

Abstract
Antioxidant signaling/communication is among the most important cellular defense and survival pathways, and the importance of redox signaling and homeostasis in aging has been well-documented. Intracellular levels of glutathione (GSH), a very important endogenous antioxidant, both govern and are governed by the Nrf2 pathway through expression of genes involved in its biosynthesis, including the subunits of the rate-limiting enzyme (glutamate cysteine ligase, GCL) in GSH production, GCLC and GCLM. Mice homozygous null for the Gclm gene are severely deficient in GSH compared to wild-type controls, expressing approximately 10% of normal GSH levels. To compensate for GSH deficiency, Gclm null mice have upregulated redox-regulated genes, and, surprisingly, are less susceptible to certain types of oxidative damage. Furthermore, young Gclm null mice display an interesting lean phenotype, resistance to high fat diet-induced diabetes and obesity, improved insulin and glucose tolerance, and decreased expression of genes involved in lipogenesis. However, the persistence of this phenotype has not been investigated into old age, which is important in light of studies which suggest aging attenuates antioxidant signaling, particularly in response to exogenous stimuli. In this work, we addressed whether aging compromises the favorable phenotype of increased antioxidant activity and improved glucose homeostasis observed in younger Gclm null mice. We present data showing that under basal conditions and in response to cadmium exposure (2 mg/kg, dosed once via intraperitoneal injection), the phenotype previously described in young (<6 months) Gclm null mice persists into old age (24+ months). We also provide evidence that transcriptional activation of the Nrf2, AMPK, and PPARγ pathways underlie the favorable metabolic phenotype observed previously in young Gclm null mice.
Keywords: Glutathione, NRF2, Diabetes, Glucose homeostasis, Insulin resistance, Aging
Graphical abstract
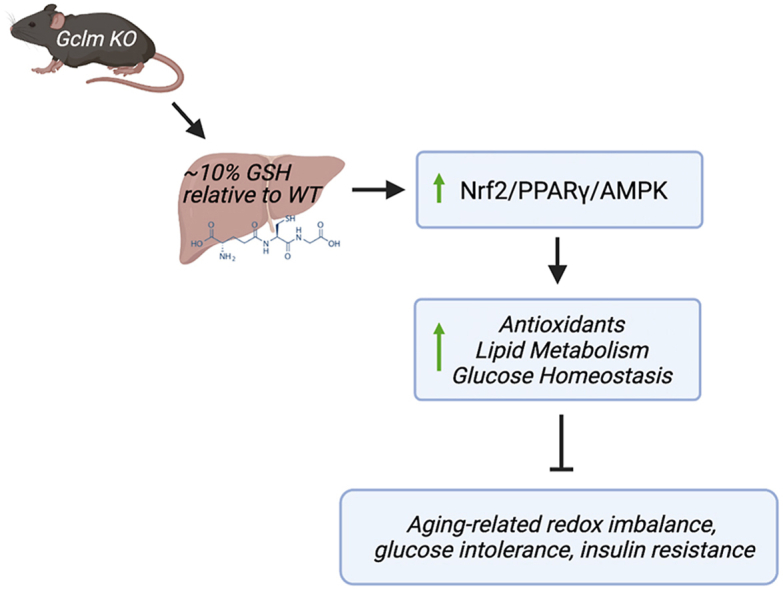
Highlights
-
•
Young Gclm-/- mice have lower body weight, more insulin sensitivity, and glucose tolerance relative to young Gclm+/+ mice.
-
•
The improved glucose homeostasis in young Gclm-/- mice under basal conditions and cadmium treatment are maintained in old age.
-
•
Gclm-/- mice have increased Nrf2-regulated genes and beta oxidation; and decreased gluconeogenesis and fatty acid synthesis.
1. Introduction
Type II diabetes mellitus (T2DM) is an increasingly important public health problem [1]. T2DM is often attributable to insulin resistance resulting from impaired insulin secretion and/or action, and is associated with many co-morbidities including cerebral vascular disease and stroke, peripheral neuropathies, coronary artery disease, retinopathy, and nephropathy, among other complications [[2], [3], [4]]. A link between redox homeostasis and T2DM is supported by many studies [[5], [6], [7]]. Activity of the redox-responsive transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), a master regulator of antioxidant response, is decreased in the nuclear fraction of peripheral blood mononuclear cells (PBMCs) in patients with T2DM, and studies in mice have demonstrated that mild Nrf2 overexpression or Nrf2 activation via small molecule stimulators (e.g., sulforaphane) can prevent T2DM onset [8,9]. Hyperglycemia, a clinical characteristic of T2DM, is associated with reactive oxygen species (ROS) and advanced glycation end-product (AGE) production, creating a feedback loop of increasing oxidative stress [[10], [11], [12], [13], [14]]. While the referenced studies did not explicitly identify which specific ROS are generated in a hyperglycemic state (since the fluorescent probes used are not that specific for particular oxidants), the results of these studies nonetheless support the conclusion that hyperglycemia is associated with ROS generation. Because oxidative stress is believed to contribute significantly to TD2M progression, it follows that reducing oxidative insult via activation of antioxidant pathways could potentially prevent or delay the onset of TD2M. Animal models and clinical studies aimed at assessing the role of Nrf2 and its regulator Keap1 (Kelch-like erythroid cell-derived protein with CNC homology [ECH]-associated protein 1) and other antioxidant systems in T2DM have yielded mixed results, and strongly suggest a delicate balance in antioxidant activity and crosstalk with other signaling pathways is needed to maintain normal glucose homeostasis. Regardless, redox homeostasis remains an attractive therapeutic target for T2DM.
Central to cellular redox homeostasis is the thiol-based, redox buffering glutathione (GSH) system. GSH is a tripeptide comprised of the amino acids glutamate, cysteine, and glycine, and is synthesized de novo using a two-step, ATP-dependent enzymatic process [15]. The first and rate-limiting step involves the conjugation of glutamate with cysteine by the enzyme glutamate-cysteine ligase (GCL; also known as γ-glutamylcysteine synthetase) to form gamma-glutamylcysteine (γ-GC) [15,16]. GCL is comprised of two subunits: GCLC and GCLM. GCLC provides all the catalytic activity for GCL, while GCLM allows for more efficient and rapid γ-GC formation by lowering the Km for glutamate and ATP, while also increasing the Ki for GSH feedback inhibition [16]. The second enzymatic step of GSH biosynthesis is the conjugation of glycine to γ-GC by the enzyme glutathione synthetase (GS). GSH can also regulate its own production through negative feedback inhibition of GCL activity when cellular GSH levels are sufficient.
Homozygous deletion of the Gclc gene in mice results in embryonic lethality [17]. However, while mice with homozygous deletion of the Gclm gene (Gclm null mice) are healthy and viable, young Gclm null mice still display low levels of GSH in their tissues (∼10–20%) compared to their Gclm wild-type litter mates [18]. Given GSH's central role in cellular redox homeostasis and antioxidant defense, one would expect Gclm null mice to be sensitive to pathophysiological conditions involving environmental and oxidative stress. While this holds true for some stressors which specifically require the GSH molecule for detoxification, such as acetaminophen-induced liver injury [18], Gclm null mice are unexpectedly resistant to many stressors, including ozone, diesel exhaust, certain nanoparticle exposures, methionine and choline-deficient (MCD) diet-induced nonalcoholic steatohepatitis (NASH)-like liver injury, 2,3,7,8-tetrachlorodibenzodioxin (TCDD)-induced steatosis, and dietary alcohol exposure [[19], [20], [21], [22], [23], [24], [25]]. In contrast, Gclm heterozygous mice are more sensitive than Gclm wild-type and null mice to some of the aforementioned stressors.
Previously published studies by colleagues at the University of Cincinnati, the University of Colorado, and Yale University, who have also developed and investigated a Gclm null mouse model that closely mirrors the phenotypes of our Gclm null mice, showed that young (∼10 weeks) Gclm null mice exhibit improved measures of glucose homeostasis relative to Gclm wild-type mice, as well as a lean phenotype which is resistant to HFD-induced obesity and development of T2DM [[26], [27], [28]]. Another study examining Gclm null mice found they display reduced hepatic glycogen storage [29]. This finding stands in contrast to mice with hepatocyte-specific Txnrd1-knockdown (another model of chronic thiol insufficiency), which display increased hepatic glycogen storage [30]. Derivation methods notwithstanding, all current Gclm null mouse models display increased activity of the oxidative stress-responsive and metabolic reprogramming transcription factor Nrf2 and subsequently, increased expression of Nrf2-regulated genes [18,20,27,31].
The lean phenotype observed in Gclm null mice likely results from a switch in hepatic energy utilization in response to GSH deficiency. Gclm null mice have fewer circulating free fatty acids relative to their wild-type counterparts (unpublished data, TJK, U of Washington), while in the liver, Gclm null mice display decreased expression of genes involved in fatty acid synthesis and gluconeogenesis, and increased expression of genes involved in glycogenesis, lipid catabolism, and lipid trafficking [20,22,27,32]. The dysregulation of these pathways relative to WT mice may be central to the T2DM- and obesity-resistant phenotype observed in young Gclm null mice. Notably, Nrf2, the most highly upregulated pathway in Gclm null mice [19,20,22,27] is believed to have a protective effect in delaying T2DM and metabolic dysfunction, including impaired glucose homeostasis, and mounting evidence suggests that Nrf2 is an important mediator between redox/cytoprotective defense and intermediary metabolism [8,33]. Further evidence as to the importance of Nrf2 in modulating glucose utilization is provided by the observation that mice with diminished methionine sulfoxide reductase B3 expression (another enzyme important in combating oxidative molecular damage) have upregulated Nrf2 in a compensatory manner and are resistant to high fat diet-induced insulin intolerance [34,35].
Accordingly, we surmised that compensatory hepatic expression of genes involved in cellular defense and energy pathways contribute significantly to the improved metabolic state observed in Gclm null mice. If such cytoprotective activities decline with age as has been previously reported [36,37], we expect that Gclm null mice would show functional signs of metabolic dysfunction (i.e. increased fasting blood glucose levels, impaired glucose tolerance, and decreased insulin sensitivity) relative to wild-type mice by 24+ months of age. Furthermore, because cadmium (Cd) has been shown to inhibit Nrf2 signaling [38] and because there is evidence that Cd may be a ‘diabetogen’ [[39], [40], [41]], we challenged mice with Cd to determine if such treatment could oppose Nrf2-mediated protection afforded by chronic Gclm deficiency.
The overarching aim of this study was to determine if the improved parameters of glucose and metabolic homeostasis observed in young Gclm null mice relative to wild-type mice (insulin sensitivity, glucose tolerance, lean phenotype) are maintained through old age and in response to cadmium treatment. We hypothesized that Gclm null mice would be unable to maintain a favorable metabolic phenotype into old age (24+ months) compared to wild-type and heterozygous mice, as previous research has shown that aging can result in decreased antioxidant expression and activity [36,37].
2. Materials and methods
2.1. Reagents
All reagents were purchased from Life Technologies (Carlsbad, CA) and/or Sigma-Aldrich (Saint Louis, MO), unless otherwise noted.
2.2. Animals
All animal experiments were performed in accordance with the National Institutes of Health Guide for the Use and Care of Laboratory Animals [42], and with the approval of the University of Washington Institutional Animal Care and Use Committee (IACUC; protocol #2384-08). We made all efforts to minimize animal distress and suffering. Gclm null mice were derived by homologous recombination techniques in mouse embryonic stem (ES) cells, as previously described [18]. The mice were back-crossed onto the C57BL/6J genetic background for at least 7 generations. Eight cohorts of male Gclm wild-type, heterozygous, and null mice were established representing four ages (10–13 weeks, 6 months, 18 months, and 24+ months) and two treatments (saline and 2 mg/kg cadmium chloride, intraperitoneal (i.p.) injection), with an n of 5–6 for each age, genotype, and treatment.
2.3. Treatments
One week following baseline glucose tolerance tests (GTT) and insulin tolerance tests (ITT), performed as previously described [27,29], mice were intraperitoneally injected with either 0.9% sterile saline, or 2 mg/kg cadmium chloride (CdCl2) in sterile saline (0.9% sodium chloride injection, USP; injection volume of 100–200 μl). Due to the dearth of toxicology studies examining acute heavy metal treatments, particularly cadmium, in aged mice, we performed a small dose-escalation study to determine an appropriate sublethal dose using 20 month-old Gclm mice. We sought to select an acute dose which also allowed us to perform functional tests of glucose homeostasis following dosing and prior to sacrifice. Based on our internal dose escalation study, and literature review of LD50 values for oral CdCl2 administration in different strains of laboratory mice ranging from 5 to 183 mg/kg [[43], [44], [45]], a 2 mg/kg CdCl2 dose of cadmium chloride was chosen. Ideally, a chronic dosing regimen would have been used to interrogate the effect of cadmium exposure. However, mice in some of the cohorts had already aged significantly, which precluded us from beginning a chronic dosing protocol.
Twenty-four hours after CdCl2 (or saline control) injection, a GTT was administered and after another 24 h, an ITT was administered. Mice were then sacrificed the following morning (∼66–72 h post-dosing). The aforementioned tests and timepoints were designed and chosen to maximize the data collected from each mouse without causing undo harm, while minimizing the number of mice needed.
2.4. Necropsies
Animals were sacrificed by placing them under CO2 narcosis, followed by cervical dislocation. Fresh blood was obtained via heart puncture and collected in plasma separator tubes, placed on ice, centrifuged at 8000 rpm (5939×g) on an Eppendorf 5415D microcentrifuge equipped with a F45-24-11 rotor for 10 min at room temperature, then stored at −20 °C. Following blood collection, tissues were dissected and either flash frozen in liquid nitrogen, fixed in 10% neutral buffered formalin, and/or embedded in OCT (Optimal Cutting Temperature) embedding compound (Sakura Finetek, Torrence, CA).
2.5. Assessment of glucose homeostasis
2.5.1. Blood glucose measurements
For each cohort, baseline data for functional measures of glucose homeostasis (GTT and ITT) were assessed. Prior to GTT, mice were fasted for 8 h from 6 a.m. to 2 p.m. At the time of testing, baseline blood glucose levels were taken, immediately followed by i.p. injection of 1.5 g glucose/kg body weight (D-(+)-Glucose, Sigma G8270, solubilized and sterile filtered in 0.9% sodium chloride for injection, USP) after which blood glucose was measured at 15, 30, 45, 60 and 120 min post-injection (Fig. 1); the glucometer and glucose strips were purchased from a retail pharmacy (Walgreen's, Inc., Seattle, WA).
Fig. 1.

(A) Overall design schematic for functional tests of glucose homeostasis, and (B) daily experimental schematic for blood glucose reading and plasma collection.
For 6 and 24+ month old Gclm wild-type and null mice, blood was collected at the time of baseline measurement for fasting plasma insulin analysis. The following day, mice were fasted for 4–6 h in preparation for an ITT (8–10 a.m. to 2 p.m.). Baseline blood glucose levels were measured, followed by IP injection of 1 U human insulin/kg body weight (Humulin R, Eli-Lilly, solubilized in 0.9% sodium chloride for injection, USP). Blood glucose was monitored at 15, 30, 45, 60 and 120 min post-insulin administration (Fig. 1).
2.5.2. Fasting plasma insulin
Plasma was collected from 6 to 24+ month Gclm wild-type and null mice by saphenous vein puncture. Plasma was separated from cellular components by centrifugation in heparin-coated tubes, then stored at −20 °C. Fasted plasma insulin levels were measured using the Mouse Ultrasensitive Insulin ELISA kit (ALPCO Diagnostics, Salem, NH).
2.6. Statistical analyses
Data are expressed as the mean and SEM, unless otherwise noted. Data were analyzed by one-, two-, or three-way ANOVA, followed by a Bonferroni post hoc test, or a Student's t-test (for pair-wise comparisons) using a statistical software package (Prism, GraphPad, Inc., San Diego, CA). Differences yielding a p-value of less than 0.05 were considered statistically significant.
2.7. High-throughput transcriptome analysis
Transcriptional analyses of liver RNA samples were performed using the TempO-Seq S1500 panel (BioSpyder Technologies), which interrogates expression of approximately 3000 genes (Table S1). Briefly, the TempO-Seq platform contains a set of paired detector oligos for each transcript that bind to the target transcript sequence. The bound oligos are then ligated, PCR amplified, and sequenced using a NextSeq sequencer (Illumina, San Diego, CA). Sequenced reads were aligned to the set of detector oligo sequences using the STAR aligner, allowing for two mismatches. Counts per gene were read into R (r-project.org) and analyzed using the limma-voom pipeline from the Bioconductor limma package [46]. Briefly, the counts per gene are normalized by library size (total counts per sample, in millions) and converted to log scale, resulting in log counts/million (logCPM). Observation and sample weights are then estimated from the data to account for the mean-variance dependence of the logCPM values and sample quality, respectively, and used in a conventional weighted analysis of variance model. Comparisons between groups were made using empirical Bayes adjusted contrasts [47]. The resulting p-values were adjusted for multiplicity using false discovery rate (FDR), and genes were selected at an FDR <0.05 [48]. Heatmaps were generated in R using the ComplexHeatmap package [49].
2.8. RT- PCR for measurement of metallothionein gene expression
Fluidigm GE Dynamic Array 96.96 plates (Fluidigm, Inc., South San Francisco, CA) were primed on the Fluidigm IFC Controller prior to loading. Four μl of each 10X assay mix were loaded onto each assay well and 5 μl of each Sample Pre-Mix were loaded onto each sample well of the GE Dynamic Array (Fluidigm). Assays were run in triplicate. The GE Dynamic Array was loaded onto the IFC Controller before thermocycling with the following profile: 98 °C for 40s followed by 40 cycles, consisting of 95 °C for 10s and 60 °C for 40s. Data were collected using the Fluidigm BioMark Data Collection Software 2.1.1 and analyzed using the Fluidigm Real-Time PCR Analysis Software 1.1.0.
3. Results
3.1. Body weight
The first parameter of metabolic health examined in this study was body weight. Gclm null mice display a leaner phenotype throughout old age compared to Gclm wildtype and heterozygous mice (Fig. 2). At 18 and 24+ months, this difference was statistically significant by two-way ANOVA, and the data also suggest that Gclm null mice display less variability in weight at 18 and 24+ months compared to wildtype and heterozygous mice. This suggests that the downregulation of fatty acid synthesis observed previously in young Gclm null mice [27] is likely maintained through 24+ months.
Fig. 2.

Body weights of Gclm WT and null mice at 12 weeks, and at 6, 18, and 24+ months (n=12–24 mice/group). Data analyzed using two-way ANOVA with Bonferroni corrections (n=12–23/genotype); *, p < 0.05; **, p < 0.001; ***, p < 0.0001. Data are shown as mean values ± the standard error of the mean (SEM).
3.2. Glucose homeostasis
Next, we examined functional parameters of glucose homeostasis—glucose tolerance and insulin sensitivity. At baseline, Gclm null mice were significantly more glucose tolerant than wild-type mice at 6 and 24+ months of age by AUC; this observation was maintained following saline and cadmium administration (Fig. 3). Regarding insulin sensitivity, Gclm null mice were significantly more sensitive than wild-type mice at baseline through 24+ months (Fig. 4). Insulin sensitivity among saline treated WT and null mice was not statistically different using two-way ANOVA at 6 or 24+ months when quantified by AUC. However, the general trend of null mice being more insulin sensitive than wild-type mice though old age remained in the ITT curves. Interestingly, at 24+ months, cadmium-treated Gclm null mice were significantly more insulin sensitive than their wild-type counterparts, presumably owing to increased insulin resistance in Cd-treated 24+ month WT mice. Overall, functional measurements of glucose homeostasis in Gclm heterozygous mice mirrored the results observed in wild-type mice across age and treatment (Supplemental Figs. 2–5).
Fig. 3.

Measurements of glucose tolerance in Gclm WT and null mice at 6 and 24+ months at baseline or following saline/cadmium treatment (n=4–18 mice/group). Area under the curve (AUC) data analyzed by two-way ANOVA with post-hoc Bonferroni correction for multiple comparisons: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Error bars represent the SEM.
Fig. 4.

Measurements of insulin tolerance/sensitivity in Gclm WT and null mice at 6 and 24+ months at baseline or following saline/cadmium treatment (n=4–18 mice/group). Area under the curve (AUC) data analyzed by two-way ANOVA with post-hoc Bonferroni correction for multiple comparisons: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Error bars represent the SEM.
Surprisingly, cadmium treatment did not have a significant effect on either GTT or ITT, though wild-type mice trend toward glucose intolerance and insulin resistance at 24+ months relative to young mice following cadmium administration (Fig. 3, Fig. 4). In contrast, the data do not suggest any changes to GTT or ITT in 24+ months Gclm null mice following administration of cadmium, aside from increased variability in both GTT and ITT AUC, though this was observed in all genotypes (Fig. 3, Fig. 4; Figs. S2–S5).
To complement GTT and ITT assessments, we measured fasting plasma insulin levels in Gclm wild-type and null mice at 6 and 24+ months of age, following saline or cadmium treatment (Fig. 5). High circulating insulin in a fasted state is a marker of insulin resistance [50]. At 24+ months, wild-type mice displayed a statistically significant increase in fasted insulin levels following saline administration (Fig. 5A). In comparison, Gclm null mice did not display statistically significant changes to fasted plasma insulin at 24+ months (Fig. 5B). Cadmium treatment exacerbated the trend seen in saline-treated wild-type mice, but did not have an effect in Gclm null mice (Fig. 5A and B, panels i-iv). Three-way ANOVA revealed statistically significant interactions between age and genotype (p = 0.008), age and treatment (p =0.005), genotype and treatment (p = 0.007), as well as age x genotype x treatment (p = 0.02). To supplement the fasting insulin measurements, homeostatic model assessment of insulin resistance (HOMA-IR), an index of insulin resistance integrating fasted insulin with fasted blood glucose, was also calculated (Fig. S6). Statistical analysis of HOMA-IR suggests that Gclm wild-type mice are insulin resistant by 24+ months of age, while Gclm null mice remain insulin sensitive. These results indicate the cadmium dosing regimen may have not have been sufficient to produce effects in GTT or ITT tests, instead eliciting more subtle effects on glucose homeostasis, such as increased fasted insulin. A previous study using young mice reported a similar effect [51]. Using a higher acute cadmium dose or a chronic exposure protocol would likely produce different results in functional measures of glucose homeostasis.
Fig. 5.

Measurements of plasma insulin levels in selected cohorts of mice (n=5–6 mice/group). (A) * = Three-Way ANOVA for genotype and treatment (Bonferroni): *p < 0.05, **p < 0.001, ***p < 0.0001. (B) Comparisons of plasma insulin levels by genotype (panels i-iv) and age (panels v-viii). * = Student's t-test: *p < 0.05, **p < 0.001, ***p < 0.0001. Error bars represent the SEM.
3.3. Transcriptome analysis
To elucidate changes at the molecular level which may be driving improved functional measures of glucose homeostasis in Gclm null mice, hepatic expression of targets important in cellular redox, defense, and energy homeostasis were examined. Transcriptomic expression of genes central to fatty acid metabolism, gluconeogenesis, 5′ adenosine monophosphate-activated protein kinase (AMPK), and peroxisome proliferator-activated receptor gamma (PPARγ) signaling was measured using BioSpyder's TempO-Seq multiplex platform (https://www.biospyder.com/; Fig. 6, Fig. 7).
Fig. 6.

Hierarchical clustering of canonical Nrf2-regulated genes. Shading is based on the fold induction of mRNA of each gene compared with the young (6 month), saline-treated wild-type mice. Colors represent log fold change in expression relative to control (n=3–4/genotype and treatment). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 7.

Hierarchical clustering of energy homeostasis targets. Expression for genes involved in fatty acid trafficking and synthesis, gluconeogenesis, insulin sensitivity, PPARγ, and AMPK are included. Shading is based on the fold induction of mRNA of each gene compared with the young (6 month), saline-treated wild-type mice; colors represent log fold change in expression relative to control (n=3–4/genotype). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Relative to young (6 month-old) saline-treated wild-type mice, Gclm null mice show strong upregulation of many Nrf2-regulated gene transcripts through 24+ months (Fig. 6). While transcript levels of Nfe2l2 (Nrf2) were only modestly increased in null mice, positive regulators of Nrf2 signaling Crebbp, Maff, Prkca, and Sqstm1 [[52], [53], [54]] were all upregulated in both young and old Gclm null mice (Fig. 7). Cadmium administration did not have a significant effect on Nrf2 target transcripts in null mice, potentially suggesting that robust basal Nrf2 activity in Gclm null mice is sufficient to protect against acute cadmium treatment (Fig. 6). Interestingly, in contrast, aged Gclm wild-type mice displayed slight repression of multiple CYPs and GSTs following cadmium treatment, indicating Nrf2 activation may be impaired in these mice compared to their younger counterparts (Fig. 6).
Fatty acid synthesis is unequivocally decreased in Gclm null mice through old age relative to young (6 month old), saline-treated wild-type mice, as evidenced by significant downregulation of fatty acid synthase (Fasn), fatty acid binding protein 5 (Fabp5), and elongation of very long chain fatty acids protein 5 (Elovl5; Fig. 7), complemented by improved fatty acid trafficking (Cd36; Fig. 7), enhanced expression of beta oxidation enzymes 17-β-hydroxysteroid dehydrogenase 10 (Hsd17b10) and long chain acyl-CoA dehydrogenase (Acadl; Fig. 7), and increased expression of a critical enzyme in the production of NADPH equivalents, Me1 (Fig. 7). These results align with prior observations of decreased circulating free fatty acids in young Gclm null mice relative to wild-type (unpublished data). Importantly, Gclm null mice have shifted away from gluconeogenesis, as evidenced by decreased expression of gluconeogenic targets glucose-6-phophatase (G6pc) and phosphoenolpyruvate carboxykinase 2 (Pck2; Fig. 7).
Expression of AMPK and PPAR, two pathways central to lipid and glucose metabolism which display significant crosstalk with Nrf2, was also assessed. Multiple AMPK subunits, as well as PPARγ, were upregulated in the young and old saline-treated Gclm null mice relative to young saline-treated wild-type. Many of their downstream targets [[55], [56], [57], [58], [59]] were also dysregulated accordingly (e.g., increased Pfkl, Me1, Nr1h3, decreased Elavl1, Hmgcs2) (Fig. 7). Furthermore, two targets negatively associated with T2DM, Irs1 (a direct indictor of insulin sensitivity [60]) and Wfs1 are increased in null mice, and expression of Ptpn1, a gene whose product (tyrosine-protein phosphatase non-receptor type 1) represses insulin signaling and is positively associated with development of T2DM and metabolic syndrome [61], is significantly decreased in null mice (Fig. 7). These results not only confirm previous observation of metabolic shift(s) in young Gclm null mice [31], but also for the first time provide evidence that these molecular level changes in Gclm null mice are conserved into old age. As a result, aged Gclm null mice may be better adapted to challenge with many oxidative and metabolic stressors than aged wild-type mice.
3.4. Metallothionein gene expression
Finally, the resistance of Gclm null mice to Cd-related decrements in glucose homeostasis led us to investigate the expression of targets involved in metal binding and detoxification. GSH is known to be an important line of defense against cadmium toxicity, and in vitro studies consistently show that low glutathione levels leave cells susceptible to cadmium-induced oxidative damage [62,63]. In addition, metallothioneins (MTs), low-molecular weight, heavy metal-binding proteins, are induced following cadmium exposure [64]. MT-I and MT-II are the main MTs expressed in liver, and MT-I/MT-II null mice are sensitive to acute Cd-induced hepatotoxicity and chronic Cd-induced nephrotoxicity, highlighting the importance of these proteins in mediating cadmium toxicity [65]. However, MT induction requires transcriptional activation by Nrf2/Keap1 to accumulate in protective amounts, meaning that sufficient MT expression is not achieved until hours after exposure. Thus, glutathione and metallothioneins act as complementary defense mechanisms against cadmium toxicity; GSH acts as an initial defense, and MTs act as a second-stage defense [63,65].
Given that MTs are also Nrf2-regulated, and that they play an important role in cadmium detoxification, we examined MT expression in the livers of young and old Gclm wild-type and null mice by qRT-PCR. Our results indicate that hepatic Mt1/2 mRNA transcripts are increased in old, but not young, Gclm null mice following cadmium administration, relative to Gclm wild-type (Fig. 8). Furthermore, the interaction of age, genotype, and treatment was statistically significant by three-way ANOVA (p = 0.034; Fig. 8). While these preliminary data suggest Gclm null mice may be able to more effectively induce MTs in response to cadmium exposure as a consequence of increased Nrf2/Keap1 activity, future research will be necessary to fully elucidate the importance of MT expression/induction in these mice.
Fig. 8.

Hepatic mRNA expression of Mt1/Mt2 using qRT-PCR in the livers of 6 and 24+ month old saline and CdCl2-treated Gclm wild-type and null mice. Given Mt1 and Mt2 are coordinately regulated by Nrf2, expression data from these isoforms were averaged. Expression values are reported as means and SEM (n = 3–4 for all groups), normalized to β-actin mRNA expression. Data analyzed by Three-Way ANOVA for age, treatment, and genotype using 6-month WT saline as control, with Bonferroni correction for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001.
4. Discussion
In this study, we assessed two functional parameters to examine general glucose homeostasis in Gclm wild-type and null mice: glucose tolerance and insulin sensitivity. During these tests, mice are challenged with a bolus of either glucose (GTT) or insulin (ITT), and blood glucose is periodically measured to see how mice respond to each challenge (i.e. how quickly blood glucose levels return to baseline). Impaired glucose tolerance and insulin sensitivity are associated with a T2DM phenotype and general metabolic dysfunction. Previous work [27] demonstrated that young Gclm null mice are more insulin sensitive and glucose tolerant than WT mice, a trend we have replicated here. However, this is the first study to examine how these findings are affected with age and in response to an exogenous toxicant challenge (cadmium).
Unexpectedly, our results contradict the hypothesis that age would have a significant effect on the ability of Gclm null mice to maintain improved insulin sensitivity and glucose tolerance relative to WT mice. Recent evidence points to an important role for Nrf2/Keap1 in maintaining metabolic homeostasis and preventing T2DM-like phenotypes, and the data presented here are consistent with Nrf2 activity not declining significantly with age in Gclm null mice [66,67]. The idea that Gclm null mice are protected from metabolic dysfunction with age is further supported by the finding that WT mice have increased fasted plasma insulin in old age, while null mice maintain low fasted insulin with age. High circulating insulin in a fasted state is indicative of insulin resistance, as it suggests that beta-cells within pancreatic islets need to produce and export more insulin to the periphery in order to maintain normal blood glucose [50]. Additionally, increased Nrf2 activity and subsequent induction of metallothioneins may also in part explain why cadmium had an effect on fasted plasma insulin in old Gclm wild-type, but not old Gclm null mice.
A potential explanation for the improved glucose homeostasis observed in Gclm null mice is through Nrf2-mediated modulation of intracellular energy pathways, such as the AMPK and PPARγ. AMPK is an energy sensor that maintains homeostasis and interacts with several cellular metabolic pathways, including Nrf2. Activation of the AMPK axis has myriad effects, ultimately resulting in suppressed hepatic lipogenesis and gluconeogenesis [68], and such changes are known to occur in the livers of Gclm null mice exposed to normal diets and to alcohol [20]. Transcriptional-level investigation suggests that Gclm null mice display modestly increased AMPK subunit and downstream target expression, including the rate-limiting gluconeogenic enzyme glucose-6-phosphatase (G6pc), and mitochondrial phosphoenolpyruvate carboxykinase (PEPCK, Pck2) [69,70]. Furthermore, PPARγ, a master regulator of adipogenesis, lipid homeostasis, and glucose metabolism [71] is highly upregulated in the livers of Gclm null mice. PPARγ stimulation improves glucose tolerance and insulin sensitivity in T2DM patients and animal models through promotion of fatty acid storage and modulation of adipocyte-secreted hormone expression [72,73]. Consequently, our findings support recent work which has revealed significant crosstalk between the Nrf2, AMPK, and PPARγ pathways, with Nrf2 acting as an “interface between redox and intermediary metabolism” [66,74,75].
Importantly, however, the improved metabolic phenotype observed in Gclm null mice is likely not only attributable to differences in gluconeogenesis and lipogenesis in the liver. Tissues such as muscle, adipose, and pancreas also likely contribute to the increased glucose tolerance, insulin sensitivity, and increased basal metabolic rate Gclm null mice display. The role(s) of these tissues in glucose signaling warrant future investigation.
Our data challenge the prevailing literature regarding glucose homeostasis changes with age in laboratory mice. Numerous studies have reported increasing insulin resistance and declining glucose tolerance concurrent with altered insulin secretion as characteristics of aging [76]. However, notable exceptions have been documented in both humans and experimental animals [[77], [78], [79], [80]]. A study from 1988 which examined glucose homeostasis in male C57BL/6J mice (the same background strain as the Gclm mice used here) came to the following conclusion: “the findings that glucose tolerance [does] not deteriorate with age, coupled with the lack of evidence for impaired beta cell responsiveness to glucose in old males, suggest that deterioration in glucose homeostasis is not an inevitable consequence of aging in the mouse” [81]. The results of a more recent study went further, showing glucose tolerance improving with age in this strain [76]. However, as part of the same study, these authors found that insulin sensitivity declined with age. In summary, age-related decrements to glucose homeostasis in C57BL/6J mice are not unequivocal, and thus, while our results run contrary to the general consensus, they are not without precedent.
While metabolic adaptation in Gclm null mice, notably enhanced Nrf2 activity, may provide an explanation for why Gclm null mice are resistant to age-induced declines in glucose homeostasis, it remains unresolved as to why no appreciable changes to functional testing of glucose tolerance and insulin sensitivity with age were observed in Gclm wild-type (and heterozygous) mice. These mice were derived from a C57BL/6J background many years ago, and even with backcrossing, it is possible that unknown factors, genetic, epigenetic, or otherwise, may have become fixed over time. It is also important to consider the study presented here used male mice exclusively; sex-related differences in aging phenotypes may be significant in Gclm mice. Furthermore, there may be environmental differences that could alter gut microbiota in mice housed at the University of Washington compared to other institutions. Nonetheless, we did observe significant changes to fasted plasma insulin levels with age in wild-type mice, indicating that age-related declines in glucose homeostasis are indeed occurring, even if they were not observed by GTT or ITT; alternative dosing protocols or exposure to compound(s) with more direct effects on metabolism may provide a clearer picture of the age-related changes to glucose homeostasis observed here.
The adaptations afforded by deletion of Gclm (e.g., increased Nrf2 signaling) are likely the basis for protection to exposure(s) to certain stressors. However, GSH levels are not completely depleted in Gclm null mice, so they still have some capacity to detoxify some agents using GSH directly, or for example, through glutathione S-transferase (GST)-mediated conjugation. Moreover, the molecular adaptations observed in Gclm null mice are likely in response to chronically reduced GSH levels, including during gestation and early life. While increased antioxidant buffering capacity through Nrf2 stimulation may confer positive effects on glucose tolerance and aging, the specific constellation of changes seen in Gclm null mice might be difficult to replicate by decreasing Gclm expression only in adulthood. In fact, without the complementary modulation of metabolic signaling (e.g., AMPK, and PPARγ) observed in Gclm null mice, constitutive overexpression of Nrf2 in adults may in fact be detrimental, owing to the so-called “dark side” of Nrf2, in which promotion and pro-diabetic shifts in metabolism are observed [82,83].
It is also important to recognize that despite a wealth of evidence suggesting that perturbations to GSH and other thiols (e.g., thioredoxin, cysteine) impact insulin signaling, parsing the precise role of particular antioxidant molecules in glucose homeostasis is complicated by the complementary, and at times, redundant, nature of these antioxidant systems [84]. For example, it has been demonstrated that chronic treatment of adult mice on a high-fat diet with the GCL inhibitor buthionine sulfoximine (BSO) produced a lean phenotype, an effect that might be due to low levels of circulating cysteine [85]. However, in another study, supplementation with glycine to increase GSH synthesis in a rat model of sucrose-induced insulin resistance resulted in improved insulin sensitivity, suggesting that GSH is specifically important for modulating insulin responsiveness [86]. Regardless, under a state of normal GCL expression, GSH likely remains a very important defense against age-induced decrements in redox and glucose homeostasis, particularly when aged adults are faced with certain challenges (e.g., inflammation, infection, ischemia/reperfusion injury, xenobiotic detoxification), where resilience depends on maintaining a “healthy” redox state.
To conclude, our mouse model of chronic glutathione deficiency—Gclm null mice—in which redox- and energy homeostasis-related genes are highly upregulated presumably to compensate for low GSH levels, maintain a favorable metabolic state into old age. In contrast, Gclm wild-type mice, which do not have similar compensatory redox/antioxidant gene upregulation under basal conditions, display increased markers of insulin resistance with age. Because aging is a known driver of general metabolic dysfunction in mammals, the results of this study were somewhat surprising. Though future study will be required to further elucidate the precise mechanisms driving the favorable metabolic phenotype in Gclm null mice, our work strongly suggests that Gclm null mice are able to resist age-induced metabolic dysfunction through protective compensatory upregulation of Nrf2, AMPK, and PPARγ pathways, lending credence to and augmenting the significant body of research supporting the idea that altered redox homeostasis contributes to T2DM development and progression [[87], [88], [89]]. The role of redox homeostasis in T2DM is more nuanced than turning a redox “switch” on or off, and persistent, adaptive responses to redox imbalance from early in life are likely important. Our research supports this idea, as Gclm null mice compensate for redox imbalance (low GSH levels) from birth, displaying lifelong modification of antioxidant activity. Further research examining early-life, chronic interventions to modulate redox status and their effect(s) on the development of metabolic dysfunction later in life are increasingly pertinent to a society in which the incidence of T2DM and other metabolic diseases continues to increase [1].
Funding
This work was supported by National Institutes of Health grants P30ES007033 and T32AG000057, and the University of Washington Department of Environmental and Occupational Health Sciences Sheldon D. Murphy Fund.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgements
We wish to thank members of the Kavanagh laboratory, and Drs. Karin E. Bornfeldt, David L. Eaton, Gary F. Merrill, Peter S. Rabinovitch, and Edward E. Schmidt for their encouragement and helpful suggestions.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102213.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.CDC . Centers for Disease Control and Prevention, US Department of Health and Human Services; 2020. National Diabetes Statistics Report, 2020. [Google Scholar]
- 2.Ahmad L.A., Crandall J.P. Type 2 diabetes prevention: a review. Clin. Diabet. 2010;28(2):53–59. [Google Scholar]
- 3.DeFronzo R.A., Ferrannini E., Groop L., Henry R.R., Herman W.H., Holst J.J., Hu F.B., Kahn C.R., Raz I., Shulman G.I., et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Prim. 2015;1 doi: 10.1038/nrdp.2015.19. [DOI] [PubMed] [Google Scholar]
- 4.Kahn S.E., Cooper M.E., Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383(9922):1068–1083. doi: 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhakkiyalakshmi E., Sireesh D., Rajaguru P., Paulmurugan R., Ramkumar K.M. The emerging role of redox-sensitive nrf2-keap1 pathway in diabetes. Pharmacol. Res. 2015;91:104–114. doi: 10.1016/j.phrs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Hayden M.R., Sowers J.R. Redox imbalance in diabetes. Antioxidants Redox Signal. 2007;9(7):865–867. doi: 10.1089/ars.2007.1640. [DOI] [PubMed] [Google Scholar]
- 7.Rehman K., Akash M.S.H. Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus: how are they interlinked? J. Cell. Biochem. 2017;118(11):3577–3585. doi: 10.1002/jcb.26097. [DOI] [PubMed] [Google Scholar]
- 8.Dieter B.P. Dysregulation of nrf2 signaling in diabetes: an opportunity for a multitarget approach. J. Diabetes Metabol. 2014;6(1) [Google Scholar]
- 9.Jiménez-Osorio A.S., Picazo A., González-Reyes S., Barrera-Oviedo D., Rodríguez-Arellano M.E., Pedraza-Chaverri J. Nrf2 and redox status in prediabetic and diabetic patients. Int. J. Mol. Sci. 2014;15(11):20290–20305. doi: 10.3390/ijms151120290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman E.A. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care. 1999;22(Suppl 2):B65–B71. [PubMed] [Google Scholar]
- 11.Schulze P.C., Yoshioka J., Takahashi T., He Z., King G.L., Lee R.T. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J. Biol. Chem. 2004;279(29):30369–30374. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- 12.Tan A.L., Forbes J.M., Cooper M.E. Age, rage, and ros in diabetic nephropathy. Semin. Nephrol. 2007;27(2):130–143. doi: 10.1016/j.semnephrol.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Yao D., Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (rage) and rage ligands. Diabetes. 2010;59(1):249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu T., Robotham J.L., Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U. S. A. 2006;103(8):2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meister A., Anderson M.E. Glutathione. Annu. Rev. Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 16.Franklin C.C., Backos D.S., Mohar I., White C.C., Forman H.J., Kavanagh T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspect. Med. 2009;30(1–2):86–98. doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalton T.P., Dieter M.Z., Yang Y., Shertzer H.G., Nebert D.W. Knockout of the mouse glutamate cysteine ligase catalytic subunit (gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun. 2000;279(2):324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- 18.McConnachie L.A., Mohar I., Hudson F.N., Ware C.B., Ladiges W.C., Fernandez C., Chatteron-Kirchmeier S., White C.C., Pierce R.H., Kavanagh T.J. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol. Sci. 2007;99(2):628–636. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y., Krishan M., Nebert D.W., Shertzer H.G. Glutathione-deficient mice are susceptible to tcdd-induced hepatocellular toxicity but resistant to steatosis. Chem. Res. Toxicol. 2012;25(1):94–100. doi: 10.1021/tx200242a. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y., Singh S., Matsumoto A., Manna S.K., Abdelmegeed M.A., Golla S., Murphy R.C., Dong H., Song B.J., Gonzalez F.J., et al. Chronic glutathione depletion confers protection against alcohol-induced steatosis: implication for redox activation of amp-activated protein kinase pathway. Sci. Rep. 2016;6 doi: 10.1038/srep29743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole T.B., Coburn J., Dao K., Roqué P., Chang Y.C., Kalia V., Guilarte T.R., Dziedzic J., Costa L.G. Sex and genetic differences in the effects of acute diesel exhaust exposure on inflammation and oxidative stress in mouse brain. Toxicology. 2016;374:1–9. doi: 10.1016/j.tox.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haque J.A., McMahan R.S., Campbell J.S., Shimizu-Albergine M., Botta D., Bammler T.K., Beyer R.P., Montine T.J., Yeh M.M., Kavanagh T.J., et al. Attenuated progression of diet-induced steatohepatitis in glutathione-deficient mice. Lab. Invest. 2010;90:1704–1717. doi: 10.1038/labinvest.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson E., Wesselkamper S.C., Shertzer H.G., Leikauf G.D., Dalton T.P., Chen Y. Glutathione deficient c57bl/6j mice are not sensitized to ozone-induced lung injury. Biochem. Biophys. Res. Commun. 2010;396(2):407–412. doi: 10.1016/j.bbrc.2010.04.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McConnachie L.A., Botta D., White C.C., Weldy C.S., Wilkerson H.W., Yu J., Dills R., Yu X., Griffith W.C., Faustman E.M., et al. The glutathione synthesis gene gclm modulates amphiphilic polymer-coated cdse/zns quantum dot-induced lung inflammation in mice. PLoS One. 2013;8(5) doi: 10.1371/journal.pone.0064165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weldy C.S., White C.C., Wilkerson H.-W., Larson T.V., Stewart J.A., Gill S.E., Parks W.C., Kavanagh T.J. Heterozygosity in the glutathione synthesis gene gclm increases sensitivity to diesel exhaust particulate induced lung inflammation in mice. Inhal. Toxicol. 2011;23(12):724–735. doi: 10.3109/08958378.2011.608095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalton T.P., Chen Y., Schneider S.N., Nebert D.W., Shertzer H.G. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic. Biol. Med. 2004;37(10):1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 27.Kendig E.L., Chen Y., Krishan M., Johansson E., Schneider S.N., Genter M.B., Nebert D.W., Shertzer H.G. Lipid metabolism and body composition in gclm(-/-) mice. Toxicol. Appl. Pharmacol. 2011;257(3):338–348. doi: 10.1016/j.taap.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y., Dieter M.Z., Chen Y., Shertzer H.G., Nebert D.W., Dalton T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 29.Lavoie S., Steullet P., Kulak A., Preitner F., Do K.Q., Magistretti P.J. Glutamate cysteine ligase-modulatory subunit knockout mouse shows normal insulin sensitivity but reduced liver glycogen storage. Front. Physiol. 2016;7:142. doi: 10.3389/fphys.2016.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iverson S.V., Eriksson S., Xu J., Prigge J.R., Talago E.A., Meade T.A., Meade E.S., Capecchi M.R., Arner E.S., Schmidt E.E. A txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radic. Biol. Med. 2013;63:369–380. doi: 10.1016/j.freeradbiomed.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y., Manna S.K., Golla S., Krausz K.W., Cai Y., Garcia-Milian R., Chakraborty T., Chakraborty J., Chatterjee R., Thompson D.C., et al. Glutathione deficiency-elicited reprogramming of hepatic metabolism protects against alcohol-induced steatosis. Free Radic. Biol. Med. 2019;143:127–139. doi: 10.1016/j.freeradbiomed.2019.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y., Golla S., Garcia-Milian R., Thompson D.C., Gonzalez F.J., Vasiliou V. Hepatic metabolic adaptation in a murine model of glutathione deficiency. Chem. Biol. Interact. 2019;303:1–6. doi: 10.1016/j.cbi.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siewert S., González I., Santillán L., Lucero R., Ojeda M.S., Gimenez M.S. Downregulation of nrf2 and ho-1 expression contributes to oxidative stress in type 2 diabetes mellitus: a study in juana koslay city, san luis, Argentina. J. Diabetes Mellitus. 2013;3(2):71–78. [Google Scholar]
- 34.Cha H.N., Woo C.H., Kim H.Y., Park S.Y. Methionine sulfoxide reductase b3 deficiency inhibits the development of diet-induced insulin resistance in mice. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwak G.H., Kim K.Y., Kim H.Y. Methionine sulfoxide reductase b3 deficiency stimulates heme oxygenase-1 expression via ros-dependent and nrf2 activation pathways. Biochem. Biophys. Res. Commun. 2016;473(4):1033–1038. doi: 10.1016/j.bbrc.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H., Davies K.J.A., Forman H.J. Oxidative stress response and nrf2 signaling in aging. Free Radic. Biol. Med. 2015;88(Pt B):314–336. doi: 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H., Liu H., Davies K.J.A., Sioutas C., Finch C.E., Morgan T.E., Forman H.J. Nrf2-regulated phase ii enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012;52(9):2038–2046. doi: 10.1016/j.freeradbiomed.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C., Zhu Y., Lu Z., Guo W., Tumen B., He Y., Chen C., Hu S., Xu K., Wang Y., et al. Cadmium induces acute liver injury by inhibiting nrf2 and the role of nf-κb, nlrp3, and mapks signaling pathway. Int. J. Environ. Res. Publ. Health. 2019;17(1) doi: 10.3390/ijerph17010138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al Doghaither H., Elmorsy E., Al-Ghafari A., Ghulam J. Roles of oxidative stress, apoptosis, and inflammation in metal-induced dysfunction of beta pancreatic cells isolated from cd1 mice. Saudi J. Biol. Sci. 2021;28(1):651–663. doi: 10.1016/j.sjbs.2020.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz G.G., Il'yasova D., Ivanova A. Urinary cadmium, impaired fasting glucose, and diabetes in the nhanes iii. Diabetes Care. 2003;26(2):468–470. doi: 10.2337/diacare.26.2.468. [DOI] [PubMed] [Google Scholar]
- 41.Xiao L., Li W., Zhu C., Yang S., Zhou M., Wang B., Wang X., Wang D., Ma J., Zhou Y., et al. Cadmium exposure, fasting blood glucose changes, and type 2 diabetes mellitus: a longitudinal prospective study in China. Environ. Res. 2021;192 doi: 10.1016/j.envres.2020.110259. [DOI] [PubMed] [Google Scholar]
- 42.NRC . 2011. Update of the Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 43.ATSDR . U.S. Department of Health and Human Services, Public Health Serivce, Agency for Toxic Substances and Disease Registry; Atlanta, GA: 2011. Cadmium. [Google Scholar]
- 44.Basinger M.A., Jones M.M., Holscher M.A., Vaughn W.K. Antagonists for acute oral cadmium chloride intoxication. J. Toxicol. Environ. Health. 1988;23(1):77–89. doi: 10.1080/15287398809531095. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Z., Hyun J.S., Satsu H., Kakuta S., Shimizu M. Oral exposure to cadmium chloride triggers an acute inflammatory response in the intestines of mice, initiated by the over-expression of tissue macrophage inflammatory protein-2 mrna. Toxicol. Lett. 2006;164(2):144–154. doi: 10.1016/j.toxlet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 46.Law C.W., Chen Y., Shi W., Smyth G.K. Voom: precision weights unlock linear model analysis tools for rna-seq read counts. Genome Biol. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 48.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57(1):289–300. [Google Scholar]
- 49.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 50.McAuley K.A., Williams S.M., Mann J.I., Walker R.J., Lewis-Barned N.J., Temple L.A., Duncan A.W. Diagnosing insulin resistance in the general population. Diabetes Care. 2001;24(3):460–464. doi: 10.2337/diacare.24.3.460. [DOI] [PubMed] [Google Scholar]
- 51.Li X., Li M., Xu J., Zhang X., Xiao W., Zhang Z. Decreased insulin secretion but unchanged glucose homeostasis in cadmium-exposed male c57bl/6 mice. J. Toxicol. 2019;2019 doi: 10.1155/2019/8121834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee D.H., Park J.S., Lee Y.S., Han J., Lee D.K., Kwon S.W., Han D.H., Lee Y.H., Bae S.H. Sqstm1/p62 activates nfe2l2/nrf2 via ulk1-mediated autophagic keap1 degradation and protects mouse liver from lipotoxicity. Autophagy. 2020;16(11):1949–1973. doi: 10.1080/15548627.2020.1712108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Z., Chin Y.E., Zhang D.D. Acetylation of nrf2 by p300/cbp augments promoter-specific dna binding of nrf2 during the antioxidant response. Mol. Cell Biol. 2009;29(10):2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tonelli C., Chio I.I.C., Tuveson D.A. Transcriptional regulation by nrf2. Antioxidants Redox Signal. 2018;29(17):1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chawla A., Boisvert W.A., Lee C.H., Laffitte B.A., Barak Y., Joseph S.B., Liao D., Nagy L., Edwards P.A., Curtiss L.K., et al. A ppar gamma-lxr-abca1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 2001;7(1):161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 56.Deleye Y., Cotte A.K., Hannou S.A., Hennuyer N., Bernard L., Derudas B., Caron S., Legry V., Vallez E., Dorchies E., et al. Cdkn2a/p16ink4a suppresses hepatic fatty acid oxidation through the ampkα2-sirt1-pparα signaling pathway. J. Biol. Chem. 2020;295(50):17310–17322. doi: 10.1074/jbc.RA120.012543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim J., Yang G., Kim Y., Ha J. Ampk activators: mechanisms of action and physiological activities. Exp. Mol. Med. 2016;48 doi: 10.1038/emm.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsusue K., Haluzik M., Lambert G., Yim S.H., Gavrilova O., Ward J.M., Brewer B.J., Reitman M.L., Gonzalez F.J. Liver-specific disruption of ppargamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 2003;111(5):737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W., Fan J., Yang X., Fürer-Galban S., Lopez de Silanes I., von Kobbe C., Guo J., Georas S.N., Foufelle F., Hardie D.G., et al. Amp-activated kinase regulates cytoplasmic hur. Mol. Cell Biol. 2002;22(10):3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kubota N., Kubota T., Itoh S., Kumagai H., Kozono H., Takamoto I., Mineyama T., Ogata H., Tokuyama K., Ohsugi M., et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metabol. 2008;8(1):49–64. doi: 10.1016/j.cmet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A.L., Normandin D., Cheng A., Himms-Hagen J., Chan C.C., et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1b gene. Science. 1999;283(5407):1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 62.Gaubin Y., Vaissade F., Croute F., Beau B., Soleilhavoup J., Murat J. Implication of free radicals and glutathione in the mechanism of cadmium-induced expression of stress proteins in the a549 human lung cell-line. Biochim. Biophys. Acta. 2000;1495(1):4–13. doi: 10.1016/s0167-4889(99)00149-4. [DOI] [PubMed] [Google Scholar]
- 63.Ochi T., Otsuka F., Takahashi K., Ohsawa M. Glutathione and metallothioneins as cellular defense against cadmium toxicity in cultured Chinese hamster cells. Chem. Biol. Interact. 1988;65(1):1–14. doi: 10.1016/0009-2797(88)90026-9. [DOI] [PubMed] [Google Scholar]
- 64.Kennette W., Collins O.M., Zalups R.K., Koropatnick J. Basal and zinc-induced metallothionein in resistance to cadmium, cisplatin, zinc, and tertbutyl hydroperoxide: studies using mt knockout and antisense-downregulated mt in mammalian cells. Toxicol. Sci. 2005;88(2):602–613. doi: 10.1093/toxsci/kfi318. [DOI] [PubMed] [Google Scholar]
- 65.Klaassen C.D., Liu J. Metallothionein transgenic and knock-out mouse models in the study of cadmium toxicity. J. Toxicol. Sci. 1998;23(Suppl 2):97–102. doi: 10.2131/jts.23.supplementii_97. [DOI] [PubMed] [Google Scholar]
- 66.Hayes J.D., Dinkova-Kostova A.T. The nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39(4):199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 67.Uruno A., Furusawa Y., Yagishita Y., Fukutomi T., Muramatsu H., Negishi T., Sugawara A., Kensler T.W., Yamamoto M. The keap1-nrf2 system prevents onset of diabetes mellitus. Mol. Cell Biol. 2013;33(15):2996–3010. doi: 10.1128/MCB.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu J., Donepudi A.C., Moscovitz J.E., Slitt A.L. Keap1-knockdown decreases fasting-induced fatty liver via altered lipid metabolism and decreased fatty acid mobilization from adipose tissue. PLoS One. 2013;8(11) doi: 10.1371/journal.pone.0079841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koo S.H., Flechner L., Qi L., Zhang X., Screaton R.A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P., et al. The creb coactivator torc2 is a key regulator of fasting glucose metabolism. Nature. 2005;437(7062):1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 70.Lochhead P.A., Salt I.P., Walker K.S., Hardie D.G., Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes pepck and glucose-6-phosphatase. Diabetes. 2000;49(6):896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- 71.Lee Y.K., Park J.E., Lee M., Hardwick J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018;2(4):209–215. doi: 10.1016/j.livres.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Picard F., Auwerx J. Ppar(gamma) and glucose homeostasis. Annu. Rev. Nutr. 2002;22:167–197. doi: 10.1146/annurev.nutr.22.010402.102808. [DOI] [PubMed] [Google Scholar]
- 73.Rangwala S.M., Lazar M.A. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol. Sci. 2004;25(6):331–336. doi: 10.1016/j.tips.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 74.Lee C. Collaborative power of nrf2 and ppar. Oxid. Med. Cell. Longev. 2017;2017 doi: 10.1155/2017/1378175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mo C., Wang L., Zhang J., Numazawa S., Tang H., Tang X., Han X., Li J., Yang M., Wang Z., et al. The crosstalk between nrf2 and ampk signal pathways is important for the anti-inflammatory effect of berberine in lps-stimulated macrophages and endotoxin-shocked mice. Antioxidants Redox Signal. 2014;20(4):574–588. doi: 10.1089/ars.2012.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oh Y.S., Seo E.H., Lee Y.S., Cho S.C., Jung H.S., Park S.C., Jun H.S. Increase of calcium sensing receptor expression is related to compensatory insulin secretion during aging in mice. PLoS One. 2016;11(7) doi: 10.1371/journal.pone.0159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bourey R.E., Kohrt W.M., Kirwan J.P., Staten M.A., King D.S., Holloszy J.O. Relationship between glucose tolerance and glucose-stimulated insulin response in 65-year-olds. J. Gerontol. 1993;48(4):M122–M127. doi: 10.1093/geronj/48.4.m122. [DOI] [PubMed] [Google Scholar]
- 78.Broughton D.L., James O.W., Alberti K.G., Taylor R. Peripheral and hepatic insulin sensitivity in healthy elderly human subjects. Eur. J. Clin. Invest. 1991;21(1):13–21. doi: 10.1111/j.1365-2362.1991.tb01352.x. [DOI] [PubMed] [Google Scholar]
- 79.Chakraborty G., Thumpayil S., Lafontant D.E., Woubneh W., Toney J.H. Age dependence of glucose tolerance in adult kk-ay mice, a model of non-insulin dependent diabetes mellitus. Lab Anim. (NY) 2009;38(11):364–368. doi: 10.1038/laban1109-364. [DOI] [PubMed] [Google Scholar]
- 80.Pacini G., Valerio A., Beccaro F., Nosadini R., Cobelli C., Crepaldi G. Insulin sensitivity and beta-cell responsivity are not decreased in elderly subjects with normal ogtt. J. Am. Geriatr. Soc. 1988;36(4):317–323. doi: 10.1111/j.1532-5415.1988.tb02358.x. [DOI] [PubMed] [Google Scholar]
- 81.Leiter E.H., Premdas F., Harrison D.E., Lipson L.G. Aging and glucose homeostasis in c57bl/6j male mice. Faseb. J. 1988;2(12):2807–2811. doi: 10.1096/fasebj.2.12.3044905. [DOI] [PubMed] [Google Scholar]
- 82.Dodson M., de la Vega M.R., Cholanians A.B., Schmidlin C.J., Chapman E., Zhang D.D. Modulating nrf2 in disease: timing is everything. Annu. Rev. Pharmacol. Toxicol. 2019;59:555–575. doi: 10.1146/annurev-pharmtox-010818-021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu P., Dodson M., Li H., Schmidlin C.J., Shakya A., Wei Y., Garcia J.G.N., Chapman E., Kiela P.R., Zhang Q.Y., et al. Non-canonical nrf2 activation promotes a pro-diabetic shift in hepatic glucose metabolism. Mol. Metabol. 2021;51 doi: 10.1016/j.molmet.2021.101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prigge J.R., Eriksson S., Iverson S.V., Meade T.A., Capecchi M.R., Arnér E.S., Schmidt E.E. Hepatocyte dna replication in growing liver requires either glutathione or a single allele of txnrd1. Free Radic. Biol. Med. 2012;52(4):803–810. doi: 10.1016/j.freeradbiomed.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Elshorbagy A.K., Jernerén F., Scudamore C.L., McMurray F., Cater H., Hough T., Cox R., Refsum H. Exploring the lean phenotype of glutathione-depleted mice: thiol, amino acid and fatty acid profiles. PLoS One. 2016;11(10) doi: 10.1371/journal.pone.0163214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.El-Hafidi M., Franco M., Ramírez A.R., Sosa J.S., Flores J.A.P., Acosta O.L., Salgado M.C., Cardoso-Saldaña G. Glycine increases insulin sensitivity and glutathione biosynthesis and protects against oxidative stress in a model of sucrose-induced insulin resistance. Oxid. Med. Cell. Long. 2018 doi: 10.1155/2018/2101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 88.Ceriello A., Testa R. Antioxidant anti-inflammatory treatment in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S232–S236. doi: 10.2337/dc09-S316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaneto H., Kajimoto Y., Miyagawa J., Matsuoka T., Fujitani Y., Umayahara Y., Hanafusa T., Matsuzawa Y., Yamasaki Y., Hori M. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes. 1999;48(12):2398–2406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.