

Abstract
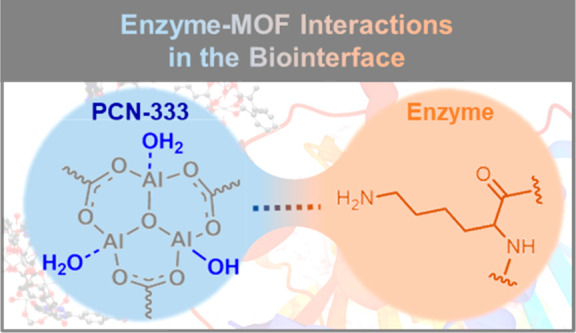
The immobilization of enzymes in metal–organic frameworks (MOFs) with preserved biofunctionality paves a promising way to solve problems regarding the stability and reusability of enzymes. However, the rational design of MOF-based biocomposites remains a considerable challenge as very little is known about the state of the enzyme, the MOF support, and their host–guest interactions upon immobilization. In this study, we elucidate the detailed host–guest interaction for MOF immobilized enzymes in the biointerface. Two enzymes with different sizes, lipase and insulin, have been immobilized in a mesoporous PCN-333(Al) MOF. The dynamic changes of local structures of the MOF host and enzyme guests have been experimentally revealed for the existence of the confinement effect to enzymes and van der Waals interaction in the biointerface between the aluminum oxo-cluster of the PCN-333 and the -NH2 species of enzymes. This kind of host–guest interaction renders the immobilization of enzymes in PCN-333 with high affinity and highly preserved enzymatic bioactivity.
Keywords: metal−organic frameworks, enzyme immobilization, solid-state NMR, biointerface interaction, biocatalysis
Introduction
Metal–organic frameworks (MOFs) are a class of extended network materials constructed from a bottom-up building block approach by connecting metal-based clusters and organic ligands.1−3 MOFs are well-known for their high degree of crystallinity, remarkable surface areas and pore volumes, and structural tunability.4,5 These design features have motivated researchers from a broad range of scientific and engineering disciplines to explore the fundamental and applied aspects of MOF materials.6 Recently, the area of enzyme/MOF biocomposites has experienced considerable growth due to their potential applications in biotechnology.7,8 Biomolecules can interact with MOFs via a wide variety of interactions including covalent attachment, coordination bonds, hydrogen bonding, π–π and hydrophobic interactions, and electrostatic attraction.9 The most widely employed strategy to synthesize enzyme/MOF biocomposites is to infiltrate the biomacromolecule within the MOF pore network.10 Due to their modular construction and chemical mutability,11 MOFs allow for their pore environment to be optimized to house specific proteins to a level of precision that is not attainable for other porous materials,12,13 such as silica matrixes,14 organic polymers,15 and DNA-based compartments.16 To date, these MOF-based biocomposites have been explored for applications in biocatalysis,7 biomedicine,17 controlled release and drug delivery,18 biopreservation,19 and cell and virus manipulation.8
However, the majority of studies, nowadays, have focused on the synthesis and performance characteristics of enzyme/MOF biocomposites;20,21 little is known about the fundamental molecular interactions between the MOF and enzyme that govern the immobilization process. When an enzyme molecule is in contact with a MOF support, its structure, orientation, and conformational mobility can be modified, and this typically leads to a change in its native functionality such as a significant loss of catalytic activity.22 Thus, to maintain the activity of enzymes immobilized within MOF pores a detailed understanding of the host–guest interactions is necessary. A number of characterization techniques have been employed to examine how MOF pore environment influences the structure of an enzyme,23 including, circular dichroism (CD),24 electron paramagnetic resonance (EPR),25,26 diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS),21 and Raman spectroscopy.27 In recent years, methodological developments have led solid-state nuclear magnetic resonance (ssNMR) to be one of the most powerful tools for determining the local structure and dynamics of various materials, including proteins, catalysts, biomaterials, pharmaceuticals, metal–organic materials, and so on.28−30 For example, Fragai et al.31 showed that the three-dimensional structures of the enzymes were retained after immobilization in a peptide-templated silica gel. Indeed, ssNMR spectroscopy has been applied as a general approach to study MOFs and their interactions with small guest species32 (e.g., H2,33 xenon,34 CO,35 CO2,36,37 nitric oxide,38 H2O,39 hydrocarbon,40 aromatics,41,42 etc.), providing complementary information for host–guest interaction.43 For example, with the help of ssNMR, the coordination of methanol to the open metal sites in [Cu3–xZnx(btc)2]n (btc = 1,3,5-benzenetricarboxylate) has been proven and unveiled in detail at an atomic level.44 In another study, the interactions between the Cr of the host MOF (Cr-MIL-101) and guest molecules (e.g., water, 2-aminopyridine, 3-aminopyridine, and diethylamine) have been revealed by a combination of density functional theory (DFT) calculations and 13C–1H rotational-echo double-resonance (REDOR) NMR.45 In addition, ssNMR is advantageous to probe the host–guest interaction in other porous materials such as zeolites,46,47 amorphous silica–alumina,48 and mesoporous organic materials.49,50 However, ssNMR has not yet been employed to examine the atomic-level host–guest interactions in MOF-based biocatalysts.
In this work, we envision that comprehensive microscopy, ssNMR, and reaction study can provide data that will advance our fundamental understanding of the host–guest interactions in the enzyme–MOF biointerface allowing for these systems to be optimized for enhanced enzyme preservation and protection. Lipase and insulin were loaded in a mesoporous MOF, PCN-333(Al), to afford lipase@PCN-333 and insulin@PCN-333 biocatalysts. The routine characterizations (X-ray diffraction (XRD) and 77 K N2 gas adsorption analysis) combined with scanning electron microscopy (SEM), confocal laser scanning microscopy (CLSM), and enzymatic tests have confirmed the successful immobilization of the targeted biocatalyst (lipase and insulin) in PCN-333. Furthermore, multinuclei 1D and 2D ssNMR and their multiple-quantum, double-resonance, and relaxation ssNMR measurements were employed to elucidate the local chemical environment and host–guest interactions between immobilized enzymes and the framework, PCN-333. The reaction results showed that the discovered host–guest interaction is able to confine the lipase inside PCN-333 while preserving its enzymatic bioactivity after immobilization.
Results and Discussion
PCN-333 has been reported to exhibit high porosity and exceptional chemical stability. These physical characteristics endow PCN-333 as an excellent platform for biomolecule immobilization. PCN-333 has been synthesized using a variety of metal precursors to realize metal nodes based on aluminum (Al), iron (Fe), and scandium (Sc).51 In this work, the Al version of PCN-333, PCN-333(Al), was selected for enzyme immobilization to facilitate the application of ssNMR techniques. The PCN-333 framework consists of [Al3(μ3-O)(OH)(H2O)2]6– clusters (Al-oxo clusters) linked by tridentate TATB (4,4′,4″-s-triazine-2,4,6-triyl-tribenzoate) ligands, which form supertetrahedra (ST). These ST are connected via vertices to form a three-dimensional mtn network. This network topology affords two types of mesopores: a dodecahedral cage measuring 42 Å and a hexacaidecahedral cage measuring 55 Å (Figure 1b). PCN-333 was prepared and activated according to the literature procedure reported by Zhou et al.51 The permanent porosity of PCN-333 was assessed by performing 77 K N2 gas adsorption isotherms. Application of the Brunauer–Emmett–Teller (BET) equation to the isotherm data afforded a surface area of 4198 m2 g–1 (Figure S1), which is consistent with the reported values.51 Recent publications have reported the successful immobilization of different enzymes within the pore network of PCN-333, including horseradish peroxidase (HRP),51 cytochrome c (Cyt c),51 microperoxidase-11 (MP-11),51 catalase (CAT),52 superoxide dismutase (SOD),52 and alcohol dehydrogenase (ADH).53 In this study, we have selected two commercially available enzymes of different sizes for the immobilization experiments: lipase (3.0 × 4.0 × 5.0 nm3)54 and insulin (1.3 × 1.3 × 3.4 nm3)55 (Figure 1c). Lipase is a commercially important enzyme that catalyzes hydrolysis reactions,56 and insulin is an important medical hormone responsible for regulating the concentration of glucose in blood plasma.57 We note that both biomolecules, in their active conformation (in crystallographic dimensions), are smaller than that of the largest mesopore (ø = 5.5 nm) in PCN-333 (Figure 1b) and, thus, in principle, can infiltrate the MOF pores.
Figure 1.

(a) Simulated (gray) and experimental PXRD patterns of PCN-333 (black), lipase@PCN-333 (blue), and insulin@PCN-333 (red). (b) Pore size distributions of PCN-333 (black), lipase@PCN-333 (blue), and insulin@PCN-333 (red). Pore size distributions were calculated using NLDFT model based on the N2 adsorption isotherms measured at 77 K (Figure S1). The salient pore features of PCN-333 are shown as insets. (c) Molecular structure of insulin (PDB 2A3G, insulin from bovine pancreas) and lipase (PDB 1OIL, lipase from Pseudomonas cepacia).
To immobilize the selected enzymes, PCN-333 crystals were soaked in an aqueous solution of lipase or insulin for 1 h at 4 °C (see Experimental Section for details). Control experiments confirmed the integrity of PCN-333 under these immobilization conditions (Figure S5). The resulting MOF-based biocatalysts were washed with water three times to remove the excess or loosely absorbed enzyme species on the MOF surface, affording biocatalysts denoted as lipase@PCN-333 and insulin@PCN-333. The loadings of lipase and insulin in lipase@PCN-333 and insulin@PCN-333 (Ploading10) were calculated to be 26.9 and 50.0 mg g–1, respectively (Bradford assay,58Figure S2). The values of loading efficiency (YP10) were then determined to be 53.8% and 100% for lipase and insulin, respectively. To determine if PCN-333 retained bulk crystallinity subsequent to the infiltration process, we performed powder X-ray diffraction (PXRD) measurements. Figure 1a shows that the PXRD patterns of both the lipase and insulin biocomposites are essentially identical to pristine PCN-333. The porosity of the biocomposites was examined by carrying out N2 77 K gas adsorption isotherms. As anticipated, the total N2 uptakes for lipase@PCN-333 and insulin@PCN-333 decrease significantly as compared to that of PCN-333 (Figure S1). In addition, the normalized pore size distributions calculated via the nonlinear density function theory (NLDFT) show a disappearance of mesoporosity in lipase@PCN-333 and insulin@PCN-333 (Figure 1b). These results can be rationalized by the presence of large biomolecules (lipase or insulin) within the mesopores of PCN-333. Attempts to characterize structural features of the encapsulated biomolecules within the enzyme@PCN-333 samples using attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) were unsuccessful due to the interference of PCN-333 background peaks (Figure S3).
We then applied scanning electron microscopy (SEM) to ascertain the crystal size and morphology of PCN-333 and the enzyme@PCN-333 biocomposites. Close inspection of the micrographs shows that PCN-333, lipase@PCN-333, and insulin@PCN-333 samples were dispersed octahedral crystals (∼300 nm in diameter, Figure S4). The morphological homogeneity and crystal size triggered us to visualize the spatial distribution of lipase within PCN-333 using confocal laser microscopy (CLSM). In order to visualize the lipase within the biocomposite crystals using CLSM, a fluorescent tag needs to be attached to the enzyme. Fluorescein-tagged lipase (FITC-lipase) was prepared and used to synthesize FITC-lipase@PCN-333. The synthesis and washing protocol for FITC-lipase@PCN-333 were analogous to those of lipase@PCN-333. CLSM images (Figure S6) show that the FITC-lipase molecules were present throughout FITC-lipase@PCN-333 crystals; however, due to the resolution of this technique, we are unable to differentiate between signal from surface-absorbed FITC-lipase and encapsulated FITC-lipase. Supplementary experiments with HRP@PCN-333 confirmed the successful infiltration/encapsulation of the biomolecules within the mesopores of PCN-333 (Figures S13–S17).
To assess the enzymatic performance of the biocomposite, we measured the catalytic hydrolysis of p-nitrophenyl butyrate (PNPB) using free FITC-lipase and FITC-lipase@PCN-333 biocatalyst (Figure 2a). A noteworthy result is that FITC-lipase@PCN-333 showed comparable activity to that of free FITC-lipase (Figure 2b). Approximately 91.6% of the bioactivity (calculated based on the slope of the reaction kinetics) of FITC-lipase was retained upon immobilization in FITC-lipase@PCN-333.
Figure 2.
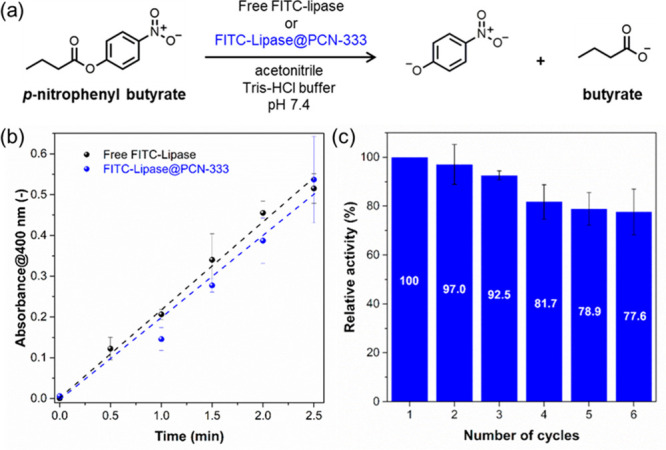
(a) Biocatalytic hydrolysis of p-nitrophenyl butyrate (PNPB). (b) Catalytic activity of FITC-lipase and FITC-lipase@PCN-333 biocomposite. The amount of FITC-lipase available was the same for each experiment (25 μg based on FITC-lipase). (c) Cycling runs for the hydrolysis of 4-nitrophenyl butyrate in the presence of FITC-lipase@PCN-333 after a reaction time of 2 min. Experiments were performed in triplicate with standard error bars shown for each run.
Given that the catalytic activity of the immobilized lipase is preserved for FITC-lipase@PCN-333 (Figure 2b), we sought to examine recyclability, an important performance factor for practical applications. The relative bioactivity of FITC-lipase@PCN-333 biocomposites was calculated by comparing the relative activity with that of the first run. We successfully cycled FITC-lipase@PCN-333 six times with a 22.4% reduction in enzymatic activity in the sixth cycle (Figure 2c). This observed reduction is actually due to enzyme leaching from the FITC-lipase@PCN-333 samples during the washing procedure between cycles (Figure S7 and Table S1–S2). Interpreting these results, it is concluded that the lipase biomolecules retained their bioactivity and are confined in PCN-333 framework in relatively high affinity.
To elucidate the interactions between the enzyme and PCN-333, we studied PCN-333, lipase@PCN-333, and insulin@PCN-333 using ssNMR techniques. A unique characteristic of MOFs is that their structural flexibility enables their molecular architectures to exhibit local motions under external stimuli.59,60 Upon pore-filling with large biomolecules, we anticipate a subtle difference in the local flexibility of the PCN-333 framework. Solid-state NMR has been proven to be a powerful tool to investigate the molecular dynamics in a variety of materials by applying relaxation or diffusion spectroscopy.61−63 In this work, in order to understand the structural flexibility of the framework, the 1H spin–lattice relaxation (1H T1) was measured by applying the inversion–recovery pulse sequence. For 1H T1 analysis, as the magnetization of 1H nuclei can rapidly diffuse throughout the entire spin network of the solid, a relatively uniform T1 value for all protons in the material is expected. In addition, due to the strong homonuclear dipolar–dipolar interactions and the high natural abundance of the 1H nuclei, the 1H T1 value of the whole set of 1H peaks can be utilized to represent the molecular dynamics of the entire network. Therefore, the peaks across the whole spectrum in the 1H spectra obtained via the application of the inversion–recovery pulse sequence were selected to acquire the relaxation curves, and the 1H T1 values are obtained accordingly (Figure S8 and Table 1).
Table 1. Molecular Dynamics Measurements for PCN-333, Lipase@PCN-333, and Insulin@PCN-333 via 1H and 27Al Relaxation Spectroscopy.
| nuclei | materials | T1 (ms) |
|---|---|---|
| 1H | PCN-333 | 936 |
| insulin@PCN-333 | 2323 | |
| lipase@PCN-333 | 2342 | |
| 27Al | PCN-333 | 14.92 |
| insulin@PCN-333 | 11.23 | |
| lipase@PCN-333 | 11.05 |
In general, increasing mobility in a solid will result in reduced 1H spin–lattice relaxation times.64 As shown in Table 1, after enzyme immobilization, lipase@PCN-333 and insulin@PCN-333 show a much longer spin–lattice relaxation time compared to PCN-333 (T1 = 2.342, 2.323, and 0.936 s for insulin@PCN-333, lipase@PCN-333, and PCN-333, respectively, Table 1 and Figure S8a). These results indicate that the mobility of protons in lipase@PCN-333 and insulin@PCN-333 decreases as compared to those in PCN-333. Since these 1H T1 values reflect the flexibility of the entire network, a longer T1 value indicates a reduction in local framework flexibility upon enzyme immobilization. This effect is expected and can be rationalized by the confinement of the large enzyme biomolecules in lipase@PCN-333 and insulin@PCN-333.65 A similar phenomenon has been observed in the case of benzene loaded UiO-66(Zr), where a decrease in the internal dynamics of π-flips of the terephthalate ligands was observed.41
T1 relaxation times of 27Al were also measured with the purpose of comparing the local mobility of the Al sites in different PCN-333 samples. Interestingly, as shown in Table 1 and Figure S8b, after enzyme immobilization, a decrease in the 27Al T1 relaxation time is observed for both lipase@PCN-333 and insulin@PCN-333. The decrease, instead of increase, in the 27Al T1 relaxation times after enzyme immobilization clearly demonstrates that the local flexibility of Al centers in the PCN-333 framework has a different dynamic behavior compared with the entire framework (Table 1).64 This observation points to the possible existence of interaction between the Al-oxo cluster and enzymes, which relax the existing coordination bonds of Al-oxo cluster in PCN-333 and contributed to a lower 27Al T1 value.
In order to further illustrate the structural change of PCN-333 after enzyme immobilization in detail and understand the nature of the host–guest interactions at the atomic scale, one-dimensional and two-dimensional ssNMR spectroscopies were conducted to investigate the local chemical structure of the PCN-333 biocomposites. The pristine PCN-333 (Figure 3) was first analyzed by ssNMR to establish a reference point. By application of 1H and 13C magic-angle spinning (MAS) NMR spectroscopy, all the proton and carbon species in PCN-333 (Figure 3) were successfully assigned. The broad 1H signal at 7–9 ppm is partially attributed to the coordinated water on the trimeric Al-oxo cluster (Al···OH2, denoted as H4 in Figure 3a), which shows a higher ppm value in comparison with that of the free H2O (δ1H = ca. 5 ppm). This assignment is supported by recent ssNMR studies.66,67 In one study of MIL-110, Haouas et al. reported a higher 1H chemical shift value for the coordinated water (δ1H = 5.8 ppm) than that of the free water (δ1H = 4.4 ppm) in the framework.66 In another case, the coordinated water (Al–H2O) in mineral alunite signaled at a lower chemical field shift in the 1H NMR spectrum (δ1H ≈ 7.4 ppm) in comparison with the free H2O (δ = 4.8 ppm).67 In addition, these broad 1H peaks (δ1H = 7–9 ppm) are associated with the aromatic C–H species (H1 and H2) of the TATB ligand (Figure 3a,b).68 The 1H signal at δ1H = 2.4 ppm is assigned to the proton of the coordinated hydroxyl (H3, Figure 3a) species in the Al-oxo cluster (Al3(μ3-O)(OH)(H2O)2).69 A broad 1H signal at around 5 ppm is assigned as the free H2O molecule.38,69 These assignments are consistent with an ssNMR study of MIL-100(Al).69 However, no proton signal was identified in the region of ∼2 ppm for MIL-100(Al), as an extra-framework btc ligand instead of a hydroxyl group is present in MIL-100(Al) to balance the charge of the Al-oxo cluster. In another study, in the absence of extra-framework btc ligand in MIL-100(Al), hydroxyl protons (−OH) peaks appear at δ1H = 1.5 and 0.3 ppm.38Figure 3c (gray line) shows the 13C CP MAS NMR spectra of the PCN-333 samples. The aromatic C2 and C5 of the TATB ligand can be observed at δ13C = 138 ppm, while C3 and C4 sit at δ13C = 129 ppm. Deconvolution of the broad peak at 170–174 ppm confirms the existence of the C1 (δ13C = 173 ppm) and C6 species (δ13C = 171 ppm) (Figure 3d and Table S3). Other small peaks originating from the residue solvent were also observed (N,N′-dimethylformamide (DMF), Figure 3b,c).70
Figure 3.

(a) Molecular representation of PCN-333(Al). (b) 1H one-pulse MAS NMR spectra. (c) 13C cross-polarization MAS NMR spectra of PCN-333 (black), lipase@PCN-333 (blue), and insulin@PCN-333 (red). Asterisks represent sidebands; filled circles indicate the residue solvent signals (DMF). (d) Deconvolution of 13C peaks in the region of 150–200 ppm.
After enzyme immobilization, a drastic change in the 1H spectra can be observed for enzyme@PCN-333 as compared to that of PCN-333 (Figure 3b). In lipase@PCN-333 and insulin@PCN-333, a sharp 1H signal emerges at 5 ppm (Figure 3b). The sharp 1H signal (δ1H = 5 ppm) can also be identified in 1H MAS NMR spectra of free enzymes (Figure S9). Compared to the broad water proton signal in PCN-333 (δ1H ≈ 5 ppm, Figure 3b), the sharp 1H peak centered at 5 ppm for lipase@PCN-333 and insulin@PCN-333 can be assigned to the -NH2 species (Figure 3b) in enzyme biomolecules69 and the free H2O in the biocomposites. In the 13C cross-polarization (CP) MAS NMR spectra (Figure 3c), no distinguishable changes are observed for the aromatic carbon species (C2, C3, C4, and C5) of the TATB ligand before and after enzyme immobilization. 13C NMR signals originating from carbonyl species (C=O) in enzyme molecules can be found at 173 ppm,71 which overlaps with the C1 13C CP NMR signal from PCN-333 (Figure 3d). In addition, only tiny 13C resonance peaks originating from the aliphatic carbon of immobilized protein molecules are also detected in Figure 3c (δ13C = 20–40 ppm) for lipase@PCN-333 and insulin@PCN-333.71 The coexistence of 1H and 13C signals of the PCN-333 framework and the guest biomolecule in enzyme@PCN-333 samples confirms the successful immobilization of lipase and insulin. This observation is in agreement with the characterization data in Figure 1, Figure S1, and Figure S6. The absence of new carbon species in the 13C CP MAS NMR spectra for PCN-333 biocomposites samples implies that there are no covalent host–guest interactions between the MOF and the enzyme biomolecules. However, after enzyme immobilization, the signal related to the carboxylate carbon (C6) shifts to a higher field (δ13C = 169 ppm) for both PCN-333 biocomposite samples (see Figure 3d and Table S3 for details). These chemical shifts of the carboxylate C6 species are reported to be an indicative probe for studying the variation in the coordination environment of metal nodes in a metal–organic complex.72 For example, Ke et al. studied the ssNMR shift of carboxylates carbon of benzoate in [Cu2(C6H5CO2)4]n to determine the nature of the interaction between the Cu-paddle wheel and urea.73 The authors claimed that upon coordination of urea to the open Cu sites, the chemical signal of the carboxylate carbons in the benzoate ligand shifted to high-fields.73 In the present study, a high-field shift in 13C signal of carboxylate carbons (C6) is also observed in enzyme@PCN-333 samples (from 171 ppm in PCN-333 to 169 ppm in lipase@PCN-333 and insulin@PCN-333, Figure 3d and Table S3). The high-field shift of C6 is consistent with the literature, indicating a change of the local chemical environment of the Al-oxo clusters in PCN-333 and possible host–guest interaction between the Al-oxo clusters of host PCN-333 and the guest biomolecules. This result is also in good agreement with the information obtained from the 27Al T1 ssNMR relaxation measurements (Table 1 and Figure S8).
To reveal the existence of interaction between the PCN-333 and the immobilized biomolecules, 27Al MAS NMR technique74,75 was applied to characterize the local chemical environment of Al nuclei in PCN-333 samples. In the 27Al MAS NMR spectrum, only one broad peak is observed in the region of 8–15 ppm for PCN-333 (Figure 4a and Figure S10). The 27Al peak position indicates the presence of hexa-coordinated Al3+ (AlVI) cations51,76 in PCN-333. After enzyme immobilization, the width of the 27Al peak decreased (Figure S10, as measured by the values of the full width at half-maximum (fwhm), Table S4). This difference in the shape of 27Al spectra further confirms the change in the chemical environment of the Al centers in PCN-333 after enzyme immobilization. These results are consistent with the information obtained via 27Al NMR relaxation spectra (Table 1, Figure S8b). Since 27Al is a quadrupolar nucleus, the strong second-order quadrupolar interaction can lead to broad peaks in the 1D ssNMR spectra and hinder the resolution of subspecies.77 To obtain detailed information on the Al-oxo cluster, two-dimensional multiple-quantum 27Al MAS NMR spectroscopy (2D 27Al MQ MAS NMR) was applied to resolve the different Al species (Figure S11a–c). For PCN-333, two contour centers for AlVI species with different isotropic chemical shifts are observed (Figure S11a and Table S5), denoted as Ala (δiso27Al = 2.7 ppm) and Alb (δiso27Al = 10.0 ppm). Based on the parameters obtained from 2D 27Al MQ MAS NMR spectra and eqs 1–4 (see Experimental Section for details), the anisotropic 1D 27Al MAS NMR spectra were deconvoluted (Figure 4a). As the peak area of the Ala is approximately double that of the Alb species (Alb/Ala = 0.48), the Ala and Alb species are assigned to Al···OH2 and AlOH groups, respectively, in the Al-oxo cluster (Al3(μ3-O)(OH)(H2O)2). The identification of the two isotropic resonances in the spectra is consistent with the observation reported by Loiseau et al.,78 who also observed two AlVI species originated from AlOH and Al···OH2 groups in MIL-110(Al). Importantly, this deconvolution result also matches well with the crystallographic data.51 After biomolecule loading, no new Al species is observed (Figure S11a–c). Both Ala and Alb species remain at similar isotropic chemical shifts with slightly increased quadrupole coupling constant (CQCC) (Table S5). The nonshifted Ala and Alb species in peak position confirm the absence of strong coordination interaction at the biointerface. The change in CQCC values of the Al sites points to only a slight distortion in the symmetry of the Al coordination environment (Table S5), which is not strong enough to cause a severe change in the electric field gradients on the Al sites. The small change in quadrupolar coupling constant further confirms the appearance of a new weak interaction between the Al-oxo cluster of the PCN-333 and the biocatalysts. In order to visualize and reveal the nature of the weak interaction between the Al-oxo cluster of the PCN-333 and the biocatalysts in-depth, 1H/27Al transfer of population in double-resonance (TRAPDOR) NMR technique was applied to study the correlation between the Al atoms in PCN-333 and their neighboring protons.79Figure 4b shows the amplified 1H/27Al TRAPDOR NMR spectra of PCN-333 samples, in which both Al···OH2 (δ1H = 7–8 ppm) and AlOH (δ1H = 2.4 ppm) can be observed. This observation further confirmed the proton assignment in the 1H spectra (Figure 3b). Interestingly, a strong correlation between the -NH2 (narrow signal δ1H = 5 ppm) and Al nuclei was observed for lipase@PCN-333 and insulin@PCN-33 (Figure 4b). Since the physisorbed water molecule is extremely dynamic compared to other molecules, the majority of the 1H–27Al dipolar couplings between the water molecules and Al-oxo cluster are averaged away. The absence of a sharp peak at δ1H = 5 ppm in PCN-333 rules out the possibility that the 1H–27Al correlation observed at 5 ppm for enzyme@PCN-333 samples was derived from the signal between the exchangeable water molecule and Al sites of PCN-333 (Figure 4b and Figure S12). Thus, the identified correlation can be assigned to the 1H–27Al dipolar couplings between the -NH2 group of the enzymes (δ1H = 5 ppm) and Al nuclei of PCN-333. This result clearly indicates the presence of proximity and host–guest interaction between the -NH2 group of the immobilized biocatalyst and the Al-oxo cluster of the PCN-333. Since no shift of peak position is observed for the -NH2 group in the 1H/27Al TRAPDOR spectra, there is no strong coordination, hydrogen bonds, or covalent interactions between the Al centers and the -NH2 functionalities. Thus, with weak van der Waals interaction between the Al sites and -NH2 sites, the biocatalysts were confined inside the cavities of the PCN-333 and immobilized on the PCN-333 crystal surface in relatively high affinity. In addition, since the signal intensity of the -NH2 is much stronger than those of the AlOH and Al···OH2 groups, we conclude that the interaction between the enzyme and PCN-333 is primarily governed by the interactions between the Al-oxo clusters of the host PCN-333 and the -NH2 moieties of the guest biocatalysts.
Figure 4.

(a) Deconvolution of 27Al MAS NMR spectra of PCN-333 samples. (b) 1H/27Al TRAPDOR difference spectra of PCN-333 samples (10 times amplified). (c) Schematic illustration of enzyme–PCN-333 interaction in the biointerface.
All experimental data are consistent with the hypothesis that the confined biocatalysts in PCN-333 are predominantly governed by the interactions between the Al sites of PCN-333 and the -NH2 functionalities of biocatalysts. The strong 1H–27Al correlation undoubtedly suggests that the host–guest interactions are with -NH2 functionalities to Al-oxo cluster (δ1H = 5.0 ppm in 1H/27Al TRAPDOR spectra). We speculate that surface-exposed lysine or arginine residues of lipase and insulin play a key role in the enzyme–MOF interaction (Figure 4c). Meanwhile, the overall dynamic change (T1 of 1H NMR) of enzyme@PCN-333 implies the existence of the confinement effects for immobilized enzymes in MOFs.
Conclusion
In conclusion, enzyme biomolecules (lipase and insulin) have been successfully immobilized in PCN-333 MOF and maintained high biocatalytic activity. The change of both the relaxation times and chemical shifts in ssNMR studies indicates that the immobilization of proteins affects both the flexibility and local structure of PCN-333. After enzyme immobilization, the overall framework flexibility was decreased (increase in 1H T1 values) due to the confinement effects to enzymes from pores, while the 27Al spin–lattice relaxation time for enzyme@PCN-333 showed a different trend due to the interaction between the Al-oxo cluster in PCN-333 and the biocatalysts. Analysis of the enzyme@PCN-333 revealed that a distinct signal related to the -NH2 group of the biocatalyst in the 1H ssNMR spectra is concomitant with a subtle chemical shift of carboxylate 13C spectral feature that is associated with a change of the coordination environment in the Al sites of PCN-333. These data suggest the dominant interactions between the biomolecules and the Al oxo-cluster of PCN-333. Via both 1H/27Al TRAPDOR NMR and multiple quantum 27Al MAS NMR, the immobilization mechanism was further revealed, where the spectroscopic data support the existence of weak interactions between the Al atoms in MOF and the -NH2 sites on the biocatalysts via van der Waals forces. After combining all the experimental evidence, we, for the first time, have experimentally visualized the immobilization of enzymes in PCN-333 and deduced the possible host–guest interaction mode between the MOF’s Al oxo-cluster and the biocatalyst’s -NH2 sites as well as the pore confinement effect. This work shed light on the complex relationship between enzyme and MOF support in the biointerface. In addition, this fundamental knowledge will help to guide the design of other supported enzymatic systems with optimum catalytic activity and stability.
Experimental Section
Syntheses
All chemicals and solvents were purchased from commercial sources and used as received without further purification.
Ligand Synthesis
4,4′,4″-s-triazine-2,4,6-triyl-tribenzoate (TATB) was synthesized according to the literature.51
PCN-333 Synthesis
PCN-333 was synthesized according to the literature.51 The as synthesized MOF materials were then washed sequentially with DMF (3 × 2 mL) and ethanol (3 × 2 mL) over 2 days. Thereafter, the PCN-333 was collected by centrifugation and dried in vacuum at room temperature. The dried PCN-333 was stored in a sealed vial at 4 °C before further usage and characterization.
Enzyme@PCN-333 Synthesis
Enzyme@PCN-333 samples were synthesized by mixing lipase (1 mL of 5 mg mL–1 lipase aqueous solution) or insulin (1 mL of 5 mg mL–1 insulin stock solution) with the as-synthesized PCN-333 (100 mg) at 4 °C for 1 h. The insulin stock solution was prepared by dissolving 5 mg of insulin in 1 mL of diluted HCl solution (pH 3.0, 20 mM). Thereafter, the solid was recovered by centrifugation and washed with H2O (3 × 1 mL) to remove the excess biomolecule. All supernatants were collected. The concentration of biomolecule in the supernatant was determined by the Bradford assay. The amount of biomolecule immobilized in PCN-333 was calculated by the difference between the concentration used in the synthesis and that in the collected supernatant.
Fluorescein-Tagged Lipase (FITC-Lipase)
A solution of fluorescein isothiocyanate (FITC, 0.005 M, 0.0003 mmol) in aqueous carbonate/bicarbonate buffer solution (60 μL, 0.1 M, pH 9.2) was added to a solution of lipase (10 mg, Sigma-Aldrich 62288 lipase from Pseudomonas cepacia, 30 units mg–1) in aqueous carbonate/bicarbonate buffer solution (1.94 mL, 0.1 M, pH 9.2) at 0 °C. The solution was covered with aluminum foil and stirred at 0 °C for 4 h. The reaction solution was then filtered through Sephadex G-25M, PD-10 column (GE Healthcare, 17085101), which had been equilibrated with water, eluting with aqueous carbonate/bicarbonate buffer solution (0.1 M, pH 9.2). The fractions containing FITC-lipase were concentrated through a Pall corporation Microsep Advance 10K membrane by centrifugation (4000 rpm; multiple batches were required ∼20 min). The concentrated FITC-lipase was washed twice with deionized water and combined to give a final volume of 2 mL, which was stored at 4 °C, protected from light, until required. The stock solution was assumed to contain 5 mg mL–1 of lipase.
FITC-Lipase@PCN-333 Synthesis
A solution of aqueous FITC-lipase (100 μL, 5 mg mL–1) was added to PCN-333 powder (10 mg) at 0 °C. The suspension was stirred at 0 °C for 1 h. The suspension was transferred to an Eppendorf tube, with deionized water and centrifuged (6000 rpm, 2 min). The supernatant was removed, the pellet was resuspended in deionized water and centrifuged as above (×3) to remove the excess FITC-lipase. The pellet was suspended in deionized water (1 mL) to afford a concentration of 0.5 mgFITC-lipase mL–1 with an assumption of 100% FITC-lipase immobilization in PCN-333. The complete immobilization of FITC-lipase was supported by the absence of fluorescence signal (from the FITC tag in the FITC-lipase) in the supernatant from the immobilization mixture. The suspension was stored at 4 °C, protected from light, until required.
Characterization
Gas Sorption
N2 sorption isotherms were measured on an autosorb iQ (Quantachrome INSTRUMENTS Inc.). Approximately 50 mg of the powdered solid was loaded into a glass analysis tube and outgassed under dynamic vacuum at 60 °C for 12 h. N2 adsorption and desorption isotherms were measured at 77 K, and data were analyzed using Brunauer–Emmett–Teller (BET) analysis models80 to determine the surface area. The pore size distribution calculations were carried out using the ASiQwin software DFT package (Quantachrome INSTRUMENTS Inc.).
Powder X-ray Diffraction (PXRD)
PXRD measurements were performed on a PANalytical XPert Pro powder diffractometer fitted with a solid-state PIXcel detector (45 kV, 40 mA, 1° divergence and antiscatter slits, and 0.3 mm receiver and detector slits) using Cu Kα (λ = 1.5406 Å) radiation.
Catalytic Assay of FITC-lipase and FITC-lipase@PCN-333
The catalytic activity of the free FITC-lipase or FITC-lipase@PCN-333 was measured according to a modified protocol from Sigma-Aldrich. UV–vis measurements were recorded on a Shimadzu UV-3600i Plus UV–vis–NIR spectrophotometer between the wavelengths of 600 and 300 nm. The enzymatic activity of free FITC-lipase or FITC-lipase@PCN-333 was calculated from the change of absorbance at 400 nm. In a specific experiment with free FITC-lipase, an aqueous solution of FITC-lipase (5 μL, 5 mgFITC-lipase mL–1) and a solution of 4-nitrophenyl butyrate in acetonitrile (10 μL, 50 mM, 0.5 mmol) were added sequentially into an aqueous Tris-HCl buffer solution (985 μL, 50 mM, pH 7.4) at room temperature. The reaction was left to stand at room temperature for the allocated reaction time before being transferred to a 1 mL cuvette for UV–vis measurement. The absorbance at 400 nm was recorded and used to measure the enzymatic activity of the materials.
For experiments with FITC-lipase@PCN-333, a suspension of FITC-lipase@PCN-333 in deionized water (50 μL, 0.5 mgFITC-lipase mL–1) was added to a solution of 4-nitrophenyl butyrate in acetonitrile (10 μL, 50 mM, 0.5 mmol) in aqueous Tris-HCl buffer solution (940 μL, 50 mM, pH 7.4) at room temperature. The reaction mixture was left to stand at room temperature before being centrifuged (13 500 RPM, 30 s) before the allocated time. The supernatant was transferred into a 1 mL cuvette, and the absorbance at 400 nm was measured.
Recycling Catalytic Activity of FITC-lipase@PCN-333
Recycling experiments of FITC-lipase@PCN-333 were completed as per the experimental protocol of the catalytic activity of FITC-lipase@PCN-333 mentioned above. Absorbance at 400 nm was collected after a reaction time of 2 min. Between each run, the FITC-lipase@PCN-333 pellet was resuspended in deionized water, centrifuged (13 500 RPM, 30 s) and the supernatant removed (× 2). The next cycle was started upon the addition of a mixture of the 4-nitrophenyl butyrate and Tris-HCl buffer solutions cited above.
Solid-State Magic Angle Spinning NMR Experiments
Prior to the 1H, 13C, and 27Al MAS NMR experiments, all samples were fully hydrated by exposure to the atmosphere for a week. The single pulse MAS NMR experiments for 1H, 13C, and 27Al were performed on a Bruker Avance III 500WB spectrometer at a resonance frequency of 400.1 MHz using a 4 mm MAS rotor. For 1H single-pulse MAS NMR, spinning rate of 12 kHz, 64 scans were recorded for the spectra after single-pulse π/2 excitation with repetition times of 20 s. 13C and 27Al MAS NMR investigations were conducted at a resonance frequency of 125.7 and 130.3 MHz, respectively. Noteworthy, a spin rate of 10 kHz has been applied for 13C MAS NMR experiments with the purpose of avoiding the overlap of the spinning sidebands from the peaks, while the 12 kHz spinning rate was applied for 27Al MAS NMR measurements. Five thousand scans were recorded to obtain 13C CP MAS NMR spectra with a contact time of 4 ms and a repetition time of 4 s. Single-pulse 27Al MAS NMR spectra were recorded after a single pulse π/6 excitation with a repetition time of 0.5 s. In order to obtain readable 27Al MAS NMR spectra, 200 scans have been recorded. The two-dimensional (2D) 27Al MQ spectra were acquired using the three-pulse z-filter MQ MAS pulse sequence.81 The spectra were sheared with TopSpin2.1 xfshear program. The repetition time for the 2D 27Al MQ experiments was 1 s, and 5000 scans were recorded. Parameters of isotropic chemical shift (δiso), quadrupolar constant (CQCC), and asymmetric parameter (η) have been obtained by applying eqs 1–4 for the 2D MQ MAS spectra77 with the purpose to obtain information for the decomposition of 27Al MAS NMR spectra:
| 1 |
| 2 |
| 3 |
| 4 |
The values of I (nuclear spin of the quadrupolar nucleus) and Vo (Larmor frequency of the quadrupolar nucleus) were 5/2 and 104.3 MHz, respectively. SOQE represents the second order quadrupolar effect parameter, and δF1 and δF2 are the centers of gravity of the MQ MAS signal in the F1 and F2 dimension.
Spin–lattice relaxation times (T1) were determined via 1H and 27Al detection applying inversion recovery and saturation recovery, with a list of t1 delay of 10, 5, 4, 3, 2, 1, 0.5, 0.25, 0.1, and 0.01 s for 1H T1 value measurements and 120, 100, 80, 40, 20, 10, 5, 2, 1, 0.5, 0.2, 0.1, 0.05, and 0.02 ms for 27Al T1 value measurements. In the 1H T1 experiment, a 180° pulse was applied first to get a fully inverted Mz. A fully recovered equilibrium magnetization (Mz > 99%) can be achieved after a long recycle delay (D1 = 50 s). Another 90° pulse was then applied to flip the magnetization. For the 27Al T1 measurement, a 90° pulse was first applied to saturate the system; before the second 90° pulse, a 2 s recycling delay was applied for the magnetization recovery. The corresponding spectra were recorded as a 2D data where the t1 delay was varied (t1 delay list). The 1H T1 and 27Al T1 spectral deconvolutions were applied to obtain readable information using the TopSpin and DMFit software.82 By application of eq 5, the corresponding T1 values were obtained. In eq 5, Mt represents the original magnetization length, t is the applied t1 delay, and M represents the magnetization recovery after a specific t1 delay.82
| 5 |
Acknowledgments
This work was supported by the Australian Research Council under the Discovery Projects Schemes (DP180104010, DP170103531, and DP200102411). W.Y. gratefully acknowledges the support of University of Sydney International Scholarship Strategic (USydIS-Strategic). The authors thank Dr. Nicole Cordina for her assistance in the NMR technique support. The authors acknowledge the facilities and the scientific and technical assistance of Sydney Analytical, a core research facility at The University of Sydney. The authors acknowledge the instruments and scientific and technical assistance of Microscopy Australia at Adelaide Microscopy, The University of Adelaide, a facility that is funded by the University and State and Federal Governments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00226.
Full analytic details, including SEM images, CLSM images, gas sorption isotherms, and ssNMR data (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Dedication
Dedicated to Professor Jianzhong Chen on the occasion of his 70th birthday.
Supplementary Material
References
- Furukawa H.; Cordova K. E.; O’Keeffe M.; Yaghi O. M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- Zhou H.-C.; Kitagawa S. Metal-organic frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415. 10.1039/C4CS90059F. [DOI] [PubMed] [Google Scholar]
- Maurin G.; Serre C.; Cooper A.; Férey G. The new age of MOFs and of their porous-related solids. Chem. Soc. Rev. 2017, 46, 3104–3107. 10.1039/C7CS90049J. [DOI] [PubMed] [Google Scholar]
- Kitagawa S. Porous crystalline materials: closing remarks. Faraday Discuss. 2017, 201, 395–404. 10.1039/C7FD90042B. [DOI] [PubMed] [Google Scholar]
- Rungtaweevoranit B.; Diercks C. S.; Kalmutzki M. J.; Yaghi O. M. Spiers Memorial Lecture: Progress and prospects of reticular chemistry. Faraday Discuss. 2017, 201, 9–45. 10.1039/C7FD00160F. [DOI] [PubMed] [Google Scholar]
- Dincă M.; Long J. R. Chem. Rev. 2020, 120, 8037–8038. 10.1021/acs.chemrev.0c00836. [DOI] [PubMed] [Google Scholar]
- Doonan C.; Riccò R.; Liang K.; Bradshaw D.; Falcaro P. Metal–organic frameworks at the biointerface: synthetic strategies and applications. Acc. Chem. Res. 2017, 50, 1423–1432. 10.1021/acs.accounts.7b00090. [DOI] [PubMed] [Google Scholar]
- Riccò R.; Liang W.; Li S.; Gassensmith J. J.; Caruso F.; Doonan C.; Falcaro P. Metal–organic frameworks for cell and virus biology: a perspective. ACS Nano 2018, 12, 13–23. 10.1021/acsnano.7b08056. [DOI] [PubMed] [Google Scholar]
- Lian X.; Fang Y.; Joseph E.; Wang Q.; Li J.; Banerjee S.; Lollar C.; Wang X.; Zhou H.-C. Enzyme–MOF (metal–organic framework) composites. Chem. Soc. Rev. 2017, 46, 3386–3401. 10.1039/C7CS00058H. [DOI] [PubMed] [Google Scholar]
- Liang W.; Wied P.; Carraro F.; Sumby C. J.; Nidetzky B.; Tsung C.-K.; Falcaro P.; Doonan C. J. Metal–Organic Framework-Based Enzyme Biocomposites. Chem. Rev. 2021, 121, 1077–1129. 10.1021/acs.chemrev.0c01029. [DOI] [PubMed] [Google Scholar]
- Guillerm V.; Kim D.; Eubank J. F.; Luebke R.; Liu X.; Adil K.; Lah M. S.; Eddaoudi M. A supermolecular building approach for the design and construction of metal–organic frameworks. Chem. Soc. Rev. 2014, 43, 6141–6172. 10.1039/C4CS00135D. [DOI] [PubMed] [Google Scholar]
- Jia F.; Narasimhan B.; Mallapragada S. Materials-based strategies for multi-enzyme immobilization and co-localization: a review. Biotechnol. Bioeng. 2014, 111, 209–222. 10.1002/bit.25136. [DOI] [PubMed] [Google Scholar]
- Küchler A.; Yoshimoto M.; Luginbühl S.; Mavelli F.; Walde P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409. 10.1038/nnano.2016.54. [DOI] [PubMed] [Google Scholar]
- Herron N.; Corbin D. R.. Inclusion Chemistry with Zeolites: Nanoscale Materials by Design; Springer Science & Business Media: 1995; Vol. 6. [Google Scholar]
- Bezerra C. S.; de Farias Lemos C. M. G.; de Sousa M.; Gonçalves L. R. B. Enzyme immobilization onto renewable polymeric matrixes: past, present, and future trends. J. Appl. Polym. Sci. 2015, 132, 42125. 10.1002/app.42125. [DOI] [Google Scholar]
- Kong G.; Xiong M.; Liu L.; Hu L.; Meng H.-M.; Ke G.; Zhang X.-B.; Tan W. DNA origami-based protein networks: from basic construction to emerging applications. Chem. Soc. Rev. 2021, 50, 1846. 10.1039/D0CS00255K. [DOI] [PubMed] [Google Scholar]
- Horcajada P.; Gref R.; Baati T.; Allan P. K.; Maurin G.; Couvreur P.; Ferey G.; Morris R. E.; Serre C. Metal–organic frameworks in biomedicine. Chem. Rev. 2012, 112, 1232–1268. 10.1021/cr200256v. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zheng M.; Xie Z. Nanoscale metal–organic frameworks for drug delivery: a conventional platform with new promise. J. Mater. Chem. B 2018, 6, 707–717. 10.1039/C7TB02970E. [DOI] [PubMed] [Google Scholar]
- Sun H.; Li Y.; Yu S.; Liu J. Metal-organic frameworks (MOFs) for biopreservation: From biomacromolecules, living organisms to biological devices. Nano Today 2020, 35, 100985. 10.1016/j.nantod.2020.100985. [DOI] [Google Scholar]
- Liang W.; Ricco R.; Maddigan N. K.; Dickinson R. P.; Xu H.; Li Q.; Sumby C. J.; Bell S. G.; Falcaro P.; Doonan C. J. Control of structure topology and spatial distribution of biomacromolecules in protein@ ZIF-8 biocomposites. Chem. Mater. 2018, 30, 1069–1077. 10.1021/acs.chemmater.7b04977. [DOI] [Google Scholar]
- Liang W.; Xu H.; Carraro F.; Maddigan N. K.; Li Q.; Bell S. G.; Huang D. M.; Tarzia A.; Solomon M. B.; Amenitsch H.; et al. Enhanced activity of enzymes encapsulated in hydrophilic metal–organic frameworks. J. Am. Chem. Soc. 2019, 141, 2348–2355. 10.1021/jacs.8b10302. [DOI] [PubMed] [Google Scholar]
- Hanefeld U.; Gardossi L.; Magner E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. 10.1039/B711564B. [DOI] [PubMed] [Google Scholar]
- Bolivar J. M.; Eisl I.; Nidetzky B. Advanced characterization of immobilized enzymes as heterogeneous biocatalysts. Catal. Today 2016, 259, 66–80. 10.1016/j.cattod.2015.05.004. [DOI] [Google Scholar]
- Sun C.; Cheng Y.; Pan Y.; Yang J.; Wang X.; Xia F. Efficient polymerase chain reaction assisted by metal–organic frameworks. Chem. Sci. 2020, 11, 797–802. 10.1039/C9SC03202A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Jiménez-Ángeles F.; Qiao B.; Krzyaniak M. D.; Sha F.; Kato S.; Gong X.; Buru C. T.; Chen Z.; Zhang X.; et al. Insights into the Enhanced Catalytic Activity of Cytochrome c When Encapsulated in a Metal–Organic Framework. J. Am. Chem. Soc. 2020, 142, 18576–18582. 10.1021/jacs.0c07870. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Jiménez-Ángeles F.; Qiao B.; Krzyaniak M. D.; Sha F.; Kato S.; Gong X.; Buru C. T.; Chen Z.; Zhang X.; Gianneschi N. C.; Wasielewski M. R.; Olvera de la Cruz M.; Farha O. K. Insights into the Enhanced Catalytic Activity of Cytochrome c When Encapsulated in a Metal–Organic Framework. J. Am. Chem. Soc. 2020, 142, 18576–18582. 10.1021/jacs.0c07870. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Han S.; Li X.; Zhang Z.; Ma S. Why does enzyme not leach from metal–organic frameworks (MOFs)? Unveiling the interactions between an enzyme molecule and a MOF. Inorg. Chem. 2014, 53, 10006–10008. 10.1021/ic501062r. [DOI] [PubMed] [Google Scholar]
- Ashbrook S. E.; Griffin J. M.; Johnston K. E. Recent advances in solid-state nuclear magnetic resonance spectroscopy. Annu. Rev. Anal. Chem. 2018, 11, 485–508. 10.1146/annurev-anchem-061417-125852. [DOI] [PubMed] [Google Scholar]
- Brunner E.; Lutz K. Solid-state NMR in biomimetic silica formation and silica biomineralization. Handbook of biomineralization: biomimetic and bioinspired chemistry 2007, 2, 19–38. 10.1002/9783527619443.ch26. [DOI] [Google Scholar]
- Molugu T. R.; Lee S.; Brown M. F. Concepts and methods of solid-state NMR spectroscopy applied to biomembranes. Chem. Rev. 2017, 117, 12087–12132. 10.1021/acs.chemrev.6b00619. [DOI] [PubMed] [Google Scholar]
- Ravera E.; Cerofolini L.; Martelli T.; Louka A.; Fragai M.; Luchinat C. 1 H-detected solid-state NMR of proteins entrapped in bioinspired silica: a new tool for biomaterials characterization. Sci. Rep. 2016, 6, 27851. 10.1038/srep27851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertmer M.Solid-state NMR of small molecule adsorption in metal–organic frameworks (MOFs). In Annual Reports on NMR Spectroscopy; Atta ur R., Ed.; Academic Press: 2020; Vol. 101, Chapter 1, pp 1–64. [Google Scholar]
- Lucier B. E. G.; Zhang Y.; Lee K. J.; Lu Y.; Huang Y. Grasping hydrogen adsorption and dynamics in metal–organic frameworks using 2H solid-state NMR. Chem. Commun. 2016, 52, 7541–7544. 10.1039/C6CC03205B. [DOI] [PubMed] [Google Scholar]
- Ueda T.; Kurokawa K.; Eguchi T.; Kachi-Terajima C.; Takamizawa S. Local Structure and Xenon Adsorption Behavior of Metal–Organic Framework System [M2(O2CPh)4(pyz)]n (M = Rh and Cu) As Studied with Use of Single-Crystal X-ray Diffraction, Adsorption Isotherm, and Xenon-129 NMR. J. Phys. Chem. C 2007, 111, 1524–1534. 10.1021/jp065321x. [DOI] [Google Scholar]
- Wong Y. T. A.; Babcock T. K.; Chen S.; Lucier B. E. G.; Huang Y. CO Guest Interactions in SDB-Based Metal–Organic Frameworks: A Solid-State Nuclear Magnetic Resonance Investigation. Langmuir 2018, 34, 15640–15649. 10.1021/acs.langmuir.8b02205. [DOI] [PubMed] [Google Scholar]
- Kong X.; Scott E.; Ding W.; Mason J. A.; Long J. R.; Reimer J. A. CO2 Dynamics in a Metal–Organic Framework with Open Metal Sites. J. Am. Chem. Soc. 2012, 134, 14341–14344. 10.1021/ja306822p. [DOI] [PubMed] [Google Scholar]
- Forse A. C.; Milner P. J.; Lee J.-H.; Redfearn H. N.; Oktawiec J.; Siegelman R. L.; Martell J. D.; Dinakar B.; Porter-Zasada L. B.; Gonzalez M. I.; Neaton J. B.; Long J. R.; Reimer J. A. Elucidating CO2 Chemisorption in Diamine-Appended Metal–Organic Frameworks. J. Am. Chem. Soc. 2018, 140, 18016–18031. 10.1021/jacs.8b10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. H.; Barth B.; Hartmann M.; Haase J. r.; Bertmer M. Nitric oxide adsorption in MIL-100 (Al) MOF studied by solid-state NMR. J. Phys. Chem. C 2018, 122, 12723–12730. 10.1021/acs.jpcc.8b01725. [DOI] [Google Scholar]
- Gul-E-Noor F.; Jee B.; Pöppl A.; Hartmann M.; Himsl D.; Bertmer M. Effects of varying water adsorption on a Cu 3 (BTC) 2 metal–organic framework (MOF) as studied by 1 H and 13C solid-state NMR spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 7783–7788. 10.1039/c0cp02848g. [DOI] [PubMed] [Google Scholar]
- Li J.; Li S.; Zheng A.; Liu X.; Yu N.; Deng F. Solid-state NMR Studies of Host–Guest Interaction between UiO-67 and Light Alkane at Room Temperature. J. Phys. Chem. C 2017, 121, 14261–14268. 10.1021/acs.jpcc.7b04611. [DOI] [Google Scholar]
- Khudozhitkov A. E.; Jobic H.; Kolokolov D. I.; Freude D.; Haase J. r.; Stepanov A. G. Probing the guest-mediated structural mobility in the UiO-66 (Zr) framework by 2H NMR spectroscopy. J. Phys. Chem. C 2017, 121, 11593–11600. 10.1021/acs.jpcc.7b03259. [DOI] [Google Scholar]
- Khudozhitkov A. E.; Arzumanov S. S.; Kolokolov D. I.; Stepanov A. G. Mobility of Aromatic Guests and Isobutane in ZIF-8 Metal–Organic Framework Studied by 2H Solid State NMR Spectroscopy. J. Phys. Chem. C 2019, 123, 13765–13774. 10.1021/acs.jpcc.9b03081. [DOI] [Google Scholar]
- Torres-Huerta A.; Galicia-Badillo D.; Aguilar-Granda A.; Bryant J. T.; Uribe-Romo F. J.; Rodríguez-Molina B. Multiple rotational rates in a guest-loaded, amphidynamic zirconia metal–organic framework. Chem. Sci. 2020, 11, 11579–11583. 10.1039/D0SC04432F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.; Li S.; Chu Y.; Xiao Y.; Xu J.; Deng F. Host–Guest Interaction between Methanol and Metal–Organic Framework Cu3–xZnx(btc)2 as Revealed by Solid-State NMR. J. Phys. Chem. C 2019, 123, 24062–24070. 10.1021/acs.jpcc.9b06332. [DOI] [Google Scholar]
- Wittmann T.; Mondal A.; Tschense C. B. L.; Wittmann J. J.; Klimm O.; Siegel R.; Corzilius B.; Weber B.; Kaupp M.; Senker J. Probing Interactions of N-Donor Molecules with Open Metal Sites within Paramagnetic Cr-MIL-101: A Solid-State NMR Spectroscopic and Density Functional Theory Study. J. Am. Chem. Soc. 2018, 140, 2135–2144. 10.1021/jacs.7b10148. [DOI] [PubMed] [Google Scholar]
- Xiao D.; Xu S.; Brownbill N. J.; Paul S.; Chen L.-H.; Pawsey S.; Aussenac F.; Su B.-L.; Han X.; Bao X.; Liu Z.; Blanc F. Fast detection and structural identification of carbocations on zeolites by dynamic nuclear polarization enhanced solid-state NMR. Chem. Sci. 2018, 9, 8184–8193. 10.1039/C8SC03848A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Chu Y.; Xu J.; Wang Q.; Qi G.; Gao P.; Zhou X.; Deng F. Extra-Framework Aluminum-Assisted Initial C–C Bond Formation in Methanol-to-Olefins Conversion on Zeolite H-ZSM-5. Angew. Chem., Int. Ed. 2018, 57, 10197–10201. 10.1002/anie.201805609. [DOI] [PubMed] [Google Scholar]
- Omegna A.; van Bokhoven J. A.; Prins R. Flexible Aluminum Coordination in Alumino–Silicates. Structure of Zeolite H–USY and Amorphous Silica–Alumina. J. Phys. Chem. B 2003, 107, 8854–8860. 10.1021/jp030094+. [DOI] [PubMed] [Google Scholar]
- Liu N.; Higashi K.; Kikuchi J.; Ando S.; Kameta N.; Ding W.; Masuda M.; Shimizu T.; Ueda K.; Yamamoto K.; Moribe K. Molecular-Level Understanding of the Encapsulation and Dissolution of Poorly Water-Soluble Ibuprofen by Functionalized Organic Nanotubes Using Solid-State NMR Spectroscopy. J. Phys. Chem. B 2016, 120, 4496–4507. 10.1021/acs.jpcb.6b00939. [DOI] [PubMed] [Google Scholar]
- Alonso B.; Marichal C. Solid-state NMR studies of micelle-templated mesoporous solids. Chem. Soc. Rev. 2013, 42, 3808–3820. 10.1039/C2CS35368G. [DOI] [PubMed] [Google Scholar]
- Feng D.; Liu T.-F.; Su J.; Bosch M.; Wei Z.; Wan W.; Yuan D.; Chen Y.-P.; Wang X.; Wang K.; et al. Stable metal-organic frameworks containing single-molecule traps for enzyme encapsulation. Nat. Commun. 2015, 6, 5979. 10.1038/ncomms6979. [DOI] [PubMed] [Google Scholar]
- Lian X.; Erazo-Oliveras A.; Pellois J.-P.; Zhou H.-C. High efficiency and long-term intracellular activity of an enzymatic nanofactory based on metal-organic frameworks. Nat. Commun. 2017, 8, 2075. 10.1038/s41467-017-02103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps J.; Chen H.; Donovan C.; Dominguez D.; Morgan S.; Weidman B.; Fan C.; Beyzavi M. H. Catalytic Activity, Stability, and Loading Trends of Alcohol Dehydrogenase Enzyme Encapsulated in a Metal–Organic Framework. ACS Appl. Mater. Interfaces 2020, 12, 26084. 10.1021/acsami.0c06964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. K.; Song H. K.; Shin D. H.; Hwang K. Y.; Suh S. W. The crystal structure of a triacylglycerol lipase from Pseudomonas cepacia reveals a highly open conformation in the absence of a bound inhibitor. Structure 1997, 5, 173–185. 10.1016/S0969-2126(97)00177-9. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Li P.; Modica J. A.; Drout R. J.; Farha O. K. Acid-resistant mesoporous metal–organic framework toward oral insulin delivery: Protein encapsulation, protection, and release. J. Am. Chem. Soc. 2018, 140, 5678–5681. 10.1021/jacs.8b02089. [DOI] [PubMed] [Google Scholar]
- Domínguez de Maria P.; Sánchez-Montero J. M.; Sinisterra J. V.; Alcántara A. R. Understanding Candida rugosa lipases: an overview. Biotechnol. Adv. 2006, 24, 180–196. 10.1016/j.biotechadv.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Alberti K. G. M. M.; Zimmet P. Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diabet. Med. 1998, 15, 539–553. . [DOI] [PubMed] [Google Scholar]
- Bradford N. A rapid and sensitive method for the quantitation microgram quantities of a protein isolated from red cell membranes. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Xiao B.; Fletcher A. J.; Thomas K. M.; Bradshaw D.; Rosseinsky M. J. Hysteretic adsorption and desorption of hydrogen by nanoporous metal-organic frameworks. Science 2004, 306, 1012–1015. 10.1126/science.1101982. [DOI] [PubMed] [Google Scholar]
- Martinez-Bulit P.; Stirk A. J.; Loeb S. J. Rotors, motors, and machines inside metal–organic frameworks. Trends Chem. 2019, 1, 588–600. 10.1016/j.trechm.2019.05.005. [DOI] [Google Scholar]
- Marangoni A.; Al-Abdul-Wahid M.; Nicholson R.; Roma A.; Gravelle A.; De Souza J.; Barbut S.; Spagnuolo P. Water immobilization by glass microspheres affects biological activity. Sci. Rep. 2018, 8, 9744. 10.1038/s41598-018-28123-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohwy M.; Jaroniec C.; Reif B.; Rienstra C.; Griffin R. Local structure and relaxation in solid-state NMR: accurate measurement of amide N– H bond lengths and H– N– H bond angles. J. Am. Chem. Soc. 2000, 122, 3218–3219. 10.1021/ja9913737. [DOI] [Google Scholar]
- Lewandowski J. R. Advances in solid-state relaxation methodology for probing site-specific protein dynamics. Acc. Chem. Res. 2013, 46, 2018–2027. 10.1021/ar300334g. [DOI] [PubMed] [Google Scholar]
- Bloembergen N.; Purcell E. M.; Pound R. V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679. 10.1103/PhysRev.73.679. [DOI] [Google Scholar]
- Okada K.; Hirai D.; Kumada S.; Kosugi A.; Hayashi Y.; Onuki Y. 1H NMR relaxation study to evaluate the crystalline state of active pharmaceutical ingredients containing solid dosage forms using time domain NMR. J. Pharm. Sci. 2019, 108, 451–456. 10.1016/j.xphs.2018.09.023. [DOI] [PubMed] [Google Scholar]
- Haouas M.; Volkringer C.; Loiseau T.; Férey G.; Taulelle F. The Extra-Framework Sub-Lattice of the Metal–Organic Framework MIL-110: A Solid-State NMR Investigation. Chem. - Eur. J. 2009, 15, 3139–3146. 10.1002/chem.200801856. [DOI] [PubMed] [Google Scholar]
- Grube E.; Nielsen U. G. The stoichiometry of synthetic alunite as a function of hydrothermal aging investigated by solid-state NMR spectroscopy, powder X-ray diffraction and infrared spectroscopy. Phys. Chem. Miner. 2015, 42, 337–345. 10.1007/s00269-014-0724-7. [DOI] [Google Scholar]
- Tang Y.; Huang H.; Li J.; Xue W.; Zhong C. IL-induced formation of dynamic complex iodide anions in IL@ MOF composites for efficient iodine capture. J. Mater. Chem. A 2019, 7, 18324–18329. 10.1039/C9TA04408F. [DOI] [Google Scholar]
- Jiang Y.; Huang J.; Marx S.; Kleist W.; Hunger M.; Baiker A. Effect of dehydration on the local structure of framework aluminum atoms in mixed linker MIL-53 (Al) materials studied by solid-state NMR spectroscopy. J. Phys. Chem. Lett. 2010, 1, 2886–2890. 10.1021/jz1010835. [DOI] [Google Scholar]
- Tay H. M.; Rawal A.; Hua C. S-Mg 2 (dobpdc): a metal–organic framework for determining chirality in amino acids. Chem. Commun. 2020, 56, 14829–14832. 10.1039/D0CC05539E. [DOI] [PubMed] [Google Scholar]
- Varghese S.; Halling P. J.; Häussinger D.; Wimperis S. High-Resolution Structural Characterization of a Heterogeneous Biocatalyst Using Solid-State NMR. J. Phys. Chem. C 2016, 120, 28717–28726. 10.1021/acs.jpcc.6b11575. [DOI] [Google Scholar]
- Brunner E.; Rauche M. Solid-state NMR spectroscopy: an advancing tool to analyse the structure and properties of metal–organic frameworks. Chem. Sci. 2020, 11, 4297–4304. 10.1039/D0SC00735H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z.; Jamieson L. E.; Dawson D. M.; Ashbrook S. E.; Bühl M. NMR chemical shifts of urea loaded copper benzoate. A joint solid-state NMR and DFT study. Solid State Nucl. Magn. Reson. 2019, 101, 31–37. 10.1016/j.ssnmr.2019.04.004. [DOI] [PubMed] [Google Scholar]
- Ehresmann J. O.; Wang W.; Herreros B.; Luigi D.-P.; Venkatraman T.; Song W.; Nicholas J. B.; Haw J. F. Theoretical and experimental investigation of the effect of proton transfer on the 27Al MAS NMR line shapes of zeolite– adsorbate complexes: an independent measure of solid acid strength. J. Am. Chem. Soc. 2002, 124, 10868–10874. 10.1021/ja012336u. [DOI] [PubMed] [Google Scholar]
- Ganapathy S.; Gore K.; Kumar R.; Amoureux J.-P. Multinuclear (27Al, 29Si, 47, 49Ti) solid-state NMR of titanium substituted zeolite USY. Solid State Nucl. Magn. Reson. 2003, 24, 184–195. 10.1016/S0926-2040(03)00044-4. [DOI] [PubMed] [Google Scholar]
- Xu M.; Shen Y.; Wang L.; Gong C.; Chen S. A Novel H2O2 Biosensor Based On the composite of MP-11 encapasulated in PCN-333 (Al)-Graphene Oxide. Int. J. Electrochem. Sci. 2017, 12, 10390–10401. 10.20964/2017.11.45. [DOI] [Google Scholar]
- Wang Z.; Jiang Y.; Yi X.; Zhou C.; Rawal A.; Hook J.; Liu Z.; Deng F.; Zheng A.; Hunger M.; et al. High population and dispersion of pentacoordinated AlV species on the surface of flame-made amorphous silica-alumina. Sci. Bull. 2019, 64, 516–523. 10.1016/j.scib.2019.04.002. [DOI] [PubMed] [Google Scholar]
- Volkringer C.; Popov D.; Loiseau T.; Guillou N.; Ferey G.; Haouas M.; Taulelle F.; Mellot-Draznieks C.; Burghammer M.; Riekel C. A microdiffraction set-up for nanoporous metal–organic-framework-type solids. Nat. Mater. 2007, 6, 760–764. 10.1038/nmat1991. [DOI] [PubMed] [Google Scholar]
- Wilke C.; Chang P. Correlation of diffusion coefficients in dilute solutions. AIChE J. 1955, 1, 264–270. 10.1002/aic.690010222. [DOI] [Google Scholar]
- Brunauer S.; Emmett P. H.; Teller E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. 10.1021/ja01269a023. [DOI] [Google Scholar]
- Amoureux J.-P.; Fernandez C.; Steuernagel S. ZFiltering in MQMAS NMR. J. Magn. Reson., Ser. A 1996, 123, 116. 10.1006/jmra.1996.0221. [DOI] [PubMed] [Google Scholar]
- Massiot D.; Fayon F.; Capron M.; King I.; Le Calvé S.; Alonso B.; Durand J. O.; Bujoli B.; Gan Z.; Hoatson G. Modelling one-and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. 10.1002/mrc.984. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.