

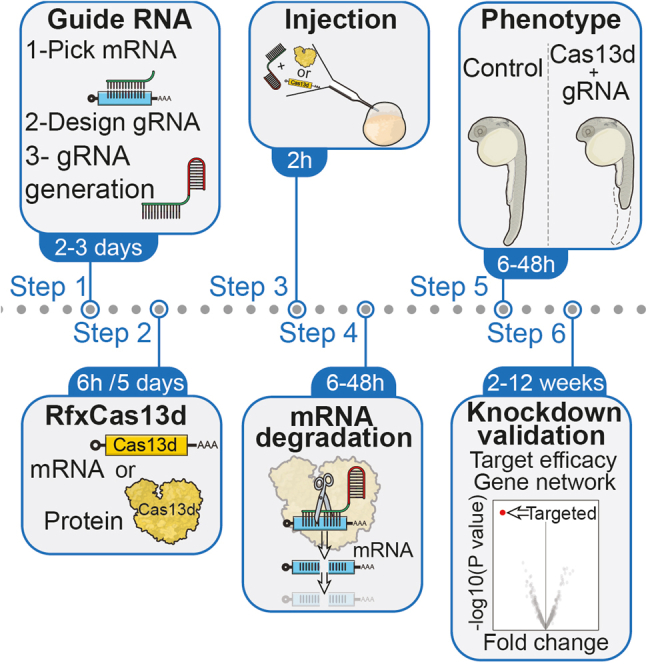
Summary
CRISPR-Cas systems have been used to induce DNA mutagenesis for gene function discovery. However, the development of tools to eliminate RNAs provides complementary and unique approaches to disrupt gene expression. Here, we present a workflow to perform specific, efficient, and cost-effective mRNA knockdown in zebrafish embryos using our in vivo optimized CRISPR-RfxCas13d (CasRx) system. Although the described protocol focuses on mRNA knockdown in zebrafish embryos, it can also be applied to other vertebrates.
For complete details on the use and execution of this protocol, please refer to Kushawah et al. (2020).
Subject areas: Biotechnology and bioengineering, CRISPR, Developmental biology, Gene Expression, Genetics, Model Organisms, Molecular Biology
Graphical abstract
Highlights
-
•
CRISPR-RfxCas13d optimized protocol for RNA targeting in zebrafish embryos
-
•
Step-by-step protocol from gRNA design to mRNA targeting and validation
-
•
Two alternative approaches including using RfxCas13d mRNA or purified protein
-
•
Different strategies for mRNA knockdown validation
CRISPR-Cas systems have been used to induce DNA mutagenesis for gene function discovery. However, the development of tools to eliminate RNAs provides complementary and unique approaches to disrupt gene expression. Here, we present a workflow to perform specific, efficient, and cost-effective mRNA knockdown in zebrafish embryos using our in vivo optimized CRISPR-RfxCas13d (CasRx) system. Although the described protocol focuses on mRNA knockdown in zebrafish embryos, it can also be applied to other vertebrates.
Before you begin
Despite the importance of the maternal-to-zygotic transition (MZT), the role of individual maternal mRNAs during early embryogenesis remains largely unknown. Zebrafish is an ideal system to study this early developmental transition but reliable knockdown strategies, to evaluate maternal RNA function have remained elusive. Knockdown reagents such as morpholino or antisense oligonucleotides have shown limited success, but factors of cost, toxicity, and recent concerns regarding specificity, have precluded their use in functional and systematic screenings. The CRISPR-Cas system has been extremely useful for the generation of mutants. However, understanding the role of different maternal factors in early development during MZT would require what are known as maternal (M) or maternal-zygotic (MZ) mutants that would have no maternal contribution. M and MZ mutants are still much more complicated to generate than zygotic mutants and might also be sterile as genes have an essential function for the female germ line, which makes the study of MZT more challenging. As an alternative, we have recently optimized the CRISPR-RfxCas13d system to efficiently and specifically target maternal RNAs in animal embryos. The protocol below describes the specific steps for zebrafish (Danio rerio) embryos. However, the efficiency of this protocol has been also tested for other animal embryos such as medaka (Oryzias latipes), killifish (Nothobranchius furzeri), and mouse (Mus musculus) (Kushawah et al., 2020.). First, we describe the design of gRNAs and their production as well as production and purification of protein and mRNA for RfxCas13d. Finally, we detail the microinjection of the CRISPR-RfxCas13d system into zebrafish embryos. This protocol is optimized for one-cell stage zebrafish embryo microinjection. Therefore, a microinjection station is required.
In summary, our optimized system emerges as a powerful technology to knockdown mRNA in vertebrates allowing to quickly and elucidate not only the role of the maternal mRNAs but also early expressed developmental genes. In addition, this tool could be used to target non-coding RNAs expressed during MZT. Moreover, the adaptation of the system to generate transgenic animals can be performed but it will imply further optimizations since toxicity has been observed in flies expressing RfxCas13d constitutively (Buchman et al., 2020).
Experimental model
All experiments involving zebrafish at CABD conform national and European Community standards for the use of animals in experimentation and were approved by the Ethical committees from the University Pablo de Olavide, CSIC and the Andalusian Government. Zebrafish wild type strains AB/Tübingen (AB/Tu) were maintained and bred under standard conditions (Westerfield, 1995). Wild-type zebrafish embryos were obtained through natural mating of AB/Tu zebrafish of mixed ages (5–18 months). Selection of mating pairs was random from a pool of 20 males and 20 females. Zebrafish embryos were staged in hours post-fertilization (hpf) as described (Kimmel et al., 1995). Zebrafish experiments at Stowers Institute were done according to the IACUC approved guidelines. Zebrafish embryos for microinjections were coming from random parents (AB, TF and TLF, 6–25 months old) mating from 4 independent strains of a colony of 500 fish. The embryos were pooled from random 24 males and 24 females for each set of experiments.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli DH5α | New England Biolabs | C2987I |
| E. coli Rosetta™(DE3) | NOVAGEN Healthcare | 70954-3 |
| Chemicals, peptides, and recombinant proteins | ||
| LB broth (LB Lennox) | Condalab | 1231 |
| Bacteriological american agar | Condalab | 1802 |
| Chloramphenicol | Sigma-Aldrich | C0378-5G |
| Kanamycin A sulfate | Sigma-Aldrich | K4000-5G |
| IPTG | Protein Ark | GEN-S-02122-25G |
| Precision Plus Protein Standard All Blue | Bio-Rad Laboratories | 161-0373 |
| UltraPure Tris | Invitrogen | 15504-020 |
| HCl | Scharlau | AC0741 |
| Polyethylene glycol 8000 (PEG) | Sigma-Aldrich | P2139 |
| Dimethyl sulfoxide (DMSO) | PanReac | 361954 |
| MgSO4·7H2O | Sigma-Aldrich | M7774 |
| 2-[4-(2-hydroxyethyl) piperazin-1-yl] ethanesulfonic acid (HEPES) | Fisher Scientific | BP310-500 |
| KOH | Merck | 105032 |
| KCl | Sigma-Aldrich | P3911-500G |
| DTT | Thermo Scientific | R0862 |
| Glycerol | Fisher Scientific | BP229-4 |
| Imidazole | Sigma-Aldrich | 56748-250G |
| TURBO-DNase | Ambion | AM2238 |
| Sodium Acetate | Sigma-Aldrich | 23424-28-4 |
| NaCl | Sigma-Aldrich | 7647-14-5 |
| KCl | Sigma-Aldrich | 7447-40-7 |
| NaHCO3 | Sigma-Aldrich | 144-55-8 |
| Tween20 | Sigma-Aldrich | P1379 |
| SDS | GE Healthcare | 17-1313-01 |
| Bromophenol Blue | GE Healthcare | 17-1329-01 |
| 10% TGX Stain-FreeTM FastCastTM Acrylamide Solutions | Bio-Rad Laboratories | 1610183 |
| Methylene blue | Sigma-Aldrich | 122965-43-9 |
| Tris Low EDTA | Sigma-Aldrich | T2194-100ML |
| Ethidium bromide | Sigma-Aldrich | 1239-45-8 |
| Restriction Enzymes | New England Biolabs | NEB |
| Q5® High-Fidelity DNA Polymerase | New England Biolabs | M0491 |
| Gel Loading Dye, Purple (6×) | New England Biolabs | B7025 |
| Gel Loading Buffer II (6×) | Thermo Fisher Scientific | 8546G |
| InstantBlue Coomassie Protein Stain | Abcam | ISB1L |
| Agarose D1 Low EEO | Condalab | 8010 |
| Critical commercial assays | ||
| AmpliScribe-T7-Flash transcription | Epicenter | ASF3507 |
| Qubit™ RNA BR Assay Kit | Thermo Fisher Scientific | Q10210 |
| mMachine T3 | Ambion | AM1348 |
| mMachine SP6 | Ambion | AM1340 |
| RNeasy Mini Kit (Qiagen) | Qiagen | 74104 |
| RC-DC™ Protein Assay Kit | Bio-Rad Laboratories | 5000122 |
| 30K Amicon Ultra-15 | Merck Millipore | UFC9030 |
| Favorprep Gel/PCR purification kit | Favorgen | FAGCK 001 |
| PureLink PCR Purification Kit | Thermo Fisher Scientific(invitrogen) | K310001 |
| Other | ||
| Incubator shaker | New Brunswick | Innova 42 |
| Horizontal laminar flow cabinet | Cruma | HZ-2 |
| Spectrophotometer | Shimadzu | UV-1280 |
| Centrifuge (for microtubes) | Eppendorf | 5415-R |
| Centrifuge (for Falcon tubes) | Eppendorf | 5804-R |
| Centrifuge (for big volumes) | Thermo Scientific | Sorvall Lynx 6000 |
| Thermoblock | Labnet International | Accublock D1100 |
| pH-meter | Hach | SensION+ PH3 |
| pH probe | Hach | 5014 |
| Sonicator | Branson | SFX 550 |
| 3/4" High-Gain Horn | Branson | 101-147-035R |
| 250 mL Rosette Glass Cooling Cell | Branson | 201-123-003 |
| ÄKTA Pure 25 L1 (Liquid Chromatography automated system) | GE Healthcare | 29-0148-31 |
| Absorbance detector U9-M (Liquid Chromatography automated system) | GE Healthcare | 28-9340-89 |
| Fraction collector F9-R (Liquid Chromatography automated system) | GE Healthcare | 29-0113-62 |
| His-Trap FF 1 mL column | GE Healthcare | 17-5319-01 |
| HiPrep 16/60 Sephacryl S-200 HR column | GE Healthcare | 17-1166-01 |
| SuperLoop 10 mL | GE Healthcare | 18-1113-81 |
| Amicon Ultra-15 (Centrifugal Filter Units) | Merck Millipore | UFC903024 |
| Qubit® 3.0 Fluorometer | Life technology | Q33216 |
| NanodropTM 2000 | Thermo Scientific | ND-2000 |
| Microinjector | Narishige | IM-300 |
| Stereomicroscope | Leica Microsystems | KL300-LED |
| Forceps Dumont #55 | Cibertec | 11255-20 |
| Acrodisc syringe filters 0.45 μm | Pall corporation | 4654 |
| 50 mL Erlenmeyer flasks | Eulabor | EFN3-050-012 |
| 2 L Erlenmeyer flasks | Eulabor | EFN3-2K0-001 |
| Petri dishes | Fisher Scientific | 12644785 |
| 50 mL centrifuge tubes | VWR | 525-1113 |
| 500 mL centrifuge tubes | Thermo Scientific | 3141-0500 |
| Clark Capillary Glass (1.0 OD × 0.58 ID × 100 L mm) | Panlab (Harvard Apparatus) | W3-30-0019 |
| Experimental models: Organisms/strains | ||
| Zebrafish | Stowers/CABD | Strains AB/Tübingen (AB/Tu), AB, TF and TLF |
| Recombinant DNA | ||
| pT3TS-RfxCas13d | Kushawah et al. (2020) | Addgene plasmid #141320 |
| pT3TS-RfxCas13d-NLS | Kushawah et al. (2020) | Addgene plasmid #141321 |
| pET28B-RfxCas13d-His | Kushawah et al. (2020) | Addgene plasmid #141322 |
| Oligonucleotides | ||
| Universal primer | Kushawah et al. (2020) | 5′TAATACGACTCACTATAGGAACCCCTACC AACTGGTCGGGGTTTGAAAC |
| Universal primer with Stem loop disruption | Wessels et al. (2020) | 5′TAATACGACTCACTATAGGTACCCCTACC AACTGGTCGGGGTTTGAAAC |
| tbxta_4 | Kushawah et al. (2020) | 5′AGCACCCGTATCTTTCAGCAAAGTTTCAA ACCCCGACCAGTT |
| tbxta_5 | Kushawah et al. (2020) | 5′GTGAGAGATACTCCAGCTTGAGGTTTCAA ACCCCGACCAGTT |
| tbxta_6 | Kushawah et al. (2020) | 5′TCGGTCCTGCTGGATTTTGTGGGTTTCAA ACCCCGACCAGTT |
| Tbxta_Fw | Kushawah et al. (2020) | 5′CGCAAGACTTCCGGAAGAGT |
| Tbxta_Rv | Kushawah et al. (2020) | 5′GAACCACAGAGCTGCTCCAT |
| Software and algorithms | ||
| RNAfold software | RNAfold web server | http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi |
| Unicorn (for chromatography system) | GE Healthcare | 7.1.0.378 |
| Image Lab Standard Edition (for SDS-PAGE analysis) | Bio-Rad Laboratories | 6.0.1.34 |
| Mammalian cell culture-based algorithm to predict CRISPR-RfxCas13d activity | Sanjana Lab (NYU, NY, USA) | https://cas13design.nygenome.org/ |
| Deposited data | ||
| Original data | This paper | Stowers Original Data Repository https://www.stowers.org/research/publications/libpb-1653 |
Materials and equipment
Here we review the material specifically needed for RfxCas13d protein purification and E3 buffer:
| Solution | Composition | Storage |
|---|---|---|
| Liquid LB | 2% (w/v) LB in ddH2O (autoclaved) | 25°C, 1 month |
| LB-Agar-Chl plates | 2% (w/v) agar, 15 μg/mL Chloramphenicol in liquid LB (autoclaved) | 4°C, 1 month |
| LB-Agar-Chl-Kan plates | 2% (w/v) agar, 15 μg/mL Chloramphenicol, 25 μg/mL Kanamycin A in liquid LB (autoclaved) | 4°C, 1 month |
| TSS Buffer | See below | 4°C, 12 months |
| 15 mg/mL Chloramphenicol | 1.5% (w/v) Chloramphenicol in ethanol | −20°C, 6 months |
| 25 mg/mL Kanamycin A | 2.5% (w/v) kanamycin sulfate in ddH2O | −20°C, 6 months |
| 1 M IPTG | 23.83% (w/v) IPTG in ddH2O | −20°C, 6 months |
| 1 M DTT | 15.4% (w/v) DTT in ddH2O | −20°C, 1 month |
| Tris·HCl 20 mM pH 7.5 | 0.24% (w/v) Tris in ddH2O, neutralized with HCl | 25°C, 6 months |
| HEPES·KOH 1M pH 7.5 | 23.83% (w/v) HEPES in ddH2O, neutralized with KOH | 25°C, 6 months |
| Lysis/Binding Buffer | See below | 25°C, 6 months |
| Elution Buffer | See below | 25°C, 6 months |
| Dialysis Buffer | See below | 25°C, 6 months |
| E3 buffer | See below | 25°C, 1 month |
TSS 2× Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| LB broth (LB Lennox) | 20 g/L | 0.2 g |
| Polyethylene glycol 8000 (PEG) | 200 g/L | 2 g |
| Dimethyl sulfoxide (DMSO) | 10% | 1 mL |
| MgSO4·7H2O | 100 mM | 246.5 mg |
| ddH2O | n/a | Up to 10 mL |
| Total | n/a | 10 mL |
Note: Adjust pH to 6.5
Lysis/Binding Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES·KOH 1M pH 7.5 | 50 mM | 25 mL |
| KCl | 500 mM | 18.64 g |
| DTT | 1 mM | 77.1 mg |
| Glycerol | 10% (v/v) | 50 mL |
| Imidazole | 10 mM | 0.34 g |
| ddH2O | n/a | Up to 500 mL |
| Total | n/a | 500 mL |
Note: Filter through 0.45 μm PES
Elution Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES·KOH 1M pH 7.5 | 50 mM | 5 mL |
| KCl | 500 mM | 3.73 g |
| DTT | 1 mM | 15.4 mg |
| Glycerol | 10% (v/v) | 10 mL |
| Imidazole | 500 mM | 3.40 g |
| ddH2O | n/a | Up to 100 mL |
| Total | n/a | 100 mL |
Note: Filter through 0.45 μm PES
Dialysis Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES·KOH 1M pH 7.5 | 50 mM | 25 mL |
| KCl | 250 mM | 9.32 g |
| DTT | 1 mM | 77.1 mg |
| Glycerol | 10% (v/v) | 10 mL |
| ddH2O | n/a | Up to 100 mL |
| Total | n/a | 500 mL |
Note: Filter through 0.45 μm PES
E3 buffer 60× buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 300 mM | 34.8 g |
| KCl | 10.2 mM | 1.6 g |
| CaCl2-2H2O | 20 mM | 5.8 g |
| MgCl2-6H2O | 20 mM | 10 mL |
| ddH2O | n/a | Up to 2 L |
| Total | n/a | 2 L |
Note: Adjust the pH to 7.2 with NaOH. Autoclave. To make a 1× E3 buffer, dilute 16.5 mL of the 60× stock to 1 L. Next, add 100 μL of 1% methylene blue (Sigma-Aldrich) as a fungicide.
Note: All reagents were purchased from commercial suppliers, stored under vendor indicated conditions, and employed without further purification.
Note: ddH2O is always autoclaved.
CRITICAL: Hazard Note-
Kanamycin – Harmful if inhaled or ingested. Potential skin irritant. Wear gloves. Chloramphenicol – Harmful if inhaled. Potential skin irritant. Wear gloves. IPTG – Harmful if swallowed or inhaled. Potential skin irritant. Wear gloves. DTT – Harmful if swallowed or inhaled. Potential skin irritant. Wear gloves.
Step-by-step method details
mRNA target sequence
Timing: 10 min
-
1.Select your target mRNA sequence in Ensembl Genome Browser (https://www.ensembl.org/):
-
a.Select: Zebrafish.
-
b.Search for a specific gene or genomic coordinates (e.g.,: tbxta, Chromosome 19: 14,187,540–14,191,592; GRCz11),
-
c.Click on your transcript of interest.
-
d.Select: cDNA (left sidebar).
-
e.Select: Download cDNA sequence in FASTA format. This includes 5′ and 3′ UTRs.
-
a.
Note: The transcript selection could be a problematic situation due to different splicing versions; therefore, we recommend selecting the longest transcript and then choose exons that are shared in all splice variants. If RNAseq data is available, it is convenient to select exons where RNA is clearly detected. And the targeted sequence can be analyzed to rule out potential mutations; however, as it will be discussed later that this step is not required.
gRNAs design
So far, there is not a systematic method or algorithm to design CRISPR-RfxCas13d gRNA to be injected into animal embryos. Two alternative methods have been tested with variable gRNA activity in vivo. First, gRNA design can be based on target accessibility according to the data in mammalian cell culture where gRNA activity is highly associated with target accessibility based on the three-dimensional conformation of the mRNA targets (Abudayyeh et al., 2017; Kushawah et al., 2020)) (option 1). Alternatively, there is a new approach for gRNA design according to an analysis in mammalian cell culture experiments that also proposes new gRNAs optimizations (Wessels et al., 2020). This gRNA design tool efficiently predicts CRISPR-RfxCas13d activity in mammalian cell culture experiments where gRNA and RfxCas13d expression is driven by constitutive promoters (Wessels et al., 2020) (option 2). Additionally, the tool also predicts potential off-targets in silico (0, 1, 2 mismatches between gRNA and RNA target). While this algorithm can efficiently predict CRISPR-RfxCas13d in mammalian cell culture, it has not been systematically tested in animal embryos. Therefore, in this protocol, we detail the different steps for the gRNAs design for both methods. Since none of the methods ensures that selected gRNAs are highly efficient in vivo, we recommend designing 3 gRNAs for each gene in the coding sequence region where the targeting is generally more efficient (Kushawah et al., 2020; Wessels et al., 2020) with 50 nt of distance between them to avoid potential competition between gRNAs to access the target.
Note: We highly recommend designing and using an additional set of 3 gRNAs that can help to validate and confirm potential phenotypes and rule out off-target effects (see mRNA knockdown validation section).
Note: If you choose to follow option 1 (gRNAs design based on target accessibility), proceed to step 2 and then follow steps 4–75. If you want to use option 2 (gRNAs design based on Wessels et al., 2020) proceed to step 3 and then follow to steps 4–75.
Option 1 – Target accessibility in silico
-
2.To find the most convenient mRNA region to be targeted, an in silico analysis is performed using RNAfold software (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi) (Lorenz et al., 2011):
-
a.Copy your cDNA sequence and paste it into the box.
-
b.Select the following options: minimum free energy (MFE) and partition function, avoid isolated base pairs, interactive RNA secondary structure plot, RNA secondary structure plots with reliability annotation (Partition function folding only) and Mountain plot.
-
c.Select: Proceed.
-
d.Select: color by base-pairing probability.
-
e.Search for protospacers of 22–23 nt with higher accessibility in silico (low base-pairing probability from minimum free energy predictions), blue or green regions, that belong to the Coding Sequence (CDS) and select 3 different protospacer spaced out across the transcript with a minimum distance of 50 nt between them to avoid an overlap between gRNAs, whenever possible (Figure 1).
Table 1.
Protospacer sequences (23 nt) of the gRNAs used in Figures 2A and 2B (gNTL)gRNA name Protospacer sequence 5′–3′ tbxta_1 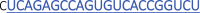
tbxta_2 
tbxta_3 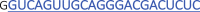 Bold and blue sequence indicates the protospacer sequence of 22 nt (WT).
Bold and blue sequence indicates the protospacer sequence of 22 nt (WT).
-
a.
Note: Recently, spacers of 23 nt and with a disruption in the first base-pair of the gRNA proximal stem loop have been shown to enhance the mRNA targeting in mammalian cell culture (Wessels et al., 2020). We have tested this enhanced gRNA version with a spacer of 23 nt and the stem loop disruption in zebrafish embryos (Figure 2A) and we have also observed a slightly higher efficiency (Figure 2B) that can be translated in higher phenotype penetrance (Figure 2C). However, we have found that these optimized gRNAs are frequently in vitro transcribed at lower efficiency (likely due to the modification in the stem loop disruption) potentially limiting their use (Figure 2D). For this reason, we normally generate gRNAs containing a spacer of 22–23 nt with the wild-type stem loop by in vitro transcription (IVT) and we preferentially order chemically synthesized gRNAs when they have the stem loop disruption and a spacer of 23 nt. In this protocol, we will detail the generation of gRNA by IVT for the CRISPR-RfxCas13d system with the 22 nt spacer and the wild-type stem loop structure.
Figure 1.

Result of the minimum free energy prediction for tbxta mRNA
The regions in green or blue show a lower base-pairing probability in silico, the 3 protospacer of 22 or 23 nt selected for the tbxta mRNA depletion are marked with black lines.
Figure 2.

Enhanced gRNAs are functional in vivo
(A) Schematic representation of the WT-gRNA with 22 nt of binding site and the optimized gRNA, SLD-gRNA, with the Stem loop disruption and 23 nt of binding sequence, both modifications are highlighted in red.
(B) qRT-PCR analysis showing the levels of tbxta mRNA in zebrafish embryos at 5 h post fertilization (hpf). Embryos were injected with 3 ng of RfxCas13d protein (Cas13d) and 300 pg of 3 gRNAs (see Table 1) targeting the ORF of tbxta mRNA. Different in vitro transcribed gRNAs were used: while gNTL-WT has the wild-type stem loop and a spacer of 22 nt, gNTL-SLD has the stem loop disruption described in Wessels et al. (2020) and a spacer of 23 nt. Results are shown as the averages ± standard deviation of the mean from two independents experiments with two biological replicates (n = 10 embryos) per experiment ∗∗∗ p< 0.001, ∗∗∗∗ p< 0.0001; (One-way anova).
(C) No-tail phenotype severity after knockdown of tbxta mRNA in similar conditions described in panel (B). Tbxta is involved in the formation of the notochord and the posterior part of the zebrafish embryo. Zebrafish embryo phenotypes were classified at 28 h post fertilization. Class I: Short tail (least extreme). Class II: Absence of notochord and short tail (medium level). Class III: Absence of notochord and extremely short tail (most extreme). ∗∗ p< 0.01; (Chi-Square). Number of embryos evaluated (n) is shown for each condition. Results from 2 independent experiments.
(D) In vitro transcription efficiency of different gRNAs formulations. Results are shown as the averages ± standard deviation of the mean from 11 RNA quantifications, after in vitro transcription of gRNAs differing in the stem loop formulation: wild-type (gRNA-WT) or with the stem loop disruption (gRNA-SLD). ∗ p< 0.05; (two tailed t test).
(E) Phenotypes obtained after injection of the Cas13d and the gRNAs (WT or optimized) targeting tbxta in zebrafish embryos (Lateral views). Levels of mosaicism compared to wild type (WT) were evaluated at 28 hpf. Class I: Short tail (least extreme). Class II: Absence of notochord and short tail (medium level). Class III: Absence of notochord and extremely short tail (most extreme). Scale bar, 0.25 mm.
Option 2 – Mammalian cell culture-based algorithm
-
3.Search for https://cas13design.nygenome.org/ to the gRNA design according to Wessels et al. (2020).
-
a.Select the model organism and select your model of interest.
-
b.Introduce the transcript ID of your interest target gene. (Also, you can search the transcript ID in the table using the gene symbol or gene name).
-
c.Introduce the Ensembl transcript ID in the search box.
-
d.Select the top 3 gRNAs targeting the Coding Sequence since gRNAs targeting UTR are less efficient (Wessels et al., 2020; Kushawah et al., 2020).
-
a.
Note: Selected gRNAs using this approach will have spacers of 23 nt.
-
4.
The DNA template to generate the gRNA is produced by fill-in-PCR with a universal primer and a specific primer.
The universal primer ( ) contains the T7 promoter (in pink) and the processed direct repeat for RfxCas13d crRNAs (in blue) preceded by 5′GG. Specific primer (
) contains the T7 promoter (in pink) and the processed direct repeat for RfxCas13d crRNAs (in blue) preceded by 5′GG. Specific primer ( ) contains the binding specific sequence (in green) and 20 nt of the repeat sequence in reverse complement orientation (in blue) (Figure 3).Note: The “5-GG” in the universal primer is required for the efficient in vitro transcription reaction mediated by T7 RNA polymerase.Note: Spacers can have 23 nt with the wild type stem loop but we did not systematically compare the efficiency of this formulation to others discussed here.Note: If a gRNA with the stem loop disruption wants to be generated, the universal would be:
) contains the binding specific sequence (in green) and 20 nt of the repeat sequence in reverse complement orientation (in blue) (Figure 3).Note: The “5-GG” in the universal primer is required for the efficient in vitro transcription reaction mediated by T7 RNA polymerase.Note: Spacers can have 23 nt with the wild type stem loop but we did not systematically compare the efficiency of this formulation to others discussed here.Note: If a gRNA with the stem loop disruption wants to be generated, the universal would be: Note: For example, injection of the mRNA encoding for RfxCas13d with gRNA targeting tbxta mRNA in zebrafish embryos should compromise notochord formation as well as a lack of posterior structures (no-tail phenotype). We selected 3 protospacers (22 nt) for the targeting of tbxta after the in-silico analysis:
Note: For example, injection of the mRNA encoding for RfxCas13d with gRNA targeting tbxta mRNA in zebrafish embryos should compromise notochord formation as well as a lack of posterior structures (no-tail phenotype). We selected 3 protospacers (22 nt) for the targeting of tbxta after the in-silico analysis: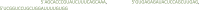 (in this case we selected them based on option 1 corresponding to regions with low probability of base pairing in Figure 1). To generate the DNA Template, we order the following primers:
(in this case we selected them based on option 1 corresponding to regions with low probability of base pairing in Figure 1). To generate the DNA Template, we order the following primers:-
a.Universal primer: This sequence contains the T7-promoter and the processed direct repeat for RfxCas13d preceded by 5′GG:
 Specific primers: This sequence contains 22nt of the specific protospacer and 20 nt of the repeat sequence in reverse complement orientation. Example primers for targeting tbxta mRNA:
Specific primers: This sequence contains 22nt of the specific protospacer and 20 nt of the repeat sequence in reverse complement orientation. Example primers for targeting tbxta mRNA:Name Sequence 5′–3′ tbxta_4 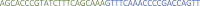
tbxta_5 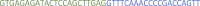
tbxta_6 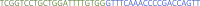 Note: gRNAs oligos can be ordered through companies such as IDT (www.idtdna.com). Oligos can be purchased as desalted and in minimal amounts.Alternatives: gRNAs can be ordered as chemically synthesized directly through companies such as Synthego (https://www.synthego.com/company). In that case, it is not necessary to include the 5′-GG, and the gRNA sequence is the correspond tail (processed direct repeat, Figure 5) of RfxCas13d gRNAs (
Note: gRNAs oligos can be ordered through companies such as IDT (www.idtdna.com). Oligos can be purchased as desalted and in minimal amounts.Alternatives: gRNAs can be ordered as chemically synthesized directly through companies such as Synthego (https://www.synthego.com/company). In that case, it is not necessary to include the 5′-GG, and the gRNA sequence is the correspond tail (processed direct repeat, Figure 5) of RfxCas13d gRNAs ( ) plus the reverse-complement of the 22–23 nt protospacer sequence (in green). Indeed, we mainly order chemically synthesized enhanced gRNAs (mentioned above) containing the stem loop disruption and a spacer of 23 nt (Wessels et al., 2020).Example of enhanced chemically synthesized gRNAs for targeting tbxta mRNA:
) plus the reverse-complement of the 22–23 nt protospacer sequence (in green). Indeed, we mainly order chemically synthesized enhanced gRNAs (mentioned above) containing the stem loop disruption and a spacer of 23 nt (Wessels et al., 2020).Example of enhanced chemically synthesized gRNAs for targeting tbxta mRNA:Name Sequence 5′–3′ (RNA) tbxta_synthego_4 
tbxta_synthego_5 
tbxta_synthego_6  CRITICAL: Currently, the design of gRNAs for RfxCas13d for targeting in vivo has some limitations, since there is not a validated strategy predictions model to design and select efficient gRNAs for in vivo approaches, and we have found gRNAs with low activity. To increase the probability of having a high level of depletion, we use 3–4 gRNAs together targeting the same RNA. In addition to using 3–4 gRNAs, we also recommend using individual gRNAs that have been tested to show higher mRNA depletion (see mRNA knockdown validation section), to minimize potential off-target effects and to provide reproducible results independent of the guide used.Note: We have observed that chemically synthesized gRNAs, either with wild type or disrupted stem loop, enhance the targeting of maternally provided in zebrafish embryos (Kushawah et al., 2020). That could be related with the highly purity and quality of these gRNAs and their more robust production since in vitro transcription may have byproducts and frequently show variability in the gRNA production among different DNA templates for distinct gRNAs (Figure 2D).
CRITICAL: Currently, the design of gRNAs for RfxCas13d for targeting in vivo has some limitations, since there is not a validated strategy predictions model to design and select efficient gRNAs for in vivo approaches, and we have found gRNAs with low activity. To increase the probability of having a high level of depletion, we use 3–4 gRNAs together targeting the same RNA. In addition to using 3–4 gRNAs, we also recommend using individual gRNAs that have been tested to show higher mRNA depletion (see mRNA knockdown validation section), to minimize potential off-target effects and to provide reproducible results independent of the guide used.Note: We have observed that chemically synthesized gRNAs, either with wild type or disrupted stem loop, enhance the targeting of maternally provided in zebrafish embryos (Kushawah et al., 2020). That could be related with the highly purity and quality of these gRNAs and their more robust production since in vitro transcription may have byproducts and frequently show variability in the gRNA production among different DNA templates for distinct gRNAs (Figure 2D).
-
a.
Figure 3.

Schematic representation of the fill-in-PCR to generate the DNA template
The Universal Primer contains the T7 promoter followed by 5′GG and the processed direct repeat for RfxCas13d gRNA, meanwhile, the Specific Primer contains the repeat sequence in reverse complement orientation to hybrid with the universal primer and the specific binding sequence of 22 nt. The product of the Fill-in-PCR is the DNA template that contains the T7 promoter with the 2 “GG” (19 nt), the processed direct repeat (30 nt) and the specific binding site sequence (22nt).
Figure 5.

Schematic representation of the in vitro transcription (IVT)
The DNA template generated (step 5) contains the T7 promoter followed by 5′GG and the processed direct repeat for RfxCas13d gRNA and the specific binding sequence of 22 nt. The overnight IVT product is the sgRNA for RfxCas13d system with the tail (in blue) and 22 nt of binding site to anneal with the mRNA target.
DNA template generation
-
5.Anneal universal primer and specific primer.
-
a.Mix the following reagents:
Reagents Amount Universal Primer (100 μM) 2.5 μL Specific Primer (100 μM) 2.5 μL dNTPs (40mM) 0.5 μL Buffer 5× 5 μL Q5® High-Fidelity DNA Polymerase (New England Biolabs #M0491) 0.25 μL DNase/RNase free water 14.25 μL Total 25 μL -
b.Enter the following program in a thermocycler:
Cycle step Temperature Time Cycles Initial denaturation 98°C 30 s 1 Denaturation 98°C 10 s 30 Annealing 60°C 30 s Extension 72°C 30 s Final extension 72°C 2 min 1 Note: If several gRNAs are used to target one gene in the same injection, the specific gRNA primers can be mixed in equimolar concentrations and then use 5 μL of this mix (100 μM of the total gRNA primer content) in the PCR reaction and a single in vitro transcription reaction is performed in the next step. Alternatively, each gRNA can be generated individually and be purified together in the same column (considering the maximal amount of RNA that each column can purify). Finally, gRNAs can be individually produced, purified and quantified and mixed at equal concentrations to generate the pool.Note: To increase the amount of DNA template, several replicates of the PCR can be performed and then mixed to carry out a single purification.Note: Since each gRNA-DNA template will be different, annealing temperature could be modified. Negative controls with universal or specific primers alone can be run to ensure that PCR bands are correct.Alternatives: Another DNA Polymerase could be used to generate the DNA -template, in that case, follow the protocol according to the manufacturer’s directions. -
c.Run PCR products (5 μL) plus 1 μL of Loading Dye (6×) in a 2% agarose gel, in TAE Buffer 1×. A single specific band of less than 100 nt is expected if the Fill-in-PCR has been successful (Figure 4).
-
a.
-
6.Purify the rest of the PCR product (at least 20 μL) using PCR/DNA Purification Kit (FavorPrep GEL/ PCR Purification Kit; FAGCK 001). Follow the manufacturer’s instructions. Briefly:
-
a.Transfer up to 100 μL of PCR product to a microcentrifuge tube (not provided) and add 5 volumes of FADF Buffer, mix briefly by vortexing.Note: For example, for 100 μL of PCR product, add 500 μL of FADF Buffer.Note: The maximum volume of PCR product is 100 μL. Do not exceed this limit. If a PCR product is more than 100 μL, divide it into multiple tubes.
-
b.Place a FADF column into a collection tube.
-
c.Transfer the sample mixture to the FADF Column. Centrifuge at 11,000 g for 30 s, then discard the flow-through.
-
d.Add 750 μL of wash buffer (ethanol added) to the FADF column. Centrifuge at 11,000 g for 30 s, and then discard the flow-through.Note: Make sure that ethanol (95%–100%) has been added to the wash buffer, when it is being opened first.
-
e.To remove residual ethanol, centrifuge at 18,000g for 3 min at room temperature (22°C–25°C). Place the FADF column in a new microcentrifuge tube (not provided).
-
f.Add 40 μL of nuclease free water to the center of the FADF membrane column. Stand the FADF Column for 1 min at room temperature.CRITICAL: For effective elution, make sure that the elution solution is dispensed onto the membrane center and is absorbed completely.CRITICAL: Elution should be performed with nuclease free water to avoid RNase contamination.Note: To work with a more concentrated template 30 μL of water can be used to elute, but we do not recommend using less than 30 μL because this could reduce the yield of DNA elution.
-
g.Centrifuge at full speed (∼18,000g) for 1 min to elute the DNA.
 Pause point: DNA template product can be kept at −20°C for several months.
Pause point: DNA template product can be kept at −20°C for several months.
-
a.
Figure 4.

Example of the gRNA template PCR amplification run in a 2% agarose gel.
-
7.
Quantify the purified PCR template by a spectrophotometer (Nanodrop).
Note: A minimum concentration of 50 ng/μL is enough to carry out the in vitro transcription reaction. Ideally, we use a total of 500–1000 ng (80–160 ng/μL) per in vitro transcription reaction (see below).
Alternatives: Another purification kit for PCR products can be also tested and used. In that case, follow the specific protocol provided in the kit.
In vitro gRNA transcription
-
8.gRNA for RfxCas13d system generation using AmpliScribe T7 Flash Transcription Kit (Epicentre, Lucigen # ASF3257) (Figure 5).
-
a.Mix the following reagents:CRITICAL: Setup IVT reaction at room temperature while keeping the enzyme solution in a −20°C cooler.
Reagents Amount gRNA DNA Template (From step 6) 6.3 μL ATP (100 mM) 1.8 μL CTP (100 mM) 1.8 μL GTP (100 mM) 1.8 μL UTP (100 mM) 1.8 μL DTT (100 mM) 2 μL AmpliScribe-T7 Flash 10× – Reaction Buffer 2 μL RiboGuard RNase Inhibitor 0.5 μL AmpliScribe T7-Flash Enzyme Solution 2 μL -
b.Incubate at 37°C for 12–16 h (overnight).
-
c.Day 2: Add 1 μL of TURBO DNase (2 U/μL).
-
d.Incubate at 37°C for 20 min.
-
a.
-
9.gRNA Precipitation.
-
a.Add 80 μL of nuclease free water to the IVT product.
-
b.Add 10 μL of 3M sodium acetate pH 5.2 and mix by vortexing.
-
c.Add 300 μL RNA-grade 95%–100% ethanol and mix by vortexing.
-
d.Incubate at least for 1 h at −80°C, alternatively incubate 5–6 h at −20°C.
-
e.Centrifuge at 16,100g for 30 min at 4°C.Note: A white pellet should be observed after centrifugation.
-
f.Discard the supernatant carefully and add 750 μL 75% ethanol to rinse without pipetting.
-
g.Centrifugate at 16,100g for 5 min at 4°C.
-
h.Repeat steps f–g.
-
i.Remove the supernatant carefully and dry the pellet 5 min to evaporate the ethanol.
-
j.Resuspend in 50 μL of nuclease free water. Make several aliquots with 10 μL and store in nuclease-free 1.5 mL tubes at −80°C.Alternatives: Despite we have not tested it, gRNAs could also be purified using small RNA purification kits.
-
a.
-
10.
gRNA quantification using the Qubit RNA BR (Broad-Range) assay kit (ThermoFisher, #Q10210)
-
11.Check the gRNA integrity by running an aliquot in a 2% agarose gel in 1× TAE Buffer. A single specific gRNAs band of 72 nt should be seen as shown in Figure 6.
-
a.Mix 1 μL of sgRNA, 4 μL Nuclease free water and 1 μL of Gel Loading Buffer II (Invitrogen, #8546G).
-
b.Incubate the mix at 70°C for 10 min and place it on ice.
-
c.Run the gel at 100 V for 20 min.
-
a.
Note: To avoid RNase contamination, run the gel in a new 1× TAE Buffer and clean the tray and the comb properly with RNase away before use. The electrophoresis system can be cleaned with water and soap and rinsed with abundant water before use.
Note: When the in vitro transcription reaction worked successfully, expected yield of gRNAs was approximately between 300 and 1000 ng/μL.
Note: See troubleshooting section problem 1 for possible solutions to low gRNA production
Figure 6.

Analyses of gRNA integrity using agarose gel electrophoresis
gRNA produced in the IVT reaction should look like a single sharp band without degradation and located below the 100 bp line of the molecular weight marker.
mRNA RfxCas13d production
There are two alternative approaches to use the RfxCas13d system: The mRNA encoding Rfx-Cas13d or the purified Rfx-Cas13d protein that enhances the mRNA targeting for maternal products (Kushawah et al., 2020), see details below. If you choose the use of mRNA-RfxCas13d follow steps 12–15 and then proceed to steps 61–75. If you want to use the RfxCas13d protein follow steps 16–75.
-
12.
Order the pT3TS-RfxCas13d-HA plasmid (Figure 7) from Addgene (Plasmid #141320, http://www.addgene.org/141320/) needed to perform the in vitro transcription of RfxCas13d (Human codon-optimized).
Note: This plasmid is owned by Moreno-Mateos Lab (CABD, Seville, Spain) and Bazzini Lab (Stowers Institute for Medical Research, Kansas City, MO, USA).
-
13.RfxCas13d linearization.
-
a.Mix the following reagents for a 30 μL reaction.
Reagents Amount 2–3 μg plasmid Variable 10× CutSmart Buffer 3 μL XbaI 1 μL Nuclease free water Up to 20 μL Alternatives: Other enzymes could be used to linearize the plasmid. Use enzymes with unique restriction sites after 3′UTR, such as Kpn1, Acc65l, SfiI, PstI, or vSbpf1. -
b.Incubate the reaction for 2 h at 37°C.
-
c.Run 2–3 μL of the reaction in a 1% agarose gel in 1× TAE Buffer. A single specific band of 6.1 kb should be observed.
-
d.Purify the linearized plasmid using a FavorPrep Gel/PCR Purification Kit (FavorPrep GEL/ PCR Purification Kit; FAGCK 001). Follow the manufacturer’s protocol as in step 6.
-
e.Quantify the linearized plasmid concentration using a spectrophotometer/fluorometer (nanodrop/Qubit).Note: An aliquot of the purified linearized plasmid could be run in a 1% agarose gel in 1× TAE Buffer to verify the efficiency of the purification kit.Pause point: The purified linearized plasmid can be stored at −20°C for several months.Alternatives: Another purification kit for PCR products can be also tested and used. In that case, follow the specific protocol provided in the kit.
-
a.
-
14.RfxCas13d in vitro transcription.Note: The following protocol is based on the mMESSAGE mMACHINE™ T3 Transcription Kit (#ThermoFisher, Ambion, AM1348)
-
a.Mix the following reagents:
Reagents Amount Linear template DNA 6 μL NTP/CAP (2×) 10 μL Reaction Buffer (10×) 2 μL Enzyme Mix (T3) 2 μL CRITICAL: Setup the IVT reaction at room temperature while keeping the enzyme at −20°C in a benchtop cooler because the spermidine in the 10× reaction buffer can precipitate if the buffer is chilled, which can cause the reaction to fail.Note: 6 μL of reaction should contain up to 1 μg of linear DNA template. -
b.Incubate the reaction for 2 h at 37°C.
-
c.Add 1 μL TURBO DNase (2 U/μL).
-
d.Incubate for 20 min at 37°C.Pause point: Stop the reaction by freezing the tube in liquid nitrogen and store it at −80°C, or continue with the mRNA purification immediately, step 15.
-
a.
-
15.Purify the mRNA of RfxCas13d using RNeasy Mini Kit (Qiagen, # 74104). All the reagents required are provided in the kit.
-
a.Add 80 μL Nuclease free water to the IVT sample from step 14.
-
b.Add 350 μL Buffer RLT and mix well.
-
c.Add 250 μL 96%–100% ethanol and mix by pipetting. Do not centrifugate. Proceed immediately to the next step.
-
d.Transfer the sample to a RNeasy Mini Spin Column placed in a 2 mL collection tube (provided in the kit). Centrifuge for 15 s at 8000g. Discard the flowthrough.
-
e.Add 500 μL Buffer RPE to the column and centrifugate for 2 min at 8000g. Discard carefully the flowthrough.
-
f.Place the RNeasy Mini Spin Column in a new 2 mL collection tube and centrifugate at maximum speed to eliminate any possible carryover of buffer RPE.
-
g.Place the RNeasy Mini Spin Column in a new 1.5 mL collection tube and add 30 μL of Nuclease free water to the spin column membrane and centrifugate at 8000g to elute the RNA.
-
h.Add the elute from the previous step to the used column and centrifugate again for 1 min at 8000g to elute the RNA.
-
i.Quantify the mRNA concentration using a spectrophotometer/fluorometer (nanodrop/Qubit). The mRNA concentration used to be between 500 and 1000 ng/μL.
-
j.Make several aliquots of 5–10 μL in nuclease-free 1.5 mL tubes and store at −80°C.
-
a.
Alternatives: RfxCas13d-HA mRNA version with NLS can be produced as described above (Plasmid #141320, http://www.addgene.org/141321/). RfxCas13d-NLS can be useful to target mRNAs that are exclusively or preferentially localized in the nucleus such as some lncRNAs, pri-miRNAs, etc. Although we have not systematically tested this RfxCas13d version, we have observed that the targeting efficiency of Rfx-Cas13d-NLS may be lower in zebrafish embryos compared to RfxCas13d without NLS (Kushawah et al., 2020). However, this is not due to a lower in vivo translation (Kushawah et al., 2020) or a lack of robust nuclear localization (data not shown) that could potentially reduce RNA targeting activity in the nucleus.
Note: See troubleshooting section problem 2 for potential solutions related to low RfxCas13d mRNA production.
Figure 7.

Map of the pT3TS-RfxCas13d-HA plasmid
Preparation of RfxCas13d protein expressing bacterial colonies
The RfxCas13d protein could be purified using Histidine tag and directly injected in zebrafish embryos at 1-cell stage. It has been demonstrated that the use of RfxCas13d protein enhances the maternal mRNA targeting in zebrafish embryos (Kushawah et al., 2020).
In this step, the E. coli Rosetta DE3 pRARE strain is chemically transformed (Chung et al., 1989). Other transformation methods or E. coli strains can be employed to afford RfxCas13d producing bacteria (i.e., we tested other E. coli BL21 DE3 based strains with positive induction results). However, induction parameters (temperature, IPTG concentration, and incubation time) have been optimized for this strain and chemical transformation.
-
16.Prepare the following items:
-
a.Autoclaved 25 mL of liquid LB broth (Sambrook et al., 1989); 2 × 50 mL Erlenmeyer flasks; 2 × 1.5 mL microcentrifuge tubes.
-
b.Petri dish with LB-Agar-Chloramphenicol; 2 × Petri dish with LB-Agar-Chloramphenicol-Kanamycin A.
-
c.TSS 2× buffer (see materials and equipment for recipes).
-
a.
Note: Antibiotic final concentrations are: 15 μg/mL for Chloramphenicol; 25 μg/mL for Kanamycin A for both the agar plates and LB.
-
17.
Streak the E. coli Rosetta DE3 pRARE on a LB petri dish with Chloramphenicol 15 μg/mL.
-
18.
Incubate the Petri dish at 37°C (16–18 h).
-
19.
Pick up a single colony from the previous culture and inoculate 10 mL of liquid LB-Chloramphenicol in a 50 mL Erlenmeyer flask.
-
20.
Cap and incubate the flask at 37°C (16–18 h) while shaking at 180 rpm.
-
21.
Next morning, dilute 100 μL of the previous culture with 10 mL of liquid LB-Chloramphenicol into a new 50 mL Erlenmeyer flask.
-
22.
Cap and incubate the flask at 37°C while shacking up at 180 rpm until an OD600 of 0.3–0.4 is reached (2 h approx.)
-
23.
At OD600 = 0.3–0.4, spin 2× 1 mL of the culture into microcentrifuge tubes (1 mL per tube) at 16000 × g for 30 s at 4°C.
-
24.
Remove the supernatants and resuspend the pellets in 75 μL of cold LB. Keep the tubes on ice for 5 min.
-
25.
Add 75 μL of cold TSS 2× buffer to each tube and mix well with a pipette. Keep the tubes on ice for 5 min.
-
26.
Add 0.2–1 μg of plasmid pET-28b-RfxCas13d-His (Kushawah et al., 2020) (Addgene #141322) to one of the tubes and mix well with a pipette. The other tube is a negative control. Keep the tubes on ice for 30 min.
-
27.
Incubate the tubes at 42°C for 40 s in a thermo block. Keep both tubes on ice for 1 min.
-
28.
Add 1 mL of LB to each tube and incubate at 37°C for 1.5 h.
-
29.
Centrifuge at 16,000 × g for 1 min at room temperature.
-
30.
Remove the supernatant without touching the pellet and resuspend them in 100 μL of LB.
-
31.
Spread the transformed bacteria onto a selective LB-Agar-Chloramphenicol-Kanamycin A petri dish and incubate at 37°C (16–18 h).
RfxCas13d protein expression
In this step, RfxCas13d protein production will be induced in cultures of previously transformed bacteria. Here we describe the protocol for a 500 mL culture but, if the appropriate equipment is available, it can be scaled up to bigger volumes (e.g.,: in our lab, we have handled up to 2 L of culture at once).
-
32.Prepare the following items:
-
a.Autoclaved: 520 mL of liquid LB broth; 2 × 50 mL Erlenmeyer flasks; 2 L Erlenmeyer flask.Solutions:
-
b.Isopropyl Beta -D-1 thiogalactopyranoside (IPTG) solution 1M; Tris·HCl solution 20 mM pH 7.5 (see materials and equipment for recipes).
-
c.A 10% poly-acrylamide gel.
-
a.
-
33.
Pick up a single colony from the plate of Rosetta cells previously transformed with pET28b-RfxCas13d and inoculate 10 mL of liquid LB-Chloramphenicol-Kanamycin A in a 50 mL Erlenmeyer flask.
-
34.
Cap and incubate the flask at 37°C (16–18 h) while shaking at 180 rpm.
-
35.
Next morning, dilute 5 mL of the culture in 500 mL of liquid LB-Chloramphenicol-Kanamycin A in a 2 L Erlenmeyer flask.
-
36.
Cap and incubate the flask at 37°C while shaking at 180 rpm.
-
37.
Measure the culture OD600 periodically (at least every 30 min) using a spectrophotometer and LB as reference blank.
-
38.At OD600 = 0.45–0.55 (annotate the data. It takes approx. 3 h):
-
a.Transfer 1 mL of the culture into a 1.5 mL microcentrifuge tube.
-
b.Spin at 16,000 × g for 1 min at 4°C, completely remove the supernatant and store the pellet at −20°C (this will be the pre-induction reference).
-
c.Add 50 μL of 1M IPTG to the Erlenmeyer flask containing the bacterial culture (final IPTG concentration = 0.1 mM) and continue the incubation at 37°C while shaking at 180 rpm for 3 h.
-
a.
-
39.
Measure the culture OD600 at this point and annotate it.
The OD600 should be between 1.5 and 2.15:-
a.Transfer 1 mL of the culture to a 1.5 mL microcentrifuge tube.
-
b.Spin the tube at 16,000 × g for 1 min at 4°C, completely remove the supernatant and store the pellet at −20°C (this will be the post-induction reference).
-
a.
-
40.
Centrifuge the 500 mL culture at 8,000 × g for 15 min at 4°C.
-
41.
Discard the supernatant and resuspend the pellet in 20 mL of Tris·HCl 20 mM (pH 7.5).
-
42.
Centrifuge at 8,000 × g for 15 min at 4°C.
-
43.
Discard the supernatant and store the pellet at −80°C.
Figure 8.

SDS-PAGE of pre and post-induction references
Performed in a 10% polyacrylamide gel and run at 200 V for 45 min. Stained with InstantBlue® Coomassie Protein Stain (Abcam) following manufacturer protocol. M is the molecular weight ladder (Precision Plus Protein Standard All Blue – BioRad). RfxCas13d (113 kDa) is highlighted with a black arrow. Pre-I (pre–induction) and Post-I (post–induction).
RfxCas13d protein purification
RfxCas13d protein (113 kDa) is purified from the frozen cell pellets prepared in the previous step. The pellets are lysed by sonication and the protein purified by two consecutive chromatography steps (Ni-affinity chromatography followed by size exclusion chromatography).
-
44.Prepare the following items:
-
a.Buffers: 300 mL of lysis/binding buffer; 100 mL of elution buffer; 500 mL of dialysis buffer (see materials and equipment for recipes).
-
b.4 × 10% polyacrylamide gels (prepared freshly right before use or stored up to 7 days at 4°C).
-
c.Prepare the chromatography system by properly washing and equilibrating the whole system (including columns) with the corresponding buffer, as well as preparing necessary recipes and microtubes for collecting eluates.
-
a.
-
45.
Thaw the bacterial pellet from the previous step on ice for 10 min.
-
46.
Resuspend the pellet in 50 mL of lysis/binding buffer.
-
47.
Place the cell suspension into a cooling cell in an ice bath and sonicate it with a horn sonicator displaying the following settings: Pulse program, 3 s ON, 10 s OFF, 120 cycles, 30% amplitude.
Note: Previous settings are optimized for our sonicator (Branson SFX 550) with its “Rosette Glass Cooling Cell” and the “High Gain Horn” (see materials and equipment for details). If other equipment is employed, settings might need to be adapted accordingly.
-
48.
Centrifuge the sonicated mixture at 10,000 × g for 15 min at 4°C.
-
49.
Filter the supernatant through a 0.45 μm PES filter. Save 10 μL of the filtrate as “input reference” for SDS-PAGE, stored on ice.
-
50.
Flow the lysate through a His-Trap FF column, pre-equilibrated with lysis/binding buffer, at 1 mL/min and 8°C. Monitor the UV absorbance at 280 nm and collect the flow through in a clean container. Take 10 μL of the output as “flow through reference” for SDS-PAGE, store on ice.
-
51.
Wash with 5 column volumes of lysis/binding buffer and collect the eluate in a separate container. Take 10 μL of the output as “wash reference” for SDS-PAGE, store on ice.
-
52.
Flow elution buffer at 1 mL/min and 8°C to recover bound proteins and collect 1.5 mL fractions of the eluate until the 280 nm UV absorbance line reaches the baseline.
-
53.
Take 5 μL aliquots of all elution fractions and run them in SDS-PAGE electrophoresis, together with input, flow through and wash references previously collected (A typical example is shown in Figure 9B).
-
54.
Put together all fractions corresponding to the previous elution peak (F4, F5 and F6 in this example, for a total volume of 4.5 mL) and load the mixture into a SuperLoop. Save 5 μL of the mixture as “input reference” for SDS-PAGE, stored on ice.
-
55.
Flow the sample on a HiPrep 16/60 Sephacryl S-200 HR column, pre-equilibrated with dialysis buffer, at 1 mL/min and 8°C. Monitor the UV absorbance at 280 nm.
-
56.
Keep eluting the sample with a dialysis buffer at 1 mL/min and collect 2 mL fractions of the eluate until the 280 nm UV absorbance line reaches the baseline.
-
57.
Take 10 μL aliquots of all elution fractions and run them in SDS-PAGE electrophoresis, together with input reference previously collected (a typical example is shown in Figure 10B).
-
58.
Put together all elution fractions with a significant amount of RfxCas13d (in Figure 10B example, F11 to F15) and concentrate the mixture in a centrifugal filter with 30 KDa molecular weight cut-off. Spin the sample at 4,000 × g at 4°C until the volume reduces to about 1 mL.
-
59.
Quantify protein concentration as described in step 60 and stop concentrating when sample reaches ∼3 mg/mL. Make 5 μL aliquots (∼200 1.5 mL tubes) and store them at −80°C. We recommend single use 5 μL aliquots. Additional thaw-frozen cycles can reduce RfxCas13d activity. We recommend to quickly store the aliquots at −80°C and not keep them at 4°C more than the needed time to make the aliquots. Storage at 4°C or −20°C will negatively affect RfxCas13d activity.
Optional: Although it is not strictly necessary, for the sake of quality control and reproducibility, we strongly recommend the quantification of the sample.
-
60.
Quantify the amount of purified RfxCas13d by Bradford densitometry on SDS-PAGE gel employing BSA as reference. RfxCas13d samples must be diluted in a dialysis buffer in a 1 to 20 ratio, loading 2–6 μL of the diluted sample in the gel. With this protocol, we usually obtain a sample concentration of ∼3 mg/mL (a typical example is shown in Figure 11).
Alternatives: Other quantification methods (e.g.: Lowry or BCA based, UV absorption) can be employed instead of the Bradford densitometry. Particularly, we have also quantified RfxCas13d with the RC-DC Protein Assay Kit II (Bio-Rad, #5000122).
Note: See troubleshooting section problem 3 for potential solutions to low RfxCas13d protein production.
Figure 9.

Affinity chromatography purification of RfxCas13d
(A) Chromatogram from RfxCas13d (113 kDa) Histidine affinity chromatography. Blue line (left): absorption at 280 nm; green line (right): % of elution buffer; both vs eluted volume.
(B) Expansion of the elution peak from previous chromatogram, showing the fractions collected.
(C) SDS-PAGE of input (In – step 49), flow through (Out – step 50), wash (W - step 51) and peak fractions (F4, F5, F5 and F7) references. Performed in a 10% polyacrylamide gel and run at 200 V for 45 min. Stained with InstantBlue® Coomassie Protein Stain (Abcam) following manufacturer protocol. M is the molecular weight ladder (Precision Plus Protein Standard All Blue – BioRad).
Figure 10.

Size Exclusion chromatography purification of RfxCas13d
(A) Chromatogram from RfxCas13d (113 kDa) size exclusion chromatography. Absorption at 280 nm vs eluted volume.
(B) SDS-PAGE of input (In – step 54) and elution fractions of main peak (F7 to F23 – step 57) references. Performed in a 10% polyacrylamide gel and run at 200 V for 45 min. Stained with InstantBlue® Coomassie Protein Stain (Abcam) following manufacturer protocol. M is the molecular weight ladder (Precision Plus Protein Standard All Blue – BioRad).
Figure 11.

SDS-PAGE of 3 different (known concentration) dilutions of BSA and 3 different (unknown concentration) dilutions of RfxCas13d (113 kDa)
Performed in a 10% polyacrylamide gel and run at 200 V for 45 min. Stained with InstantBlue® Coomassie Protein Stain (Abcam) following manufacturer protocol. M is the molecular weight ladder (Precision Plus Protein Standard All Blue – BioRad). The calibration curve and final quantification were performed with the Image Lab software (Bio-Rad).
Microinjection of CRISPR-RfxCas13d system in zebrafish embryos
-
61.
Day 1. Zebrafish Breeding. Separate adult zebrafish of at least 3–4 months old (male/female) in 1 L breeding tanks, using a separating barrier, in the afternoon. We recommend separating 5 males and 5 females in group mating, in a ratio of 1:1.
Note: Females differ from males by having a bigger belly size, lighter silverish stripes and by the presence of a membrane in the anal fin .
Note: See troubleshooting section problem 4 for possible solutions to low zebrafish embryo production.
-
62.
Prepare several agarose plates at 1% of agarose in E3 medium for the maintenance of zebrafish embryos. Plates for microinjection are prepared by adding 2% agarose to E3 medium and placing a plastic mold (teeth down) to make the different lines where the embryos will be placed, while the agarose is still hot (before solidifying). Store the plates upside down at 4°C 1 week as maximum. To avoid potential medium contamination, we recommend not to reuse the plates.
-
63.
Day 2. Needle calibration. In the morning, thaw the RfxCas13d mRNA/protein (300 ng/μL and 3 μg/μL, respectively) and gRNAs on ice. Pipette 2 μL of the RfxCas13d mRNA/protein and load into the needle (Harvard, apparatus, 1.0 OD × 0.58 ID × 100 L mm #30-0019 Capillaries GC100F-10) and calibrate the needle using a micrometer calibration slide microscope with 0.01 mm ruler. Using a microinjector (Narishige IM-300) and a stereomicroscope (Leica KL300) and with the help of forceps open the needle end to an adequate size opening. Calibrate the system in order to obtain a bubble of 0.01mm that corresponds to an injection volume of 0.5 nL.
Note: RfxCas13d mRNA could be diluted in Nuclease-free water meanwhile the RfxCas13d protein must be diluted using the dialysis buffer (see materials and equipment for recipes).
Note: Needles need to be prepared from glass capillaries with the Flaming/Brown Micropipette Puller (or similar horizontal pullers) and the following conditions: Pull (150), Vel (70), Time (80), Heat (590). Heat temperature is calculated with the Run-Test function that results in the RAMP value, Heat = RAMP value + 30. These conditions may change between different Micropipette Pullers and should be adjusted to the specific model.
Note: Microinjector pressure should be between 2.0 and 10.0 psi. We recommend a pressure of 6.0–7.0 psi depending on the needle pore size.
Note: RfxCas13d protein calibration could be more complicated due to glycerol content that can clog the needle. We recommend widened needle pore size and inject with a lower pressure (3.0–5.0 psi). If the needle is clogged during the injection, we suggest opening more of the needle and recalibrating it again.
-
64.
When the zebrafish facility lights turn on in the morning, remove the separating barriers between males and females from the breeding tank. Wait until eggs are laid and collect them from the tank using a strainer. This typically takes about 5–10 min but it can vary.
-
65.
Dechorionate the embryos using pronase at 1 mg/mL in E3. Incubate the embryos in 3 mL of pronase solution for approximately 2 min.
Note: Touch a few embryos with the forceps to assure whether the pronase solution is causing a proper dechorionation, if the solution has worked optimality the corium will be removed when is touched with the forceps. We highly recommend to dechorionate the embryos. This will facilitate that the RfxCas13d mRNA/protein and gRNAs are injected into the cell and not in the yolk, which enhances the mRNA targeting. Alternatively, chorionated embryos can be injected but it is important to inject always into the cell.
-
66.
Carefully wash the embryos several times (5–7 washes approximately) with new E3 medium in a 250 mL glass beaker and carefully remove the dechorionated embryos with a glass pipette. Repeat this step until most of the embryos are dechorionated.
-
67.
Place 60–70 embryos in each row of the microinjection plate (prepared the day before) with a glass pipette and add a thin overlay of E3 medium to the plate.
Note: You can flame the glass pipette tip just enough to polish it and create a smooth surface that improves embryo survival during manipulations.
-
68.
Inject each one-cell stage embryo with 1 nL of mix of RfxCas13d purified protein (3.0 μg/L) or mRNA (200–300 ng/μL) and 300–900 ng/μL (individual or mix gRNA final concentration) of gRNA (optimal conditions tested) using the microinjector stereomicroscope. This provides 3 ng of protein or 200–300 pg of mRNA and 300–900 pg of gRNAs per embryo. RfxCas13d protein or mRNA can be mixed in one tube with gRNAs at concentrations described above or to maximize the reagents concentrations two injections can also be performed, 1 nL for RfxCas13d (protein or mRNA) and 1 nL for gRNAs. This can be done when it is not possible to have the optimized concentrations described before in one solution. We have not observed differences between two approaches
-
69.
Transfer the embryos to 1% agarose coated plates with E3-medium and keep them at 28°C.
Note: To enhance the mRNA targeting with the RfxCas13d system we strongly recommend injecting directly 1 nL in the cell of one-cell-stage embryos and we suggest to inject at least 60–70 embryos for each condition.
Note: Inject 60–70 embryos with RfxCas13d protein or mRNA without gRNA as a control for the early development. These embryos must have a normal development such as a non-injected embryo. gRNAs alone can be also injected as a second control. An additional control could be to inject RfxCas13d mRNA or protein plus a gRNA targeting a sequence that is absent from the Zebrafish Genome.
Note: As a positive control injecting 3 ng of RfxCas13d purified protein or 200 pg RfxCas13d mRNA and 300 pg of in vitro transcribed gNTL-ctr (tbxta-4, stem loop disrupted and spacer of 23 nt version, Protospacer: 5′CUUUGCUGAAAGAUACGGGUGCT) per embryo. This will generate 80%–90% of zebrafish embryos with extreme no-tail phenotype.
Embryos care
-
70.
Keep the injected embryos in an incubator at 28°C in 1% agarose plates with E3 medium.
-
71.
Remove the unfertilized egg and dead embryos from the plate at 2 hpf.
-
72.
Repeat step 71 at 4 and 6 hpf. If the experiment goes beyond the first 7–8 hpf we recommend to transfer all viable embryos to a new 1% agarose plate with E3 fresh medium prewarm at 28°C
Note: During these cleaning steps E3 medium will be also removed from the plate. Thus, fresh E3 medium prewarm at 28°C can be added to the plate if needed
Note: See troubleshooting section problem 5 for possible solutions to percentage of zebrafish embryo survival.
mRNA knockdown validation
Different alternatives and complementary methods are proposed here to validate the efficiency of the mRNA knockdown.
-
73.
Employ a mix of different gRNAs targeting the same mRNA to validate the possible phenotype, decrease in mRNA levels and off-target effects obtained with the RfxCas13d system.
Alternatives: Perform mRNA knockdown experiments using new gRNAs or individual gRNAs from the mix injected independently. The use of new gRNAs could confirm the developmental and molecular phenotype in an independent experiment. As an alternative, gRNAs from the mix could be injected independently to confirm the mRNA knockdown and the phenotype but also to analyze what gRNA has the highest efficiency. This single and highly efficient gRNA can be used for future experiments instead of the mix. This can help to reduce potential off-targets from the mix where more gRNAs are used
-
74.
Analyze the mRNA relative levels or your target gene by qPCR analysis or ideally by RNA-seq analysis that will show the whole transcriptome.
-
75.
Inject a mRNA version resistant to gRNA (with mutations not altering the amino acid sequence) to rescue the phenotype. A titration will be required to ensure that overexpression does not cause any developmental defect. We recommend injecting different concentrations (from 10 to 200 pg per embryo, for example) of resistant mRNA to evaluate overexpression effects and phenotype rescue.
Note: See troubleshooting section problem 6 for possible solutions to low or no mRNA depletion.
Expected outcomes
The CRISPR-RfxCas13d system is a powerful technique for mRNA depletion in vertebrate embryos. Either mRNA coding for RfxCas13d or purified RfxCas13d protein efficiently and specifically induces mRNA degradation. The purified protein leads to faster mRNA target depletion but the protein purification requires more specialized equipment and resources than using RfxCas13 mRNA. Specific outcomes have been detailed in each section showing examples and figures.
Limitations
In our hands the CRISPR-RfxCas13d system has been shown to be very efficient in animal embryos, despite a variable activity among different gRNAs. However, this can be bypassed by using several (3–4) gRNAs targeting the same mRNA, which usually results in a high level of depletion. Although a tool to predict CRISPR-RfxCas13d activity in mammalian cell culture models is available (Wessels et al., 2020), it has not been systematically tested in vivo with in vitro transcribed or chemically synthesized gRNAs together with mRNA or purified protein RfxCas13d (Kushawah et al., 2020). Indeed, significant differences in efficiency have been shown for other CRISPR-Cas prediction tools when distinct approaches and models are used (Moreno-Mateos et al., 2015; Haeussler et al., 2016). Therefore, an algorithm able to predict CRISPR-RfxCas13d activity in zebrafish and other animal embryos would be helpful to select the most efficient gRNAs in vivo.
On the other hand, RfxCas13d mRNA is less convenient to eliminate maternally provided RNA in zebrafish embryos than using the purified protein (Kushawah et al., 2020). Purified protein injections accelerate RNA degradation and increase the penetrance of the phenotypes (Kushawah et al., 2020). However, as with other RNA knock-down approaches, CRISPR-RfxCas13d cannot maternally target the provided protein in animal embryos.
Troubleshooting
Most problems with this system arise from RNA/protein degradation. To verify if that is the case, check the purity and integrity of in vitro transcribed gRNAs and RfxCas13d mRNA and RfxCas13d protein (agarose and acrylamide gels). Include positive controls of gRNAs previously shown to be efficient, in the embryo injections. We suggest using tbxta gRNAs as a control to measure the toxicity and quantify the phenotype (Figure 2A) (Kushawah et al., 2020). In addition, to test earliest knockdown activity of RfxCas13d, we suggest using nanog gRNAs as a positive control (Kushawah et al., 2020).
Potential problems and detailed explanation of solutions are listed below.
Problem 1
Low gRNA concentration (expected yield between 300 and 1000 ng/μL) (steps 4–9).
Potential solution
Cas13d system efficiency could be severely affected due to a low gRNA concentration. Here, we describe potential solutions to increase the gRNA concentration:
The ideal DNA template concentration used for an IVT reaction should be between 500 and 1000 ng. A Low yield of fill-in PCR could be caused by a wrong primer’s selection or wrong PCR settings. We suggest reviewing the sequence of the primers used in the fill-in-PCR (step 4) and check all the reagents and PCR programs as suggested in the protocol or follow the manufacturer’s instructions for other DNA polymerases different from Q5® High-Fidelity DNA Polymerase (New England Biolabs #M0491) (step 5). Also, several replicates of the PCR can be performed and then mixed to increase the amount of DNA template.
gRNA production efficiency may vary along different DNA templates because of their sequence or secondary structures. However, inefficient in vitro transcription could be due to 10× reaction buffer precipitation. Therefore, it is critical to set up the IVT reaction at room temperature or heat the tube to 37°C for 5 min and mix thoroughly to resuspend the precipitate (step 8).
gRNA degradation during IVT reaction is not frequent but it can be possible. To avoid RNA degradation, clean the laboratory bench and pipettes with RNase away and make sure all products and reagents used are RNase-free (step 8). Also, make sure all the traces of ethanol have been eliminated from the tube and allow the RNA pellet to dry up to 5–7 min in the last step of gRNA precipitation (step 9).
Problem 2
Low RfxCas13d mRNA production (step 13–14).
Potential solution
Inefficient mRNA synthesis can be caused due to a low mRNA plasmid linearization or a low efficiency of the in vitro transcription reaction.
There are some possible solutions to optimize mRNA synthesis:
To increase the amount of the IVT template, more than one plasmid linearization reaction can be performed and purified together (step 13).
Similarly, as we described for gRNA production, RfxCas13d mRNA inefficient in vitro transcription could be due to 10× reaction buffer precipitation for this reason it is critical to setup the IVT reaction at room temperature or heat the tube to 37°C for 5 min and mix thoroughly to resuspend the precipitate (step 14).
To avoid RNA degradation, clean all the surfaces and pipettes with RNase away and make sure all products and reagents used are RNase-free.
Problem 3
Low RfxCas13d protein production (steps 16–56).
Potential solution
Several problems could be noticed during RfxCas13d protein purification. Here we describe possible solution to problems related with bacterial transformation (steps 16–31), wrong protein binding to HisTrap columns (steps 49–50) or protein purification after size exclusion chromatography (SEC) (steps 54–56)
The presence of bacteria in the negative control plate or the absence of no transformed bacteria indicates problems related with the transformation protocol. We suggest checking that the proper antibiotics solutions are being added or prepare fresh antibiotics solutions and to check that buffers correct composition or prepare fresh buffers (steps 16–31).
If the protein is not binding or poorly binding to HisTrap column, it could be due to several problems. Some of the potential solutions for such problems are described below (steps 32–50):
Wrong protein induction: Check the IPTG concentration or use fresh IPTG stock (step 32, see materials and equipment for recipes).
Excessive lysis: Check that protein is in the soluble fraction (After step 49)
Sample too viscous: Dilute the sample with lysis buffer (After step 49).
Too high loading flow rate: Reduce flow rate during sample loading (step 50).
Wrong buffer pH: Check and adjust buffer pH (step 50–52, see materials and equipment for recipes).
Clogged column: Filter sample and buffers through 0.45 μm PES filter (In steps 50–52).
Saturated resin: Clean and restore the resin according to the manufacturer instructions. Use a bigger column or reduce the amount of sample loaded. (In steps 50–52).
If RfxCas13d protein is not efficiently eluted from HisTrap column check and adjust buffer pH and the imidazole concentration (See materials and equipment for recipes) or prepare fresh buffer.
Also, increasing the washing volume can reduce the presence of contaminants in the elution fraction (step 51).
Poor protein purification could be related to inadequate settings in the SEC system (steps 54–56), we indicate the following potential issues and solutions:
Inadequately packed column: Perform a column performance test and repack if necessary (step 55).
Inadequate system settings: Minimize system tubing (diameter and length) (step 55–56).
Excessive sample volume: Check column’s recommendations and reduce sample volume (step 55–56).
Inadequate flow rate: Optimize flow rate. We suggest increasing the flow rate if peaks are too wide or reduce if the sample eluted too soon (step 55–56).
The observation of several bands in the final SDS-PAGE could suggest protein degradation (step 60), to avoid protein degradation, keep the samples on ice (or refrigerated at 4°C) during the whole purification process. Also, it is crucial to dilute the samples with a protein dialysis buffer because RfxCas13d protein precipitates in distilled water.
Problem 4
Low zebrafish embryo production (step 61).
Potential solution
Low zebrafish embryo production could be related to wrong male and female separation or by an inadequate water parameter in the zebrafish facility. Check water parameters such as temperature, pH, conductivity, noise, light-dark cycle, fish density in breeding tanks and other factors such as severe inbreeding or old age in the fish that could also affect the embryo production (step 61).
Problem 5
A low percentage of embryo survival (step 62–72).
Potential solution
Low percentage of embryo survival could affect the quality and robustness of the experiments. Here, we suggest a possible solution to increase the percentages of survival embryos until 24hpf. (Steps 62–72):
Review that the incubator temperature is at 28°C, before you start your experiments.
When using dechorionated embryos, long pronase incubation and/or pronase concentration can lead to low embryo viability. We strongly recommend using the correct concentration of pronase solution for a given time as indicated in the protocol (step 65).
Inefficient embryo manipulation using a broken glass pipette may reduce embryo viability. Use new and flamed glass pipette tip as indicated in the protocol. Keep the plates clean during the whole experiment and keep on removing dead embryos every 2 h and transfer the embryos to a new 1% agarose coated plates with E3 medium till 7–8 h post fertilization and for 24 h incubations at 28°C (step 72).
Problem 6
Low or no RNA depletion (step 74).
Potential solution
If low or no mRNA depletion is observed after following the protocol without any issue, this might be due to the lack of gRNAs activities or technical problems related with the microinjection procedures:
Check that the gRNAs are properly designed either by the option 1 or option 2 as described in this protocol and review that gRNAs are targeting an isoform of the mRNA that is present in the embryo.
Adjust the horizontal puller conditions to generate needles with an optimal pore size (that could variate between different models) and adjust the microinjection pressure to obtain the correct bubble size when calibrating the needles (step 63).
Inefficient mRNA targeting can be due to a low gRNA concentration. Make sure that the gRNA quantification has been done using Qubit RNA Broad Range Assay because the use of nanodrop will overestimate the gRNA concentration (step 10). In addition, an increased amount of gRNAs can be injected to the zebrafish embryos (step 68).
Make sure that the RfxCas13d mRNA or protein and gRNAs are injected into the animal pole of one cell stage embryo. This will increase the mRNA targeting. Phenol red solution may help to visualize the injected material into the cell (step 68).
If the inefficient mRNA targeting persists, use a new combination of 3–4 gRNAs. Also, check how many times gRNAs have been frozen and thawed (avoid more than 3–4 cycles).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Miguel A. Moreno-Mateos (mamormat@upo.es).
Materials availability
Plasmids used in this study have been deposited to Addgene, pT3TS-RfxCas13d-HA (#141320), pT3TS-RfxCas13d-NLS-HA (#141321) and pET-28b-RfxCas13d-His (#141322).
Acknowledgments
This work was supported by Ramon y Cajal (RyC-2017-23041), PGC2018-097260-B-I00 grant and MDM-2016-0687 program (Spanish Ministerio de Ciencia, Innovación y Universidades, and European Union), Universidad Pablo de Olavide (UPO) Research and the Springboard programs from UPO and CABD, respectively (M.A.M.-M.). This study was supported by the Stowers Institute for Medical Research. A.A.B. was awarded a Pew Innovation Fund and the US National Institutes of Health (R01 GM136849). This work was performed as part of the research for the obtainment of a G.dS.P degree., Graduate School of the Stowers Institute for Medical Research. The CABD is an institution funded by Pablo de Olavide University, Consejo Superior de Investigaciones Científicas (CSIC), and Junta de Andalucía. L.H.-H. is a recipient of a predoctoral fellowship from Ministerio de Ciencia. We thank Alejandra Cano for their technical assistance.
Author contributions
Conceptualization, L.H.-H., A.A.B., and M.A.M.-M.; methodology, L.H.-H. with the contribution of A.D.-M., L.T.-G., G.K., I.M.-S., and G.dS.P.; resources, A.A.B. and M.A.M.-M.; writing – original draft, L.H.-H. and A.D.-M.; writing – review & editing, G.K., G.dS.P., L.T.-G., A.A.B., and M.A.M.-M.; supervision, A.A.B. and M.A.M.-M.; project, A.A.B. and M.A.M.-M; funding acquisition, A.A.B. and M.A.M.-M.
Declaration of interests
The authors declare no conflict of interest.
Contributor Information
Luis Hernandez-Huertas, Email: lherhue@upo.es.
Ariel A. Bazzini, Email: arb@stowers.org.
Miguel A. Moreno-Mateos, Email: mamormat@upo.es.
Data and code availability
Software resources used in this study http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi and https://cas13design.nygenome.org/ are freely available.
Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/libpb-1653.
References
- Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J., Verdine V., Cox D.B.T., Kellner M.J., Regev A., et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman A.B., Brogan D.J., Sun R., Yang T., Hsu P.D., Akbari O.S. Programmable RNA targeting using CasRx in flies. CRISPR J. 2020;3:164–176. doi: 10.1089/crispr.2020.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.T., Niemela S.L., Miller R.H. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution (recombinant DNA) Biochemistry. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kushawah G., Hernandez-Huertas L., Abugattas-Nuñez del Prado J., Martinez-Morales J.R., DeVore M.L., Hassan H., Moreno-Sanchez I., Tomas-Gallardo L., Diaz-Moscoso A., Monges D.E., et al. CRISPR-Cas13d induces efficient mRNA knockdown in animal embryos. Dev. Cell. 2020;54:805–817.e7. doi: 10.1016/j.devcel.2020.07.013. [DOI] [PubMed] [Google Scholar]
- Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.D., Fernandez J.P., Mis E.K., Khokha M.K., Giraldez A.J. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.R., Maniatis T. 2nd ed. Cold Spring Harbor Laboratory Press; 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Wessels H.H., Méndez-Mancilla A., Guo X., Legut M., Daniloski Z., Sanjana N.E. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 2020;38:722–727. doi: 10.1038/s41587-020-0456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. Third Edition. The University of Oregon Press; 1995. The Zebrafish Book. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Software resources used in this study http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi and https://cas13design.nygenome.org/ are freely available.
Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at https://www.stowers.org/research/publications/libpb-1653.