

Abstract
The anion [P4O11]2−, employed as its bis(triphenylphosphine)iminium (PPN) salt, is shown herein to be a versatile reagent for nucleophile tetraphosphorylation. Treatment under anhydrous conditions with an alkylamine base and a nucleophile (HNuc1), such as an alcohol (neopentanol, cyclohexanol, 4-methylumbelliferone, and Boc-Tyr-OMe), an amine (propargylamine, diethylamine, morpholine, 3,5-dimethylaniline, and isopropylamine), dihydrogen phosphate, phenylphosphonate, azide ion, or methylidene triphenylphosphorane, results in nucleophile substituted tetrametaphosphates ([P4O11Nuc1]3−) as mixed PPN and alkylammonium salts in 59% to 99% yield. Treatment of the resulting functionalized tetrametaphosphates with a second nucleophile (HNuc2), such as hydroxide, a phenol (4-methylumbelliferone), an amine (propargylamine and ethanolamine), fluoride, or a nucleoside monophosphate (uridine monophosphate, deoxyadenosine monophosphate, and adenosine monophosphate), results in ring opening to linear tetraphosphates bearing one nucleophile on each end ([Nuc1(PO3)3PO2Nuc2]4−). When necessary, these linear tetraphosphates are purified by reverse phase or anion exchange HPLC, yielding triethylammonium or ammonium salts in 32% to 92% yield from [PPN]2[P4O11]. Phosphorylation of methylidene triphenylphosphorane as Nuc1 yields a new tetrametaphosphate-based ylide ([Ph3PCHP4O11]3−, 94% yield). Wittig olefination of 2’,3’-O-isopropylidene-5’-deoxy-5’-uridylaldehyde using this ylide results in a 3’-deoxy-3’,4’-didehydronucleotide derivative, isolated as the triethylammonium salt in 54% yield.
Graphical Abstract
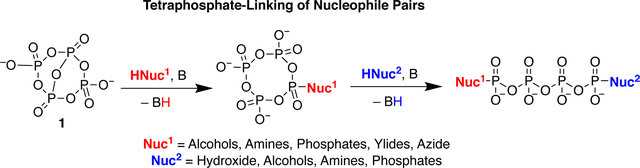
Introduction
Oligophosphates permeate biology, with roles in energy transfer,1 signaling,2 phosphate storage,3 and enzymatic regulation.4,5 However, the syntheses of such molecules are complex, historically requiring iterative phosphorylation procedures6 or tedious separation of complex mixtures.7 While enzymatic methods are utilized for the preparation of canonical compounds such as 5’-adenosine triphosphate (ATP), these procedures typically cannot be applied to non-natural nucleotides, which are essential to medical and biochemical research.8 In recent years, significant progress has been made to efficiently introduce oligophosphate groups in a single operation.9–14 With the greater availability of these compounds, increased research has been conducted on the roles of oligophosphates in biology, such as elucidating the human PolyP-ome.15,16 A dominant motif in these new oligophosphorylation procedures has been the introduction of a triphosphate chain from readily available trimetaphosphate.10–13,17
Activated forms of trimetaphosphate have been used in conjunction with monophosphate substrates to synthesize various tetraphosphates, such as Taylor’s work on the synthesis of nucleoside 5’-tetraphosphates (Figure 1).11,12 More recently, Jessen reported the synthesis of bifunctional tetraphosphates from a P(III) phosphoramidite reagent and various phosphate nucleophiles.9 Jessen’s methodology utilizes an oxidation step to form a cyclic trimetaphosphate that is subsequently ring opened with another nucleophile (Figure 1). The redox-neutral synthesis of tetraphosphate derivatives from an activated tetrametaphosphate reagent would be highly efficient, resulting in a simplified procedure and allowing phosphorylation of oxidizable substrates. For example, utilizing a tetrametaphosphate based reagent allows synthesis of nucleoside tetraphosphate derivatives from nucleosides rather than first requiring synthesis of the corresponding nucleoside monophosphate and subsequent treatment with a trimetaphosphate based reagent. We recently demonstrated this chemistry by introducing an activated tetrametaphosphate reagent ([PPN]2[P4O11], [PPN]2[1])18 and reporting the synthesis of nucleoside tetra- and pentaphosphates.19 Herein, we expand the reaction scope of this reagent to a range of C, N, and O nucleophiles (Figure 1). Furthermore, we adapt and expand the existing ring opening chemistry of substituted trimetaphosphate derivatives9–14 to the synthesized tetrametaphosphate derivatives. Mixing and matching the two nucleophiles in these reactions leads to a variety of terminally disubstituted tetra- and pentaphosphates.
Figure 1.
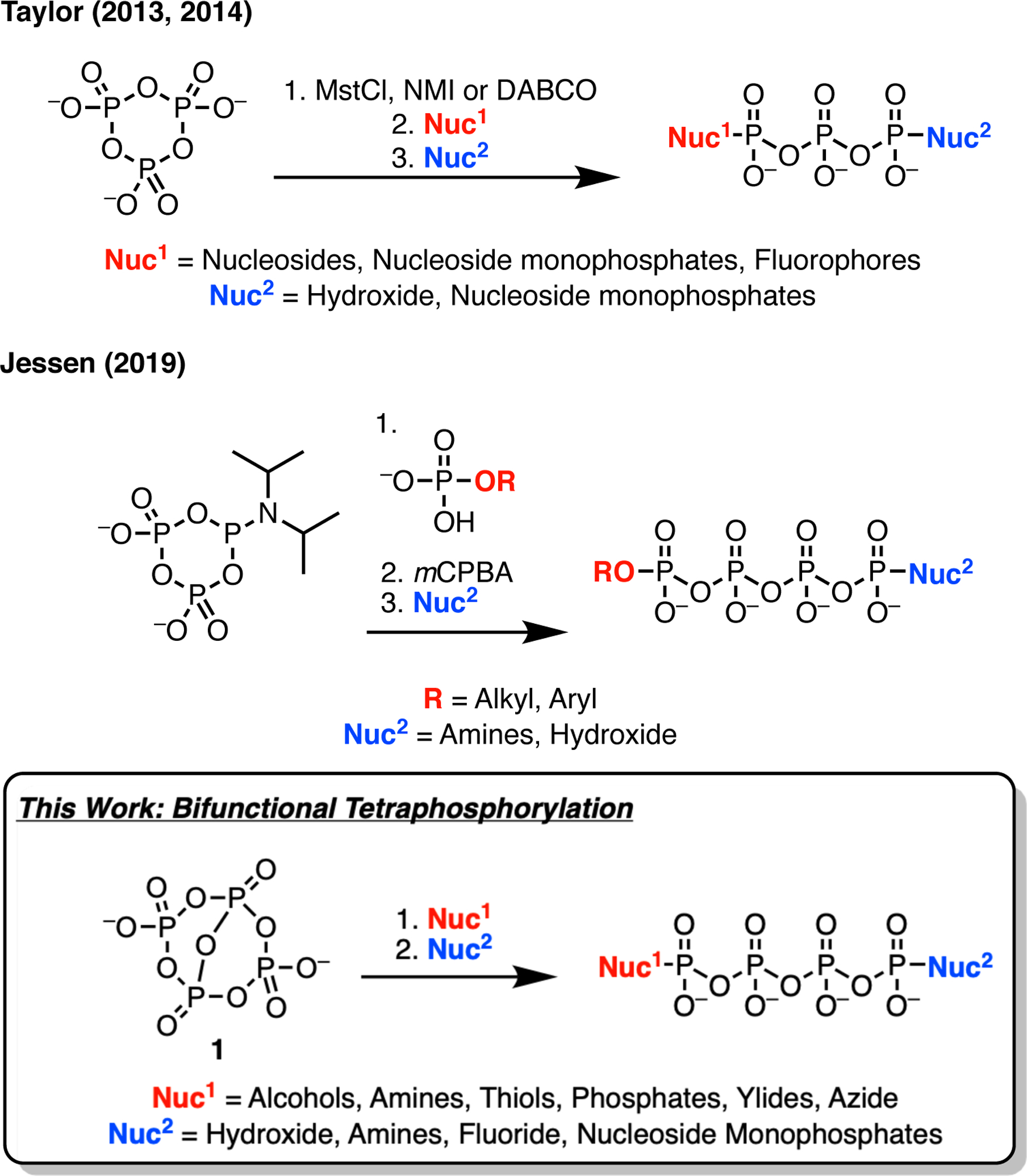
Representative syntheses of bifunctional tetraphosphates from Taylor,11,12 Jessen,9 and this work on the tetraphosphorylation of a diverse range of nucleophiles to make mono- and disubstituted tetraphosphates.
Results
Synthesis of Substituted Metaphosphates ([P4O11Nuc1]3−)
Under anhydrous conditions, 1 reacts rapidly and cleanly with many nucleophiles. Salts of the products, substituted metaphosphates, are isolated easily and in high yield by precipitation from the reaction mixture with diethyl ether. We have previously reported the treatment of anion 1 with methanol18 and nucleosides.19 We therefore treated anion 1 with various alcohols in the presence of triethylamine, to prevent side reactions by scavenging the acidic proton that is produced. In this manner, mixed PPN and triethylammonium salts of anions 2 and 3 were obtained by treating anion 1 with neopentanol, a hindered primary alcohol, and cyclohexanol (Figure 2A). Likewise, anion 1 reacts rapidly with phenol derivatives such as 4-methylumbelliferone, a common fluorophore, in the presence of triethylamine. The resulting mixed PPN and triethylammonium salt of anion 4 was isolated in 91% yield by precipitation from acetonitrile with diethyl ether. The isolated alcohol derivatives of tetrametaphosphate formed oils upon precipitation rather than crystalline material, and removing solvent under vacuum yielded amorphous solids.
Figure 2.
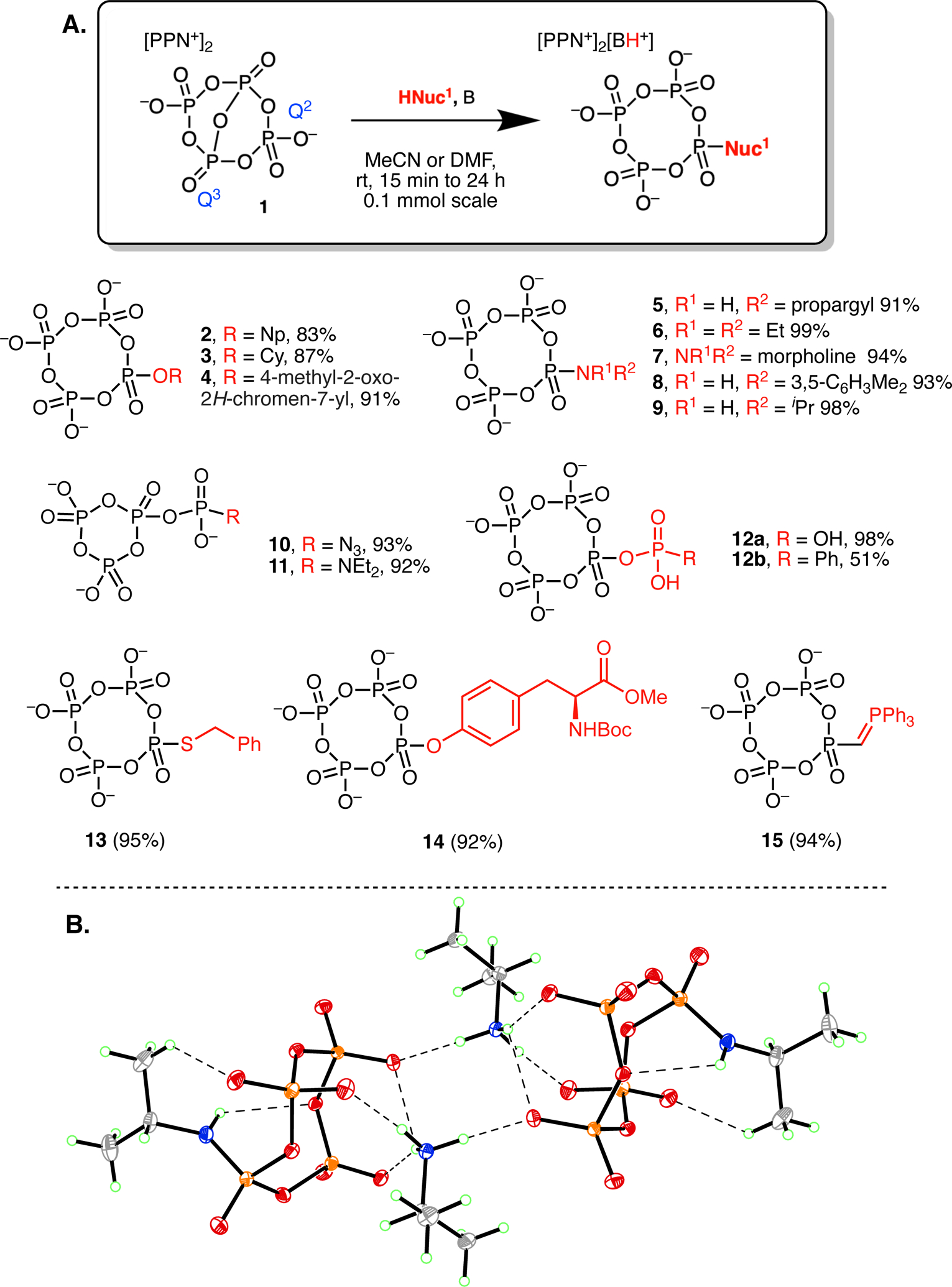
A. Synthesis of anions 2 to 15 from anion 1 B. X-ray crystal structure of [PPN]2[H3NiPr][9] with thermal ellipsoids set at 50% probability, the PPN counterions omitted for clarity, and hydrogen bonds denoted by dashed lines (P = orange, O = red, N = blue, C = grey, H = green).
Anion 1 also reacts readily with primary and secondary amines resulting in tetrametaphosphate phosphoramidate derivatives containing a P–N bond. Near quantitative reactivity was observed between anion 1 and propargylamine, diethylamine, morpholine, 3,5-dimethylaniline, and isopropylamine resulting in crystalline compounds 5-9 (Figure 2). In the case of alkyl amine substrates, a second equivalent of amine is consumed by the acidic proton that is generated to yield a mixed PPN and alkylammonium salt.
Compounds 5-9 were obtained as crystalline salts, such as [PPN]2[H3NiPr][9], which was crystallographically characterized. This structure is similar to that of amine functionalized trimetaphosphates we have previously reported,10 with planar phosphoramidate nitrogens (Figure 2B). Furthermore, these phosphoramidate N-H groups are found to engage in intramolecular hydrogen bonds that likely help to lock in the observed conformation. The acidic isopropylammonium N-H groups are hydrogen bonded to basic phosphate oxygens on two molecules of 9 resulting in a dimeric structure in the solid state. The resulting dimeric tetraanion is such that it presents four lipophilic isopropyl groups on its surface in a manner that may aid solubility and ability to crystallize.
Anion 1 is also a suitable reagent for tetraphosphorylation of inorganic nucleophiles. Treatment with tetrabutylammonium azide ([TBA][N3]) cleanly forms compound 10 in 24 hours. Relatedly, phosphoazidate analogs of nucleoside 5’-triphosphates have been used as photolabile affinity labels, extruding N2 under irradiation and covalently attaching to a protein binding site.20 Interestingly, anion 10 is an isomer of tetrametaphosphate consisting of an azidophosphate substituted trimetaphosphate. The isomer observed in 10 ostensibly results from nucleophile attack at a Q2 phosphorus rather than attack at a Q3 phosphorus (Figure 2A).21 Interestingly, adding DMAP (4-dimethylaminopyridine) to 1 promotes formation of this isomer, allowing phosphorylation of diethylamine to give 11.
Recalling our previous work on functionalized trimetaphosphates,10 the parent monophosphate substituted tetrametaphosphate can be obtained by treatment of anion 1 with [PPN][H2PO4], immediately leading to the PPN salt of anion 12a (Figure 2A). Similarly, treatment of anion 1 with the TBA salt of monohydrogen phenylphosphonate22 yields anion 12b.
Treatment of 1 with benzylmercaptan and triethylamine yields anion 13 in manner similar to that of the alcohol substrates. Organothiophosphates have widespread applications and typically must be prepared from a P(V) species, as is the case here.23 In a P(III) based synthesis, such as the oligophosphorylation methodology employed by Jessen,9,14 the oxidation of P(III) to P(V) would also be expected to oxidize the sulfur atom, preventing the isolation of species with an oxidized phosphorus and reduced sulfur.
We also sought to achieve tetraphosphorylation of an amino acid side chain as oligophosphorylation of protein side chains has recently been discovered as a post-translational modification.5,24 Tyrosine was chosen due to the UV absorbance of the aromatic ring and the good nucleophilicity of phenolate under basic conditions. Protection of the amine moiety was found to be necessary for an efficient reaction due to the improved solubility of protected amino acids and inhibition of side reactivity at the amine position. Accordingly, treatment of 1 with Boc-Tyr-OMe (N-(tert-butoxycarbonyl)-L-tyrosine methyl ester) in acetonitrile in the presence of triethylamine provides anion 14.
Synthesis of Linear Tetraphosphate Derivatives ([Nuc1(PO3)3−PO2Nuc2]4–)
In one-pot reactions, we synthesized a variety of disubstituted tetraphosphates ([Nuc1(PO3)3PO2Nuc2]4−) by in situ generation of [P4O11Nuc1]3− species followed by treatment with a second nucleophile. When necessary, the resulting compounds were purified by RP-HPLC with a triethylammonium (TEA) acetate buffer or AX-HPLC with ammonium bicarbonate buffer, giving TEA or ammonium salts after lyophilization. Ring opening of these metaphosphate derivatives selectively gives linear tetraphosphate derivatives as opposed to branched phosphates, a result that has been investigated computationally by Jessen for trimetaphosphate derivatives.14 This result is logical as ring opening reactions that would give rise to a highly charged terminal −[PO3]2− group should be disfavored compared to those that produce a terminal −[PO3R]−group. Ring opening reactions that have been demonstrated for trimetaphosphate derivatives9,14 appear to be generally applicable to the tetrametaphosphate derivatives presented here, albeit often with increased reaction times, suggesting that larger metaphosphate rings are more difficult to ring open.
Ring opening of substituted metaphosphates with hydroxide or water gives monofunctionalized tetraphosphate products. In a prototypical example, treatment of the phenolic tetrametaphosphate intermediate 4 with aqueous tetrabutylammonium hydroxide ([TBA][OH]) yields a phenolic linear tetraphosphate (16) and replaces the PPN cations with water soluble TBA. HPLC purification, facilitated by the strong UV absorbance of this umbelliferone-derived substrate, provided the TEA salt in 49% yield from [PPN]2[1] (Figure 3A). The ring opening reaction is not perfectly efficient, necessitating HPLC purification and resulting in a lower yield. Additionally, cation exchange from water insoluble PPN salts to soluble TBA or sodium salts for HPLC purification involves precipitation and filtration steps which may diminish yields. In contrast, ring opening of thiol derivative 13 with [TBA][OH] proceeds cleanly, and the TBA salt of 17 was isolated without HPLC purification. The moderate 71% yield of 17 is likely a result of the cation exchange from PPN to TBA. Tyrosine derivative 14 is also cleanly ring opened to 18 with saponification of the methyl ester upon introduction of [TBA][OH]. Protection of the carboxylate moiety as well as having a base stable amine protecting group such as Boc were necessary for this reaction to proceed. Treatment of 1 with either Boc-Tyr-OH or Fmoc-Tyr-OMe and subsequent treatment with [TBA][OH] yielded only tetrametaphosphate instead of the desired linear tetraphosphate derivative. Additionally, the P–N bond in 10 is remarkably water tolerant, and this compound can be converted accordingly to a linear phosphate as well. Cation exchange and dissolution of 10 in neutral water results in formation of compound 19 in 24 hours with only minor hydrolytic cleavage of the azide group (Figure 3A).
Figure 3.
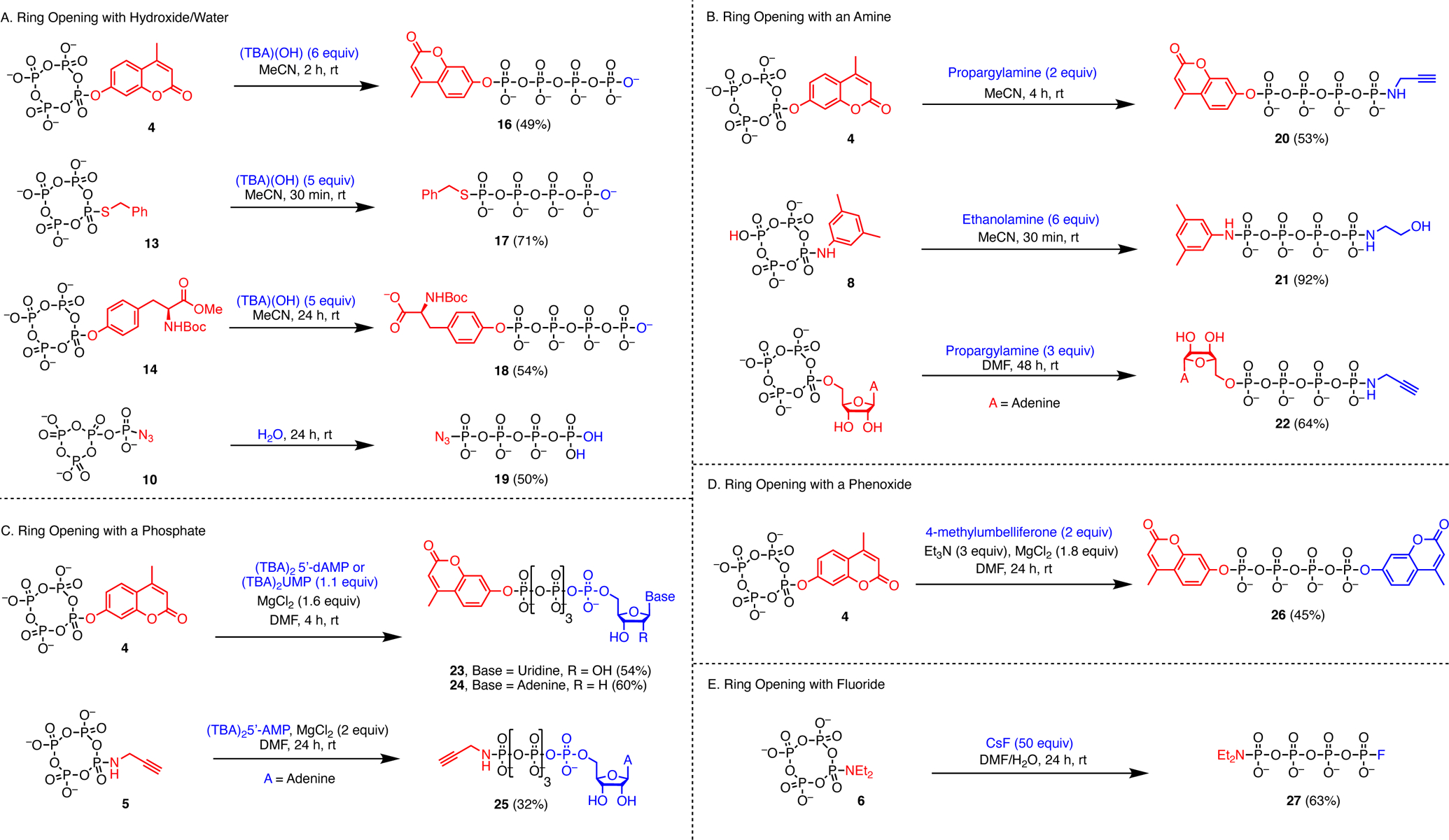
Synthesis of terminally disubstituted linear tetra- and pentaphosphates (16–27). The reported yields are for one-pot reactions from 1.
Ring opening is also possible with primary amines, and in a one-pot reaction, a disubstituted tetraphosphate bearing a 4-methylumbelliferone-derived group at one end and a propargylamine-derived phosphoramidate group at the other was synthesized via intermediate 4 and purified by HPLC, providing the TEA salt of anion 20 in 62% yield from [PPN]2[1] (Figure 3A). Similar alkyne substituted tetraphosphates have been functionalized further through cycloaddition “click” chemistry.9 We found that amine substituted tetrametaphosphate species are also amenable to ring opening with a second nucleophile. In a one-pot reaction from [PPN]2[1], treating intermediate 8 with ethanolamine rapidly and cleanly leads to a disubstituted tetraphosphate bearing two different phosphoramidate groups at the termini (21, Figure 3B). Other phosphoramidate P-N bond containing compounds such as anion 20 are stable in neutral water at room temperature for days. However, anion 21 was found to hydrolyze rapidly in room temperature neutral water, demonstrating the variable water stability of phosphoramidates.25 However, anion 21 forms cleanly and was isolated as a mixed PPN and hydroxyethylammonium salt in 92% yield from [PPN]2[1] without HPLC purification. Additionally, treatment of 1 with adenosine and subsequent ring opening with propargylamine results in a terminally amino functionalized adenosine tetraphosphate (22) after HPLC purification.
Previous syntheses of tetraphosphates have utilized the reaction of substituted trimetaphosphates with nucleoside monophosphates in the presence of MgCl2 as a promoter.9,11,12 In an analogous fashion, treatment of substituted tetrametaphosphate intermediate 4 with a nucleoside monophosphate and MgCl2 yields ϵ-fluorophore labelled nucleoside pentaphosphates 23 and 24 in 5 hours. These compounds were isolated as their TEA salts in 54% and 60% yield respectively after HPLC purification in a one-pot reaction from 1 (Figure 3C). It has been shown that although γ-fluorophore labelled nucleoside triphosphates are poor substrates for DNA and RNA polymerases, ϵ-labelled nucleoside pentaphosphates exhibit significantly higher activity.26,27 Therefore, related compounds have been used in high-throughput DNA sequencing, and single nucleotide polymorphism (SNP) assays.26–30 The sole previous synthesis of ϵ-fluorophore labelled nucleoside pentaphosphates is low yielding and tedious, requiring expensive nucleoside triphosphates (NTP) and taking 7 days to complete.26 This facile synthesis of such compounds may facilitate their use in such biochemical studies. MgCl2 promoted ring opening with nucleoside monophosphates is also amenable to amine substituted tetrametaphosphates. Treatment of 5 with AMP and MgCl2 gives the ammonium salt of pentaphosphate derivative 25 in 32% yield from 1 after AX-HPLC purification (Figure 3C).
MgCl2 promoted ring opening is also applicable to nucleophiles other than phosphates. Treating intermediate 4 with an additional equivalent of 4-methylumbelliferone in the presence of triethyamine, yielded the symmetric tetraphosphate 26, isolated as its TEA salt in 45% yield from [PPN]2[1] after HPLC purification (Figure 3A).
Conditions for ring opening trimetaphosphate derivatives with cesium fluoride were adapted from Jessen14 to achieve ring opening of amine derivative 5 to fluoro substituted anion 27. In contrast to similar trimetaphosphate derivatives which are ring opened in minutes under these conditions,14 this reaction reached completion in approximately 24 hours.
Phosphorylation of Methylenetriphenylphosphorane and Wittig Olefination Chemistry
In a previous report, we detailed the triphosphorylation of the phosphorus ylide methylene triphenylphosphorane as a new route to oligophosphate analogues containing nonhydrolyzable P–C bonds.10 Anion 1 similarly reacted with methylene triphenylphosphorane31 (2 equiv) to give the mixed PPN and methyltriphenylphosphonium salt of tetrametaphosphate based ylide 15. This new ylide effected Wittig olefination of paraformaldehyde, yielding vinyl tetrametaphosphate 28 (Figure 4). Hydrolysis of 15 led to methyl tetrametaphosphate derivative 29. In order to synthesize a nucleotide analogue, 15 was treated with 2’,3’-O-isopropylidene-5’-deoxy-5’-uridylaldehyde,32 a reaction which surprisingly resulted in removal of the isopropylidene protecting group and dehydration to a 3’-deoxy-3’,4’-didehydronucleoside derivative whose formula and structure we confirmed by 2D-NMR spectroscopy and ESI-MS. Ring opening of this metaphosphate derivative with [TBA][OH] yielded the linear form 30, which was purified by HPLC to give the TEA salt. A number of syntheses of 3’,4’-didehydronucleosides have been reported,33,34 including from Wittig chemistry.35,36 Some of these derivatives have found biological utility, including as a tumor suppressor.33,34,37 Furthermore, nucleosides containing a 5’-(E)-vinylphosphonate moiety have been used to synthesize modified single-stranded small interfering RNAs (ss-siRNAs) that bind to Argonaute-2 (Ago2), an enzyme responsible for cleaving mRNA.38,39
Figure 4.
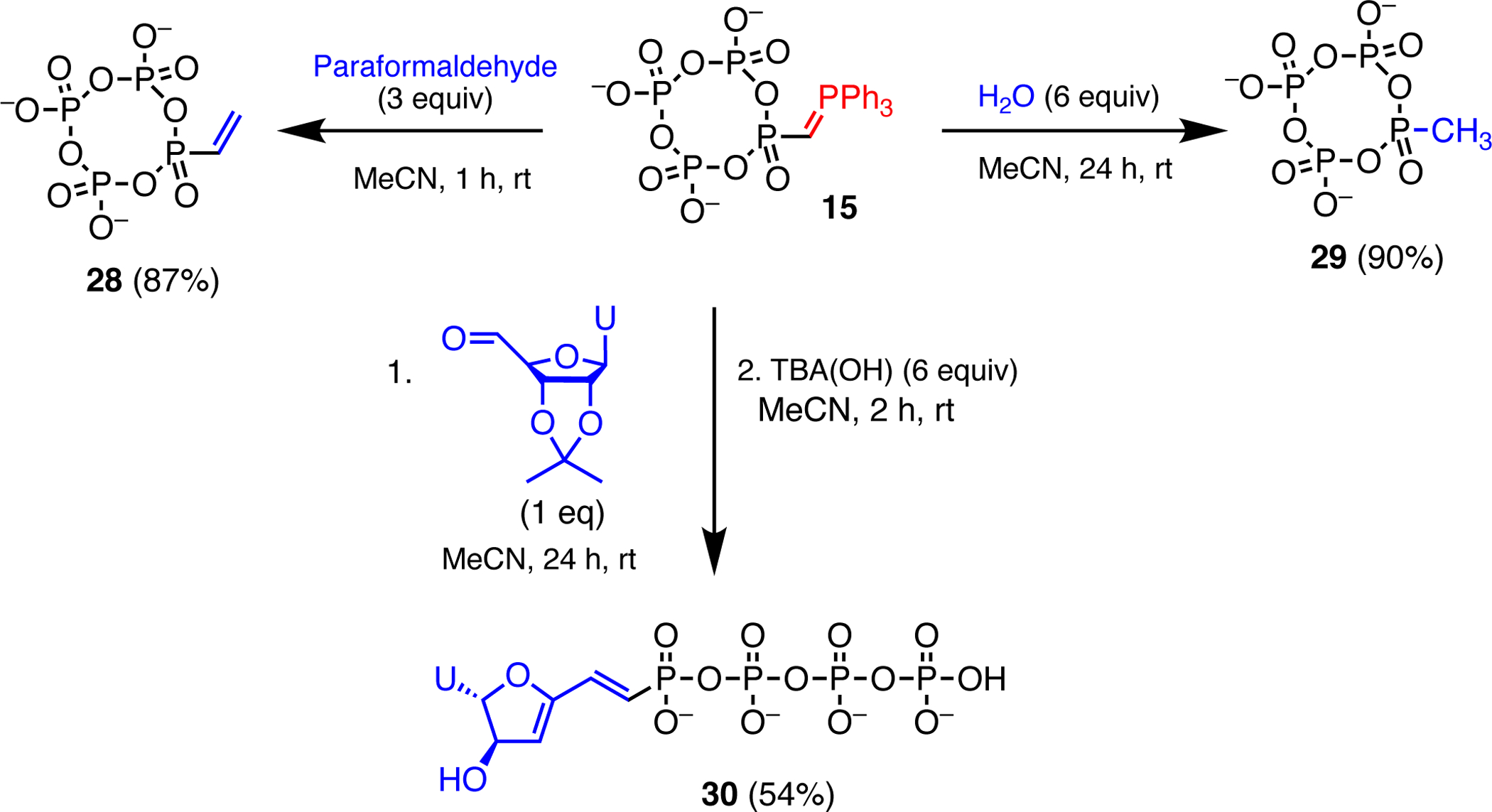
Synthesis of tetrametaphosphates 28–29, and a linear 3’-deoxy-3’,4’-didehydronucleoside derivative 30 upon treatment of 15 with 2’,3’-O-isopropylidene-5’-deoxy-5’-uridylaldehyde and subsequent ring opening with [TBA][OH].
Discussion
NMR Characterization of Oligophosphates
The phosphates synthesized in this study are characterized by multinuclear NMR and ESI-MS, which together give definitive assignment of these species. The sensitivity of 31P{1H} NMR and the highly diagnostic chemical shift of different phosphate environments facilitate rapid identification of these species from crude 31P{1H} NMR spectra in non-deuterated solvents (Figure 5). Orthophosphate and its esters appear in the range 5 to −5 ppm, pyrophosphate and other terminal linear phosphates from −5 to −15 ppm, bridging linear phosphates as well as metaphosphates from −15 to −25 ppm, and ultraphosphates appear from −25 to −40 ppm.40 While the chemical shifts of terminal phosphates are highly sensitive to changes in pH, the other groups are almost invariant to pH. Furthermore, aliphatic alcohol derived phosphoesters show almost no chemical shift change from that of the unfunctionalized phosphate, while phenol derived phosphoesters show an upfield shift by ~5 ppm. Aliphatic phosphoramidates show a downfield shift of ~10 ppm, and aryl phosphoramidates show a downfield shift of ~2 ppm. In combination with 31P−31P coupling patterns inherent in oligophosphates, the species in Figure 5 can be assigned readily even without the 31P−1H coupling information gained from 31P NMR.
Figure 5.
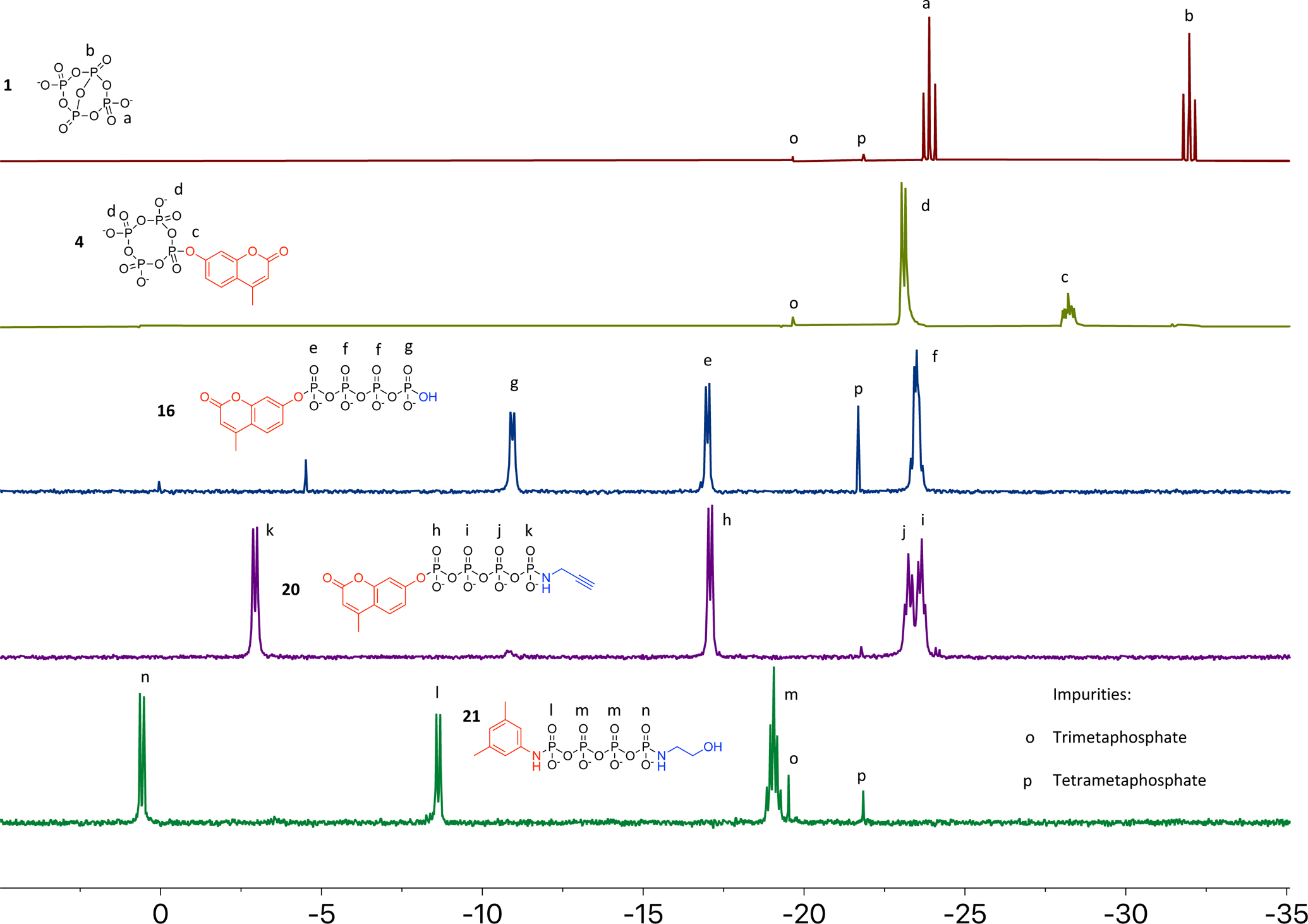
31P{1H} NMR spectra of selected compounds. Functionalized phosphates exhibit highly diagnostic chemical shifts according to their substituents. In conjunction with 2D NMR experiments and MS data, this affords definitive assignment of the isolated compounds. The chemical shifts of unfunctionalized terminal phosphates are, however, highly pH dependent.
Limitations of Reagent [PPN]2[1]
Anion 1 is an active reagent for the tetraphosphorylation of a wide range of anhydrous nucleophiles, but it is highly moisture sensitive. Adventitious water reacts to form dihydrogen tetrametaphosphate ([P4O12H2]2−)18 which is difficult to separate from [P4O11Nuc1]3− compounds. Therefore, when possible, stock solutions of nucleophiles in solvents such as acetonitrile and dimethylformamide were stored over 4Å molecular sieves for at least 12 hours before being utilized. The phosphorylation procedures in this report were carried out under a dry N2 atmosphere in a glovebox, although we have also reported the use of 1 with Schlenk procedures.19
Some substrates were found to be unsuitable for this tetraphosphorylation protocol. No reactivity was observed between anion 1 and stoichiometric tert-butanol, a tertiary alcohol, at ambient or elevated temperatures. Similarly, no reactivity was observed with adamantanethiol, a tertiary thiol. These results suggest an upper limit on the steric bulk of Nuc1.
Additionally, some substrates upon treatment with anion 1 give reversible addition. In acetonitrile, treatment with TBA acetate generates acetyl tetrametaphosphate nearly quantitatively by 31P{1H} NMR (See SI). However, primarily tetrametaphosphate or anion 1 is isolated upon work up. We obtained similar results with TBA bisulfate, and these observations could be explained by equilibria that favor the products in acetonitrile. Reagent 1 reacts with nucleophiles to give an intramolecular leaving group that is part of the metaphosphate ring, and it is thus well poised to reversibly expel weak nucleophiles. Attempting to precipitate putative equilibrium species ([P4O11Nuc1]3−) with diethyl ether results in precipitation of anion 1. The more soluble nucleophile remains in solution, driving the equilibria towards the reactants. In contrast, phosphorylation of substrates such as neutral alcohols and primary or secondary amines results in a proton transfer step to an external base. The favorability of this proton transfer step is likely key to the irreversibility of these reactions.
We have also been unable to identify conditions for ring opening compounds 12a and 12b to linear pentaphosphate derivatives. Treatment of 12a and 12b with hydroxide or amines instead results in cleavage to tetrametaphosphate and a monophosphate derivative, indicating that tetrametaphosphate is a better leaving group than the linear phosphate that would result from ring opening. No reactivity was observed between these compounds and nucleoside monophosphates in the presence of MgCl2. Furthermore, substituted tetrametaphosphate derivatives such as 4 did not undergo a reaction with the TBA salt of monohydrogen phenylphosphonate ([TBA][HPO3Ph]) even in the presence of MgCl2. This difference in reactivity between [TBA][HPO3Ph]22 and [TBA]2[AMP]45 is likely due to the difference in charge. [TBA]2[AMP], the TBA salt of a dianion, is readily isolated and soluble in solvents such as acetonitrile and dimethylformamide, possibly due to stabilization of the deprotonated phosphate group through hydrogen bonding interactions to the sugar moiety. However, we were unable to isolate [TBA]2[PO3Ph] in organic solvents. The lower charge of monohydrogen [TBA][HPO3Ph] may correspond to lower nucleophilicity.
Phosphorylation Mechanism and Isomerism
Treatment of anion 1 with most nucleophiles yields a substituted tetrametaphosphate derivative. However, we have observed reversible addition of some nucleophiles as well as rare formation of another product isomer (10 and 11), prompting a computational investigation of the reaction mechanism. The reaction coordinate was calculated for the addition of inorganic azide to 1, leading to both a substituted tetrametaphosphate derivative via nucleophilic attack at a Q3 phosphorus or the observed orthophosphoryl-trimetaphosphate isomer, 10, via nucleophilic attack at a Q2 phosphorus (Figure 6). Geometries were optimized and frequency calculations performed at the B3LYP-D3BJ/ma-def2-TZVP(-f) level of theory41–43 with an acetonitrile CPCM solvation model in Orca 4.2.44 The resulting DFT thermochemistry data for this reaction is presented at 298.15 K. For azide, the orthophosphoryl-trimetaphosphate isomer (10) is thermodynamically downhill from the starting material by 2.0 kcal/mol while the tetrametaphosphate isomer (iso-10 is uphill by 2.4 kcal/mol, explaining the observed selectivity. However, this energy difference is small. The energies of the possible products are close enough to that of the starting materials to explain the reversible addition of some substrates. A transition state was also located corresponding to conversion between the two isomers without the intermediacy of 1, TS3. However, this transition state is thermodynamically uphill from the starting materials by 31.1 kcal/mol, indicating that this direct interconversion is unfavorable. In the case of protic substrates, the transfer of an acidic proton to an amine scavenger after phosphorylation results in an irreversible reaction. In reactions where no proton transfer occurs, the phosphorylation step is reversible and solubility differences between phosphates and the substrate can be exploited to regenerate 1.
Figure 6.
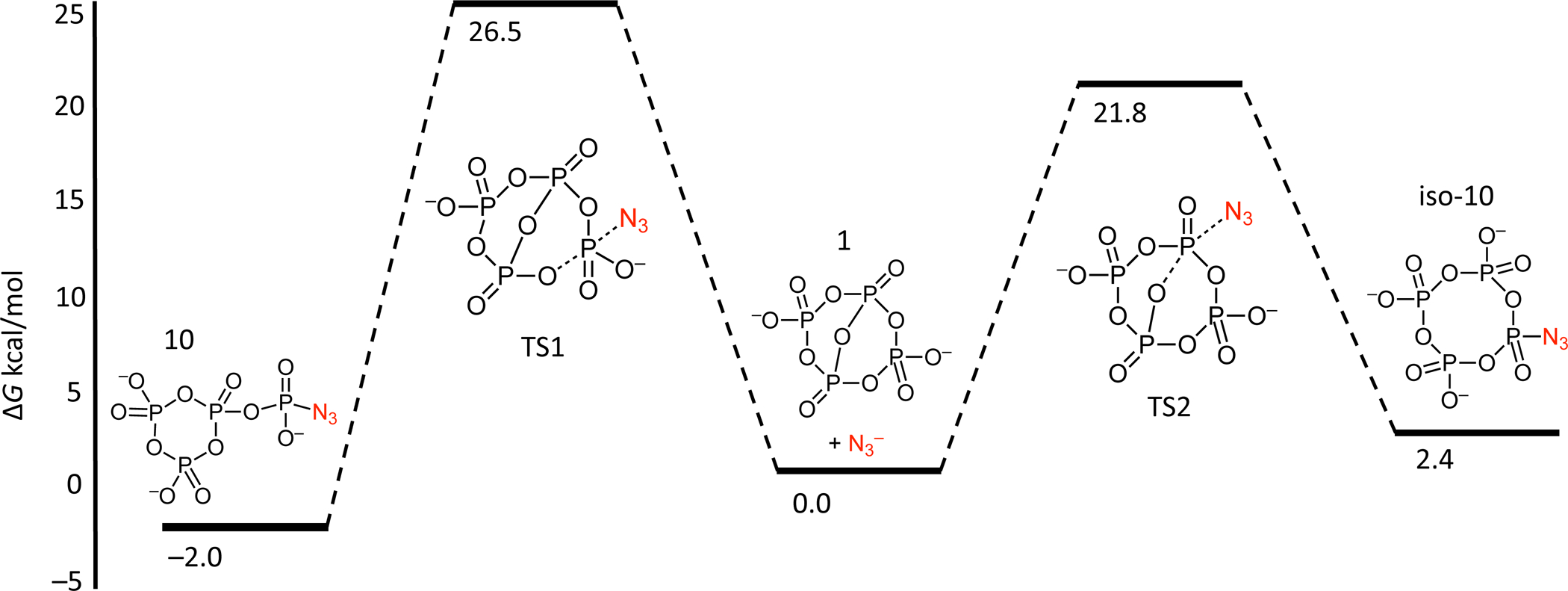
Reaction coordinate for treatment of 1 with inorganic azide at 298.15 K. Geometries and energies were calculated at the B3LYP-D3BJ/ma-def2-TZVP(-f) level of theory41–43 with an acetonitrile CPCM solvation model in Orca 4.2.44
Notably, the thermodynamic barrier for forming the tetrametaphosphate isomer is less than the barrier to form the thermodynamically preferred orthophosphoryl-trimetaphosphate isomer. Therefore, the irreversibility of reactions involving a proton transfer (phosphorylation of alcohols, primary amines, etc.) suggests that many of our isolated tetrametaphosphate derivatives may have been trapped as the kinetic product rather than proceeding to the thermodynamically prefered orthophosphoryl-trimetaphosphate isomer. We therefore sought to selectively synthesize a derivative of the orthophosphoryl-trimetaphosphate isomer by exploiting the reversible addition of a neutral, aprotic nucleophile. Accordingly, treating 1 with DMAP reversibly forms an adduct of the desired orthophosphoryl-trimetaphosphate isomer. Subsequent addition of diethylamine results in phosphorylation and irreversible proton transfer to give the isolable orthophosphoryl-trimetaphosphate isomer 11 (Figure 2A) in contrast to the tetrametaphosphate isomer 6 that is obtained in the absence of DMAP. Such orthophosphoryl-trimetaphosphate derivatives and their ring opening reactions have been well studied as intermediates in the synthesis of oligophosphates.9,45
Conclusion
We have demonstrated that anion 1 is an effective tetraphosphorylation reagent for C, N, and O nucleophiles. This reagent therefore joins the nascent body of literature detailing oligophosphate syntheses to meet the growing demand for such compounds in biochemical applications. Furthermore, this reagent extends beyond most previous metaphosphate based phosphorylation reagents, which have focused on trimetaphosphate derivatives. In contrast to such reported methods, we are able to synthesize linear tetraphosphate derivatives without requiring a phosphate nucleophile by utilizing tetrametaphosphate as the starting material. Furthermore, anion 1 has allowed us to easily obtain ϵ-modified nucleoside 5’-pentaphosphates, a class of compounds that have heretofore been difficult and time-consuming to synthesize, via a one-pot reaction sequence. Having demonstrated the scope of the reactivity of anion 1 towards simple nucleophiles and nucleoside derivatives, we are now exploring tetraphosphorylation of complex biologically relevant molecules to enable the study of pressing biochemical questions concerning the roles and utility of oligophosphates.
Supplementary Material
Acknowledgement
Research reported in this publication was supported by the National Institutes of Health under award number R01GM130936. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank the Raines lab for allowing us to use their lyophilizer, Paul Varava and Miller Tan for their early contributions to this tetraphosphorylation chemistry, Yanfeng Jiang for refinement of the crystal structure of 9, Peter Müller for providing graphic 2B, and the Ibn Khaldun Fellowship for Saudi Arabian Women for supporting Dr. Norah Alhokbany.
Footnotes
Supporting Information Available
The Supporting Information is available free of charge online: https://pubs.acs.org/doi/10.1021/jacs.0c11884.
Synthetic details, spectra, and crystallographic details Structure of 9: CCDC 1991326
References
- (1).Knowles JR Enzyme-catalyzed phosphoryl transfer reactions. Annu. Rev. Biochem 1980, 49, 877–919. [DOI] [PubMed] [Google Scholar]
- (2).Jankowski V; Tölle M; Vanholder R; Schönfelder G; van der Giet M; Henning L; Schlüter H; Paul M; Zidek W; Jankowski J Uridine adenosine tetraphosphate: a novel endothelium-derived vasoconstrictive factor. Nat. Med 2005, 11, 223–227. [DOI] [PubMed] [Google Scholar]
- (3).Harold FM Inorganic polyphosphates in biology: structure, metabolism, and function. Microbiol. Mol. Biol. Rev 1966, 30, 772–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bhandari R; Saiardi A; Ahmadibeni Y; Snowman AM; Resnick AC; Kristiansen TZ; Molina H; Pandey A; Werner JK; Juluri KR Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc. Natl. Acad. Sci. USA 2007, 104, 15305–15310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Azevedo C; Livermore T; Saiardi A Protein polyphosphorylation of lysine residues by inorganic polyphosphate. Mol. Cell 2015, 58, 71–82. [DOI] [PubMed] [Google Scholar]
- (6).Roy B; Depaix A; Perigaud C; Peyrottes S Recent trends in nucleotide synthesis. Chem. Rev 2016, 116, 7854–7897. [DOI] [PubMed] [Google Scholar]
- (7).Khorana H Carbodiimides. Part V.1 A Novel Synthesis of Adenosine Di-and Triphosphate and P1, P2-Diadenosine-5’-pyrophosphate. J. Am. Chem. Soc 1954, 76, 3517–3522. [Google Scholar]
- (8).Bala S; Liao J-Y; Ngor AK; Yik E; Chaput JC P(V) Reagents for the Scalable Synthesis of Natural and Modified Nucleoside Triphosphates. J. Am. Chem. Soc 2019, 141, 13286–13289. [DOI] [PubMed] [Google Scholar]
- (9).Singh J; Steck N; De D; Hofer A; Ripp A; Captain I; Keller M; Wender PA; Bhandari R; Jessen HJ A Phosphoramidite Analogue of Cyclotriphosphate Enables Iterative Polyphosphorylations. Angew. Chem. Int. Ed 2019, 58, 3928–3933. [DOI] [PubMed] [Google Scholar]
- (10).Shepard SM; Cummins CC Functionalization of Intact Trimetaphosphate: A Triphosphorylating Reagent for C, N, and O Nucleophiles. J. Am. Chem. Soc 2019, 141, 1852–1856. [DOI] [PubMed] [Google Scholar]
- (11).Mohamady S; Taylor SD Synthesis of nucleoside tetraphosphates and dinucleoside pentaphosphates via activation of cyclic trimetaphosphate. Org. Lett 2013, 15, 2612–2615. [DOI] [PubMed] [Google Scholar]
- (12).Mohamady S; Taylor SD Synthesis of nucleoside 5’-tetraphosphates containing terminal fluorescent labels via activated cyclic trimetaphosphate. J. Org. Chem 2014, 79, 2308–2313. [DOI] [PubMed] [Google Scholar]
- (13).Mohamady S; Taylor SD Synthesis of nucleoside triphosphates from 2’−3’-protected nucleosides using trimetaphosphate. Org. Lett 2016, 18, 580–583. [DOI] [PubMed] [Google Scholar]
- (14).Singh J; Ripp A; Haas TM; Qiu D; Keller M; Wender PA; Siegel JS; Baldridge KK; Jessen HJ Synthesis of Modified Nucleoside Oligophosphates Simplified: Fast, Pure, and Protecting Group Free. J. Am. Chem. Soc 2019, 141, 15013–15017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Azevedo C; Singh J; Steck N; Hofer A; Ruiz FA; Singh T; Jessen HJ; Saiardi A Screening a protein array with synthetic biotinylated inorganic polyphosphate to define the human PolyP-ome. ACS Chem. Bio 2018, 13, 1958–1963. [DOI] [PubMed] [Google Scholar]
- (16).Fernandes-Cunha GM; McKinlay CJ; Vargas JR; Jessen HJ; Waymouth RM; Wender PA Delivery of Inorganic Polyphosphate into Cells Using Amphipathic Oligo-carbonate Transporters. ACS Cent. Sci 2018, 4, 1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bezold D; Dürr T; Singh J; Jessen HJ Cyclotriphosphate: A Brief History, Recent Developments, and Perspectives in Synthesis. Chem. Eur. J 2020, 2298–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jiang Y; Chakarawet K; Kohout AL; Nava M; Marino N; Cummins CC Dihydrogen Tetrametaphosphate, [P4O12H2]2−: Synthesis, Solubilization in Organic Media, Preparation of its Anhydride [P4O11]2− and Acidic Methyl Ester, and Conversion to Tetrametaphosphate Metal Complexes via Protonolysis. J. Am. Chem. Soc 2014, 136, 11894–11897. [DOI] [PubMed] [Google Scholar]
- (19).Shepard SM; Windsor I; Raines RT; Cummins CC Nucleoside Tetra-and Pentaphosphates Prepared Using a Tetraphosphorylation Reagent are Potent Inhibitors of Ribonuclease A. J. Am. Chem. Soc 2019, 141, 18400–18404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chladek S; Quiggle K; Chinali G; Kohut III J; Ofengand J Synthesis and properties of nucleoside 5’-phosphoazidates derived from guanosine and adenosine nucleotides: effect on elongation factors G and Tu dependent reactions. Biochemistry 1977, 16, 4312–4319. [DOI] [PubMed] [Google Scholar]
- (21).Q2 phosphorus atoms are those that are bound to two other phosphate groups, and Q3 phosphorus atoms are those that are bound to three other phosphate groups 46.
- (22).Nakamura T; Yonemura S; Nabeshima T Synthesis of per (5-N-carboxamide-5-dehydroxylmethyl)-β-cyclodextrins and their selective recognition ability utilizing multiple hydrogen bonds. Chem. Comm 2019, 55, 3872–3875. [DOI] [PubMed] [Google Scholar]
- (23).Jones DJ; O’Leary EM; O’Sullivan TP Modern Synthetic Approaches to Phosphorus-Sulfur Bond Formation in Organophosphorus Compounds. Adv. Synth. Catal 2020, 362, 2801–2846. [Google Scholar]
- (24).Azevedo C; Desfougères Y; Jiramongkol Y; Partington H; Trakansuebkul S; Singh J; Steck N; Jessen HJ; Saiardi A Development of a yeast model to study the contribution of vacuolar polyphosphate metabolism to lysine polyphosphorylation. J. Biol. Chem 2020, 295, 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Benkovic SJ; Sampson EJ Structure-reactivity correlation for the hydrolysis of phosphoramidate monoanions. J. Am. Chem. Soc 1971, 93, 4009–4016. [Google Scholar]
- (26).Sood A; Kumar S; Nampalli S; Nelson JR; Macklin J; Fuller CW Terminal phosphate-labeled nucleotides with improved substrate properties for homogeneous nucleic acid assays. J. Am. Chem. Soc 2005, 127, 2394–2395. [DOI] [PubMed] [Google Scholar]
- (27).Kumar S; Sood A; Wegener J; Finn PJ; Nampalli S; Nelson JR; Sekher A; Mitsis P; Macklin J; Fuller CW Terminal phosphate labeled nucleotides: synthesis, applications, and linker effect on incorporation by DNA polymerases. Nucleosides Nucleotides Nucleic Acids 2005, 24, 401–408. [DOI] [PubMed] [Google Scholar]
- (28).Eid J; Fehr A; Gray J; Luong K; Lyle J; Otto G; Peluso P; Rank D; Baybayan P; Bettman B Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [DOI] [PubMed] [Google Scholar]
- (29).Sims PA; Greenleaf WJ; Duan H; Xie XS Fluorogenic DNA sequencing in PDMS microreactors. Nat. Methods 2011, 8, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kore AR; Yang B; Srinivasan B Efficient synthesis of terminal 4-methylumbelliferyl labeled a potential probe for homogenous fluorescent assay. Tetrahedron Lett 2014, 55, 4822–4825. [Google Scholar]
- (31).Transue WJ; Yang J; Nava M; Sergeyev IV; Barnum TJ; McCarthy MC; Cummins CC Synthetic and spectroscopic investigations enabled by modular synthesis of molecular phosphaalkyne precursors. J. Am. Chem. Soc 2018, 140, 17985–17991. [DOI] [PubMed] [Google Scholar]
- (32).Michailidou F; Chung C.-w.; Brown MJ; Bent AF; Naismith JH; Leavens WJ; Lynn SM; Sharma SV; Goss RJ Pac13 is a Small, Monomeric Dehydratase that Mediates the Formation of the 3’-Deoxy Nucleoside of Pacidamycins. Angew. Chem. Int. Ed 2017, 56, 12492–12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Petrová M; Buděšínskỳ M; Rosenberg I Straightforward synthesis of 3’-deoxy-3’,4’-didehydronucleoside-5’-aldehydes via 2’,3’-O-orthoester group elimination: a simple route to 3’,4’-didehydronucleosides. Tetrahedron Lett 2010, 51, 6874–6876. [Google Scholar]
- (34).Petrová M; Buděšínskỳ M; Zborníková E; Fiedler P; Rosenberg I A Ferrier-Type Allylic Rearrangement of 3’-Deoxy-3’, 4’-didehydronucleosides Mediated by DMF Dimethyl Acetal: Direct Access to 4’-Alkoxy-2’, 3’-didehydro-2’, 3’-dideoxynucleosides. Org. Lett 2011, 13, 4200–4203. [DOI] [PubMed] [Google Scholar]
- (35).Jun-Dong Z; Li-He Z Application of Wittig Reaction to Adenosine Derivatives. Synthesis 1990, 1990, 909–911. [Google Scholar]
- (36).Lyga JW; Secrist JA III Synthesis of chain-extended and C-6’functionalized precursors of the nucleoside antibiotic sine-fungin. J. Org. Chem 1983, 48, 1982–1988. [Google Scholar]
- (37).Torrance CJ; Agrawal V; Vogelstein B; Kinzler KW Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat. Biotechnol 2001, 19, 940–945. [DOI] [PubMed] [Google Scholar]
- (38).Lima WF; Prakash TP; Murray HM; Kinberger GA; Li W; Chappell AE; Li CS; Murray SF; Gaus H; Seth PP; Swayze EE; Crooke ST Single-stranded siRNAs activate RNAi in animals. Cell 2012, 150, 883–894. [DOI] [PubMed] [Google Scholar]
- (39).Schirle NT; Kinberger GA; Murray HF; Lima WF; Prakash TP; MacRae IJ Structural analysis of human Argonaute-2 bound to a modified siRNA guide. J. Am. Chem. Soc 2016, 138, 8694–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Costello AJ; Glonek T; Myers TC; Van Wazer JR Structure and properties of the condensed phosphates. XVIII. Ring-chain and other equilibriums in organic solvents. Inorg. Chem 1974, 13, 1225–1230. [Google Scholar]
- (41).Lee C; Yang W; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- (42).Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (43).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (44).Neese F The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- (45).Mohamady S; Taylor SD Synthesis of Nucleoside Tetraphosphates and Dinucleoside Pentaphosphates via Activation of Cyclic Trimetaphosphate. Org. Lett 2013, 15, 2612–2615. [DOI] [PubMed] [Google Scholar]
- (46).Kirkpatrick RJ; Brow RK Nuclear magnetic resonance investigation of the structures of phosphate and phosphate-containing glasses: a review. Solid State Nucl. Magn. Reson 1995, 5, 9–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.