

Abstract
Synthetic, cationic random nylon-3 polymers (β-peptides) show promise as inexpensive antimicrobial agents less susceptible to proteolysis than normal peptides. We have used superresolution, single-cell, time-lapse fluorescence microscopy to compare the effects on live Escherichia coli cells of four such polymers and the natural antimicrobial peptides LL-37 and cecropin A. The longer, densely charged monomethyl-cyclohexyl (MM-CH) copolymer and MM homopolymer rapidly traverse the outer membrane and the cytoplasmic membrane. Over the next ∼5 min, they locally rigidify the chromosomal DNA and slow the diffusive motion of ribosomal species to a degree comparable to LL-37. The shorter dimethyl-dimethylcyclopentyl (DM-DMCP) and dimethyl-dimethylcyclohexyl (DM-DMCH) copolymers, and cecropin A are significantly less effective at rigidifying DNA. Diffusion of the DNA-binding protein HU and of ribosomal species is hindered as well. The results suggest that charge density and contour length are important parameters governing these antimicrobial effects. The data corroborate a model in which agents having sufficient cationic charge distributed across molecular contour lengths comparable to local DNA-DNA interstrand spacings (∼6 nm) form a dense network of multivalent, electrostatic “pseudo-cross-links” that cause the local rigidification. In addition, at times longer than ∼30 min, we observe that the MM-CH copolymer and the MM homopolymer (but not the other four agents) cause gradual coalescence of the two nucleoid lobes into a single dense lobe localized at one end of the cell. We speculate that this process involves coacervation of the DNA by the cationic polymer, and may be related to the liquid droplet coacervates observed in eukaryotic cells.
Significance
Certain sequence-random, synthetic cationic copolymers disrupt bacterial membranes, a mode of antibacterial action reminiscent of the action of natural antimicrobial peptides. We use single-cell fluorescence microscopy to directly compare the symptoms induced in Escherichia coli during the attack of several β-peptide polymers and the natural antimicrobial peptides LL-37 and cecropin A. Agents with sufficient contour length and positive-charge density locally dramatically attenuates the jiggling motion of the chromosomal DNA. The suggested mechanism is noncovalent, electrostatic cross-linking across nearby DNA stands. Once the membrane is breached, the bacterial cytoplasm with its preponderance of anionic biopolymers attracts a very high, damaging concentration of polycations. Such mechanistic studies may inform the design of new antimicrobial agents.
Introduction
Widespread infection by antibiotic-resistant bacteria necessitates the design and development of new antimicrobial agents that target resistant pathogens (1, 2, 3). Natural antimicrobial peptides (AMPs; also called host-defense peptides), part of the host innate immune system of plants and animals, are promising agents in this context. AMPs comprise an ancient class of short- and medium-sized polypeptides (typically <40 amino acids) that exhibit broad-spectrum antibacterial activity against both Gram-negative and Gram-positive bacteria (2). Most AMPs are amphipathic and highly cationic in physiological conditions. They are attracted to and permeabilize the negatively charged membranes of bacteria but leave the zwitterionic eukaryotic membranes intact (2). Permeabilization of the bacterial outer membrane (OM) and cytoplasmic membrane (CM), with the concomitant loss of the proton motive force and critical small molecules, has long been recognized as a primary cause of AMP activity. However, mounting evidence suggests that some AMPs may kill bacteria via non-membrane-permeabilization mechanisms that disrupt specific intracellular processes (2,4, 5, 6).
Recently several groups have shown that membrane permeabilization leads to a massive influx of certain cationic AMPs into the bacterial cytoplasm. The threshold concentrations of bound peptide molecules per cell needed to kill bacteria varies from 7 × 106 to 2 × 108, depending on the peptide and measuring method (1,6, 7, 8). We suggest that the preponderance of large, polyanionic species (chromosomal DNA, mRNA, rRNA, and ribosomes) renders the bacterial cytoplasm highly attractive to polycationic AMPs. Presumably the key driving force for AMP absorption is Coulombic attraction between polycations and polyanions enhanced by the entropically favorable concomitant release of small cations (mostly K+) to the cell surround (6).
We have used single-cell fluorescence microscopy to study the effects of AMPs on live Escherichia coli cells in real time (5,6,9,10). At concentrations comparable to the minimal inhibitory concentration (MIC), the human antimicrobial peptide LL-37 (+6 net charge, ∼10.4 nm in backbone contour length) permeabilizes both the OM and CM of E. coli to globular proteins and to smaller species on a timescale of several minutes (6). We estimated that on average each E. coli cell subsequently absorbs ∼108 LL-37 copies, corresponding to a mean concentration of ∼90 mM averaged over the entire cell volume (cell membranes, periplasm, and cytoplasm) (6).
We also tracked the motion of the DNA locus Right 2 with 30-nm localization accuracy before and after CM permeabilization by LL-37 (6). The local jiggling motion of Right2 essentially freezes within 1 min of CM permeabilization. The chromosomal DNA meshwork has apparently rigidified, at least locally. We speculated that the high concentration of absorbed polycationic LL-37 forms a dense, fluctuating network of noncovalent, electrostatic “pseudo-cross-links” within the chromosomal DNA. In addition, single-particle tracking showed that the average diffusion coefficient of ribosomal species decreases by a factor of 2, whereas motion of a substantial subpopulation essentially halts. Some of the 70S-polysomes are frozen in place. The damage inflicted on bacterial cells by AMPs goes well beyond membrane permeabilization. The parallel occurrence of multiple damage pathways helps explain why bacteria develop resistance to AMPs very slowly.
Modifications of natural AMP sequences have been actively investigated for their potential use as antimicrobial agents. However, such α-peptides suffer from the high cost of synthesis of a defined sequence and from susceptibility to proteolytic degradation. Substantial effort has been devoted to the development of synthetic analogs of natural AMPs that utilize different polymeric backbones. We have previously reported several sequence- and stereorandom nylon-3 polymers (β-peptide backbone) that mimic natural AMPs in antibacterial ability while minimizing hemolysis (11, 12, 13, 14, 15). Such sequence-random copolymers are much easier to synthesize than peptides, and they also resist proteolysis.
The primary purpose of this study is to test whether, like LL-37, the nylon-3 polymers can rigidify the chromosomal DNA and diminish ribosome mobility after penetration of the CM.
By testing polymers of varying composition and mean chain length, we can begin to understand what chemical and physical properties enable these effects. Typical DNA-DNA nearest interstrand distances in E. coli are ∼6 nm (16). We reasoned that the ideal pseudo-cross-linking agent should have sufficient backbone contour length to span at least a 6-nm gap and sufficient cationic charge density at both ends to enable strong electrostatic binding to DNA. Accordingly, we have studied two chemically well-defined AMPs (LL-37 and cecropin A) and four polymer mixtures with different charge densities and mean backbone contour lengths as described in Table 1. Our study mainly focuses on one of the polymers called MM-CH, which is copolymerized from monomethyl β-lactam (MM) and cyclohexyl β-lactam (CH) monomers, mixed in a 65:35 molar ratio. The MM subunit after deprotection has an ammonium group, which has a positive charge at the physiological pH. The CH subunit is hydrophobic. The results reveal the multipronged nature of the attack of MM-CH on E. coli and support formation of a dense meshwork of DNA-DNA pseudocrosslinks shortly after the polymer gains access to the cytoplasm. Cecropin A, similar to LL-37 in length and with the same +6 net charge, has its positive charges concentrated toward the N-terminus and is less effective in DNA rigidification. The two longest, most densely charged polymers (MM-CH and MM homopolymer) are comparable to LL-37 in their degree of DNA rigidification. The two shorter, densely charged polymers (dimethyl-dimethylcyclopentyl (DM-DMCP) and dimethyl-dimethylcyclohexyl (DM-DMCH)) are less effective. The rigidification of DNA and the cytoplasm greatly inhibits the essential motion of ribosomal species and of the DNA-binding protein HU.
Table 1.
Comparison of physical and chemical properties of AMPs and polymers
| Peptide/polymera | Sequence/structure | Contour lengthb | Net charge | Net charge density | Rigidify DNA? | Coalesce DNA lobes? |
|---|---|---|---|---|---|---|
| MM-CH | 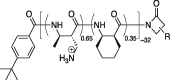 |
11.5 nm | Avg +21 | +1.83 e/nm | Strong | Yes |
| MM homopolymer | 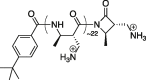 |
7.9 nm | Avg +22 | +2.78 e/nm | Strong | Yes |
| LL-37 | LLGDFFRKSKEKIG | 10.4 nm | +6 | +0.58 e/nm | Strong | No |
| KEFKRIVQRIKDFL | ||||||
| RNLVPRTES | ||||||
| Cecropin A | KWKLFKKIEKVG | 10.4 nm | +6 | +0.58 e/nm | Less effective | No |
| QNIRDGIIKAGPAV | ||||||
| AVVGQATQIAK | ||||||
| DM-DMCP | 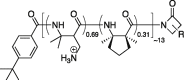 |
4.7 nm | Avg +9 | +1.91 e/nm | Less effective | No |
| DM-DMCH | 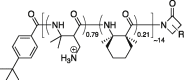 |
5.0 nm | Avg +11 | +2.2 e/nm | Less effective | No |
Naming convention specifies the side chains of the cationic and hydrophobic monomers used, with the cationic side chain first and the hydrophobic side chain second, separated by a hyphen.
The contour length is estimated by multiplying N-to-N spatial distance by the degree of polymerization. The N-to-N spatial distance between two neighboring residues was measured with PyMol based on the crystal structures of poly-α and poly-β-peptides. The length of the end groups are neglected. The relative lengths between species can be compared. The actual end-to-end distance of the polymer will generally be shorter than the counterlength reported here. For example, we estimated LL-37 has a length of 5.7 nm in an α-helix conformation.
For the MM-CH copolymer and MM homopolymer, at longer times (>30 min) after the onset of polymer flow we discovered a surprising additional effect on the gross DNA morphology. In our relatively fast growth condition at 30°C (45 min doubling time), the chromosomal DNA normally exhibits two distinct, axially separate nucleoid lobes. We found that MM-CH copolymer and MM homopolymer can cause gradual coalescence of the two lobes into a single lobe of much higher apparent density localized at one end of the cell. The other four agents showed no such effect. We speculate that this new effect may be related to the well-known polymer phenomenon of coacervation, the condensation of DNA in solution phase by a sufficient concentration of a sufficiently long positively charged polymer (17, 18, 19, 20).
Materials and methods
Nylon-3 polymers were prepared in a nitrogen-purged glove box at room temperature using previously reported methods (13). Live E. coli cells are immobilized on a polylysine-coated coverslip in a microfluidic channel that enables us to begin flowing the antimicrobial agent at known concentration at time t = 0. An EMCCD camera images the cells in real time. The strains described in Table S2 enable fluorescence imaging of green fluorescent protein (GFP) that has been exported to the periplasm; of ribosomal species labeled by the photoswitchable protein S2-mEos2 or S2-Dendra2; of the DNA locus Right2 labeled by ParB-GFP binding to an engineer parS site; or of the global distribution of chromosomal DNA labeled by the dye Sytox Orange or by the nonspecific DNA-binding protein HU-PAmCherry. Additional details are provided in each subsection of Results and in the Supporting materials and methods.
Results
Osmotic effects and CM permeabilization by MM-CH copolymer
The MIC results for the different polymers and AMPs against different bacterial strains are shown in Table 2. Throughout this study, beginning at time t = 0, we flow 2× MIC of each agent to ensure that the vast majority of the bacterial cells are affected by the antimicrobial agent on the imaging timescale of 30–90 min. First we carried out a detailed study of the effects of copolymer MM-CH on E. coli using the strain JCW10 that expresses GFP from a plasmid and exports it to the periplasm (Table S2, WT-ppGFP) (11). We included 5 nM Sytox Orange in the flowing medium.
Table 2.
MIC for different bacterial strains with different agents
| Agent | VH1000 | JCW154 | MSG192 | MG1655 | JCW10 | SM7 | MDG196 |
|---|---|---|---|---|---|---|---|
| MM-CH copolymer (μg/mL) | 25 | 25 | 25 | 25 | 25 | 25 | 25 |
| MM homopolymer (μg/mL) | 31 | 31 | 31 | 31 | 31 | 31 | 31 |
| LL-37 (μM) | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Cecropin A (μM) | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| DM-DMCP copolymer (μg/mL) | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | – | – |
| DM-DMCH copolymer (μg/mL) | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | – | – |
Note that MIC is measured in μΜ for the two chemically defined AMPs, but in μg/mL for the polymers, which lack a well-defined molecular weight. From the known molecular weights of the natural AMPs, for LL-37 we can convert MIC = 4 μM = 18 μg/mL; for cecropin A, MIC = 2 μM = 8 μg/mL. When expressed in μg/mL, all the MIC values lie in a fairly narrow range from 8 to 31 μg/mL.
For these 90-min long, three-channel experiments, in each 12-s cycle, we interleave a phase contrast image (enabling measurement of tip-to-tip cell length), a GFP image (providing total GFP fluorescence intensity and GFP spatial distribution), and a Sytox Orange image (determining when the dye gains access to the cytoplasm and binds to the chromosomal DNA to become fluorescent).
Results for a representative cell (Video S1) are shown in Fig. 1, A–C. Before flowing MM-CH at the concentration of 50 μg/mL (image at t = −4 min), the GFP channel shows the characteristic periplasmic “halo” distribution (Fig. 1, A and B). Within one 12-s camera frame of the onset of flow, the apparent cell length begins to shrink. Several minutes, later it partially recovers, as observed before (11). Until t = 2.2 min, the periplasmic GFP gradually accumulates at the two cell endcaps (Fig. 1, A and B). We interpret this as an osmotic effect known as “plasmolysis” (21). A large concentration of highly cationic MM-CH along with its counterions has translocated across the OM to the periplasm, leading to an apparent osmotic upshift that draws water from the cytoplasm to the periplasm. To accommodate the cytoplasmic volume decrease and to conserve the surface area of the CM, the CM collapses its endcaps and creates more periplasmic volume at the two cell poles, between the CM and OM. The periplasmic GFP redistributes to the cell poles (Fig. 1, A and B). A more detailed explanation can be found in (11). At this stage, the periplasmic GFP has not leaked into the surroundings, indicating that MM-CH has traversed the OM without permeabilizing it to GFP.
Figure 1.

Effects of copolymer MM-CH on a representative E. coli cell that exports GFP to the periplasm (Video S1). Flow of 2× MIC of MM-CH in EZRDM with 5 nM of the DNA stain Sytox Orange begins at t = 0. Images acquired at 12 s per frame. (A) Fluorescence snapshots of GFP (left) and Sytox Orange (right). (B) Projected axial (top) and transverse (bottom) GFP intensity profiles at the times indicated. (C) Cell length (black), total GFP intensity (green) and Sytox Orange intensity (orange) versus time. Note: scales for GFP intensity and cell length are very different. Over the time interval t = 2–60 min, GFP intensity decreases by 50%, but cell length decreases by only 11%. (D) Cumulative distribution function of the fraction of cells for which CM permeabilization has occurred versus time for LL-37, MM-CH copolymer, and MM homopolymer, each at 2X MIC.
Effects of copolymer MM-CH on a representative E. coli cell that exports GFP to the periplasm. Phase contrast image (left), fluorescence snapshots of GFP (middle) and Sytox Orange (right) are shown. Flow of 2× MIC of MM-CH in EZRDM with 5 nM of the DNA stain Sytox Orange begins at t = 0. Images were acquired at 12 s per frame for 90 min.
From t = 2.2 to 2.4 min, the total GFP intensity decreases by ∼20%; this loss of GFP intensity indicates that the OM has become permeabilized to GFP. The apparent cell length partially recovers, probably due to the initial osmoprotection response of the cell (active import of K+) (11). At t = 2.4 min the CM becomes permeabilized to GFP and the GFP intensity pattern abruptly changes to that of a filled cytoplasm (Fig. 1, A and B). The rate of loss of total GFP intensity decreases. Evidently either the OM or the CM or both membranes quickly reseal(s), much as observed earlier for melittin (10).
From t = 2.4 to 85 min, GFP intensity gradually decreases by ∼50%. This decrease is due in part to photobleaching. At these longer times, we infer that the cell envelope is nearly impermeable to GFP and other such globular proteins. The behavior described here is quite general. Across several experiments, all 37 cells that successfully expressed GFP exhibited similar behavior.
Sytox Orange intensity starts to increase at t 3 min for this cell, then increases almost linearly for ∼60 min and finally levels off from 60 to 80 min, likely due to saturation of the available DNA-binding sites. Both OM and CM have become permeable to the small dye molecule and remain so even after the transient events of high permeability to GFP. Over the same 80-min time window, cell length gradually decreases by ∼11%. This may be due to gradual loss of small osmolytes and a corresponding decrease in cytoplasmic osmolality. We define the onset of Sytox Orange intensity as the time of CM permeabilization (CMP; Table 3). In Fig. 1 D, we compare the cumulative distribution function of CMP times for MM-CH, MM homopolymer, and LL-37. In all three cases, essentially all cells have had their CM permeabilized within 25 min after treatment begins. The MM homopolymer permeabilizes the CM slightly more slowly than MM-CH, perhaps because it lacks the hydrophobic CH subunit.
Table 3.
Timing and extent of CM permeabilization
| Cell treatment | Fraction of cells permeabilized within 1 h | Mean time to CM permeabilization (min) |
|---|---|---|
| MM-CH copolymer | 100% | 6.8 ± 6.2 |
| MM homopolymer | 98.3% | 8.3 ± 5.9 |
| LL-37 | 100% | 8.2 ± 4.3 |
Local rigidification of the chromosomal DNA by MM-CH
To characterize the local dynamics of chromosomal DNA before and after MM-CH treatment, we tracked the DNA locus Right 2 labeled by the fusion protein ParB-GFP (strain JCW154, Table S2). In this strain, the fluorescent ParB-GFP protein expressed from a plasmid polymerizes specifically at the parS site engineered into the chromosome near the locus Right 2 (22). The resulting bright puncta can be tracked for some 600 frames with an exposure time of 50 ms/frame without extensive photobleaching. For comparison, we also include the results for normally growing cells (6), growing cells treated with carbonylcyanide-m-chlorophenylhydrazone (CCCP) + 2-deoxyglucose (which depletes ATP by the dissipation of the proton motive force and prevention of glycolysis) (23), and cells after chemical fixation by formaldehyde (6).
At 1 s/frame, we can monitor locus position with ∼30 nm accuracy for 600 frames = 10 min. We first calculate the mean-square displacement (MSD) averaged over trajectories obtained within different 10-min time windows after the onset of MM-CH flow. For each time window, we estimate an apparent diffusion coefficient Dapp from the slope of the linear least-squares fit to the first 10 experimental points of the MSD plot. A plot of Dapp versus the central time for each window gives a sense of the gradual slowing of the DNA local jiggling motion over time (Fig. S1). For example, the MM-CH curve shows that the copolymer attenuates the DNA loci motion by more than a factor of 10 over 45 min (from Dapp = 2.0 × 10−4 to 1.7 × 10−5 μm2/s), and that most of the attenuation occurs over the first 10 min (Table 3). In addition, all of the polymers and AMPs permeabilize the CM of most cells within 25 min. We designate cells 25 min after injection of the antibacterial agent as “polymer- or AMP-treated cells.” Rigidification of the DNA occurs on the same timescale as CM permeabilization (Figs. 1 D and S1). This is consistent with our earlier study of LL-37, in which the freezing of DNA loci motion occurred within 1 min of the time of CM permeabilization ((6); Fig. 1 D).
By combining data at times greater than 25 min from all the trajectories including all experiments, for each agent we calculate the final MSD plot after treatment (Fig. 2, A and B). These MSD plots show clear negative curvature for treated cells as well as for normally growing cells (6,24). Such curvature is the signature of subdiffusive motion, as expected for the local dynamics of a small segment within a large, confined polymer. The degree of curvature is less for the agents that cause the greatest attenuation of DNA motion. To enable semiquantitative comparisons on the 10-s timescale, for each MSD curve we compute an apparent diffusion coefficient Dapp from the slope of the linear least-squares fit to the first 10 experimental points. Numerical results (Table 4) are: Dapp = (2.0 ± 0.2) × 10−4 μm2/s for normal growing cells; (4.6 ± 0.2) × 10−5 μm2/s for cells after CCCP treatment; (7.6 ± 2.1) × 10−6 μm2/s for fixed cells; (1.7 ± 0.1) × 10−5 μm2/s for cells 25 min after treating MM-CH.
Figure 2.

Effect of AMPs and polymers on DNA and protein diffusive motion. (A) MSD versus lag time for DNA loci under various treatments, obtained from videos taken at 1 s/camera frame for 600 frames. Data for treated cells obtained at least 25 min after beginning of flow of antimicrobial agent. Left: MSD covering 100 s. Right: expanded view of the first 10 s. Apparent diffusion coefficient Dapp obtained from linear fit to first 10 points. Numerical results are in Table 4. (B) HU-PAmCherry MSD versus lag time with trajectories taken at 30 ms/frame in normal growth and after polymer. The apparent diffusion coefficient Dapp is obtained by linear fitting of the first three data points. The numerical results are: (0.098 ± 0.004) μm2/s for cells in normal growth (data from (24)); (0.034 ± 0.001) μm2/s for cells after MM-CH; (0.045 ± 0.002) μm2/s for cells after MM homopolymer. (C) Ribosome MSD versus lag time from S2-mEos2 trajectories taken at 30 ms/frame in after the different treatments as shown. The apparent diffusion coefficient Dapp is obtained by linear fitting of the first three data points. Numerical results are in Table 4.
Table 4.
Apparent diffusion coefficients of DNA loci and ribosomal species after 25 min of polymer or peptide treatment
| Cell treatment | Dapp (μm2/s) for DNA loci (1 s/frame) | Dapp (μm2/s) for ribosomal species (30 ms/frame) |
|---|---|---|
| Normal growtha | (2.0 ± 0.2) × 10−4 | 0.042 ± 0.001 |
| CCCP + 2-deoxyglucosea | (4.6 ± 0.2) × 10−5 | 0.028 ± 0.001 |
| Fixed cellsa | (7.6 ± 2.1) × 10−6 | – |
| MM-CH | (1.7 ± 0.1) × 10−5 | 0.010 ± 0.001 |
| MM homopolymer | (1.2 ± 0.3) × 10−5 | 0.0099 ± 0.0004 |
| LL-37 | (1.0 ± 0.1) × 10−5 | 0.018 ± 0.001 |
| Cecropin A | (3.1 ± 0.4) × 10−5 | 0.023 ± 0.001 |
| DM-DMCP | (5.2 ± 0.4) × 10−5 | 0.020 ± 0.001 |
| DM-DMCH | (6.3 ± 0.4) × 10−5 | 0.013 ± 0.001 |
From slope of MSD plots. See text for additional information.
Data from (6) included here for comparison.
Attenuation of ribosome and HU diffusion by MM-CH
To further explore the effect of MM-CH on cytoplasm, we tracked the diffusive motion of single fluorescently labeled ribosomal particles before and after polymer treatment at 30 ms/frame. These experiments use the strain MDG196, in which the chromosomal DNA is altered to append a photoconvertible mEos2 protein to the C-terminus of the ribosomal protein S2 (Table S2). In effect, we are tracking 30S ribosomal subunits, which may occur as either free 30S subunits or 30S subunits incorporated into translating 70S ribosomes, including 70S-polysomes.
We analyzed only those trajectories that last six steps or longer and truncated the longer trajectories at six steps. In Fig. 2 C, we compare MSD plots for ribosomes (S2-mEos2) in different conditions. The apparent diffusion coefficient Dapp is obtained by linear fitting of the first three data points. The numerical results (Table 4) are: (0.042 ± 0.001) μm2-s−1 for cells in normal growth; (0.028 ± 0.001) μm2-s−1 for cells after CCCP treatment (6); (0.010 ± 0.001) μm2-s−1 for cells after MM-CH treatment. MM-CH treatment diminishes the motion of ribosomal particles by a factor of 4. A separate experiment tracking the ribosomal protein S2–Dendra2 showed similar results (Fig. S2).
In the gated diffusion model of Chow and Skolnick (16), motion of the nonspecific DNA-binding protein HU would provide another probe of local DNA dynamics. We tracked single copies of HU-PAmCherry in the strain SM7. MSD plots for HU after MM-CH treatment are quite linear on a 180-ms timescale (Fig. 2 B). In this case, we estimate an apparent diffusion coefficient from the slope of the linear least-squares fit to the first three experimental points. The numerical results are Dapp = (0.098 ± 0.004) μm2/s in normal growth conditions; (0.034 ± 0.001) μm2-s−1 for cells after MM-CH; (0.045 ± 0.002) μm2-s−1 for cells after MM homopolymer. MM-CH and MM homopolymer have a similar effect as LL-37 (6) in reducing the mean HU diffusion coefficient.
Coalescence of two nucleoid lobes induced by MM-CH at long times
In the initial experiments incorporating Sytox Orange into the flow, we sometimes noticed a surprising change in the intensity distribution between the two nucleoid lobes at longer times. To more incisively explore the effects of MM-CH on nucleoid morphology, we conducted a set of experiments in which cells were prestained with 500 nM Sytox Orange for 10 min before imaging (25) and Sytox Orange was omitted from the flow. In these experiments, the total Sytox Orange fluorescence intensity inside each cell is approximately constant during the time of imaging. As before, 2× MIC of MM-CH flowed beginning at t = 0. Images of Sytox Orange fluorescence were acquired at 1 min per frame to minimize photobleaching over 90 min.
Sytox Orange fluorescence snapshots of a representative E. coli cell during the attack of MM-CH are shown in Fig. 3 A. Sytox Orange integrated intensity versus time for the entire cell and for the two individual lobes labeled a and b are shown in Fig. 3 B. The total intensity within the cell remains almost constant during the ∼1.5 h of imaging. However, at t1 = 26 min the left nucleoid lobe a begins to merge into the right nucleoid lobe b, as shown by the decrease in intensity in region a and the simultaneous increase in intensity in region b. We denote this time t1 to mark the beginning of phase 1, coalescence of the two nucleoid lobes. This process continued until the combined nucleoid reached its most compact state at t2 = 39 min for this cell (the beginning of phase 2). The nucleoid remained in this compact state for ∼20 min, then began to expand at t3 = 57 min (the beginning of phase 3). The left nucleoid lobe intensity continues to increase until the final state is reached at t4 = 73 min (beginning of phase 4). This final detected stage is slightly different from the initial stage in that the intensity of lobe a remains somewhat lower than the intensity of lobe b. The projected axial Sytox Orange intensity profile along the cell long axis at different times is shown in Fig. 3 C.
Figure 3.

Effects of MM-CH on nucleoid morphology at long times. Chromosomal DNA was stained with 500 nM Sytox Orange for 10 min before imaging. Flow of 2× MIC MM-CH began at t = 0. Images acquired at 1 min per frame. (A) Sytox Orange fluorescence snapshots of single E. coli cell at different times. (B) Sytox Orange intensity versus time for the same cell shown in (A). Total Sytox Orange intensity from the entire cell (yellow curve), from the left nucleoid lobe (red curve, region a as in A), and from the right nucleoid lobe (blue curve, region b as in A). Total intensity from the entire cell is scaled by a factor of 1.6. Labeled times are t1 (beginning of coalescence of nucleoid lobes), t2 (beginning of phase 2, the nucleoid reaches its most compact state), t3 (beginning of phase 3, partial recovery), and t4 (nucleoids have reached the apparent final state). (C) Projected axial Sytox Orange intensity profile along the cell long axis (arrowhead in A) at different times. Left nucleoid lobe merged to the other one and partially recovered at later times. (D) The timeline of events t1–t4 for 43 cells from four experiments. (E) Phase contrast, Sytox Orange fluorescence and ribosome S2-YFP snapshots of single E. coli cell at different times after flowing MM-CH.
We observed 61 cells from four different experiments; 43 cells showed a similar slow nucleoid coalescence/expansion phenomenon. The timeline of events t1 through t4 for these 43 cells is shown in Fig. 3 D. The cells exhibit strong heterogeneity in the occurrence and timing of different events.
For most cells, the coalescence begins ∼30 min after flowing MM-CH, much later than the osmotic effect and the membrane disruption events described above. Not all cells exhibit phase 3 and phase 4 during our 90-min imaging timescale. In phase 2, the diminished nucleoid lobe retains some residual intensity for some cells, as in Fig. 3 A, whereas for other cells, it completely disappears.
In phase 4, the diminished nucleoid lobe recovers to different degrees compared with the other lobe. An additional 18 cells from the four experiments did not exhibit such a nucleoid morphology change.
The analysis includes cells of all lengths. Both the cells undergoing DNA coalescence and cells not undergoing DNA coalescence include both septating cells and nonseptating cells of different lengths.
We repeated this experiment using the bacterial strain MSG192, in which the ribosomal protein S2 is labeled with YFP. We found that when the DNA lobes merged at one end of the cell, the ribosomes moved to the other end of the cell, where the DNA has become much less dense (Fig. 3 E, Video S2). The strong anticorrelation of the DNA and the ribosome spatial distributions that occurs in normal growth persists after the coalescence. Details are provided in Supporting materials and methods and Fig. S3.
Phase contrast image (left), Sytox Orange fluorescence (middle) and ribosome S2-YFP fluorescence (right) snapshots of single E. coli cell at different times during the attack of copolymer MM-CH are shown. Cells with ribosomal S2 subunit labeled with YFP were stained with 500 nM Sytox Orange for 10 min before imaging. 2× MIC MM-CH flowed beginning at t = 0. Images were acquired at 1 min per frame for 90 min.
We also carried out superresolution imaging of the nonspecific DNA-binding protein HU-PAmCherry. The results (Fig. S4) corroborate the widefield Sytox Orange results showing coalescence of the two nucleoid lobes at longer times for many cells. At t = 25 min after MM-CH addition, for cells in the length range 4.0–4.3 μm, we observed 17 cells showing two symmetric nucleoid lobes and 22 cells showing asymmetric nucleoid lobes. In addition, the superresolution HU-PAmCherry images enable us to compare the nucleoid radius of untreated cells with that of cells treated with 2X MIC MM-CH for 25 min, as described earlier (24). Details are provided in Supporting materials and methods.
For untreated cells, <rnucleoid> = 0.29 ± 0.04 μm (33 cells with cell length in the range 4.0–4.3 μm) compared with <rnucleoid> = 0.22 ± 0.05 μm (17 cells with two nucleoid lobes and cell length in the range 4.0–4.3 μm) for cells after treatment with MM-CH. Nucleoid lengths are similar under both conditions. The apparent nucleoid volume has shrunk by a factor of ∼1.7 (the square of the ratio of radii) due to MM-CH treatment (Fig. S5). This may be a signature of coacervation, as discussed below.
Upper limit on the total number of charges that MM-CH brings into the cell
One of the most important parameters in the coacervation of polymer mixtures is the ratio of the total concentration of positive charges on the polycations to the total concentration of negative charges on the polyanions (18,19,26). Global DNA rigidification by electrostatic cross-linking also requires adsorption of a large number of polycationic molecules. To explore the plausibility of coacervation as well as rigidification of DNA by MM-CH, we estimate an upper limit on the amount of MM-CH per bacterial cell using a similar method as reported before (6, 7, 8). We incubated various concentrations of MM-CH polymer with various total cell counts (colony-forming units, CFU per mL) in PBS solution for 60 min in the test tube. Then we aliquoted each sample into 96-well plates supplied with EZ rich, defined medium (EZRDM) and incubated for 24 h. For each MM-CH concentration and for each initial cell count, we measured the optical density (OD) at 595 nm before and after 24-hr incubation of the cells with MM-CH. The change in optical density is defined as ΔOD = ODt = 24 h − ODt = 0.
If the OD does not increase for 24-h, then that amount of MM-CH was large enough to kill the initial number of cells. In this way, we can measure the minimal bulk MM-CH concentration required to kill a particular initial number of cells. For example, the MM-CH concentration of 50 μg/mL (as used in the flow experiments) is sufficient to kill 4 × 107 CFU/mL (Fig. S6). If it were the case that all of the MM-CH were absorbed by the bacterial cells, then on average each cell would have absorbed ∼2.6 × 109 positive charges. Because some of the MM-CH may remain in solution under these conditions, this number provides an upper bound to the actual number of positive charges absorbed. Details of the calculation are provided in Supporting materials and methods.
In the earlier work on LL-37, we used fluorescence and GC/MS to measure the fraction of LL-37 bound to the cells. For a bulk concentration of 20 μM, we estimated that an average of ∼2 × 108 LL-37 copies were absorbed per cell. Net positive charge of LL-37 at neutral pH is +6, so the net positive charge absorbed per cell was ∼1.2 × 109, comparable with the estimated upper bound on MM-CH uptake.
Recovery experiments
To explore whether or not the short-term DNA rigidification and longer-term DNA coalescence are related to bacterial growth inhibition, we conducted a series of recovery experiments. In these experiments, we flow 2× MIC of MM-CH in EZRDM across plated WT cells for 5 or 10 min and then change the flow to EZRDM without MM-CH for an additional 90 min. Phase contrast imaging provides an estimate of cell length versus time to test for possible recovery of cell growth. We observed 37 cells after 5-min exposure to MM-CH and 26 cells after 10-min exposure. In all 63 cases, the brief exposure was sufficient to completely halt apparent cell growth for at least 90 min of observation time by phase contrast imaging. However, for 5-min exposure to MM-CH, all 37 cells retained two separated nucleoid lobes (no DNA morphology change). For 10-min exposure to MM-CH, 18 of the 26 cells eventually showed the DNA coalescence behavior described above, even though the medium had been changed to EZRDM.
Comparison of LL-37, cecropin A, and other synthetic copolymers
Finally, we tested the other agents in Table 1 for their ability to locally rigidify the chromosome and to induce coalescence of the two chromosomal DNA lobes. The results provide some insight into what features of an antimicrobial agent enable these effects. From the DNA locus tracking data taken at times greater than 25 min of treatment by each agent, we obtained the MSD plots shown in Fig. 2 A. The apparent diffusion coefficients Dapp from linear fits to the first 10 data points are collected in Table 4. LL-37, MM-CH copolymer, and MM homopolymer rapidly rigidify local chromosomal motion almost as effectively as chemical fixation by formaldehyde. Cecropin A and the two shorter copolymers DM-DMCH and DM-DMCP reduced the DNA loci motion by a factor of 3–7 compared with normally growing cell. This is comparable to the effect of ATP depletion by the CCCP treatment. The diffusion coefficients of ribosomes under different treatments are also included in Table 4. The antimicrobial agents all slowed ribosome diffusion more than did ATP depletion.
Coalescence of the two nucleoid lobes on a 90-min timescale was induced only by MM-CH and by MM homopolymer. The natural AMPs LL-37 and cecropin A and the two shorter copolymers DM-DMCP and DM-DMCH did not cause coalescence on a 90-min timescale.
Discussion
Short-term effects of cationic polymers and peptides
This study has compared the effects on E. coli of the six antimicrobial agents whose chemical compositions, contour lengths, and linear charge densities are given in Table 1. At 2× MIC, the long, highly charged copolymer MM-CH (11.5-nm mean contour length, mean charge +21, mean linear charge density +1.8 e/nm) penetrates both the OM and CM remarkably quickly. Once MM-CH has permeabilized both the CM and the OM to small molecules, there occurs a massive influx of highly positively charged copolymer into the cytoplasm. After gaining access to the cytoplasm, MM-CH causes several striking changes in the chromosomal DNA. On a timescale of ∼5–10 min, the local jiggling motion of the DNA polymer is greatly attenuated, an effect we call rigidification of the nucleoid (6). On a similar timescale, the apparent volume of each of the two separate nucleoid lobes decreases by an estimated factor of ∼1.7. On a much longer timescale of ∼30 min, in many of the cells one nucleoid lobe slowly migrates to coalesce with the other lobe, forming a dense, single-lobed nucleoid that strongly excludes ribosomal species. In many cases, this coalescence partially reverses on the timescale 60–90 min after initiation of copolymer flow. All of these same events were caused by the somewhat shorter (7.9 nm), but even more densely charged MM homopolymer (+22, +2.8 e/nm).
In contrast, the shorter DM-DMCP and DM-DMCH copolymers were less effective in rigidifying the DNA and did not cause large-scale coalescence of nucleoid lobes. DM-DMCP and DM-DMCH have similar charge densities as MM-CH, but different chemical compositions and also substantially shorter mean contour lengths (4.7 and 5.0 nm, respectively). The natural AMP LL-37 was as effective as MM-CH in locally rigidifying the DNA. The natural AMP cecropin A was less effective. Neither AMP caused large-scale nucleoid lobe coalescence.
As before (6), we suggest two underlying causes of the attenuation of the DNA jiggling motion. First, permeabilization of the CM and concomitant reduction in the transmembrane potential will slow or halt ATP production and presumably enable leakage of existing ATP to the cell surround.
This reduces the motor driven contribution to the jiggling motion of the DNA, as shown by the CCCP assay and observed earlier by others (27,28). This effect likely occurs for all six agents studied here. Second, we suggest that additional local rigidification of the DNA can be caused by “electrostatic pseudo-cross-linking” of nearby DNA strands within the dense nucleoid. Here, we envision transient, noncovalent binding of long polycationic species to release nearby K+ counterions and hinder the relative motion of adjacent DNA strands. The coarse-grained simulations of Chow and Skolnick (16) suggest typical gaps between nearby DNA strands of ∼6 nm. We suggest that longer molecules with high positive-charge density should accomplish this cross-linking more effectively. Accordingly, the MM-CH copolymer and MM homopolymer are quite long and densely charged. LL-37 is long but less densely charged. However, LL-37 has positive Lys and Arg residues at either end of the chain, enabling positive charges to span substantial gaps between DNA stands. Cecropin A concentrates its charges at its N-terminal half, rendering it less able to span large gaps with its positive charge.
The same pseudo-cross-linking effect may also explain the threefold decrease in the mean diffusion coefficient of HU (Fig. 2 B) induced by MM-CH on the 25-min timescale. The HU trajectories presumably involve an average over time spent diffusing “freely” (not bound to DNA) within the nucleoid and time spent bound to DNA. The coarse-grained simulation model of Chow and Skolnick (16) suggests that thermal motion of the DNA strands enables hopping of the transcription repressor protein LacI from one DNA cage within the nucleoid to an adjacent cage, a process they called “gated diffusion.” It seems plausible that the observed rigidification of the DNA would hinder gated diffusion. A similar reduction in HU diffusion is induced by MM homopolymer (Fig. 2 B) and by LL-37, the two agents that are most effective at DNA rigidification.
All six agents decrease the mean diffusion coefficient of ribosomal species 25 min after treatment by a factor the varies from 2 to 4 (Fig. 2 C), all larger effects than CCCP treatment. Each mean diffusion coefficient is an average over an unknown distribution of ribosomal species, including 30S subunits, 70S complete ribosomes, 70S polysomes, and potentially agglomerations of such ribosomal species induced by the polycationic agent (29). Pseudo-cross-linking by polycationic agents may still occur between these negatively charged ribosome species to slow diffusion of ribosomes.
Coacervation?
We have no clear understanding of the intriguing coalescence of the two nucleoid lobes induced at t ∼30 min, observed only for the longer, densely charged MM-CH copolymer and by the MM homopolymer, not for the other four agents. We tentatively suggest that this behavior is related to the well-known phenomenon of associative phase separation or coacervation (18,26,30). In certain equilibrium solution phase conditions, sufficiently long and densely charged polycations at sufficiently high concentration are able to condense DNA from its natural elongated, swollen structure into a compact structure via coacervation (26). There is in essence a phase separation between a polycation/polyanion rich phase and the remaining polymer-depleted portion of the solution.
A detailed study in vitro (26) found that 92-mer poly-L-lysine induces gradual compaction of DNA to a shrunken coil structure as the relative concentration of poly-L-lysine increases. The compaction was greatest when the ratio of total positive charges on the polycations to total negative charges on DNA reached one (17,26). The coacervation mechanism did not occur for short poly-L-lysine (n < 9) (17,26). Small cations can compact DNA, but only at much higher concentrations (26).
The E. coli cytoplasm is dense with polyanionic species to which the polycationic MM-CH copolymer or MM homopolymer should bind strongly (31). As an upper limit, we estimated that on average, each cell may absorb as many as ∼109 positive charges carried by the MM-CH copolymer. For a typical total cell volume of ∼1.9 μm3, this corresponds to an average concentration of ∼0.9 M of polymeric positive charge, a very large number. Under our growth conditions we estimate that cytoplasmic polyanionic species (chromosome, ribosome, transfer RNA (tRNA), mRNA) provide ∼2 × 108 negative charges (∼200 mM); these are compensated primarily by K+ (Supporting materials and methods). It is plausible that sufficient MM-CH is absorbed within the cytoplasm to cause coacervation with the chromosomal DNA.
Before the addition of MM-CH, the chromosomal DNA is already highly compacted due to pairwise binding by a variety of nucleoid-associated proteins (17,19,32, 33, 34); segregative phase separation due to DNA-ribosome repulsion (19,35,36); configurational entropy of the confined DNA polymer to avoid the cell boundaries (37); neutralization of DNA charges by multivalent ions and some DNA-binding proteins; macromolecular crowding (17,30,38,39); and DNA supercoiling (17,33,40,41). Nevertheless, as MM-CH gains access to the cytoplasm we observe significant radial shrinkage of the two nucleoid lobes, with the mean radius decreasing from 0.29 to 0.22 nm (for cells in 4–4.3 μm length range, Fig. S5). The nucleoid volume decreases by a factor of ∼1.7. This is consistent with the suggestion that MM-CH causes coacervation of the chromosomal DNA before the longer-term coalescence of the two nucleoid lobes. In contrast, in the earlier study LL-37 did not contract the nucleoid volume. LL-37 is likely insufficiently positive and too short to induce this effect.
The coacervation hypothesis may also provide a partial explanation for the observed slow migration of one nucleoid lobe to coalesce with the other lobe. In solution phase, coacervate droplets have been observed to coalesce with each other over time, perhaps to minimize surface tension with the surrounding phase (42,43). Similar behavior of liquid droplets has been observed in eukaryotic cells (44, 45, 46). We lack a good rationale for the slow reversal from the one-lobed, coalesced condition back toward the original two-lobed condition. On the 90-min timescale of a video (Video S1, Video S2), this occurs in roughly half of the that showed coalescence. Concentrations of the myriad species within the cytoplasm continue to change as the concentration of absorbed polymer increases. The coacervation process occurs at a narrow concentration ratio range for polycations and polyanions (18,43,47,48).
Conclusions
To date, all of our time-resolved, single-cell mechanistic studies have used linear, polycationic antibacterial agents, including both AMPs of defined sequence and sequence-random polymers (6,9, 10, 11,49). Does permeabilization of the bacterial membranes by these agents cause the halting of growth? Our experience supports the conclusion that there is a threshold concentration above which permeabilization of the OM occurs; below that concentration cells continue to grow, albeit sometimes more slowly. Permeabilization of the CM then occurs rapidly and eventually growth halts in a seemingly irreversible fashion. CM permeabilization destroys the transmembrane potential and rapidly causes loss to the cell surround of small molecules such as ATP and perhaps of globular proteins and tRNA. These same cationic agents may also interfere with a wide variety of cellular processes including cell envelope biosynthesis, DNA replication and transcription, and protein synthesis (2,4,5). A surprising effect that is beginning to seem quite general is the massive uptake of polycationic agents once the OM and CM have been breached (6, 7, 8). A bacterial cytoplasm exposed to the surround appears to be a sponge that soaks up polycations to very high internal concentrations (108–109 positive charges). Again, we suggest that the key driving force is strong, noncovalent, multivalent electrostatic binding between polycations and polyanions (DNA, ribosomes, mRNA, tRNA, and even globular proteins) (19,31). Nature has designed a bacterial cytoplasm that is remarkably susceptible to damage by polycationic agents. At least for linear, polycationic agents, our view is that membrane permeabilization appears to be a necessary first step leading to a cascade of downstream events, many occurring on a timescale of a few minutes or less. With so many effects likely occurring on a similar timescale, it is difficult to attribute the halting of growth (and apparent killing of cells) to any single event, but membrane permeabilization is the necessary triggering event.
This work on cationic polymers highlights several new effects: rigidification of the DNA and attenuation of ribosome and protein diffusion on a short timescale of several minutes and coalescence of the two nucleoid lobes at longer times of 30 min or more. The recovery experiments show that the cells absorb sufficient MM-CH copolymer during the first 5 min of exposure to prevent subsequent growth for at least 90 min after the flow had switched to fresh EZRDM without copolymer. These same cells do not subsequently show DNA coalescence or gross content leakage (no white band on phase contrast image). Evidently sufficient MM-CH enters the cells in 5 min to induce enough damage to halt growth. In contrast, after a 10 min exposure to MM-CH followed by 90 min in fresh EZRDM, cells eventually show both nucleoid coalescence and complete membrane disruption. The additional polymer absorbed over the interval from 5 to 10 min seems essential to drive the long-term nucleoid coalescence. Such coalescence is evidently not necessary to halt growth—only a fraction of the cells exhibited this behavior. In addition, all six of the tested agents halt the growth of E. coli, but some agents cause the long-term coalescence of the two nucleoid lobes and others do not. It also seems unlikely that DNA rigidification is necessary to halt growth. The agents exhibit widely varying degrees of DNA rigidification and diffusion attenuation.
Finally, we find it remarkable that the sequence-random, cationic polymers studied here are so readily able to traverse the OM and CM of E. coli. This result seems incompatible with models of membrane disruption that posit formation of structured pores, either toroidal or barrel-stave.
Author contributions
Y.Z. and M.M. carried out the microscopy studies. L.L., L.A.R., and S.H.G. designed, synthesized, and characterized the polymer samples. Y.Z. and J.C.W. planned the microscopy studies and wrote the article, with help from L.L. and S.H.G.
Acknowledgments
J.C.W. thanks the Wisconsin Alumni Research Foundation for a faculty fellowship. We thank Prof. Arun Yethiraj, Dr. Tyler Lytle, and Dr. Sonisilpa Mohapatra for helpful discussions.
This work was supported by the National Institutes of Health (grants R01-GM094510 to J.C.W. and R33-AI121684 to S.H.G.) and by the National Science Foundation (grant MCB-1512946 to J.C.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Editor: Antje Pokorny Almeida.
Footnotes
Yanyu Zhu’s present address is Department of Bioengineering, Stanford University, Stanford, California 94305.
Lei Liu’s present address is Department of Chemistry, University of Chicago, Chicago, Illinois 60637.
Mainak Mustafi’s present address is Department of Biochemistry and Molecular Biophysics, Columbia University, New York, New York 10032.
Leslie A. Rank’s present address is Johnson & Johnson Consumer Health, Skillman, New Jersey 08558.
Yanyu Zhu, Lei Liu, and Mainak Mustafi contributed equally to this work.
Supporting material can be found online at https://doi.org/10.1016/j.bpj.2021.10.037.
Supporting material
References
- 1.Savini F., Bobone S., et al. Stella L. From liposomes to cells: filling the gap between physicochemical and microbiological studies of the activity and selectivity of host-defense peptides. Peptide Science. 2018;110:e24041. [Google Scholar]
- 2.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 3.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 4.Scocchi M., Mardirossian M., et al. Benincasa M. Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr. Top. Med. Chem. 2016;16:76–88. doi: 10.2174/1568026615666150703121009. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y., Weisshaar J.C., Mustafi M. Long-term effects of the proline-rich antimicrobial peptide Oncocin112 on the Escherichia coli translation machinery. J. Biol. Chem. 2020;295:13314–13325. doi: 10.1074/jbc.RA120.013587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Y., Mohapatra S., Weisshaar J.C. Rigidification of the Escherichia coli cytoplasm by the human antimicrobial peptide LL-37 revealed by superresolution fluorescence microscopy. Proc. Natl. Acad. Sci. USA. 2019;116:1017–1026. doi: 10.1073/pnas.1814924116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roversi D., Luca V., et al. Stella L. How many antimicrobial peptide molecules kill a bacterium? The case of PMAP-23. ACS Chem. Biol. 2014;9:2003–2007. doi: 10.1021/cb500426r. [DOI] [PubMed] [Google Scholar]
- 8.Starr C.G., He J., Wimley W.C. Host cell interactions are a significant barrier to the clinical utility of peptide antibiotics. ACS Chem. Biol. 2016;11:3391–3399. doi: 10.1021/acschembio.6b00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi H., Rangarajan N., Weisshaar J.C. Lights, camera, action! Antimicrobial peptide mechanisms imaged in space and time. Trends Microbiol. 2016;24:111–122. doi: 10.1016/j.tim.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z., Choi H., Weisshaar J.C. Melittin-induced permeabilization, re-sealing, and re-permeabilization of E. coli membranes. Biophys. J. 2018;114:368–379. doi: 10.1016/j.bpj.2017.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi H., Chakraborty S., et al. Weisshaar J.C. Single-cell, time-resolved antimicrobial effects of a highly cationic, random nylon-3 copolymer on live Escherichia coli. ACS Chem. Biol. 2016;11:113–120. doi: 10.1021/acschembio.5b00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mowery B.P., Lee S.E., et al. Gellman S.H. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 2007;129:15474–15476. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 13.Mowery B.P., Lindner A.H., et al. Gellman S.H. Structure-activity relationships among random nylon-3 copolymers that mimic antibacterial host-defense peptides. J. Am. Chem. Soc. 2009;131:9735–9745. doi: 10.1021/ja901613g. [DOI] [PubMed] [Google Scholar]
- 14.Liu R., Chen X., et al. Gellman S.H. Tuning the biological activity profile of antibacterial polymers via subunit substitution pattern. J. Am. Chem. Soc. 2014;136:4410–4418. doi: 10.1021/ja500367u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rank L.A., Agrawal A., et al. Gellman S.H. Diverse impacts on prokaryotic and eukaryotic membrane activities from hydrophobic subunit variation among nylon-3 copolymers. ACS Chem. Biol. 2021;16:176–184. doi: 10.1021/acschembio.0c00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow E., Skolnick J. DNA internal motion likely accelerates protein target search in a packed nucleoid. Biophys. J. 2017;112:2261–2270. doi: 10.1016/j.bpj.2017.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joyeux M. Compaction of bacterial genomic DNA: clarifying the concepts. J. Phys. Condens. Matter. 2015;27:383001. doi: 10.1088/0953-8984/27/38/383001. [DOI] [PubMed] [Google Scholar]
- 18.van der Gucht J., Spruijt E., et al. Cohen Stuart M.A. Polyelectrolyte complexes: bulk phases and colloidal systems. J. Colloid Interface Sci. 2011;361:407–422. doi: 10.1016/j.jcis.2011.05.080. [DOI] [PubMed] [Google Scholar]
- 19.Joyeux M. In vivo compaction dynamics of bacterial DNA: a fingerprint of DNA/RNA demixing? Curr. Opin. Colloid Interface Sci. 2016;26:17–27. [Google Scholar]
- 20.Estevez-Torres A., Baigl D. DNA compaction: fundamentals and applications. Soft Matter. 2011;7:6746–6756. [Google Scholar]
- 21.Sochacki K.A., Shkel I.A., et al. Weisshaar J.C. Protein diffusion in the periplasm of E. coli under osmotic stress. Biophys. J. 2011;100:22–31. doi: 10.1016/j.bpj.2010.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espeli O., Mercier R., Boccard F. DNA dynamics vary according to macrodomain topography in the E. coli chromosome. Mol. Microbiol. 2008;68:1418–1427. doi: 10.1111/j.1365-2958.2008.06239.x. [DOI] [PubMed] [Google Scholar]
- 23.Cavari B.Z., Avi-Dor Y. Effect of carbonyl cyanide m-chlorophenylhydrazone on respiration and respiration-dependent phosphorylation in Escherichia coli. Biochem. J. 1967;103:601–608. doi: 10.1042/bj1030601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y., Mustafi M., Weisshaar J.C. Biophysical properties of Escherichia coli cytoplasm in stationary phase by superresolution fluorescence microscopy. MBio. 2020;11 doi: 10.1128/mBio.00143-20. e00143-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakshi S., Choi H., et al. Weisshaar J.C. Nonperturbative imaging of nucleoid morphology in live bacterial cells during an antimicrobial peptide attack. Appl. Environ. Microbiol. 2014;80:4977–4986. doi: 10.1128/AEM.00989-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akitaya T., Seno A., et al. Yoshikawa K. Weak interaction induces an ON/OFF switch, whereas strong interaction causes gradual change: folding transition of a long duplex DNA chain by poly-L-lysine. Biomacromolecules. 2007;8:273–278. doi: 10.1021/bm060634j. [DOI] [PubMed] [Google Scholar]
- 27.Weber S.C., Spakowitz A.J., Theriot J.A. Nonthermal ATP-dependent fluctuations contribute to the in vivo motion of chromosomal loci. Proc. Natl. Acad. Sci. USA. 2012;109:7338–7343. doi: 10.1073/pnas.1119505109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parry B.R., Surovtsev I.V., et al. Jacobs-Wagner C. The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell. 2014;156:183–194. doi: 10.1016/j.cell.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chongsiriwatana N.P., Lin J.S., et al. Barron A.E. Intracellular biomass flocculation as a key mechanism of rapid bacterial killing by cationic, amphipathic antimicrobial peptides and peptoids. Sci. Rep. 2017;7:16718. doi: 10.1038/s41598-017-16180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lerman L.S. A transition to a compact form of DNA in polymer solutions. Proc. Natl. Acad. Sci. USA. 1971;68:1886–1890. doi: 10.1073/pnas.68.8.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schavemaker P., Smigiel W., Poolman B. Ribosome surface properties may impose limits on the nature of the cytoplasmic proteome. Elife. 2017;6:e30084. doi: 10.7554/eLife.30084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song D., Loparo J.J. Building bridges within the bacterial chromosome. Trends Genet. 2015;31:164–173. doi: 10.1016/j.tig.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Wang X., Montero Llopis P., Rudner D.Z. Organization and segregation of bacterial chromosomes. Nat. Rev. Genet. 2013;14:191–203. doi: 10.1038/nrg3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W., Li G.W., et al. Zhuang X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science. 2011;333:1445–1449. doi: 10.1126/science.1204697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krotova M.K., Vasilevskaya V.V., et al. Khokhlov A.R. DNA compaction in a crowded environment with negatively charged proteins. Phys. Rev. Lett. 2010;105:128302. doi: 10.1103/PhysRevLett.105.128302. [DOI] [PubMed] [Google Scholar]
- 36.Yoshikawa K., Hirota S., et al. Yoshikawa Y. Compaction of DNA induced by like-charge protein: opposite salt-effect against the polymer-salt-induced condensation with neutral polymer. J. Phys. Chem. Lett. 2010;1:1763–1766. [Google Scholar]
- 37.Mondal J., Bratton B.P., et al. Weisshaar J.C. Entropy-based mechanism of ribosome-nucleoid segregation in E. coli cells. Biophys. J. 2011;100:2605–2613. doi: 10.1016/j.bpj.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asakura S., Oosawa F. On interaction between 2 bodies immersed in a solution of macromolecules. J. Chem. Phys. 1954;22:1255–1256. [Google Scholar]
- 39.Teif V.B., Bohinc K. Condensed DNA: condensing the concepts. Prog. Biophys. Mol. Biol. 2011;105:208–222. doi: 10.1016/j.pbiomolbio.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Benza V.G., Bassetti B., et al. Lagomarsino M.C. Physical descriptions of the bacterial nucleoid at large scales, and their biological implications. Rep. Prog. Phys. 2012;75:076602. doi: 10.1088/0034-4885/75/7/076602. [DOI] [PubMed] [Google Scholar]
- 41.Postow L., Hardy C.D., et al. Cozzarelli N.R. Topological domain structure of the Escherichia coli chromosome. Genes Dev. 2004;18:1766–1779. doi: 10.1101/gad.1207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Priftis D., Farina R., Tirrell M. Interfacial energy of polypeptide complex coacervates measured via capillary adhesion. Langmuir. 2012;28:8721–8729. doi: 10.1021/la300769d. [DOI] [PubMed] [Google Scholar]
- 43.Sing C.E. Development of the modern theory of polymeric complex coacervation. Adv. Colloid Interface Sci. 2017;239:2–16. doi: 10.1016/j.cis.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Deshpande S., Brandenburg F., et al. Dekker C. Spatiotemporal control of coacervate formation within liposomes. Nat. Commun. 2019;10:1800. doi: 10.1038/s41467-019-09855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Astoricchio E., Alfano C., et al. Pastore A. The wide world of coacervates: from the sea to neurodegeneration. Trends Biochem. Sci. 2020;45:706–717. doi: 10.1016/j.tibs.2020.04.006. [DOI] [PubMed] [Google Scholar]
- 46.Shin Y., Brangwynne C. Liquid phase condensation in cell physiology and disease. Science. 2017;357:eaaf4382. doi: 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- 47.Lytle T.K., Sing C.E. Transfer matrix theory of polymer complex coacervation. Soft Matter. 2017;13:7001–7012. doi: 10.1039/c7sm01080j. [DOI] [PubMed] [Google Scholar]
- 48.Singh A.N., Yethiraj A. Driving force for the complexation of charged polypeptides. J. Phys. Chem. B. 2020;124:1285–1292. doi: 10.1021/acs.jpcb.9b09553. [DOI] [PubMed] [Google Scholar]
- 49.Sochacki K.A., Barns K.J., et al. Weisshaar J.C. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc. Natl. Acad. Sci. USA. 2011;108:E77–E81. doi: 10.1073/pnas.1101130108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of copolymer MM-CH on a representative E. coli cell that exports GFP to the periplasm. Phase contrast image (left), fluorescence snapshots of GFP (middle) and Sytox Orange (right) are shown. Flow of 2× MIC of MM-CH in EZRDM with 5 nM of the DNA stain Sytox Orange begins at t = 0. Images were acquired at 12 s per frame for 90 min.
Phase contrast image (left), Sytox Orange fluorescence (middle) and ribosome S2-YFP fluorescence (right) snapshots of single E. coli cell at different times during the attack of copolymer MM-CH are shown. Cells with ribosomal S2 subunit labeled with YFP were stained with 500 nM Sytox Orange for 10 min before imaging. 2× MIC MM-CH flowed beginning at t = 0. Images were acquired at 1 min per frame for 90 min.