

Abstract

The rational design of high-performance catalysts is hindered by the lack of knowledge of the structures of active sites and the reaction pathways under reaction conditions, which can be ideally addressed by an in situ/operando characterization. Besides the experimental insights, a theoretical investigation that simulates reaction conditions—so-called operando modeling—is necessary for a plausible understanding of a working catalyst system at the atomic scale. However, there is still a huge gap between the current widely used computational model and the concept of operando modeling, which should be achieved through multiscale computational modeling. This Perspective describes various modeling approaches and machine learning techniques that step toward operando modeling, followed by selected experimental examples that present an operando understanding in the thermo- and electrocatalytic processes. At last, the remaining challenges in this area are outlined.
Keywords: Heterogeneous catalysis, Global optimization, Operando modeling, Ab initio molecular dynamics, Machine learning
Introduction
Heterogeneous catalysis is at the heart of the chemical industry, where a high-performance catalyst with a fast conversion of the reactants and a high selectivity toward targeted products is greatly desired.1−3 However, the rational design of catalysts is hindered by the lack of molecular-level knowledge about the structures of active sites and the reaction pathways under reaction conditions. It has been observed that, during the reaction, most heterogeneous catalysts undergo a structural reconstruction accompanied by a varying catalytic performance.4 Moreover, the concept of “dynamic fluxionality” reminds us to see the active structure as a collection of many structures that dynamically interconvert with a low energy barrier. It is therefore significant to characterize the changing surface morphology, short-lived intermediates, and stepwise reaction kinetics under the reaction conditions, which can be addressed by the fast-developing operando characterization.
In the definition, operando refers to the real-time measurement of a catalyst at its working place under real reaction conditions, with the simultaneous online analysis of catalyst performance.5 With the establishment of structure/composition-performance correlations, the operando characterizations provide a dynamic insight into the atomic and electronic structures of heterogeneous catalysts under working conditions, which help to deeply understand the interfacial behavior and the catalytic mechanism.6 As a comparison, the ex situ characterization presents the pre- and postchemical states, which may not truly reflect the dynamic nature of the heterogeneous catalysts during the reaction.4
For a complete understanding of a working catalyst system at an atomic scale, a theoretical investigation that can realistically simulate operando conditions is necessary. However, the operando conditions have been significantly simplified in the current widely used computational model. A typical example is the periodic slab model for a heterogeneous catalysis, which is created by cutting a crystal through a specific crystalline plane and adding a vacuum in the direction orthogonal to the surface. However, its validity can be expected only when the active structure of a catalyst system is known (or correctly guessed) and is not dramatically affected by the reaction environment.7 It was reported that, without an explicit consideration of the solvent effect, a density functional theory (DFT) calculation outputs a result contradictory to the experimentally observed high activity of Ni single atom over graphene.8
The interaction between the environment and the reaction intermediate species plays an important role in the exploration of the catalytic mechanism, which should not be ignored.1 It gives rise to the concept of operando modeling that describes the catalyst behavior in an experimentally spatiotemporal scale under true reaction conditions.7 A schematic depiction of the operando modeling is illustrated in Figure 1. Compared with the simplified periodic slab model, operando modeling further considers the temperature, pressure, and/or solvent effect on the active structure of the catalyst. Beyond that, given that catalysts can undergo a constant structural change during the reaction, a long-term, large-scale simulation is expected to track this dynamic process. With numbers of intermediates and reaction pathways being visited, a large reaction network is thus possible to be established, providing a way to fully reveal the catalytic mechanism under the operando condition.
Figure 1.
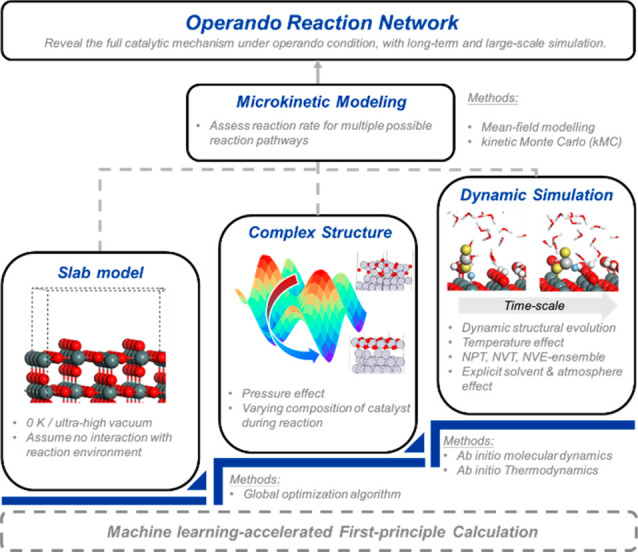
Schematic depiction of the concept of operando modeling. The ML technique significantly accelerates the first-principle calculation and makes a long-term, large-scale, and accurate simulation possible. The operando modeling is aimed to reveal the full catalytic mechanism under operando conditions, within which the catalyst behavior should be described in an experimentally spatiotemporal scale under true reaction conditions with both thermodynamic and kinetic aspects.
Because of the overwhelming complexity, a comprehensive operando modeling cannot be achieved by a single computational method (e.g., DFT calculations).4,7Operando modeling should be achieved through a multiscale computational modeling, with multifaceted physical and chemical methodologies being involved. The most stable structure of a heterogeneous catalyst under operando conditions can be obtained by global optimization (GO) techniques, which can also help to study the dynamic fluxionality of the catalyst. Ab initio thermodynamics (AITD) can be utilized to assess the thermodynamic stabilities under the varying reaction conditions, while ab initio molecular dynamics (AIMD) can identify the dynamic interfacial structure of the heterogeneous catalyst. With the numbers of intermediates and reaction pathways being visited, kinetic modeling is required to explain the stepwise kinetics to build up the reaction network. Considering the huge computational cost of the above method, the machine learning (ML) technique can serve as a surrogate model to accelerate the time-consuming simulation by orders of magnitude.
This Perspective describes the current progress and remaining challenges in this area. Beginning with the brief introduction of the operando technique, this Perspective will illustrate how the above-mentioned computational techniques link with each other, achieve the concept of operando modeling, and help clarify the underlying mechanism in a heterogeneous catalysis. In the end, we will present how a joint effort by experimental and theoretical studies can be made toward an atomistic understanding of the thermo- and electrocatalytic processes.
Operando Characterization
The term operando was coined by Eric Gaigneaux, Gerhard Mestl, Miguel A. Bañares, and Bert M. Weckhuysena at the 220th ACS National Meeting in 2000 and first appeared in the catalytic literature in 2002 with several publications putting the idea of operando into practice.9−14 Recently, various operando techniques, including optical spectroscopies (Raman, IR, etc.), X-ray-based characterizations (X-ray diffraction (XRD), X-ray absorption near edge structure (XANES), etc.), microscopies (scanning/transmission electron microscopes (S/TEM), scanning tunneling microscopy (STM), etc.), Mössbauer spectroscopy, and other methods have been successfully implemented to track the dynamic behavior of a catalyst under reaction conditions. Table 1 briefly lists some representative operando techniques with their main function.
Table 1. Representative Operando Techniques for Atomic-Scale Studies of Surface Catalysis.
| operando technique | function |
|---|---|
| infrared (IR) spectroscopy | Monitor chemisorbed species on the catalyst |
| Raman spectroscopy | Monitor the intermediates’ formation |
| X-ray diffraction (XRD) | Monitor the crystal structural change and phase transitions. |
| Mössbauer spectroscopy | Clarify the chemical state and spin state (for specific elements) |
| X-ray absorption near edge structure (XANES) spectroscopy | Monitor electronic/oxidation states of the target atoms. |
| extended X-ray absorption fine structure (EXAFS) spectroscopy | Provide information about the coordination environments of target atoms, including their coordination number and the bond distance. |
| X-ray photoelectron spectroscopy (XPS) | Provide information about elemental composition, chemical and electronic state information on the catalyst. |
| X-ray emission spectroscopy (XES) | Clarify the local electronic structure and bonding configuration of the absorbing atom. |
| nuclear magnetic resonance (NMR) spectroscopy | Observe the chemical components and their interactions with active sites. |
| electron paramagnetic resonance (EPR) spectroscopy | Monitor the evolution of redox reactions. |
| scanning/transmission electron microscopes (S/TEM) | Provide sub-Ångström spatial resolution with compositional information and electronic structure. |
| atomic force microscopy (AFM) | Detect evolution in surface morphology and surface potential. |
| scanning electrochemical microscopy (SECM) | Monitor the electrochemical activity, kinetics, and adsorbate coverages. |
| differential electrochemical mass spectrometry (DEMS) | Detect the reaction products and adsorbates for studying kinetics. |
| electrochemical quartz crystal microbalance (EQCM) | Monitor mass change on the catalyst electrode. |
It is worth noting that it is difficult to achieve an in-depth and comprehensive understanding of a catalytic mechanism through only a single operando technique. When multiple operando techniques are combined, the dynamic catalytic behavior can be identified from complementary aspects.4 For example, Zakharov et al.16 combined the operando XANES, extended X-ray absorption fine structure (EXAFS), and S/TEM measurements to characterize all-metal species present in a silica-supported Pt catalyst (0–5 nm), while EXAFS alone cannot achieve this characterization due to a poor spatial resolution. Weckhuysen and co-workers have developed several combined operando techniques to get a full insight into the reactions. Figure 2 presented their combined operando XRD and UV–vis setup for studying the lattice expansion of zeolite catalysts caused by the formation of hydrocarbon species. The evolution of the hydrocarbon pool was measured using operando UV–vis spectroscopy, and the resulting zeolite lattice expansion was measured using operando XRD.15 More combined operando instruments developed by Weckhuysen and co-workers included luminescence thermometry/Raman spectroscopy to simultaneously obtain local thermal and chemical information, X-ray powder diffractometry (XRPD)/Raman spectroscopy that monitors the simultaneous evolution of phases and various formed species during the reaction, etc.17−21
Figure 2.
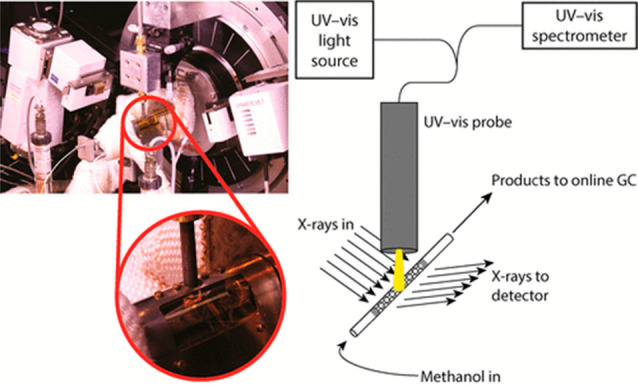
Schematic of the combined operando XRD/UV–vis setup showing the X-ray diffractometer with the mounted capillary; in the middle of the capillary, the spot of the UV–vis light source can be seen. Reprinted with permission from ref (15) . Copyright 2018 American Chemical Society.
Modeling Strategies for Realistic Simulation
Prediction of Surface Structure for Heterogeneous Catalyst under Operando Conditions
The actual surface structure of the catalyst mostly is the lowest energy-point on the potential energy surface (PES), the so-called global minimum (GM). Finding the GM of a working catalyst constitutes a GO problem. During the GO process, metastable structures of the catalyst can be obtained. Note that a recent study by Sun and Sautet shows which of some metastable structures might contribute more to the total activity than the global minimum structures.22 Because of their importance, the controllable synthesis of metastable structures has received extensive attention in the experiment.23,24
In the past decade, there has been significant progress in developing GO algorithms and software packages for chemical structure optimization. The frequently used GO algorithms and their representative software packages are listed in Table 2.
Table 2. Frequently used GO algorithm for chemical structure optimization and their representative software package.
| GO algorithm | software package |
|---|---|
| genetic algorithm | USPEX,25 XtalOpt,26 BCGA,27,28 BPGA,29 GA module of ASE,30,31 HAGA,32 GOFEE33 |
| differential evolution | PDECO34 |
| covariance matrix adaptation evolution strategy (CMA-ES) | Clinamen35 |
| stochastic surface walking (SSW) | LASP36 |
| particle swarm optimization | CALYPSO37 |
| artificial bee colony | NWPEsSe,38 ABCluster39 |
| basin and minima hopping | TGMin,40 Basin and Minima Hopping module of ASE31 |
| Bayesian optimization | BOSS,41 BASC42 |
As this Perspective especially focuses on operando modeling that highlights the characterization of the catalyst surface, the following content is about the interface structure of the heterogeneous catalyst. Owing to the periodicity of the crystal and the presence of strong covalent bonds to the underlying support, the optimization of the surface structure is more geometrically restricted than that of free particles.7 For the application of GO algorithms in free particles like metal clusters and nanoalloys, we refer the reader to some recent reviews.43−45
The existence of a support can make the catalyst structure very different from that in the gas phase because of the strong metal–support interaction.46 Davis et al.28 performed the Birmingham parallel genetic algorithm (BPGA) to reveal that the existence of the MgO(100) support could suppress the spin of Au–Ir clusters, causing a huge difference in the gas-phase and MgO-supported structure. For the metal–organic framework (MOF)-supported systems, Vilhelmsen et al.47 investigated MOF-supported Au, Pd, and Au–Pd clusters and concluded that Pd could bind more tightly with the MOF support than Au. Compared with the aromatic ring, the open metal site in the MOF did not provide much tighter binding sites, which provided an understanding of how the local environment effects the catalyst structure. It is worth noting that whether the support has a significant effect rests on the nature of the support and catalysts.46 For example, Pt13 catalysts in the gas phase hold a similar structure with that upon a graphene surface,48 but the structure significantly changes when on a CeO2 surface.49
GO algorithms can provide insights into the defective structure, like steps, vacancies, and adgrowths,7 which commonly occur in realistic conditions. Some examples include the exploration of the ⟨111⟩ and ⟨001⟩ step edges of the TiO2(110) surface50 and the oxidized edge of graphene sheets on the Ir(111) surface.33 Arrigoni et al.35 have developed a GO software package, named Clinamen, that helps to discover the low-energy defective structure, and its capabilities have been demonstrated in an optimization of TiO2 anatase with oxygen vacancies. Moreover, the catalyst structure can be different when deposited on the defective surfaces and the stoichiometric surface. After confirming the Fs defect could profoundly modify the absorption features,51 Fortunelli and co-workers have presented series of GO studies for Au,52,53 Ag,54,55 Pd–Ag56 clusters adsorbed on the Fs-defected MgO(100) surface.
For alloy catalysts, the catalytic properties can be dramatically different with even a small change in composition. The GO algorithm plays a role generally when the number of atoms is correctly guessed. For the catalyst system in which the composition of the GM remains unclear, the GO method needs to be modified to search over composition space and to perform fixed-stoichiometry searches at many compositions.
Upon exposure to oxygen, reactant gas, and additional adsorbates, the supported catalysts can undergo a structural change. As shown in Figure 3, Sierka et al.57 showed that the one-dimensional (1D) and two-dimensional (2D) silica structures deposited upon a Mo(112) surface are subject to the oxygen pressure and amount of deposited Si atoms.32 Later the same technique was applied in a search of the stable structure of p(1 × 2)-, p(1 × 3)-,32 and O(2 × 3)-58 on a Mo(112) surface, with full support from experimental observations. Moreover, Liu et al.59 applied GA to identify surface phases of Pt, Pt3Ni, and Pt3Au surfaces with high oxygen coverages and concluded that the existence of oxygen created oxide skins with a different morphology. The adsorbed GM is the foundation of the further analysis of catalytic behavior. For example, after obtaining the GM of the fully CO-saturated alumina-supported Pt10, Yin et al.60 found that this structure can coadsorb O2 at the interface, which is beneficial to catalytic activity through the OOCO mechanism. In the case of the Ag/Pt catalyst, a complete CO → CO2 reaction catalytic cycle was drawn after the GM was obtained, as the OOCO mechanism still got supported.
Figure 3.
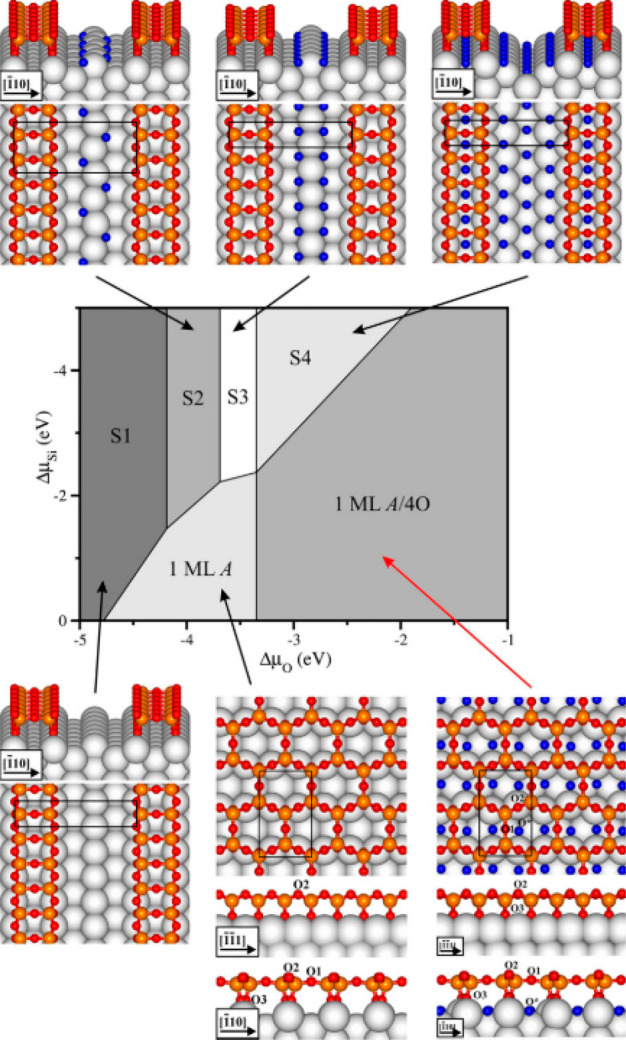
Calculated phase diagram of 1D and 2D crystalline silica over a Mo(112) substrate as a function of ΔμO and ΔμSi chemical potentials. Reprinted with permission from ref (73). Copyright 2010 Elsevier.
The GM obtained from the GO algorithm for catalysts with different sizes can help to understand the size-evolution trend in the catalyst growth mechanism. As shown in Figure 4a, Wang et al.61 revealed that the CeO2(111)-supported Pdn (n = 1–21) catalysts first grow on the base layer and then on the second layer to undermine the metal–support interactions. For the Cun (n = 1–10) catalysts on a ZnO(1010) surface, Cu atoms prefer an adsorption between the Zn and O or a direct adsorption to an O atom, and the interface of Cun (n > 5) and ZnO(1010) presents a continuum between Cu(111) and (110) structures.62 Moreover, the GO algorithm can optimize nanosized particle systems. Di Valentin and co-workers63−67 have performed GO algorithms to model nanosize TiO2 particles with water to study their photoexcitation processes, proton transfer mechanism, etc.
Figure 4.
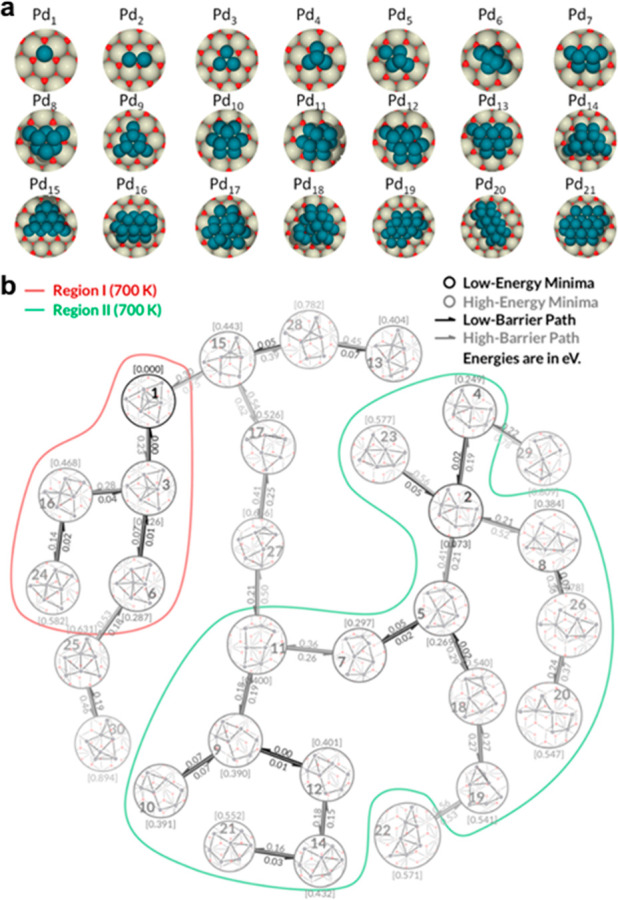
(a) GM of Pdn catalyst (n = 1–21) over CeO2(111). Reprinted with permission from ref (61). Copyright 2020 American Chemical Society. (b) Minimal energy paths of Pt7 on Al2O3 obtained using a bipartite matching algorithm, showing the picture of the fluxional behavior of Pt7. Reprinted with permission from ref (75). Copyright 2018 American Chemical Society.
Not only the GM but also some metastable isomers of the catalyst obtained during the GO process can play a significant role in building the reaction network. Fang et al.68 selected some metastable isomers to build the whole reconstruction process of a Au(100) surface induced by the adsorption of CO. Earlier, Fortunelli and co-workers69,70 have developed kinetics-driven Reactive Global Optimization (RGO) to study a reaction network for supported catalysts, in which reaction rates are calculated during the GO process and are used to guide the GO. The RGO has been successfully applied to study the propylene partial oxidation by MgO(100)-supported Ag3 and the CO oxidation by MgO(100)-supported AgxAu3–x.71,72 However, it suffers a great cost of searching.38
In some cases, metastable isomers may even lead to a higher activity. It was shown that the catalytically relevant sites of the Mo catalyst over γ-Al2O3 do not correspond to its GM.74 And for Pt13, the highest activity toward methane activation corresponds to its second lowest-lying isomer, rather than GM.22 Furthermore, many metastable isomers are energetically accessible, and thermodynamic equilibration between them is kinetically possible. It reminds us not to see the active structure as a single GM but as a statistical ensemble representation, a collection of many structures that dynamically interconvert with a lower energy barrier.46 This concept is the so-called “dynamic fluxionality”, which implies that the single structure obtained from the GO algorithm represents the most abundant isomer in the ensemble of structures.44
An experimental observation of the dynamic fluxionality under an operando condition has been reported on a silica-supported Pd for ethylene hydrogenation,76 ceria-supported Pt for water gas shift,77 Al2O3-supported Cu oxide,78 etc. Recently, Alexandrova and co-workers have presented series of works that contribute to the deeper theoretical understanding of dynamic fluxionality. Using the GO technique, they revealed the highly fluxional behavior of Pt7 over Al2O3,75 Cu oxide at a high temperature and different O2 pressures,79 and hexagonal boron nitride in the oxidative dehydrogenation of propane.80 They found that dynamic fluxionality may break the scaling relationships,81 cause non-Arrhenius behavior,82 or accelerate the Ostwald ripening of a supported catalyst.83 Moreover, a bipartite matching algorithm was introduced to connect low-lying isomers obtained from the GO technique and to build the whole picture of the fluxional behavior,75 as shown in Figure 4b. More discussion about dynamic fluxionality can be seen in their recently published review.2,46,84
Dynamic Modeling of Reaction Environment
The structure of the catalyst also depends on the realistic chemical environment (solvent, electrochemical potential, and temperature). Indeed, GO has been used to identify the GM of water clusters85,86 and microsolvated ions87 and to reveal explicitly solvated transition state88,89 and reaction mechanisms.90 However, it more represents a static snapshot of what a stable structure looks like, so a GO is seldom applied to operando descriptions of surface systems with an explicit description of a chemical environment46 when a dynamic interaction between catalyst and environment matters.
Solvents play a crucial role in electrocatalytic reactions. To mimic operando conditions and illustrate the active surface structure, an explicit consideration of the solvent is necessary. The AIMD technique can identify the dynamic interfacial structure of heterogeneous catalysts with an explicit solvent. Ren et al.49 used the AIMD to study the CeO2-supported Pt13 catalyst in an aqueous phase, whose structure is significantly different from that in the gas phase, as shown in Figure 5a. Greeley and co-workers have presented a series of works using the AIMD to study the solvation effects on the catalytic mechanism, which revealed that an explicit solvent molecule can interact with a defect and active site, transfer the electron, alter the adsorption behavior of catalytic spices, and so on.91−95 The AIMD can also help to unravel the atomistic structures of electric double layers (EDL) and to understand the dynamic behavior of catalytic species at the interface, a field in which Cheng and co-workers have presented a series of significant works recently.96−100 More examples include the explicit description of a mixture solvent, like supercritical CO2/H2O.101 Similarly, the transition state may suffer a strong explicit solvent effect. Herron et al.102 used AIMD to study the methanol electrooxidation on Pt(111) and reveal that a water solvation reduces the barriers for both the C–H and O–H bonds of methanol, while the effect is more pronounced for a C–H bond activation. Moreover, the dynamics of electrons can be also modeled by AIMD as shown in Figure 5b. Selcuk et al.65,103 thus found that the behavior of excess electrons on anatase surfaces depended strongly on the nature of the exposed facet, the environment, and the character of the electron donor during the photocatalytic reaction.
Figure 5.
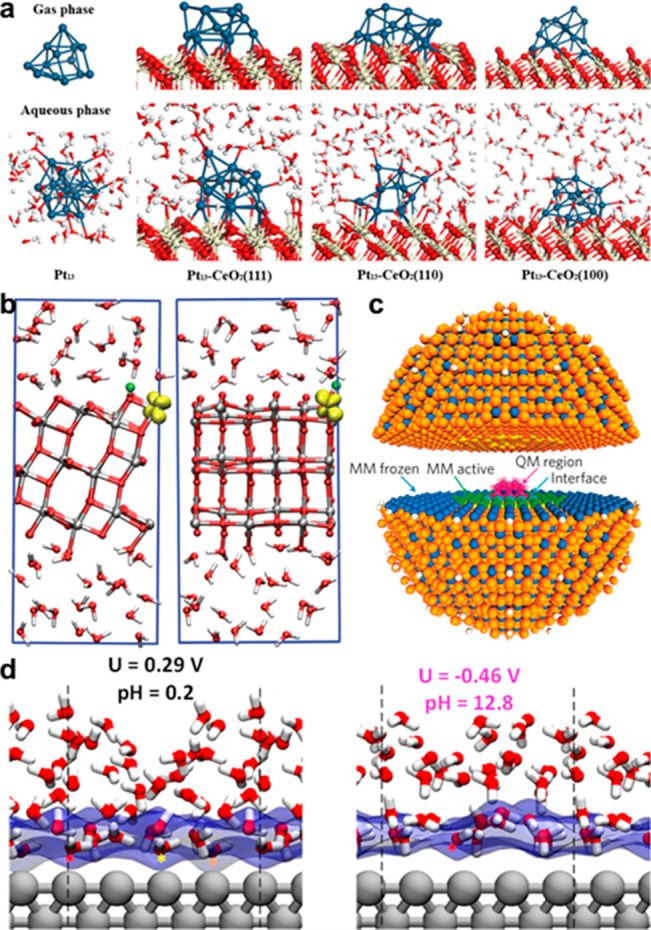
(a) Electron density difference plots for the optimized CeO2-supported Pt13 cluster in the gas and aqueous phases. Reprinted with permission from ref (49). Copyright 2018 American Chemical Society. (b) Hydroxylated anatase (101) slab immersed in water; the eex– is shown as a yellow iso-surface plot. Reprinted from ref (103). Copyright 2018 Nature Portfolio. (c) Graphic of the QM/MM cluster used for rutile in the positive charge state. The cluster is divided into hemispheres to highlight the different regions in the model. Hole density iso-surfaces are shown in the QM region. Reprinted from ref (105). Copyright 2018 Nature Portfolio. (d) The snapshots with atomic details of the interfaces are shown at U = +0.29 V and at U = −0.46 V. The first layer of water is highlighted by a plot of the van der Waals surface of oxygen as transparent blue. Reprinted with permission from ref (106). Copyright 2018 American Chemical Society.
Another simulation method is the hybrid quantum mechanics/molecular mechanics (QM/MM) approach, which retains an ab initio consideration at the interface and leaves other surrounding molecules for the force field.104 As shown in Figure 5c, Scanlon et al.105 found the band alignment of −0.4 eV for the anatase–rutile phase junction by using QM/MM, which explains the robust separation of photoexcited charge carriers between the two phases. Moreover, to investigate the pH-dependent hydrogen oxidation reaction (HOR) and hydrogen evolution reaction (HER) performance, Cheng et al.106 calculated the hydrogen binding energy (HBE) as a descriptor and simulated the full solvent water/Pt(100) interface using a QM/MD approach at an applied voltage from +0.29 to −0.46 V, which is equivalent to pH from 0.2 to 12.8 at 0.3 V applied voltage. The snapshots with atomic details of the interfaces were shown in Figure 5d. The results revealed that the pH-dependent HBE was mainly due to the distinct water adsorption under different pH values, and its changing trend was very close to the experimental observation.
In the thermo-catalytic processes, the above-mentioned methods can also be applied to observe dynamic structural changes induced by the reaction atmosphere. He et al.107 used AIMD to study the structure of CeO2-supported Au19 under a CO atmosphere. Being consistent with our experimental observations, the AIMD result confirmed that Au19 undergoes a layer-to-three-dimensional transition after an exposure to a CO atmosphere, caused by an extraction and motion of Au atoms in the form of gold–carbonyl species. Besides the solvent/atmosphere effect, the catalyst structure can change significantly under a high temperature/pressure. The temperature/pressure effect can also be described in an ab initio calculation. With different temperatures, the ab initio calculation revealed that the supported catalyst undergoes a different adsorption behavior,108 solvent interaction,109 and reactive pathway.110 Besides AIMD, AITD is routinely employed in this case. Originally introduced by Reuter and co-workers, AITD highlights the thermodynamic stabilities (of intermediates, active sites, etc.) under the varying reaction conditions, which are currently widely used in heterogeneous systems like reactive surfaces and nanoporous spaces.111,112 Senftle et al.113 applied AITD to assess the stability of CeO2-supported Pd7Ox catalysts for a methane conversion, as a function of temperature and oxygen pressure. The stability of intermediates can also be analyzed by AITD to confirm the proposed mechanism under the operando conditions. Using the AITD analysis, Li et al.114 analyzed the stability of the potential molybdenum (oxy)carbide species form during the methane dehydroaromatization and finally concluded that the binuclear [Mo2C2]2+ site is a more likely candidate for the active sites. A surface Pourbaix diagram can be obtained through AITD to study the stable surface phases under different potential/pH conditions.115 Moreover, combining GO methods with AITD is a powerful strategy to construct structural models. After obtaining a series of GM of Cu(111)-supported ZnyOx catalysts through GO, Reichenbach et al.116 performed AITD to further assess, and ensure, the thermodynamic stabilities of these structures under different temperatures and O2 pressures.
In the experiment, the reaction intermediates detected by operando techniques comprehensively understand the reaction mechanisms. With an explicit description of the reaction environment, the computational mechanisms obtained from an ab initio simulation can better explain the experimental observation from an atomistic view. As a representative example, Li and co-workers have presented series of works using AIMD to systemically study the dynamic catalytic behavior of a supported Au single-atom catalyst upon exposure to a reactant gas, including its formation, size effect, and reaction mechanism for CO oxidation,107,117−120 which provide insights into the existing experimental finding. Moreover, they have applied a similar strategy to identify intermediates and to study the catalytic mechanism of a Pt single-atom catalyst for a CO oxidation and Fe for a nonoxidative conversion of methane.121,122
The inconsistency between the experimental result and the output of an ab initio calculation is sometimes due to the failing capture of rare events (like chemical reactions) and the limited time-scale of simulation. For a complex reaction mechanism with multiple reaction channels, an enhanced sampling technique can be applied to facilitate the crossing of energy barriers and extend the sampling time scales of ab initio calculation, like umbrella sampling, meta-dynamics, and so on.123 Goddard and co-workers124−127 have presented a series of works that combine meta-dynamics to identify the complex reaction pathway for a CO2 reduction reaction (CRR) and CO reduction reactions (CORR) on a Cu (100) surface with an explicit solvent. The intermediates captured from a picosecond-level simulation can successfully interpret the experimental operando spectrum.5 For nanoporous catalysts, Speybroeck and co-workers used molecular dynamics (MD) techniques with enhanced sampling to describe their phase stability/transformation, active site, and reaction kinetics,94,128−130 together with the diffusion behavior and nucleation process of solvent/reactant inside the nanoporous cages.131−133
To fully reveal the reaction network, microkinetic modeling is required to assess the reaction rate for multiple possible reaction pathways and eventually explain the macroscopic kinetics under operando conditions.134 Taking the H2 oxidation over Ru as an example, the microkinetic modeling was successfully applied to simulate the local oscillation frequency and to support the local reaction kinetics observed from imaging microscopies.135−138 Two widely utilized modeling methods in this field are mean-field microkinetic modeling and kinetic Monte Carlo (kMC).139 An emerging alternative is a hybrid method called extended phenomenological kinetics (XPK), which is proposed by Xu and co-workers and is specially designed to describe complex catalytic kinetics under operando conditions.140 With enhanced accuracy and efficiency, XPK has been successfully described as a complex reaction network on the surface, including syngas conversion,141 formic acid decomposition,142 and CO oxidation.143 Besides, some automatic techniques has been applied to automatically generate microkinetic mechanisms for the surface catalytic process like graph theory-driven software Reaction Mechanism Generator (RMG) for an oxidative coupling over a Pt surface and the combustion of methane over a Ni surface,144,145 CatNet for a syngas conversion over Rh(111),146,147 stochastic surface walking (SSW) for a water gas shift reaction over Cu(111),148 and the artificial force induced reaction (AFIR) method for a CO oxidation over Pt(111).149
After a full reaction network is constructed, it is of greater importance to determine the optimal pathway based on the calculated kinetics. The practice developed by Xiao and co-workers is worth considering, which is to introduce an energy optimization to determine the pathway with minimal ΔG-limiting energy.150 As illustrated in Figure 6, the optimal pathway of NO2 conversion over anatase TiO2(101) was studied in their recent work, which helped to clarify the activity and selectivity of NO2 conversion over different active sites under photo and dark conditions.151
Figure 6.
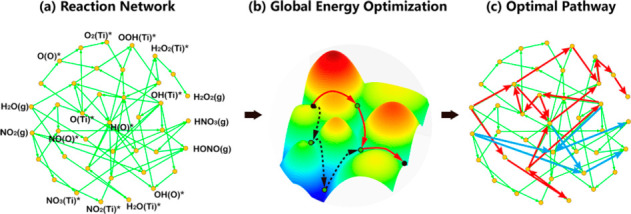
(a) The complete reaction network of NO2 conversion with stable intermediates marked. (b) The algorithm of determining the pathways from global energy optimization. (c) The optimal reaction pathway over the perfect anatase TiO2(101) surface (blue, NO2 evolutional paths; red, H2O evolutional paths). Reprinted with permission from ref (151). Copyright 2021 American Chemical Society.
Machine Learning for Accelerating Heterogeneous Catalysis Research
Recently, the data-driven ML technique emerged as a useful tool and surrogate model to accelerate the time-consuming simulation. Machine learning potentials (MLP), which directly learns the potential surface from ab initio calculations, have been developed to act as a cheap energy calculator with a high accuracy (within a few meV/atom for energies, ∼0.1 eV/Å for forces) while maintaining the large speedup (with several orders of magnitude faster). Once a successful MLP has been derived, it has the potential to be used efficiently on much larger systems than the one on which it was trained, significantly lowering the computational cost for the simulation of large-scale systems.152
In recent years, an increasing number of MLP has been published, including a variety of neural network potentials (NNP), graph networks, Gaussian approximation potentials, and many others.153 This Perspective especially focuses on the application of the ML technique in simulating realistic catalyst systems under operando conditions. For a comprehensive review of the technical details of ML techniques that are widely used in materials science, we refer readers to a few recent reviews.153,154
Coupling the ML technique with the GO provides significant advantages in the acceleration of searches. Performing a search on the ML model provides a cheap energy calculator without requiring expensive DFT calculations. It is worth noting that the ML model fitted to DFT data can show better accuracy than the empirical potential for a GO search.155 Liu and co-workers combined a neural network (NN) with their own developed SSW global optimization method to develop the SSW-NN method as implemented in the LASP code,156,157 which can explore complex reactions systems unbiasedly and automatedly. A more detailed introduction and application of the LASP code on heterogeneous catalyst systems can be seen in the review.36,158−160 ML can also be combined well with a population-based GO method like GA to formulate an active learning framework, that is, on-the-fly training.161 Kolsbjerg et al.162 showed that, with an active learning framework, the number of DFT calculations required to obtain the GM of MgO-support Pt13 was significantly reduced from 8900 to 260. An ML model trained during the GO can be further utilized to search for the transition barrier and then to build up the complete image of a Pt13 structural transformation, which is extremely time-consuming at the DFT level. Further, the Bayesian concepts can be introduced to accelerate the GO search not only by reducing the number of required DFT calculations but also by balancing “exploration” (probing uncertain regions of configuration space) and “exploitation” (exhausting the local region known to have low-energy structures).163 Hammer and co-workers have utilized Bayesian concepts in the context of Gaussian processes to develop the open-source GA-based software package GOFEE33 for the surface system, which outperforms by 2 orders of magnitude a DFT-based GA in reducing computational cost. Other ML methods, like cluster analysis, have also been used to increase the structural diversity by suppressing similar structures164 and to bias searching toward lower energy basins after clustering and characterizing local atomic environments,165 both of which accelerate the GO search for surface structure.
The ML-accelerated GO can be applied to predict phase diagrams for alloy catalysts with an unfixed composition or in the situation that the catalyst composition changes during the reaction. Hajinazar et al.155 used an NN-accelerated GO to obtain the phase diagram of ternary Cu–Pd–Ag nanoalloys, which showed an encouraging agreement between the NN and the DFT methods. By utilizing the SSW-NN method, Ma et al.166 established the phase diagram of a ZnCr oxide catalyst for a syngas-to-methanol conversion and then identified an active Zn3Cr3O8 phase containing [ZnO6] octahedra in bulk that is experimentally known for high activity. Moreover, for a catalyst under operando conditions, the compositions and the total number of atoms can change dynamically. The SSW-NN method was also utilized to study the operando formation of PdHx during an acetylene semihydrogenation and identified the Pd4H3 phase as the most responsible for the deep hydrogenation to ethane at high H2 pressures.167 Besides, the temperature effect can be included in a predicting phase diagram by using an ML descriptor for the temperature-dependent contribution Gδ(T) to the Gibbs energy G(T).168,169
The understanding of reaction intermediates and catalysts’ active sites has been classically studied by operando techniques, and the determination of active sites is crucial for clarifying the reaction mechanism. However, the correct modeling of these species remains a huge challenge due to the enormous number of possible adsorption configurations. Using NNP as a surrogate model, Ulissi et al.170 discover an unexplored active site of Ni/Ga intermetallic catalysts with the best thermodynamics for CO reduction and step-like kinetic behavior. The number of ab initio calculations was reduced by an order of magnitude in this study. This high-throughput framework was then applied to accelerate the discovery of CO2 electrocatalysts, resulting in the identification of Cu–Al catalysts with the highest Faradaic efficiency.171 Not only the thermodynamics for active sites, the evaluation of kinetic properties like reaction rate can also be accelerated by ML, even in a microstructure with a large size.172 Apart from directly modeling the structure, ML can be trained using experimental spectra like XANES or Raman spectroscopy and then directly predict the local atomic environment of reaction intermediates.173−176
The overwhelming complexity of reaction networks limits the modeling of the experimental behavior in surface catalytic reactions at the DFT level, which can be addressed by MLP. A typical reaction network contains not only the relevant intermediates but also transition states that connect between intermediates. A nudged elastic band (NEB) calculation is the most popular method for transition-state searching, which can also be accelerated by MLP.139,177,178 It is worth noting that, when preparing a training data set that is used to build MLP for reaction networks, the imbalance between the number of reactants and product configurations versus the number of transition state structure needs to be considered.179 Ulissi et al.180 performed Gaussian process models that are trained on-the-fly, to iteratively optimize reaction networks of the syngas reaction over Rh(111) under the guidance of uncertainty. Moreover, Kang et al.158 used SSW-NN to study the reaction network of the water gas shift reaction over Cu(111). An optimal network was built after 375 000 minima were sampled and more than 10 000 reaction pairs were collected, which is almost impossible for a DFT calculation. The final reaction network was shown in Figure 7, revealing the lowest-energy pathway for this reaction.
Figure 7.
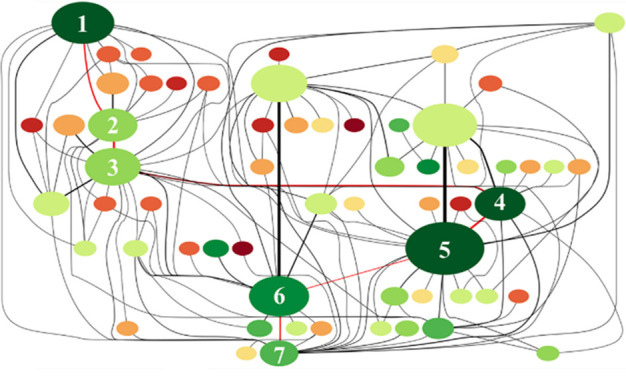
Reaction network for water gas shift reaction (WGSR) on Cu(111). The system starts from two CO and two H2O on the Cu(111) surface. The key intermediates along the WGSR lowest-energy pathway are marked by red lines, e.g., (1) 2CO+2H2O; (2) 2CO + H2O + OH + H; (3) COOH + CO + H2O + H; (4) HCOOH + CO+H2O; (5) HCOO+CO+H2O + H; (6) CO2+CO+H2O+H+H; (7) CO2+CO+H2O + H2. The color of the circles from dark green to dark red indicates the energy from low to high; the area of the circle represents the frequency of the state encountered during the search; the width of the line corresponds to the occurrence number of the transformation in simulation. Reprinted with permission from ref (158). Copyright 2021 Elsevier.
Using MLP can significantly extend the spatial and time scale of atomistic simulations, providing more opportunities to simulate large-scale catalyst systems while maintaining a high accuracy.139,183 Cheng et al.181 used NNP to perform molecular dynamics simulation (NN-MD) and to describe the realistic and dynamic oxide-derived copper (OD-Cu) surface models during CO2 electro-reduction to C2+ products, as shown in Figure 8a,b. After scanning over 150 surface sites, three square-like sites for C2+ products were identified finally, providing fundamental insights into the origin of activity and selectivity over Cu-based catalysts. In this study, the error of *CO and *COCO adsorption energy between the NNP and DFT calculation is as low as 0.08 and 0.13 eV, respectively. The ML-enhanced large-scale modeling also enables simulations of complex systems such as electrolytes or solid–liquid interfaces. Natarajan et al.184 investigated the behavior of interfacial water molecules at the low-index Cu(111), (100), and (110) surfaces, using a model containing 128 explicit water. Quaranta et al.185 even showed that MLP can well capture the surface reactions such as proton transfer, taking the water-ZnO(1010) interface as an example. Furthermore, Artrith et al.182 used NNP with Monte Carlo (MC) simulations for bimetallic Au/Cu nanoalloys with up to 3915 atoms (∼6 nm). With the consideration of explicit solvent molecules, the different preferred structures in a vacuum and aqueous solution were further revealed. The simulation results are in agreement with an experimental EXAFS analysis. The same method was also used to model the surface structure after an interaction with solvent, as depicted in Figure 8c.
Figure 8.
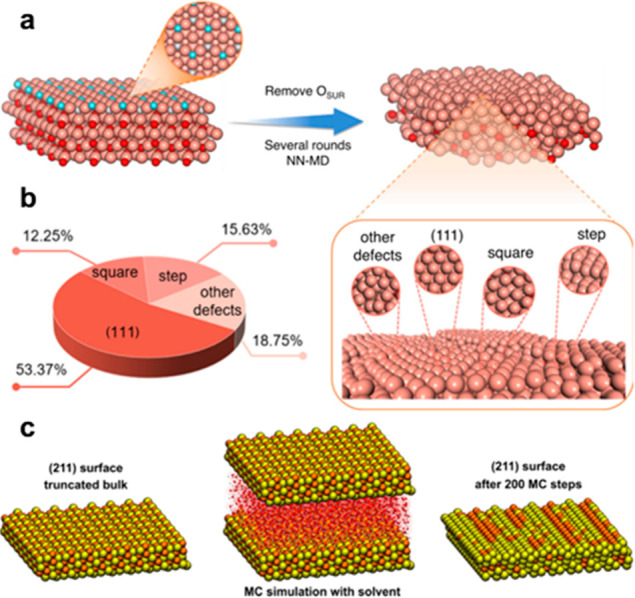
(a) Illustration of the procedure to construct an OD-Cu model with an NN-MD simulation. (b) Proportions of different surface structures of the OD-Cu model. Reprinted from ref (181) under a Creative Commons CC BY License. (c) Snapshots from the MC simulation of a large (211) surface slab model with solvent, containing in total ∼6500 atoms. (left) Initial truncated-bulk structure. (center) The same structure of water including the hydration shell of water molecules. (right) The optimized composition after 200 MC steps. Reprinted with permission from ref (182). Copyright 2014 American Chemical Society.
Besides, ML can accelerate enhanced sampling simulations to predict the long-time-scale surface reaction by using an ab initio calculation. Jiang and co-workers186−191 have presented a series of works that fit NN to construct a high-dimensional reactive PES for AIMD simulations. A simulation lasting tens of picoseconds was achieved in their studies, which required a tremendous computational cost without ML. The gas-surface reaction dynamics (nonadiabatic effect, dissociative/adsorptive behavior, etc.) of the surface system, like CO2 on Ni(100), H2O on Pt(110), etc., were revealed, without losing accuracy (∼1 meV/atom root-mean-square error (RMSE) for energy and ∼10 meV/Å for atomic force).
Experimental Operando Application
Thermocatalytic Processes
A typical feature of thermocatalysis is that catalytic species are exposed to an atmosphere that contains reactants, with high reaction temperatures. The catalytic reactions are driven by heat, and the catalyst undergoes a continuous structural change during the thermocatalysis. Thus, clarifying the dynamic evolution of thermocatalyst is essential for the understanding of overall catalytic performance. According to the different driving factors of dynamic evolution, we divide the section into two parts: atmosphere and temperature.
Atmosphere-Induced Dynamic Evolution
The gas–solid interface is a focus of the thermocatalysis process. When passed through such an interface, reactants will continuously absorb and react on the surface, which will also induce the changing morphologies or composition of catalyst. The CO oxidation reaction is generally treated as a model system for studying the gas–solid interface, and Au is a typical catalyst for studying the dynamic features of a catalyst. Wang et al.118 investigated the formation process of a Au single-atom catalyst induced by the adsorption of CO molecules. As shown in Figure 9a, AIMD showed that the adsorption of CO enhanced the reconstruction of Au particles and generated more low-coordination sites to bind with CO. Besides, CO would diffuse on the support surface that was accompanied by one Au atom formed by the strong interaction between Au and CeO2.
Figure 9.
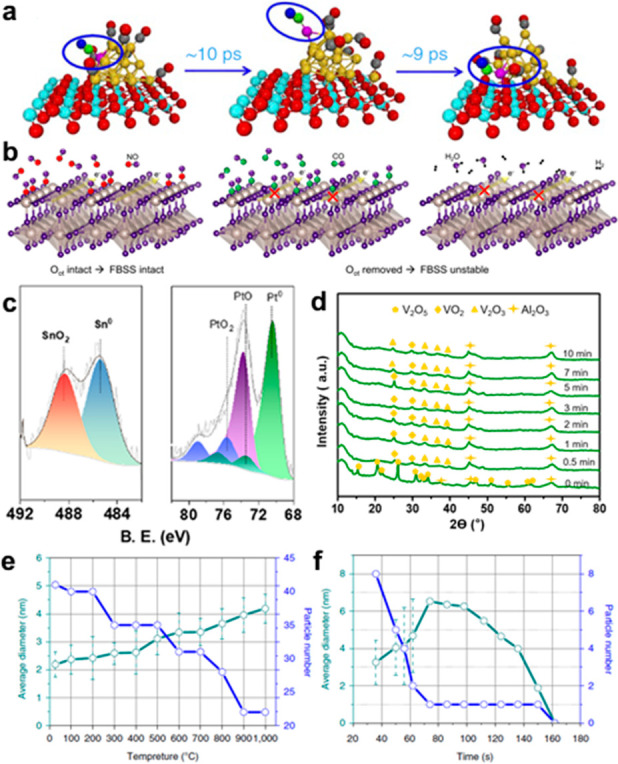
(a) Selected snapshots of the MD trajectory for Au20/CeO2 with a circled Au-CO unit to show the diffusion process. Reprinted from ref (118) under a Creative Commons CC BY License. (b) FBSS state induced by different absorbates on RuO2. Reprinted from ref (193). Copyright 2021 American Chemical Society. (c) Quasi in situ XPS of Pt–Sn/SBA-15 reduced by H2. Reprinted from refs (195) and (196). Copyright 2021 American Chemical Society. (d) XRD pattern of Mo-doped VOx/Al2O3 at 500 °C during the dehydrogenation step for 0–10 min. Continuous phase variation from V2O5 to V2O3 via VO2 was observed due to the induction by a lattice evolution under the propane stream. Reprinted from ref (197). Copyright 2021 American Chemical Society. (e) Average diameter and number of particles as a function of heating temperatures. (f) Average diameter and number of particles vs pyrolyzing time at 1000 °C. Reprinted from ref (198). Copyright 2018 Nature Portfolio.
For the CO oxidation that uses nanoparticles, Albinsson et al.192 pointed out the importance of bridging the gap between local and averaging characterization methods. With operando plasmonic nanoimaging, they revealed that both surface and bulk oxidation states changed by the CO oxidation of a Cu catalyst. For the larger particle, where a periodic surface calculation is appropriate, Jovic et al.193 focused on the RuO2 surface and its unique change of electronic structure during the CO adsorption process by operando angle-resolved photoemission spectroscopy. They pointed out the flat-band surface state (FBSS) promoted catalytic charge transfer processes and the corresponding CO oxidation reaction (Figure 9b). Nguyen et al.194 further considered the function of O2 reactant during a CO oxidation, and they found O2 would produce a missing row reconstruction on a Rh(110) surface, while the origin Rh(110) structure was retained even under one monolayer of CO coverage.
H2 is another typical composition of the atmosphere during the thermocatalytic process, acting as a reactant or product. The morphology of metal nanoparticles will be prominently affected by H2, as Johnson et al.199 pointed out that H2 would lead to a phase transformation in Pd nanoparticles. According to in situ XRD patterns, the α-phase disappeared while the β-phase was generated after the catalyst was exposed to H2, which strengthened the adsorption of hydrogen on Pd nanoparticles. But the situation on Pt nanoparticles is quite different. The existence of the H atom will lead to the sintering and growth of Pt particles rather than to the formation of another phase. Song et al.200 showed that Pt–H interactions could weaken the adhesion of Pt on MoS2 and accelerate particle migration and coalescence. Introducing Au to form a Pt–Au alloy could inhibit sintering prominently by decreasing the interaction with Pt atoms. Liu et al.201 draw a similar conclusion after studying a Pt species on MCM-22 zeolite; they showed how highly dispersed Pt was reduced and agglomerated into clusters in the reductive atmosphere, while an oxidative atmosphere had an opposite function. A Pt–Sn alloy is another important system for the dehydrogenation of propane. Combining in situ XPS, TEM, and DFT calculations, Wang et al.195 revealed the phenomena of migration of Sn atoms from the inner core to the surface during a reduction induced by H2 (Figure 9c) and the formation of the Pt–Sn alloy surface structure.
The redox reactions under a reductive/oxidative atmosphere also induce a dynamic structural change of catalyst, together with a changing valence state. Zichittella et al.196 utilized operando EPR spectroscopy to quantify the oxidation state of representative CrPO4 and EuOCl catalysts during a propane oxychlorination reaction. They found the number of redox-active centers (Cr2+ or Eu2+) can be directly correlated with the space-time yield of propylene. Chen et al.197 investigated the reduction process of Mo-doped vanadia (Figure 9d) by XRD. They observed the phase transformation process from V2O5 to VO2, V2O3 under a reductive propane atmosphere, which explained the change of propane dehydrogenation performance. Koch et al.202 considered the application of perovskites in an alkane oxidation, showing that steam added to the feed could increase the selectivity of the partial oxidation product propylene. By analyzing the surface of the working catalyst, they found Mn in a low oxidation state (2+/3+), an increased concentration of hydroxyl groups, and a higher abundance of adsorbed activated oxygen species on the catalyst surface is the origin of the better performance of the two-dimensional MnOx surface phase.
Temperature-Induced Dynamic Evolution
Temperature is another factor that induces the dynamic evolution of the catalyst. In a thermocatalytic process, the catalyst will experience a heating-up process under reaction conditions. The results from an in situ environmental TEM performed by Wei et al.198 show that the particle size of Pd will increase with rising temperature under 900 °C. When the temperature rises to 1000 °C, a N dopant could help anchor the Pd atoms and form Pd single atoms, as shown in Figure 9e,f.
Adjusting the reaction temperature can alter the catalytic performance in some cases. Passos et al.203 reported how heating versus cooling cycles influence the single nanoparticle elastic energy landscape. The highly compressive and tensile strain distribution was accordingly changed with temperature and so was the CO oxidation performance. Another important application of changing the temperature is the thermal annealing process. Kozlovskiy et al.204 investigated the phase transformation process of FeCo-Fe2CoO4/Co3O4 nanocomposites that was induced by an annealing. Increasing the annealing temperature can help the catalyst keep a crystal structure with fewer deformations and distortions and can release its lattice oxygen more easily to act as an oxidant. The kMC model proposed by Zhou et al.205 can simulate the structural evolution during annealing, which may boost the further development of research in the annealing process.
(Photo) Electrochemical Catalytic Processes
It has been well-known that most catalysts would undergo a structural reconfiguration during the electrochemical reaction. Here we summarize the combination of theoretical calculations and operando characterization to track this information from three aspects, including (i) the reconstruction of the catalysts (phase, morphology, etc.) under actual operating conditions (pH values, potential, etc.), (ii) the determination of the oxidation states of the active centers, and (iii) the recording of the reaction intermediates.
Reconstruction of the Catalysts during the Reaction
Because of the unique electrochemical environment, the structural evolution of the catalysts can be affected by pH values and potential.206 Currently, many studies have used the DFT calculation to understand the interfacial structures at the atomic scale. Hansen et al.207 constructed surface Pourbaix diagrams based on the computational hydrogen electrode (CHE) approach, which described the most stable surface structures of Pt(111), Ag(111), and Ni(111) in an aqueous environment as a function of pH values and potential for an oxygen reduction reaction (ORR). As mentioned above, an explicit solvent model is also adopted. Zhao et al.8 included the effect of surface charge and hydrogen bonding by using AIMD with enhanced sampling and successfully clarified the experimentally observed high activity and selectivity of Ni single-atom anchored in graphene, which a previous DFT calculation without an explicit consideration cannot.
By the operando characterization, the changing catalyst surface structure under different pH values can be observed. Pi et al.208 showed that IrNix nanoparticles exhibited distinct composition-segregated features under acidic and alkaline conditions during the oxygen evolution reaction (OER). Coincidentally, by combining operando XAFS and photoelectron spectroscopic measurements, Cao et al.206 visualized a chameleon-like structural self-optimization of Co9S8 supported by a single-walled carbon nanotube under neutral/alkaline conditions during the OER. They identified that the precatalyst self-optimized into CoOOH with residual S species under alkaline conditions, while the oxygenated CoS was formed under neutral conditions. Gao et al.209 utilized the photothermal effect to enhance the OER activity of NiFe2O4 nanoparticles. Operando Raman spectroscopy revealed that the photothermal effect facilitated the surface reconstruction into high-active oxyhydroxides, and a lower kinetics barrier under applied anodic potentials was confirmed by DFT.
The operando characterization can also help to explore how the potential affects the reconstruction of the catalysts. Dionigi et al.210 utilized operando wide-angle X-ray scattering (WAXS) to study the phase-transition process of NiFe and CoFe layered double hydroxides as OER electrocatalysts under applied anodic potentials and found that they all transformed from the as-prepared α-phase to the active γ-phase. Moreover, these measured results were in excellent agreement with the DFT calculations and AIMD simulations, indicating that the operando scattering analysis can accurately identify the catalytically active phases. Using operando XRD, Reikowski et al.211 studied the relationship between the potential-dependent structure of thin Co3O4(111) and CoOOH(001) films and the electrochemical current. It was found that CoOOH(001) could maintain good stability at a wide OER current densities range, while they observed the fast and fully reversible structural changes on Co3O4(111). Su et al.212 reported a NiFe Prussian blue analogue as OER electrocatalyst and revealed that Ni(OH)2 was the active species. Through operando X-ray spectroscopy, the in situ generated Ni(OH)2 was detected to transform into NiOOH2–x, which contained Ni4+, under an applied potential. The amount of Ni4+ was a function of the applied potential.
Determination of the Oxidation States of the Active Sites
Identifying the oxidation states of the active site under reaction conditions plays a crucial role in the electrocatalyst design. For CRR, copper has been proved a promising electrocatalyst for converting CO2 to C2 products among all of the explored metals.213,214 To improve the catalytic performance of Cu-based catalysts, the relationship between the oxidation states of Cu and catalytic properties has been extensively studied using operando characterization. Chou et al.215 found that the CRR mechanism is related to the oxidation state distribution on a Cu surface by employing in situ surface-enhanced infrared absorption spectroscopy (SEIRAS) and in situ soft X-ray absorption spectroscopy (XAS). As shown in Figure 10a, the results showed that C1 hydrocarbon products could be obtained on a Cu(I) dominant surface, whereas a Cu(0) dominant surface inhibits hydrocarbon formation. The selectivity of ethylene was further enhanced on the surface with the coexistence of Cu(I) and Cu(0). Likewise, by employing operando seconds-resolved XAS, Lin et al.216 reported an oxide-derived Cu electrocatalyst with a steady chemical state of half Cu(0) and half Cu(I), which could produce C2H5OH with a considerably high selectivity in a wide potential range. By employing DFT calculations, it was revealed that the equal numbers of Cu and Cu(I) on the top layer of Cu2O could facilitate the coupling of dual carbon monoxide to form the *OCCO intermediate, which improved the CRR selectivity toward C2 products. Zhou et al.217 correlated the oxidation state of copper with the preference for the electrosynthesis of C2 hydrocarbon and utilized boron to tune the average copper valence state at +0.35, which was confirmed by in situ XANES, achieving a high Faradaic efficiency for C2 hydrocarbons of ∼80%.
Figure 10.
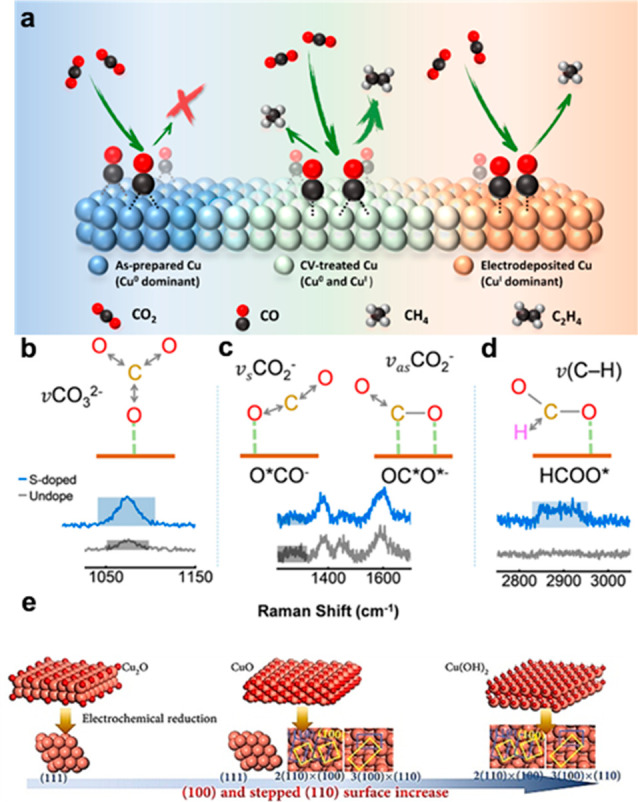
(a) Schematic illustration of the electrochemical CRR on the Cu Surface. Reprinted from ref (215). Copyright 2020 American Chemical Society. (b–d) Absorption structure of the intermediate with relative Raman spectra. Reprinted from ref (220). Copyright 2020 American Chemical Society. (e) Illustration of the preparation of Cu(OH)2-derived/Cu foil, CuO-derived/Cu foil, and Cu2O-derived/Cu foil. Reprinted from ref (222). Copyright 2020 Wiley.
The oxidation state of single-atom active sites is difficult to determine during an electrochemical reaction. By using operando XAFS, Cao et al.218 revealed that the coordination-unsaturated single Co1–N4 site transformed into a high-oxidization HO-Co1–N2 moiety with a hydroxyl adsorption under an alkaline HER. A DFT calculation demonstrated that this highly oxidized Co site could enhance the HER activity. In contrast, Fang et al.219 revealed that the single Pt atom tended to release from the nitrogen–carbon support and exhibited a close-to-zero valence state during the HER, which optimized the adsorption energies of the reactants.
Detection of the Reaction Intermediates
Using some operando characterization, such as Fourier transform infrared spectroscopy (FTIR) and Raman spectroscopy, the formation of the adsorbed species on a catalyst surface can be recorded under the actual operating conditions in aqueous media and obtain a deeper understanding of the reaction mechanism.4 To enhance the CO2-to-formate selectivity in CRR, Pan et al.220 employed in situ Raman spectroscopy to monitor the vibration models of CO2– intermediates on Cu-based electrocatalysts. It was found that introducing the S atom can change the adsorption state from coexisting O*CO– and OC*O*– to the dominating OC*O*–, resulting in a high selectivity and activity toward formate (Figure 10b–d). Furthermore, to improve the CRR selectivity toward C2 products, Chen et al.221 reported a controllable boundary-rich copper catalyst (GB-Cu) with the high C2 selectivity of 70%. In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and DFT calculation revealed that the key intermediate *CO was binding more strongly on the Cu surface in the presence of a large number of grain boundaries, which promote the C–C coupling. In addition, Zhong et al.222 synthesized a series of Cu catalysts with different exposed surfaces in a controlled manner by modifying the structures of precursors (Cu(OH)2, CuO, and Cu2O). By employing in situ ATR-SEIRAS, in situ Raman spectroscopy, and DFT calculations together, the facet effect of copper crystals toward the C2 selectivity/activity was investigated at the atomic level (Figure 10e). The results unveiled that the stepped Cu(110) and Cu(100) are crucial, which facilitated the CO adsorption and promoted a CO dimerization. Guo et al.223 utilized in situ Raman spectroscopy to explore the HER mechanism during photocatalytic processes on MoS2xSe2(1–x) nanosheets. They demonstrated that H atoms were bonded to active S and Se atoms and a guideline to directly assess the HER performance owing to the exponential relationship between the number of reactive electrons and the Raman intensity of intermediate species.
The large noises in the spectrum make the operando FTIR and Raman spectroscopy limited in identifying the atomistic structure of the reactive intermediates.124 A theoretical calculation is an effective tool providing an atomistic understanding, but sometimes the results do not match with the experimental observation due to an unrealistic modeling. To solve this issue, Cheng et al.124 utilized QM/MD with an explicit solvent and applied a potential at 298 K to resemble the operando experimental condition and identified reactive intermediates in CRR. The results showed that the value of the predicted C–O stretch of *HOC–COH and the C–C stretch of *C–COD is consistent with that of the experimental peak. In addition, to explore how to improve the hydrocarbon product selectivity and reaction rates, they126 further performed AIMD to calculate the CRR pathways and kinetics on the water/Cu(100) interface with five layers of the explicit water at pH 7. It was found that hydrocarbon products could be obtained selectively and efficiently with an applied potential greater than −0.60 V (RHE) and that accelerating the CO dimerization was an efficient way to improve the reaction rates.
Operando methods are also commonly used in photocatalysis to explore the reaction mechanism. Xu et al.224 combined operando NMR spectroscopy, DFT calculations, and AIMD simulations to understand the dynamic behaviors of the methanol–water cluster intermediates on the surfaces of rutile-TiO2 in a photocatalytic reaction. The results demonstrated that the motions of the methanol–water clusters govern the number of methanol molecules that reach the surface of the photocatalyst per unit time and, in turn, determine the yields of methanol-reforming products.
Challenges and Opportunities
The union of operando characterization with the modeling strategies for a realistic reaction represents an exciting area for new insight into heterogeneous catalysis. Here, we discuss a few of the most recent advances, challenges, and opportunities in this area: the more accurate description of a reaction environment, the further consideration of a highly dynamic fluxionality of catalyst structure, along with the deeper incorporation of the ML technique.
Improved Operando Techniques with High Spatiotemporal Resolutions
Despite the fast-developing operando techniques, the gap between experimental and theoretical spatiotemporal windows still exists, which can result in a difference between experimental results and theoretical prediction. Currently, operando microscopy, like liquid-cell S/TEM, can achieve a nanometer and even an atomic-scale spatial resolution, while attosecond spectroscopy even enables a visualization of the electron motion in atoms, molecules, and solids.152 ,225 ,226 In heterogeneous catalysis, theoretical investigations generally provide an atomistic model of reaction intermediates whose lifetime is approximately a few hundred femtoseconds or even much shorter.227 However, simultaneously obtaining such high temporal and spatial resolutions remains highly challenging.5 It implies that current operando techniques may only capture the (quasi)stable states of reaction intermediates.228 Not to mention, heterogeneous catalysts have a completely different structure with their specificities and reaction environments that cause even more complex heterogeneities in spatiotemporal windows. In a word, to further combine the experimental and theoretical techniques, the development of novel operando techniques with high spatiotemporal resolutions is desired.
Dynamic Fluxionality of Catalyst Structure
The concept of dynamic fluxionality reminds us to see the active structure under operando conditions as an ensemble representation of many structures that dynamically interconvert with a lower energy barrier.46
The Unclear Importance of Each Isomer
The possible number of dynamically accessible isomers is large, which even grows exponentially as the number of atoms increases.75 Unfortunately, there is no a priori knowledge of which isomers play a more important role than others in the dynamic reaction.46 The GM is believed to be the most abundant isomer in an ensemble of structures at low temperatures. However, some metastable structures might contribute more to the total activity than the most stable ones.22 The situation can be further complicated with varying reaction environments (temperature, adsorbates coverage, etc.).84 Boltzmann statistics have revealed that the populations of different isomers can change with temperatures; thus, different ensemble-averaged properties are expected.229 In short, the dynamic fluxionality somehow leaves the true active structure in doubt.
The Interconversion Pattern of Possible Isomers
The ensemble representation of active structure implies that the actual reaction mechanism might be not one but many, with slightly different mechanisms and rates.2 However, it is even unclear whether the catalyst undergoes an interconversion concurrently with the reaction step or sequentially with it, which should depend on the reaction energy barrier and cluster interconversion barrier.84 Things can become extremely complicated if the reaction and interconversion happen concurrently, as the transition state of such a process is almost impossible to obtain.46 Under operando conditions, as the catalyst undergoes continuous structural changes, the complexity to accurately describe its dynamic reaction mechanism is daunting.
In Alexandrova and co-workers’ recent review,2,46 they proposed that a full theoretical description of a dynamic catalyst system calls for the all-around development of correlated electronic structure methods, efficient PES sampling, statistical mechanics, and accelerated dynamics. As for the experiment, it is expected for operando tools with the per-site resolution and the ability to find the minority sites that govern the catalytic activity and the combination of multiple operando techniques to build a coherent picture.46,228
The Accurate Description of the Reaction Environment
The active structure of the catalyst highly depends on the reaction environment (solvent, electrochemical potential, and temperature). However, realistic simulations of surface catalytic reactions under operando conditions remain a grand challenge from many perspectives, and continuous efforts are required.
The Consideration of Coverage Effect
The coverage effect can be a decisive factor that causes deviations between simulations and experimental measurables, especially for the high-coverage system with a surface nonuniformity and strong self-/cross-interactions of adsorbates. As is well-known, the kMC method can be used to study how the surface coverage affects the reaction kinetics by quantitatively altering rate coefficients, whose calculated turnover frequencies (TOF) and product selectivity show good agreement with the experimental data.230−232 However, it is noteworthy that the catalyst can undergo a different structural change with different coverage, which can be analyzed with the help of GO techniques as mentioned above. To fully illustrate the coverage effect, a combinational view of both thermodynamics and kinetics is expected.
Constant Potential in Electrocatalytic Modeling
The explicit consideration further raises problems of how to keep a constant applied potential in electrocatalytic modeling, which is significant especially for electron-transfer reactions.233 With an explicit solvent, the applied potential can be modulated by charges or extra hydrogen atoms. In this case, a constant potential is hard to maintain, as the work function (which needs to be fixed to keep the potential constant) is sensitive to the coordinates of an explicitly treated solvent.104 A constant potential model with numerous explicit solvent molecules in a continuum dielectric may be a potential solution for this issue.234 Also, Chan and co-workers have presented series of works to extend the constant charge model to the constant potential model, using methods like charge extrapolation, effective charge, etc.235−238 However, a generally accepted constant potential method is currently not yet available.104,239
Take Advantage of the Machine Learning Technique
As mentioned above, the data-driven ML technique significantly accelerates the time-consuming simulation. For the further development and integration of ML technique on operando modeling, the following points are worth attention.
Lack of Benchmark for Heterogeneous Catalysis
As a data-driven technique, the most important strategy for the application of MLP in heterogeneous catalysis is the development of an open-source data set. Not only for training but a benchmark data set is also critical to establishing a consensus on testing and reporting newly developed ML models, and will ultimately improve data veracity in this field.240 For the prediction of molecular properties, today’s benchmark is the QM9 data set containing DFT-computed properties for ∼134 000 molecules with up to nine heavy atoms.241,242 However, a universally recognized benchmark is still lacking in the study of heterogeneous catalysis. Moreover, another concern is that, currently, operando experiments have not been explored fully, causing a lack of refined experimental data to guide the development of ML methods.243
Lack of a Large Data Set of Heterogeneous Catalysis
The data sets for heterogeneous catalysis are relatively difficult to collect due to the structural complexity and higher computational cost, and the number remains small.244 The currently available data sets include the Catalysis Hub, Open Catalyst 2020 (OC20) Data set, CMR project, etc.244−247 Except for the limited number of data sets, the data set’s chemical diversity also limits the generalizability of ML predictions.248 One more thing to take care of is the inconsistency of data sets, as they may obtain from different levels of theory.104 A potential solution to the limited numbers of the data set is a hybrid approach based on an active learning framework with an uncertainty quantification, where training data are generated on-the-fly whenever needed.249
Next-Generation MLP
In Behler’s recent review, he pointed out that the next generation of MLP focus on the consideration of global charge distribution. The current construction of MLP depends on the local environments inside a cutoff sphere. However, in some cases, the atomic charge can strongly relate to structural changes very far away, of which current MLP fails to produce an accurate result.153 Another challenging area is the interpretability of MLP. Some well-developed interpretable ML models include TinNet,250 SISSO,251 and iGAM,252 which have been applied to study the surface alloy and doped catalysts, like its prediction of the structural stability, to study adsorption behavior, to screen high-performance catalysts, etc.252−256 The advantage of these methods is the physical transparency, as the obtained ML models are the explicit and analytic functions of input physical quantities, which can shine some light on the underlying structure–property relationship. Moreover, the transferability of MLP may be improved by active learning that can achieve high accuracy and data efficiency with fixed training data.257
Path to a Long-Term and Large-Scale Simulation
There is certainly a need for even larger and longer simulations at heterogeneous interfaces on the experimental scales, particularly with ab initio accuracy. Advances in both methods and software/hardware were pivotal to be developed successfully. For example, the MLP software DeePMD-kit was applied in the millisecond-level MD simulation of 100 million atoms water and Cu system.258 Moreover, the ability of a graphics processing unit (GPU)-based architecture to improve computational efficiency was demonstrated by simulating a massive 200 million atoms and was adopted in the popular MD software like GROMACS and NAMD.152,259,260 With the help of these emerging techniques, the concept of hybrid machine learning/molecular mechanics (ML/MM) methods (proposed by Aspuru-Guzik) may be achieved before long, which can substantially accelerate the QM/MM simulations for the substantially larger systems and longer time scales, from microscopic (nm/ns) to macroscopic (μm/μs) systems.249 The development of a facile multiscale modeling method with the integration of ML still requires a continuous effort in the coming years.
Summary and Outlook
The fast-developing operando characterization allows the real-time detection of the dynamic structure of the catalyst, reaction intermediates, and catalytic products, which is helpful to precisely understand the catalytic mechanism and rational design of high-performance catalysts. For a complete understanding of a working catalyst system at an atomic scale, a theoretical investigation that simulates operando conditions is a must, which can be achieved through a multiscale computational modeling approach. In this Perspective, we describe various modeling approaches and machine learning techniques that step toward operando modeling. GO mostly helps in finding the most stable structure of the heterogeneous catalyst, while ab initio techniques can include the dynamic effect of the reaction environment. Microkinetic modeling can help explain the stepwise kinetics and build up the full reaction network, while the ML technique can serve as a surrogate model to accelerate the time-consuming simulation by orders of magnitude.
The real active intermediate under the interaction with the reaction environment species plays an important role in the exploration of the catalytic mechanism, which should not be ignored. We hope that this short Perspective can provide a useful guide to how to model the catalyst behavior on an experimentally spatiotemporal scale under true reaction conditions. Indeed, there are still significant challenges on the way to operando modeling: the limited spatiotemporal resolutions of operando techniques, the huge computational cost of ab initio calculation for a long-time/large-scale simulation, the unclear extremely dynamic nature of the catalyst, etc. However, considering the fast progress in recent years, especially the huge improvement of computational efficiency with the help of the ML technique, we can expect that, soon, it will become a new normal to describe the catalyst behavior in a spatiotemporal manner under true reaction conditions, with an affordable computational cost. With structure–activity relationships and reaction mechanisms being revealing, the rational design of highly efficient heterogeneous catalysts is expected to be greatly promoted.
Acknowledgments
This work is supported by the National Science Foundation of China (22121004, U1862207, and 2212100031) and the Program of Introducing Talents of Discipline to Universities (No. BP0618007).
Glossary
Abbreviations
- AITD
ab initio thermodynamics
- AIMD
ab initio molecular dynamics
- BPGA
Birmingham parallel genetic algorithm
- CRR
CO2 reduction reaction
- CORR
CO reduction reactions
- EXAFS
X-ray absorption fine structure
- EPR
electron paramagnetic resonance
- GA
genetic algorithm
- GO
global optimization
- GM
global minimum
- HER
hydrogen evolution reaction
- IR
infrared
- kMC
kinetic Monte Carlo
- ML
machine learning
- MLP
machine learning potentials
- MOF
metal–organic framework
- NNP
neural network potentials
- NEB
nudged elastic band
- OER
oxygen evolution reaction
- PES
potential energy surface
- QM/MM
quantum mechanics/molecular mechanics
- RGO
reactive global optimization
- RMG
reaction mechanism generator
- S/TEM
scanning/transmission electron microscopes
- SSW
stochastic surface walking
- XRD
X-ray diffraction
- XANES
X-ray absorption near edge structure
- XPS
X-ray photoelectron spectroscopy
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Li L.; Chang X.; Lin X.; Zhao Z.-J.; Gong J. Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 2020, 49 (22), 8156–8178. 10.1039/D0CS00795A. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Zandkarimi B.; Alexandrova A. N. Ensembles of Metastable States Govern Heterogeneous Catalysis on Dynamic Interfaces. Acc. Chem. Res. 2020, 53 (2), 447–458. 10.1021/acs.accounts.9b00531. [DOI] [PubMed] [Google Scholar]
- Du H.; Fan J.; Miao C.; Gao M.; Liu Y.; Li D.; Feng J. Recent Advances in Constructing Interfacial Active Catalysts Based on Layered Double Hydroxides and Their Catalytic Mechanisms. Trans. Tianjin Univ. 2021, 27 (1), 24–41. 10.1007/s12209-020-00277-1. [DOI] [Google Scholar]
- Zhu Y.; Wang J.; Chu H.; Chu Y.-C.; Chen H. M. In Situ/Operando Studies for Designing Next-Generation Electrocatalysts. ACS Energy Lett. 2020, 5 (4), 1281–1291. 10.1021/acsenergylett.0c00305. [DOI] [Google Scholar]
- Meirer F.; Weckhuysen B. M. Spatial and temporal exploration of heterogeneous catalysts with synchrotron radiation. Nature Reviews Materials 2018, 3 (9), 324–340. 10.1038/s41578-018-0044-5. [DOI] [Google Scholar]
- Choi J. I. J.; Kim T.-S.; Kim D.; Lee S. W.; Park J. Y. Operando Surface Characterization on Catalytic and Energy Materials from Single Crystals to Nanoparticles. ACS Nano 2020, 14 (12), 16392–16413. 10.1021/acsnano.0c07549. [DOI] [PubMed] [Google Scholar]
- Grajciar L.; Heard C. J.; Bondarenko A. A.; Polynski M. V.; Meeprasert J.; Pidko E. A.; Nachtigall P. Towards operando computational modeling in heterogeneous catalysis. Chem. Soc. Rev. 2018, 47 (22), 8307–8348. 10.1039/C8CS00398J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Liu Y. Unveiling the Active Structure of Single Nickel Atom Catalysis: Critical Roles of Charge Capacity and Hydrogen Bonding. J. Am. Chem. Soc. 2020, 142 (12), 5773–5777. 10.1021/jacs.9b13872. [DOI] [PubMed] [Google Scholar]
- Weckhuysen B. M. Snapshots of a working catalyst: possibilities and limitations of in situ spectroscopy in the field of heterogeneous catalysis. Chem. Commun. (Cambridge, U. K.) 2002, (2), 97–110. 10.1039/b107686h. [DOI] [PubMed] [Google Scholar]
- Weckhuysen B. M. Determining the active site in a catalytic process: Operando spectroscopy is more than a buzzword. Phys. Chem. Chem. Phys. 2003, 5 (20), 4351–4360. 10.1039/b309650p. [DOI] [Google Scholar]
- Bañares M. A. Operando methodology: combination of in situ spectroscopy and simultaneous activity measurements under catalytic reaction conditions. Catal. Today 2005, 100 (1–2), 71–77. 10.1016/j.cattod.2004.12.017. [DOI] [Google Scholar]
- Guerrero-Pérez M. O.; Bañares M. A. Operando Raman study of alumina-supported Sb–V–O catalyst during propane ammoxidation to acrylonitrile with on-line activity measurement. Chem. Commun. (Cambridge, U. K.) 2002, (12), 1292–1293. 10.1039/b202556f. [DOI] [PubMed] [Google Scholar]
- Bañares M. A.; Wachs I. E. Molecular structures of supported metal oxide catalysts under different environments. J. Raman Spectrosc. 2002, 33 (5), 359–380. 10.1002/jrs.866. [DOI] [Google Scholar]
- Bañares M. A.; Guerrero-Pérez M. O.; Fierro J. L. G.; Cortez G. G. Raman spectroscopy during catalytic operations with on-line activity measurement (operando spectroscopy): a method for understanding the active centres of cations supported on porous materials. J. Mater. Chem. 2002, 12 (11), 3337–3342. 10.1039/B204494C. [DOI] [Google Scholar]
- Goetze J.; Yarulina I.; Gascon J.; Kapteijn F.; Weckhuysen B. M. Revealing Lattice Expansion of Small-Pore Zeolite Catalysts during the Methanol-to-Olefins Process Using Combined Operando X-ray Diffraction and UV–vis Spectroscopy. ACS Catal. 2018, 8 (3), 2060–2070. 10.1021/acscatal.7b04129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zakharov D.; Zhao S.; Tappero R.; Jung U.; Elsen A.; Baumann P.; Nuzzo R. G.; Stach E. A.; Frenkel A. I. Complex structural dynamics of nanocatalysts revealed in Operando conditions by correlated imaging and spectroscopy probes. Nat. Commun. 2015, 6 (1), 7583. 10.1038/ncomms8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman T.; Geitenbeek R. G.; Wondergem C. S.; van der Stam W.; Weckhuysen B. M. Operando Nanoscale Sensors in Catalysis: All Eyes on Catalyst Particles. ACS Nano 2020, 14 (4), 3725–3735. 10.1021/acsnano.9b09834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman T.; Geitenbeek R. G.; Whiting G. T.; Weckhuysen B. M. Operando monitoring of temperature and active species at the single catalyst particle level. Nat. Catal. 2019, 2 (11), 986–996. 10.1038/s41929-019-0352-1. [DOI] [Google Scholar]
- Qian Q.; Vogt C.; Mokhtar M.; Asiri A. M.; Al-Thabaiti S. A.; Basahel S. N.; Ruiz-Martínez J.; Weckhuysen B. M. Combined Operando UV/Vis/IR Spectroscopy Reveals the Role of Methoxy and Aromatic Species during the Methanol-to-Olefins Reaction over H-SAPO-34. ChemCatChem 2014, 6 (12), 3396–3408. 10.1002/cctc.201402714. [DOI] [Google Scholar]
- Cats K. H.; Weckhuysen B. M. Combined Operando X-ray Diffraction/Raman Spectroscopy of Catalytic Solids in the Laboratory: The Co/TiO2 Fischer–Tropsch Synthesis Catalyst Showcase. ChemCatChem 2016, 8 (8), 1531–1542. 10.1002/cctc.201600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paalanen P. P.; van Vreeswijk S. H.; Weckhuysen B. M. Combined In Situ X-ray Powder Diffractometry/Raman Spectroscopy of Iron Carbide and Carbon Species Evolution in Fe(-Na–S)/α-Al2O3 Catalysts during Fischer–Tropsch Synthesis. ACS Catal. 2020, 10 (17), 9837–9855. 10.1021/acscatal.0c01851. [DOI] [Google Scholar]
- Sun G.; Sautet P. Metastable Structures in Cluster Catalysis from First-Principles: Structural Ensemble in Reaction Conditions and Metastability Triggered Reactivity. J. Am. Chem. Soc. 2018, 140 (8), 2812–2820. 10.1021/jacs.7b11239. [DOI] [PubMed] [Google Scholar]
- Tappan B. A.; Barim G.; Kwok J. C.; Brutchey R. L. Utilizing Diselenide Precursors toward Rationally Controlled Synthesis of Metastable CuInSe2 Nanocrystals. Chem. Mater. 2018, 30 (16), 5704–5713. 10.1021/acs.chemmater.8b02205. [DOI] [Google Scholar]
- Zou Y.; Sun C.; Gong W.; Yang X.; Huang X.; Yang T.; Lu W.; Jiang J. Morphology-Controlled Synthesis of Hybrid Nanocrystals via a Selenium-Mediated Strategy with Ligand Shielding Effect: The Case of Dual Plasmonic Au–Cu2–xSe. ACS Nano 2017, 11 (4), 3776–3785. 10.1021/acsnano.6b08641. [DOI] [PubMed] [Google Scholar]
- Glass C. W.; Oganov A. R.; Hansen N. USPEX—Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175 (11–12), 713–720. 10.1016/j.cpc.2006.07.020. [DOI] [Google Scholar]
- Lonie D. C.; Zurek E. XtalOpt: An open-source evolutionary algorithm for crystal structure prediction. Comput. Phys. Commun. 2011, 182 (2), 372–387. 10.1016/j.cpc.2010.07.048. [DOI] [Google Scholar]
- Johnston R. L. Evolving better nanoparticles: Genetic algorithms for optimizing cluster geometries. Dalton Trans. 2003, (22), 4193–4207. 10.1039/b305686d. [DOI] [Google Scholar]
- Davis J. B. A.; Horswell S. L.; Johnston R. L. Application of a Parallel Genetic Algorithm to the Global Optimization of Gas-Phase and Supported Gold–Iridium Sub-Nanoalloys. J. Phys. Chem. C 2016, 120 (7), 3759–3765. 10.1021/acs.jpcc.5b10226. [DOI] [Google Scholar]
- Davis J. B. A.; Shayeghi A.; Horswell S. L.; Johnston R. L. The Birmingham parallel genetic algorithm and its application to the direct DFT global optimization of IrN(N = 10–20) clusters. Nanoscale 2015, 7 (33), 14032–14038. 10.1039/C5NR03774C. [DOI] [PubMed] [Google Scholar]
- Vilhelmsen L. B.; Hammer B. A genetic algorithm for first principles global structure optimization of supported nano structures. J. Chem. Phys. 2014, 141 (4), 044711. 10.1063/1.4886337. [DOI] [PubMed] [Google Scholar]
- Hjorth Larsen A.; Jørgen Mortensen J.; Blomqvist J.; Castelli I. E.; Christensen R.; Dułak M.; Friis J.; Groves M. N.; Hammer B.; Hargus C.; Hermes E. D.; Jennings P. C.; Bjerre Jensen P.; Kermode J.; Kitchin J. R.; Leonhard Kolsbjerg E.; Kubal J.; Kaasbjerg K.; Lysgaard S.; Bergmann Maronsson J.; Maxson T.; Olsen T.; Pastewka L.; Peterson A.; Rostgaard C.; Schiøtz J.; Schütt O.; Strange M.; Thygesen K. S.; Vegge T.; Vilhelmsen L.; Walter M.; Zeng Z.; Jacobsen K. W. The atomic simulation environment—a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29 (27), 273002. 10.1088/1361-648X/aa680e. [DOI] [PubMed] [Google Scholar]
- Sierka M.; Todorova T. K.; Sauer J.; Kaya S.; Stacchiola D.; Weissenrieder J.; Shaikhutdinov S.; Freund H.-J. Oxygen adsorption on Mo(112) surface studied by ab initio genetic algorithm and experiment. J. Chem. Phys. 2007, 126 (23), 234710. 10.1063/1.2743427. [DOI] [PubMed] [Google Scholar]
- Bisbo M. K.; Hammer B. Efficient Global Structure Optimization with a Machine-Learned Surrogate Model. Phys. Rev. Lett. 2020, 124 (8), 086102. 10.1103/PhysRevLett.124.086102. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Jiang X.; Li J.; Li S.; Wang L. PDECO: Parallel differential evolution for clusters optimization. J. Comput. Chem. 2013, 34 (12), 1046–1059. 10.1002/jcc.23235. [DOI] [PubMed] [Google Scholar]
- Arrigoni M.; Madsen G. K. H. Evolutionary computing and machine learning for discovering of low-energy defect configurations. npj Comput. Mater. 2021, 7 (1), 71. 10.1038/s41524-021-00537-1. [DOI] [Google Scholar]
- Huang S. D.; Shang C.; Kang P. L.; Zhang X. J.; Liu Z. P. LASP: Fast global potential energy surface exploration. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2019, 9 (6), e1415 10.1002/wcms.1415. [DOI] [Google Scholar]
- Wang Y.; Lv J.; Zhu L.; Ma Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183 (10), 2063–2070. 10.1016/j.cpc.2012.05.008. [DOI] [Google Scholar]
- Zhang J.; Glezakou V.-A.; Rousseau R.; Nguyen M.-T. NWPEsSe: An Adaptive-Learning Global Optimization Algorithm for Nanosized Cluster Systems. J. Chem. Theory Comput. 2020, 16 (6), 3947–3958. 10.1021/acs.jctc.9b01107. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Dolg M. ABCluster: the artificial bee colony algorithm for cluster global optimization. Phys. Chem. Chem. Phys. 2015, 17 (37), 24173–24181. 10.1039/C5CP04060D. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zhao Y. F.; Zhang Y. Y.; Li J. TGMin: An efficient global minimum searching program for free and surface-supported clusters. J. Comput. Chem. 2018, 1105–1112. 10.1002/jcc.25649. [DOI] [PubMed] [Google Scholar]
- Todorović M.; Gutmann M. U.; Corander J.; Rinke P. Bayesian inference of atomistic structure in functional materials. npj Comput. Mater. 2019, 5, 35. 10.1038/s41524-019-0175-2. [DOI] [Google Scholar]
- Carr S.; Garnett R.; Lo C.. BASC: Applying Bayesian Optimization to the Search for Global Minima on Potential Energy Surfaces. In Proceedings of The 33rd International Conference on Machine Learning, Maria Florina B.; Kilian Q. W., Eds.; Proceedings of Machine Learning Research; MLResearch Press, 2016; Vol. 48, pp 898–907.
- Jäger M.; Schäfer R.; Johnston R. L. First principles global optimization of metal clusters and nanoalloys. Adv. Phys.: X 2018, 3 (1), 1516514. 10.1080/23746149.2018.1516514. [DOI] [Google Scholar]
- Zhang J.; Glezakou V. A. Global optimization of chemical cluster structures: Methods, applications, and challenges. Int. J. Quantum Chem. 2021, 121 (7), e26553 10.1002/qua.26553. [DOI] [Google Scholar]
- Khatun M.; Majumdar R. S.; Anoop A. A Global Optimizer for Nanoclusters. Front. Chem. 2019, 7, 644. 10.3389/fchem.2019.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandkarimi B.; Alexandrova A. N. Surface-supported cluster catalysis: Ensembles of metastable states run the show. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2019, 9 (6), e1420 10.1002/wcms.1420. [DOI] [Google Scholar]
- Vilhelmsen L. B.; Walton K. S.; Sholl D. S. Structure and Mobility of Metal Clusters in MOFs: Au, Pd, and AuPd Clusters in MOF-74. J. Am. Chem. Soc. 2012, 134 (30), 12807–12816. 10.1021/ja305004a. [DOI] [PubMed] [Google Scholar]
- Lim D.-H.; Wilcox J. Mechanisms of the Oxygen Reduction Reaction on Defective Graphene-Supported Pt Nanoparticles from First-Principles. J. Phys. Chem. C 2012, 116 (5), 3653–3660. 10.1021/jp210796e. [DOI] [Google Scholar]
- Ren Z.; Liu N.; Chen B.; Li J.; Mei D. Theoretical Investigation of the Structural Stabilities of Ceria Surfaces and Supported Metal Nanocluster in Vapor and Aqueous Phases. J. Phys. Chem. C 2018, 122 (9), 4828–4840. 10.1021/acs.jpcc.7b10208. [DOI] [Google Scholar]
- Martinez U.; Vilhelmsen L. B.; Kristoffersen H. H.; Stausholm-Møller J.; Hammer B. Steps on rutile TiO2(110): Active sites for water and methanol dissociation. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84 (20), 205434. 10.1103/PhysRevB.84.205434. [DOI] [Google Scholar]
- Barcaro G.; Causà M.; Fortunelli A. A comparison between the absorption properties of the regular and F s -defected MgO (100) surface. Theor. Chem. Acc. 2007, 118 (4), 807–812. 10.1007/s00214-007-0324-4. [DOI] [Google Scholar]
- Barcaro G.; Fortunelli A. Small Au clusters on a defective MgO(100) surface. Chem. Phys. Lett. 2008, 457 (1–3), 143–147. 10.1016/j.cplett.2008.03.077. [DOI] [Google Scholar]
- Barcaro G.; Fortunelli A. Rotational Invariance and Double Frustration in the Structures of Gold Clusters Growing around the Fs-Defected MgO (100) Surface. J. Phys. Chem. B 2006, 110 (42), 21021–21027. 10.1021/jp064519e. [DOI] [PubMed] [Google Scholar]
- Barcaro G.; Aprà E.; Fortunelli A. Structure of Ag Clusters Grown on Fs-Defect Sites of an MgO(1 0 0) Surface. Chem. - Eur. J. 2007, 13 (22), 6408–6418. 10.1002/chem.200601796. [DOI] [PubMed] [Google Scholar]
- Barcaro G.; Fortunelli A. Magic silver cluster on a MgO(100) terrace with defects. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 76 (16), 165412. 10.1103/PhysRevB.76.165412. [DOI] [Google Scholar]
- Barcaro G.; Fortunelli A. A Magic Pd-Ag Binary Cluster on the Fs-Defected MgO(100) Surface. J. Phys. Chem. C 2007, 111 (30), 11384–11389. 10.1021/jp072263m. [DOI] [Google Scholar]
- Sierka M.; Todorova T. K.; Kaya S.; Stacchiola D.; Weissenrieder J.; Lu J.; Gao H.; Shaikhutdinov S.; Freund H.-J.; Sauer J. Interplay between theory and experiment in the quest for silica with reduced dimensionality grown on a Mo(112) surface. Chem. Phys. Lett. 2006, 424 (1–3), 115–119. 10.1016/j.cplett.2006.04.072. [DOI] [Google Scholar]
- Kaya S.; Weissenrieder J.; Stacchiola D.; Todorova T. K.; Sierka M.; Sauer J.; Shaikhutdinov S.; Freund H. J. Formation of one-dimensional molybdenum oxide on Mo(112). Surf. Sci. 2008, 602 (21), 3338–3342. 10.1016/j.susc.2008.09.010. [DOI] [Google Scholar]
- Liu S.; Zong J.; Zhao Z.-J.; Gong J. Exploring the initial oxidation of Pt, Pt3Ni, Pt3Au (111) surfaces: a genetic algorithm based global optimization with density functional theory. Green Chem. Eng. 2020, 1 (1), 56–62. 10.1016/j.gce.2020.09.006. [DOI] [Google Scholar]
- Negreiros F. R.; Halder A.; Yin C.; Singh A.; Barcaro G.; Sementa L.; Tyo E. C.; Pellin M. J.; Bartling S.; Meiwes-Broer K.-H.; Seifert S.; Sen P.; Nigam S.; Majumder C.; Fukui N.; Yasumatsu H.; Vajda S.; Fortunelli A. Bimetallic Ag-Pt Sub-nanometer Supported Clusters as Highly Efficient and Robust Oxidation Catalysts. Angew. Chem., Int. Ed. 2018, 57 (5), 1209–1213. 10.1002/anie.201709784. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Su Y.-Q.; Hensen E. J. M.; Vlachos D. G. Finite-Temperature Structures of Supported Subnanometer Catalysts Inferred via Statistical Learning and Genetic Algorithm-Based Optimization. ACS Nano 2020, 14 (10), 13995–14007. 10.1021/acsnano.0c06472. [DOI] [PubMed] [Google Scholar]
- Paleico M. L.; Behler J. Global optimization of copper clusters at the ZnO(1010) surface using a DFT-based neural network potential and genetic algorithms. J. Chem. Phys. 2020, 153 (5), 054704. 10.1063/5.0014876. [DOI] [PubMed] [Google Scholar]
- Selli D.; Fazio G.; Di Valentin C. Modelling realistic TiO2 nanospheres: A benchmark study of SCC-DFTB against hybrid DFT. J. Chem. Phys. 2017, 147 (16), 164701. 10.1063/1.4994165. [DOI] [PubMed] [Google Scholar]
- Selli D.; Fazio G.; Di Valentin C. Using Density Functional Theory to Model Realistic TiO2 Nanoparticles, Their Photoactivation and Interaction with Water. Catalysts 2017, 7 (12), 357. 10.3390/catal7120357. [DOI] [Google Scholar]
- Shirai K.; Fazio G.; Sugimoto T.; Selli D.; Ferraro L.; Watanabe K.; Haruta M.; Ohtani B.; Kurata H.; Di Valentin C.; Matsumoto Y. Water-Assisted Hole Trapping at the Highly Curved Surface of Nano-TiO2 Photocatalyst. J. Am. Chem. Soc. 2018, 140 (4), 1415–1422. 10.1021/jacs.7b11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi C.; Datteo M.; Kaviani M.; Selli D.; Di Valentin C. Unraveling Dynamical and Light Effects on Functionalized Titanium Dioxide Nanoparticles for Bioconjugation. J. Phys. Chem. C 2019, 123 (15), 10130–10144. 10.1021/acs.jpcc.9b01385. [DOI] [Google Scholar]
- Soria F. A.; Di Valentin C. Reactive molecular dynamics simulations of hydration shells surrounding spherical TiO2 nanoparticles: implications for proton-transfer reactions. Nanoscale 2021, 13 (7), 4151–4166. 10.1039/D0NR07503E. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Gong X. Genetic algorithm aided density functional theory simulations unravel the kinetic nature of Au(100) in catalytic CO oxidation. Chin. Chem. Lett. 2019, 30 (6), 1346–1350. 10.1016/j.cclet.2018.12.025. [DOI] [Google Scholar]
- Negreiros F. R.; Aprà E.; Barcaro G.; Sementa L.; Vajda S.; Fortunelli A. A first-principles theoretical approach to heterogeneous nanocatalysis. Nanoscale 2012, 4 (4), 1208–1219. 10.1039/C1NR11051A. [DOI] [PubMed] [Google Scholar]
- Halder A.; Curtiss L. A.; Fortunelli A.; Vajda S. Perspective: Size selected clusters for catalysis and electrochemistry. J. Chem. Phys. 2018, 148 (11), 110901. 10.1063/1.5020301. [DOI] [PubMed] [Google Scholar]
- Lei Y.; Mehmood F.; Lee S.; Greeley J.; Lee B.; Seifert S.; Winans R. E.; Elam J. W.; Meyer R. J.; Redfern P. C.; Teschner D.; Schlogl R.; Pellin M. J.; Curtiss L. A.; Vajda S. Increased Silver Activity for Direct Propylene Epoxidation via Subnanometer Size Effects. Science 2010, 328 (5975), 224–228. 10.1126/science.1185200. [DOI] [PubMed] [Google Scholar]
- Negreiros F. R.; Sementa L.; Barcaro G.; Vajda S.; Aprá E.; Fortunelli A. CO Oxidation by Subnanometer AgxAu3–x Supported Clusters via Density Functional Theory Simulations. ACS Catal. 2012, 2 (9), 1860–1864. 10.1021/cs300275v. [DOI] [Google Scholar]
- Sierka M. Synergy between theory and experiment in structure resolution of low-dimensional oxides. Prog. Surf. Sci. 2010, 85 (9–12), 398–434. 10.1016/j.progsurf.2010.07.004. [DOI] [Google Scholar]
- Handzlik J.; Sautet P. Active sites of olefin metathesis on molybdena-alumina system: A periodic DFT study. J. Catal. 2008, 256 (1), 1–14. 10.1016/j.jcat.2008.02.016. [DOI] [Google Scholar]
- Zhai H.; Alexandrova A. N. Local Fluxionality of Surface-Deposited Cluster Catalysts: The Case of Pt7 on Al2O3. J. Phys. Chem. Lett. 2018, 9 (7), 1696–1702. 10.1021/acs.jpclett.8b00379. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Li Y.; Liu D.; Liu J.; Liu Y.-M.; Zakharov D. N.; Wu Q.; Orlov A.; Gewirth A. A.; Stach E. A.; Nuzzo R. G.; Frenkel A. I. Multimodal Study of the Speciations and Activities of Supported Pd Catalysts During the Hydrogenation of Ethylene. J. Phys. Chem. C 2017, 121 (34), 18962–18972. 10.1021/acs.jpcc.7b06270. [DOI] [Google Scholar]
- Li Y.; Kottwitz M.; Vincent J. L.; Enright M. J.; Liu Z.; Zhang L.; Huang J.; Senanayake S. D.; Yang W.-C. D.; Crozier P. A.; Nuzzo R. G.; Frenkel A. I. Dynamic structure of active sites in ceria-supported Pt catalysts for the water gas shift reaction. Nat. Commun. 2021, 12, 914. 10.1038/s41467-021-21132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandkarimi B.; Sun G.; Halder A.; Seifert S.; Vajda S.; Sautet P.; Alexandrova A. N. Interpreting the Operando XANES of Surface-Supported Subnanometer Clusters: When Fluxionality, Oxidation State, and Size Effect Fight. J. Phys. Chem. C 2020, 124 (18), 10057–10066. 10.1021/acs.jpcc.0c02823. [DOI] [Google Scholar]
- Sun G.; Alexandrova A. N.; Sautet P. Structural Rearrangements of Subnanometer Cu Oxide Clusters Govern Catalytic Oxidation. ACS Catal. 2020, 10 (9), 5309–5317. 10.1021/acscatal.0c00824. [DOI] [Google Scholar]
- Zhang Z.; Jimenez-Izal E.; Hermans I.; Alexandrova A. N. Dynamic Phase Diagram of Catalytic Surface of Hexagonal Boron Nitride under Conditions of Oxidative Dehydrogenation of Propane. J. Phys. Chem. Lett. 2019, 10 (1), 20–25. 10.1021/acs.jpclett.8b03373. [DOI] [PubMed] [Google Scholar]
- Zandkarimi B.; Alexandrova A. N. Dynamics of Subnanometer Pt Clusters Can Break the Scaling Relationships in Catalysis. J. Phys. Chem. Lett. 2019, 10 (3), 460–467. 10.1021/acs.jpclett.8b03680. [DOI] [PubMed] [Google Scholar]
- Zandkarimi B.; Alexandrova A. N. Can Fluxionality of Subnanometer Cluster Catalysts Solely Cause Non-Arrhenius Behavior in Catalysis?. J. Phys. Chem. C 2020, 124 (36), 19556–19562. 10.1021/acs.jpcc.0c04136. [DOI] [Google Scholar]
- Zandkarimi B.; Poths P.; Alexandrova A. N. When Fluxionality Beats Size Selection: Acceleration of Ostwald Ripening of Sub-Nano Clusters. Angew. Chem., Int. Ed. 2021, 60, 11973. 10.1002/anie.202100107. [DOI] [PubMed] [Google Scholar]
- Zhai H.; Alexandrova A. N. Fluxionality of Catalytic Clusters: When It Matters and How to Address It. ACS Catal. 2017, 7 (3), 1905–1911. 10.1021/acscatal.6b03243. [DOI] [Google Scholar]
- Bandow B.; Hartke B. Larger Water Clusters with Edges and Corners on Their Way to Ice: Structural Trends Elucidated with an Improved Parallel Evolutionary Algorithm. J. Phys. Chem. A 2006, 110 (17), 5809–5822. 10.1021/jp060512l. [DOI] [PubMed] [Google Scholar]
- Alexandrova A. N. H·(H2O)nClusters: Microsolvation of the Hydrogen Atom via Molecular ab Initio Gradient Embedded Genetic Algorithm (GEGA). J. Phys. Chem. A 2010, 114 (48), 12591–12599. 10.1021/jp1092543. [DOI] [PubMed] [Google Scholar]
- Basdogan Y.; Groenenboom M. C.; Henderson E.; De S.; Rempe S. B.; Keith J. A. Machine Learning-Guided Approach for Studying Solvation Environments. J. Chem. Theory Comput. 2020, 16 (1), 633–642. 10.1021/acs.jctc.9b00605. [DOI] [PubMed] [Google Scholar]
- Yang H.; Wong M. W. Automatic Conformational Search of Transition States for Catalytic Reactions Using Genetic Algorithm. J. Phys. Chem. A 2019, 123 (47), 10303–10314. 10.1021/acs.jpca.9b09543. [DOI] [PubMed] [Google Scholar]
- Ludwig T.; Gauthier J. A.; Brown K. S.; Ringe S.; Nørskov J. K.; Chan K. Solvent–Adsorbate Interactions and Adsorbate-Specific Solvent Structure in Carbon Dioxide Reduction on a Stepped Cu Surface. J. Phys. Chem. C 2019, 123 (10), 5999–6009. 10.1021/acs.jpcc.8b11571. [DOI] [Google Scholar]
- Basdogan Y.; Keith J. A. A paramedic treatment for modeling explicitly solvated chemical reaction mechanisms. Chem. Sci. 2018, 9 (24), 5341–5346. 10.1039/C8SC01424H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowski B. C.; Bates J. S.; Gounder R.; Greeley J. Defect-Mediated Ordering of Condensed Water Structures in Microporous Zeolites. Angew. Chem. 2019, 131 (46), 16574–16578. 10.1002/ange.201908151. [DOI] [PubMed] [Google Scholar]
- Deshpande S.; Greeley J. First-Principles Analysis of Coverage, Ensemble, and Solvation Effects on Selectivity Trends in NO Electroreduction on Pt3Sn Alloys. ACS Catal. 2020, 10 (16), 9320–9327. 10.1021/acscatal.0c01380. [DOI] [Google Scholar]
- Bates J. S.; Bukowski B. C.; Greeley J.; Gounder R. Structure and solvation of confined water and water–ethanol clusters within microporous Brønsted acids and their effects on ethanol dehydration catalysis. Chem. Sci. 2020, 11 (27), 7102–7122. 10.1039/D0SC02589E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieme J.; Lejaeghere K.; Kresse G.; Van Speybroeck V. Tuning the balance between dispersion and entropy to design temperature-responsive flexible metal-organic frameworks. Nat. Commun. 2018, 9, 4899. 10.1038/s41467-018-07298-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; Zhang C.; Cheng J. Origin of Asymmetric Electric Double Layers at Electrified Oxide/Electrolyte Interfaces. J. Phys. Chem. Lett. 2021, 12 (19), 4616–4622. 10.1021/acs.jpclett.1c00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le J.-B.; Chen A.; Li L.; Xiong J.-F.; Lan J.; Liu Y.-P.; Iannuzzi M.; Cheng J. Modeling Electrified Pt(111)-Had/Water Interfaces from Ab Initio Molecular Dynamics. JACS Au 2021, 1 (5), 569–577. 10.1021/jacsau.1c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le J.-B.; Fan Q.-Y.; Li J.-Q.; Cheng J. Molecular origin of negative component of Helmholtz capacitance at electrified Pt(111)/water interface. Sci. Adv. 2020, 6 (41), eabb1219 10.1126/sciadv.abb1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.-Y.; Le J.-B.; Wang Y.-H.; Chen S.; Yang Z.-L.; Li J.-F.; Cheng J.; Tian Z.-Q. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 2019, 18 (7), 697–701. 10.1038/s41563-019-0356-x. [DOI] [PubMed] [Google Scholar]
- Le J.-B.; Cheng J. Modeling electrified metal/water interfaces from ab initio molecular dynamics: Structure and Helmholtz capacitance. Current Opinion in Electrochemistry 2021, 27, 100693. 10.1016/j.coelec.2021.100693. [DOI] [Google Scholar]
- Le J.; Iannuzzi M.; Cuesta A.; Cheng J. Determining Potentials of Zero Charge of Metal Electrodes versus the Standard Hydrogen Electrode from Density-Functional-Theory-Based Molecular Dynamics. Phys. Rev. Lett. 2017, 119, 016801. 10.1103/PhysRevLett.119.016801. [DOI] [PubMed] [Google Scholar]
- Lee M.-S.; Peter McGrail B.; Rousseau R.; Glezakou V.-A. Structure, dynamics and stability of water/scCO2/mineral interfaces from ab initio molecular dynamics simulations. Sci. Rep. 2015, 5, 14857. 10.1038/srep14857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron J. A.; Morikawa Y.; Mavrikakis M. Ab initio molecular dynamics of solvation effects on reactivity at electrified interfaces. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (34), E4937–E4945. 10.1073/pnas.1604590113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcuk S.; Selloni A. Facet-dependent trapping and dynamics of excess electrons at anatase TiO2 surfaces and aqueous interfaces. Nat. Mater. 2016, 15 (10), 1107–1112. 10.1038/nmat4672. [DOI] [PubMed] [Google Scholar]
- Li Q.; Ouyang Y.; Lu S.; Bai X.; Zhang Y.; Shi L.; Ling C.; Wang J. Perspective on theoretical methods and modeling relating to electro-catalysis processes. Chem. Commun. (Cambridge, U. K.) 2020, 56 (69), 9937–9949. 10.1039/D0CC02998J. [DOI] [PubMed] [Google Scholar]
- Scanlon D. O.; Dunnill C. W.; Buckeridge J.; Shevlin S. A.; Logsdail A. J.; Woodley S. M.; Catlow C. R. A.; Powell M. J.; Palgrave R. G.; Parkin I. P.; Watson G. W.; Keal T. W.; Sherwood P.; Walsh A.; Sokol A. A. Band alignment of rutile and anatase TiO2. Nat. Mater. 2013, 12 (9), 798–801. 10.1038/nmat3697. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Wang L.; Merinov B. V.; Goddard W. A. Explanation of Dramatic pH-Dependence of Hydrogen Binding on Noble Metal Electrode: Greatly Weakened Water Adsorption at High pH. J. Am. Chem. Soc. 2018, 140 (25), 7787–7790. 10.1021/jacs.8b04006. [DOI] [PubMed] [Google Scholar]
- He Y.; Liu J.-C.; Luo L.; Wang Y.-G.; Zhu J.; Du Y.; Li J.; Mao S. X.; Wang C. Size-dependent dynamic structures of supported gold nanoparticles in CO oxidation reaction condition. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (30), 7700–7705. 10.1073/pnas.1800262115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bučko T.; Benco L.; Hafner J.; Ángyán J. G. Monomolecular cracking of propane over acidic chabazite: An ab initio molecular dynamics and transition path sampling study. J. Catal. 2011, 279 (1), 220–228. 10.1016/j.jcat.2011.01.022. [DOI] [Google Scholar]
- Schnur S.; Groß A. Challenges in the first-principles description of reactions in electrocatalysis. Catal. Today 2011, 165 (1), 129–137. 10.1016/j.cattod.2010.11.071. [DOI] [Google Scholar]
- Gomes J.; Head-Gordon M.; Bell A. T. Reaction Dynamics of Zeolite-Catalyzed Alkene Methylation by Methanol. J. Phys. Chem. C 2014, 118 (37), 21409–21419. 10.1021/jp502804q. [DOI] [Google Scholar]
- Kuliaev P. O.; Pidko E. A. Operando Modeling of Multicomponent Reactive Solutions in Homogeneous Catalysis: from Non-standard Free Energies to Reaction Network Control. ChemCatChem 2020, 12 (3), 795–802. 10.1002/cctc.201901911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter K.; Scheffler M. First-Principles Atomistic Thermodynamics for Oxidation Catalysis: Surface Phase Diagrams and Catalytically Interesting Regions. Phys. Rev. Lett. 2003, 90 (4), 046103. 10.1103/PhysRevLett.90.046103. [DOI] [PubMed] [Google Scholar]
- Senftle T. P.; van Duin A. C. T.; Janik M. J. Methane Activation at the Pd/CeO2 Interface. ACS Catal. 2017, 7 (1), 327–332. 10.1021/acscatal.6b02447. [DOI] [Google Scholar]
- Li G.; Vollmer I.; Liu C.; Gascon J.; Pidko E. A. Structure and Reactivity of the Mo/ZSM-5 Dehydroaromatization Catalyst: An Operando Computational Study. ACS Catal. 2019, 9 (9), 8731–8737. 10.1021/acscatal.9b02213. [DOI] [Google Scholar]
- Griesser C.; Li H.; Wernig E.-M.; Winkler D.; Shakibi Nia N.; Mairegger T.; Götsch T.; Schachinger T.; Steiger-Thirsfeld A.; Penner S.; Wielend D.; Egger D.; Scheurer C.; Reuter K.; Kunze-Liebhäuser J. True Nature of the Transition-Metal Carbide/Liquid Interface Determines Its Reactivity. ACS Catal. 2021, 11 (8), 4920–4928. 10.1021/acscatal.1c00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach T.; Walter M.; Moseler M.; Hammer B.; Bruix A. Effects of Gas-Phase Conditions and Particle Size on the Properties of Cu(111)-Supported ZnyOx Particles Revealed by Global Optimization and Ab Initio Thermodynamics. J. Phys. Chem. C 2019, 123 (51), 30903–30916. 10.1021/acs.jpcc.9b07715. [DOI] [Google Scholar]
- Wang Y.-G.; Cantu D. C.; Lee M.-S.; Li J.; Glezakou V.-A.; Rousseau R. CO Oxidation on Au/TiO2: Condition-Dependent Active Sites and Mechanistic Pathways. J. Am. Chem. Soc. 2016, 138 (33), 10467–10476. 10.1021/jacs.6b04187. [DOI] [PubMed] [Google Scholar]
- Wang Y.-G.; Mei D.; Glezakou V.-A.; Li J.; Rousseau R. Dynamic formation of single-atom catalytic active sites on ceria-supported gold nanoparticles. Nat. Commun. 2015, 6, 6511. 10.1038/ncomms7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.-C.; Tang Y.; Wang Y.-G.; Zhang T.; Li J. Theoretical understanding of the stability of single-atom catalysts. Natl. Sci. Rev. 2018, 5 (5), 638–641. 10.1093/nsr/nwy094. [DOI] [Google Scholar]
- Liu J.-C.; Wang Y.-G.; Li J. Toward Rational Design of Oxide-Supported Single-Atom Catalysts: Atomic Dispersion of Gold on Ceria. J. Am. Chem. Soc. 2017, 139 (17), 6190–6199. 10.1021/jacs.7b01602. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Wang Y.-G.; Li J. Theoretical Investigations of Pt1@CeO2 Single-Atom Catalyst for CO Oxidation. J. Phys. Chem. C 2017, 121 (21), 11281–11289. 10.1021/acs.jpcc.7b00313. [DOI] [Google Scholar]
- Liu Y.; Liu J. C.; Li T. H.; Duan Z. H.; Zhang T. Y.; Yan M.; Li W. L.; Xiao H.; Wang Y. G.; Chang C. R.; Li J. Unravelling the Enigma of Nonoxidative Conversion of Methane on Iron Single-Atom Catalysts. Angew. Chem., Int. Ed. 2020, 59 (42), 18586–18590. 10.1002/anie.202003908. [DOI] [PubMed] [Google Scholar]
- Wang A.-h.; Zhang Z.-c.; Li G.-h. Advances in enhanced sampling molecular dynamics simulations for biomolecules. Chin. J. Chem. Phys. 2019, 32 (3), 277–286. 10.1063/1674-0068/cjcp1905091. [DOI] [Google Scholar]
- Cheng T.; Fortunelli A.; Goddard W. A. Reaction intermediates during operando electrocatalysis identified from full solvent quantum mechanics molecular dynamics. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (16), 7718–7722. 10.1073/pnas.1821709116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T.; Xiao H.; Goddard W. A. Reaction Mechanisms for the Electrochemical Reduction of CO2 to CO and Formate on the Cu(100) Surface at 298 K from Quantum Mechanics Free Energy Calculations with Explicit Water. J. Am. Chem. Soc. 2016, 138 (42), 13802–13805. 10.1021/jacs.6b08534. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Xiao H.; Goddard W. A. Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (8), 1795–1800. 10.1073/pnas.1612106114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum Y.; Cheng T.; Goddard W. A.; Ager J. W. Electrochemical CO Reduction Builds Solvent Water into Oxygenate Products. J. Am. Chem. Soc. 2018, 140 (30), 9337–9340. 10.1021/jacs.8b03986. [DOI] [PubMed] [Google Scholar]
- Nastase S. A. F.; Cnudde P.; Vanduyfhuys L.; De Wispelaere K.; Van Speybroeck V.; Catlow C. R. A.; Logsdail A. J. Mechanistic Insight into the Framework Methylation of H-ZSM-5 for Varying Methanol Loadings and Si/Al Ratios Using First-Principles Molecular Dynamics Simulations. ACS Catal. 2020, 10 (15), 8904–8915. 10.1021/acscatal.0c01454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailleul S.; Dedecker K.; Cnudde P.; Vanduyfhuys L.; Waroquier M.; Van Speybroeck V. Ab initio enhanced sampling kinetic study on MTO ethene methylation reaction. J. Catal. 2020, 388, 38–51. 10.1016/j.jcat.2020.04.015. [DOI] [Google Scholar]
- Lamaire A.; Wieme J.; Hoffman A. E. J.; Van Speybroeck V. Atomistic insight in the flexibility and heat transport properties of the stimuli-responsive metal–organic framework MIL-53(Al) for water-adsorption applications using molecular simulations. Faraday Discuss. 2021, 225, 301–323. 10.1039/D0FD00025F. [DOI] [PubMed] [Google Scholar]
- Bailleul S.; Rogge S. M. J.; Vanduyfhuys L.; Van Speybroeck V. Insight into the Role of Water on the Methylation of Hexamethylbenzene in H-SAPO-34 from First Principle Molecular Dynamics Simulations. ChemCatChem 2019, 11 (16), 3993–4010. 10.1002/cctc.201900618. [DOI] [Google Scholar]
- Cnudde P.; Redekop E. A.; Dai W.; Porcaro N. G.; Waroquier M.; Bordiga S.; Hunger M.; Li L.; Olsbye U.; Van Speybroeck V. Experimental and Theoretical Evidence for the Promotional Effect of Acid Sites on the Diffusion of Alkenes through Small-Pore Zeolites. Angew. Chem., Int. Ed. 2021, 60 (18), 10016–10022. 10.1002/anie.202017025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Rogge S. M. J.; Lamaire A.; Vandenbrande S.; Wieme J.; Siviour C. R.; Van Speybroeck V.; Tan J.-C. High-rate nanofluidic energy absorption in porous zeolitic frameworks. Nat. Mater. 2021, 20 (7), 1015–1023. 10.1038/s41563-021-00977-6. [DOI] [PubMed] [Google Scholar]
- Lukashuk L.; Yigit N.; Rameshan R.; Kolar E.; Teschner D.; Hävecker M.; Knop-Gericke A.; Schlögl R.; Föttinger K.; Rupprechter G. Operando Insights into CO Oxidation on Cobalt Oxide Catalysts by NAP-XPS, FTIR, and XRD. ACS Catal. 2018, 8 (9), 8630–8641. 10.1021/acscatal.8b01237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeininger J.; Suchorski Y.; Raab M.; Buhr S.; Grönbeck H.; Rupprechter G. Single-Particle Catalysis: Revealing Intraparticle Pacemakers in Catalytic H2 Oxidation on Rh. ACS Catal. 2021, 11 (15), 10020–10027. 10.1021/acscatal.1c02384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchorski Y.; Datler M.; Bespalov I.; Zeininger J.; Stöger-Pollach M.; Bernardi J.; Grönbeck H.; Rupprechter G. Surface-Structure Libraries: Multifrequential Oscillations in Catalytic Hydrogen Oxidation on Rhodium. J. Phys. Chem. C 2019, 123 (7), 4217–4227. 10.1021/acs.jpcc.8b11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchorski Y.; Datler M.; Bespalov I.; Zeininger J.; Stöger-Pollach M.; Bernardi J.; Grönbeck H.; Rupprechter G., Visualizing catalyst heterogeneity by a multifrequential oscillating reaction. Nat. Commun. 2018, 9 ( (1), ). 10.1038/s41467-018-03007-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchorski Y.; Zeininger J.; Buhr S.; Raab M.; Stöger-Pollach M.; Bernardi J.; Grönbeck H.; Rupprechter G. Resolving multifrequential oscillations and nanoscale interfacet communication in single-particle catalysis. Science 2021, 372 (6548), 1314–1318. 10.1126/science.abf8107. [DOI] [PubMed] [Google Scholar]
- Xu J.; Cao X.-M.; Hu P. Perspective on computational reaction prediction using machine learning methods in heterogeneous catalysis. Phys. Chem. Chem. Phys. 2021, 23, 11155–11179. 10.1039/D1CP01349A. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Wang H.; Su N. Q.; Duan S.; Shen T.; Xu X. Beyond Mean-Field Microkinetics: Toward Accurate and Efficient Theoretical Modeling in Heterogeneous Catalysis. ACS Catal. 2018, 8 (7), 5816–5826. 10.1021/acscatal.8b00943. [DOI] [Google Scholar]
- Chen Z.; Wang H.; Liu Z.; Xu X. Dynamic and Intermediate-Specific Local Coverage Controls the Syngas Conversion on Rh(111) Surfaces: An Operando Theoretical Analysis. ACS Catal. 2021, 11 (7), 3830–3841. 10.1021/acscatal.0c05070. [DOI] [Google Scholar]
- Ding C.; Shen T.; Yang Y.; Xu X. Involvement of the Unoccupied Site Changes the Kinetic Trend Significantly: A Case Study on Formic Acid Decomposition. ACS Catal. 2020, 10 (9), 5153–5162. 10.1021/acscatal.0c00361. [DOI] [Google Scholar]
- Wang H.; Shen T.; Duan S.; Chen Z.; Xu X. Bistability for CO Oxidation: An Understanding from Extended Phenomenological Kinetics Simulations. ACS Catal. 2019, 9 (12), 11116–11124. 10.1021/acscatal.9b03407. [DOI] [Google Scholar]
- Goldsmith C. F.; West R. H. Automatic Generation of Microkinetic Mechanisms for Heterogeneous Catalysis. J. Phys. Chem. C 2017, 121 (18), 9970–9981. 10.1021/acs.jpcc.7b02133. [DOI] [Google Scholar]
- Blondal K.; Jelic J.; Mazeau E.; Studt F.; West R. H.; Goldsmith C. F. Computer-Generated Kinetics for Coupled Heterogeneous/Homogeneous Systems: A Case Study in Catalytic Combustion of Methane on Platinum. Ind. Eng. Chem. Res. 2019, 58 (38), 17682–17691. 10.1021/acs.iecr.9b01464. [DOI] [Google Scholar]
- Gu T.; Wang B.; Chen S.; Yang B. Automated Generation and Analysis of the Complex Catalytic Reaction Network of Ethanol Synthesis from Syngas on Rh(111). ACS Catal. 2020, 10 (11), 6346–6355. 10.1021/acscatal.0c00630. [DOI] [Google Scholar]
- Wang B.; Chen S.; Zhang J.; Li S.; Yang B. Propagating DFT Uncertainty to Mechanism Determination, Degree of Rate Control, and Coverage Analysis: The Kinetics of Dry Reforming of Methane. J. Phys. Chem. C 2019, 123 (50), 30389–30397. 10.1021/acs.jpcc.9b08755. [DOI] [Google Scholar]
- Zhang X.-J.; Shang C.; Liu Z.-P. Stochastic surface walking reaction sampling for resolving heterogeneous catalytic reaction network: A revisit to the mechanism of water-gas shift reaction on Cu. J. Chem. Phys. 2017, 147 (15), 152706. 10.1063/1.4989540. [DOI] [PubMed] [Google Scholar]
- Sugiyama K.; Sumiya Y.; Takagi M.; Saita K.; Maeda S. Understanding CO oxidation on the Pt(111) surface based on a reaction route network. Phys. Chem. Chem. Phys. 2019, 21 (26), 14366–14375. 10.1039/C8CP06856A. [DOI] [PubMed] [Google Scholar]
- Guo C.; Fu X.; Long J.; Li H.; Qin G.; Cao A.; Jing H.; Xiao J. Toward computational design of chemical reactions with reaction phase diagram. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2021, 11 (5), e1514 10.1002/wcms.1514. [DOI] [Google Scholar]
- Guo P.; Fu X.; Deák P.; Frauenheim T.; Xiao J. Activity and Mechanism Mapping of Photocatalytic NO2 Conversion on the Anatase TiO2(101) Surface. J. Phys. Chem. Lett. 2021, 12, 7708–7716. 10.1021/acs.jpclett.1c02263. [DOI] [PubMed] [Google Scholar]
- Van Speybroeck V.; Vandenhaute S.; Hoffman A. E. J.; Rogge S. M. J. Towards modeling spatiotemporal processes in metal–organic frameworks. Trends in Chemistry 2021, 3 (8), 605–619. 10.1016/j.trechm.2021.04.003. [DOI] [Google Scholar]
- Behler J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev. 2021, 121 (16), 10037–10072. 10.1021/acs.chemrev.0c00868. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Lei Y.-K.; Zhang Z.; Chang J.; Li M.; Han X.; Yang L.; Yang Y. I.; Gao Y. Q. A Perspective on Deep Learning for Molecular Modeling and Simulations. J. Phys. Chem. A 2020, 124 (34), 6745–6763. 10.1021/acs.jpca.0c04473. [DOI] [PubMed] [Google Scholar]
- Hajinazar S.; Sandoval E. D.; Cullo A. J.; Kolmogorov A. N. Multitribe evolutionary search for stable Cu–Pd–Ag nanoparticles using neural network models. Phys. Chem. Chem. Phys. 2019, 21 (17), 8729–8742. 10.1039/C9CP00837C. [DOI] [PubMed] [Google Scholar]
- Huang S.-D.; Shang C.; Kang P.-L.; Liu Z.-P. Atomic structure of boron resolved using machine learning and global sampling. Chem. Sci. 2018, 9 (46), 8644–8655. 10.1039/C8SC03427C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-D.; Shang C.; Zhang X.-J.; Liu Z.-P. Material discovery by combining stochastic surface walking global optimization with a neural network. Chem. Sci. 2017, 8 (9), 6327–6337. 10.1039/C7SC01459G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P.-L.; Liu Z.-P. Reaction prediction via atomistic simulation: from quantum mechanics to machine learning. iScience 2021, 24 (1), 102013. 10.1016/j.isci.2020.102013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S.; Shang C.; Liu Z.-P. Heterogeneous catalysis from structure to activity via SSW-NN method. J. Chem. Phys. 2019, 151 (5), 050901. 10.1063/1.5113673. [DOI] [Google Scholar]
- Kang P.-L.; Shang C.; Liu Z.-P. Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration. Acc. Chem. Res. 2020, 53 (10), 2119–2129. 10.1021/acs.accounts.0c00472. [DOI] [PubMed] [Google Scholar]
- Jennings P. C.; Lysgaard S.; Hummelshøj J. S.; Vegge T.; Bligaard T. Genetic algorithms for computational materials discovery accelerated by machine learning. npj Comput. Mater. 2019, 5, 46. 10.1038/s41524-019-0181-4. [DOI] [Google Scholar]
- Kolsbjerg E. L.; Peterson A. A.; Hammer B. Neural-network-enhanced evolutionary algorithm applied to supported metal nanoparticles. Phys. Rev. B: Condens. Matter Mater. Phys. 2018, 97 (19), 195424. 10.1103/PhysRevB.97.195424. [DOI] [Google Scholar]
- Jørgensen M. S.; Larsen U. F.; Jacobsen K. W.; Hammer B. Exploration versus Exploitation in Global Atomistic Structure Optimization. J. Phys. Chem. A 2018, 122 (5), 1504–1509. 10.1021/acs.jpca.8b00160. [DOI] [PubMed] [Google Scholar]
- Jørgensen M. S.; Groves M. N.; Hammer B. Combining Evolutionary Algorithms with Clustering toward Rational Global Structure Optimization at the Atomic Scale. J. Chem. Theory Comput. 2017, 13 (3), 1486–1493. 10.1021/acs.jctc.6b01119. [DOI] [PubMed] [Google Scholar]
- Sørensen K. H.; Jørgensen M. S.; Bruix A.; Hammer B. Accelerating atomic structure search with cluster regularization. J. Chem. Phys. 2018, 148 (24), 241734. 10.1063/1.5023671. [DOI] [PubMed] [Google Scholar]
- Ma S.; Huang S.-D.; Liu Z.-P. Dynamic coordination of cations and catalytic selectivity on zinc–chromium oxide alloys during syngas conversion. Nat. Catal. 2019, 2 (8), 671–677. 10.1038/s41929-019-0293-8. [DOI] [Google Scholar]
- Li X.-T.; Chen L.; Wei G.-F.; Shang C.; Liu Z.-P. Sharp Increase in Catalytic Selectivity in Acetylene Semihydrogenation on Pd Achieved by a Machine Learning Simulation-Guided Experiment. ACS Catal. 2020, 10 (17), 9694–9705. 10.1021/acscatal.0c02158. [DOI] [Google Scholar]
- Bartel C. J.; Millican S. L.; Deml A. M.; Rumptz J. R.; Tumas W.; Weimer A. W.; Lany S.; Stevanović V.; Musgrave C. B.; Holder A. M. Physical descriptor for the Gibbs energy of inorganic crystalline solids and temperature-dependent materials chemistry. Nat. Commun. 2018, 9, 4168. 10.1038/s41467-018-06682-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary J. M.; Holder A. M.; Musgrave C. B. Computationally Predicted High-Throughput Free-Energy Phase Diagrams for the Discovery of Solid-State Hydrogen Storage Reactions. ACS Appl. Mater. Interfaces 2020, 12 (43), 48553–48564. 10.1021/acsami.0c13298. [DOI] [PubMed] [Google Scholar]
- Ulissi Z. W.; Tang M. T.; Xiao J.; Liu X.; Torelli D. A.; Karamad M.; Cummins K.; Hahn C.; Lewis N. S.; Jaramillo T. F.; Chan K.; Nørskov J. K. Machine-Learning Methods Enable Exhaustive Searches for Active Bimetallic Facets and Reveal Active Site Motifs for CO2 Reduction. ACS Catal. 2017, 7 (10), 6600–6608. 10.1021/acscatal.7b01648. [DOI] [Google Scholar]
- Zhong M.; Tran K.; Min Y.; Wang C.; Wang Z.; Dinh C.-T.; De Luna P.; Yu Z.; Rasouli A. S.; Brodersen P.; Sun S.; Voznyy O.; Tan C.-S.; Askerka M.; Che F.; Liu M.; Seifitokaldani A.; Pang Y.; Lo S.-C.; Ip A.; Ulissi Z.; Sargent E. H. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 2020, 581 (7807), 178–183. 10.1038/s41586-020-2242-8. [DOI] [PubMed] [Google Scholar]
- Núñez M.; Vlachos D. G. Multiscale Modeling Combined with Active Learning for Microstructure Optimization of Bifunctional Catalysts. Ind. Eng. Chem. Res. 2019, 58 (15), 6146–6154. 10.1021/acs.iecr.8b04801. [DOI] [Google Scholar]
- Trummer D.; Searles K.; Algasov A.; Guda S. A.; Soldatov A. V.; Ramanantoanina H.; Safonova O. V.; Guda A. A.; Copéret C. Deciphering the Phillips Catalyst by Orbital Analysis and Supervised Machine Learning from Cr Pre-edge XANES of Molecular Libraries. J. Am. Chem. Soc. 2021, 143 (19), 7326–7341. 10.1021/jacs.0c10791. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Chen C.; Chen Y.; Ong S. P. Random Forest Models for Accurate Identification of Coordination Environments from X-Ray Absorption Near-Edge Structure. Patterns 2020, 1 (2), 100013. 10.1016/j.patter.2020.100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankine C. D.; Madkhali M. M. M.; Penfold T. J. A Deep Neural Network for the Rapid Prediction of X-ray Absorption Spectra. J. Phys. Chem. A 2020, 124 (21), 4263–4270. 10.1021/acs.jpca.0c03723. [DOI] [PubMed] [Google Scholar]
- Hu W.; Ye S.; Zhang Y.; Li T.; Zhang G.; Luo Y.; Mukamel S.; Jiang J. Machine Learning Protocol for Surface-Enhanced Raman Spectroscopy. J. Phys. Chem. Lett. 2019, 10 (20), 6026–6031. 10.1021/acs.jpclett.9b02517. [DOI] [PubMed] [Google Scholar]
- Peterson A. A. Acceleration of saddle-point searches with machine learning. J. Chem. Phys. 2016, 145 (7), 074106. 10.1063/1.4960708. [DOI] [PubMed] [Google Scholar]
- Koistinen O.-P.; Dagbjartsdóttir F. B.; Ásgeirsson V.; Vehtari A.; Jónsson H. Nudged elastic band calculations accelerated with Gaussian process regression. J. Chem. Phys. 2017, 147 (15), 152720. 10.1063/1.4986787. [DOI] [PubMed] [Google Scholar]
- Yang M.; Bonati L.; Polino D.; Parrinello M.. Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water. Catal. Today 2021. 10.1016/j.cattod.2021.03.018 [DOI] [Google Scholar]
- Ulissi Z. W.; Medford A. J.; Bligaard T.; Nørskov J. K. To address surface reaction network complexity using scaling relations machine learning and DFT calculations. Nat. Commun. 2017, 8, 14621. 10.1038/ncomms14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D.; Zhao Z.-J.; Zhang G.; Yang P.; Li L.; Gao H.; Liu S.; Chang X.; Chen S.; Wang T.; Ozin G. A.; Liu Z.; Gong J. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 2021, 12, 395. 10.1038/s41467-020-20615-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artrith N.; Kolpak A. M. Understanding the Composition and Activity of Electrocatalytic Nanoalloys in Aqueous Solvents: A Combination of DFT and Accurate Neural Network Potentials. Nano Lett. 2014, 14 (5), 2670–2676. 10.1021/nl5005674. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Chen Y.; Cheng T.; Wang L.-W.; Goddard W. A. Identification of the Selective Sites for Electrochemical Reduction of CO to C2+ Products on Copper Nanoparticles by Combining Reactive Force Fields, Density Functional Theory, and Machine Learning. ACS Energy Lett. 2018, 3 (12), 2983–2988. 10.1021/acsenergylett.8b01933. [DOI] [Google Scholar]
- Natarajan S. K.; Behler J. Neural network molecular dynamics simulations of solid–liquid interfaces: water at low-index copper surfaces. Phys. Chem. Chem. Phys. 2016, 18 (41), 28704–28725. 10.1039/C6CP05711J. [DOI] [PubMed] [Google Scholar]
- Quaranta V.; Hellström M.; Behler J. Proton-Transfer Mechanisms at the Water–ZnO Interface: The Role of Presolvation. J. Phys. Chem. Lett. 2017, 8 (7), 1476–1483. 10.1021/acs.jpclett.7b00358. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhou X.; Jiang B. Bridging the Gap between Direct Dynamics and Globally Accurate Reactive Potential Energy Surfaces Using Neural Networks. J. Phys. Chem. Lett. 2019, 10 (6), 1185–1191. 10.1021/acs.jpclett.9b00085. [DOI] [PubMed] [Google Scholar]
- Hu C.; Zhang Y.; Jiang B. Dynamics of H2O Adsorption on Pt(110)-(1 × 2) Based on a Neural Network Potential Energy Surface. J. Phys. Chem. C 2020, 124 (42), 23190–23199. 10.1021/acs.jpcc.0c07182. [DOI] [Google Scholar]
- Zhu L.; Zhang Y.; Zhang L.; Zhou X.; Jiang B. Unified and transferable description of dynamics of H2 dissociative adsorption on multiple copper surfaces via machine learning. Phys. Chem. Chem. Phys. 2020, 22 (25), 13958–13964. 10.1039/D0CP02291H. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Zhou X.; Zhou L.; Zhang Y.; Luo X.; Guo H.; Jiang B. Constructing High-Dimensional Neural Network Potential Energy Surfaces for Gas–Surface Scattering and Reactions. J. Phys. Chem. C 2018, 122 (3), 1761–1769. 10.1021/acs.jpcc.7b12064. [DOI] [Google Scholar]
- Kolb B.; Luo X.; Zhou X.; Jiang B.; Guo H. High-Dimensional Atomistic Neural Network Potentials for Molecule–Surface Interactions: HCl Scattering from Au(111). J. Phys. Chem. Lett. 2017, 8 (3), 666–672. 10.1021/acs.jpclett.6b02994. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Zhang Y.; Guo H.; Jiang B. Towards bridging the structure gap in heterogeneous catalysis: the impact of defects in dissociative chemisorption of methane on Ir surfaces. Phys. Chem. Chem. Phys. 2021, 23 (7), 4376–4385. 10.1039/D0CP06535H. [DOI] [PubMed] [Google Scholar]
- Albinsson D.; Boje A.; Nilsson S.; Tiburski C.; Hellman A.; Strom H.; Langhammer C. Copper catalysis at operando conditions-bridging the gap between single nanoparticle probing and catalyst-bed-averaging. Nat. Commun. 2020, 11 (1), 4832. 10.1038/s41467-020-18623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovic V.; Consiglio A.; Smith K. E.; Jozwiak C.; Bostwick A.; Rotenberg E.; Di Sante D.; Moser S. Momentum for Catalysis: How Surface Reactions Shape the RuO2 Flat Surface State. ACS Catal. 2021, 11 (3), 1749–1757. 10.1021/acscatal.0c04871. [DOI] [Google Scholar]
- Nguyen L.; Liu L.; Assefa S.; Wolverton C.; Schneider W. F.; Tao F. F. Atomic-Scale Structural Evolution of Rh(110) during Catalysis. ACS Catal. 2017, 7 (1), 664–674. 10.1021/acscatal.6b02006. [DOI] [Google Scholar]
- Wang J.; Chang X.; Chen S.; Sun G.; Zhou X.; Vovk E.; Yang Y.; Deng W.; Zhao Z.-J.; Mu R.; Pei C.; Gong J. On the Role of Sn Segregation of Pt-Sn Catalysts for Propane Dehydrogenation. ACS Catal. 2021, 11 (8), 4401–4410. 10.1021/acscatal.1c00639. [DOI] [Google Scholar]
- Zichittella G.; Polyhach Y.; Tschaggelar R.; Jeschke G.; Perez-Ramirez J. Quantification of Redox Sites during Catalytic Propane Oxychlorination by Operando EPR Spectroscopy. Angew. Chem., Int. Ed. 2021, 60 (7), 3596–3602. 10.1002/anie.202013331. [DOI] [PubMed] [Google Scholar]
- Chen S.; Zeng L.; Mu R.; Xiong C.; Zhao Z. J.; Zhao C.; Pei C.; Peng L.; Luo J.; Fan L. S.; Gong J. Modulating Lattice Oxygen in Dual-Functional Mo-V-O Mixed Oxides for Chemical Looping Oxidative Dehydrogenation. J. Am. Chem. Soc. 2019, 141 (47), 18653–18657. 10.1021/jacs.9b09235. [DOI] [PubMed] [Google Scholar]
- Wei S.; Li A.; Liu J. C.; Li Z.; Chen W.; Gong Y.; Zhang Q.; Cheong W. C.; Wang Y.; Zheng L.; Xiao H.; Chen C.; Wang D.; Peng Q.; Gu L.; Han X.; Li J.; Li Y. Direct observation of noble metal nanoparticles transforming to thermally stable single atoms. Nat. Nanotechnol. 2018, 13 (9), 856–861. 10.1038/s41565-018-0197-9. [DOI] [PubMed] [Google Scholar]
- Johnson N. J. J.; Lam B.; MacLeod B. P.; Sherbo R. S.; Moreno-Gonzalez M.; Fork D. K.; Berlinguette C. P. Facets and vertices regulate hydrogen uptake and release in palladium nanocrystals. Nat. Mater. 2019, 18 (5), 454–458. 10.1038/s41563-019-0308-5. [DOI] [PubMed] [Google Scholar]
- Song B.; Yang T. T.; Yuan Y.; Sharifi-Asl S.; Cheng M.; Saidi W. A.; Liu Y.; Shahbazian-Yassar R. Revealing Sintering Kinetics of MoS2-Supported Metal Nanocatalysts in Atmospheric Gas Environments via Operando Transmission Electron Microscopy. ACS Nano 2020, 14 (4), 4074–4086. 10.1021/acsnano.9b08757. [DOI] [PubMed] [Google Scholar]
- Liu L.; Zakharov D. N.; Arenal R.; Concepcion P.; Stach E. A.; Corma A. Evolution and stabilization of subnanometric metal species in confined space by in situ TEM. Nat. Commun. 2018, 9 (1), 574. 10.1038/s41467-018-03012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch G.; Hävecker M.; Teschner D.; Carey S. J.; Wang Y.; Kube P.; Hetaba W.; Lunkenbein T.; Auffermann G.; Timpe O.; Rosowski F.; Schlögl R.; Trunschke A. Surface Conditions That Constrain Alkane Oxidation on Perovskites. ACS Catal. 2020, 10 (13), 7007–7020. 10.1021/acscatal.0c01289. [DOI] [Google Scholar]
- Passos A. R.; Rochet A.; Manente L. M.; Suzana A. F.; Harder R.; Cha W.; Meneau F. Three-dimensional strain dynamics govern the hysteresis in heterogeneous catalysis. Nat. Commun. 2020, 11 (1), 4733. 10.1038/s41467-020-18622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovskiy A. L.; Kenzhina I. E.; Zdorovets M. V. FeCo– Fe2CoO4/Co3O4 nanocomposites: Phase transformations as a result of thermal annealing and practical application in catalysis. Ceram. Int. 2020, 46 (8), 10262–10269. 10.1016/j.ceramint.2020.01.019. [DOI] [Google Scholar]
- Wang Zhou X.; Karnesky R. A.; Yang N.; Yee J. K. Kinetic Monte Carlo simulations of structural evolution during anneal of additively manufactured materials. Comput. Mater. Sci. 2020, 179, 109605. 10.1016/j.commatsci.2020.109605. [DOI] [Google Scholar]
- Cao D.; Liu D.; Chen S.; Moses O. A.; Chen X.; Xu W.; Wu C.; Zheng L.; Chu S.; Jiang H.; Wang C.; Ge B.; Wu X.; Zhang J.; Song L. Operando X-ray spectroscopy visualizing the chameleon-like structural reconstruction on an oxygen evolution electrocatalyst. Energy Environ. Sci. 2021, 14 (2), 906–915. 10.1039/D0EE02276D. [DOI] [Google Scholar]
- Hansen H. A.; Rossmeisl J.; Nørskov J. K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni(111) surfaces studied by DFT. Phys. Chem. Chem. Phys. 2008, 10 (25), 3722–3730. 10.1039/b803956a. [DOI] [PubMed] [Google Scholar]
- Pi Y.; Shao Q.; Zhu X.; Huang X. Dynamic Structure Evolution of Composition Segregated Iridium-Nickel Rhombic Dodecahedra toward Efficient Oxygen Evolution Electrocatalysis. ACS Nano 2018, 12 (7), 7371–7379. 10.1021/acsnano.8b04023. [DOI] [PubMed] [Google Scholar]
- Gao L.; Cui X.; Wang Z.; Sewell C. D.; Li Z.; Liang S.; Zhang M.; Li J.; Hu Y.; Lin Z. Operando unraveling photothermal-promoted dynamic active-sites generation in NiFe2O4 for markedly enhanced oxygen evolution. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (7), e2023421118 10.1073/pnas.2023421118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionigi F.; Zeng Z.; Sinev I.; Merzdorf T.; Deshpande S.; Lopez M. B.; Kunze S.; Zegkinoglou I.; Sarodnik H.; Fan D.; Bergmann A.; Drnec J.; Araujo J. F.; Gliech M.; Teschner D.; Zhu J.; Li W. X.; Greeley J.; Cuenya B. R.; Strasser P. In-situ structure and catalytic mechanism of NiFe and CoFe layered double hydroxides during oxygen evolution. Nat. Commun. 2020, 11 (1), 2522. 10.1038/s41467-020-16237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikowski F.; Maroun F.; Pacheco I.; Wiegmann T.; Allongue P.; Stettner J.; Magnussen O. M. Operando Surface X-ray Diffraction Studies of Structurally Defined Co3O4 and CoOOH Thin Films during Oxygen Evolution. ACS Catal. 2019, 9 (5), 3811–3821. 10.1021/acscatal.8b04823. [DOI] [Google Scholar]
- Su X.; Wang Y.; Zhou J.; Gu S.; Li J.; Zhang S. Operando Spectroscopic Identification of Active Sites in NiFe Prussian Blue Analogues as Electrocatalysts: Activation of Oxygen Atoms for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2018, 140 (36), 11286–11292. 10.1021/jacs.8b05294. [DOI] [PubMed] [Google Scholar]
- Takahashi I.; Koga O.; Hoshi N.; Hori Y. Electrochemical reduction of CO2 at copper single crystal Cu(S)-[n(111)×(111)] and Cu(S)-[n(110)×(100)] electrodes. J. Electroanal. Chem. 2002, 533 (1–2), 135–143. 10.1016/S0022-0728(02)01081-1. [DOI] [Google Scholar]
- Hori Y.; Takahashi I.; Koga O.; Hoshi N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A: Chem. 2003, 199 (1–2), 39–47. 10.1016/S1381-1169(03)00016-5. [DOI] [Google Scholar]
- Chou T. C.; Chang C. C.; Yu H. L.; Yu W. Y.; Dong C. L.; Velasco-Velez J. J.; Chuang C. H.; Chen L. C.; Lee J. F.; Chen J. M.; Wu H. L. Controlling the Oxidation State of the Cu Electrode and Reaction Intermediates for Electrochemical CO2 Reduction to Ethylene. J. Am. Chem. Soc. 2020, 142 (6), 2857–2867. 10.1021/jacs.9b11126. [DOI] [PubMed] [Google Scholar]
- Lin S. C.; Chang C. C.; Chiu S. Y.; Pai H. T.; Liao T. Y.; Hsu C. S.; Chiang W. H.; Tsai M. K.; Chen H. M. Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction. Nat. Commun. 2020, 11 (1), 3525. 10.1038/s41467-020-17231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Che F.; Liu M.; Zou C.; Liang Z.; De Luna P.; Yuan H.; Li J.; Wang Z.; Xie H.; Li H.; Chen P.; Bladt E.; Quintero-Bermudez R.; Sham T. K.; Bals S.; Hofkens J.; Sinton D.; Chen G.; Sargent E. H. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 2018, 10 (9), 974–980. 10.1038/s41557-018-0092-x. [DOI] [PubMed] [Google Scholar]
- Cao L.; Luo Q.; Liu W.; Lin Y.; Liu X.; Cao Y.; Zhang W.; Wu Y.; Yang J.; Yao T.; Wei S. Identification of single-atom active sites in carbon-based cobalt catalysts during electrocatalytic hydrogen evolution. Nat. Catal. 2019, 2 (2), 134–141. 10.1038/s41929-018-0203-5. [DOI] [Google Scholar]
- Fang S.; Zhu X.; Liu X.; Gu J.; Liu W.; Wang D.; Zhang W.; Lin Y.; Lu J.; Wei S.; Li Y.; Yao T. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 2020, 11 (1), 1029. 10.1038/s41467-020-14848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z.; Wang K.; Ye K.; Wang Y.; Su H.-Y.; Hu B.; Xiao J.; Yu T.; Wang Y.; Song S. Intermediate Adsorption States Switch to Selectively Catalyze Electrochemical CO2 Reduction. ACS Catal. 2020, 10 (6), 3871–3880. 10.1021/acscatal.9b05115. [DOI] [Google Scholar]
- Chen Z.; Wang T.; Liu B.; Cheng D.; Hu C.; Zhang G.; Zhu W.; Wang H.; Zhao Z.-J.; Gong J. Grain-Boundary-Rich Copper for Efficient Solar-Driven Electrochemical CO2 Reduction to Ethylene and Ethanol. J. Am. Chem. Soc. 2020, 142 (15), 6878–6883. 10.1021/jacs.0c00971. [DOI] [PubMed] [Google Scholar]
- Zhong D.; Zhao Z. J.; Zhao Q.; Cheng D.; Liu B.; Zhang G.; Deng W.; Dong H.; Zhang L.; Li J.; Li J.; Gong J. Coupling of Cu(100) and (110) Facets Promotes Carbon Dioxide Conversion to Hydrocarbons and Alcohols. Angew. Chem., Int. Ed. 2021, 60 (9), 4879–4885. 10.1002/anie.202015159. [DOI] [PubMed] [Google Scholar]
- Guo S.; Li Y.; Tang S.; Zhang Y.; Li X.; Sobrido A. J.; Titirici M. M.; Wei B. Monitoring Hydrogen Evolution Reaction Intermediates of Transition Metal Dichalcogenides via Operando Raman Spectroscopy. Adv. Funct. Mater. 2020, 30 (35), 2003035. 10.1002/adfm.202003035. [DOI] [Google Scholar]
- Xu B.-B.; Zhou M.; Ye M.; Yang L.-Y.; Wang H.-F.; Wang X. L.; Yao Y.-F. Cooperative Motion in Water–Methanol Clusters Controls the Reaction Rates of Heterogeneous Photocatalytic Reactions. J. Am. Chem. Soc. 2021, 143 (29), 10940–10947. 10.1021/jacs.1c02128. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Xiong Y.; Zeng R.; Lu X.; Krumov M.; Huang X.; Xu W.; Wang H.; DiSalvo F. J.; Brock J. D.; Muller D. A.; Abruña H. D. Operando Methods in Electrocatalysis. ACS Catal. 2021, 11 (3), 1136–1178. 10.1021/acscatal.0c04789. [DOI] [Google Scholar]
- Hassan M. T. Attomicroscopy: from femtosecond to attosecond electron microscopy. J. Phys. B: At., Mol. Opt. Phys. 2018, 51 (3), 032005. 10.1088/1361-6455/aaa183. [DOI] [Google Scholar]
- Dou J.; Sun Z.; Opalade A. A.; Wang N.; Fu W.; Tao F. Operando chemistry of catalyst surfaces during catalysis. Chem. Soc. Rev. 2017, 46 (7), 2001–2027. 10.1039/C6CS00931J. [DOI] [PubMed] [Google Scholar]
- Li J.; Gong J. Operando characterization techniques for electrocatalysis. Energy Environ. Sci. 2020, 13 (11), 3748–3779. 10.1039/D0EE01706J. [DOI] [Google Scholar]
- Zhai H.; Alexandrova A. N. Ensemble-Average Representation of Pt Clusters in Conditions of Catalysis Accessed through GPU Accelerated Deep Neural Network Fitting Global Optimization. J. Chem. Theory Comput. 2016, 12 (12), 6213–6226. 10.1021/acs.jctc.6b00994. [DOI] [PubMed] [Google Scholar]
- Xie W.; Xu J.; Ding Y.; Hu P. Quantitative Studies of the Key Aspects in Selective Acetylene Hydrogenation on Pd(111) by Microkinetic Modeling with Coverage Effects and Molecular Dynamics. ACS Catal. 2021, 11 (7), 4094–4106. 10.1021/acscatal.0c05345. [DOI] [Google Scholar]
- Yao Z.; Guo C.; Mao Y.; Hu P. Quantitative Determination of C–C Coupling Mechanisms and Detailed Analyses on the Activity and Selectivity for Fischer–Tropsch Synthesis on Co(0001): Microkinetic Modeling with Coverage Effects. ACS Catal. 2019, 9 (7), 5957–5973. 10.1021/acscatal.9b01150. [DOI] [Google Scholar]
- Chen Z.; Liu Z.; Xu X. Coverage-Dependent Microkinetics in Heterogeneous Catalysis Powered by the Maximum Rate Analysis. ACS Catal. 2021, 11 (15), 9333–9344. 10.1021/acscatal.1c01997. [DOI] [Google Scholar]
- Van den Bossche M.; Skúlason E.; Rose-Petruck C.; Jónsson H. Assessment of Constant-Potential Implicit Solvation Calculations of Electrochemical Energy Barriers for H2 Evolution on Pt. J. Phys. Chem. C 2019, 123 (7), 4116–4124. 10.1021/acs.jpcc.8b10046. [DOI] [Google Scholar]
- Tian Z.; Priest C.; Chen L. Recent Progress in the Theoretical Investigation of Electrocatalytic Reduction of CO2. Advanced Theory and Simulations 2018, 1 (5), 1800004. 10.1002/adts.201800004. [DOI] [Google Scholar]
- Chan K.; Nørskov J. K. Electrochemical Barriers Made Simple. J. Phys. Chem. Lett. 2015, 6 (14), 2663–2668. 10.1021/acs.jpclett.5b01043. [DOI] [PubMed] [Google Scholar]
- Chan K.; Nørskov J. K. Potential Dependence of Electrochemical Barriers from ab Initio Calculations. J. Phys. Chem. Lett. 2016, 7 (9), 1686–1690. 10.1021/acs.jpclett.6b00382. [DOI] [PubMed] [Google Scholar]
- Gauthier J. A.; Dickens C. F.; Heenen H. H.; Vijay S.; Ringe S.; Chan K. Unified Approach to Implicit and Explicit Solvent Simulations of Electrochemical Reaction Energetics. J. Chem. Theory Comput. 2019, 15 (12), 6895–6906. 10.1021/acs.jctc.9b00717. [DOI] [PubMed] [Google Scholar]
- Kristoffersen H. H.; Chan K. Towards constant potential modeling of CO-CO coupling at liquid water-Cu(1 0 0) interfaces. J. Catal. 2021, 396, 251–260. 10.1016/j.jcat.2021.02.023. [DOI] [Google Scholar]
- Magnussen O. M.; Groß A. Toward an Atomic-Scale Understanding of Electrochemical Interface Structure and Dynamics. J. Am. Chem. Soc. 2019, 141 (12), 4777–4790. 10.1021/jacs.8b13188. [DOI] [PubMed] [Google Scholar]
- Medford A. J.; Kunz M. R.; Ewing S. M.; Borders T.; Fushimi R. Extracting Knowledge from Data through Catalysis Informatics. ACS Catal. 2018, 8 (8), 7403–7429. 10.1021/acscatal.8b01708. [DOI] [Google Scholar]
- Strieth-Kalthoff F.; Sandfort F.; Segler M. H. S.; Glorius F. Machine learning the ropes: principles, applications and directions in synthetic chemistry. Chem. Soc. Rev. 2020, 49 (17), 6154–6168. 10.1039/C9CS00786E. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Lin L.; Jia Q.; Cheng Z.; Jiang Y.; Guo Y.; Ma J. Transferable Multilevel Attention Neural Network for Accurate Prediction of Quantum Chemistry Properties via Multitask Learning. J. Chem. Inf. Model. 2021, 61 (3), 1066–1082. 10.1021/acs.jcim.0c01224. [DOI] [PubMed] [Google Scholar]
- Toyao T.; Maeno Z.; Takakusagi S.; Kamachi T.; Takigawa I.; Shimizu K.-i. Machine Learning for Catalysis Informatics: Recent Applications and Prospects. ACS Catal. 2020, 10 (3), 2260–2297. 10.1021/acscatal.9b04186. [DOI] [Google Scholar]
- Chanussot L.; Das A.; Goyal S.; Lavril T.; Shuaibi M.; Riviere M.; Tran K.; Heras-Domingo J.; Ho C.; Hu W.; Palizhati A.; Sriram A.; Wood B.; Yoon J.; Parikh D.; Zitnick C. L.; Ulissi Z. Open Catalyst 2020 (OC20) Dataset and Community Challenges. ACS Catal. 2021, 11 (10), 6059–6072. 10.1021/acscatal.0c04525. [DOI] [Google Scholar]
- Winther K. T.; Hoffmann M. J.; Boes J. R.; Mamun O.; Bajdich M.; Bligaard T. Catalysis-Hub.org, an open electronic structure database for surface reactions. Sci. Data 2019, 6, 75. 10.1038/s41597-019-0081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis D. D.; Hummelshoj J. S.; Nestorov S.; Greeley J.; Dulak M.; Bligaard T.; Norskov J. K.; Jacobsen K. W. The Computational Materials Repository. Comput. Sci. Eng. 2012, 14 (6), 51–57. 10.1109/MCSE.2012.16. [DOI] [Google Scholar]
- Draxl C.; Scheffler M. NOMAD: The FAIR concept for big data-driven materials science. MRS Bull. 2018, 43 (9), 676–682. 10.1557/mrs.2018.208. [DOI] [Google Scholar]
- Glavatskikh M.; Leguy J.; Hunault G.; Cauchy T.; Da Mota B. Dataset’s chemical diversity limits the generalizability of machine learning predictions. J. Cheminf. 2019, 11, 69. 10.1186/s13321-019-0391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friederich P.; Häse F.; Proppe J.; Aspuru-Guzik A. Machine-learned potentials for next-generation matter simulations. Nat. Mater. 2021, 20 (6), 750–761. 10.1038/s41563-020-0777-6. [DOI] [PubMed] [Google Scholar]
- Wang S.-H.; Pillai H. S.; Wang S.; Achenie L. E. K.; Xin H. Infusing theory into deep learning for interpretable reactivity prediction. Nat. Commun. 2021, 12, 5288. 10.1038/s41467-021-25639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang R.; Curtarolo S.; Ahmetcik E.; Scheffler M.; Ghiringhelli L. M. SISSO: A compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. Phys. Rev. Mater. 2018, 2 (8), 083802. 10.1103/PhysRevMaterials.2.083802. [DOI] [Google Scholar]
- Esterhuizen J. A.; Goldsmith B. R.; Linic S. Theory-Guided Machine Learning Finds Geometric Structure-Property Relationships for Chemisorption on Subsurface Alloys. Chem. 2020, 6 (11), 3100–3117. 10.1016/j.chempr.2020.09.001. [DOI] [Google Scholar]
- Andersen M.; Levchenko S. V.; Scheffler M.; Reuter K. Beyond Scaling Relations for the Description of Catalytic Materials. ACS Catal. 2019, 9 (4), 2752–2759. 10.1021/acscatal.8b04478. [DOI] [Google Scholar]
- Han Z.-K.; Sarker D.; Ouyang R.; Mazheika A.; Gao Y.; Levchenko S. V. Single-atom alloy catalysts designed by first-principles calculations and artificial intelligence. Nat. Commun. 2021, 12, 1833. 10.1038/s41467-021-22048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Andersen M.; Reuter K. Data-Driven Descriptor Engineering and Refined Scaling Relations for Predicting Transition Metal Oxide Reactivity. ACS Catal. 2021, 11 (2), 734–742. 10.1021/acscatal.0c04170. [DOI] [Google Scholar]
- Bartel C. J.; Sutton C.; Goldsmith B. R.; Ouyang R.; Musgrave C. B.; Ghiringhelli L. M.; Scheffler M. New tolerance factor to predict the stability of perovskite oxides and halides. Sci. Adv. 2019, 5 (2), eaav0693 10.1126/sciadv.aav0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubatiuk T.; Isayev O. Development of Multimodal Machine Learning Potentials: Toward a Physics-Aware Artificial Intelligence. Acc. Chem. Res. 2021, 54 (7), 1575–1585. 10.1021/acs.accounts.0c00868. [DOI] [PubMed] [Google Scholar]
- Lu D.; Wang H.; Chen M.; Lin L.; Car R.; E W.; Jia W.; Zhang L. 86 PFLOPS Deep Potential Molecular Dynamics simulation of 100 million atoms with ab initio accuracy. Comput. Phys. Commun. 2021, 259, 107624. 10.1016/j.cpc.2020.107624. [DOI] [Google Scholar]
- Phillips J. C.; Hardy D. J.; Maia J. D. C.; Stone J. E.; Ribeiro J. V.; Bernardi R. C.; Buch R.; Fiorin G.; Hénin J.; Jiang W.; McGreevy R.; Melo M. C. R.; Radak B. K.; Skeel R. D.; Singharoy A.; Wang Y.; Roux B.; Aksimentiev A.; Luthey-Schulten Z.; Kalé L. V.; Schulten K.; Chipot C.; Tajkhorshid E. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153 (4), 044130. 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Páll S.; Zhmurov A.; Bauer P.; Abraham M.; Lundborg M.; Gray A.; Hess B.; Lindahl E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153 (13), 134110. 10.1063/5.0018516. [DOI] [PubMed] [Google Scholar]