

Abstract
Unidentified peaks remain a major problem in untargeted metabolomics by LC-MS/MS. Confidence in peak annotations increases by combining MS/MS matching and retention time. We here show how retention times can be predicted from molecular structures. Two large, publicly available data sets were used for model training in machine learning: the Fiehn hydrophilic interaction liquid chromatography data set (HILIC) of 981 primary metabolites and biogenic amines, and the RIKEN plant specialized metabolome annotation (PlaSMA) database of 852 secondary metabolites that uses reversed-phase liquid chromatography (RPLC). Five different machine learning algorithms have been integrated into the Retip R package: the random forest, Bayesian-regularized neural network, XGBoost, light gradient-boosting machine (LightGBM), and Keras algorithms for building the retention time prediction models. A complete workflow for retention time prediction was developed in R. It can be freely downloaded from the GitHub repository (https://www.retip.app). Keras outperformed other machine learning algorithms in the test set with minimum overfitting, verified by small error differences between training, test, and validation sets. Keras yielded a mean absolute error of 0.78 min for HILIC and 0.57 min for RPLC. Retip is integrated into the mass spectrometry software tools MS-DIAL and MS-FINDER, allowing a complete compound annotation workflow. In a test application on mouse blood plasma samples, we found a 68% reduction in the number of candidate structures when searching all isomers in MS-FINDER compound identification software. Retention time prediction increases the identification rate in liquid chromatography and subsequently leads to an improved biological interpretation of metabolomics data.
Graphical Abstract
In untargeted metabolomics by liquid chromatography–tandem mass spectrometry (LC-MS/MS), usually less than 20% of all deconvoluted metabolic peak clusters are identified, even if method blanks are subtracted and molecular adducts are considered.2 LC-MS/MS peaks are best annotated by matching their mass spectra, retention times, and accurate masses against libraries of authentic standards of compounds that have been acquired under identical conditions.3 However, such libraries are small in comparison to the swaths of natural products and synthetic chemicals.4 With 102 million chemicals deposited in PubChem, even the largest mass spectral libraries such as NIST17 or Wiley cover only a few percent of the known chemosphere.5 Therefore, nontargeted LC-MS/MS analyses should additionally use computational predictions for physicochemical properties such as MS/MS spectra and retention times.6
LC retention times (RT) should be predictable because they depend on physical interactions of molecular structures with solvents and chromatographic particles. In liquid chromatography, reversed-phase (RP) or hydrophilic interaction chromatography (HILIC) are most commonly used. However, the large variety of column chemistries, solvents, buffers, and the vast chemical diversity of small molecules make prediction models difficult to transfer between methods. In addition, accuracy in predictions and comparisons of computational models have not always been thoroughly tested. One of the largest retention time prediction sets (2,157 reference compounds) utilized ensemble methods for artificial neural networks to predict RPLC retention times on the Agilent Zorbax C18 column,7 showing it could reduce structure isomer hit lists when used prior to in silico MS/MS prediction software.
In another report on reversed-phase methods, 1,383 synthetic chemicals were used to build kernel-based partial least-squares models to predict RPLC retention times for the Waters BEH C18 column using both molecular fingerprints and calculated (structure-derived) ordinary molecular descriptors such as AlogP.8 Correlations of predicted and experimental retention times in external validation sets was found at Q2 = 0.84, but confidence intervals were not given in terms of average deviation of retention time predictions. In another report on reversed-phase retention prediction, 10 different public data sets were analyzed, and 1,117 compounds were utilized for retention time prediction and artificial neural network (ANN) approaches.9 Here, the average absolute accuracy of retention predictions was found around 1 ± 0.5 min for all methods, giving a good target value for our efforts in predictions. A recent report compared seven machine learning algorithms on 36 distinct, public metabolomics data sets to gain insight into LC-MS retention prediction.10 Interestingly, the authors found that different machine learning algorithms performed to a varying accuracy depending on the type of analyses and analytical protocols. Here, approaches such as the PredRet database may serve as a repository for models to transfer retention times between two chromatographic systems.11 All such retention time models are restricted to the tested chromatographic methods. Predictions regularly perform poorly outside the trained situations, due to the high selectivity of LC methods that rely on specific solvents, pH, ionic strengths, and stationary phases. Compared to RPLC retention time prediction, HILIC predictions have been significantly less studied. In two consecutive papers, Taraji et al. investigated the use of quantitative structure-retention relationships for HILIC predictions on a database of pharmaceutical compounds.12 With genetic algorithms selecting molecular descriptors, retention mean absolute errors ranged from 13 to 50 s for five different HILIC columns, but usability for an extended chemical complexity was not further explored.13
Here, we present the first report on applying a set of machine learning algorithms on both reversed phase and HILC liquid chromatography retention time predictions. We cover five of the best-performing machine learning algorithms, utilizing public data sets from the MassBank of North America and the RIKEN institute in Japan. We took explicit precautions to separate training, testing, and external validation sets to report final prediction errors that would be used in a compound identification workflow. We present R-based code as open source to enable users to apply this method to other chromatographic conditions. We also integrated RT prediction into MS-FINDER software as complete package (http://prime.psc.riken.jp/).
METHODS
Experimental Methods.
We utilized three publicly available LC-MS spectra libraries for retention time prediction model development and validation. For development of the HILIC data set we utilized the MassBank of North America (MoNA) database (http://massbank.us/). The HILIC data set contained a total of 970 compounds, including MS/MS spectra and retention time, with methods given in the MoNA database as using a Waters Acquity UPLC-BEH Amide column (150 mm × 2.1 mm; 1.7 μm) coupled to an Acquity UPLC BEH Amide VanGuard precolumn (5 × 2.1 mm2; 1.7 μm). The column was maintained at 45 °C with a flow rate of 0.4 mL/min. The mobile phases consisted of (A) water with ammonium formate (10 mM) and formic acid (0.125%) and (B) acetonitrile:water (95:5, v/v) with ammonium formate (10 mM) and formic acid (0.125%). The separation was conducted under the following gradient: 0 min 100% B; 0–2 min 100% B; 2–7.7 min 70% B; 7.7–9.5 min 40% B; 9.5–10.25 min 30% B; 10.25–12.75 min 100% B; 12.75–17 min 100% B.14 We used the “Pathogen Box” (https://www.mmv.org/mmv-open) data set measured on the same Waters BEH Amide HILIC column as the external validation set. Chemical diversity and mass spectra can be downloaded from the MoNA database. For the RPLC data set, we used the RIKEN plant specialized metabolome annotation (PlaSMA) database,15 created with fully labeled 13C plants and enriched metabolites, which were measured with a Waters Acquity ultraperformance liquid chromatography (UPLC) ethylene-bridged hybrid (BEH) C18 column (100 mm × 2.1 mm; 1.7 μm particle diameter), maintained at 40 °C. The mobile phases consisted of (A) water including 0.1% formic acid and solvent (B) acetonitrile including 0.1% formic acid. The separation was conducted under the following gradient: 0.5% (B) at 0 min; 0.5% (B) at 0.1 min; 80% (B) at 10 min; 99.5% (B) at 10.1 min; 99.5% (B) at 12.0 min; 0.5% (B) at 12.1 min, isocratic until 15.0 min. A flow gradient was employed from 0.3 to 0.4 mL/min.15 For the case study, we utilized public data sets containing BioRec (now BioIVT) human blood plasma MS/MS data downloaded from http://metabolomicsworkbench.org with accession ID ST001154. Compounds were annotated using the Fiehnlab HILIC experimental MS/MS spectral library. These 143 identified metabolites (“true positives”) were used as “test set” for HILIC retention time prediction and removed from the overall HILIC libraries downloaded from MoNA. Residual HILIC library entries were used as training set molecules.
Computational Methods.
Structure Standardization and Cleaning.
Compound structures were curated with the ChemAxon standardizer to remove salts and metal-containing compounds. Simplified Molecular Input Line Entry System (SMILES) codes that were not compatible with the R platform-based Chemistry Development Kit16 (rCDK) and ChemAxon and OpenBabel toolkits17 were excluded or reformatted. Whenever possible, we also used the structure-data file (*.sdf) format to avoid conversion errors.
Calculating Chemical Compound Descriptors.
Chemical descriptors are more interpretable than structure fingerprints. We utilized multiple descriptor packages, including the CDK,16 Padel,18 Dragon 7, and alvaDesc (Kode Chemoinformatics srl, Italy) as well as ChemAxon. For the final implementation, we used CDK descriptors that can be publicly distributed as a software package in rCDK. The initial SMILES data processing was implemented as a parsing function in the Retip function getCD(). This code relies on the “‘rcdk’” package (version 3.4.7.1),19 an R interface to the CDK.20 Compounds that failed during descriptor calculation were automatically removed. In total, 286 chemical descriptors were computed for each library compound. After the SMILES code was exported from different libraries, we generated 2D coordinates based on the connectivity data. For our current version of Retip, we utilized 2D-based descriptors due to the computational overhead for 3D-optimized structures. SMILES codes were converted to the ChemAxon Extended SMILES (*.cxsmiles) format to store additional atom properties and coordinates. Explicit hydrogens were added for correct representations and accurate atomic and enhanced atomic partition coefficient (Alog and XlogP, respectively) calculations. Finally, we added ChemAxon (https://chemaxon.com/) pK values, including the acidic (pKa1, pKa2) and basic (pKb1 and pKb2) pK values, because initial investigations showed that these descriptors may improve HILIC predictions. The alvaDesc descriptor software was used for visual inspections molecular weight histograms, logP distributions, and multivariate inspections (principal component analysis, PCA).
Machine Learning Models.
Retention time prediction utilizing chemical descriptors can be described as a regression problem. We utilized root-mean-square errors (RMSEs) as a loss function for resulting regression models by calculating the minimized residuals between observed and predicted values. We used correlation R2 values between observed and predicted retention times to indicate linear relationships and for global generalization of the prediction set. For all models except for Keras, 10-fold cross-validation was employed. In Keras, the internal function validation_split was set to 0.2, instead.
Parameter tuning is an essential step for good model performance. Tuning parameters can include random searches or grid searches of the parameter space. We used five independent regression models including parameter tuning: (1) XGBoost21 performs gradient-boosting for regression and classification problems. We implemented automatic grid search tuning for the parameters nrounds, max_depth, and eta, while the fixed parameters were gamma, colsample_bytree, subsample, and min_child_weight. (2) Keras is a high-abstraction layer available for GPU and CPU processing for deep learning and neural networks, using TensorFlow, the Microsoft Cognitive Toolkit, and Theano libraries. Data were centered and scaled. We automatically tuned the dense_unit, epochs, and dropout parameters; other parameters such as batch_size and learning_rate were manually tuned. (3) The light gradient-boosting machine (LightGBM)22 is known for its high efficiency and low RAM usage. It can efficiently process millions of rows in parallel. For parameter optimization, Retip automatically searches for the optimal nrounds parameter based on the best iter value in the cross-validation model. Other model values, such as regressions L1 and L2 regularization, the learning rate, eval_freq, metric, early_stopping_rounds, maximum depth, maximum leaf, and maximum bin were manually tuned to identify the best values and deal with overfitting. (4) The random forest (RF) algorithm23 is one of the most popular algorithms in machine learning. We tuned the mtry parameter, which describes the number of variables that are sampled as candidates for each split. (5) We tuned the number of neurons for the Bayesian-regularized neural network (BRNN),24 an algorithm that uses Bayesian regularization for feedforward neural networks.
Retip R Package Functions.
Functions of the Retip package are explained in detail in the online R package documentation and the GitHub-hosted Web site (https://www.retip.app/). Retip enables a complete workflow from experimental retention time data to a final deployable prediction model. The prepare.wizard() function activates the parallel computation inside Retip. The getCD() function is utilized to compute chemical descriptors. The cesc() function is needed to center and scale the data set, especially for neural network predictions. The chem.space() function plots molecules based on chemical similarity in principal components analysis. Two libraries can be superimposed, for example, a training and a test compound library.25 The proc.data() function handles non-existent values and low-variance columns. Machine learning models use fitting functions with parameter optimizations (i.e., fit.rf, fit.brnn, fit.keras, fit.xgboost, and fit.lightgbm). The get.score() function calculates model statistics, including RMSE, R2, MAE, and 95% confidence intervals. The plot.model() function plots retention time error distributions. The RT.spell() function predicts the retention times of user-uploaded models. The prep.mona() function allows integration with the freely available MoNA interface. The add.rt.mona() function was employed to add RT information in the mass search format spectral files (*.msp) that can be utilized with National Institute of Standards and Technology (NIST)-compatible MS/MS search software.
Integration with Independent Mass Spectrometry Software.
To integrate retention time prediction results into independent software packages, we provide the RT.export() function in the Retip R package. This function enables the use of retention time filters in packages such as MS-DIAL, MS-FINDER, the Agilent MassHunter Suite, and Waters and ThermoFisher Scientific software. The R package documentation with example data sets is provided at the Retip GitHub code repository (https://www.retip.app). Retip supports the open-source software packages MS-DIAL and MS-FINDER with.msp formated MS/MS spectra. For MS-FINDER, use of retip was newly developed for scoring or filtering structure candidates by retention time similarity using Gaussian functions26 and RT tolerances.
Retip Package Versions and Hardware.
Retip was built as a package in R (3.5.3) using R Studio 1.1.143. The R dependencies are caret (6.0–81), ggplot2 (3.1.0), rcdk (3.4.7.1), doParallel (1.0.14), keras (2.2.4.9), stringi (1.4.3), xgboost (0.82.1), brnn (0.7), and lightgbm (2.2.3). The hardware employed was an HP Zbook 15 G5 mobile workstation with an Intel(R) Xeon(R) E-2186 M CPU at 2.90 GHz with 6 cores, 12 logical processors, and 64 GB of RAM and running Windows 10 Pro 64-bit.
RESULTS
Compound Libraries Used for Retention Time Machine Learning Models.
We have developed a generic workflow to utilize experimental liquid chromatography retention time data to train and test machine learning models. Such models are then deployed on large libraries of chemical structures (Figure 1). We have implemented this workflow as user-friendly, freely available R-package Retip.app. Specifically, we showcase the usability and performance of the Retip workflow by applying it to two complementary LC methods for metabolomics, using hydrophilic interaction chromatography data (HILIC) and reversed-phase liquid chromatography data (RP). Experimental retention times, chemical structures, and MS/MS data files were downloaded from https://massbank.us/ and the https://www.metabolomicsworkbench.org/ (for HILIC data) and from http://plasma.riken.jp/ (for RP data). Experimental retention time data were divided into training, test, and external validation sets. For the HILIC database, we divided the set into 880 compounds for training and 143 compounds for testing (see Methods). An additional 377 compounds from the “Pathogen Box” data set were utilized as an external validation set, including exclusively drug candidate compounds for potential use against rare diseases (https://www.mmv.org/mmv-open/pathogen-box). All HILIC compounds were detected between 0.9 and 10.3 min retention times with a total run time of 12 min. The RIKEN library entirely consisted of plant specialized metabolites, whereas the HILIC library included mostly general metabolites, but also some drug compounds. For RPLC retention time predictions, the RIKEN RP library15 was randomly divided into 398 compounds for training, 96 compounds for testing, and 358 compounds for further validation.15 All RP compounds were detected between 1.5 and 10.4 min retention times. Using chemical descriptors, we assessed the degree of structure similarities between training, test, and validation sets for correct use of machine learning methods.
Figure 1.
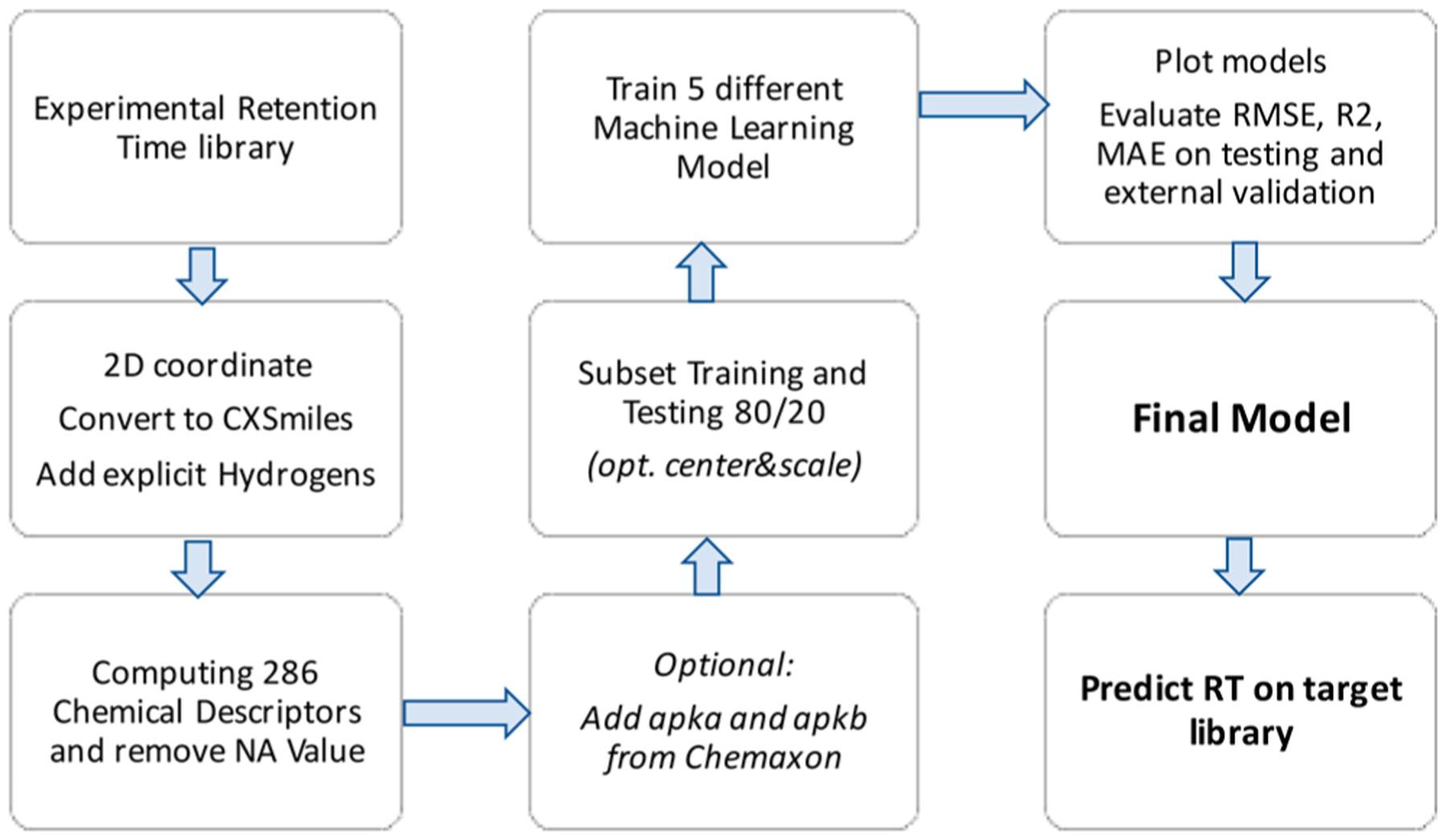
Workflow for predicting LC-retention times from experimental retention time libraries.
For both HILIC and RP libraries, chemical space distributions were very similar to a total of 41% variance explained by the first two principal components for HILIC, and 69% variance explained for the RP compound sets (Supporting Information Figure S1). For HILIC, the validation set was a little outside the chemical space of training and test data, as it only consisted of drug-like chemicals (Supporting Information Figure S1). Conversely, the validation set for RP compounds consisted of true positives from one extract of a specific plant (Ophiorrhiza pumila) that had lower confidence in compound identification, yet identical distribution as the training set across the chemical space. Both chemical distributions indicate high enough structural overlaps to allow the training models to be applied on the test and validation sets (Supporting Information Figure S1).
Accuracy of Retention Time Machine Learning Models on Average Mean Errors.
The training and testing splits were made in the ratio of 80:20, respectively, with the caret package in R27 using XlogP as the index. Each model was trained with 10-fold cross-validation. Several training sessions were executed to find tuning parameters that work best with libraries with 300–1,000 compounds. The fine-tuning set was automated to adapt the model to the final user library. The root-mean-square error (RMSE), the coefficient of determination (R2), and the mean absolute error (MAE) were calculated to compare the predicted and experimental retention times in the test data set. We investigated five common machine learning models (XGboost, BRNN, Random Forest, LightGBM, and Keras) and tested if any model would outperform others in a drastic manner for accuracy (Table 1). On the basis of MAE accuracy, training models performed better than test data sets for all models (Table 1). All data are given in Supporting Information Tables S1–S6. For HILIC models, mean absolute errors almost doubled from 0.54 ± 0.22 min in training data to 0.99 ± 0.13 min in test models. With errors of 0.72 ± 0.11 min, HILIC validation data showed that the machine learning methods were not largely overfitted and could indeed be used on chemicals that showed modest structural deviation from the training and test sets. For reversed phase data, we found a 67% increase in mean absolute errors from 0.31 ± 0.16 to 0.51 ± 0.30 min in test models. As expected, validation MAE errors slightly increased to 0.74 ± 0.04 min (Table 1) because chemical diversity in validation sets can be expected to be slightly different from training sets (Supporting Information Figure S1).
Table 1.
Mean Absolute Errors (min) for RT Predictions
| HILIC | reversed phase LC | |||||
|---|---|---|---|---|---|---|
| training | test | validation | training | test | validation | |
| XGBoost | 0.38 | 1.02 | 0.64 | 0.25 | 0.48 | 0.68 |
| BRNN | 0.37 | 1.05 | 0.60 | 0.43 | 0.51 | 0.76 |
| RF | 0.85 | 1.11 | 0.68 | 0.23 | 0.51 | 0.75 |
| LightGBM | 0.39 | 0.99 | 0.86 | 0.12 | 0.49 | 0.72 |
| Keras | 0.70 | 0.78 | 0.82 | 0.50 | 0.57 | 0.80 |
Next, we investigated differences in mean absolute errors across the machine learning models, to define which model might work best for structure/retention time predictions. Here, we used the difference between [test-training] errors and the difference between [validation-training] errors. Large error differences to training errors indicated that machine learning methods might overfit the training data. Here, Keras outperformed the other four models with average MAE values of 0.14 min error differences across test and validation predictions in both HILIC and RP chromatography (Table 1). In comparison, XGBoost showed less robustness in MAE differences across HILIC and RP test and validation data with an average of 0.39 min error differences, BRNN with 0.33 min errors, RF with 0.22 min errors, and LightGBM with average 0.51 min error differences (Table 1). We argue that this robustness criterion is even more important than overall errors in test and validation sets because any set of chemicals may have intrinsic biases that could lead to slightly different values if other molecules were to be tested.
Distribution of Retention Time Predictions.
On average, the five machine learning models yielded ±1 min MAE values on the HILIC test data set (Table 1). We therefore investigated how many true positive chemicals would be covered in ±1 min retention time windows and how symmetrical error distributions were represented. Figure 2 gives violin plots of error distributions and percentages of true positive coverage within the retention time windows. RP models gave highly symmetrical errors, whereas, for HILIC models, absolute predictions errors were slightly skewed toward positive errors. Across all machine learning models, test data covered more true positive compounds within ±1 min RT windows for RP models (average 87%) than for HILIC models (average 65%). Yet, for validation data, both RP and HILIC models showed 76% average true positive coverage (Figure 2). While all models showed some extreme outliers, ±1 min appeared to be a practical retention error window as it included the clear majority of true positive structures in both test and validation data sets. Importantly, Keras machine learning models showed robust performance for both test and validation data and in both RP and HILIC chromatography, supporting the notion of little overfitting during model training. A similar result was obtained when plotting predicted versus experimental retention times for both HILIC and RP data sets (Supporting Information Figure S2). Here, the Keras model achieved linear correlations R2 ≥ 0.9 in training and test data with a slight reduction to R2 = 0.75 in HILIC validation and R2 = 0.87 in RP validation data sets (Supporting Information Figure S2). While overall, Keras models showed the best robustness, for other classes of molecules or other chromatographic conditions, other machine learning models might outperform Keras. We therefore propose using Retip to enable selecting the best model by using five machine learning models simultaneously.
Figure 2.
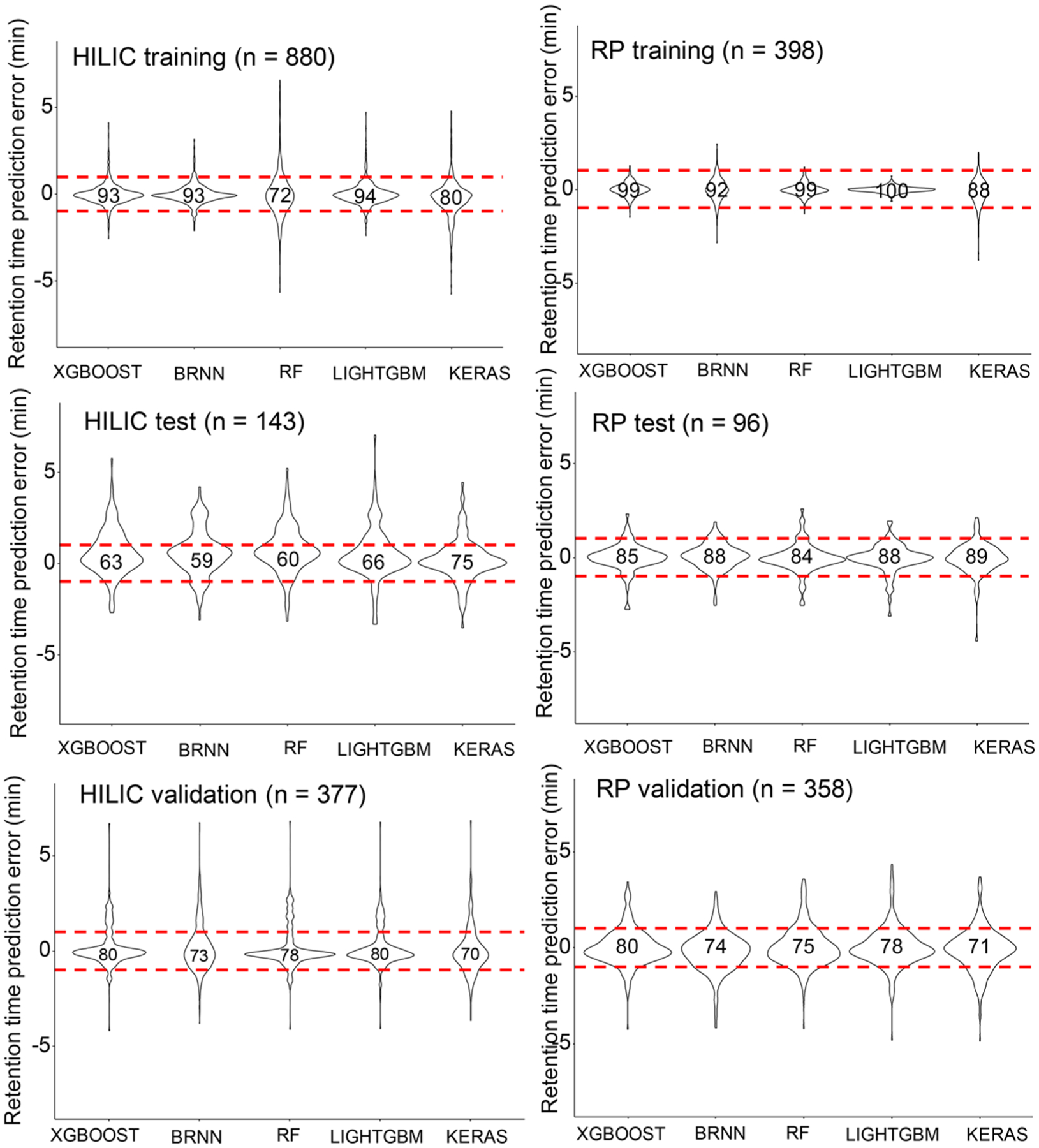
Violin plots for HILIC and RPLC prediction errors by five machine learning models. The number of independent compounds is given in parentheses. Upper panels: training data. Middle panels: test data. Lower panels: external validation data. The numbers in the violin plots show the percentage of compounds within ±1 min retention time windows, given by red dotted lines.
Impact of Chemical Descriptors on RT Predictions.
Next, we investigated the most important chemical descriptors in model building. Although the HILIC retention mechanism includes electrostatic interactions with stationary phase functional groups in addition to dipole–dipole interactions, the most predictive chemical descriptor was found to be the octanol/water partition coefficient XLogP (100%) and ALogP (98%), followed in order of decreasing importance by number of atoms in the largest π chain (87.9%), number of nonrotatable bonds (60.77%), Kier and Hall κ molecular shape indices 2 (45.9%), and a pKa (42.7%). For RP liquid chromatography, the Keras model again found the lipophilicity descriptor XlogP (100%) as the most important predictive chemical descriptor, followed by Basic Group Count (90.6%), Total Polar Surface Area Efficiency (58.9%), Valence Path 0 (30.6%), and the Moreau–Broto autocorrelation ATS.C2 (29.4%). Differences between chemical descriptors for RP and HILIC models were found in pH-related descriptors such as the pKa and shape-related descriptors, which improved the prediction accuracy in the HILIC prediction model.
Software Implementation and Application.
The Retip package provides a complete workflow for retention time prediction as a free software package in R-language. To include practical solutions for metabolomics scientists, we used the Keras model in Retip to predict HILIC and RP retention times for 320,919 compounds as part of 18 publicly available small-molecule databases such as HMDB, DrugBank, FooDB, and the STOFF-IDENT repository.28 These RT predictions are now included in the MS-FINDER program28 version 3.24 or higher for downloads to rank and exclude chemical structures in compound ID studies. Similarly, Retip-predicted retention times can be used with the MS-DIAL software version 3.90 and higher to exclude MS/MS compound annotations with high spectra similarity scores but that are likely false positive matches. Users can also utilize the Retip package to predict retention times for their own chromatographic methods. In MS-DIAL, users can set retention time windows as a component of the total compound identification confidence score. As example, we removed a range of false positive MS/MS compound annotations from mouse plasma metabolomics data1 on the basis of retention time predictions (Table 2). For several compounds, experimental MS/MS spectra were found with high similarities to library spectra but with RT errors far exceeding the ±1 min limits (Table 2). Such false positive, high-similarity MS/MS spectra may be caused by in-source fragmentation in the electrospray ionization process. Hence, relying on MS/MS spectra alone is misleading and can be addressed by ranking likely candidate structures using retention time windows.
Table 2.
Examples for Eliminating False Positive MS/MS Compound Annotations in Human BioIVT Plasma by Predicting HILIC Retention Time Using Retip
| false positive MS/MS spectrum | MS/MS score | retention time obsd (min) | retention time predicted (min) | Δ (retention time) (obsd – pred) (min) | true retention time HILIC library (min) |
|---|---|---|---|---|---|
| carnitine | 981 | 9.6 | 7.6 | 2.0 | 7.7 |
| nicotinamide | 758 | 7.5 | 2.6 | 4.9 | 1.8 |
| pipecolic acid | 876 | 9.5 | 7.1 | 2.4 | 7.7 |
| indole-3-carboxylic acid | 656 | 7.9 | 1.8 | 6.1 | 1.4 |
| amantadine | 626 | 1.4 | 4.9 | 3.5 | 4.8 |
| mepirizole | 735 | 6.0 | 1.1 | 4.9 | 1.0 |
| N-methylproline | 878 | 9.6 | 7.3 | 2.3 | 7.0 |
| phenethylamine | 761 | 1.5 | 5.0 | 3.5 | 4.7 |
Application of Retention Time Predictions with the External MS-FINDER Software.
For unknown molecules that lack matches to experimental MS/MS spectral libraries, MS-FINDER software can be used to predict MS/MS spectra. However, in silico MS/MS predictions may yield a large set of false positive compound annotations. Such risk is smaller if false positive candidate structures can be removed by filters such as retention time. As example, we used all 5,036 deconvoluted MS/MS spectra from a recently published HILIC plasma study.1 MS-FINDER calculated 2,002 elemental formulas totaling 8,685 chemical structures that had at least one structure matched to its built-in structure repository of 320,919 compounds (Table 3). Using retention time filtering to exclude candidate structures that exceeded ±1 min retention time search windows, on average 68% of all candidate structures were excluded as likely false positives. For example, 580 formulas had only one candidate structure in MS-FINDER, but 48% of these structures could be removed as unlikely due to large RT differences (Table 3). Similarly, 87 formulas in the data set comprised 7 isomers each, giving rise to 609 potential candidates; 84% of these structures were ranked as unlikely due to high RT errors, leaving only 98 candidate structures worthy for in-depth validation. These examples show that the application of retention time modeling can have a high impact in workflows in metabolomics compound annotations.
Table 3.
Reduction in Candidate Chemical Search Space for Blood Plasma Experimental MS/MS Spectra1
| without RT filtering | with ±1 min RT filtering | |||
|---|---|---|---|---|
| no. of isomers | no. of formulas | no. of isomers | no. of formulas | no. of isomers |
| 1 | 580 | 580 | 300 | 300 |
| 2 | 281 | 562 | 152 | 304 |
| 3 | 241 | 723 | 93 | 279 |
| 4 | 155 | 620 | 48 | 192 |
| 5 | 102 | 510 | 21 | 105 |
| 6 | 87 | 522 | 32 | 192 |
| 7 | 87 | 609 | 14 | 98 |
| 8 | 47 | 376 | 9 | 72 |
| 9 | 37 | 333 | 9 | 81 |
| 10 | 385 | 3850 | 125 | 1250 |
| total | 2002 | 8685 | 803 | 2873 |
DISCUSSION
Adding retention time predictions to workflows in LC-MS-based untargeted metabolomics can improve peak annotations.29–31 The performance of our machine learning models and resulting prediction accuracies were consistent with previously described retention time models,30,32,33 but the R-package Retip is the first software to predict retention times for small molecules in both HILIC and reversed-phase chromatography. Our predictions showed about 3-fold less error compared to previous models with a total median 10.8% error relative to absolute retention times (previously 35% error34), or a total median 3.4% error relative to the total run time (previously 12% error35). Despite previous efforts, to date no chromatographic processing tool actively employs retention time predictions for metabolome-wide compound annotations. Using MS-FINDER 3.30, we here show that Retip models of predicted retention times can be included in independent software applications. Such predictions are only valid for a specific set of chromatographic parameters, including pH, temperature, buffer conditions, chromatographic columns, injection conditions, solvent compositions, and gradients. It was beyond the scope of our study to test if retention time predictions could be adjusted for small changes in chromatographic methods.
For retention time predictions, compounds for training and prediction must have similar chemical diversities. Hence, if Retip is used for predictions outside the chemical landscape of the training set, results may be more erroneous. The prediction accuracy is generally limited by the number of compounds used for training and their chemical diversity. For example, complex, high molecular weight lipids were largely excluded in our study such as triacylglycerols or phosphatidylcholines. Yet, our methods covered a large number of hydrophilic and semipolar compounds and are therefore highly useful for common metabolomics studies. It is interesting to note that previous models30,32,33 found the octanol/water partition coefficient to be a highly important predictor for RPLC and HILIC molecule/column interactions, similar to our investigations. The fact that none of the current models yield validation MAEs better than ±1 min in retention prediction lets us assume that molecular descriptors themselves are not sufficiently complex to be used in such predictions, for example, by using 3D descriptors instead of 2D structures. Moreover, molecular descriptors by themselves seem to fail to fully explain the complex interactions between LC buffer systems (including ionic strengths), microenvironments (such as static aqueous phases in HILIC), and adsorption and desorption processes in LC stationary phases.
Within our trained and tested chemical space, our prediction models were robust as demonstrated by large external validation sets. Yet, these validations showed that both HILIC and reversed-phase LC retention time predictions had mean absolute errors of about 1 min, which we used as thresholds for excluding false positives. Such 1 min windows excluded about 25% true positives in our models. Yet, if we had used other thresholds (such as including 95% of all true positives), then we had yielded a much-reduced power for excluding false positives. Ultimately, users may use different sensitivity/selectivity ratios for their applications (different settings for false discovery rates), depending on their context of research. For today’s typical UPLC methods with 12 min run times, 1 min prediction errors preclude discriminating closely eluting compounds. Therefore, we strongly propose to develop multitiered compound identification software that includes LC retention time predictions as method-specific filter along with accurate mass precursors, MS/MS spectra, dedicated chemical structure libraries, and additional orthogonal parameters such as ion-mobility-derived collisional cross-section values.36,37
CONCLUSIONS
We have developed Retip.app as a novel, freely available, R-based retention time prediction tool using machine learning algorithms to facilitate peak annotations in mass spectrometry based metabolomics. We achieved reasonably accurate retention time predictions for both HILIC and RPLC methods that we showed to be useful for excluding unlikely structure candidates in untargeted MS/MS library spectral matching and in silico MS/MS compound annotations. This exclusion of false positive candidate structures will have a large impact on metabolomics confidence scoring schemas. Yet, outliers in the test and validation results of our machine learning models showed that there are still many chemicals that were not well represented by the training models. Similarly, the descriptor space underlying these retention time models showed that lipophilicity had a large weight in the model training, although, especially for HILIC methods, additional mechanisms are known to affect experimental retention times. Hence, predictions are not yet adequate for narrow retention time search windows but are useful to disclose likely in-source fragmentation MS/MS spectra and to rank positional isomers in compound identification workflows.
Supplementary Material
ACKNOWLEDGMENTS
We thank the contributors from MassBank and MassBank of North America for providing public access to the mass spectra. P.B. specially thanks Veronica Cirino, Luigi Lucini, and Giuseppe Colla for useful discussions.
Funding
This study was funded by the U.S. National Institutes of Health Award No. NIH U2C ES030158. Funding for NGAlab was provided by ATENS—Agrotecnologías Naturales SL.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05765.
(Table S1–S6) Worksheets listing S1-HILIC and S4-RP training data, S2-HILIC and S5-RP test data, S3-HILIC and S6 validation data (XLSX)
(Figure S1) Principal component analyses on 2D-chemical descriptors to show chemical diversity in training and validation sets used for predicting LC-retention times on HILIC- and reversed-phase LC methods; (Figure S2) scatter plot to visualize prediction errors for Keras machine learning for HILIC- and reversed-phase LC methods (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.9b05765
All data and software for this article may be accessed, for commercial and noncommercial use, and source code and data are available on the Retip GitHub page at https://github.com/PaoloBnn/Retip
Contributor Information
Paolo Bonini, NGAlab, La Riera de Gaia, Tarragona 43762, Spain;.
Tobias Kind, West Coast Metabolomics Center, UC Davis Genome Center, University of California, Davis, Davis, California 95616, United States.
Hiroshi Tsugawa, RIKEN Center for Sustainable Resource Science, Yokohama 230-0045, Japan; RIKEN Center for Integrative Medical Sciences, Yokohama 230-0045, Japan;.
Dinesh Kumar Barupal, West Coast Metabolomics Center, UC Davis Genome Center, University of California, Davis, Davis, California 95616, United States.
Oliver Fiehn, West Coast Metabolomics Center, UC Davis Genome Center, University of California, Davis, Davis, California 95616, United States;.
REFERENCES
- (1).Barupal DK; Zhang Y; Shen T; Fan S; Roberts BS; Fitzgerald P; Wancewicz B; Valdiviez L; Wohlgemuth G; Byram G; Choy YY; Haffner B; Showalter MR; Vaniya A; Bloszies CS; Folz JS; Kind T; Flenniken AM; McKerlie C; Nutter LMJ; Lloyd KC; Fiehn O Metabolites 2019, 9 (5), 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Seitzer PM; Searle BC J. Proteome Res 2019, 18 (2), 791–796. [DOI] [PubMed] [Google Scholar]
- (3).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Metabolomics 2007, 3 (3), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cui Y; Balshaw DM; Kwok RK; Thompson CL; Collman GW; Birnbaum LS Environ. Health Perspect 2016, 124 (8), A137–A140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Blaženović I; Kind T; Ji J; Fiehn O Metabolites 2018, 8 (2), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Blaženović I; Kind T; Ji J; Fiehn O Metabolites 2018, 8 (2), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Samaraweera MA; Hall LM; Hill DW; Grant DF Anal. Chem 2018, 90 (21), 12752–12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Falchi F; Bertozzi SM; Ottonello G; Ruda GF; Colombano G; Fiorelli C; Martucci C; Bertorelli R; Scarpelli R; Cavalli A; Bandiera T; Armirotti A Anal. Chem 2016, 88 (19), 9510–9517. [DOI] [PubMed] [Google Scholar]
- (9).Barron LP; McEneff GL Talanta 2016, 147, 261–70. [DOI] [PubMed] [Google Scholar]
- (10).Bouwmeester R; Martens L; Degroeve S Anal. Chem 2019, 91 (5), 3694–3703. [DOI] [PubMed] [Google Scholar]
- (11).Stanstrup J; Neumann S; Vrhovšek U Anal. Chem 2015, 87 (18), 9421–8. [DOI] [PubMed] [Google Scholar]
- (12).Taraji M; Haddad PR; Amos RI; Talebi M; Szucs R; Dolan JW; Pohl CA J. Chromatogr A 2017, 1486, 59–67. [DOI] [PubMed] [Google Scholar]
- (13).Taraji M; Haddad PR; Amos RIJ; Talebi M; Szucs R; Dolan JW; Pohl CA J. Chromatogr A 2017, 1507, 53–62. [DOI] [PubMed] [Google Scholar]
- (14).Blaženović I; Kind T; Sa MR; Ji J; Vaniya A; Wancewicz B; Roberts BS; Torbašinović H; Lee T; Mehta SS; Showalter MR; Song H; Kwok J; Jahn D; Kim J; Fiehn O Anal. Chem 2019, 91 (3), 2155–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tsugawa H; Nakabayashi R; Mori T; Yamada Y; Takahashi M; Rai A; Sugiyama R; Yamamoto H; Nakaya T; Yamazaki M; Kooke R; Bac-Molenaar JA; Oztolan-Erol N; Keurentjes JJB; Arita M; Saito K Nat. Methods 2019, 16 (4), 295–298. [DOI] [PubMed] [Google Scholar]
- (16).Willighagen EL; Mayfield JW; Alvarsson J; Berg A; Carlsson L; Jeliazkova N; Kuhn S; Pluskal T; Rojas-Chertó M; Spjuth O; Torrance G; Evelo CT; Guha R; Steinbeck CJ Cheminf. 2017, 9 (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).O’Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR J. Cheminf 2011, 3 (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yap CW J. Comput. Chem 2011, 32 (7), 1466–74. [DOI] [PubMed] [Google Scholar]
- (19).Guha RJ Stat. Soft 2007, 18 (5), 16. [Google Scholar]
- (20).Steinbeck C; Hoppe C; Kuhn S; Floris M; Guha R; Willighagen EL Curr. Pharm. Des 2006, 12 (17), 2111–20. [DOI] [PubMed] [Google Scholar]
- (21).Chen T; Guestrin C, XGBoost: A Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM: San Francisco, CA, USA, 2016; pp 785–794, DOI: 10.1145/2939672.2939785. [DOI] [Google Scholar]
- (22).Ke G; Meng Q; Finley T; Wang T; Chen W; Ma W; Ye Q; Liu T-Y LightGBM: A highly efficient gradient boosting decision tree. Advances in Neural Information Processing Systems 30 (NIPS 2017); Neural Information Processing Systems Foundation, 2017; pp 3146–3154. [Google Scholar]
- (23).Liaw A; Wiener M R News 2002, 2 (3), 18–22. [Google Scholar]
- (24).Pérez-Rodríguez P; Gianola D; Weigel KA; Rosa GJ; Crossa JJ Anim. Sci 2013, 91 (8), 3522–31. [DOI] [PubMed] [Google Scholar]
- (25).Weaver S; Gleeson MP J. Mol. Graphics Modell 2008, 26 (8), 1315–26. [DOI] [PubMed] [Google Scholar]
- (26).Tsugawa H; Cajka T; Kind T; Ma Y; Higgins B; Ikeda K; Kanazawa M; VanderGheynst J; Fiehn O; Arita M Nat. Methods 2015, 12 (6), 523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kuhn MJ Stat. Soft 2008, 28 (5), 26. [Google Scholar]
- (28).Tsugawa H; Kind T; Nakabayashi R; Yukihira D; Tanaka W; Cajka T; Saito K; Fiehn O; Arita M Anal. Chem 2016, 88 (16), 7946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Creek DJ; Jankevics A; Breitling R; Watson DG; Barrett MP; Burgess KE Anal. Chem 2011, 83 (22), 8703–10. [DOI] [PubMed] [Google Scholar]
- (30).Cao M; Fraser K; Huege J; Featonby T; Rasmussen S; Jones C Metabolomics 2015, 11 (3), 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wen Y; Amos RIJ; Talebi M; Szucs R; Dolan JW; Pohl CA; Haddad PR Anal. Chem 2018, 90 (15), 9434–9440. [DOI] [PubMed] [Google Scholar]
- (32).Hall LM; Hall LH; Kertesz TM; Hill DW; Sharp TR; Oblak EZ; Dong YW; Wishart DS; Chen MH; Grant DF J. Chem. Inf. Model 2012, 52 (5), 1222–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Eugster PJ; Boccard J; Debrus B; Bréant L; Wolfender JL; Martel S; Carrupt PA Phytochemistry 2014, 108, 196–207. [DOI] [PubMed] [Google Scholar]
- (34).Creek DJ; Jankevics A; Breitling R; Watson DG; Barrett MP; Burgess KE Anal. Chem 2011, 83 (22), 8703–8710. [DOI] [PubMed] [Google Scholar]
- (35).Aalizadeh R; Nika M-C; Thomaidis NS J. Hazard. Mater 2019, 363, 277–285. [DOI] [PubMed] [Google Scholar]
- (36).Colby SM; Thomas DG; Nuñez JR; Baxter DJ; Glaesemann KR; Brown JM; Pirrung MA; Govind N; Teeguarden JG; Metz TO; Renslow RS Anal. Chem 2019, 91 (7), 4346–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Blaženović I; Shen T; Mehta SS; Kind T; Ji J; Piparo M; Cacciola F; Mondello L; Fiehn O Anal. Chem 2018, 90 (18), 10758–10764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.