

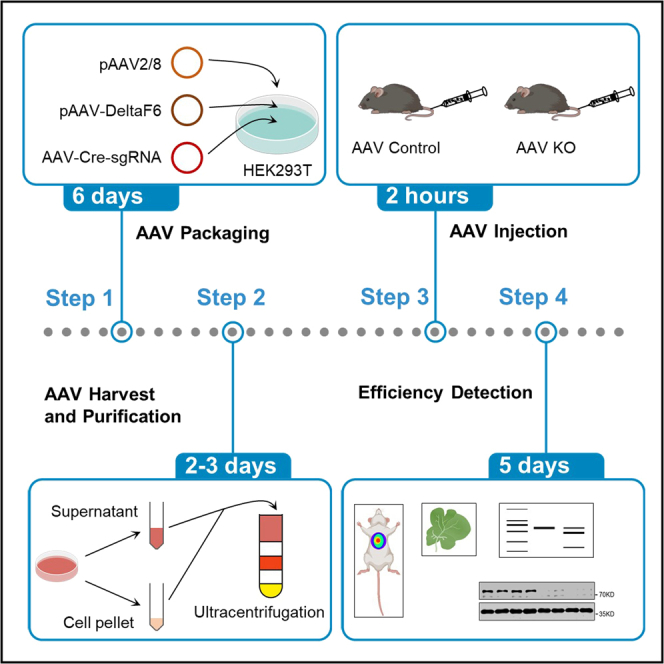
Summary
We provide a protocol for gene editing in mouse hepatocytes in vivo using the CRISPR-Cas9 technology via AAV delivery. This protocol describes the construction of AAV plasmids, AAV packaging, injection, and the detection of in vivo knockout efficiency. Using this protocol, we can get up to 1014 AAV and knock out genes in hepatocytes efficiently within 15 days. Moreover, we describe an optimized protocol to simultaneously target two genes via AAV delivery of CRISPR-Cas9 materials in the liver.
For complete details on the use and execution of this profile, please refer to Wei et al. (2020).
Subject areas: Metabolism, Model Organisms, Molecular Biology, CRISPR
Graphical abstract
Highlights
-
•
Gene editing in hepatocytes in vivo using CRISPR-Cas9 technology
-
•
Dual gene editing in vivo using optimized protocol
-
•
High titer of AAV can be produced using this protocol
-
•
Multiple techniques to detect AAV infection efficiency
We provide a protocol for gene editing in mouse hepatocytes in vivo using the CRISPR-Cas9 technology via AAV delivery. This protocol describes the construction of AAV plasmids, AAV packaging, injection, and the detection of in vivo knockout efficiency. Using this protocol, we can get up to 1014 AAV and knock out genes in hepatocytes efficiently within 15 days. Moreover, we describe an optimized protocol to simultaneously target two genes via AAV delivery of CRISPR-Cas9 materials in the liver.
Before you begin
Preparation of Rosa26-LSL-Cas9 knock-in mice
Timing: n/a
-
1.
The Rosa26-LSL (loxP-stop-loxP)-Cas9 knock-in mice harbor a Cas9 transgene expression cassette in the Rosa26 locus (Platt et al., 2014), with an EGFP cDNA also linked via P2A to Cas9. Thus, the expression of Cas9 and EGFP is induced after exposing to the CRE recombinase. With this, introducing a liver cell specific-Cre and sgRNA into the mouse can specifically knock out the target gene in hepatocytes.
Preparation of AAV constructs
-
2.sgRNA design and synthesis.
-
a.sgRNA design. sgRNAs can be designed using the online CRISPR design tools. For example, Broad Institute CRISPick (Doench et al., 2016; Sanson et al., 2018). Online calculation of sgRNA obtained through algorithm optimization may have a higher probability of obtaining sgRNA with higher editing efficiency and lower off-target efficiency. At the same time, online calculation will also give potential off-target sites and probability, so we can choose suitable sgRNAs. You can also design sgRNAs manually as previously described (Qiu and Ding, 2021).Note: If you do not have an sgRNA sequence that efficiently targets the gene of interest, you need to design at least three sgRNAs and screen for efficient ones as previously described (Qiu and Ding, 2021). And it is recommended that at least two different sgRNAs are applied in all experimental procedures, with careful evaluation of the sgRNA off-target effects (Ding et al., 2014).
-
b.sgRNA oligonucleotides synthesis. Synthesis of two sgRNA oligonucleotides as follows:sgRNA-F: 5′-ACCGNNNNNNNNNNNNNNNNNNNN- 3′sgRNA-R: 3′-CNNNNNNNNNNNNNNNNNNNNCAA-5′Note: The polyN sequence represents the 20 bp sgRNA sequence that targets the gene of interest.
-
a.
-
3.Preparation of AAV-Cre-sgRNA.
-
a.Construct the AAV-Cre-sgRNA plasmid (Figure 1).
-
i.Delete the GFP expression cassette between two inverted terminal repeats (ITRs) of pAAV-GFP.
-
ii.The CRE expression cassette was inserted between the two ITRs. The Cre gene is driven by a liver-specific TBG promoter, which is also linked via a T2A sequence to a Luciferase gene to facilitate the in vivo imaging of CRE-expressing cells later.
-
iii.A U6 driven sgRNA expression cassette was inserted after the Cre expression cassette. The sequence containing two Sap I sites was added before the sgRNA scaffold sequence, where the sgRNA targeting sequence will be inserted for each gene. The sequence containing two Sap I sites is as follows:5′-ACCGGAAGAGCAAGCTCTTCGGTT-3′3′-TGGCCTTCTCGTTCGAGAAGCCAA-5′
-
i.
-
b.Digest 1 μg AAV-Cre-sgRNA plasmid with Sap I restriction enzyme for 1 h at 37°C.
Reagent Amount AAV-Cre-sgRNA 1 μg Sap I 1 μL CutSmart Buffer 5 μL ddH2O n/a Total 50 μL -
c.Run the Sap I digestion products on a 1% agarose gel.
-
d.Cut the 7399 bp DNA band. Extract DNA from the gel using a gel extraction kit.
-
e.Denature the pair of oligos from step 2b at 95°C for 5 min, then slowly cool the solution 0.1°C/s to 25°C in a thermocycler.
Reagent Amount sgRNA-F (100 μM) 1 μL sgRNA-R (100 μM) 1 μL Annealing buffer (10×) 5 μL ddH2O n/a Total 50 μL -
f.Set up the ligation reaction and incubate at 16°C for 30 min.
Reagent Amount Digested plasmid from 3d 50 ng Annealed oligos from 3e 2 μL Ligation High (premixed T4 ligase) 3 μL ddH2O n/a Total 9 μL -
g.Set up a ligation reaction without annealed oligos as a negative control and incubate at 16°C for 30 min.
-
h.Transform all the ligation products form step f into Stbl3 chemically competent cells. Transform all the products form step g into Stbl3 competent cell as a negative control.Note: It is recommended to transform the AAV plasmids into Stbl3 competent cells to reduce potential homologous recombination through the ITRs.
-
i.Add 100 μL LB medium to the competent cells, then incubate in a 37°C shaker for 1 h.
-
j.Spread competent cells evenly on an LB plate containing 100 μg/mL ampicillin.
-
k.Incubate the LB plates at 37°C for 12–16 h.
-
l.Assess the plate for colony growth. Typically, multiple colonies should grow on the plate, and the negative control plate should not have colony growth.
-
m.Pick three single colonies with pipette tips and put them into different 15 mL centrifuge tubes.
-
n.Add 2 mL of LB medium containing 100 μg/mL ampicillin to each centrifuge tube and incubate in a shaker at 37°C for 6 h.
-
o.Isolate plasmid DNA from the bacterial cultures using a plasmid miniprep kit.
-
p.Use U6 sequencing primer U6-F to identify whether the 20 bp sgRNA oligonucleotides sequence is inserted correctly into the AAV-Cre-sgRNA vector.Optional: Preparation of AAV-Cre and AAV-mCherry-sgRNA1-sgRNA2 plasmids.The AAV-Cre-sgRNA only targets one gene. If you want to simultaneously knockout two genes, you can construct the AAV-Cre and AAV-mCherry-sgRNA1-sgRNA2 plasmid (Figure 2). In the AAV-Cre plasmid, the sgRNA cassette was removed from the AAV-Cre-sgRNA construct via a PCR strategy. The AAV-mCherry-sgRNA1-sgRNA2 was inserted into an mCherry expressing cassette driven by the TBG promoter and two sgRNA cassettes with one driven by the mouse U6 promoter, the other sgRNA driven by the human U6 promoter. With these two AAV constructs, two different genes can be simultaneously and equally targeted in vivo via the CRISPR/Cas 9 technology. In addition, the co-expression of the two fluorescent proteins can indicate the targeting efficiency—the mCherry expression indicates the successful delivery and expression of sgRNAs, while the GFP expression demonstrates the delivery of the AAV-Cre virus and expression of Cas9.
-
a.
Figure 1.

Schematic of the AAV-Cre-sgRNA vector
Figure 2.

Schematic of the AAV-Cre and AAV-mCherry-sgRNA1-sgRNA2
Preparation of buffer and solution
-
4.Preparation of polyethylenimine solution.
-
a.Add 1 g polyethylenimine (PEI) to 1 L ddH2O.
-
b.Stir to dissolve the PEI.
-
c.Add 1 M NaOH to adjust pH to 7.0.
-
d.Sterilize the solution using a 0.22 μm sterile filter.
-
e.Store at −20°C after aliquoting.
-
a.
-
5.Preparation of luciferin solution.
-
a.Prepare a 15 mg/mL fluorescein stock solution with sterile DPBS
-
b.Sterilize with 0.2 μm filter membrane.
-
c.After aliquoting, store at −80°C and protect from light.
-
a.
-
6.
Preparation of 10× DNA annealing buffer.
Note: Refer to Materials and equipment.
-
7.
Preparation of lysis buffer.
Note: Refer to Materials and equipment.
-
8.
Preparation of iodixanol solutions of different concentrations.
Note: Refer to Materials and equipment.
Design qPCR primers for AAV titer determination
-
9.
Choose one gene on the AAV vector to design qPCR primers for AAV titer determination. The primers used in this protocol are Cre-F and Cre-R.
Preparation of −80°C alcohol bath
-
10.
Pour 300 mL ethanol into a 500 mL enamel cup and put it at −80°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Sap I | New England Biolabs | Cat# R0569L |
| T7 Endonuclease I (T7EI) | New England Biolabs | Cat# M0302L |
| Ligation High | TOYOBO | Cat# LGK-101 |
| Polyethylenimine (PEI) | Polysciences | Cat# 23966-1 |
| Benzonase Nuclease | Merck | Cat# 70746-3 |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | Cat# D1556 |
| DNase I Amplification Grade | Sigma-Aldrich | Cat# AMPD1-D5307 |
| 10× Reaction Buffer | Sigma-Aldrich | Cat# AMPD1-R6273 |
| Proteinase K | TIANGEN | Cat# RT403 |
| D-Luciferin, Potassium Salt | YEASEN | Cat# 40902ES01 |
| Critical commercial assays | ||
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat# A25742 |
| TIANprep Mini Plasmid Kit | TIANGEN | Cat# DP103 |
| EndoFree Maxi Plasmid Kit | TIANGEN | Cat# DP117 |
| TIANamp Genomic DNA Kit | TIANGEN | Cat# DP304-03 |
| Gel Extraction Kit | CWBIO | Cat# CW2302M |
| Deposited data | ||
| AAV-Cre-sgRNA | This paper | Mendeley Data: https://doi.org/10.17632/y5g3kffbg9.1 |
| AAV-Cre | This paper | Mendeley Data: https://doi.org/10.17632/y5g3kffbg9.1 |
| AAV-mCherry-sgRNA1-sgRNA2 | This paper | Mendeley Data: https://doi.org/10.17632/y5g3kffbg9.1 |
| Experimental models: Cell lines | ||
| HEK293T | Chinese National Collection of Authenticated Cell Cultures | Cat# GNHu17 |
| Experimental models: Organisms/strains | ||
| Rosa26-LSL-Cas9 knock-in mouse | The Jackson Laboratory | Stock No: 024857 |
| Oligonucleotides | ||
| sgRNA-F | CACCGNNNNNNNNNNNNNNNNNNNN | N/A |
| sgRNA-R | AAACNNNNNNNNNNNNNNNNNNNNC | N/A |
| U6-F | GACTATCATATGCTTACCGT | N/A |
| Cre-F | TGAAGACCATCCAACAGCAC | N/A |
| Cre-R | CAAAGTCAGTGCGTTCAAAGG | N/A |
| Recombinant DNA | ||
| pAAV2/8 | Gift from James M. Wilson | Addgene plasmid # 112864 |
| pAdDeltaF6 | Gift from James M. Wilson | Addgene plasmid # 112867 |
| pAAV-GFP | Cell Biolabs | Cat# AAV-400 |
| Other | ||
| Centrifugal Filter | Merck Millipore | Cat# UFC910096 |
| 0.22 μm Sterile Filter | Merck Millipore | Cat# SLGPR33RB |
| Centrifuge Bottle | BECKMAN COULTER | Cat# 355618 |
| Optima XPN Ultracentrifuge | BECKMAN COULTER | Model No: XPN-100 - IVD |
| QuantStudio 6 Flex Real-Time PCR System | Thermo Fisher Scientific | Cat# 4485697 |
| IVIS Lumina II In Vivo Imaging System | PerkinElmer | Model No: Lumina II |
| Laser Scanning Microscopes | OLYMPUS | Model No: FV1200 |
Materials and equipment
10× DNA annealing buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl (pH8.0) | 400 mM | 2.4227 g |
| NaCl | 500 mM | 1.4611 g |
| MgCl2 | 100 mM | 0.4761 g |
| ddH2O | n/a | n/a |
| Total | n/a | 50 mL |
Store at 4°C for up to 1 year
Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl (pH8.0) | 20 mM | 0.1211 g |
| NaCl | 150 mM | 0.4383 g |
| ddH2O | n/a | n/a |
| Total | n/a | 50 mL |
Store at 4°C for up to 1 year
17% iodixanol
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PBS | 1 × | 5 mL |
| MgCl2 (1 M) | 1 mM | 0.05 mL |
| KCl (1 M) | 2.5 mM | 0.125 mL |
| NaCl (5 M) | 1 M | 10 mL |
| OptiPrep | n/a | 12.5 mL |
| ddH2O | n/a | 22.325 mL |
| Total | n/a | 50 mL |
Store at 4°C for up to 1 year
25% iodixanol
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PBS | 1 × | 5 mL |
| MgCl2 (1 M) | 1 mM | 0.05 mL |
| KCl (1 M) | 2.5 mM | 0.125 mL |
| OptiPrep | n/a | 20.83 mL |
| ddH2O | n/a | 23.995 mL |
| Total | n/a | 50 mL |
Store at 4°C for up to 1 year
40% iodixanol
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PBS | 1 × | 5 mL |
| MgCl2 (1 M) | 1 mM | 0.05 mL |
| KCl (1 M) | 2.5 mM | 0.125 mL |
| OptiPrep | n/a | 33.33 mL |
| ddH2O | n/a | 11.495 mL |
| Total | n/a | 50 mL |
Store at 4°C for up to 1 year
60% iodixanol
| Reagent | Final concentration | Amount |
|---|---|---|
| MgCl2 (1 M) | 1 mM | 0.05 mL |
| KCl (1 M) | 2.5 mM | 0.125 mL |
| OptiPrep | n/a | 50 mL |
| Total | n/a | 50.175 mL |
Store at 4°C for up to 1 year
Step-by-step method details
Production of AAV
-
1.Preparation of HEK293T cells.
-
a.Seed HEK293T cells into a 10 cm dish. Cells were cultured in DMEM supplemented with penicillin-streptomycin and 10% FBS at 37°C in a 5% CO2 incubator.
-
b.When the cells reach 80% confluence, split cell to 5 10 cm dishes.
-
c.Pass 5 dishes of HEK293T cells to 25 dishes, when the cells reach 80% confluency.
-
a.
Note: 25 dishes of HEK293T cells can pack 1013–1014 viruses. You can use the appropriate amount of 293T cells for virus packaging according to your needs.
-
2.Plasmids transfection.
-
a.When the cell density reaches 80%–90%, prepare for transfection.
-
b.Preheat PEI solution and DMEM at 37°C.
-
c.Add the pAAV2/8, pAdDeltaF6, and AAV-Cre-sgRNA to DMEM, pipetting up and down a few times to mix.
Reagent Amount pAAV2/8 10 μg pAdDeltaF6 10 μg AAV-Cre-sgRNA 10 μg PEI 90 μL DMEM n/a Total 1 mL -
d.Add 90 μL PEI (the ratio of PEI volume (mL) to the amount of total DNA (mg) is 3:1) to the Plasmid/DMEM mixture.
-
e.Vortex the mixture. Place at 20°C–24°C for 10 min.
-
f.Gently add the mixture dropwise to the cell culture medium and shake gently to mix.Note: The amount of reagent used in step 2 is for transfecting 1 dish of HEK293T cells.Optional: The AAV-Cre-sgRNA plasmid can be replaced with the pAAV-GFP plasmid, and one dish of HEK293T cells can be transfected at the same time to test the transfection efficiency. 24 h after transfection, GFP-positive cells should be greater than 80%. Troubleshooting 1
-
a.
-
3.
After 10–12 h of plasmids transfection, replace the medium with fresh culture medium.
Harvest of AAV
After 60 h of plasmid transfection, harvest the AAV from the culture medium and HEK293T cells.
-
4.Harvest the AAV from the culture medium.
-
a.Collect all the culture medium in several 50 mL centrifuge tubes.
-
b.Centrifuge at 3000 g for 10 min to remove cell debris.
-
c.Transfer all the supernatant to new centrifuge tubes.
-
d.Pipette 15 mL of supernatant into the centrifugal filter.
-
e.Centrifuge at 3000 g for 30 min at 4°C.
-
f.Remove the bottom fraction. Collect the concentrated supernatant in the filter into a new 50 mL centrifuge tube.
-
g.Repeat steps d-f, until all the supernatant is concentrated.
-
a.
CRITICAL: All supernatant should be concentrated to less than 7 mL, otherwise one centrifuge bottle will not be enough to hold all the concentrated supernatants.
-
5.Isolation of the AAV from HEK293T cells.
-
a.Add 1–2 mL DMEM into one dish and resuspend the HEK293T cells with a pipette by pipetting up and down several times.
-
b.Transfer the suspension to a 50 mL centrifuge tube.
-
c.In the same way, transfer the remaining 24 dishes of HEK293T cells to the 50 mL centrifuge tube.
-
d.Centrifuge at 3000 g for 15 min at 4°C
-
e.Discard the supernatant and resuspend the cell pellet in 3 mL lysis buffer.
-
f.Freeze the resuspended cells in an alcohol bath at −80°C for 30 min
-
g.Quickly transfer the frozen cells to a 37°C water bath for 20 min.
-
h.Repeat steps f to g.
-
i.Put the thawed cell suspension in the −80°C alcohol bath.
-
a.
Purification of AAV
-
6.
Quickly transfer the frozen cells from step 5 to a 37°C water bath for 20 min.
-
7.
Mix the concentrated supernatant from step 4 and the freeze-thaw cell suspension, add 1 M MgCl2 to a final MgCl2 concentration of 1 mM.
-
8.
Add Benzonase to the mixture to a final Benzonase concentration of 25 U/mL, mix well and incubate at 37°C for 30 min.
-
9.
Centrifuge at 3000 g for 20 min at 4°C.
-
10.
Preparation of discontinuous iodixanol gradients (Figure 3). Slowly add 3.5 mL 60%, 3.5 mL 40%, 4 mL 25%, 4 mL 17% iodixanol to the centrifuge bottle from bottom to top.
-
11.
Add the supernatant from step 9 onto the top of the 17% layer of iodixanol gradients very slowly and gently.
-
12.
Use Beckman Optima XPN-100-IVD ultracentrifuge, 70Ti rotor – centrifugation at 60000 rpm (max rcf ≈ 370000 g) for 2 h at 4°C, acceleration 6, deceleration 9.
-
13.
Using a 5 mL syringe and a 20-gauge needle to collect the 40% layer of iodixanol gradients.
-
14.
Transfer the 40% iodixanol to a centrifugal filter.
-
15.
Fill up the centrifugal filter with PBS. The volume after filling is about 15 mL.
-
16.
After mixing upside down, centrifuge at 3000 g for 30 min at 4°C. Troubleshooting 2
-
17.
Remove the bottom fraction.
-
18.
Repeat step 15 to 17 twice.
-
19.
Collect the remaining virus suspension in the filter into a 1.5 mL centrifuge tube, add PBS to dilute to about 1 mL. Troubleshooting 3
-
20.
After pipetting and mixing, aliquot 100 μL virus into each tube and freeze at −80°C. Aspirate 5 μL and prepare for AAV titer measurement later.
Pause point: Store at −80°C until titer measurement or AAV injection.
Figure 3.

Illustration of iodixanol gradients before centrifugation
The liquid in the centrifuge bottle from bottom to top is 60%, 40%, 25%, 17% iodixanol, and unpurified AAV.
qPCR for AAV genome titer measurement
-
21.
Prepare standards. Dilute the AAV-Cre-sgRNA plasmid at a ratio of 1:10 into 7 gradients. The highest starting concentration is recommended to be around 10 μg/mL.
-
22.Preparation of viral genomic DNA
-
a.Incubate 5 μL virus with DNase I at 37°C for 10 min to remove residual DNA in the solution.
Reagent Amount Virus 5 μL DNase I 5 μL 10× Reaction Buffer 5 μL ddH2O 35 μL Total 50 μL -
b.Incubate the mixture at 95°C for 10 min to denature DNase I
-
c.Add 1 μL proteinase K and incubate at 37°C for 10 min to digest the virus protein coat.
Reagent Amount From step b 50 μL Proteinase K 1 μL 10× Reaction Buffer 5 μL ddH2O 44 μL Total 50 μL -
d.Inactivate proteinase K at 95°C for 10 min.
-
a.
-
23.
Set up the qPCR reactions.
| Reagent | Amount |
|---|---|
| PowerUp SYBR Green Master Mix (2×) | 5 μL |
| Cre-F | 0.4 μL |
| Cre-R | 0.4 μL |
| DNA template | 1 μL |
| Nuclease-free water | 3.2 μL |
| Total | 10 μL |
-
24.
Set up and run the real-time PCR instrument.
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Activation | 50°C | 2 min | 1 |
| 95°C | 10 min | ||
| Denaturation | 95°C | 15 s | 40 |
| Annealing/ Extension | 60°C | 1 min | |
-
25.Calculate the titer of the virus
-
a.The number of copies N of single-stranded DNA per microliter in each standard can be approximated using the following formula from the concentration C (μg/μL) and the length L (bp) of the standard of the plasmid.Note: The average relative molecular mass of each deoxyribonucleoside is approximately 330. The length of the plasmid standard is L. Therefore, the relative molecular mass of the standard product is 2 × 330 × L. 6.02 × 1023 is Avogadro constant. So, the copy number of 2 × 330 × L g standard plasmid is about 6.02 × 1023.
-
b.Calculate the number of copies of single-stranded DNA in each standard
-
c.The Ct value of each standard product as the independent variable x, and the logarithm of the copy number with base 10 as the dependent variable y, to fit the linear regression equation (Figure 4). Troubleshooting 4
-
d.The Ct value of the viral genome is substituted into the linear regression equation, and the value y is calculated. Troubleshooting 5
-
e.10 to the power of y is the number of copies of the viral genome.
-
f.Because the virus is diluted 20 times in step 22, the concentration of the virus stock solution needs to be multiplied by 20.
-
a.
Figure 4.

An example of linear regression equation curve for standard products
The abscissa is the Ct value of the standard product. The ordinate is the logarithm of the standard DNA copy number with the base ten. The linear regression equation is y = −0.3076x + 11.242. The coefficient of determination (R2) is 0.9983.
Injection of AAV
-
26.
Remove the AAV from −80°C and dissolve it at 37°C.
-
27.
Dilute the virus titer with sterile PBS to 1 × 109/μL.
-
28.
Inject 250 μL of virus per mouse via tail vein injection.
Efficiency detection
-
29.In vivo bioluminescence imaging to detect whether the virus successfully infects the animal livers.Note: The AAV-Cre-sgRNA vector contains the luciferase gene, so in vivo bioluminescence imaging can be used to detect whether the virus successfully infects liver cells and expresses the viral genes.
-
a.1–2 weeks after virus injection, the virus infection is detected by in vivo imaging.
-
b.Each mouse was injected intraperitoneally with a luciferin/body weight concentration of 150 mg/kg.
-
c.After 10–15 min, when the light signal reaches the strongest stable plateau, perform the imaging analysis.
-
a.
-
30.Detect the green fluorescence of mouse liver tissue to determine whether the mouse successfully expresses the Cas9 protein.Note: In the Rosa26-LSL-Cas9 animals, the EGFP protein is co-expressed with Cas9 via a P2A linker sequence. Therefore, by observing the green fluorescence of the mouse liver, we can judge whether expression of the Cas9 protein in liver cells is successfully induced.
-
a.Detect the green fluorescence of the whole liver.
-
i.The mice were anesthetized by intraperitoneal injection of 4% chloral hydrate at a dose of 10 mL/kg.
-
ii.Open the abdominal cavity of the mouse and remove the liver.
-
iii.The liver was washed to remove residual blood in PBS.
-
iv.Put the liver in a 6 cm culture dish.
-
v.Observe the green fluorescence of the liver with a fluorescence microscope.
-
i.
-
b.Detect the green fluorescence of frozen sections of the liver.
-
i.Cut the liver tissue block with a thickness of less than 5 mm.
-
ii.Place the tissue block onto a cryomold.
-
iii.Cover the entire tissue block with the OCT compound.
-
iv.Slowly put the bottom of the mold into liquid nitrogen until the entire tissue block is completely frozen.
-
v.Transfer the frozen tissue block to a cryotome cryostat. Cut sections at a thickness of 4 μm.
-
vi.Observe the green fluorescence of liver slices with a fluorescence microscope.
-
i.
-
a.
-
31.Use T7E1 assay to detect genome editing efficiency.
-
a.Design primers to amplify mouse genome sites targeted by the sgRNA.Note: We recommend that the length of the PCR product should be between 600 bp–1000 bp. Long PCR products will increase the difficulty of amplification. The distance from the cut site should be greater than 150 bp. Otherwise, it will be difficult to separate the undigested DNA band and the longer digested fragment through DNA gel, and the shorter digested fragment will also be blurry on the DNA gel.
-
b.Extract genomic DNA from mouse liver.
-
c.Use the primers to amplify the genomic sequence targeted by sgRNA.
-
d.Run the PCR products on a 1% agarose gel. Cut the target DNA band and extract the DNA using a gel extraction kit.
-
e.Denature the DNA at 95°C for 5 min, then slowly cool the solution 0.1°C/s to 25°C in a thermocycler.
Reagent Amount DNA product from step 31b 500 ng NEBuffer 2 (10×) 2 μL ddH2O n/a Total 20 μL -
f.Digest the annealed DNA fragments with 0.5 μL T7 Endonuclease I (T7EI) enzyme at 37°C for 30 min.
-
g.Run samples on a 1% agarose gel.
-
h.Determine the cleavage efficiency by quantifying gel bands without overexposing them. A high-efficiency sgRNA will result in obvious cleavage bands, whereas the negative controls should have no cleavage band.
-
a.
-
32.
Use Western blot to detect whether the target protein in mouse liver has been successfully knocked out.
Expected outcomes
After the qPCR for AAV genome titer measurement, the total titer of the viral genome you can get is about 1014. The coefficient of determination (R2) of the standard linear regression equation should be greater than 0.98 and the amplification efficiency of primers should be between 90%–110%. This will ensure that your calculated titer is more accurate. One weeks after AAV injection, you can observe significant fluorescence intensity in mouse liver through in vivo imaging (Figure 5A). Three weeks after AAV injection, the liver DNA sequence targeted by sgRNA will generate obvious cleavage bands after T7E1 enzyme digestion (Figure 5B). If cutting bands are observed in the control group, it may indicate that there are natural heterozygous mutations (e.g., single nucleotide polymorphism, SNP) in the amplified DNA sequence, which can also be recognized by the T7E1 enzyme. Also, in some occasional situations, the inactivation of the T7E1 enzyme can also lead to the failure to detect digested fragments. Sanger sequencing can be performed on the amplified DNAs to directly determine whether the site has been edited. We can observe the green fluorescence of the liver with a fluorescence microscope (Figure 5C). We can also observe the green fluorescence of hepatocytes in frozen sections of the liver with a fluorescence microscope (Figure 5D). The target protein expression level will be significantly reduced, 3 weeks after injection of AAV carrying CRE and sgRNA (Figure 5E).
Figure 5.

The anticipated outcomes of the protocol
(A) In vivo bioluminescence imaging of mice one week after administration with 0, 5 × 1010, 1 × 1011 AAV-Cre-sgRNA AAVs.
(B) Gel image of liver DNA T7E1 assay after 3 weeks of infection with AAVs that does not carry (AAV-Control) and carries sgRNA (AAV-sgRNA).
(C) The images of GFP expression in fresh liver tissues from mice upon two weeks’ infection with 0 or 1 × 1011 AAV-Cre-sgRNA AAVs. Scale bars represent 1 mm. (D) The images of GFP expression in frozen liver sections from mice upon two weeks’ infection with 0 or 1 × 1011 AAV-Cre-sgRNA AAVs. Scale bars represent 250 μm.
(E) Western analysis of protein A expression in liver tissues from animals after 3 weeks’ infection with AAVs not carrying (AAV-Control) or carrying sgRNA (AAV-sgRNA).
Limitations
Due to the upper limit of cargo size in AAV packaging, it is difficult to express the spCas9 using AAV vectors. Therefore, our in vivo knockout system must use the Cas9 transgenic mice.
An sgRNA that can efficiently target the gene of interest will need to be screened out and inserted into the AAV vector. Usually we will need to first verify the activity of different sgRNAs on the cell line in vitro, and select the most efficient sgRNA for AAV packaging. However, due to the significant differences in cell types infected, as well as the delivery method adopted between the in vivo and in vitro experiments, some sgRNAs which work in vitro in cell lines may not work as well in vivo in liver cells.
Knocking out of the target gene directly in liver in vivo through the CRISPR system may also have potential off-target effects. Different sgRNAs targeting the same gene can be applied to exclude potential phenotypic changes due to off-target effects. Direct PCR and sequencing the potential off-target sites or even whole genome sequencing can be applied to evaluate the off-target effects.
We use qPCR to quantify the number of viral genomic DNA to determine the titer of AAV. However, this may overestimate the title of active AAVs, as some AAVs may not have infectious activity despite having the AAV genome. In addition, freezing and thawing can also affect the AAV activity. Therefore, the number of active AAV viruses we obtain should be lower than the titer we determined by qPCR.
Troubleshooting
Problem 1
Few GFP-positive cells. Plasmid transfection efficiency is low.
Potential solution
Plasmids transfection should be performed when the HEK293T cells reach 80%–90% confluency, otherwise the transfection efficiency will be greatly reduced.
After the frozen PEI solution is thawed, there will be some precipitate. Ensure that your PEI solution has been fully dissolved.
HEK293T cells are easily contaminated by mycoplasma. Mycoplasma contamination will reduce the transfection efficiency of cells. Make sure your cells are clean by testing cell supernatant with a mycoplasma detection kit.
Plasmids extraction with an endotoxin-free plasmid maxiprep kit can improve the transfection efficiency.
Problem 2
The liquid in the centrifugal filter is difficult to be filtered by centrifugation.
Potential solution
The 40% iodixanol and PBS in the centrifugal filter should be thoroughly mixed. Otherwise, the density of the 40% iodixanol at the bottom is too high to be centrifuged.
Problem 3
The virus solution is too turbid.
Potential solution
Centrifuge the virus solution at 3000 g for several minutes. Collect the virus supernatant and discard the pellet.
Problem 4
The coefficient of determination (R2) of the standard product linear regression equation is less than 0.95. A point deviates from the linear regression curve.
Potential solution
The standard was not sufficiently mixed when diluted. Ensure that each standard is thoroughly mixed when diluted.
The highest concentration of standard is recommended to be around 10 μg/mL. A too high concentration of DNA will not be able to obtain accurate Ct value through qPCR.
Problem 5
qPCR amplification curve of virus sample is not smooth. Did not detect the Ct value of virus samples.
Potential solution
The concentration of the viral genome was too high. Dilute the viral genome 10 times or 100 times before qPCR.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Qiurong Ding (qrding@sibs.ac.cn).
Materials availability
All unique materials generated from this study are available from the lead contact with a complete Materials Transfer Agreement.
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (2017YFA0102800, 2017YFA0103700), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16030402), the National Natural Science Foundation of China (31971063), Program of Shanghai Academic/Technology Research Leader (20XD1424500), Shanghai Science and Technology Commission (21140901700, 20dz1101400). We would like to thank Konglun Pan and Yifan Bu from Institutional Center for Shared Technologies and Facilities of SINH, CAS for technical assistance.
Author contributions
Conceptualization, Y.C. and Q.D.; Investigation, Y.C.; Writing – Original Draft, Y.C.; Writing – Review & Editing, Y.C. and Q.D.; Funding Acquisition, Q.D.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Yanhao Chen, Email: chenyanhao@sibs.ac.cn.
Qiurong Ding, Email: qrding@sibs.ac.cn.
Data and code availability
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/y5g3kffbg9.1.
References
- Ding Q., Strong A., Patel K.M., Ng S.L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt R.J., Chen S.D., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M., et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y., Ding Q. Optimized protocol for gene editing in adipocytes using CRISPR-Cas9 technology. STAR Protoc. 2021;2:100307. doi: 10.1016/j.xpro.2021.100307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson K.R., Hanna R.E., Hegde M., Donovan K.F., Strand C., Sullender M.E., Vaimberg E.W., Goodale A., Root D.E., Piccioni F., Doench J.G. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-07901-8. ARTN 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.D., Tian C., Zhao Y.X., Liu X.J., Liu F., Li S., Chen Y.H., Qiu Y., Feng Z.H., Chen L.L., et al. MRG15 orchestrates rhythmic epigenomic remodelling and controls hepatic lipid metabolism. Nat. Metab. 2020;2:447. doi: 10.1038/s42255-020-0203-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/y5g3kffbg9.1.