

Abstract
Over the last few decades, an increasing number of vertebrate taxa have been identified that undergo programmed genome rearrangement, or programmed DNA loss, during development. In these organisms, the genome of germ cells is often reproducibly different from the genome of all other cells within the body. Although we clearly have not identified all vertebrate taxa that undergo programmed genome loss, the list of species known to undergo loss now represents ~10% of vertebrate species, including several basally diverging lineages. Recent studies have shed new light on the targets and mechanisms of DNA loss and their association with canonical modes of DNA silencing. Ultimately, expansion of these studies into a larger collection of taxa will aid in reconstructing patterns of shared/independent ancestry of programmed DNA loss in the vertebrate lineage, as well as more recent evolutionary events that have shaped the structure and content of eliminated DNA.
Keywords: programmed genome rearrangement, programmed DNA loss, genome, vertebrate, evolution
1. INTRODUCTION
In many species, the genome of each cell is essentially identical to the genome of every other cell in that organism’s body. But is this the rule or the exception in vertebrates? Over the last several decades, a growing number of species have been identified that show variation in genome content across cell types, often reproducibly. These reproducible differences are broadly termed programmed genome rearrangements and may take the form of developmentally programmed chromosome losses, segmental losses, translocations, or other recombination events (Figure 1). At some level, all vertebrates undergo some sort of programmatic rearrangement in the context of generating diversity among immune receptors: immunoglobulins in gnathostomes and variable lymphocyte receptors in agnathans (lampreys and hagfish) (1–3). However, rearrangements occurring at larger genomic and developmental scales have also been observed in several species, including representatives of many of the most deeply diverging vertebrate lineages (Figure 2).
Figure 1.

Integration of programmed DNA elimination with development. Germline-specific chromosomes/segments are shown in red. Functions encoded on these genes are limited to the germline and irreversibly silenced in somatic cells.
Figure 2.
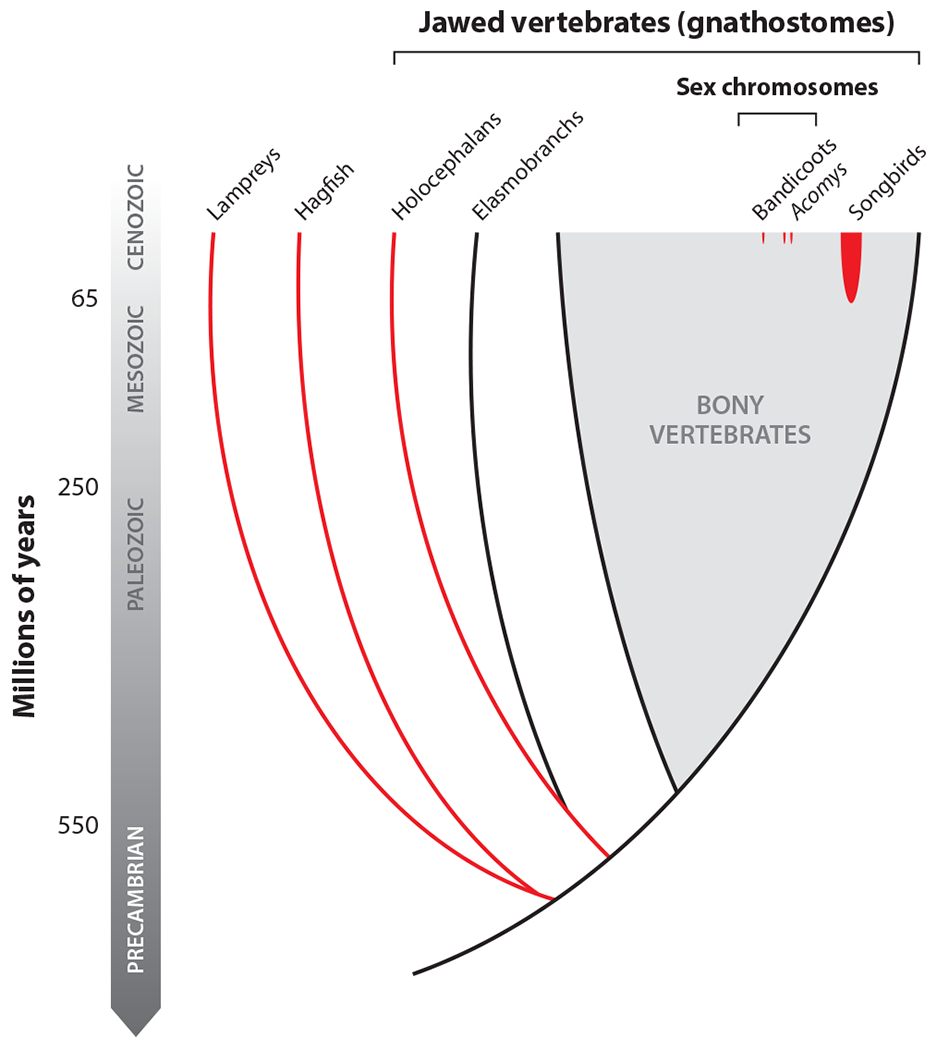
A phylogeny of the vertebrates, highlighting taxa known to undergo programmed DNA elimination (red); unknown taxa/lineages appear in black or gray. Divergence dates are taken from References 14, 38, and 119.
The understanding that genomes can potentially vary between cell types in animals is far from new. Large structural differences between germline and somatic cell lineages were observed in parasitic Ascaris roundworms as early as 1887 (4), wherein the physical structure of the genome and chromosome numbers were observed to change as cell lineages differentiated to give rise to the precursors of somatic tissues. In these roundworms, the original genome structure of the one-cell zygote is inherited only by the germline. These observations, and others in Ascaris, contributed to our understanding that chromosomes serve as the units of inheritance and evolutionary/developmental continuity of the germline. Similarly, differentiation of germline and somatic genomes was observed in single-celled ciliates in the nineteenth century (5, 6), and the role of programmed rearrangement in defining these differences has been resolved to ever-increasing accuracy over the last century (7–11). The understanding that in a single cell there existed a silent germline nucleus and an active somatic nucleus was fundamental to revealing the histone code (12–14). Importantly, these early observations have also added to our ever-growing understanding of the astonishing diversity of gene-regulatory mechanisms that have been shaped over the last two billion years of eukaryotic evolution.
One feature that distinguishes programmed rearrangements (particularly deletions) from other forms of epigenetic silencing is that deletions are effectively irreversible with respect to cell lineage. Nearly every other mode of silencing must be actively maintained to achieve continuous repression and can also be either intentionally reversed later in development to permit expression or potentially misexpressed in the context of disease (i.e., cancer). As such, it might be expected that the targets of programmed deletion will be enriched in a subset of genes that are both permanently dispensable to the cell lineage undergoing elimination and pose some liability if misexpressed in somatic tissues at later developmental stages. The irreversible nature of eliminations may also explain why PGRs have a patchy distribution across lineages. If eliminations act to restrict cell fate, then they might be selectively disadvantageous in species that rely on adult dedifferentiation of somatic cell lineages (i.e., taxa with extensive regenerative capacities) or might not be expected to persist with rapid generation times wherein opportunities for the development of cancer or similar diseases are limited within an individual’s reproductive life span.
The species that are currently known to undergo programmed DNA elimination (Figure 2) are likely only a subset of the species that undergo similar changes in DNA content, and we are still in the early stages of understanding key details related to the evolutionary origins and diversification of germline-specific genes, the functions of those genes, and the molecular mechanisms that mediate the reproducible loss from somatic cells. However, significant progress has been made in several of these areas, including the discovery of new taxa that undergo PGR and the broadening of these discoveries within their larger evolutionary clades. However, it is clear that much remains to be learned. Below, we discuss taxa that have thus far been identified as undergoing PGR and what is known regarding the mechanisms and genetic outcomes in each taxon. We then outline questions regarding the deeper evolutionary origins and diversification of PGR that are raised, given the increasing number of taxa found to undergo PGR, and finally we lay out some of the major challenges and future research directions that extend from studies performed thus far.
2. PROGRAMMED REARRANGEMENT IN HAGFISH
The first vertebrates known to undergo extensive programmed DNA elimination were the hagfish. In addition to lampreys (discussed in more detail below), extant species of hagfish represent the living vestiges of the vertebrate chordate lineages that split before the evolution of jaws, paired appendages, and other features that are shared by the vast majority of living vertebrates. Consensus phylogenies place hagfish as sister species to the lampreys (collectively known as cyclostomes), although the degree of statistical support for cyclostome monophyly and the ability of molecular phylogenetics to address the questions are a subject of debate. Regardless of the precise topology of the ancestral vertebrate tree, both lampreys and hagfish provide critical comparative perspective on the evolution of vertebrate genomes and genome biology (15).
The initial discovery of programmed elimination in hagfish derived from cytogenetic studies that were performed in an attempt to reconcile conflicting reports regarding chromosome numbers for Eptatretus burgeri (16–18). In this initial study, metaphases were observed from 25 individuals (7 females and 18 males) in spermatogonia, primary spermatocytes, and a panel of four somatic tissues (blood, liver, gill, and kidney) (17). Spermatogonia and the primary spermatocytes were observed to have a haploid chromosome number of 26, whereas all male and female somatic tissues had a haploid chromosome number of 18. This suggested that 8 pairs of chromosomes were missing from somatic tissues. To further quantify the differences between tissues, Feulgen densitometry was used to estimate the relative difference in DNA content between germline and somatic tissues. These measurements revealed that the size of the genome in germ cells was ~26% larger than the genome of erythrocytes, or by extension that these eliminated chromosomes comprised ~21% of the germline genome. Because the reduced number of chromosomes and reduced DNA content in somatic cells were consistent across a wide range of somatic cell types, it was proposed that these germline-specific chromosomes were selectively eliminated during embryogenesis from cell populations that would eventually give rise to the somatic tissues in the adult.
Following on these observations, somatic and germline karyotypes were examined for another hagfish, Myxine garmani. In M. garmani, somatic tissues also possess a reduced number of chromosomes relative to the germline, although the details regarding the eliminated material differ slightly from those observed in E. burgeri. In M. garmani, the haploid chromosome number for somatic tissues (liver, gill, and blood) was 7, whereas the germline (testis) contained two additional large homologous chromosomes that are approximately twice as long as the next largest pair of chromosomes. These chromosomes were estimated to comprise ~30% of the genome. As in studies of E. burgeri, these observations were interpreted as evidence that this pair of large chromosomes is eliminated from somatic progenitor cells during early embryogenesis. To date, eight hagfish species spanning two families (Eptatretidae and Myxinidae) have been shown to eliminate a fraction of their genome from somatic progenitor cells (17, 19–24; summarized in Table 1). Thus, programmed DNA elimination occurs in all species of hagfish examined to date. From these studies, it is evident that there exists substantial interspecies variation in the number of chromosomes and proportion of the genome that are selected for elimination from somatic tissues. As such, programmed DNA elimination likely represents an ancestral, and evolutionarily dynamic, feature of early development shared by hagfish species.
Table 1.
A summary of the chromosome numbers found in the germline and soma across eight species of hagfish, the number of chromosomes eliminated, and the proportion of the genome encompassed by germline-specific DNA
| Species name | Germline haploid chromosome number | Somatic haploid chromosome number | Chromosome pairs eliminateda | % of Genome eliminated | References |
|---|---|---|---|---|---|
| Eptatretus stoutii | 24 | 17 | 7a | 52.8 | 22, 23 |
| Eptatretus burgeri | 26 | 18 | 8 | 21 | 17 |
| Eptatretus okinoseanus (A) | 27 | 17 | 10 | 45.4 | 20, 21, 23 |
| Eptatretus okinoseanus (B) | 27 | 17 | 10a | 55.1 | |
| Eptatretus cirrhatus (A) | 36 | 17 | 19a | 48.7 | 23 |
| Eptatretus cirrhatus (B) | 40 | 17 | 23a | 54.6 | |
| Myxine glutinosa | 22 | 14 | 8 | 43.5 | |
| Myxine garmani | 8 | 7 | 1a | 30 | 19, 20 |
| Paramyxine sheni | 33–48 | 17 | 16–31a | 70+ | 24, 33 |
| Paramyxine atami | 24 | 17 | 7 | 40 | 20 |
These species have been reported to eliminate terminal regions of chromosomes in addition to entire chromosomes.
2.1. Heterochromatin Is Selectively Eliminated from Somatic Cells
A common thread among descriptions of programmed DNA elimination in hagfish is the presence of C-banded chromatin on the germline-specific chromosomes. In nearly all species of hagfish examined thus far, heterochromatic chromosomes are present exclusively in the germline (testes) and eliminated from somatic tissues (25), with the exception of M. garmani, which retains C-positive chromatin in terminal positions within three of its seven somatically retained chromosome pairs (19). In a broad sense, this mirrors observations of the eliminated chromosomes in zebra finch (as discussed below), wherein the germline-restricted chromosome (GRC) was found to be highly heterochromatic in testes, though notably not in oocytes (26, 27).
In general, C-band-positive chromosomes were found to make up a majority of the DNA that is eliminated in hagfish, and in some hagfish, the number of C-band-positive chromosomes found in the germline is equal to the number of chromosomes that are eliminated from somatic tissues. For example, in E. burgeri, the number of C-band-positive chromosome pairs in spermatogonia is eight, which exactly matches the number of eliminated chromosome pairs. Given that C-band-positive chromatin is completely absent from somatic cells, the heterochromatic chromosomes are presumed to be the targets of programmed elimination in this species. In other species, there are fewer C-band-positive chromosomes than eliminated chromosomes, suggesting that some C-band-negative chromosomes, in addition to heterochromatic chromosomes, are eliminated. This is observed in Eptatretus stoutii, in which seven pairs of chromosomes are eliminated from the somatic cell line, but only four of those are C-band positive (22).
Eliminated chromatin may also comprise heterochromatic fragments of chromosomes in addition to chromosomes that are heterochromatic over most of their length. In Eptatretus okinoseanus, individuals belonging to two subtypes, A and B, can be cytogenetically characterized based on the quantity and distribution of heterochromatin in their spermatogonial karyotypes (20, 21). For both types, the haploid chromosome number in spermatogonia is 27, whereas the haploid chromosome number in somatic cells is 17. In type A individuals, all of the heterochromatic chromatin is localized to 10 pairs of C-band-positive chromosomes that are eliminated from the somatic cells during embryogenesis. In contrast, most of the germline chromosomes from type B individuals are C-band positive in their terminal regions in addition to the 10 pairs of heterochromatic chromosomes observed in type A. Because heterochromatin is not observed in somatic cells for either type, this was taken as evidence that both types eliminate 10 pairs of C-band-positive chromosomes, and that type B individuals also excise and remove the terminal heterochromatic regions from their chromosomes. Heterochromatin removal from the terminal ends of chromosomes has also been reported to occur in E. stoutii (22). In spermatogonial metaphases from this species, there are 4 pairs of fully heterochromatic chromosomes and approximately 13 pairs of chromosomes that are heterochromatic only in their terminal regions. Because no heterochromatin is observed in somatic metaphases, it was inferred that these terminal ends must be excised and removed along with the rest of the eliminated material.
The only species of hagfish identified so far that retains any heterochromatin in the somatic cells is M. garmani (19). In spermatogonial metaphases from this species, heterochromatin is localized to the central regions in the pair of the two largest chromosomes, as well as to the terminal ends of three smaller pairs of chromosomes. C-banding in somatic metaphases revealed that heterochromatin was also found in the terminal regions of three pairs of chromosomes, suggesting that, unlike patterns observed in E. okinoseanus and E. stoutii, these terminal regions are not excised but rather retained in the soma. Further studies are clearly needed to resolve the distribution and nature of C-band-positive heterochromatin in hagfish testes, soma, and especially ovaries to more precisely understand why C-band-positive heterochromatin is apparently targeted for complete removal in some species but retained in others. Of note, in the broader context of vertebrate DNA elimination, studies of C-band heterochromatin in hagfish suggest a link between canonical chromatin-based DNA silencing pathways and DNA elimination in a manner that is at least superficially similar to those described for lamprey and zebra finch.
2.2. Eliminated DNA Contains Highly Repetitive Elements
Building on the observation that most hagfish species undergo programmed DNA elimination, subsequent studies shifted their focus toward investigating the nature of the eliminated material at the molecular level in an attempt to shed light on the possible biological consequences of chromosome elimination (28–33). These attempts have resulted in the discovery of multiple families of highly repetitive sequences that are restricted to the hagfish germline. Naturally, these findings have led to questions about what these sequences may encode, whether they are necessary for elimination to occur, and what their biological role is, if any, in the context of the germline and its development.
Initial studies in E. okinoseanus (types A and B), E. burgeri, and M. garmani used restriction enzyme analysis of somatic and germline DNA in an attempt to identify sequences that were enriched in germline (28). These studies led to the discovery of several germline-specific bands upon visualization via gel electrophoresis. In E. okinoseanus, a total of three germline-specific bands from two restriction enzymes (two from BamHI and one from DraI) were identified in both type A and B. However, no germline-specific bands were generated from the E. burgeri or M. garmani digests with these enzymes. When these E. okinoseanus bands were radiolabeled and hybridized against somatic and germline DNA, the BamHI fragments hybridized exclusively to BamHI-digested germline DNA, and the DraI fragments hybridized exclusively to DraI-digested germline DNA. No hybridization signals were observed in the somatic DNA digests for any of the fragments, suggesting that these fragments are exclusive to the germline genome and somatically eliminated. Partial digests of germline DNA hybridized with BamHI fragments revealed a pattern of regularly spaced bands at increasing multiples of 90 bp. This was interpreted as evidence that the BamHI fragments are clustered in tandem arrays of this 90-bp repeat in the E. okinoseanus germline. Furthermore, no hybridization signals for any of these fragments were found in the germline or somatic DNA digests for E. burgeri or M. garmani. Interestingly, when the BamHI and DraI fragments were used as probes against one another, no signal was detected in the germline DNA from either type of E. okinoseanus, suggesting that fragments generated from BamHI and DraI represent two unique repetitive sequences. Sanger sequencing on the two BamHI fragments revealed that both were approximately 95 bp in length and were highly homologous to one another, containing minor single-nucleotide variations and small indels. Thus, the BamHI fragments are grouped together into a single family designated EEEo1 (for Eliminated Element of E. okinoseanus 1). The DraI fragment is approximately 85 bp in length and appears to consist of a single family of sequences, with minor variations present in terms of the length and nucleotide sequence. As such, this single family of sequences comprises the major component of the DraI fragment and was designated EEEo2 (for Eliminated Element of E. okinoseanus 2) (28).
Following the restriction enzyme analysis and hybridization experiments, EEEo1 and EEEo2 fragments were then used as probes for fluorescence in situ hybridization (FISH), which was carried out on somatic and germline metaphases in both types of E. okinoseanus (28). Fluorescent signals for EEEo1 and EEEo2 were observed in the germline metaphases for both types of E. okinoseanus, but no signals were observed in either of the somatic metaphases. Moreover, for both types, the signals were localized to several germline-specific chromosomes that were C-band positive along most of their length. The signals also showed some differences in their quantity and distribution patterns among germline chromosomes. Fluorescent signals for EEEo1 in type A individuals tended to be more numerous, and the signals were concentrated on discreet points in C-band-positive chromosomes. In type B individuals, there was less signal observed overall, and the signals formed a much more diffuse pattern along the length of the chromosomes. As mentioned previously, most of the chromosomes in germline metaphases from individuals of type B, but not type A, had C-band-positive chromatin at their terminal regions. However, no signal for EEEo1 or EEEo2 was observed at the terminal regions of any of the chromosomes for type A (which does not undergo terminal deletion) or type B (which does undergo terminal deletion). Therefore, EEEo1 and EEEo2 appear to be markers of germline-specific chromosomes but apparently not germline-specific termini.
Following up on these findings, 10 more germline-restricted repetitive families have been identified in 4 different species of hagfish; EEPa1 in Paramyxine atami, EEEc1–3 in Eptatretus cirrhatus, EEEb1–2 in E. burgeri, and EEPs1–jl4 in Paramyxine sheni (29–31). Many of these repetitive families were later found to be present in other hagfish, suggesting that they existed in the common ancestor of the two or more species in which they were found and conserved as germline-restricted chromatin after speciation occurred (29–33). EEPa1 provided the first direct evidence that germline-restricted repetitive elements in hagfish may be shared between species, as EEPa1 hybridizes with germline DNA from E. okinoseanus and E. burgeri but not with somatic DNA from either species (29). Sequence comparisons between EEPa1 and both EEEo1 and EEEo2 revealed that these sequences are not homologous to one another and that they represent distinct sequences that are present in E. okinoseanus, P. atami, and E. burgeri. A DNA fragment isolated from E. burgeri using the same restriction digest approaches discussed above was Sanger sequenced and compared with the consensus sequence for EEPa1 from P. atami, revealing that the P. atami and E. burgeri fragments share a 92% sequence identity and are thus highly homologous (29). Despite their high similarity to one another, the hybridization signal seen in the E. burgeri germline using EEPa1 as a probe was weak, even after prolonged exposure. This was most likely due to proportional differences in the copy number in the repeating units between both species, suggesting that the EEPa1-like sequence found in E. burgeri underwent fewer rounds of amplification than that found in P. atami (29).
2.3. Terminal Heterochromatin
Many hagfish species, including E. okinoseanus, E. cirrhatus, E. stoutii, M. garmani, and P. sheni, possess heterochromatin in the terminal ends of their chromosomes in germline metaphases (20, 22, 28, 30, 33). With the exception of M. garmani, these heterochromatic termini are absent from chromosomes in the somatic tissues of all hagfish studied to date, suggesting that they are eliminated simultaneously with entire heterochromatic chromosomes. Moreover, their distribution across meiotic chromosomes seems to have a distinctive pattern, in that they appear to be found almost exclusively on C-band-negative chromosomes, as is the case in E. okinoseanus and P. sheni (in the hagfish literature, reports of C-band-negative chromosomes in germline cells often refer to chromosomes that possess heterochromatin only at their terminal ends). These C-band-negative meiotic chromosomes are thought to be the same chromosomes that are retained in somatic tissues, as evidenced by the observation that the number of C-band-negative chromosomes with heterochromatic termini in germline metaphases often matches the number of C-band-negative chromosomes found in the somatic tissues. In P. sheni, for example, germline metaphases have anywhere between 33 and 48 pairs of chromosomes, of which 17 are C-band negative. Somatic metaphases, in contrast, have exactly 17 pairs of chromosomes, suggesting that these are somatic chromosomes (33). In addition, further studies have shown that many repetitive DNA families are enriched in these heterochromatic chromosome ends (30, 33). In P. sheni, the repetitive DNA families EEPs1, EEps2, EEPs4, and EEEo2 were all found to accumulate at the ends of C-band-negative chromosomes, and these elements were likewise absent from their corresponding locations in somatic chromosomes (33). In addition, FISH experiments in both types of E. cirrhatus showed that EEEc2 is present at the terminal ends of 3–5 pairs of C-band-negative chromosomes (30).
Another question regarding these heterochromatic termini concerns the mechanisms by which they are excised and removed from the chromosomes. Kojima et al. (33) proposed two models whereby the removal of terminal heterochromatin could be achieved. First, subtelomeric heterochromatin could be excised via chromosomal breakage without telomere loss. The elimination of internal, heterochromatic DNA sequences is a well-known process in ciliates, although whether these internal sequences contain repetitive elements is still an active area of research (34). The second model involves the removal of both the subtelomeric chromatin and the distal telomeres, followed by de novo addition of a new telomere cap. The process of chromosomal breakage and de novo addition of telomeres to the newly formed chromosomes is well characterized in nematodes (35). Ultimately, Kojima et al. (33) considered it more likely that chromosome fragmentation followed by subsequent telomere addition is the more probable mechanism employed by hagfish, because the subtelomeric repeats may be too long to simply be excised, although direct empirical evidence to support this model does not yet exist.
2.4. Evolution of Repetitive Sequences
For most of the repetitive hagfish families identified thus far, repetitive sequences show high degrees of sequence similarity within species but low sequence similarity between species, consistent with the action of concerted evolution (30, 31). Concerted evolution—driven by processes such as gene conversion, unequal crossing over, and DNA amplification—leads to the homogenization of repetitive DNA sequences within a species but allows for rapid divergence after speciation has occurred (32, 36, 37). Analyses of the repetitive family EEEo2 provide a more detailed perspective on the evolution of tandemly repetitive sequences on germline-specific chromosomes (32). The EEEo2 repeat family is present in five species of the family Eptatretidae (E. okinoseanus, E. cirrhatus, E. burgeri, P. atami, and P. sheni), suggesting it was present in the common ancestor of this family and retained after speciation. EEEo2 was found to be present at different copy numbers in each of the species examined. The numbers of copies per diploid germline genome are particularly high for E. okinoseanus (7.2–8.2 × 106), E. cirrhatus (1.5–2.2 × 107), and P. sheni (6.3–7.3 × 107), whereas the copy number for E. burgeri is much lower (2.2 × 104). These findings indicate that EEEo2 sequences were amplified frequently in three species but only rarely in E. burgeri. Hybridization experiments showed that neither EEEo1–2, EEPs1–4, EEc1–3, nor EEPa1 were present in any of the species from the family Myxinidae (M. garmani and Myxine glutinosa), suggesting that these repetitive elements were either lost entirely from Myxinidae or originated and retained in the eptatretid lineage after these two families split. Similar to EEEo2, EEEb2 has been found in six hagfish species, E. okinoseanus, E. cirrhatus, E. burgeri, E. stoutii, P. atami, and P. sheni, indicating that this repeat was also present in the last common ancestor of the family Eptatretidae.
Concerted evolution of repeated sequences is thought to provide a strong homogenizing force within species, whereas rates of sequence heterogeneity tend to be much higher between species (36, 37). However, this is not always the case, as the homogenization of repetitive sequences relies on several factors, including population size, natural selection strength, mutation frequency, and repeat copy number. For example, the averaged intraspecific divergences (sequence variability within a species) of EEEo2 in E. okinoseanus type A (1.5%) and type B (2.2%), and E. cirrhatus type A (1.2%) and type B (2.1%), are much lower than in E. burgeri (20.4%) (32). This suggests that the homogenizing forces of concerted evolution in E. burgeri are relatively weak. This could be due to the relatively low copy number of EEEo2 in E. burgeri, as intraspecific divergence tends to increase as copy number decreases (32). In contrast, the averaged intraspecific divergences of the repeat families EEEb1 and EEEb2 discovered in E. burgeri are only 7.9% and 12.5%, respectively (31). Consistent with the predicted effects of copy number on intraspecific divergence, EEEb1 is present in 1.9 × 107 copies per diploid germline genome, whereas EEEb2 is present in 2.7 × 104 copies, which reflects its higher sequence divergence (31). Thus, repeat families within species can be subject to evolutionary forces of differing strength, resulting in substantial differences in the observed sequence homogeneity. Clearly, further studies will be needed to understand fully the intricacies of concerted and other modes of evolution in the repetitive DNA families and possible functions of these repeats within the hagfish germline genome.
3. PROGRAMMED REARRANGEMENT IN LAMPREYS
One of the more recent discoveries of programmed genome rearrangement involves extensive DNA losses that occur in the sea lamprey and several other lamprey species. Lampreys are jawless vertebrates and are living representatives of an ancient lineage that separated from the common ancestor of all jawed vertebrates approximately 500 million years ago and are generally thought to be a sister group to the hagfish (38, but see 39). The initial discovery of programmed genome rearrangement in sea lamprey stemmed from efforts to improve a nascent genome assembly of the species and a knowledge of previous reports of programmed DNA elimination in hagfish. The first germline-enriched sequence was identified via Southern blotting (40). A set of computationally predicted interspersed repetitive elements were radiolabeled and hybridized to blood and sperm DNA from the same individual. These hybridizations revealed bands that were stronger in the germline or apparently unique to the germline. A fragment corresponding to one dense ~10-kb band (called Germ1) was cloned, labeled, and hybridized to both somatic and germline chromosome spreads. The resulting hybridizations showed several strong signals (N = 8) in germline meiotic metaphase I and yielded only a pair of signals in mitotic metaphase cells isolated from gill epithelia. Notably, the sea lamprey karyotype is complex and consists of 84 (somatic) or 96 (germline) pairs of small acrocentric chromosomes that are nearly indistinguishable from one another (40, 41) (Figure 3). Sequencing of this fragment revealed that it was homologous to the ribosomal DNA repeats over much of its length, and alignment to an existing somatic Sanger genome shotgun sequencing data set identified that a 1.2-kb region (termed the somatically rare region) was present at approximately tenfold-lower copy number than the rest of the fragment. Subsequently, this region was identified as corresponding to a derived expansion within the 3′ untranslated region of an R2 retrotransposon (42).
Figure 3.
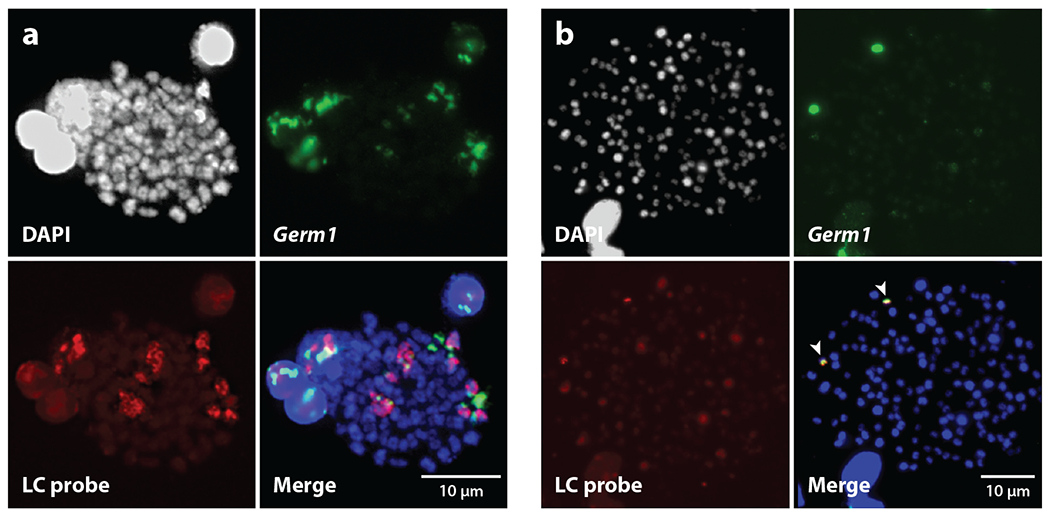
Differences between lamprey germline and somatic karyotypes. (a) A meiotic metaphase I chromosome spread. (b) A mitotic metaphase spread generated from a 16-day-old embryo. Chromosomes were stained with DAPI and labeled with fluorescence in situ hybridization probes to the Germ1 repeat (green) and a laser capture (LC; red) probe, generated from eliminated chromatin (41, 120). Both Germ1 and LC probes hybridize with multiple tetrads in meiotic metaphase I, whereas signals are observed on only one pair of mitotic chromosomes (indicated with arrows). Hybridization of the LC probe to somatic cells yields weaker signals that are only slightly brighter than background fluorescence, in contrast to hybridization patterns on meiotic chromosomes.
Aside from providing a handle to identify programmed rearrangements in sea lamprey, the identification of Germ1 also provided a tool for tracking elimination across tissues and over the time course of early embryogenesis. Real-time probes developed for the somatically rare region of Germ1 revealed that DNA loss was essentially completed by the third day of development and that presumptively germline-specific DNA was absent from multiple somatic tissues, including blood, liver, fin, muscle, and kidney. Moreover, the presence of this one germline-specific element begged the question of whether single-copy sequences (including genes) were also eliminated. Because initial efforts to sequence the lamprey genome used somatic tissues (43), the identification of germline-specific genes required the development of new sequence resources. Successive improvements in sampling of the germline genome through low-throughput bacterial artificial chromosome end sequencing (44), shotgun/transcriptome sequencing (45, 46), and genome assembly (47) revealed a growing and increasingly accurate list of genes that are eliminated via PGR.
3.1. Cellular and Molecular Mechanisms
In many species, embryo accessibility presents a major barrier toward understanding the mechanisms underlying PGR. Fortunately, lamprey embryos are readily accessible at all developmental stages, and thousands of simultaneously fertilized embryos can be produced via in vitro fertilization. Although the annual maturation cycle of wild lamprey populations limits embryo production to summer months, the opportunity to access large numbers of embryos has proven invaluable to efforts to understand the cellular and molecular mechanisms underlying DNA loss. Imaging of embryos that are actively undergoing DNA elimination has revealed several important features that are critical for developing models of the mechanistic underpinning of programmed genome rearrangement (Figure 4).
Figure 4.
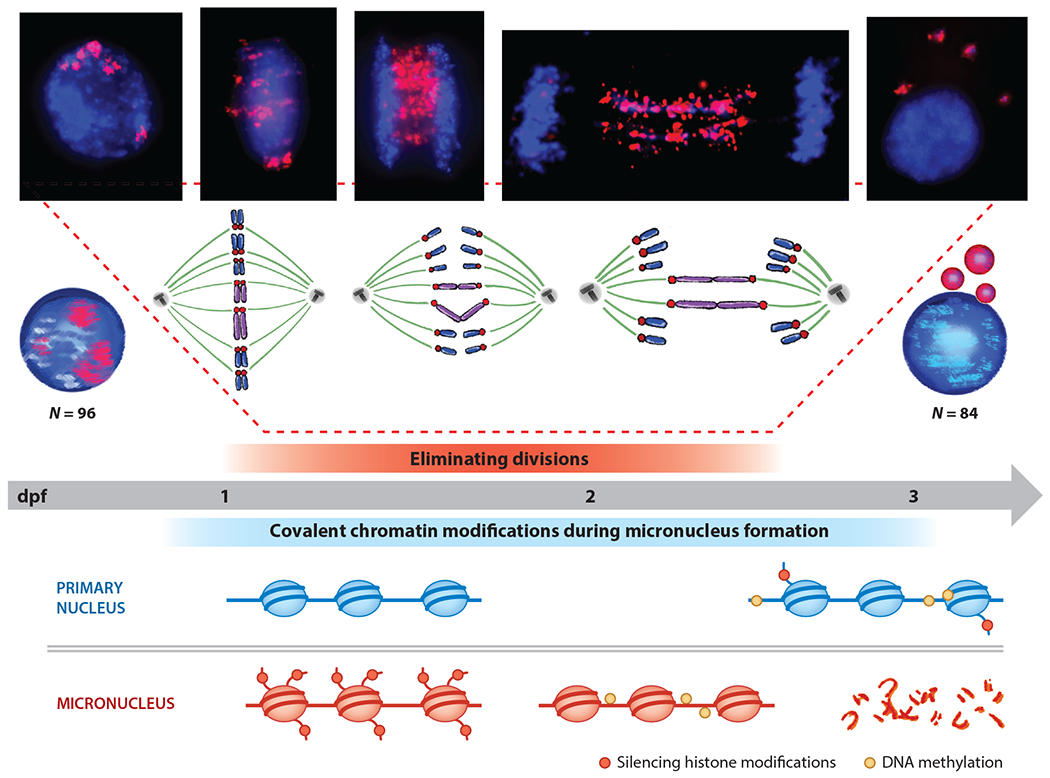
Chromosome elimination during sea lamprey development. Embryos have an initial haploid chromosome number of N = 96. Chromosome elimination is initiated after the sixth cleavage division at one day postfertilization (dpf). Presomatic cells isolate 12 whole chromosomes that are packaged into micronuclei, which initially accumulate silencing marks on histones and then 5-methylcytosine. Primary nuclei show signs of epigenetic silencing later in development, at the time that eliminated chromosomes are being degraded within micronuclei.
In sea lamprey, PGR is initiated at the start of the sixth cell division (48). At the cellular level, the first sign of elimination is the differential motion of germline-specific DNA during anaphase. Eliminated chromosomes appear to engage the spindle fibers at their centromeres, owing to the fact that regions of these chromosomes containing centromeric repeats are oriented toward the spindle poles and move away from the metaphase plane, albeit not as far as the retained chromosomes. As the centromeres of the (generally acrocentric) germline-specific chromosomes move toward the spindle poles, the distal telomeric regions remain near the original metaphase plane. Notably, patterns of hybridization along lagging chromosomes also show an antiparallel pattern, suggesting that sister chromatids interact with one another at their distal ends (41, 48). These observations indicate that much of DNA loss is mediated by manipulation of chromosome movement during anaphase, rather than by DNA recombination/breakage, as was proposed on the basis of early sequence evidence (45), though notably it does not rule out the possibility that recombination and breakage also contribute to DNA loss in lamprey, as has been seen for hagfish.
As the cell transitions into telophase, retained chromosomes recruit nuclear envelope and coalesce to form the nucleus, whereas eliminated chromatin retains a stretched morphology during early telophase. This chromatin ultimately condenses to form small, rounded micronuclei that appear to resist fusion to the primary nucleus. This may be because micronuclear envelope lacks lamin B1 protein and nuclear pore O-linked glycoprotein (48), or because eliminated chromatin is packaged in something other than nuclear envelope. At formation, micronuclei are enriched in trimethylated H3K9 and subsequently accumulate 5-methylcytosine (48). At this point in development, nuclear (retained) chromatin is essentially devoid of 5-methylcytosine; thus, the first de novo methylation events in sea lamprey appear to be targeted to the eliminated chromatin. After accumulation of methylation marks, eliminated DNA undergoes fragmentation and is ultimately degraded within the cell, such that very little of the germline-specific DNA is detectable by the fourth day of development.
In the context of PGR, many of the epigenetic modifications that are enriched in eliminated DNA are observable by immunohistochemistry only after the DNA is packaged into micronuclei. This suggests that DNA and histone methylation may act to reinforce the elimination pathway and/or inhibit transcription between the point at which DNA is slated for elimination (at anaphase) and the point at which it begins to degrade within the micronuclei. Although understanding the interplay between classical epigenetic silencing marks and PGR is likely to provide insight into the evolution of both processes and their integration with development, we anticipate that it will be necessary to look beyond covalent histone and DNA modifications to identify the upstream factors that initiate and orchestrate the early stages of elimination.
3.2. Content and Structure of Eliminated Regions
The recent sequencing and assembly of the lamprey germline genome has provided a more comprehensive view of DNA sequences that are eliminated via PGR and allowed the identification of a large number of genes that are deleted during these elimination events (47). As with earlier studies, this list of genes showed enrichment of germline functions and contains several known oncogenes and cancer/testes genes. The presence of these genes in the germline-specific chromosomes is one of the primary lines of evidence supporting the idea that programmed genome rearrangements act to mediate genetic trade-offs between germline and soma. This growing list of genes has also revealed possible connections between the regulatory logic of PGR and early epigenetic changes that contribute to defining germline versus somatic lineages in mammals. Specifically, several genes that are targeted for elimination in lamprey have gnathostome (mouse) homologs that are regulated by polycomb repressive complex in embryonic stem cells and retain either a poised or activated state in primordial germ cells (47, 49, 50). This suggests that lamprey PGR and gnathostome polycomb repressive complex have at least partially overlapping functions with respect to defining epigenetic silencing in soma versus germline.
Although the germline genome assembly was informative for identifying eliminated genes and achieved chromosome-scale scaffolding for most of the genome, the germline-specific regions remained highly fragmentary, which likely prevents the identification of some eliminated genes, complicates effective comparison of synteny between eliminated and noneliminated regions, and inhibits the identification of adjacent regulatory features that contribute to gene expression or programmed loss. In-depth analyses of repeat content, as well as ongoing improvements in sequencing and assembly pipelines (https://vertebrategenomesproject.org/), are starting to resolve these issues.
Recent in-depth analyses of repeat content in germline versus soma have improved our understanding of the chromosomal structure of the germline-specific DNA and have also identified candidate functional elements that could potentially contribute to the identification of germline-specific sequences during elimination or directly participate in interactions that are necessary for DNA loss. Recent analyses have expanded the number of known germline-enriched repeats beyond Germ1 to include several new repeats that are (or are nearly) exclusive to the germline genome (41). All of these new repeats are short (13–57-bp) satellite repeats and show much stronger enrichment in germline than Germ1, which may not be surprising given that the discovery of Germ1 was predicated on repetitive elements that were mined from available somatic genome sequencing data. The development of FISH probes for these repeats makes it possible to identify 12 germline-specific chromosomes, each of which can be individually identified on the basis of its unique distribution of repetitive elements. As the lamprey assembly improves, knowledge of the distribution of these repeats within and among germline-specific chromosomes should also allow anchoring and validation of their assemblies. With respect to the cellular basis of elimination, two of these elements seem particularly intriguing. Two elements known as Germ2 and Germ6 are found on all 12 germline-specific chromosomes and also localize to a zone of interaction during anaphase wherein eliminated chromosomes appear to maintain contact with their sister chromatids at the original metaphase plane.
As in zebra finch (discussed below), a growing appreciation of parallels between DNA loss and silencing has revealed that there are likely to be functional interactions between these mechanisms both during development and over evolutionary time. The identification of germline-specific repeats in lamprey and in particular an understanding of their location in elimination anaphases suggest that these could be used as a tool for identifying other factors that bind and dictate the movement of eliminated DNA during anaphase. Moreover, these raise the possibility that germline-specific satellites observed in hagfish might represent not only passive passengers of elimination but potentially also functionally relevant motifs that permit the identification of the DNA during the process of elimination in hagfish embryos (28–33).
3.3. Evolutionary Conservation of Programmed Rearrangements
Thus far, our understanding of evolutionary conservation of PGR (as executed by lamprey) is limited to several relatively closely related species of lamprey that occur in the northern hemisphere and which shared a common ancestor approximately 30 million years ago. Three-dimensional-FISH experiments on Pacific lamprey (Entosphenus tridentatus) embryos revealed lagging chromatin structures that are similar to those observed in similarly staged sea lamprey embryos. These also showed some cross-hybridization with germline-specific probes developed for sea lamprey. Notably, though, the amount of lagging material in Pacific lamprey appears to be smaller than that in sea lamprey, which may not be surprising given the large difference in genome size between sea lamprey and other northern-hemisphere lampreys [sea lamprey: 1.82 Gb germline, 2.31 Gb somatic (40); others: ~1.3 Gb somatic (51)]. Hybridization of sea lamprey germline-specific probes to Pacific lamprey lagging anaphases seems to indicate that the content of eliminated material has changed over the course of evolution, as the hybridization pattern of germline-specific probes shows overlap with both lagging chromatin and normally migrating chromosomes. Estimates of genome sizes of germline (~1.6 Gb) and somatic (~1.4 Gb) tissues in Lampetra morii indicate that elimination also occurs in this species (52). Efforts to characterize Germ1 in other lamprey taxa provide further evidence that PGR has existed in the lamprey lineage for at least the last 3 0 million years. Germline-enriched bands are detected for Germ1 homologs in several species of northern hemisphere lamprey, including representatives of all genera (42). As we discuss below, expanding searches for programmatic losses beyond known taxa will be critical to resolving the origins and evolution of elimination events in the vertebrate lineage.
4. A GERMLINE-RESTRICTED CHROMOSOME IN BIRDS
Within birds, the first germline-specific chromosome was discovered in the zebra finch (Taeniopygia guttata) during analyses of male and female meiotic germline (26). In the course of examining the meiotic behavior of sex chromosomes, ZW in females and ZZ in males, an additional single accessory element was discovered in all female oocytes and male gametes. This chromosome is known as the germline-restricted chromosome (GRC) and is the largest visible chromosome in the germline. The large size of this chromosome undoubtably facilitated the fortuitous discovery of the GRC in zebra finch. It was proposed that this accessory chromosome evolved from a B chromosome, a type of selfish genetic element that is present in various plant and animal species but in the case of zebra finch evolved to function and reside specifically in the germline (26, 27). In the zebra finch, the GRC is absent in all somatic tissues of females and males examined thus far. Somatic tissues assayed thus far include bone marrow, liver and skin fibroblasts, adult skin cells, and both muscle and skin cultures from embryos (26, 53).
4.1. Identification and Characterization of Germline-Restricted Chromosomes in Other Passerines
The discovery of the GRC inspired a search for similar chromosomes in other bird species. The second species to be identified as possessing a GRC was the Bengalese finch (Lonchura striata domestica), a close relative to the zebra finch (6). The GRCs of these finches are very similar, in that both are large, acrocentric, present in two copies in female and in one copy in male germline, absent from all somatic cells, and eliminated during spermatogenesis (6, 26, 27). Building on this, the recent discovery of the first protein-coding gene localized to the GRC of the zebra finch hinted at the possibility of a deeper ancestry of the GRC within the avian lineage (54, 55). Using a subtractive transcriptomic approach and RNA sequencing data from germline tissue of male and female adult birds, Biederman et al. (54) identified a single gene, α-SNAP, a member of the α-soluble N-ethylmaleimide-sensitive fusion attachment protein, that was localized to the GRC and was missing from zebra finch gene annotations. Intriguingly, these two α-SNAP genes showed substantial divergence, even at the amino acid level, and patterns of substitution that were consistent with long-term diversifying selection after an ancient autosomal duplication event led to the formation of separate germline-restricted and somatic copies (54).
This finding was rapidly followed by the discovery of GRCs in 16 species of songbirds (56). A broad comparative cytogenetic study of the germ cell karyotypes from 24 avian species representing 8 orders demonstrated that the GRC was present in all major passerine groups (56). The development of microdissected finch GRC libraries and subsequent sequencing of these libraries permitted the identification of an additional three genes (dph6, gbe1, and robo1) and multiple repetitive sequences. The majority of these repeats were long terminal repeats and long interspersed nuclear elements. However, it appears that there may not be enrichment in transposable elements or satellite repeats (as in hagfish and lamprey) in germline samples relative to soma samples (57).
Several lines of evidence indicate that the GRC likely plays a functional role in the germline, beyond the simple fact that it has been retained over evolutionary time. Early studies resolving the euchromatic state of the GRC indicate that the chromosome is transcriptionally active at least during oogenesis (27). Analysis of the GRC at the lampbrush stage also points to its active transcriptional state during oogenesis. In oocytes, transcribed regions extend laterally from otherwise condensed chromosomes, forming ornate loops that are often maximally loaded with RNA-polymerase II, to allow for the increased production of proteins in large meiotic cells (58). The presence of such loops on the GRC, in conjunction with RNA-polymerase II labels, suggests that the GRC is being actively transcribed in oocytes and contributed to germ cell biology, at least in females (56). More recently, the compilation of a growing list of GRC-specific genes has provided more detailed insight into the possible roles of this chromosome and seems to support the idea that this genetic element is not merely a parasitic selfish B chromosome but rather contributes significantly to the process of germline development not only in zebra finch but presumably in ~5,000 species of songbirds (56, 59). It is worth noting that the passerines comprise approximately 10% of known vertebrate species, meaning that at least in terms of raw numbers of taxa, programmed DNA elimination can probably no longer be considered a rare phenomenon.
The most comprehensive list of GRC-encoded genes to date was compiled using 10X single-molecule genome-sequencing technology that permits reconstruction of long haplotypes through linked read and analysis of single-nucleotide variants (SNVs) (57). This analysis revealed that the zebra finch accessory chromosome contains more than 115 genes paralogous to single-copy genes on 18 somatic autosomes and the Z chromosome. Several of these genes are amplified on the GRC, with some being represented by up to 300+ copies per gene. Among these GRC genes, transcription was detected for 6 in testes and 32 in the ovaries. As might be expected from studies of lamprey and rearranging invertebrates [e.g., roundworms (60)], the GRC is enriched in genes that are highly expressed in germline tissues of other species that are not known to possess germline-specific DNA (chicken and human). Therefore, the GRC of songbirds appears to play an important role in both female and male germline development. It seems plausible that its elimination from somatic cell lineages also aids in resolving soma-germline conflict (antagonistic pleiotropy), as has been proposed for other species that undergo programmed elimination.
4.2. Proposed Model of the Evolution of the Avian Germline-Restricted Chromosomes
The discovery of GRCs within representatives of all major passerine groups suggests that the chromosome was present in an ancestral bird that gave rise to half of all extant bird species. Cytogenetic analyses revealed that the GRC varied in size (macro-GRC or micro-GRC) and genomic content among species (56). Surprisingly, transitions between macro- and micro-GRCs seem to have changed rapidly over evolutionary time. For example, within the family Hirundinidae, pale and sand martins have macro-GRCs, but barn swallows have a micro-GRC. Despite changes in size, hybridization studies indicate that GRC content may change in a way that is more consistent with phylogenetic relationships. FISH experiments using GRC-derived probes suggest that distant species are less similar in GRC content given that the intensity of specific GRC signals was much lower than in intraspecies FISH. Notably, GRC-derived probes also cross-hybridized with A chromosomes, indicating homology of the GRCs to repeated and unique regions present in somatic genomes. These hybridizations are consistent with genomic analyses indicating that many A chromosome genes have been duplicated onto the GRC (57).
It has been proposed that the GRC formed from an ancestral selfish B microchromosome as the product of a whole-chromosome duplication (56). As such, this proto-GRC may have already contained several copies of A-chromosome genes that contributed to reproductive and developmental processes. Subsequent duplications of these and other translocated genes would presumably increase dosage of gene products deriving from these genes and permit functional diversification of these genes. Along these lines, it was found that 17 GRC-linked genes are evolving faster than their somatic paralogs, and 9 appear to be evolving under long-term purifying selection (57).
4.3. Inheritance of the Germline-Restricted Chromosomes and Elimination During Spermatogenesis
Although essentially nothing is known regarding the somatic loss of the GRC during zebra finch embryogenesis, it has been reported that the GRC is eliminated during spermatogenesis and passed to the next generation only through the female germline (27). In the male germ cells, the single GRC is regularly present in prophase, observed at early stages (leptotene–zygotene) as a densely packed body composed of a coiled chromatin filament. At this stage, the GRC lacks key components of the synaptonemal complex, unlike other chromosomes. In spermatogonial mitoses in metaphase I, the GRC is distinguishable as an unpaired heterochromatic chromosome. This chromosome is present in approximately half of meiotic metaphase II cells, as would be expected if its unpaired state results in inheritance by only one daughter cell following metaphase I. During spermatid maturation, the GRC is ejected as a dense heterochromatic body, and as a result, spermatozoa do not carry the GRC.
Available evidence suggests that the GRC is transmitted to the next generation only by the oocyte, at least in finches. During oogenesis, the GRC is present in two copies, forming a bivalent in meiotic prophase with a typical synaptonemal complex, terminal kinetochores, and two or three foci of recombination (27). This raises the question of how the germline constitution of the GRC is achieved (one copy in males versus two in females). If it is true that both male and female zygotes inherit one copy of the GRC, there should be a specific mechanism for the formation of a second copy in the female germline. This could be achieved by selective nondisjunction of GRC homologs in mitotic divisions during germline–soma differentiation during early female embryogenesis, thereby resulting in primordial germ cells that are diploid for the GRC and somatic cells lacking the GRC (27, 61). During male development, expulsion or degradation of GRC chromatids from somatic progenitors could result in its absence from somatic tissues, with a monosomic restricted chromosome in germline resulting from maintenance of the zygotic karyotype that was established at fertilization (61).
Unfortunately, analyses of meiotic divisions during early embryogenesis have yet to be performed, partly owing to the difficulty of accessing this material. Female meiotic divisions occur one to two hours before ovulation, the timing of which seems impossible to determine (62); early mitotic divisions likely occur while the egg is passing through the oviducts; and moreover the egg is telolecithal (with uneven distribution of yolk in the cytoplasm), with the embryo forming as a small disc of cytoplasm sitting atop a large yolk. Although these do not present absolute barriers toward studying the embryonic behavior of the GRC, they certainly present major challenges. As such, any discussion of the cytological events surrounding chromosome elimination in the zebra finch is largely speculative.
4.4. Variation in Germline-Restricted Chromosome Transmission and Inheritance
A recent analysis has shown that there is variation in the number of the GRCs in pale and sand martins, Riparia riparia and Riparia diluta (62). In martins, as in the zebra finch, the GRCs are usually present in two copies in female and in one copy in male pachytene cells. However, in approximately 15% of females (4/27 examined), the GRC was present in one copy with no detected mosaicism. A similar chromosome number polymorphism was discovered in pale martin males, wherein two out of nine nonmosaic males (22%) had a single copy of GRC, which forms a univalent in all examined pachytene cells. An additional seven males examined in this study were found to be mosaic for the number of GRCs: Six individuals carried two copies in a fraction (2–61%) of their germ cells, and one individual carried three GRCs in 6% of its cells. This observation is also seemingly consistent with the variation in zebra finch GRC constitution that was reported across early studies. Based on electron microscopy of the synaptonemal complex, it was proposed that both females and males possessed an asynaptic accessory axis, along with two-axis autosomal SCs and a nonequalized ZW pair, implying the presence a single GRC in the germline of both sexes. It was also assumed that the preferential segregation to the oocyte-forming nucleus takes place while GRC-free polar bodies are formed. These oocytes, being fertilized by GRC-free spermatozoa, would reestablish the presence of a single GRC in males and females (26). Considered in light of more recent studies showing synapsed GRC in oocytes (27), this could indicate either that electron microscopy images were misinterpreted or that variation in GRC number occurs at a similar frequency in zebra finch (1 out of 7) and martins.
4.5. Mechanisms of Elimination in Relation to Epigenetic Silencing
Although when and how somatic cells expel the GRC during embryogenesis are completely unknown, the cellular features of meiotic elimination of the GRC during spermatogenesis have been relatively well described in finches (6, 11, 63). Consistent with cytogenetic descriptions of a condensed and heterochromatic GRC (26), the male meiotic GRC has also been observed to accumulate several epigenetic marks that are associated with transcriptional repression, H3K9me2 and -me3, H4K20me2 and -me3, and macro H2A, and lack marks of active chromatin, H3K9ac, H4K5ac, H4K8ac, H4K12ac, H3K4me2, and H3K4me3 (6, 11, 63) (Figure 5). During oogenesis, the GRC is only partially enriched with the heterochromatic marker H3K9me3, although the signal is much less intense in comparison to the single GRC that is slated for elimination in spermatocytes (63). This difference in accumulation of heterochromatic marks mirrors the fate of the GRC in sperm (loss) versus oocytes (retention) and likely explains observed differences in the transcription of genes on the GRC, wherein only 6 GRC-linked genes were transcriptionally detected in the testes, compared with 32 in the ovaries (57).
Figure 5.
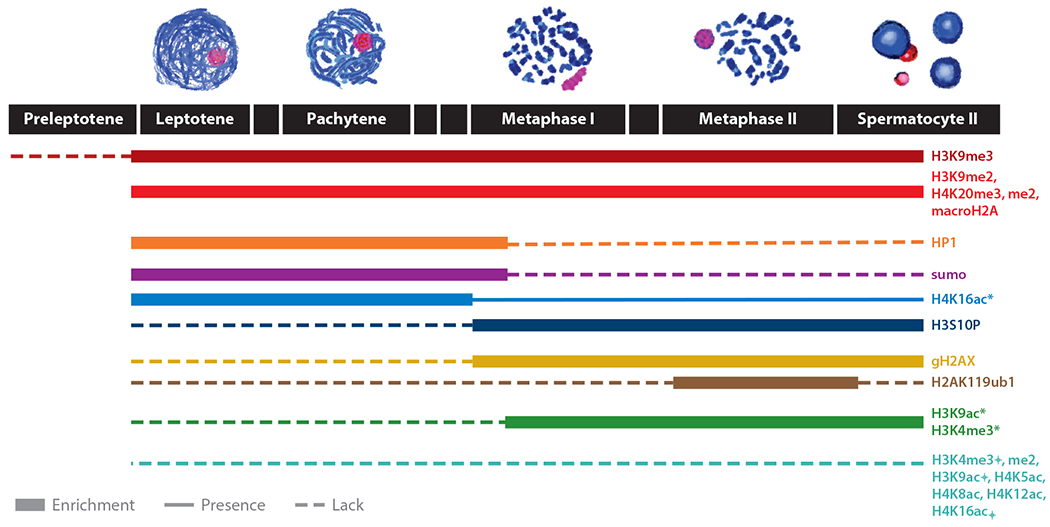
Schematic overview of epigenetic modifications that are visible on the germline-restricted chromosome during male meiosis of the zebra finch and Bengalese finch (6, 11, 63). Differing results from Schoenmakers et al. (63) (*) and Goday & Pigozzi (11) (⌖).
Other heterochromatin-related proteins are also associated with the GRC and seem to shed some light onto the transition between silencing and loss. One of these proteins is SUMO1 (small ubiquitin-related modifier-1). This protein participates in silencing and heterochromatinization of the XY-body in mouse and is present in constitutive heterochromatin in human (64, 65). SUMO1 was strongly associated with the GRC during meiotic entry from leptotene onward and remained through late pachytene (63).
Another protein found to be enriched on the male GRC is heterochromatin protein 1 (HP1). In general, the accumulation of heterochromatic di- and trimethylated H3K9 is closely associated with accumulation of HP1 (66, 67). Immunostaining revealed that anti-HP1β, though not anti-HP1α, yielded strong signal on the GRC univalent during meiotic prophase (11). Importantly, after meiosis II, HP1 is no longer observed on the GRC, whereas the GRC is still strongly enriched with H3K9me2–3 and H4K20me2–3 marks. This may be due to the accumulation of H3S10 phosphorylation (H3S10P), which could cause the release of HP1 (11). Cell cycle–dependent phosphorylation of H3S10 is related to chromosome condensation in metaphase and proper chromosome behavior during mitotic and meiotic divisions (68–71). H3S10P is detected during spermatogonial mitoses in all metaphase chromosomes, including the GRC, but the GRC is devoid of this modification during meiotic prophase and in metaphase I, whereas this same mark persists on the A chromosomes (6, 11). The observation of differential phosphorylation of H3S10 suggests the possibility that the GRC is differentially targeted by one or more kinases or phosphatases that modify the S10 residue (72). Notably, the GRC also lacks the inner centromere protein, which is known to regulate Aurora kinase B to ensure normal metaphase chromosome alignment (63, 73, 74). Such epigenetic isolation of the GRC may lead to DNA fragmentation following metaphase I, as evidenced by intense labeling of the GRC with markers of double-strand DNA breaks, including γH2AX, H2AK119 ubiquitylation, TUNEL staining, and deubiquitylation of H2A, which precede GRC expulsion (63).
In summary, the broadscale differences in GRC epigenetic status during spermatogenesis appear to be attributed to silencing of this chromosome in male meiotic germline when its expression is no longer needed, and the unique patterns of epigenetic marks may help guide the chromosome to its eventual elimination.
5. SEX CHROMOSOME ELIMINATION IN MAMMALS
Essentially all reports of chromosome elimination in mammals involve sex-chromosome elimination or mosaicism. Almost all therian mammals are characterized by a XX/XY sex-determining system with heterogametic (XY) males and homogametic (XX) females. In many cases, sex chromosome elimination appears to be an extension of dosage compensation, which is achieved by epigenetic inactivation of one of the two X chromosomes present in the female (75). In eutherian mammals, the chromosome-wide inactivation of one of the X chromosomes occurs randomly with respect to parent origin and takes place during early embryogenesis, after which point inactivation is stable and somatically heritable.
Therefore, female mammals are composed of clonal cell populations in which the maternally or paternally derived chromosome is silenced by epigenetic chromatin modifications (76). In contrast to eutherians, X inactivation in marsupials is less random and results in preferential silencing of paternally derived X chromosomes (77). However, owing to the lack of an X-inactivation center (and the absence of the gene XIST), X-linked gene expression may differ among loci, tissues, and species (78).
A noncanonical cytological manifestation of dosage compensation in the form of chromosome elimination was shown in several species of peramelid bandicoots. Cytogenetic patterns consistent with X-chromosome loss from some somatic tissues of XX females and Y-chromosome loss from somatic tissues of XY males were first observed in three Australian bandicoots: Isoodon obesulus, Isoodon macrourus, and Perameles nasuta (79). Subsequent studies of Peramelidae species showed that sex chromosomes are lost from some or all cells of the liver, spleen, bone marrow, thymus, lung, and kidney and retained in skin fibroblasts, testes, and ovarian tissues (Table 2). Remarkable variation was found in the percentage of sex chromosome loss in different tissues and individuals depending on sex and developmental stage, which might be explained partially by age-related hematopoietic activities of various tissues, including spleen, thymus, and liver (80), and by the presence of X-monosomic blood cells in other tissues, including lung and cornea (81). Additionally, in male I. macrourus, the Y chromosome has been found to be retained in a small fraction of hematopoietic tissues and peripheral blood, tissues that were previously believed to completely eliminate the Y chromosome (82). In female I. obesulus, the eliminated X chromosome is paternal in origin (82), i.e., a chromosome that normally undergoes somatic inactivation in all marsupials (77). Because the paternal X chromosome in females and Y chromosome in males are characterized by late DNA replication, and the absence of a second sex chromosome is rather mosaic, the elimination of these chromosomes is thought to be related to dosage compensation (83–90). Under this model, chromosome losses are caused by a mitotic error and should be more noticeable at later stages of development. Since the discovery of sex chromosome elimination in bandicoots, several attempts have been undertaken to search for evidence of sex chromosome loss in other marsupial species. Some evidence of somatic Y-chromosome loss from males and somatic X-chromosome loss from females has been described in two closely related species within the subfamily Hemibelideinae, formerly Petauridae: the greater glider (Petauroides volans) and the lemur-like possum (Hemibelideus lemuroides) (77, 91) (Table 2); however, it seems that chromosome elimination may not be widespread in marsupials.
Table 2.
Summary of mammalian species that have been observed to undergo reproducible (or semi-reproducible) sex chromosome loss
| Species | 2n | Eliminated chromosome | Tissue/chromosome constitution | Reference |
|---|---|---|---|---|
| Peramelid species | ||||
| Northern brown bandicoot (Isoodon macrourus) | 14 | X – females Y – males |
Hematopoetic – 13, XO Leucocytes – 13, XO Spleen – 13, XO Corneal epithelium – 14, XX (females)/13, XO (males) Intestinal epithelium – 14, XX (or XY)/13, XO Liver – 14, XX (or XY)/13, XO Thymus – 14, XX (or XY)/13, XO Lung – 14, XX (or XY)/13, XO Fibroblasts – 14, XX (or XY) Heart – 14, XX (or XY) Kidney – 14, XX (or XY) Testes – 14, XY Ovarian tissue – 14, XX |
79–81, 83 |
| Southern brown bandicoot (Isoodon obesulus) | 14 | X – females Y – males |
Bone marrow – 13, XO Leucocytes – 13, XO Spleen – 13, XO Corneal epithelium – 14, XX (or XY)/13, XO Intestinal epithelium – 13, XO Testes – 14, XY Ovarian tissue – 14, XX |
|
| Long-nosed bandicoot (Perameles nasuta) | 14 | X – females Y – males |
Bone marrow – 13, XO Leucocytes – 13, XO Spleen – 13, XO Corneal epithelium – 14, XX (or XY) Intestinal epithelium – 14, XX (or XY) Liver – 14, XX (or XY)/13, XO Thymus – 14, XX (or XY)/13, XO Lung – 14, XX (or XY)/13, XO Heart – 14, XX (or XY) Kidney – 14, XX (or XY) Testes – 14, XY Ovarian tissue – 14, XX |
|
| Eastern barred bandicoot (Perameles gunnii) | 14 | X – females Y – males |
Bone marrow – 13, XO Leucocytes – 13, XO Spleen – 13, XO Corneal epithelium – 14, XX/14, XY Testes – 14, XY Ovarian tissue – 14, XX |
|
| Common spiny bandicoot (Echymipera kalubu) | 14 | X – females Y – males |
Bone marrow – 13, XO Corneal epithelium – 14, XX (or XY) |
84 (cited in 80, 81) |
| Striped bandicoot (Peroryctes longicaudata) | 14 | X – females Y – males |
Bone marrow – 13, XO Corneal epithelium – 14, XX (or XY) |
|
| Long-nosed spiny bandicoot (Echymipera rufescens) | 14 | X – females (only one female examined) | Bone marrow – 13, XO Corneal epithelium – 14, XX |
85 (cited in 80, 81) |
| Petaurid species (78)/Subfamily: Hemibelideinae (modern classification) | ||||
| Greater glider (Petauroides volans) | 22 | Y – males | Bone marrow – 21, XO Liver (15 days old) – 22, XY Liver (35–80 days old) – 21, XO/22, XY (20–25%/75–80%) Spleen (60–100 days old) – 21, XO/22, XY (55–50%/45–50%) Skin fibroblasts – 22, XY Spermatogonia – 22, XY |
86 |
| Lemur-like possum (Hemibelideus lemuroides) | 20 | X – females Y – males |
Bone marrow – 19, XO/20, XX (66–100%/0–34%) Bone marrow – 19, XO (males) Skin fibroblasts – 20, XX or XY Spermatogonia – 20, XY |
91 |
| Eutherian mammals | ||||
| Spiny mouse (Tanzanian Acomys) | 60 | X – females Y – males |
Bone marrow – 59, XO/60, XX and 59, XO/60, XY (97%/3%) | 99 |
| Oregon meadow mouse (Microtus oregoni) | 18 | X-loss in male meiosis Nondisjunction X in female meiosis |
Male soma – 18, XY Spermatogonia – 17, YO Female soma – 17, XO Oogonia – 18, XX |
101 |
Therian mammals are generally considered to have a stricter XX/XY sex-determining system, with the XIST gene being responsible for centralized whole–X chromosome inactivation. However, unusual sex chromosome karyotypes have been described in several eutherian species, although most consist of translocation events within the rodent lineage, and only a few cases involve sex chromosome loss (92–98). To our knowledge, only one rodent species with sex chromosome mosaicism in somatic tissues (bone marrow) has been reported thus far, an unnamed species of spiny mouse (Acomys) from Tanzania (99). Both males and females are mosaic for sex chromosomes in bone marrow cells (Table 2). In this Tanzanian Acomys, one X chromosome is lost almost entirely from female hemopoietic cells, with 97% of cells possessing a single giant X chromosome, and males possess ~70% XO and 30% XY cells in somatic tissues and 100% XY cells in the germinal lineage. Patterns described for Tanzanian Acomys are reminiscent of those described for the marsupial family Hemibelideinae (82, 89), members of which also show varying scales of X-chromosome silencing from mosaicism to complete loss. Another, somewhat contrasting example of gonosomic mosaicism has been reported for the Oregon meadow mouse, Microtus oregoni (100, 101). The chromosomal complement of male somatic cells consists of 18 chromosomes with an X and a Y; however, all spermatogonial metaphases have an odd number of chromosomes (2N=17). Observations of M. oregoni spermatogenesis imply that X chromosomes are being eliminated from germ cells, resulting in two types of sperm cells: O and Y. In this species, the sperm lacking a sex chromosome is female determining because females have an odd chromosome number (17, XO) and produce XX oogonia by selective nondisjunction, which results in the production of X-bearing oocytes.
Based on observations reported thus far, it seems that sex chromosome loss in mammals is generally related to dosage compensation and the chromatin condition of silenced X chromosomes or highly heterochromatinized Y chromosomes. However, cases of complete or large-scale sex chromosome elimination from some tissues might not be explained simply by delayed DNA replication and mitotic errors. Some determining factors may play a role in sex chromosome elimination, perhaps related to covalent chromatin modifications similar to those discussed above, tissue specificity, and the sequence content of eliminated chromosomes. However, the variety of patterns of sex chromosome loss seems consistent with the idea that many may be incidental to dosage compensation and caused by mitotic errors related to late replication, in which case chromosome loss could be nearly selectively neutral. These errors might be influenced by a combination of several factors, such as age, cell-cycle rate, tissue environment, or the state of differentiation of cell lineages (81).
6. EVIDENCE FOR PROGRAMMED REARRANGEMENT IN HOLOCEPHALANS
Programmed DNA loss has also been observed to a degree in at least one representative of all extant holocephalan genera (including Hydrolagus colliei, Callorhinchus milii, Chimaera monstrosa, and Harriotta raleighana) (102). These species shared a common ancestor ~170 million years ago (14), suggesting the possibility that DNA elimination has persisted for at least this long in the holocephalan lineage. In these species, DNA loss is known to occur during male meiosis. As yet, only one study has reported male meiotic loss in species within this lineage, and among these the most in-depth description was reported for the ratfish H. colliei. In the ratfish, eliminated DNA becomes separated from the rest of the genome at meiotic metaphase I and is packaged in a double membrane. This membrane-bound material is passed to one of four spermatids at meiosis II and is eventually taken up by the Sertoli cells during later stages of spermatogenesis. This pattern of loss is superficially similar to elimination of germline-specific chromatin during spermatogenesis in zebra finch, which raises the intriguing possibility that embryonic eliminations might also occur in the holocephalans. However, essentially nothing is known regarding the embryological details of DNA elimination or the sequence of eliminated DNA in Holocephali.
7. OPEN QUESTIONS AND FUTURE DIRECTIONS
This review covers a set of vertebrates that are known to undergo programmed (or at least semi-reproducible) DNA elimination. These studies have shown that programmed elimination occurs in a fairly large number of taxa (especially birds) and that it is intimately related to other canonical forms of silencing. One fact that should also be clear is that there is still much to be learned with respect to the gene targets of programmed elimination in each lineage, the cellular mechanisms that mediate DNA loss, and the occurrence of programmatic losses across the vertebrate tree of life.
In at least two cases (lamprey and birds), it seems clear that programmed eliminations serve to control the gene content of the germline relative to somatic cell lineages to ensure that specific functions are limited to the germline. Other species, particularly hagfish, likely also use programmed elimination to mediate genetic trade-offs between germline and soma, along with other modes of epigenetic silencing. As yet, there are no published assemblies of hagfish germline DNA (although these may soon be forthcoming), but given the large amount of DNA that is eliminated in some species (up to ~70% of the genome) and the apparent deep evolutionary conservation in the lineage, it would be shocking of there were no germline-specific protein-coding genes in hagfish.
A further reason for generating germline sequence information from hagfish is that this information may illuminate the evolutionary origins of programmed elimination in the agnathans. The divergence of ancestral lamprey and hagfish lineages was nearly concurrent, in evolutionary time, with their divergence from the ancestral lineage that gave rise to all jawed vertebrates (38, 39). Sequence data from hagfish, perhaps along with resolution of events occurring in Holocephalans, should allow us to begin to test whether programmed DNA elimination has evolved multiple times in the vertebrate lineage, or whether there is evidence for shared ancestry of PGR among these taxa, which would support the somewhat more intriguing but not mutually exclusive possibility that programmed DNA elimination was a feature of the ancestral vertebrate lineage.
In addition to a need to resolve evolutionary patterns within the deepest branches of the vertebrate tree, we currently have an exceedingly limited understanding of the phylogenetic distribution of taxa that undergo programmed elimination within the jawed vertebrates. Discoveries of DNA elimination have followed parallel paths, wherein initial discoveries have relied on careful follow-up of unexpected cytogenetic observations and extension of these observations to closely related taxa. Perhaps not surprisingly, initial discoveries are often made in species wherein large differences exist between germline and soma. However, in the case of lamprey and birds, extension to other taxa has identified species in which the amount of germline-specific DNA is much smaller than in the species that were used to discover DNA elimination in the lineage. It can be argued that in the vast majority of species we do not know whether programmed elimination occurs during development. Given the relative ease of DNA sequencing and the development of computational methods for identifying germline-specific sequences or duplications, we anticipate that the rate of discovery of species with programmed DNA elimination will increase. In the case of lamprey and zebra finch, initial assemblies were generated from somatic tissues (43, 103, 104), followed by later assembly of germline genomes (47, 57), but in retrospect targeting germline DNA from the onset would have certainly facilitated the discovery, validation, and characterization of programmed elimination.
Finally, and perhaps more challenging than assaying for the presence of programmed elimination, is the characterization of the cellular mechanisms that underlie the process. Work in lamprey, finch, and several invertebrate species (60, 105–108) has shown that programmed elimination is intimately related to other forms of epigenetic silencing. These findings allow programmed elimination to be appropriately conceptualized as one of many epigenetic mechanisms and provide important perspective on the broader functionality and evolution of DNA methylation and histone-mediated silencing. It seems likely, however, that other mechanisms are required to specifically identify DNA that is slated for elimination, direct its differential motion or behavior in the cell during elimination, and regulate its stability in the germline.
Achieving these goals will require observation and manipulation of programmed elimination during the embryonic stages in which it occurs. Lamprey embryos are substantially more accessible and can be produced in large numbers during the annual lamprey spawning season (109). This has permitted an in-depth description of events that unfold during PGR and is opening the door to functional analyses by building on existing methods for gene knockdown (109–111), knockout (112, 113), and transgenesis (114, 115). Embryonic elimination events have yet to be observed in hagfish and finch, largely owing to the difficulty of obtaining embryos at the appropriate stages. It does not seem that it will be impossible to obtain appropriately staged embryos from either of these species. However, the embryology of these species clearly presents challenges, as the appropriate finch stages likely occur while the egg is still in the oviduct, and only a handful of pre-neurula hagfish embryos have been observed from collection that occurred more than a century ago (116), although later-stage hagfish embryos have been observed in the lab more recently (117, 118).
In closing, although much remains to be learned regarding the mechanisms and evolution of programmed DNA elimination in vertebrates, the three-and-a-half decades since the discovery of the first eliminating vertebrate have brought tremendous progress in understanding the large number of diverse taxa that experience programmatic DNA loss and a nascent understanding of the mechanisms by which DNA loss is achieved. Given the momentum these studies provide, we expect that the next decade will bring exponentially greater progress in these areas and a broader appreciation for the role of PGR in the evolution of vertebrate genome biology.
ACKNOWLEDGMENTS
This work was funded by grants from the National Institutes of Health (NIH) (R35GM130349) and National Science Foundation (NSF) (MCB1818012) to J.J.S. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or NSF.
Footnotes
The Annual Review of Animal Biosciences is online at animal.annualreviews.org
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Das S, Hirano M, Tako R, McCallister C, Nikolaidis N. 2012. Evolutionary genomics of immunoglobulin-encoding loci in vertebrates. Curr. Genom 13:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirano M, Das S, Guo P, Cooper MD. 2011. The evolution of adaptive immunity in vertebrates. Adv. Immunol 109:125–57 [DOI] [PubMed] [Google Scholar]
- 3.Saha NR, Smith J, Amemiya CT. 2010. Evolution of adaptive immune recognition in jawless vertebrates. Semin. Immunol 22:25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boveri T. 1887. Über differenzierung der zellkerne während der furchung des eies von Ascaris megalocephala. Anat. Anz 2:288–693 [Google Scholar]
- 5.Bütschli O. 1876. Studien über die ersten Entwicklungsvorgänge der und die Konjugation der Infusorien. Abh. Senckenberg. Naturforschenden Ges 10:1–250 [Google Scholar]
- 6.del Priore L, Pigozzi MI. 2014. Histone modifications related to chromosome silencing and elimination during male meiosis in Bengalese finch. Chromosoma 123:293–302 [DOI] [PubMed] [Google Scholar]
- 7.Scherbaum OH, Louderback AL, Jahn TL. 1958. The formation of subnuclear aggregates in normal and synchronized protozoan cells. Biol. Bull 115:269–75 [Google Scholar]
- 8.Yao MC, Gorovsky MA. 1974. Comparison of the sequences of macro- and micronuclear DNA of Tetrahymena pyriformis. Chromosoma 48:1–18 [DOI] [PubMed] [Google Scholar]
- 9.Yao MC, Gall JG. 1979. Alteration of the Tetrahymena genome during nuclear differentiation. J. Protozool 26:10–13 [Google Scholar]
- 10.Cleffmann G. 1980. Chromatin elimination and the genetic organisation of the macronucleus in Tetrahymena thermophila. Chromosoma 78:313–25 [DOI] [PubMed] [Google Scholar]
- 11.Goday C, Pigozzi MI. 2010. Heterochromatin and histone modifications in the germline-restricted chromosome of the zebra finch undergoing elimination during spermatogenesis. Chromosoma 119:325–36 [DOI] [PubMed] [Google Scholar]
- 12.Allis CD. 2018. Pursuing the secrets of histone proteins: an amazing journey with a remarkable supporting cast. Cell 175:18–21 [DOI] [PubMed] [Google Scholar]
- 13.Allis CD, Glover CVC, Bowen JK, Gorovsky MA. 1980. Histone variants specific to the transcriptionally active, amitotically dividing macronucleus of the unicellular eucaryote, Tetrahymena thermophila. Cell 20:609–17 [DOI] [PubMed] [Google Scholar]
- 14.Inoue JG, Miya M, Lam K, Tay BH, Danks JA, et al. 2010. Evolutionary origin and phylogeny of the modern holocephalans (Chondrichthyes: Chimaeriformes): a mitogenomic perspective. Mol. Biol. Evol 27:2576–86 [DOI] [PubMed] [Google Scholar]
- 15.Smith JJ, Saha NR, Amemiya CT. 2010. Genome biology of the cyclostomes and insights into the evolutionary biology of vertebrate genomes. Integr. Comp. Biol 50:130–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitada J, Tagawa M. 1975. Somatic chromosomes of three species of Cyclostomata. Chrom. Inform. Serv 18:10–12 [Google Scholar]
- 17.Kohno S, Nakai Y, Satoh S, Yoshida M, Kobayashi H. 1986. Chromosome elimination in the Japanese hagfish, Eptatretus burgeri (Agnatha, Cyclostomata). Cytogenet. Cell Genet 41:209–14 [DOI] [PubMed] [Google Scholar]
- 18.Nogusa S. 1960. A comparative study of the chromosomes in fishes with particular considerations on taxonomy and evolution. Mem. Hyogo Univ. Agricult 3:1–68 [Google Scholar]
- 19.Nakai Y, Kohno S. 1987. Elimination of the largest chromosome pair during differentiation into somatic cells in the Japanese hagfish, Myxine garmani (Cyclostomata, Agnatha). Cytogenet. Cell Genet 45:80–83 [Google Scholar]
- 20.Nakai Y, Kubota S, Kohno S. 1991. Chromatin diminution and chromosome elimination in four Japanese hagfish species. Cytogenet. Cell Genet 56:196–98 [DOI] [PubMed] [Google Scholar]
- 21.Kubota S, Nakai Y, Kuro-o M, Kohno S. 1992. Germ line-restricted supernumerary (B) chromosomes in Eptatretus okinoseanus. Cytogenet. Cell Genet 60:224–28 [DOI] [PubMed] [Google Scholar]
- 22.Kubota S, Nakai Y, Sato N, Kuro-o M, Kohno S. 1994. Chromosome elimination in northeast Pacific hagfish, Eptatretus stoutii (Cyclostomata, Agnatha). J. Hered 85:413–15 [Google Scholar]
- 23.Nakai Y, Kubota S, Goto Y, Ishibashi T, Davison W, Kohno S. 1995. Chromosome elimination in three Baltic, south Pacific and north-east Pacific hagfish species. Chromosome Res. 3:321–30 [DOI] [PubMed] [Google Scholar]
- 24.Shichiri M, Kikuma Y, Kuo C-H, Liu L-L, Kubota S, Kohno S. 1997. Chromosome elimination and germ line-restricted microchromosomes in Paramyxine sheni from Taiwan. Chromosome Sci. 1:49–53 [Google Scholar]
- 25.Kohno S, Kubota S, Nakai Y. 1998. Chromatin diminution and chromosome elimination in hagfishes. In The Biology of Hagfishes, pp. 81–100. Dordrecht, Neth.: Springer [Google Scholar]
- 26.Pigozzi MI, Solari AJ. 1998. Germ cell restriction and regular transmission of an accessory chromosome that mimics a sex body in the zebra finch, Taeniopygia guttata. Chromosome Res. 6:105–13 [DOI] [PubMed] [Google Scholar]
- 27.Pigozzi MI, Solari AJ. 2005. The germ-line-restricted chromosome in the zebra finch: recombination in females and elimination in males. Chromosoma 114:403–9 [DOI] [PubMed] [Google Scholar]
- 28.Kubota S, Kuro-o M, Mizuno S, Kohno S. 1993. Germ line-restricted, highly repeated DNA sequences and their chromosomal localization in a Japanese hagfish (Eptatretus okinoseanus). Chromosoma 102:163–73 [DOI] [PubMed] [Google Scholar]
- 29.Kubota S, Ishibashi T, Kohno S. 1997. A germline restricted, highly repetitive DNA sequence in Paramyxine atami: an interspecifically conserved, but somatically eliminated, element. Mol. Gen. Genet 256:252–56 [DOI] [PubMed] [Google Scholar]
- 30.Goto Y, Kubota S, Kohno S. 1998. Highly repetitive DNA sequences that are restricted to the germ line in the hagfish Eptatretus cirrhatus: a mosaic of eliminated elements. Chromosoma 107:17–32 [DOI] [PubMed] [Google Scholar]
- 31.Kubota S, Takano J, Tsuneishi R, Kobayakawa S, Fujikawa N, et al. 2001. Highly repetitive DNA families restricted to germ cells in a Japanese hagfish (Eptatretus burgeri): a hierarchical and mosaic structure in eliminated chromosomes. Genetica 111:319–28 [DOI] [PubMed] [Google Scholar]
- 32.Nabeyama M, Kubota S, Kohno S. 2000. Concerted evolution of a highly repetitive DNA family in Eptatretidae (Cyclostomata, Agnatha) implies specifically differential homogenization and amplification events in their germ cells. J. Mol. Evol 50:154–69 [DOI] [PubMed] [Google Scholar]
- 33.Kojima NF, Kojima KK, Kobayakawa S, Higashide N, Hamanaka C, et al. 2010. Whole chromosome elimination and chromosome terminus elimination both contribute to somatic differentiation in Taiwanese hagfish Paramyxine sheni. Chromosome Res. 18:383–400 [DOI] [PubMed] [Google Scholar]
- 34.Jahn CL, Klobutcher LA. 2002. Genome remodeling in ciliated protozoa. Annu. Rev. Microbiol 56:489–520 [DOI] [PubMed] [Google Scholar]
- 35.Muller F, Tobler H. 2000. Chromatin diminution in the parasitic nematodes Ascaris suum and Parascaris univalens. Int. J. Parasitol 30:391–99 [DOI] [PubMed] [Google Scholar]
- 36.Elder JF Jr., Turner BJ. 1995. Concerted evolution of repetitive DNA sequences in eukaryotes. Q. Rev. Biol 70:297–320 [DOI] [PubMed] [Google Scholar]
- 37.Liao D. 1999. Concerted evolution: molecular mechanism and biological implications. Am. J. Hum. Genet 64:24–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuraku S, Kuratani S. 2006. Time scale for cyclostome evolution inferred with a phylogenetic diagnosis of hagfish and lamprey cDNA sequences. Zool. Sci 23:1053–64 [DOI] [PubMed] [Google Scholar]
- 39.Thomson RC, Plachetzki DC, Mahler DL, Moore BR. 2014. A critical appraisal of the use of microRNA data in phylogenetics. PNAS 111:E3659–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith JJ, Antonacci F, Eichler EE, Amemiya CT. 2009. Programmed loss of millions of base pairs from a vertebrate genome. PNAS 106:11212–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Timoshevskiy VA, Timoshevskaya NY, Smith JJ. 2019. Germline-specific repetitive elements in programmatically eliminated chromosomes of the sea lamprey (Petromyzon marinus). Genes 10:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strange RM, Moore LL. 2019. Characterization and evolution of Germ1, an element that undergoes diminution in lampreys (Cyclostomata: Petromyzontidae). J. Mol. Evol 87:298–308 [DOI] [PubMed] [Google Scholar]
- 43.Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, et al. 2013. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet 45:415–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith JJ, Stuart AB, Sauka-Spengler T, Clifton SW, Amemiya CT. 2010. Development and analysis of a germline BAC resource for the sea lamprey, a vertebrate that undergoes substantial chromatin diminution. Chromosoma 119:381–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith JJ, Baker C, Eichler EE, Amemiya CT. 2012. Genetic consequences of programmed genome rearrangement. Curr. Biol 22:1524–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bryant SA, Herdy JR, Amemiya CT, Smith JJ. 2016. Characterization of somatically-eliminated genes during development of the sea lamprey (Petromyzon marinus). Mol. Biol. Evol 33:2337–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith JJ, Timoshevskaya N, Ye C, Holt C, Keinath MC, et al. 2018. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet 50:270–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Timoshevskiy VA, Herdy JR, Keinath MC, Smith JJ. 2016. Cellular and molecular features of developmentally programmed genome rearrangement in a vertebrate (sea lamprey: Petromyzon marinus). PLOS Genet. 12:e1006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. 2010. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26:2438–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sachs M, Onodera C, Blaschke K, Ebata KT, Song JS, Ramalho-Santos M. 2013. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep. 3:1777–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson ES, Potter IC, Atkin NB. 1975. The nuclear DNA content of lampreys. Experientia 31:912–13 [DOI] [PubMed] [Google Scholar]
- 52.Yan X, Meng W, Wu F, Xu A, Chen S, Huang S. 2016. The nuclear DNA content and genetic diversity of Lampetra morii. PLOS ONE 11:e0157494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Itoh Y, Arnold AP. 2005. Chromosomal polymorphism and comparative painting analysis in the zebra finch. Chromosome Res. 13:47–56 [DOI] [PubMed] [Google Scholar]
- 54.Biederman MK, Nelson MM, Asalone KC, Pedersen AL, Saldanha CJ, Bracht JR. 2018. Discovery of the first germline-restricted gene by subtractive transcriptomic analysis in the zebra finch, Taeniopygia guttata. Curr. Biol 28:1620–27.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith JJ. 2018. Programmed DNA elimination: keeping germline genes in their place. Curr. Biol 28:R601–R3 [DOI] [PubMed] [Google Scholar]
- 56.Torgasheva AA, Malinovskaya LP, Zadesenets KS, Karamysheva TV, Kizilova EA, et al. 2019. Germline-restricted chromosome (GRC) is widespread among songbirds. PNAS 116:11845–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kinsella CM, Ruiz-Ruano FJ, Dion-Côté AM, Charles AJ, Gossmann TI, et al. 2019. Programmed DNA elimination of germline development genes in songbirds. Nat. Commun 10:5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gall JG. 2012. Are lampbrush chromosomes unique to meiotic cells? Chromosome Res. 20:905–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hansson B. 2019. On the origin and evolution of germline chromosomes in songbirds. PNAS 116:11570–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Gao S, Mostovoy Y, Kang Y, Zagoskin M, et al. 2017. Comparative genome analysis of programmed DNA elimination in nematodes. Genome Res. 27:2001–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itoh Y, Kampf K, Pigozzi MI, Arnold AP. 2009. Molecular cloning and characterization of the germline-restricted chromosome sequence in the zebra finch. Chromosoma 118:527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malinovskaya LP, Zadesenets KS, Karamysheva TV, Akberdina EA, Kizilova EA, et al. 2020. Germline-restricted chromosome (GRC) in the sand martin and the pale martin (Hirundinidae, Aves): synapsis, recombination and copy number variation. Sci. Rep 10:1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schoenmakers S, Wassenaar E, Laven JS, Grootegoed JA, Baarends WM. 2010. Meiotic silencing and fragmentation of the male germline restricted chromosome in zebra finch. Chromosoma 119:311–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vigodner M, Morris PL. 2005. Testicular expression of small ubiquitin-related modifier-1 (SUMO-1) supports multiple roles in spermatogenesis: silencing of sex chromosomes in spermatocytes, spermatid microtubule nucleation, and nuclear reshaping. Dev. Biol 282:480–92 [DOI] [PubMed] [Google Scholar]
- 65.Metzler-Guillemain C, Depetris D, Luciani JJ, Mignon-Ravix C, Mitchell MJ, Mattei M-G. 2008. In human pachytene spermatocytes, SUMO protein is restricted to the constitutive heterochromatin. Chromosome Res. 16:761–82 [DOI] [PubMed] [Google Scholar]
- 66.Greil F, van der Kraan I, Delrow J, Smothers JF, de Wit E, et al. 2003. Distinct HP1 and Su(var)3–9 complexes bind to sets of developmentally coexpressed genes depending on chromosomal location. Genes Dev. 17:2825–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grewal SI, Jia S. 2007. Heterochromatin revisited. Nat. Rev. Genet 8:35–46 [DOI] [PubMed] [Google Scholar]
- 68.Giet R, Glover DM. 2001. Drosophila Aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol 152:669–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gurley LR, D’Anna JA, Barham SS, Deaven LL, Tobey RA. 1978. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur. J. Biochem 84:1–15 [DOI] [PubMed] [Google Scholar]
- 70.Paulson JR, Taylor SS. 1982. Phosphorylation of histones 1 and 3 and nonhistone high mobility group 14 by an endogenous kinase in HeLa metaphase chromosomes. J. Biol. Chem 257:6064–72 [PubMed] [Google Scholar]
- 71.Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD. 1998. Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. PNAS 95:7480–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sawicka A, Seiser C. 2012. Histone H3 phosphorylation—a versatile chromatin modification for different occasions. Biochimie 94:2193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li W, Wang P, Zhang B, Zhang J, Ming J, et al. 2017. Differential regulation of H3S10 phosphorylation, mitosis progression and cell fate by Aurora Kinase B and C in mouse preimplantation embryos. Protein Cell 8:662–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adams RR, Maiato H, Earnshaw WC, Carmena M. 2001. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol 153:865–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lyon MF. 1962. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet 14:135–48 [PMC free article] [PubMed] [Google Scholar]
- 76.Brockdorff N, Duthie SM. 1998. X chromosome inactivation and the Xist gene. Cell. Mol. Life Sci 54:104–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richardson BJ, Czuppon AB, Sharman GB. 1971. Inheritance of glucose-6-phosphate dehydrogenase variation in kangaroos. Nat. New Biol 230:154–55 [DOI] [PubMed] [Google Scholar]
- 78.Deakin JE, Chaumeil J, Hore TA, Marshall Graves JA. 2009. Unravelling the evolutionary origins of X chromosome inactivation in mammals: insights from marsupials and monotremes. Chromosome Res. 17:671–85 [DOI] [PubMed] [Google Scholar]
- 79.Hayman DL, Martin PG. 1965. Sex chromosome mosaicism in the marsupial genera Isoodon and Perameles. Genetics 52:1201–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Close RL. 1979. Sex chromosome mosaicism in liver, thymus, spleen and regenerating liver of Perameles nasuta and Isoodon macrourus. Aust. J. Biol. Sci 32:615–24 [Google Scholar]
- 81.Close RL. 1984. Rates of sex chromosome loss during development in different tissues of the bandicoots Perameles nasuta and Isoodon macrourus (Marsupialia: Peramelidae). Aust. J. Biol. Sci 37:53–61 [Google Scholar]
- 82.Watson CM, Margan SH, Johnston PG. 1998. Sex-chromosome elimination in the bandicoot Isoodon macrourus using Y-linked markers. Cytogenet. Cell Genet 81:54–59 [DOI] [PubMed] [Google Scholar]
- 83.Hayman DL, Margan SH. 1969. Cytogenetics of marsupials. In Comparative Mammalian Cytogenetics, ed. Benirschke K, pp. 191–217. Berlin, Heidelberg, Ger.: Springer [Google Scholar]
- 84.Hayman DL, Martin PG. 1974. Mammalia I: Monotremata and Marsupialia. In Animal Cytogenetics 4. Chordata, ed. John B, pp. 19–84. Berlin, Stuttgart, Ger.: Gebruder Borntraeger [Google Scholar]
- 85.Sharman GB. 1974. Marsupial taxonomy and phylogeny. Aust. Mammal 1:137–54 [Google Scholar]
- 86.Murray JD, McKay GM. 1979. Y chromosome mosaicism in pouch young of the marsupial, greater glider (Marsupialia: Petauridae). Chromosoma 72:329–34 [Google Scholar]
- 87.Guttenbach M, Koschorz B, Bernthaler U, Grimm T, Schmid M. 1995. Sex chromosome loss and aging: in situ hybridization studies on human interphase nuclei. Am. J. Hum. Gene 57:1143–50 [PMC free article] [PubMed] [Google Scholar]
- 88.Bukvic N, Gentile M, Susca F, Fanelli M, Serio G, et al. 2001. Sex chromosome loss, micronuclei, sister chromatid exchange and aging: a study including 16 centenarians. Mutat. Res 498:159–67 [DOI] [PubMed] [Google Scholar]
- 89.Johnston PG, Watson CM, Adams M, Paull DJ. 2002. Sex chromosome elimination, X chromosome inactivation and reactivation in the southern brown bandicoot Isoodon obesulus (Marsupialia: Peramelidae). Cytogenet. Genome Res 99:119–24 [DOI] [PubMed] [Google Scholar]
- 90.Nazarenko SA, Timoshevskii VA. 2004. Analysis of the frequency of spontaneous aneuploidy in human somatic cells using interphase cytogenetic technology. Genetika 40:195–204 [PubMed] [Google Scholar]
- 91.McKay GM, McQuade JD, Murray JD, von Sturmer SR. 1984. Sex-chromosome mosaicism in the lemur-like possum Hemibelideus lemuroides (Marsupialia: Petauridae). Aust. J. Biol. Sci 37:131–36 [Google Scholar]
- 92.Bianchi NO, Contreras JR. 1967. The chromosomes of the field mouse Akodon azarae (Cricetidae, Rodentia) with special reference to sex chromosome anomalies. Cytogenetics 6:306–13 [DOI] [PubMed] [Google Scholar]
- 93.Bianchi NO, Reig OA, Molina OJ, Dulout FN. 1971. Cytogenetics of the South American akodont rodents (Cricetidae). I. A progress report of Argentinian and Venezuelan forms. Evolution 25:724–36 [DOI] [PubMed] [Google Scholar]
- 94.Fredga K. 1983. Aberrant sex chromosome mechanisms in mammals: evolutionary aspects. Differentiation 23(Suppl.):S23–30 [DOI] [PubMed] [Google Scholar]
- 95.Vogel W, Jainta S, Rau W, Geerkens C, Baumstark A, et al. 1998. Sex determination in Ellobius lutescens: the story of an enigma. Cytogenet. Cell Genet 80:214–21 [DOI] [PubMed] [Google Scholar]
- 96.Hoekstra HE, Edwards SV 2000. Multiple origins of XY female mice (genus Akodon): phylogenetic and chromosomal evidence. Proc. Biol. Sci 267:1825–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arakawa Y, Nishida-Umehara C, Matsuda Y, Sutou S, Suzuki H. 2002. X-chromosomal localization of mammalian Y-linked genes in two XO species of the Ryukyu spiny rat. Cytogenet. Genome Res 99:303–9 [DOI] [PubMed] [Google Scholar]
- 98.Just W, Baumstark A, Suss A, Graphodatsky A, Rens W, et al. 2007. Ellobius lutescens: sex determination and sex chromosome. Sex Dev. 1:211–21 [DOI] [PubMed] [Google Scholar]
- 99.Castiglia R, Makundi R, Corti M. 2007. The origin of an unusual sex chromosome constitution in Acomys sp. (Rodentia, Muridae) from Tanzania. Genetica 131:201–7 [DOI] [PubMed] [Google Scholar]
- 100.Matthey R. 1958. New type of chromosomal sex determination in the mammals Ellobius lutescens Th. and Microtus (Chilotus) oregoni Bachm. (Muridae, Microtinae). Experientia 14:240–41 [DOI] [PubMed] [Google Scholar]
- 101.Ohno S, Jainchill J, Stenius C. 1963. The creeping vole (Microtus oregoni) as a gonosomic mosaic. I. The Oy/Xy constitution of the male. Cytogenetics 2:232–39 [DOI] [PubMed] [Google Scholar]
- 102.Stanley HP, Kasinsky HE, Bols NC. 1984. Meiotic chromatin diminution in a vertebrate, the holocephalan fish Hydrolagus collie (Chondrichthyes, Holocephali). Tissue Cell 16:203–15 [DOI] [PubMed] [Google Scholar]
- 103.Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, et al. 2010. The genome of a songbird. Nature 464:757–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Korlach J, Gedman G, Kingan SB, Chin CS, Howard JT, et al. 2017. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience 6:gix085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chalker DL, Meyer E, Mochizuki K. 2013. Epigenetics of ciliates. Cold Spring Harb. Perspect. Biol 5:a017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Escriba MC, Goday C. 2013. Histone H3 phosphorylation and elimination of paternal X chromosomes at early cleavages in sciarid flies. J. Cell Sci 126:3214–22 [DOI] [PubMed] [Google Scholar]
- 107.Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD. 2007. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 21:1530–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duharcourt S, Yao MC. 2002. Role of histone deacetylation in developmentally programmed DNA rearrangements in Tetrahymena thermophila. Eukaryot. Cell 1:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nikitina N, Bronner-Fraser M, Sauka-Spengler T. 2009. Culturing lamprey embryos. Cold Spnng Harb. Protoc 2009. 10.1101/pdb.prot5122 [DOI] [PubMed] [Google Scholar]
- 110.Sauka-Spengler T, Meulemans D, Jones M, Bronner-Fraser M. 2007. Ancient evolutionary origin of the neural crest gene regulatory network. Dev. Cell 13:405–20 [DOI] [PubMed] [Google Scholar]
- 111.Nikitina N, Sauka-Spengler T, Bronner-Fraser M. 2008. Dissecting early regulatory relationships in the lamprey neural crest gene network. PNAS 105:20083–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Square T, Romasek M, Jandzik D, Cattell MV, Klymkowsky M, Medeiros DM. 2015. CRISPR/Cas9-mediated mutagenesis in the sea lamprey Petromyzon marinus: a powerful tool for understanding ancestral gene functions in vertebrates. Development 142:4180–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.York JR, Yuan T, Zehnder K, McCauley DW. 2017. Lamprey neural crest migration is Snail-dependent and occurs without a differential shift in cadherin expression. Dev. Biol 428:176–87 [DOI] [PubMed] [Google Scholar]
- 114.Parker HJ, Sauka-Spengler T, Bronner M, Elgar G. 2014. A reporter assay in lamprey embryos reveals both functional conservation and elaboration of vertebrate enhancers. PLOS ONE 9:e85492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Parker HJ, De Kumar B, Green SA, Prummel KD, Hess C, et al. 2019. A Hox-TALE regulatory circuit for neural crest patterning is conserved across vertebrates. Nat. Commun 10:1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dean B. 1899. On the Embryology of Bdellostoma stouti: A General Account of Myxinoid Development from the Egg and Segmentation to Hatching. Jena, Ger.: Verlag von Gustav Fischer [Google Scholar]
- 117.Ota KG, Kuraku S, Kuratani S. 2007. Hagfish embryology with reference to the evolution of the neural crest. Nature 446:672–75 [DOI] [PubMed] [Google Scholar]
- 118.Pascual-Anaya J, Sato I, Sugahara F, Higuchi S, Paps J, et al. 2018. Hagfish and lamprey Hox genes reveal conservation of temporal colinearity in vertebrates. Nat. Ecol. Evol 2:859–66 [DOI] [PubMed] [Google Scholar]
- 119.Hedges SB, Marin J, Suleski M, Paymer M, Kumar S. 2015. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol 32:835–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Timoshevskiy VA, Lampman RT, Hess JE, Porter LL, Smith JJ. 2017. Deep ancestry of programmed genome rearrangement in lampreys. Dev. Biol 429:31–34 [DOI] [PMC free article] [PubMed] [Google Scholar]