Li J, Balistrieri A, Balistrieri F, Parmisano S, Sun X, Voldez-Jasso D, Shyy JY, Thistlethwaite PA, Wang J, Makino A, Yuan JX. Mouse model of experimental pulmonary hypertension: lung angiogram and right heart catheterization. Pulm Circ 11: 20458940211041512, 2021. doi: 10.1177/20458940211041512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Z, Wang Y, Long A, Feng T, Ocampo M, Chen S, Tang H, Guo Q, Minshall R, Makino A, Huang W, Chen J. Pulmonary vessel casting in a rat model of monocrotaline-mediated pulmonary hypertension. Pulm Circ 10: 2045894020922129, 2020. doi: 10.1177/2045894020922129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soliman EM, Rodrigues MA, Gomes DA, Sheung N, Yu J, Amaya MJ, Nathanson MH, Dranoff JA. Intracellular calcium signals regulate growth of hepatic stellate cells via specific effects on cell cycle progression. Cell Calcium 45: 284–292, 2009. doi: 10.1016/j.ceca.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song S, Ayon RJ, Yamamura A, Yamamura H, Dash S, Babicheva A, Tang H, Sun X, Cordery AG, Khalpey Z, Black SM, Desai AA, Rischard F, McDermott KM, Garcia JG, Makino A, Yuan JX. Capsaicin-induced Ca2+ signaling is enhanced via upregulated TRPV1 channels in pulmonary artery smooth muscle cells from patients with idiopathic PAH. Am J Physiol Lung Cell Mol Physiol 312: L309–L325, 2017. doi: 10.1152/ajplung.00357.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song S, Jacobson KN, McDermott KM, Reddy SP, Cress AE, Tang H, Dudek SM, Black SM, Garcia JG, Makino A, Yuan JX. ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells. Am J Physiol Cell Physiol 310: C99–C114, 2016. doi: 10.1152/ajpcell.00092.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mammano F, Bortolozzi M. Ca2+ imaging: principles of analysis and enhancement. In: Calcium Measurement Methods, Neuromethods, edited by Verkhratsky A, Petersen OH.. New York, NY: Springer, 2010, p. 57–80. [Google Scholar]
- 65.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284: C316–C330, 2003. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- 66.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 67.McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JX. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol 280: L870–L880, 2001. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- 68.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11–21, 2000. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 69.Goncharova EA, Simon MA, Yuan JX. mTORC1 in pulmonary arterial hypertension. At the crossroads between vasoconstriction and vascular remodeling? Am J Respir Crit Care Med 201: 1177–1179, 2020. doi: 10.1164/rccm.202001-0087ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song S, Carr SG, McDermott KM, Rodriguez M, Babicheva A, Balistrieri A, Ayon RJ, Wang J, Makino A, Yuan JX. STIM2 (stromal interaction molecule 2)-mediated increase in resting cytosolic free Ca2+ concentration stimulates PASMC proliferation in pulmonary arterial hypertension. Hypertension 71: 518–529, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xi Q, Adebiyi A, Zhao G, Chapman KE, Waters CM, Hassid A, Jaggar JH. IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ Res 102: 1118–1126, 2008. [Erratum in Circ Res 105: e1, 2009]. doi: 10.1161/CIRCRESAHA.108.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chemla D, Castelain V, Humbert M, Hebert JL, Simonneau G, Lecarpentier Y, Herve P. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest 126: 1313–1317, 2004. doi: 10.1378/chest.126.4.1313. [DOI] [PubMed] [Google Scholar]
- 73.Parasuraman S, Walker S, Loudon BL, Gollop ND, Wilson AM, Lowery C, Frenneaux MP. Assessment of pulmonary artery pressure by echocardiography—a comprehensive review. Int J Cardiol Heart Vasc 12: 45–51, 2016. doi: 10.1016/j.ijcha.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol 559: 685–706, 2004. [Erratum in J Physiol 560: 949, 2004]. doi: 10.1113/jphysiol.2004.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J 16: 1145–1150, 2002. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- 76.Boulay G. Ca2+-calmodulin regulates receptor-operated Ca2+ entry activity of TRPC6 in HEK-293 cells. Cell Calcium 32: 201–207, 2002. doi: 10.1016/s0143416002001550. [DOI] [PubMed] [Google Scholar]
- 77.Chen GL, Zeng B, Eastmond S, Elsenussi SE, Boa AN, Xu SZ. Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. Br J Pharmacol 167: 1232–1243, 2012. doi: 10.1111/j.1476-5381.2012.02058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. 2-Aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J Neurosci 24: 5177–5182, 2004. doi: 10.1523/JNEUROSCI.0934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Togashi K, Inada H, Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol 153: 1324–1330, 2008. doi: 10.1038/sj.bjp.0707675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu SZ, Zeng F, Boulay G, Grimm C, Harteneck C, Beech DJ. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br J Pharmacol 145: 405–414, 2005. doi: 10.1038/sj.bjp.0706197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allbritton NL, Oancea E, Kuhn MA, Meyer T. Source of nuclear calcium signals. Proc Natl Acad Sci USA 91: 12458–12462, 1994. doi: 10.1073/pnas.91.26.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blaustein MP. The pump, the exchanger, and the holy spirit: origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system. Am J Physiol Cell Physiol 314: C3–C26, 2018. doi: 10.1152/ajpcell.00196.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science 275: 1643–1648, 1997. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- 84.Golovina VA, Blaustein MP. Unloading and refilling of two classes of spatially resolved endoplasmic reticulum Ca2+ stores in astrocytes. Glia 31: 15–28, 2000. doi: 10.1002/(SICI)1098-1136(200007)31:1<15::AID-GLIA20>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 85.Berridge MJ. Calcium signalling and cell proliferation. Bioessays 17: 491–500, 1995. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- 86.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Rande JL, Amsellem V, Adnot S. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol 48: 568–577, 2013. doi: 10.1165/rcmb.2012-0429OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miyazaki T, Honda K, Ohata H. Requirement of Ca2+ influx- and phosphatidylinositol 3-kinase-mediated m-calpain activity for shear stress-induced endothelial cell polarity. Am J Physiol Cell Physiol 293: C1216–C1225, 2007. doi: 10.1152/ajpcell.00083.2007. [DOI] [PubMed] [Google Scholar]
- 88.Ogawa A, Firth AL, Ariyasu S, Yamadori I, Matsubara H, Song S, Fraidenburg DR, Yuan JX. Thrombin-mediated activation of Akt signaling contributes to pulmonary vascular remodeling in pulmonary hypertension. Physiol Rep 1: e00190, 2013. doi: 10.1002/phy2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 4: 64, 2014. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501, 2002. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 91.Wang Q, Wang X, Hernandez A, Hellmich MR, Gatalica Z, Evers BM. Regulation of TRAIL expression by the phosphatidylinositol 3-kinase/Akt/GSK-3 pathway in human colon cancer cells. J Biol Chem 277: 36602–36610, 2002. doi: 10.1074/jbc.M206306200. [DOI] [PubMed] [Google Scholar]
- 92.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129: 864–874, 2014. doi: 10.1161/CIRCULATIONAHA.113.004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. doi: 10.1152/ajplung.00242.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tang H, Wu K, Wang J, Vinjamuri S, Gu Y, Song S, Wang Z, Zhang Q, Balistrieri A, Ayon RJ, Rischard F, Vanderpool R, Chen J, Zhou G, Desai AA, Black SM, Garcia JGN, Yuan JX, Makino A. Pathogenic role of mTORC1 and mTORC2 in pulmonary hypertension. JACC Basic Transl Sci 3: 744–762, 2018. doi: 10.1016/j.jacbts.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest 115: 2691–2694, 2005. doi: 10.1172/JCI26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 302: C405–C411, 2012. doi: 10.1152/ajpcell.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ten Freyhaus H, Berghausen EM, Janssen W, Leuchs M, Zierden M, Murmann K, Klinke A, Vantler M, Caglayan E, Kramer T, Baldus S, Schermuly RT, Tallquist MD, Rosenkranz S. Genetic ablation of PDGF-dependent signaling pathways abolishes vascular remodeling and experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol 35: 1236–1245, 2015. doi: 10.1161/ATVBAHA.114.304864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berridge MJ. Smooth muscle cell calcium activation mechanisms. J Physiol 586: 5047–5061, 2008. doi: 10.1113/jphysiol.2008.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mauban JR, Remillard CV, Yuan JX. Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol (1985) 98: 415–420, 2005. doi: 10.1152/japplphysiol.00732.2004. [DOI] [PubMed] [Google Scholar]
- 101.Sweeney M, Yuan JX. Hypoxic pulmonary vasoconstriction: role of voltage-gated potassium channels. Respir Res 1: 40–48, 2000. doi: 10.1186/rr11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen X, Sooch G, Demaree IS, White FA, Obukhov AG. Transient receptor potential canonical (TRPC) channels: then and now. Cells 9: 1983, 2020. doi: 10.3390/cells9091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Remillard CV, Yuan JX. TRP channels, CCE, and the pulmonary vascular smooth muscle. Microcirculation 13: 671–692, 2006. doi: 10.1080/10739680600930313. [DOI] [PubMed] [Google Scholar]
- 104.Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honore E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455: 1097–1103, 2008. doi: 10.1007/s00424-007-0359-3. [DOI] [PubMed] [Google Scholar]
- 105.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 106.Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem 279: 35741–35748, 2004. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 107.Hendron E, Wang X, Zhou Y, Cai X, Goto J, Mikoshiba K, Baba Y, Kurosaki T, Wang Y, Gill DL. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium 56: 482–492, 2014. doi: 10.1016/j.ceca.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bilmen JG, Wootton LL, Godfrey RE, Smart OS, Michelangeli F. Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+-binding sites. Eur J Biochem 269: 3678–3687, 2002. doi: 10.1046/j.1432-1033.2002.03060.x. [DOI] [PubMed] [Google Scholar]
- 109.Lemonnier L, Prevarskaya N, Mazurier J, Shuba Y, Skryma R. 2-APB inhibits volume-regulated anion channels independently from intracellular calcium signaling modulation. FEBS Lett 556: 121–126, 2004. doi: 10.1016/s0014-5793(03)01387-5. [DOI] [PubMed] [Google Scholar]
- 110.Rae MG, Hilton J, Sharkey J. Putative TRP channel antagonists, SKF 96365, flufenamic acid and 2-APB, are non-competitive antagonists at recombinant human a1b2g2 GABA(A) receptors. Neurochem Int 60: 543–554, 2012. doi: 10.1016/j.neuint.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 111.Fuchs B, Rupp M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Gudermann T, Dietrich A, Weissmann N. Diacylglycerol regulates acute hypoxic pulmonary vasoconstriction via TRPC6. Respir Res 12: 20, 2011. doi: 10.1186/1465-9921-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Urban N, Hill K, Wang L, Kuebler WM, Schaefer M. Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell Calcium 51: 194–206, 2012. doi: 10.1016/j.ceca.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 113.Imai Y, Itsuki K, Okamura Y, Inoue R, Mori MX. A self-limiting regulation of vasoconstrictor-activated TRPC3/C6/C7 channels coupled to PI(4,5)P(2)-diacylglycerol signalling. J Physiol 590: 1101–1119, 2012. doi: 10.1113/jphysiol.2011.221358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA 103: 16586–16591, 2006. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ichikawa J, Inoue R. TRPC6 regulates cell cycle progression by modulating membrane potential in bone marrow stromal cells. Br J Pharmacol 171: 5280–5294, 2014. doi: 10.1111/bph.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Numaga-Tomita T, Shimauchi T, Oda S, Tanaka T, Nishiyama K, Nishimura A, Birnbaumer L, Mori Y, Nishida M. TRPC6 regulates phenotypic switching of vascular smooth muscle cells through plasma membrane potential-dependent coupling with PTEN. FASEB J 33: 9785–9796, 2019. doi: 10.1096/fj.201802811R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ogawa A, Firth AL, Yao W, Rubin LJ, Yuan JX. Prednisolone inhibits PDGF-induced nuclear translocation of NF-kappaB in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 295: L648–L657, 2008. doi: 10.1152/ajplung.90245.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tang H, Yamamura A, Yamamura H, Song S, Fraidenburg DR, Chen J, Gu Y, Pohl NM, Zhou T, Jimenez-Perez L, Ayon RJ, Desai AA, Goltzman D, Rischard F, Khalpey Z, Black SM, Garcia JG, Makino A, Yuan JX. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L846–L859, 2016. doi: 10.1152/ajplung.00050.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kurahara LH, Hiraishi K, Sumiyoshi M, Doi M, Hu Y, Aoyagi K, Jian Y, Inoue R. Significant contribution of TRPC6 channel-mediated Ca2+ influx to the pathogenesis of Crohn's disease fibrotic stenosis. J Smooth Muscle Res 52: 78–92, 2016. doi: 10.1540/jsmr.52.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, Inoue R, Mori Y, Kurose H. Ga12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem 282: 23117–23128, 2007. doi: 10.1074/jbc.M611780200. [DOI] [PubMed] [Google Scholar]
- 121.Oda S, Numaga-Tomita T, Kitajima N, Toyama T, Harada E, Shimauchi T, Nishimura A, Ishikawa T, Kumagai Y, Birnbaumer L, Nishida M. TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice. Sci Rep 7: 7511, 2017. doi: 10.1038/s41598-017-07903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun 3: 1238, 2012. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamaguchi Y, Iribe G, Nishida M, Naruse K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: linking physiology and pathophysiology. Prog Biophys Mol Biol 130: 264–272, 2017. doi: 10.1016/j.pbiomolbio.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 124.Dionisio N, Albarran L, Berna-Erro A, Hernandez-Cruz JM, Salido GM, Rosado JA. Functional role of the calmodulin- and inositol 1,4,5-trisphosphate receptor-binding (CIRB) site of TRPC6 in human platelet activation. Cell Signal 23: 1850–1856, 2011. doi: 10.1016/j.cellsig.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 125.Ortiz-Munoz G, Yu MA, Lefrancais E, Mallavia B, Valet C, Tian JJ, Ranucci S, Wang KM, Liu Z, Kwaan N, Dawson D, Kleinhenz ME, Khasawneh FT, Haggie PM, Verkman AS, Looney MR. Cystic fibrosis transmembrane conductance regulator dysfunction in platelets drives lung hyperinflammation. J Clin Invest 130: 2041–2053, 2020. doi: 10.1172/JCI129635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vemana HP, Karim ZA, Conlon C, Khasawneh FT. A critical role for the transient receptor potential channel type 6 in human platelet activation. PLoS One 10: e0125764, 2015. doi: 10.1371/journal.pone.0125764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun 3: 649, 2012. doi: 10.1038/ncomms1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med 212: 1883–1899, 2015. doi: 10.1084/jem.20150353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lindemann O, Umlauf D, Frank S, Schimmelpfennig S, Bertrand J, Pap T, Hanley PJ, Fabian A, Dietrich A, Schwab A. TRPC6 regulates CXCR2-mediated chemotaxis of murine neutrophils. J Immunol 190: 5496–5505, 2013. doi: 10.4049/jimmunol.1201502. [DOI] [PubMed] [Google Scholar]
- 130.Thippegowda PB, Singh V, Sundivakkam PC, Xue J, Malik AB, Tiruppathi C. Ca2+ influx via TRPC channels induces NF-kB-dependent A20 expression to prevent thrombin-induced apoptosis in endothelial cells. Am J Physiol Cell Physiol 298: C656–C664, 2010. doi: 10.1152/ajpcell.00456.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stewart JM. TRPV6 as a target for cancer therapy. J Cancer 11: 374–387, 2020. doi: 10.7150/jca.31640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Prikhodko V, Chernyuk D, Sysoev Y, Zernov N, Okovityi S, Popugaeva E. Potential drug candidates to treat TRPC6 channel deficiencies in the pathophysiology of Alzheimer's disease and brain ischemia. Cells 9: 2351, 2020. doi: 10.3390/cells9112351. [DOI] [PMC free article] [PubMed] [Google Scholar]