

Abstract
Background:
Multiple pathogenic genetic variants are associated with pancreatitis in patients of European (EA) and Asian ancestries, but studies on patients of African ancestry (AA) are lacking. We evaluated the prevalence of known genetic variations in African-American subjects in the US.
Methods:
We studied prospectively enrolled controls (n = 238) and patients with chronic (CP) (n = 232) or recurrent acute pancreatitis (RAP) (n = 45) in the NAPS2 studies from 2000–2014 of self-identified AA. Demographic and phenotypic information was obtained from structured questionnaires. Ancestry and admixture were evaluated by principal component analysis (PCA). Genotyping was performed for pathogenic genetic variants in PRSS1, SPINK1, CFTR and CTRC. Prevalence of disease-associated variants in NAPS2 subjects of AA and EA was compared.
Results:
When compared with CP subjects of EA (n = 862), prevalence of established pathogenic genetic variants was infrequent in AA patients with CP, overall (29 vs. 8.19%, OR 4.60, 95% CI 2.74–7.74, p < 0.001), and after stratification by alcohol etiology (p < 0.001). On PCA, AA cases were more heterogeneous but distinct from EA subjects; no difference was observed between AA subjects with and without CP-associated variants. Of 19 A A patients with CP who had pathogenic genetic variants, 2 had variants in PRSS1 (R122H, R122C), 4 in SPINK1 (all N34S heterozygotes), 12 in CFTR (2 CFTRsev, 9 CFTRBD, 1 compound heterozygote with CFTRsev and CFTRBD), and 1 in CTRC (R254W).
Conclusion:
Pathogenic genetic variants reported in EA patients are significantly less common in AA patients. Further studies are needed to determine the complex risk factors for AA subjects with pancreatitis.
Keywords: African-American, African ancestry, Blacks, Genetics, Pancreatitis, Alcohol
1. Introduction
Chronic pancreatitis (CP) is a pathologic fibro-inflammatory syndrome of the pancreas in individuals with genetic, environmental and/or other risk factors who develop persistent pathologic responses to parenchymal injury or stress that eventually results in pancreatic atrophy, fibrosis, pain syndromes, duct distortion and strictures, calcifications, pancreatic exocrine dysfunction, pancreatic endocrine dysfunction and dysplasia [1]. Clinically, most patients with CP report a history of acute pancreatitis (AP) or recurrent AP (RAP) [2,3], indicating a complex, multifactorial, progressive disease scenario. CP results in multiple complications with a significant reduction in quality of life (QOL) when compared with non-pancreatitis controls [4], as well as overall survival when compared with age- and sex-matched general population [5]. Pancreatitis represents a significant burden to the health care system, and accounts for an annual health care expenditure of over $2.8 billion in the US [6].
Since the discovery in 1996 of a germline mutation in PRSS1 as the cause of hereditary pancreatitis [7], disease-associated variations in several other pancreatitis susceptibility genes, including cystic fibrosis transmembrane conductance regulator [CFTR], serine peptidase inhibitor, Kazal type 1 [SPINK1], and chymotrypsin C [CTRC] have been identified and replicated in multiple cohorts from North America, Europe and Asia [8–17]. Testing for pathogenic variants in these genes is now performed routinely in clinical practice, especially when the cause of RAP or CP is unknown.
In epidemiologic studies, the risk of pancreatitis is noted to be 2–3 folds greater in patients of African ancestry (AA) including African-Americans, when compared with Caucasians [18–21]. AA patients also have a greater risk of readmission after an episode of pancreatitis [22]. The precise causes underlying these differences are unknown. Few studies have examined the effect of environmental and/or behavioral risk factors as potential mediators of racial differences in pancreatitis. For instance, racial differences in pancreatitis have been partially attributed to a higher prevalence of alcohol and tobacco exposure in AA patients [18–20]. In the North American Pancreatitis Study 2 (NAPS2), we found in AA patients with CP that the odds of self-reported heavy or very heavy drinking were 2.62 folds greater and odds of ever smoking were 2.97 folds greater than patients of European ancestry (EA). Similarly, the odds of physician-defined alcohol etiology and smoking as a risk factor were 4.31 and 2.54 times greater in AA patients with CP when compared to EA patients with CP [23].
Ancestral germ line genetic differences could also affect differences in disease risk and prevalence between Americans of European ancestry and African Americans [24,25]. Pathogenic genetic variants in PRSS1, SPINK1, CFTR and CTRC are clearly associated with pancreatitis in populations of EA and Asian ancestry (7–16). The effects and burden of these pathogenic genetic variants for RAP or CP in AA patients has not been reported.
We tested the hypothesis that the prevalence of pathogenic variations linked to the four most common RAP and CP susceptibility genes in patients of self-reported AA in the United States is similar to those of EA in the NAPS2 cohort. We further tested for the amounts of ancestral admixture, since previous studies have demonstrated the importance of genetics on disease risks and outcomes based on difference in the minor allele frequency [25,26]. Here, we report that RAP/CP-associated variants that are common in patients of EA are rare in patients of AA. These findings suggest that genetic testing of RAP/CP susceptibility variants that are frequent in cases of EC will likely be of lower yield in patients of AA. Further studies are needed to understand genetic and environmental risk factors for pancreatitis in subjects of AA.
2. Methods
2.1. Study population
The North American Pancreatitis Study Group has prospectively recruited carefully phenotyped controls and patients with RAP or CP from 27 US centers from 2000–2014 in a series of 3 studies: North American Pancreatitis Study 2 (NAPS2 Original), North American Pancreatitis Study 2 Continuation and Validation (NAPS2-CV), and North American Pancreatitis Study 2 Ancillary Study (NAPS2-AS) [23,27,28]. Although previous genetic studies focused on subjects of EA, this cohort also contains one of the largest number of RAP/CP patients of AA ascertained in the US [23]. RAP was defined by two or more documented episodes of AP (typical abdominal pain with elevation of serum pancreatic enzymes to more than 3 times the upper limit of normal or imaging evidence of AP), and CP was defined using definitive evidence on imaging or histology [27]. The NAPS2 studies were approved by the Institutional Review Board of each participating center, and all subjects provided informed consent prior to study enrollment. We have previously reported the prevalence of genetic variations in cases and controls of EA from the NAPS2 cohort [9,17,29–31].
2.2. Data collection
Study procedures and information collected in the NAPS2 studies have been described earlier [23,27,28]. Two detailed sets of questionnaires were used to obtain information from the study subjects and the enrolling physician. Questionnaires were completed by patients and controls with assistance of a trained research coordinator. Information was collected on demographics, personal and family history, exposure to environmental risk factors (alcohol, smoking), pain experience (presence, pattern), hospitalizations, emergency room visits, disability related to pain and pancreatitis, medication use, and quality of life. Race was self-reported. Smoking status was also self-reported and categorized as never, past or current, the amount of smoking was quantified as packs per day (<1 or ≥1) and as pack years (1–12, 12–35, ≥35). Information on self-reported average weekly alcohol intake (quantity-frequency criteria, i.e. average amount consumed on a drinking day and the number of days per month that this amount was consumed) during the maximum drinking period in life was used to categorize drinking status as (1) Abstainers: no alcohol use or <20 drinks in lifetime; (2) Light drinkers: ≤3 drinks/week; (3) Moderate drinkers: 4–7 drinks/week for females; 4–14 drinks/week for males; (4) Heavy drinkers: 8–34 drinks/week for females; 15–34 drinks/week for males; (5) Very heavy drinkers: ≥35 drinks/week. Physicians provided information on phenotypic features including history of AP, ages at the onset of AP, CP symptoms and CP diagnosis, risk factors, etiology, pain experience (presence, pattern), pain medication use, exocrine insufficiency, diabetes, details of imaging studies, treatments received and their perceived effectiveness.
2.3. Ancestral admixture analyses
A blood sample was obtained from each subject at enrollment, and peripheral blood leukocyte DNA was isolated following established protocols [9,11,30]. A major subset of subjects in the NAPS2 cohorts (174 A A Controls, 39 A A RAP, 179 A A CP, 541 EA Controls, 430 EA RAP, and 772 EA CP) were genotyped using a custom Illumina Infinium HumanExome + beadchip previously developed by the NIDDK IBD consortium [32]. These genotypes were used for the ancestral classifying using principal component analysis (PCA). The custom microarray contained ~270,000 markers and includes exonic and splice variants, ancestry informative markers, stop codon variants and MHC tag SNPs. Genotype data were called using the Illumina GenomeStudio v 2011.1 software. Quality control for each cohort was achieved by examining missingness by genotype (excluding SNPs with >10% missing data) and missingness by individual subject (excluding subjects with >20% missing genotype calls). Additional QC was achieved by examining Hardy-Weinberg Equilibrium (HWE) in controls (Chi-square test threshold P = 1e-5) and extreme heterozygosity. Following QC, 19,179 markers were removed due to excess missingness, and 3814 markers due to non-conformity with HWE. Individuals were excluded from the final data set if they had high proportion of missing genotypes (n = 376), showed discrepancies between reported and genotypic sex (n = 30) or had high heterozygosity (n = 46). Markers were LD pruned (r2 > 0.4) to produce higher quality PC estimates. After all QC and LD-pruning, the genotype call rate was 99.98% for the entire data set. In the final dataset 392 A A and 1743 Caucasian subjects were genotyped for a total 140,577 markers.
2.4. PRSS1, SPINK1, CFTR and CTRC genetic analysis
All patients were genotyped for known variants in PRSS1, SPINK1 (N34S), and CFTR genes by a combination of sequencing and Taqman® assays (Thermo-Fisher, Waltham MA) [9]. A multiplex Taqman (iPlex) was designed to capture the majority of reported pancreatitis-associated variants in CFTR (79 mutations total, see Supplementary Table 1, and LaRusch et al. for methods [9]). PCR primers were designed for CTRC gene exons 2, 3, and 7 and the corresponding intronic regions that contain most common mutations reported from Germany, India, France, and Taiwan [10,14,33,34]. We tested for R254W using Restriction Fragment Length Polymorphism (RFLP) sequencing that captured both the R254W and K247_R254del mutations [30]. All positive and random negative results were confirmed by Sanger sequencing.
2.5. Statistical analysis
SAS 9.4 (SAS Institute, Inc., Cary, NC) was used to analyze and generate summary statistics of the dataset. Descriptive statistics were calculated as proportions for categorical data and mean and standard deviation for continuous data. Demographic comparisons based on diagnosis (controls, CP, RAP) were performed using Fischer’s exact test for categorical variables and Student’s t-test for continuous variables. To characterize any genomic heterogeneity within the NAPS2 AA subjects we performed PCA using PLINK v1.9 (https://www.cog-genomics.org/plink2) to infer presence of genetic stratification in the final sample of 392 A A and 1743 Caucasian NAPS2 participants. A plot of first PC vs. the second PC was used to visualize data. Self-identified race and AA subjects carrying mutations were color-coded to aid with data visualization.
To evaluate the genetics of our cohort, we first compared the presence of any of the candidate SNP frequencies in any of the 4 genes, and then in individual genes between AA cases and controls. Next, we compared the presence of any of the candidate SNP frequencies between AA subjects and EA subjects in the NAPS2 studies using Fisher’s exact test. For assessment of presence of any SNP in the 4 genes, a two-tailed p-value <0.05 was considered statistically significant, while for assessment of individual genes, a Bonferroni correction was made for multiple comparisons.
Group and subgroup analysis used in the study are in Table 1.
Table 1.
Summary of Analyses Performed in this Study.
| Analysis Level | Subject Groups | Comparisons |
|---|---|---|
| Analysis 1 | AA Controls, RAP, CP, EA Controls, RAP, CP | PCA, to evaluate genomic heterogeneity and admixture in self-reported populations |
| Analysis 2 | AA Controls, AA RAP, AA CP | Demographics and Clinical features between Controls, RAP, and CP patients |
| Analysis 3 | AA Controls, AA RAP, AA CP | Prevalence and comparison of known pancreatitis-susceptibility mutations in PRSS1, SPINK1, CTRC and CFTR genes (individual or any) in Controls, RAP and CP patients |
| Analysis 4 | AA Controls, EA Controls, AA CP patients, EA CP patients | Prevalence and comparison of known pancreatitis-susceptibility mutations in PRSS1, SPINK1, CTRC and CFTR genes (individual or any) in Controls and CP patients of EA and AA |
AA – African ancestry; CP – chronic pancreatitis; EA – European ancestry; RAP – recurrent acute pancreatitis.
3. Results
3.1. Study population – subjects with AA
Together, the three NAPS2 studies enrolled 268 controls, 248CP and 50 RAP patients who self-identified as having AA. Of these, complete phenotype and genotype information was available for 238 controls, 232CP and 45 RAP patients (Table 2) who formed the final cohort for this study. Information on demographics, exposure to alcohol and tobacco, and etiology of pancreatitis is provided in Table 3.
Table 2.
Number of AA subjects in the NAPS2 cohort.
| Years | All NAPS2 studies | NAPS2 (Original) | NAPS2 Continuation and Validation (NAPS2-CV) | NAPS2 Ancillary Study (NAPS2-AS) |
|---|---|---|---|---|
| 2000–2014 | 2000–2006 | 2008–2012 | 2011–2014 | |
| Number of centers | 21 | 14 | 8 | 8 |
| Subjects enrolled | 566 | 123 | 63 | 380 |
| Controls | 268 (47) | 41 (33) | 1 (2) | 226 (59) |
| RAP | 50 (9) | 24 (20) | 6 (10) | 20 (5) |
| CP | 248 (44) | 58 (47) | 56 (89) | 134 (35) |
| Subjects with genetic test results* | 515 | 104 | 58 | 353 |
| Controls** | 238 (46) | 30 (29) | 1 (2) | 207 (59) |
| RAP | 45 (9) | 21 (20) | 5 (9) | 19 (5) |
| CP | 232 (45) | 53 (51) | 52 (90) | 127 (36) |
AA – African ancestry; CP – chronic pancreatitis; NAPS2 – North American Pancreatitis Study; RAP – recurrent acute pancreatitis.
Final sample size for the present study.
Excludes family controls.
Table 3.
Demographics and select characteristics in AA subjects in the NAPS2 cohort.
| Controls | RAP | CP | |
|---|---|---|---|
| Number | 238 | 45 | 232 |
| Age - mean ± sd | 47.4 ± 13.5 | 45.7 ± 13.6 | 51.9 ± 10.5 |
| Male - n (%) | 91 (38.2) | 17 (37.8) | 145 (62.5) |
| Smoking statusa - n (%) | |||
| Never | 119 (50.0) | 12 (27.3) | 28 (12.1) |
| Past | 47 (19.8) | 11 (25.0) | 57 (24.7) |
| Current | 72 (30.3) | 21 (47.7) | 146 (63.2) |
| Smokingb- n (%) | |||
| Never | 119 (50.0) | 12 (27.9) | 28 (12.4) |
| <1 pack-per-day | 98 (41.2) | 19 (44.2) | 136 (60.2) |
| 1 or more pack-per-day | 21 (8.8) | 12 (27.9) | 62 (27.4) |
| Drinking Categoryc- n (%) | |||
| Abstainer | 23 (11.4) | 7 (16.3) | 21 (9.3) |
| Light | 70 (34.7) | 9 (20.9) | 18 (8.0) |
| Moderate | 40 (19.8) | 10 (23.3) | 25 (11.1) |
| Heavy | 40 (19.8) | 4 (9.3) | 29 (12.8) |
| Very Heavy | 29 (14.4) | 13 (30.2) | 133 (58.9) |
| Etiology - n (%) | |||
| Alcohol | NA | 15 (33.3) | 178 (76.7) |
| Other | NA | 30 (66.7) | 54 (23.3) |
| Age at first attack of APd - mean ± sd | NA | 41.7 ± 15.3 | 40.1 ± 11.1 |
AA – African ancestry; CP – chronic pancreatitis; NAPS2 – North American Pancreatitis Study; RAP – recurrent acute pancreatitis.
Missing Data
(n = 2),
(n = 8),
(n = 44)
(n = 22);
Proportions/medians shown are based on effective numbers.
CP vs. controls: p < 0.001 (age, sex, smoking status, smoking ppd, drinking category); CP vs. RAP: p = 0.005 (age); p = 0.003 (sex); p = 0.04 (smoking status); p = 0.03 (smoking ppd); p = 0.001 (drinking category); p < 0.001 (etiology).
AA patients with CP were significantly (p < 0.05) more likely to be male, ever and current smokers, and very heavy drinkers when compared with controls and RAP patients. When compared with RAP patients, CP patients were significantly (p < 0.05) more likely to have physician-defined alcohol etiology (76.7% vs 33.3%, p < 0.001). About two-thirds of CP patients had at least one attack of AP. The age at the time of first-attack of AP was similar in CP and RAP patients, but the age at enrollment of CP patients was significantly higher than that of RAP patients (p = 0.005) (Table 3).
3.2. Genetics of self-reported ancestry in the NAPS2 cohort
PCA of the NAPS2 AA cases and control cohorts identified two components that corresponded to self-identified ancestry. Self-identified AA subjects showed significant heterogeneity when compared with subjects with self-identified EA. The AA cases and the few AA subjects with RAP or CP with established pathogenic variants in PRSS1, SPINK1 and/or CFTR clustered with other AA subjects (Fig. 1). Thus, it is unlikely that the pathogenic variants resulted primarily from admixture of patients with AA and EA.
Fig. 1.
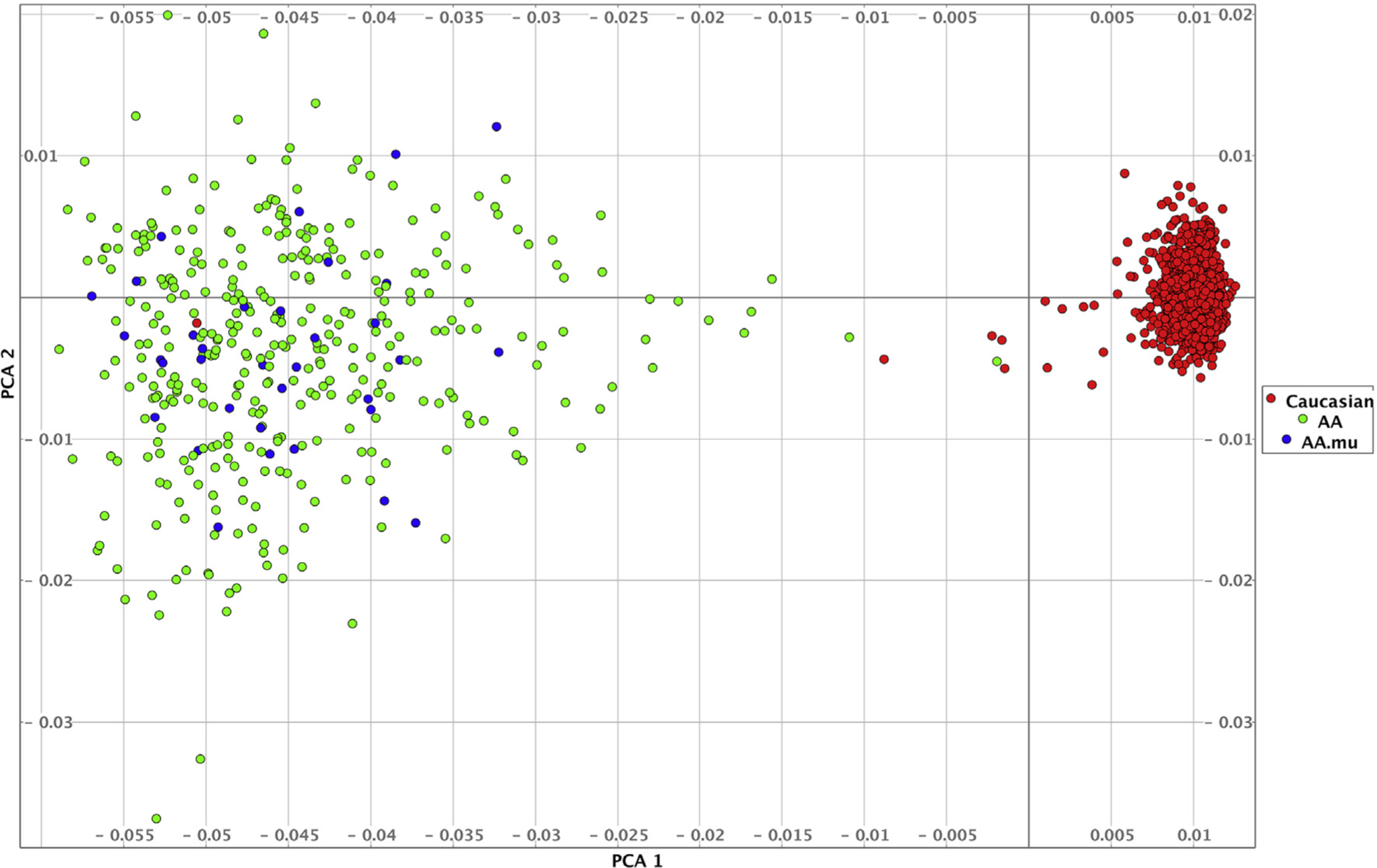
PCA plot showing PC1 (horizontal axis) v PC2 (vertical axis). Self-identified European Americans (red circles, Caucasians) cluster tightly along one axis. Self-identified patients with AA (Green circles, AA) show greater genetic heterogeneity. Blue circles (AA.mu) represent patients with AA carrying known candidate gene mutations. No systematic differences are visible between mutation carriers and non-carriers based on this analysis.
3.3. Genetic variants in AA subjects
3.3.1. AA controls
Only 16 (6.7%) controls carried a pathogenic variant linked to four CP susceptibility genes tested (Table 4). None of them had PRSS1 or SPINK1 mutations. Five (2.1%) were heterozygous carriers for severe, CF-associated pathogenic CFTR variants (CFTRsev): F508del in 4, R553X in 1 - of these 2 reported moderate and 1 heavy drinking during the maximum drinking period of life, but neither reported symptoms of pancreatitis. Additionally, 10 were heterozygous carriers for bicarbonate conductance defective CFTR variants (CFTRBD): R74W-D1270 N in 5, R75Q in 4, L997F in 1 - of them 4 reported very heavy, 1 heavy, 3 moderate, and 1 light drinking. One control was a carrier in the CTRC gene (R254W).
Table 4.
Prevalence and type of mutations identified in AA subjects in the NAPS2 cohort.
| Controls (n = 238) | CP (n = 232) | RAP (n = 45) | ||||
|---|---|---|---|---|---|---|
| n (%) | Type of mutation | n (%) | Type of mutation | n (%) | Type of mutation | |
| PRSS1 | 0 (0) | – | 2 (0.87) | R122H | 0 (0) | – |
| SPINK1 | 0 (0) | – | 4 (1.72) | N34S | 0 (0) | – |
| CFTRSEV | 5 (2.10) | F508del (4) | 3 (1.29) | F508del (1) | 0 (0) | – |
| R553X (1) | 3120 + 1 GtoA (2) | |||||
| CFTRBD | 10 (4.20) | R74W-D1270N (5) | 10 (4.31) | R74W-D1270N (7) | 5 (11.1) | R74W-D1270N (3) |
| R75Q (4) | R75Q (2) | L967S (1) | ||||
| L997F (1) | L967S (1) | I148T (1) | ||||
| CTRC | 1 (0.42) | R254W | 1 (0.4) | R254W | 0 (0) | – |
AA – African ancestry; CP – chronic pancreatitis; NAPS2 – North American Pancreatitis Study; RAP – recurrent acute pancreatitis.
3.3.2. AA patients with CP
Of CP patients, 19 (8.2%) had pathogenic genetic variants (p = 0.48 vs. controls) (Table 4). Two patients were heterozygous for the disease-causing PRSS1 variants (p.R122C and p. R122H), 4 carried the SPINK1 N34S risk variant (all were heterozygous), 3 carried severe CFTR variants (CFTRsev) (F508del in 1, 3120 + 1 GtoA in 2), 10 had CFTRBD variants (R74W-D1270 N in 7, R75Q in 2 and L967S in 1), and 1 had a pathogenic CTRCsev variant (R254W). Of the 12 patients with any CF mutation, only one was a compound heterozygote with CFTRsev 3120+1GtoA and CFTRBD L967S variants. Except for SPINK1 which showed borderline association (0.059), no other mutation showed significant difference between control subjects and CP patients.
The enrolling physician identified alcohol as the etiology in 14/19 (74%) patients with an identified mutation (vs. 164/213 [77%] with no mutations, p = 0.78). Interestingly, self-reported heavy or very heavy drinking habits were reported by 12 of these 14 patients, supporting the physician diagnosis. All patients but one (94.7%) who carried any pathogenic variants was a smoker (vs. 185/211 [87.6%] with no pathogenic variants, p = 0.59) (Table 5).
Table 5.
Select characteristics of AA CP patients who had an identified mutation/polymorphism in pancreatitis susceptibility genes.
| Patient | Gender | Physician-defined alcohol etiology | Drinking Category* | Smoking status* | Smoking amount* | Gene affected | Type of mutation |
|---|---|---|---|---|---|---|---|
| 1 | Female | Yes | Very Heavy | Never | – | PRSS1 | R122H G/A |
| 2 | Male | No | Abstainer | Current | <1 ppd | PRSS1 | R122C C/T |
| 3 | Male | Yes | Very Heavy | Current | <1 ppd | SPINK1 | N34S heterozygous |
| 4 | Male | Yes | Very Heavy | Past | <1 ppd | SPINK1 | N34S heterozygous |
| 5 | Male | Yes | Very Heavy | Current | <1 ppd | SPINK1 | N34S heterozygous |
| 6 | Female | Yes | Very Heavy | Current | >1 ppd | SPINK1 | N34S heterozygous |
| 7 | Female | No | Abstainer | Current | ≥1 ppd | CTRC | R254W |
| 8 | Male | Yes | Heavy | Current | <1 ppd | CFTRSev + CFTRBD | 3120+1GtoA + L967S |
| 9 | Female | No | Light | Past | <1 ppd | CFTR Sev | F508del |
| 10 | Male | No | Heavy | Current | <1 ppd | CFTR BD | R74W-D1270 N |
| 11 | Male | Yes | Heavy | Current | ≥1 ppd | CFTR BD | R74W-D1270 N |
| 12 | Male | Yes | Very Heavy | Current | <1 ppd | CFTR BD | R74W-D1270 N |
| 13 | Female | Yes | Heavy | Current | <1 ppd | CFTR BD | R74W-D1270 N |
| 14 | Female | Yes | Unknown | Current | ≥1 ppd | CFTR BD | R74W-D1270 N |
| 15 | Female | Yes | Very Heavy | Current | <1 ppd | CFTR BD | R74W-D1270 N |
| 16 | Female | Yes | Abstainer | Past | <1 ppd | CFTR BD | R74W-D1270 N |
| 17 | Female | No | Moderate | Past | <1 ppd | CFTR BD | R75Q |
| 18 | Female | Yes | Very Heavy | Current | <1 ppd | CFTR BD | R75Q |
| 19 | Female | Yes | Very Heavy | Current | <1 ppd | CFTR BD | 3120+1GtoA |
AA – African ancestry; CP – chronic pancreatitis.
Based on self-report.
Both CP patients who had PRSS1 mutations had onset of pancreatitis at an early age. One of them had RAP with first-attack of AP at age 15 years, and was diagnosed with CP at the time of enrollment (age 19 years). This patient gave history of AP in father and a grandparent. The second patient also had RAP with first-attack of AP at age 24 years, was diagnosed with CP at age 28 years, and enrolled at age 36 years. This patient also had a history of multiple family members with pancreatitis (grandmother with CP, father and two sisters with AP).
3.3.3. AA patients with RAP
None of the RAP subjects had a pathogenic PRSS1, SPINK1 or CTRC variant associated with pancreatitis susceptibility. Five had heterozygous pathogenic CFTRBD variants: R74W-D1270 N in 3, L967S in 1, and I148T in 1 (Table 4). There were no homozygous, compound heterozygous or multigenic combinations for CFTR variants. In 2 subjects with pathogenic variants in the CFTR gene, the enrolling physician identified alcohol as the etiology – of these one self-reported very heavy and the other moderate drinking.
3.4. Genetic variants in AA vs. EA
We have previously reported the prevalence of disease-associated variants in the four primary pancreatitis susceptibility genes among controls and pancreatitis patients of EA in the NAPS2 cohort [17]. Here we compared the prevalence of these variants between AA subjects and EA subjects in the NAPS2 cohort. The prevalence of disease-associated variants in any of the four selected genes was significantly higher among EA patients with CP than AA patients with CP (29% vs 8.18; OR 4.60 95% CI 2.74–7.74) (Table 6). Significant differences were noted in the prevalence of mutations in each of the genes among CP patients except for the CTRC gene. The prevalence trends were similar when patients were stratified by physician-defined alcohol etiology.
Table 6.
Prevalence of any known mutations in pancreatitis susceptibility genes in controls and CP patients of EA and AA in the NAPS2 Cohort.
| % | Controls | CP (all) | CP (alcohol etiology) | CP (no alcohol etiology) | ||||
|---|---|---|---|---|---|---|---|---|
| European (n = 610) | African American (n = 238) | European (n = 862) | African American (n = 232) | European (n = 362) | African American (n = 178) | European (n = 500) | African American (n-54) | |
| Mutation (1 or more) present in any of the four genes | 14.92* | 6.72 | 29.00* | 8.19 | 22.38* | 7.87 | 33.80* | 9.26 |
| Any mutation present in PRSS1 gene | 0.16 | 0.00 | 4.87* | 0.86 | 1.38 | 0.56 | 7.40 | 1.85 |
| SPINK1 (N34S) mutation present | 2.46* | 0.00 | 9.40* | 1.72 | 7.73* | 2.25 | 10.60 | 0.00 |
| Any mutation in CFTR gene: | ||||||||
| CFTR total | 12.13* | 6.30 | 17.87* | 5.17 | 14.92* | 5.06 | 20.00* | 5.56 |
| CFTR sev | 3.11 | 2.10 | 8.12* | 1.29 | 5.25 | 1.12 | 10.20 | 1.85 |
| CFTR BD | 9.34 | 4.20 | 10.21* | 4.31 | 9.67 | 4.49 | 10.60 | 3.70 |
| Any mutation in the CTRC gene | 0.33 | 0.42 | 1.28 | 0.43 | 1.38 | 0.00 | 1.20 | 1.85 |
Results for mutations are presented as proportions.
p < 0.05 for any mutation; comparisons for individual genes (p < 0.0125 – after Bonferroni adjustment for 4 comparisons).
AA – African ancestry; CP – chronic pancreatitis; EA – European ancestry; NAPS2 – North American Pancreatitis Study.
4. Discussion
CP is a complex syndrome with significant heterogeneity that is determined, in part, by genetic factors. In this large cohort, we found the prevalence of known mutations in the pancreatitis-susceptibility genes reported in many populations to be infrequent in AA patients with RAP and/or CP from North America. A PCA of the cohort demonstrated two distinct clusters of subjects that map onto self-reported ancestry of AA and EA as demonstrated in other ancestral studies [35]. In AA patients with CP, borderline significance between cases versus controls was noted only for SPINK1 N34S mutation. To our knowledge, this is the first report of pathogenic, gain-of function PRSS1 variants in any AA patient. Genetic differentiation by PCA indicated that AA patients who carry any of the known disease-associated mutations have an overall genomic background indistinguishable from those that do not carry these mutations. These findings have a direct implication on clinical practice, in that testing for the known mutations in PRSS1, SPINK1 or CFTR in AA patients who have CP or RAP will be of low yield. These mutation specific results indicate that as yet unknown genetic variants may be responsible for the etiology of pancreatitis in AA patients. Future larger scale genetic analysis will be required to determine the common pathogenic germline variants in AA patients.
Since the discovery of gain-of-function mutations in the PRSS1 gene as the cause of hereditary pancreatitis, other variations in PRSS1 and genes linked to trypsin-dependent (SPINK1, CTRC) or trypsin-independent pathway (CFTR, CPA1) have been identified in people of many geographic locations and ancestries [8–16,36,37]. The prevalence of specific pathogenic variants within a population originates through random germline mutations occurring in previous generations, and mixed or segregated based on migration and/or environmental advantages, as suggested for example for CFTR variants and cholera [38], and sickle cell disease and malaria [39].
There are no data on the prevalence of mutations in pancreatitis susceptibility genes in AA patients. We found that the prevalence of mutations in genes commonly associated with pancreatitis in EA subjects was significantly lower in AA CP patients in the United States. Among the genes tested, only SPINK1 mutations had a trend toward being overrepresented in AA patients versus AA controls (CP 1.7%, controls 0%, p = 0.059). The lack of statistical significance for an association between SPINK1 N34S high risk haplotype and CP is likely related to small sample size (type 2 error). Of note, all 4 patients with SPINK1 mutations had physician-defined alcohol etiology, while none with no alcohol etiology (n = 54) had the high risk SPINK1 N34S haplotype. However, the prevalence of SPINK1 mutation was still much lower than expected, overall or after stratification by physician-defined alcohol etiology [17,40].
Cystic Fibrosis is an autosomal recessive disorder with phenotypic variance caused by two CFTRsev variants and other modifier genes [41]. Over 2000 unique CFTR variants have been identified, and only around 100 variants are well-characterized as disease causing – either with CF or CFTR-associated disorders such as pancreatitis. Clinical pancreatitis due to CFTR mutations has been typically linked to compound, complex genotypes, i.e. presence of CFTRsev genetic variants on one allele and CFTRmild-var variants on the other allele. More recently, CFTRBD variants that alter bicarbonate but not chloride conductance have been linked to pathology in organs that utilize CFTR to transport bicarbonate, such as the pancreas [9]. The prevalence of CFTR mutations is much higher in EA populations (approximately 1 in 2500 individuals) and lower in AA populations (1 in 15,000 individuals) [41]. Many of the pathogenic CFTR variants in African Americans are attributable to admixture with European populations, since mutations such as CFTR F508del are rare in Africa [42]. These observations likely explain the lower prevalence of CF mutations in AA subjects observed in our study. Interestingly, two patients in our study were found to have the CFTRsev 3120 + 1 G > A variants of African/Arabian origin [42,43].
We report two AA patients with CP who were found to have gain-of-function PRSS1 variants. Similar to CFTR mutations, there is a possibility that the observed variants in these subjects represent admixture of the PRSS1 locus from EA individuals, or it could represent de-novo mutations. However, this does not negate the rarity of hereditary pancreatitis in AA patients. In the current study, only one CP patient and one control each had CTRC R254W.
Many patients have clear environmental risk factors or exposures, and the overall risk can be increased in the presence of genetic risk [44]. A high proportion (77.6%) of AA patients with CP were classified as alcohol etiology, and they also had a high prevalence of smoking (87.6%). While we hypothesized that genetic cofactors may contribute to this higher prevalence of disease in AA patients, the common genetic susceptibility factors observed among EA patients and those of Asian ancestry could not explain this observation in our cohort. Only 14/178 (7.8%) AA patients with CP and alcohol etiology had mutations found in EA patients when compared with 5/54 (9.3%, p = 0.78) patients without alcohol etiology. Furthermore, as previously reported, the risk of CP in very heavy alcohol drinkers of AA in the original NAPS2 study appears to be much higher than for patients of EA (OR 20.86, CI 1.38–316.13 vs OR 2.51, CI 1.46–4.34) suggesting that a potent and yet to be identified co-factor is important in AA patients [45]. Taken together, these data indicate that the genetic etiologies of RAP and/or CP in AA patients are different than those identified in EA patients. However, while the current cohort is sufficient to demonstrate that the genetic susceptibility of AA patients is different than EA patients, it is too small to conduct a genome-wide association study to identify alternative pathogenic genetic mechanisms, and especially if the nested studies of RAP/CP patients with and without alcohol/smoking are planned.
A limitation of our study is the small number of patients of AA, especially with RAP, as the recruitment patterns mirrored the case mixture of the participating institutions and because the focus of NAPS2-AS was aimed at recruiting CP patients. Thus, appropriately powered comparison of AA with RAP vs. AA with CP were not possible. In addition, our analysis focused on only four genes associated with susceptibility to CP. We cannot exclude the possibility that other high-risk genetic pathways exist between cases of AA and EA, or that other as-of-yet unidentified polymorphisms or mutations may play an important role particularly for patients with non-toxic chronic pancreatitis. Future studies should perform more detailed ancestral analyses and broaden the genetic evaluation.
In conclusion, the four most common pathogenic genetic variants associated with CP in EA patients are uncommon in AA patients. Since the risk of RAP/CP is greater in populations of AA than EA, future studies on the complex gene-environment interactions in cases of AA are needed.
Supplementary Material
Acknowledgements
This study was presented as a Poster at the Digestive Disorders Week 2016 and published in an abstract form in Gastroenterology April 2016, Vol 150, Supp 4, Page S913 as “Common Genetic Susceptibility Factors for Recurrent Acute Pancreatitis (RAP) and Chronic Pancreatitis (CP) in White patients are rare in Black patients.” The authors acknowledge the Epidemiology Data Center, Michael O’Connell, PhD Division of Gastroenterology & Hepatology at the University of Pittsburgh for data management of NAPS2 studies, Indrani Halder, PhD Division of Cardiology at the UPMC Heart and Vascular Institute for critical review of versions of the manuscript and providing helpful feedback, Kim Stello and Danielle Dwyer for genotyping and laboratory management, and other members of the NAPS2 consortium.
Grant support
This research was partly supported by the UPMC GI T32 Training Grant T32 DK063922 (AEP, JL) NIH DK061451 (DCW), DK077906 (DY), UO1 DK108327 (DC), UO1 DK108320 (CEF), U01 DK108306 (DCW, DY). This publication was made possible in part by Grant Number UL1 RR024153 and UL1TR000005 from the National Center for Research Resources, a component of the National Institutes of Health, and NIH Roadmap for Medical Research (University of Pittsburgh. PI, Steven E Reis, MD). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NCRR or NIH.
Footnotes
Affiliation of authors during patient recruitment were – Bimaljit S Sandhu (Virginia Commonwealth University, Richmond VA), John Baillie (Duke University, Durham, NC), Darwin Conwell (Brigham & Women’s Hospital, Boston, MA), Gregory A Coté (Indiana University, Indianapolis, IN), Andres Gelrud (University of Pittsburgh Medical Center, Pittsburgh, PA and University of Chicago, Chicago, IL).
Disclosure
The authors declare no relevant conflicts to disclose.
References
- [1].Whitcomb DC, Frulloni L, Garg P, et al. Chronic pancreatitis: an international draft consensus proposal for a new mechanistic definition. Pancreatology 2016;16:218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ahmed Ali U, Issa Y, Hagenaars JC, et al. Risk of recurrent pancreatitis and progression to chronic pancreatitis after a first episode of acute pancreatitis. Clin Gastroenterol Hepatol 2016;14:738–46. [DOI] [PubMed] [Google Scholar]
- [3].Sankaran SJ, Xiao AY, Wu LM, et al. Frequency of progression from acute to chronic pancreatitis and risk factors: a meta-analysis. Gastroenterology 2015;149:1490–500. e1. [DOI] [PubMed] [Google Scholar]
- [4].Amann ST, Yadav D, Barmada MM, et al. Physical and mental quality of life in chronic pancreatitis: a case-control study from the North American Pancreatitis Study 2 cohort. Pancreas 2013;42:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yadav D, Timmons L, Benson JT, et al. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol 2011;106: 2192–9. [DOI] [PubMed] [Google Scholar]
- [6].Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology 2012;143:1179–87. e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Whitcomb DC, Preston RA, Aston CE, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology 1996;110:1975–80. [DOI] [PubMed] [Google Scholar]
- [8].Witt H, Luck W, Becker M, et al. Mutation in the SPINK1 trypsin inhibitor gene, alcohol use, and chronic pancreatitis. J Am Med Assoc 2001;285:2716–7. [DOI] [PubMed] [Google Scholar]
- [9].LaRusch J, Jung J, General IJ, et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet 2014;10. e1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 2008;40:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141–5. [DOI] [PubMed] [Google Scholar]
- [12].Masamune A, Nakano E, Kume K, et al. Identification of novel missense CTRC variants in Japanese patients with chronic pancreatitis. Gut 2013;62:653–4. [DOI] [PubMed] [Google Scholar]
- [13].Masamune A, Nakano E, Kume K, et al. PRSS1 c.623G>C (p.G208A) variant is associated with pancreatitis in Japan. Gut 2014;63:366. [DOI] [PubMed] [Google Scholar]
- [14].Paliwal S, Bhaskar S, Mani KR, et al. Comprehensive screening of chymotrypsin C (CTRC) gene in tropical calcific pancreatitis identifies novel variants. Gut 2013;62:1602–6. [DOI] [PubMed] [Google Scholar]
- [15].Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med 1998;339:645–52. [DOI] [PubMed] [Google Scholar]
- [16].Cohn JA, Friedman KJ, Noone PG, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 1998;339:653–8. [DOI] [PubMed] [Google Scholar]
- [17].Larusch JSK, Yadav D, Whitcomb DC. CFTR, PRSS1, SPINK1, and CTRC mutations in the final NAPS2 cohort. Pancreatology 2015;15:S79. [Google Scholar]
- [18].Yadav D, Muddana V, O’Connell M. Hospitalizations for chronic pancreatitis in allegheny county, Pennsylvania, USA. Pancreatology 2011;11:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang AL, Vadhavkar S, Singh G, et al. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch Intern Med 2008;168:649–56. [DOI] [PubMed] [Google Scholar]
- [20].Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol 2012;107:1096–103. [DOI] [PubMed] [Google Scholar]
- [21].Lowenfels AB, Maisonneuve P, Grover H, et al. Racial factors and the risk of chronic pancreatitis. Am J Gastroenterol 1999;94:790–4. [DOI] [PubMed] [Google Scholar]
- [22].Yadav D, Lee E, Papachristou GI, et al. A population-based evaluation of readmissions after first hospitalization for acute pancreatitis. Pancreas 2014;43:630–7. [DOI] [PubMed] [Google Scholar]
- [23].Wilcox CM, Sandhu BS, Singh V, et al. Racial differences in the clinical profile, causes, and outcome of chronic pancreatitis. Am J Gastroenterol 2016;111(10):1488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Halder I, Shriver M, Thomas M, et al. A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: utility and applications. Hum Mutat 2008;29:648–58. [DOI] [PubMed] [Google Scholar]
- [25].Halder I, Matthews KA, Buysse DJ, et al. African genetic ancestry is associated with sleep depth in older african Americans. Sleep 2015;38:1185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Halder I, Kip KE, Mulukutla SR, et al. Biogeographic ancestry, self-identified race, and admixture-phenotype associations in the Heart SCORE Study. Am J Epidemiol 2012;176:146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Whitcomb DC, Yadav D, Adam S, et al. Multicenter approach to recurrent acute and chronic pancreatitis in the United States: the North American Pancreatitis Study 2 (NAPS2). Pancreatology 2008;8:520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wilcox CM, Yadav D, Ye T, et al. Chronic pancreatitis pain pattern and severity are independent of abdominal imaging findings. Clin Gastroenterol Hepatol 2015;13:552–60. quiz e28–e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schneider A, Larusch J, Sun X, et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011;140: 162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].LaRusch J, Lozano-Leon A, Stello K, et al. The common chymotrypsinogen C (CTRC) variant G60G (C.180T) increases risk of chronic pancreatitis but not recurrent acute pancreatitis in a North American population. Clin Transl Gastroenterol 2015;6. e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Whitcomb DC, LaRusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet 2012;44:1349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li D, Achkar JP, Haritunians T, et al. A pleiotropic missense variant in SLC39A8 is associated with Crohn’s disease and human gut microbiome composition. Gastroenterology 2016;151:724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Masson E, Chen JM, Scotet V, et al. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum Genet 2008;123:83–91. [DOI] [PubMed] [Google Scholar]
- [34].Chang MC, Chang YT, Wei SC, et al. Association of novel chymotrypsin C gene variations and haplotypes in patients with chronic pancreatitis in Chinese in Taiwan. Pancreatology 2009;9:287–92. [DOI] [PubMed] [Google Scholar]
- [35].Zakharia F, Basu A, Absher D, et al. Characterizing the admixed african ancestry of african Americans. Genome Biol 2009;10. R141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chandak GR, Idris MM, Reddy DN, et al. Absence of PRSS1 mutations and association of SPINK1 trypsin inhibitor mutations in hereditary and non-hereditary chronic pancreatitis. Gut 2004;53:723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Witt H, Beer S, Rosendahl J, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet 2013;45:1216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kavic SM, Frehm EJ, Segal AS. Case studies in cholera: lessons in medical history and science. Yale J Biol Med 1999;72:393–408. [PMC free article] [PubMed] [Google Scholar]
- [39].Modiano D, Luoni G, Sirima BS, et al. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature 2001;414:305–8. [DOI] [PubMed] [Google Scholar]
- [40].Masson E, Chen JM, Audrezet MP, et al. A conservative assessment of the major genetic causes of idiopathic chronic pancreatitis: data from a comprehensive analysis of PRSS1, SPINK1, CTRC and CFTR genes in 253 young French patients. PLoS One 2013;8. e73522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zvereff VV, Faruki H, Edwards M, et al. Cystic fibrosis Carrier screening in a North American population. Genet Med 2014;16:539–46. [DOI] [PubMed] [Google Scholar]
- [42].Macek M Jr, Mackova A, Hamosh A, et al. Identification of common cystic fibrosis mutations in African-Americans with cystic fibrosis increases the detection rate to 75%. Am J Hum Genet 1997;60:1122–7. [PMC free article] [PubMed] [Google Scholar]
- [43].Cabello GM, Cabello PH, Llerena JC Jr, et al. Polymorphic markers suggest a gene flow of CFTR gene from Sub-Saharan/Arabian and Mediterranean to Brazilian Population. J Hered 2006;97:313–7. [DOI] [PubMed] [Google Scholar]
- [44].Whitcomb DC. Framework for interpretation of genetic variations in pancreatitis patients. Front Physiol 2012;3:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yadav D, Hawes RH, Brand RE, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med 2009;169:1035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.