

Abstract
Objective
Methylglyoxal (MG) is a highly reactive α-oxoaldehyde that glycates proteins. MG has been linked to the development of diabetic complications: MG is the major precursor of advanced glycation end products (AGEs), a risk marker for diabetic complications in humans. Furthermore, flies and fish with elevated MG develop insulin resistance, obesity, and hyperglycemia. MG is detoxified in large part through the glyoxalase system, whose rate-limiting enzyme is glyoxalase I (Glo1). Hence, we aimed to study how Glo1 activity is regulated.
Methods
We studied the regulation and effect of post-translational modifications of Glo1 in tissue culture and in mouse models of diabetes.
Results
We show that Glo1 activity is promoted by phosphorylation on Tyrosine 136 via multiple kinases. We find that Glo1 Y136 phosphorylation responds in a bimodal fashion to glucose levels, increasing in cell culture from 0 mM to 5 mM (physiological) glucose, and then decreasing at higher glucose concentrations, both in cell culture and in mouse models of hyperglycemia.
Conclusions
These data, together with published findings that elevated MG leads to hyperglycemia, suggest the existence of a deleterious positive feedback loop whereby hyperglycemia leads to reduced Glo1 activity, contributing to elevated MG levels, which in turn promote hyperglycemia. Hence, perturbations elevating either glucose or MG have the potential to start an auto-amplifying feedback loop contributing to diabetic complications.
Keywords: Glyoxalase, Diabetes, Phosphorylation
Highlights
-
•
Phosphorylation of Glo1 on Y136 promotes its activity.
-
•
Y136 phosphorylation decreases when cells are exposed to hyperglycemia.
-
•
This results in reduced Glo1 activity when MG detoxification is needed the most.
1. Introduction
Although diabetic patients are characterized by hyperglycemia, a recent study has shown that renormalization of blood glucose has a mild impact on the progression of diabetic complications [1,2], suggesting the role of additional mechanistic factors in the development of diabetic complications. One such factor could be methylglyoxal (MG). MG is a reactive α-oxoaldehyde that is formed non-enzymatically in animal cells mainly from the triose-phosphate intermediates of glycolysis [3]. MG reacts directly and rapidly with proteins to form advanced glycation end-products (AGEs) which disrupt protein function, leading to organ damage. Multiple observations suggest MG may contribute to the development of diabetic complications. Diabetic patients have elevated levels of MG and MG-adducts [4,5]. Exposure to MG causes kidney damage [6], degenerative changes in cutaneous microvessels [7] and neuropathies [[8], [9], [10], [11], [12]]. Elevated MG could also contribute to insulin resistance and hyperglycemia. For instance, treatment of muscle cells in culture with MG induces insulin resistance [13]. Flies lacking glyoxalase 1 (Glo1), an enzyme involved in MG detoxification, have mildly elevated MG levels and develop obesity, insulin resistance, and hyperglycemia [14]. Likewise, Glo1 knockout fish developed post-prandial hyperglycemia, and when overfed, insulin resistance [15]. Hence, elevated MG can induce several diabetic phenotypes – insulin resistance, hyperglycemia, obesity, and damaged blood vessels, kidneys and neurons.
Methylglyoxal is detoxified by several different enzymes [16]. The glyoxalase system comprises two enzymes, Glyoxalase 1 (Glo1) and Glyoxalase 2, which act in succession to convert MG into d-lactate using a catalytic amount of reduced glutathione [17]. When Glo1 is knocked out either in cell culture or in mice, the activity of aldehyde dehydrogenases (ALDH) and Aldo-keto reductases (AKR) is increased, which metabolizes MG to pyruvate and hydroxyacetone, respectively [18,19]. The mechanisms regulating the activity of these enzymes are of relevance for diabetes, as they could modulate MG detoxification and consequently the progression of diabetic complications. In particular, Glo1 is the rate-limiting enzyme in the glyoxalase system and accounts for a large fraction of the MG detoxification in cells and tissues [16]. We study here whether Glo1 activity is regulated via post-translational modifications (PTMs).
A few PTMs have been reported on Glo1. TNF signaling leads to phosphorylation of Glo1 on Thr106 via activation of PKA and CaMKII [20,21]. Although this was originally reported to not affect Glo1 enzymatic activity [20], a recent study showed that it affects both the KM and Vmax of Glo1 as well as Glo1 protein stability [22]. Several cysteines of Glo1 are nitrosylated, in particular Cys138, although the functional relevance of this PTM remains unknown [23]. Finally, Glo1 has N-terminal acetylation on Ala2 which does not impact Glo1 activity and glutathionylation on Cys139 which strongly inhibits Glo1 activity in vitro [24].
We report here that Glo1 is phosphorylated on Tyr136 by multiple kinases. This phosphorylation has a bimodal response to glucose levels, increasing from no glucose to physiological levels of glucose, and then decreasing in hyperglycemic conditions. We find phosphorylation of Glo1 on Tyr136 stimulates activity. As such, Tyr136 phosphorylation represents a possible mechanistic link to how hyperglycemia negatively impacts the ability of a cell to detoxify the reactive metabolite methylglyoxal.
2. Materials & methods
2.1. Antibodies and reagents
Primary antibodies: Calnexin (Enzo #ADI-SPA-960-D), Src (Cell Signaling Technology #21080), phospho-Src (Y417) (Cell Signaling Technology #6943), phospho-FAK (Tyr925) (Cell Signaling Technology #3284), phospho-FAK (Tyr397) (D20B1) (Cell Signaling Technology #8556), FAK (Cell Signaling Technology #13009), TNK1 (C44F9) (Cell Signaling Technology #4570), Her2/ErbB2 (D8F12) XP (Cell Signaling Technology #4290), Tubulin (Sigma #T9026, 1:5000), Insulin receptor β (4B8) (Cell Signaling Technology #3025S), JAK2 (691R5) (Thermo Scientific #AHO1352), Phospho-Stat3 (Tyr705) (Cell Signaling Technology #9131), Stat3 (12H6) (Cell Signaling Technology #9139), p-Tyr Antibody (Y99) (Santa Cruz Biotechnology sc-7020), total Glo1 [14]. All antibodies were used 1:1000 unless specified otherwise. Secondary antibodies were anti-rabbit HRP (Jackson ImmunoResearch #111-035-003), anti-mouse HRP (Jackson ImmunoResearch #115-035-003), anti-guinea pig (Jackson ImmunoResearch #106-035-003).
To generate phospho-Glo1(Y136) antibody, rabbits were immunized with the peptide IAVPDV (phosphoY) SA (homoalanine) KRFC coupled to Keyhole limpet hemocyanin (KLH) by Seramun Diagnostica GmbH. For the initial immunization, Freund's complete adjuvant was used, followed by 3 booster injections with Freund's incomplete adjuvant. The collection of hyperimmune serum was executed on day 7 after the last booster injection. Serum proteins were precipitated by the addition of (NH4)2SO4 (0.452 g/2 ml) and the antibody was affinity purified using the phospho-peptide coupled to beads. The resin generation and the antibody purification were performed using the SulfoLink Immobilization Kit for Peptides following the manufacturer's instructions (Thermo Scientific # 44999).
2.2. Cell culture conditions and treatments
HeLa (ATCC #CCL-2) and HepG2 (ATCC #HB-8065) cells were cultured in DMEM (Life Technologies #41965-062) containing 25 mM glucose, 4 mM l-glutamine, 1% penicillin/streptomycin (Life Technologies #15140-122), and 10% fetal bovine serum (FBS). U-937 (DSMZ #ACC5) cells were cultured in RPMI 1640 (Life Technologies #52400-025) with 1 mM sodium pyruvate (Life Technologies #11360070), 2 mM l-glutamine (Life Technologies #25030024), 1x penicillin/streptomycin (Life Technologies #15140-122), and 10% FBS. All cell lines were maintained in 5% CO2 at 37 °C and tested for mycoplasma to exclude contamination.
The glucose experiments were performed in DMEM containing 5 mM glucose that was prepared from a combination of DMEM high glucose, no glutamine, no phenol red (Life Technologies #31053028) and DMEM no glucose, no glutamine, no phenol red (Life Technologies # A1443001). Additionally, 2 mM l-glutamine (Life Technologies #25030024) and 10% FBS were added to the media. The cells were seeded in the regular medium the day before the experiment. On the day of the experiment, incubation with either fresh high (25 mM) or low (5 mM) glucose media was performed overnight.
For treatments with inhibitors or methylglyoxal, cells were seeded the day before the experiment. The next day, the inhibitor or methylglyoxal was directly added to the well and the incubations were performed for 30 min. The concentrations used are indicated in the figures.
Generation of Glo1 knockout cells, plasmids transfections, and siRNA knockdowns
Monoclonal Glo1 knockout cell lines were generated via CRISPR-Cas9 mediated genome editing using the PX459 plasmid expressing Cas9 and sgRNAs from the website CHOP–CHOP (https://chopchop.cbu.uib.no/) [25] (sequences shown in Supplementary Table 3). The knockout cell line was confirmed by Sanger sequencing of the target genomic locus after PCR amplification and TOPO cloning (Life Technologies #450640), as well as by immunoblotting for Glo1 protein, Glo1 activity assays, and qRT-PCR.
For the tyrosine kinase CRISPR-Cas9 library, the gRNAs sequences were obtained from the Brunello and Brie library [26] and were cloned into pSpCas9(BB)-2A-Puro (PX459) V1.0 from Feng Zhang [27] (Addgene #48139).
The Glo1 coding sequence was PCR amplified from HeLa cDNA and cloned into pcDNA3.1 (+). All the Glo1 point mutations were introduced through PCR-based site-directed mutagenesis using the oligos indicated in Supplementary Table 3.
All transfections were performed using Lipofectamine 2000 Reagent (Thermo Scientific) according to the manufacturer's instruction.
siRNA-mediated knockdown was performed by reverse transfection with 15 nM pools of four siRNAs against human SRC (siGENOME, Horizon Discovery #MU-003175-03-0002) or renilla luciferase as a negative control (siGENOME, Horizon Discovery #P-002070-01-50). Three days after transfection, the cells were lysed and analyzed by immunoblotting.
2.3. Western blot analysis
For immunoblotting experiments, the cells were lysed in (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 2 mM orthovanadate, 1x Roche Complete protease inhibitor cocktail with EDTA, and 1x Roche PhosSTOP phosphatase inhibitors). The samples were clarified by centrifugation (15 min, 14,000 rpm, 4 °C). The protein concentration of each sample was measured by BCA (ThermoFisher #23225) or Bradford (Biorad #500-0006). 15–20 μg of protein per sample were loaded on SDS-PAGE gels. The proteins were blotted onto nitrocellulose membrane, blocked with PBS-T containing 5% skim milk powder, incubated overnight with primary antibodies diluted 1:1000 in 5% BSA, and subsequently incubated for 1 h with HRP-conjugated secondary antibodies diluted 1:10,000 in 5% milk. The immunoblots were developed by the addition of ECL substrates (Western Lightning Plus-ECL, PerkinElmer #NEL105001EA, or Supersignal West Femto, Thermo Fisher #34095). A ChemiDoc imager (Biorad) was used for detecting and quantifying the signal.
2.4. Generation and quantification of rAAV8 viral particles
Recombinant rAAV8 viruses were generated in HEK293T cells and purified using iodixanol gradient ultracentrifugation [28,29]: 1.8 × 108 HEK293T cells were cultured in a ten chamber CellStack (Corning, USA) with DMEM + Glutamax (Gibco, Thermo Fisher Scientific, USA) supplemented with 10% FBS and 1% penicillin G/streptomycin. After 48 h, a 1:1:1 M ratio of pAAV-D377YmPCSK9-bGHpA plasmid [30] the rep-cap AAV8 helper plasmid; an adenoviral helper plasmid was mixed and transfected using polyethylenimine (PEI) (Polysciences Inc., Warrington, PA 18976 USA). Then, 72 h after transfection, the cells were harvested in 3 ml lysis buffer and lysed by four freeze–thaw cycles. The vector particles were purified using an iodixanol (Progen, Germany) gradient consisting of four phases with decreasing density (60%, 40%, 25%, 15%) spun at 50,000×g for 135 min at 4 °C. Approximately 3 ml of the 40% phase, in which predominantly full virus particles accumulate, were recovered with a 27G needle. Finally, the vector solution dialyzed into PBS (Zeba Spin Desalting Columns 7K MWCO, Thermo Scientific, USA) and concentrated (VivaSpin 10K MWCO, Sartorius, Germany). Viral titer was quantified as genome copy numbers per ml using a qPCR SYBR-Green assay (Biorad) with primers bGHpA-fw 5′ and bGHpA-rev (sequences in Suppl. Table 3) [31].
2.5. Mouse breeding, housing conditions, and extraction of mouse tissues
All animal experiments were approved by the ethical authorities (Regierungspräsidium Karlsruhe, Germany) and performed in accordance with the German Animal Welfare Act for the care and use of laboratory animals and the rules of the regulatory authorities in Baden-Württemberg
For hypercholesterolemia and hyperglycemia (HGHCi) mice: Female wild-type C57BL/6N were obtained from Charles River Laboratories (Wilmington, MA, USA). All animal experiments complied with the ARRIVE guidelines and were carried out following the Directive 2010/63/EU guidelines. Adeno-associated viral vectors encoding the gain-of-function variant D377Y of murine PCSK9 (rAAV8-PCSK9D377Y) under control of a liver-specific promoter were delivered via a single retro-orbital sinus injection (1.0 × 1011 viral genomes/mouse). To induce chronic hypercholesterolemia the animals were fed a high-cholesterol/high-fat Paigen diet (PD, containing 16% fat, 1.25% cholesterol, and 0.5% sodium cholate). To induce chronic hyperglycemia, mice were injected with streptozotocin (STZ, 60 mg/kg, intraperitoneally, once daily for five consecutive days, freshly dissolved in 0.05 M sterile sodium citrate, pH 4.5) one week after rAAV8-PCSK9D377Y application. Blood glucose levels were monitored twice per week using Accu-Chek Aviva (Roche, USA) and maintained in a range of 300–500 mg/dl. Body weight was measured once weekly. HFD and hyperglycemia (minimum 300 mg/dl) were maintained for 6 weeks. Mice were then sacrificed by cervical dislocation. Liver tissue was shortly washed in ice-cold NaCl and immediately snap frozen in liquid nitrogen [28,29,32,33].
Diabetic heterozygous Ins2Akita/+ (Ins2Akita) mice, purchased from Jackson Laboratory (Charles River Laboratories, Germany), were bred at the animal facility of the University Hospital Mannheim, Heidelberg University. Age-matched non-diabetic homozygous Ins2+/+ littermates served as controls. Blood glucose was monitored throughout the study period via punctuation of the tail using a syringe cannula via a BG Star blood glucose meter (Sanofi-Aventis Deutschland GmbH, Germany). Insulin was occasionally administered to prevent critical weight loss. All animals were housed in groups of 3–4 animals at 21 ± 1 °C with a 12-h light–dark cycle. At the end of the experimental period, animals were killed by cervical dislocation under intraperitoneal ketamine/xylazine anesthesia. Unless stated otherwise, all reagents were obtained from Sigma–Aldrich Chemie GmbH (Seelze, Germany).
2.6. Preparation of mouse tissue lysates
Livers that were previously snap-frozen in liquid nitrogen and stored at −80 °C, were ground on dry ice using a glass homogenizer and lysed by adding (40 mM Tris pH 7.4, 1% Triton X-100, 10 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 30 mM NaF, 2x Roche Complete protease inhibitor cocktail with EDTA, and 2x Roche PhosSTOP phosphatase inhibitors). The lysates were cleared by centrifugation (15 min, 14,000 rpm, 4 °C).
2.7. RNA extraction, reverse transcription, and qPCR
RNA was extracted from cultured cells with Trizol (Life Technologies #15596018) following the manufacturer's instructions. Next, 2 μg RNA per sample were reverse-transcribed with Maxima H Minus reverse transcriptase (Thermo Fisher Scientific, #EP0753) and oligo-dT primer (Supplementary Table 3). Quantitative PCR was performed using an SYBRGreen-based master mix (primaQUANT CYBR 2x qPCR SYBRGreen Master Mix with LOW ROX, Streinbrenner, #SL-9913).
2.8. Recombinant human Glo1 protein purification
The hGlo1 coding sequence was cloned into the pEMT-11 plasmid (EMBL protein expression/purification facility, Heidelberg) and expressed in Rosetta™(DE3) Competent Cells (Novagen #70954). Protein expression was induced by the addition of 0.5 mM IPTG to a growing culture. After incubating the culture for 4 h at 37 °C, the bacteria were spun down, lysed and the recombinant protein was purified with a Ni-NTA agarose affinity resin under native conditions according to the manufacturer's instruction (Qiagen #R90101).
2.9. Glo1 activity assay
Cells were seeded in a 6-well plate the day before the experiment. Homogenization was performed with 400 μL buffer (10 mM sodium phosphate buffer pH 7.0, 0.02% Triton X-100) and sonication using a Branson sonifier for 15 s with 10% amplitude, followed by centrifugation at 14,000 rpm for 30 min at 4 °C. Substrate mix (50 mM sodium phosphate buffer pH 6.6, 20 μM methylglyoxal (Sigma M0252), 20 μM reduced l-Glutathione (Sigma G6013)) was incubated in a 96-well microtiter plate at 37 °C for 10min. Thereafter, 10 μL of the cleared lysates were added to a 190 μL substrate mix in each well. Changes in absorbance at 235 nm were then measured kinetically for 15 min using the SPECTROstar Omega plate reader (BMG LABTECH).
2.10. In vitro kinase assay
For in vitro kinase assays, human Glo1 recombinant protein was mixed with 600 ng of recombinant human kinases (ProQinase), in 1x kinase assay buffer (50 mM HEPES pH 7.4, 6 mM MgCl2, 6 mM b-glycerophosphate, 1 mM DTT), and 500 μM ATP in a 25 μL total volume and incubated for 1 h at 30 °C. The phosphorylation reaction was stopped by adding 2x Laemmli and boiling the sample for 5 min at 95 °C.
2.11. Quantifications and statistical analyses
Immunoblots were quantified using Image Lab (Biorad). Statistical analyses were performed using GraphPad Prism.
3. Results
3.1. Mutation of Tyr136 reduces Glo1 activity
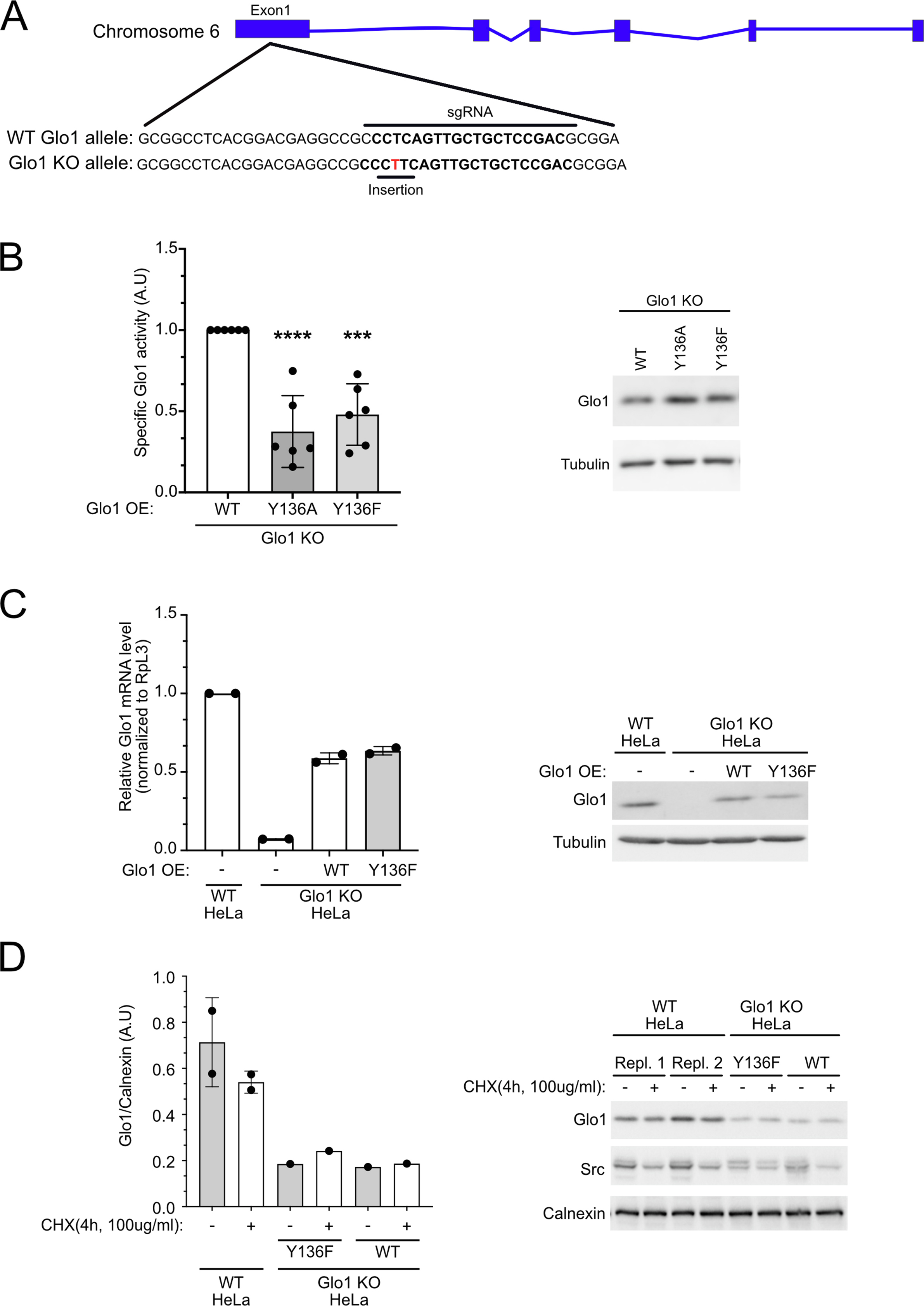
To study the regulation of human Glo1 by post-translational modifications (PTMs), we first analyzed publicly available proteomics databases and generated a list of all residues in Glo1 observed to be either phosphorylated or acetylated by mass spectrometry (Suppl. Table 1). We then systematically screened these residues by introducing mutations into Glo1 that prevent the PTMs – alanine in the case of phospho-residues and arginine for acetylated lysines – and assayed the effect on Glo1 enzymatic activity. In order to assay only the activity of mutated Glo1, we first generated Glo1 knockout HeLa cells via CRISPR/Cas9 (Suppl. Figure 1A) and then reconstituted these cells with mutant forms of Glo1. The Glo1 knockout (Glo1KO) cells had no detectable Glo1 protein or Glo1 activity (lower and upper panels in Figure 1A, respectively). Expression of wildtype Glo1 reconstituted Glo1 protein levels and activity to roughly endogenous levels (last lanes, Figure 1A). For each mutant, we calculated Glo1 specific activity by normalizing Glo1 enzymatic activity to Glo1 proteins levels and found that mutation of Tyr136 to alanine consistently reduced Glo1 specific activity (Figure 1A). Mutation of Tyr136 gave the strongest and most consistent phenotype; therefore, we focused on Tyr136 here. Since alanine is structurally different from tyrosine, we also mutated Tyr136 to phenylalanine, which only differs by one hydroxyl group. This mutation also consistently reduced Glo1 specific activity (Figure 1B). Compared to Y136A, Y136F caused a slight decrease in Glo1 activity (Suppl. Figure 1B).
Figure 1.

Regulation of Glo1 activity via tyrosine 136 phosphorylation.(A) Mutation of Glo1 Y136 to alanine reduces Glo1 activity. Glo1 knockout HeLa cells were used to express 9 different point mutants of Glo1 or the wildtype protein as a control. Glo1 protein levels were quantified by immunoblotting and imaging with a Chemidoc (lower panel) and used to normalize Glo1 activity measurements, to arrive at Glo1 specific activity (upper panel). n = 4, ∗p < 0.05, ∗∗p < 0.01 by mixed-effect ANOVA with Geisser-Greenhouse correction. (B) Mutation of Glo1 Y136 to phenylalanine reduces Glo1 activity. Glo1 protein levels and activity were quantified as in panel A. In all assays, Glo1 activity was normalized by total Glo1 protein. n = 10, ∗∗∗p < 0.001 by two-sided unpaired t-test.
Since our calculation of specific activity normalizes away possible effects of the mutation on Glo1 protein stability, we assayed this separately but found no effect: The Y136F mutation neither affected the ratio of Glo1 protein to mRNA levels in the cell (Suppl. Figure 1C) nor the stability of Glo1 protein in a cycloheximide chase experiment (Suppl. Figure 1D). In sum, we conclude that mutation of Y136 to non-phosphorylatable phenylalanine specifically reduces Glo1 enzymatic activity. A direct evidence that Y136 phosphorylation activates Glo1 activity is provided below using an in vitro kinase assay.
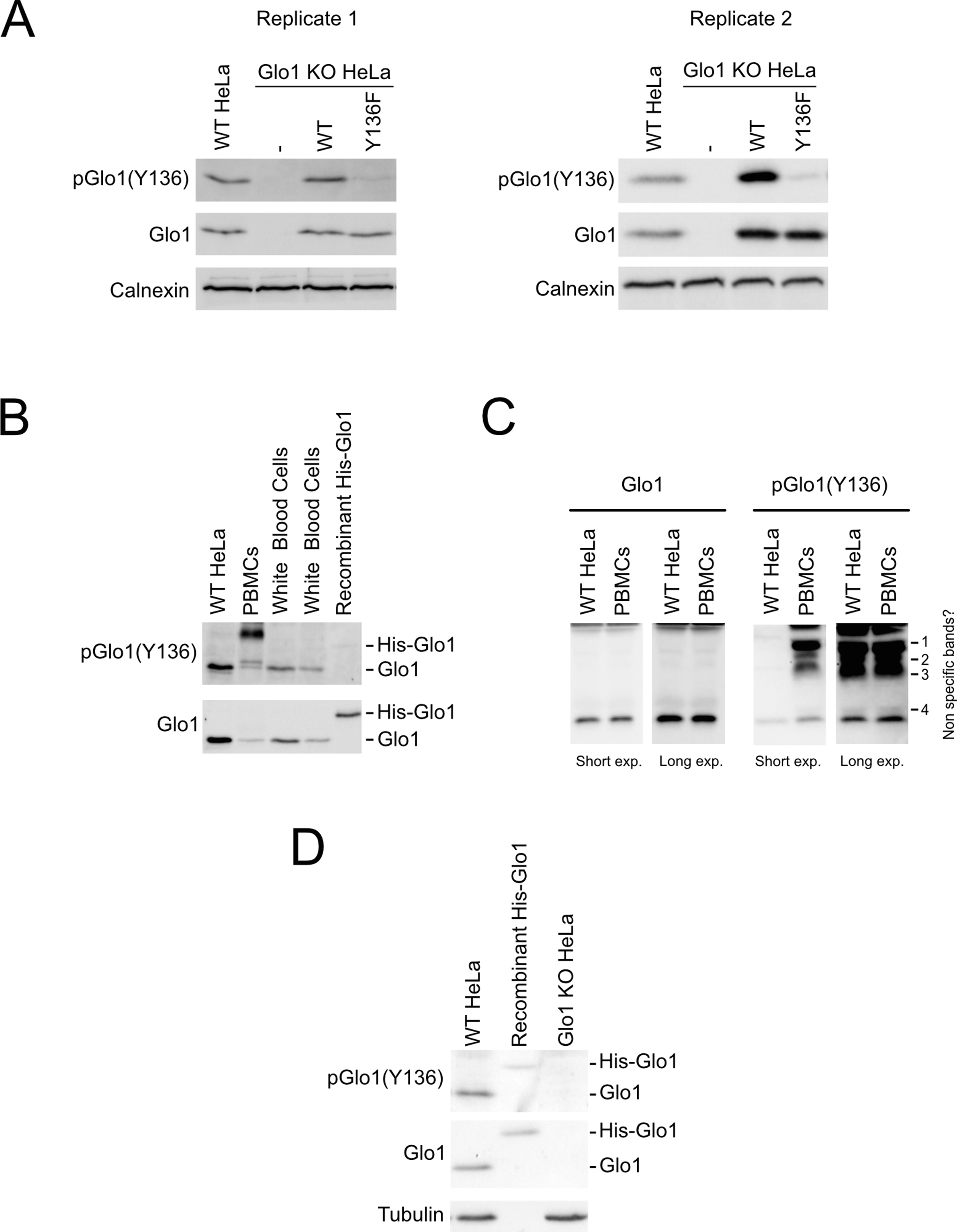
To verify whether Y136 is phosphorylated in vivo, we generated a Glo1(Y136) phospho-specific antibody. This antibody detects endogenous Glo1 in wildtype HeLa cells but not Glo1KO cells, and it detects the Y136F mutant much less strongly than wildtype Glo1 (Suppl. Figure 2A). As an additional control for phospho-specificity, this antibody gives a significantly weaker signal with recombinant His-tagged wildtype Glo1 purified from bacteria, which do not phosphorylate tyrosine residues (Suppl. Figure 2B and D). Using this antibody, we found that Glo1(Y136) is phosphorylated in several different cell types (see below) including HeLa and primary white blood cells (Suppl. Figure 2B). In the peripheral blood mononuclear cell (PBMC) sample additional bands were recognized by the pGlo1(Y136) antibody that was not detected by the total Glo1 antibody, suggesting they are unspecific (Suppl. Figure 2B and C).
3.2. Y136 is redundantly phosphorylated by multiple kinases, including Src-family kinases
We next sought to identify the kinases responsible for phosphorylating this site. To this end, we generated a library of sgRNAs with two independent sgRNA constructs targeting each of the 90 tyrosine kinases. We screened this library in an arrayed format, combining the two sgRNAs targeting one kinase together into one well, transiently selecting the transfected cells with puromycin for 3 days, and then assaying Glo1(Y136) phosphorylation by immunoblotting (Figure 2A). This led to the efficient depletion of 5 of the 6 kinases that we tested by immunoblotting (Suppl. Figure 3A). From this screen (Suppl. Figure 3B) we selected the most promising candidates for an additional round of testing (Figure 2B). Depletion of multiple different kinases led to a partial reduction in Glo1(Y136) phosphorylation. These included members of the Src family (Src, Yes1, FGR, and the related Abl1), and of the FAK, EPHA, FGFR, and VEGFR families (Figure 2B), suggesting phosphorylation of Glo1 on Y136 by multiple different kinases. In vitro kinase assays revealed that all the members of the Src family, as well as Epha5 and VEGFR3, can efficiently phosphorylate recombinant Glo1 on Y136 (Figure 2C–D). Contrastively, Fak1 could not phosphorylate Glo1(Y136) in vitro (Figure 2C, Suppl. Figure 4C), suggesting the effect of Fak1 knockdown on Glo1(Y136) phosphorylation in vivo may be indirect.
Figure 2.

Glo1 Y136 is phosphorylated by multiple different kinases including all members of the Src family.(A) Schematic diagram illustrating the tyrosine kinase CRISPR-Cas9 screen. A library of sgRNAs was generated to target each of the 90 known tyrosine kinases. HeLa cells were transfected with two sgRNAs for each kinase simultaneously. After puromycin selection, immunoblots were performed on lysates from each well using the pGlo1(Y136) antibody. (B) Knockdown of several different kinases in HeLa cells leads to a partial reduction in Glo1 Y136 phosphorylation. Tyrosine kinase knockouts were performed as in panel A, and immunoblots were done on the transiently-selected cell pools. Knockout of all tyrosine kinases is shown in Suppl. Figure 2. Shown here are two biological replicates of the promising candidates (47 of the 90 kinases) which showed a reduction in pGlo1(Y136) in the main screen. Knockouts causing a drop in the pGlo1/total Glo1 ratio in both biological replicates are indicated in blue. (C–D) Multiple different kinases can phosphorylate Glo1 Y136 in an in vitro kinase assay, including members of the Src family. (E) siRNA-mediated knockdown of Src kinase recapitulates results from the CRISPR-Cas9 screening (Suppl. Figure 3B), causing reduced Glo1(Y136) phosphorylation. A pool of 4 siRNAs was used to knock down either Src kinase or luciferase as negative control (“control”). n = 9, ∗∗∗p = 0.002 by 2-sided unpaired t-test. (F) Inhibition of the Src kinase family does not lead to a drop in Glo1 Y136 phosphorylation. HeLa cells were incubated for 30 min with either the broad range Src inhibitor Saracatinib and/or the Abl1 inhibitor Nilotinib. Inhibition of the Src kinase family was confirmed using pSrc(Y417) or pFak (Y925) antibodies.
Since members of the Src family are particularly active in phosphorylating Glo1(Y136), we tested whether they are the predominant kinases phosphorylating Glo1(Y136) in vivo. Knockdown of Src with siRNAs recapitulated the effect of Src knockout using sgRNAs on pGlo1(Y136) (Figure 2E). Since independent reagents targeting Src (sgRNA and siRNAs) cause a reduction in Glo1(Y136) phosphorylation, this indicates the effect is on-target. The reduction in Glo1 phosphorylation is partial, indicating that Src is not the only kinase phosphorylating Glo1(Y136) in vivo. To test whether inhibition of the entire Src family yields a stronger drop in Glo1(Y136) phosphorylation, we treated HeLa cells for 30 min with Saracatinib, a broad inhibitor of Src family kinases, with Nilotinib which inhibits Abl, or with a combination of the two (Figure 2F). However, these drugs did not cause a drop in Glo1(Y136) phosphorylation despite markedly reducing Fak phosphorylation as a positive control. As 30 min may be insufficient to cause a drop in Glo1 phosphorylation, we also tested Src inhibitors Saracatinib, Dasatinib, and PP2 overnight but this also could not cause a drop in pGlo1(Y136) phosphorylation (Suppl. Figure 4A and B). We do not know why we see a discrepancy between genetic and pharmacological inhibition. One option is that inhibition of the entire family of Src kinases may induce a compensatory effect that is not observed with inhibition of Src only. Alternatively, the consequences of Src inhibition depend on another parameter (e.g., growth factor stimulation as seen in [34]) that we are not controlling and differs between the genetic and pharmacological inhibition setups.
3.3. Phosphorylation of Glo1 on Y136 increases its activity
We next sought to confirm that phosphorylation of Y136 increases Glo1 activity, as predicted from the point mutations presented in Figure 1. To this end, we in vitro phosphorylated recombinant Glo1 with either Src or Lyn and found that this significantly increased Glo1 activity (Figure 3A). This increase in activity was largely blunted when Y136 was mutated to phenylalanine (Figure 3B) indicating it is largely due to the phosphorylation of Y136. There was still a residual effect of Src on Glo1(Y136F) suggesting that Src may also phosphorylate additional stimulatory sites on Glo1. Notably, in vitro phosphorylation reactions typically lead to phosphorylation of a small fraction of the substrate protein, in this case, Glo1, present in the assay. Consistent with this, we find that phosphorylated Glo1 runs higher than total Glo1 on a well-resolved 15% SDS-PAGE gel, and this corresponds to a minor fraction of total Glo1 protein because an upper band is hardly visible on the total Glo1 immunoblot (Suppl. Figure 5A). Hence, our in vitro kinase assay underestimates the magnitude of the effect of Y136 phosphorylation on Glo1 activity.
Figure 3.

Phosphorylation of Glo1 on Y136 by Src kinases increases its activity.(A–B) In vitro phosphorylation of Glo1 Y136 increases Glo1 activity. Recombinant non-phosphorylated Glo1(WT) or Glo1(Y136F) were phosphorylated in vitro with either Src (A–B) or Lyn (A) kinases and subsequently, Glo1 activity was determined. (B top panel) Immunoblot of Glo1(WT) or Glo1(Y136F) in vitro phosphorylated or not with Src kinase. n = 3 for (A), n = 5 for (B). For all panels, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001 by one-way ANOVA (multiple comparations).
3.4. Glo1(Y136) phosphorylation drops in response to hyperglycemia in cell culture
We next tested whether Glo1(Y136) phosphorylation changes in response to different stimuli. Methylglyoxal, the substrate of Glo1, is produced as a byproduct of glycolysis. Therefore, stimuli or environmental conditions that alter glycolytic rates and hence MG production could be expected to regulate Glo1 activity. This would allow a cell's MG detoxification capacity to adjust to the level of MG production. In HeLa cells, we found no stimulus that affected Glo1 phosphorylation. However, in myeloid A-937 cells and liver HepG2 cells, we found that Glo1(Y136) phosphorylation responds to glucose concentration in a bimodal fashion. As glucose in the medium increases from 0 mM to physiological 5 mM, Glo1(Y136) phosphorylation increases (Figure 4A). This effect should be cytoprotective since it leads to elevated MG detoxification when MG production increases due to increased glucose. When glucose levels increase from physiological 5 mM to hyperglycemic 25 mM, Glo1(Y136) phosphorylation shows a decrease (Figure 4B and C). As predicted, this decrease in Y136 phosphorylation was accompanied by a decrease in Glo1 activity (Figure 4D–E). This decrease in Glo1 phosphorylation and activity in 25 mM glucose was the opposite of what we expected since it indicates that cells experiencing hyperglycemia, where MG production is elevated, have reduced Glo1-dependent detoxification capacity. We tested whether Glo1 Y136 phosphorylation is affected by MG, but this was not the case (Suppl. Figure 6A), suggesting that the effects of glucose on Y136 phosphorylation are not via MG production. Y136 phosphorylation was also not affected by a serum that contains insulin (Suppl. Figure 6B).
Figure 4.

Glucose levels regulate Glo1 Y136 phosphorylation and Glo1 activity in HepG2 and U-937 cells.(A–C) Glo1 Y136 phosphorylation responds to glucose levels in a bimodal fashion, increasing from 0 mM to 5 mM glucose (A) and then decreasing when cells are cultured with a medium containing high glucose concentrations (B–C). Cells were cultured overnight in low (0–2 mM), physiological (5 mM), or hyperglycemic (25 mM) glucose concentrations. n = 4 biological replicates for (A), 5 biological replicates for (B), and 2 biological replicates for (C). Error bars = std. dev. (D–E) Glo1 activity decreases in HepG2 cells (A) or U-937 cells (B) when cells are cultured with a medium containing high glucose concentrations. Cells were treated with normal or high glucose overnight as in B–C, followed by quantification of Glo1 activity. n = 10 biological replicates for D, and 2 biological replicates for E. For all panels, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001 by two-sided unpaired t-test.
3.5. Glo1(Y136) phosphorylation drops in experimental diabetes
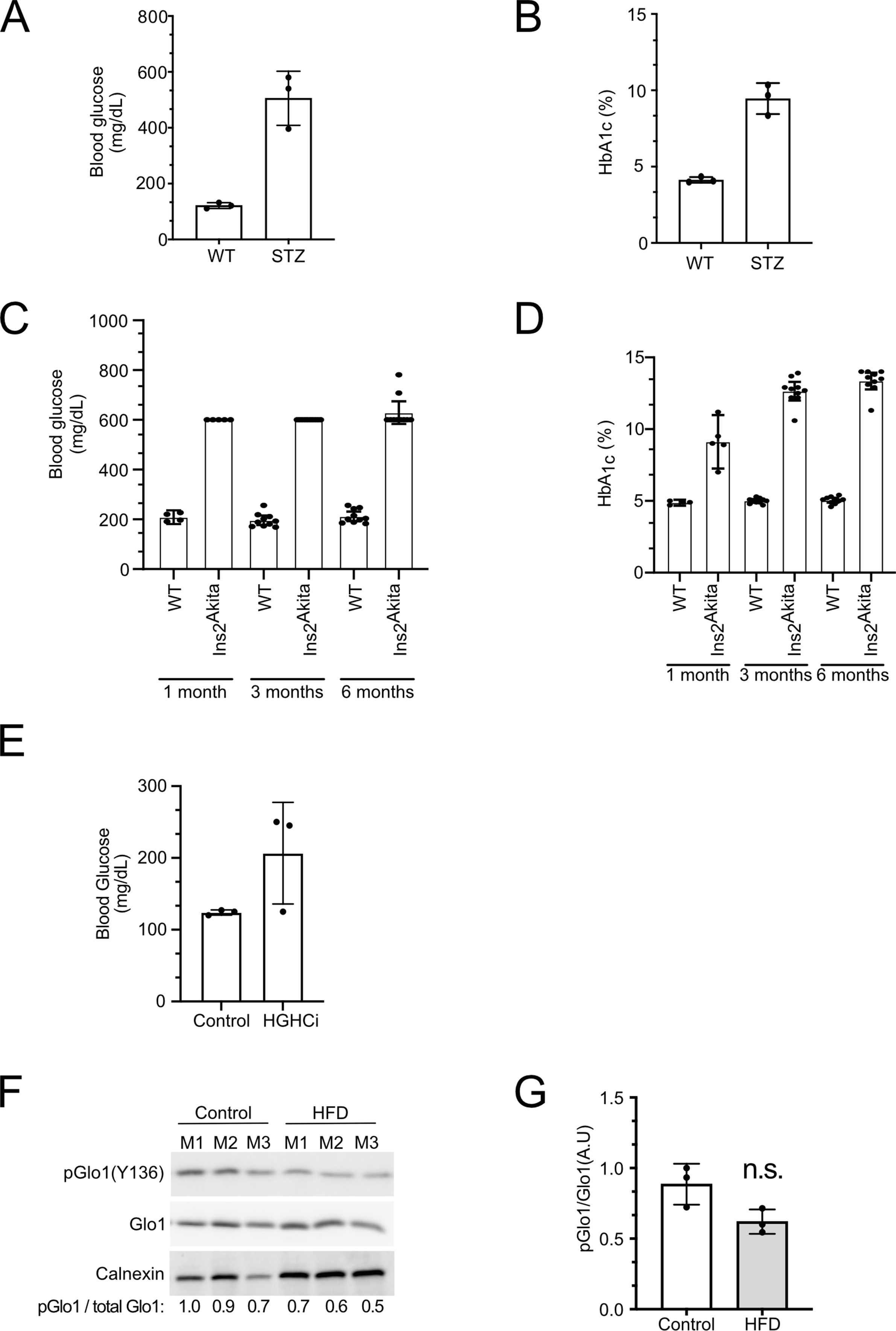
We next tested whether Glo1(Y136) phosphorylation also drops in mouse models with elevated blood glucose [[37], [38], [39]]. We and others previously observed that diabetic mice have reduced Glo1 activity, for instance in the liver and kidney, although the molecular mechanism for this observation was not known [19,40]. We found that Glo1(Y136) phosphorylation is reduced in livers of mice treated with streptozotocin, which kills pancreatic beta cells leading to reduced insulin signaling and hyperglycemia (Figure 5A and B, Suppl. Figure 7A and B). Likewise, Glo1(Y136) phosphorylation is reduced in livers of Ins2Akita mice which have dysfunctional insulin 2 genes at 3 and 6 months of age (Figure 5E–H, Suppl. Figure 7C and D). Surprisingly, at 1 month of age, Ins2Akita mice had higher, not lower pGlo (Y136) compared to controls (Figure 5C and D) despite being hyperglycemic (Suppl. Figure 7C). Although at 1 month the glycated hemoglobin (HbA1c) levels of the Ins2Akita mice are not as elevated as at 3 or 6 months (Suppl. Figure 7D), this suggests additional factors also affect phosphorylation of Glo1 on Y136. Lastly, Glo1(Y136) phosphorylation is also reduced in mice that are hypercholesterolemic and hyperglycemic, induced by expressing the D377Y variant of murine PCSK9 in the liver, injecting the animals with streptozotocin, and feeding them a high-fat diet (Figure 5I and J, Suppl. Figure 7E). A high-fat diet alone, which causes hyperglycemia, also reduces Glo1 (Y136) phosphorylation, although not significantly (Suppl. Figure 7F and G). Together with the cell-culture-based data, the above findings suggest that Glo1 (Y136) phosphorylation is responsive to glucose levels in vivo.
Figure 5.

Glo1 Y136 phosphorylation is reduced in livers of diabetic mice.(A-J) Glo1 Y136 phosphorylation quantified by immunoblotting liver extracts of control or STZ (A–B), Ins2Akita (C–H), or high-glucose high-cholesterol HGHCi mice (I–J). Ins2Akita and matched control mice were assayed at 1, 3 or 6 months of age, as indicated. Representative immunoblots are shown in (A, C, E, G, I). Quantifications of multiple biological replicates are in (B, D, F, H, J). (A) n = 3 biological replicates, (B) n = 3 biological replicates x 4 technical replicates, (C–D) n = 5 biological replicates, (E–F) n = 5 biological replicates for WT and 7 biological replicates for Ins2Akita, (G–H) n = 5 biological replicates, (I–J) n = 3 biological replicates x 2 technical replicates. For panel (I) the total-Glo1 blot is a re-hybridization of the phospho-Glo1 membrane. For all panels, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001 by two-sided unpaired t-test.
4. Discussion
Impaired Glo1 activity has been shown to have physiological effects in several systems. Glo1 knockout flies develop obesity, insulin resistance and hyperglycemia with time [14]. Glo1 knockout fish develop post-prandial hyperglycemia and become insulin resistant and hyperglycemic when overfed [15]. Glo1 knockout mice on a standard diet do not have elevated MG due to elevated compensatory MG detoxification by other enzymes [19], suggesting a second genetic or environmental ‘hit’ such as a high-sugar diet may be required to induce diabetic late complications in mice. Nonetheless, Glo1 knockdown mice on a standard diet have impaired proteostasis [41]. Thus, understanding how Glo1 activity is regulated will improve our understanding of how this enzyme can affect animal physiology and pathophysiology. We identify here phosphorylation of Glo1 on Y136 as a posttranslational modification that affects Glo1 activity. Our data indicate that loss of phosphorylation on Y136 leads to a decrease in Glo1 activity by roughly 40% (Figure 1B).
Glo1 was previously shown to have a flexible loop near that active site which opens to allow entry of substrate to the active site [24,35,36]. This flexible loop is positively charged, containing two lysine residues (Lys156, Lys158) (Figure 6A). Tyrosine 136 is positioned such that its phosphorylation, which introduces a negative charge, would be predicted to cause electrostatic attraction of the flexible loop and opening of the catalytic site. This suggests a rationale for how Y136 phosphorylation can affect Glo1 activity.
Figure 6.

Location of Y136 in Glo1 and schematic diagram illustrating the positive feedback loop between MG and glucose.(A) Glo1 Y136 is located in proximity to a flexible loop (blue) near the catalytic site of the enzyme which gates entry of substrate to Glo1. Glo1 structure was visualized using UCSF ChimeraX [43]. (B) In this study, we find that hyperglycemia impairs Glo1 activity by reducing Y136 phosphorylation. This contributes to elevated MG. From published results, elevated MG can lead to hyperglycemia.
Y136 phosphorylation responds in a bimodal fashion to glucose levels. Up to physiological glucose levels, Y136 phosphorylation increases with increasing glucose. Since increased glucose metabolism leads to elevated MG production [42], this mechanism plays a protective role, increasing Glo1 activity, and MG detoxification when elevated levels of MG are produced by cells. Glo1 is not directly activated by its substrate MG (Suppl. Figure 6A), which is perhaps too toxic, but preemptively by the nutrient, glucose, which yields MG. When glucose levels increase above 5 mM in cell culture, we find that Y136 phosphorylation drops. This is recapitulated in several diabetic mouse models. This regulation is maladaptive as it results in reduced MG detoxification when MG production is high. An earlier study showed that Glo1 activity is reduced by ∼50% in livers of STZ-induced diabetic mice [19]. The drop we see in Glo1 phosphorylation could explain this effect in part, although additional PTMs may also be at play.
We found that several diverse kinases could phosphorylate Glo1 on Y136 including all members of the Src family, Abl1, Epha5, and VEGFR3. We could not identify one kinase as the predominant one responsible for phosphorylating Y136 in cells, suggesting Y136 might be phosphorylated by multiple kinases in accordance with our in vitro kinase assay. Since MG is toxic, redundant phosphorylation of Y136 by multiple kinases might have evolved evolutionarily to avoid that MG-detoxification via Glo1 is reliant on a single signaling pathway.
Studies in the future are required to understand mechanistically the bimodal response of Y136 phosphorylation to glucose. Several kinases such as AMPK are known to be glucose-responsive, but these are serine/threonine kinases, not tyrosine kinases. One option is that one set of tyrosine kinases responds bimodally to glucose levels, first increasing and then decreasing as glucose levels increase. Alternatively, one set of kinases may increase activity up to 5 mM glucose, and another set of kinases may decrease activity at higher levels. Yet another option is that phosphatase activity may increase with hyperglycemia. These options are difficult to disentangle given that multiple kinases are involved.
Combining the data we present herewith previously published work suggests the existence of a deleterious positive feedback loop whereby hyperglycemia and MG induce each other (Figure 6B): Given that MG results from a chemical breakdown of glycolytic intermediates, elevated glucose levels are thought to lead to increased MG production [42]. Our data indicate that hyperglycemia also impairs Glo1-mediated MG detoxification. Moreover, previous studies by us and others have shown that elevated MG leads to insulin resistance and hyperglycemia [14,15]. If such a vicious cycle exists in people, it would reinforce a state of high glucose and high MG in the body once hyperglycemia or elevated MG has started, possibly contributing to diabetes and diabetic complications. It will be interesting to explore this mechanism further in the future.
Acknowledgments
We thank Julia Szendrödi for her scientific discussions and critical reading of the manuscript and the S01 and S02 core projects of the SFB1118 for support and materials. Molecular graphics and analyses were performed with UCSFChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. D.P. received funding from the University of Heidelberg's Physician-Scientist-Program. This work was funded in part by a DFG SFB1118 grant to A.A.T, H.-P. H., P.P.N., and M.F.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101406.
Conflict of interest
None.
Appendix ASupplementary data
The following are the Supplementary data to this article:
Support to main figure 1. (A) Molecular characterization Glo1 KO HeLa cells. (B) Mutation of Glo1 Y136 to phenylalanine reduces Glo1 activity somewhat less strongly than mutation to alanine. Glo1 knockout HeLa cells were transfected to express either wildtype or mutant Glo1. Glo1 protein levels were quantified by immunoblotting and imaging with a Chemidoc imaging system (right panel), and these values were used to normalize Glo1 activity measurements to arrive at Glo1 specific activity (left panel). ∗p<0.05, ∗∗p<0.01 by one-way ANOVA (multiple comparations). (C–D) Glo1 Y136F mutation does not affect the stability of Glo1 protein, assayed by comparing Glo1 mRNA to protein (C) or using a cycloheximide timecourse (D). Glo1 KO HeLa cells were transfected either with WT or Y136F Glo1 and (C) the stability of Glo1 mRNA and protein were assessed by immunoblot and Q-RT-PCR, or (D) the cells were incubated with 100 μg/ml cycloheximide for 4 h to assay Glo1 protein stability in the absence of de novo synthesis.
{kind=link}
Additional support to main figure 1. (A–D) The phospho-Glo1(Y136) antibody is specific for Glo1 and preferentially detects Glo1 when phosphorylated on Y136. (A) Extracts from wildtype HeLa cells, or Glo1 KO cells transiently transfected with either WT or Y136F Glo1 were analyzed by immunoblotting with the pGlo1(Y136) antibody. In (B) and (D) recombinant His-tagged wildtype Glo1 protein from bacteria, which lack tyrosine phosphorylation, was also used as a control for phospho-specificity of the antibody. (B–C) Lysates from different cells lines were tested by immunoblotting to assess Glo1 phosphorylation. Note that on the pGlo1(Y136) immunoblot of the PBMC sample there are 4 bands that are not detected by the total Glo1 antibody even after a long exposure (B–C) suggesting they are non-specific.
{kind=link}
sgRNA screen knocking out all 90 tyrosine kinases to assay the effect on Glo1 Y136 phosphorylation. (A) sgRNA efficiency assessed by western blot for different tyrosine kinases. Wildtype HeLa cells were simultaneously transfected with 2 sgRNA constructs for each tyrosine kinase, transiently selected with puromycin for 3 days, and then lysed to assay the efficiency of knockout in the cell pool. All antibodies we tested are shown here. For JAK2, we are unsure whether the bands are specific. (B) CRISPR-Cas9 screen, knocking out all tyrosine kinases to assay their effect on Glo1 Y136 phosphorylation. Which number corresponds to which kinase is indicated in Suppl. Table 2. Note that the total Glo1 blot is a rehybridization of the pGlo1 (Y136) blot. (The total Glo1 signal is much stronger than the phospho-Glo1 signal).
{kind=link}
Support to main figure 2. (A–B) Long-term inhibition of all Src kinase family members does not lead to a drop in Glo1 Y136 phosphorylation. HeLa cells were incubated overnight with either the broad range Src inhibitor Saracatinib (A) or PP2 (B) or the broad range Src family/Abl1 inhibitor Dasatinib (A). As a negative control for PP2 treatments, PP3 was used. Inhibition of the Src kinase family was confirmed using pSrc(Y417) or pFak(Y925) antibodies. Members of the Src kinase family are Src, Yes, Fyn, Fgr, Lck, Hck, Blk, and Lyn. (C) Fak1 protein is active, as it can phosphorylate a protein in HeLa cell lysates. HeLa cell lysates were incubated with or without recombinant Fak1, ATP or the Fak1 inhibitor PF-431396 in an in the vitro kinase assay. The activity of Fak1 was assessed by immunoblotting with pTyr antibody PY99 which detects total tyrosine phosphorylation.
{kind=link}
Support to main figure 3. (A) In vitro phosphorylation of Glo1 only leads to phosphorylation of a small fraction of the Glo1 protein present in the assay. Recombinant non-phosphorylated WT Glo1 was phosphorylated in vitro with Src kinase and then run on an immunoblot to detect total Glo1 and phospho-Glo1(Y136). The membrane was cut in two parts (black line) to hybridize the two halves with the two different antibodies, and it was then re-assembled for exposure to obtain optimal alignment. Although the phosphorylated protein runs higher on this well-resolved 15% gel, it is hardly visible in the total-Glo1 blot, indicating it only represents a minor fraction of the total protein.
{kind=link}
Glo1 Y136 phosphorylation is not affected by methylglyoxal, serum, or insulin. (A) Glo1 phosphorylation in HepG2 cells incubated for 30 min with 0, 125 or 250 μM methylglyoxal. (B) Glo1 Y136 phosphorylation in HepG2 cells treated with control medium or medium lacking serum for 1 h, or serum-starved for 1 h followed by serum add-back for 1 h.
{kind=link}
(A–B) Blood glucose (A) and HbA1c (B) levels in the STZ-treated mice used in main Figure 5A–B. (C–D) Blood glucose (C) and HbA1c (D) levels in the Ins2Akita mice used in main Figure 5C–H. (E) Blood glucose levels in the high-glucose high-cholesterol (HGHCi) mice used in Main Figure 5I–J. (F–G) Glo1 Y136 phosphorylation quantified by immunoblotting liver extracts of control or high-fat diet-fed mice. (F) Representative immunoblot, (G) quantification of 3 biological replicates.
{kind=link}
References
- 1.Demir S., Nawroth P.P., Herzig S., Ekim Ustunel B. Emerging targets in type 2 diabetes and diabetic complications. Advancement of Science. 2021 doi: 10.1002/advs.202100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eckel R.H., Bornfeldt K.E., Goldberg I.J. Cardiovascular disease in diabetes, beyond glucose. Cell Metabolism. 2021;33(8):1519–1545. doi: 10.1016/j.cmet.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramasamy R., Yan S.F., Schmidt A.M. Methylglyoxal comes of AGE. Cell. 2006;124(2):258–260. doi: 10.1016/j.cell.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Wang H., Meng Q.H., Gordon J.R., Khandwala H., Wu L. Proinflammatory and proapoptotic effects of methylglyoxal on neutrophils from patients with type 2 diabetes mellitus. Clinical Biochemistry. 2007;40(16–17):1232–1239. doi: 10.1016/j.clinbiochem.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 5.Lu J., Randell E., Han Y., Adeli K., Krahn J., Meng Q.H. Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clinical Biochemistry. 2011;44(4):307–311. doi: 10.1016/j.clinbiochem.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Golej J., Hoeger H., Radner W., Unfried G., Lubec G. Oral administration of methylglyoxal leads to kidney collagen accumulation in the mouse. Life Sciences. 1998;63(9):801–807. doi: 10.1016/s0024-3205(98)00336-1. [DOI] [PubMed] [Google Scholar]
- 7.Berlanga J., Cibrian D., Guillen I., Freyre F., Alba J.S., Lopez-Saura P., et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clinical Science. 2005;109(1):83–95. doi: 10.1042/CS20050026. [DOI] [PubMed] [Google Scholar]
- 8.Duran-Jimenez B., Dobler D., Moffatt S., Rabbani N., Streuli C.H., Thornalley P.J., et al. Advanced glycation end products in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes. 2009;58(12):2893–2903. doi: 10.2337/db09-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Loreto S., Zimmitti V., Sebastiani P., Cervelli C., Falone S., Amicarelli F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. The International Journal of Biochemistry & Cell Biology. 2008;40(2):245–257. doi: 10.1016/j.biocel.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 10.Hansen C.S., Jensen T.M., Jensen J.S., Nawroth P., Fleming T., Witte D.R., et al. The role of serum methylglyoxal on diabetic peripheral and cardiovascular autonomic neuropathy: the ADDITION Denmark study. Diabetic Medicine. 2015;32(6):778–785. doi: 10.1111/dme.12753. [DOI] [PubMed] [Google Scholar]
- 11.Eberhardt M.J., Filipovic M.R., Leffler A., de la Roche J., Kistner K., Fischer M.J., et al. Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. Journal of Biological Chemistry. 2012;287(34):28291–28306. doi: 10.1074/jbc.M111.328674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bierhaus A., Fleming T., Stoyanov S., Leffler A., Babes A., Neacsu C., et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nature Medicine. 2012;18(6):926–933. doi: 10.1038/nm.2750. [DOI] [PubMed] [Google Scholar]
- 13.Riboulet-Chavey A., Pierron A., Durand I., Murdaca J., Giudicelli J., Van Obberghen E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes. 2006;55(5):1289–1299. doi: 10.2337/db05-0857. [DOI] [PubMed] [Google Scholar]
- 14.Moraru A., Wiederstein J., Pfaff D., Fleming T., Miller A.K., Nawroth P., et al. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metabolism. 2018;27(4):926–934 e928. doi: 10.1016/j.cmet.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Lodd E., Wiggenhauser L.M., Morgenstern J., Fleming T.H., Poschet G., Buttner M., et al. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight. 2019;4(12) doi: 10.1172/jci.insight.126154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thornalley P.J. The glyoxalase system in health and disease. Molecular Aspects of Medicine. 1993;14(4):287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 17.Thornalley P.J. Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochemical Society Transactions. 2003;31(Pt 6):1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 18.Morgenstern J., Fleming T., Schumacher D., Eckstein V., Freichel M., Herzig S., et al. Loss of glyoxalase 1 induces compensatory mechanism to achieve dicarbonyl detoxification in mammalian schwann cells. Journal of Biological Chemistry. 2017;292(8):3224–3238. doi: 10.1074/jbc.M116.760132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumacher D., Morgenstern J., Oguchi Y., Volk N., Kopf S., Groener J.B., et al. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Molecular Metabolism. 2018;18:143–152. doi: 10.1016/j.molmet.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Herreweghe F., Mao J., Chaplen F.W., Grooten J., Gevaert K., Vandekerckhove J., et al. Tumor necrosis factor-induced modulation of glyoxalase I activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):949–954. doi: 10.1073/pnas.012432399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Hemptinne V., Rondas D., Toepoel M., Vancompernolle K. Phosphorylation on Thr-106 and NO-modification of glyoxalase I suppress the TNF-induced transcriptional activity of NF-kappaB. Molecular and Cellular Biochemistry. 2009;325(1–2):169–178. doi: 10.1007/s11010-009-0031-7. [DOI] [PubMed] [Google Scholar]
- 22.Morgenstern J., Katz S., Krebs-Haupenthal J., Chen J., Saadatmand A., Cortizo F.G., et al. Phosphorylation of T107 by CamKIIdelta regulates the detoxification efficiency and proteomic integrity of glyoxalase 1. Cell Reports. 2020;32(12):108160. doi: 10.1016/j.celrep.2020.108160. [DOI] [PubMed] [Google Scholar]
- 23.de Hemptinne V., Rondas D., Vandekerckhove J., Vancompernolle K. Tumour necrosis factor induces phosphorylation primarily of the nitric-oxide-responsive form of glyoxalase I. Biochemical Journal. 2007;407(1):121–128. doi: 10.1042/BJ20070379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birkenmeier G., Stegemann C., Hoffmann R., Gunther R., Huse K., Birkemeyer C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Research. 2019;47(W1):W171–W174. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 2016;34(2):184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grieger J.C., Choi V.W., Samulski R.J. Production and characterization of adeno-associated viral vectors. Nature Protocols. 2006;1(3):1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 29.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 1999;6(6):973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 30.Bjorklund M.M., Hollensen A.K., Hagensen M.K., Dagnaes-Hansen F., Christoffersen C., Mikkelsen J.G., et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circulation Research. 2014;114(11):1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 31.Jungmann A., Leuchs B., Rommelaere J., Katus H.A., Muller O.J. Protocol for efficient generation and characterization of adeno-associated viral vectors. Human Gene Therapy Methods. 2017;28(5):235–246. doi: 10.1089/hgtb.2017.192. [DOI] [PubMed] [Google Scholar]
- 32.Seehaus S., Shahzad K., Kashif M., Vinnikov I.A., Schiller M., Wang H., et al. Hypercoagulability inhibits monocyte transendothelial migration through protease-activated receptor-1-, phospholipase-Cbeta-, phosphoinositide 3-kinase-, and nitric oxide-dependent signaling in monocytes and promotes plaque stability. Circulation. 2009;120(9):774–784. doi: 10.1161/CIRCULATIONAHA.109.849539. [DOI] [PubMed] [Google Scholar]
- 33.Shahzad K., Thati M., Wang H., Kashif M., Wolter J., Ranjan S., et al. Minocycline reduces plaque size in diet induced atherosclerosis via p27(Kip1) Atherosclerosis. 2011;219(1):74–83. doi: 10.1016/j.atherosclerosis.2011.05.041. [DOI] [PubMed] [Google Scholar]
- 34.Konig H., Copland M., Chu S., Jove R., Holyoake T.L., Bhatia R. Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Research. 2008;68(23):9624–9633. doi: 10.1158/0008-5472.CAN-08-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan Y., Lu T., Lovett P.S., Creighton D.J. Evidence for a (triosephosphate isomerase-like) "catalytic loop" near the active site of glyoxalase I. Journal of Biological Chemistry. 1995;270(22):12957–12960. doi: 10.1074/jbc.270.22.12957. [DOI] [PubMed] [Google Scholar]
- 36.Cameron A.D., Ridderstrom M., Olin B., Kavarana M.J., Creighton D.J., Mannervik B. Reaction mechanism of glyoxalase I explored by an X-ray crystallographic analysis of the human enzyme in complex with a transition state analogue. Biochemistry. 1999;38(41):13480–13490. doi: 10.1021/bi990696c. [DOI] [PubMed] [Google Scholar]
- 37.Kunjathoor V.V., Wilson D.L., LeBoeuf R.C. Increased atherosclerosis in streptozotocin-induced diabetic mice. Journal of Clinical Investigation. 1996;97(7):1767–1773. doi: 10.1172/JCI118604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oyadomari S., Koizumi A., Takeda K., Gotoh T., Akira S., Araki E., et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. Journal of Clinical Investigation. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrikopoulos S., Blair A.R., Deluca N., Fam B.C., Proietto J. Evaluating the glucose tolerance test in mice. American Journal of Physiology. Endocrinology and Metabolism. 2008;295(6):E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 40.Maher P., Dargusch R., Ehren J.L., Okada S., Sharma K., Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One. 2011;6(6) doi: 10.1371/journal.pone.0021226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Queisser M.A., Yao D., Geisler S., Hammes H.P., Lochnit G., Schleicher E.D., et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59(3):670–678. doi: 10.2337/db08-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rabbani N., Thornalley P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochemical and Biophysical Research Communications. 2015;458(2):221–226. doi: 10.1016/j.bbrc.2015.01.140. [DOI] [PubMed] [Google Scholar]
- 43.Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Science. 2021;30(1):70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Support to main figure 1. (A) Molecular characterization Glo1 KO HeLa cells. (B) Mutation of Glo1 Y136 to phenylalanine reduces Glo1 activity somewhat less strongly than mutation to alanine. Glo1 knockout HeLa cells were transfected to express either wildtype or mutant Glo1. Glo1 protein levels were quantified by immunoblotting and imaging with a Chemidoc imaging system (right panel), and these values were used to normalize Glo1 activity measurements to arrive at Glo1 specific activity (left panel). ∗p<0.05, ∗∗p<0.01 by one-way ANOVA (multiple comparations). (C–D) Glo1 Y136F mutation does not affect the stability of Glo1 protein, assayed by comparing Glo1 mRNA to protein (C) or using a cycloheximide timecourse (D). Glo1 KO HeLa cells were transfected either with WT or Y136F Glo1 and (C) the stability of Glo1 mRNA and protein were assessed by immunoblot and Q-RT-PCR, or (D) the cells were incubated with 100 μg/ml cycloheximide for 4 h to assay Glo1 protein stability in the absence of de novo synthesis.
Additional support to main figure 1. (A–D) The phospho-Glo1(Y136) antibody is specific for Glo1 and preferentially detects Glo1 when phosphorylated on Y136. (A) Extracts from wildtype HeLa cells, or Glo1 KO cells transiently transfected with either WT or Y136F Glo1 were analyzed by immunoblotting with the pGlo1(Y136) antibody. In (B) and (D) recombinant His-tagged wildtype Glo1 protein from bacteria, which lack tyrosine phosphorylation, was also used as a control for phospho-specificity of the antibody. (B–C) Lysates from different cells lines were tested by immunoblotting to assess Glo1 phosphorylation. Note that on the pGlo1(Y136) immunoblot of the PBMC sample there are 4 bands that are not detected by the total Glo1 antibody even after a long exposure (B–C) suggesting they are non-specific.
sgRNA screen knocking out all 90 tyrosine kinases to assay the effect on Glo1 Y136 phosphorylation. (A) sgRNA efficiency assessed by western blot for different tyrosine kinases. Wildtype HeLa cells were simultaneously transfected with 2 sgRNA constructs for each tyrosine kinase, transiently selected with puromycin for 3 days, and then lysed to assay the efficiency of knockout in the cell pool. All antibodies we tested are shown here. For JAK2, we are unsure whether the bands are specific. (B) CRISPR-Cas9 screen, knocking out all tyrosine kinases to assay their effect on Glo1 Y136 phosphorylation. Which number corresponds to which kinase is indicated in Suppl. Table 2. Note that the total Glo1 blot is a rehybridization of the pGlo1 (Y136) blot. (The total Glo1 signal is much stronger than the phospho-Glo1 signal).
Support to main figure 2. (A–B) Long-term inhibition of all Src kinase family members does not lead to a drop in Glo1 Y136 phosphorylation. HeLa cells were incubated overnight with either the broad range Src inhibitor Saracatinib (A) or PP2 (B) or the broad range Src family/Abl1 inhibitor Dasatinib (A). As a negative control for PP2 treatments, PP3 was used. Inhibition of the Src kinase family was confirmed using pSrc(Y417) or pFak(Y925) antibodies. Members of the Src kinase family are Src, Yes, Fyn, Fgr, Lck, Hck, Blk, and Lyn. (C) Fak1 protein is active, as it can phosphorylate a protein in HeLa cell lysates. HeLa cell lysates were incubated with or without recombinant Fak1, ATP or the Fak1 inhibitor PF-431396 in an in the vitro kinase assay. The activity of Fak1 was assessed by immunoblotting with pTyr antibody PY99 which detects total tyrosine phosphorylation.
Support to main figure 3. (A) In vitro phosphorylation of Glo1 only leads to phosphorylation of a small fraction of the Glo1 protein present in the assay. Recombinant non-phosphorylated WT Glo1 was phosphorylated in vitro with Src kinase and then run on an immunoblot to detect total Glo1 and phospho-Glo1(Y136). The membrane was cut in two parts (black line) to hybridize the two halves with the two different antibodies, and it was then re-assembled for exposure to obtain optimal alignment. Although the phosphorylated protein runs higher on this well-resolved 15% gel, it is hardly visible in the total-Glo1 blot, indicating it only represents a minor fraction of the total protein.
Glo1 Y136 phosphorylation is not affected by methylglyoxal, serum, or insulin. (A) Glo1 phosphorylation in HepG2 cells incubated for 30 min with 0, 125 or 250 μM methylglyoxal. (B) Glo1 Y136 phosphorylation in HepG2 cells treated with control medium or medium lacking serum for 1 h, or serum-starved for 1 h followed by serum add-back for 1 h.
(A–B) Blood glucose (A) and HbA1c (B) levels in the STZ-treated mice used in main Figure 5A–B. (C–D) Blood glucose (C) and HbA1c (D) levels in the Ins2Akita mice used in main Figure 5C–H. (E) Blood glucose levels in the high-glucose high-cholesterol (HGHCi) mice used in Main Figure 5I–J. (F–G) Glo1 Y136 phosphorylation quantified by immunoblotting liver extracts of control or high-fat diet-fed mice. (F) Representative immunoblot, (G) quantification of 3 biological replicates.