

Abstract
Atom- or bond-level chemical properties of interest in medicinal chemistry, such as drug metabolism and electrophilic reactivity, are important to understand and predict across arbitrary new molecules. Deep learning can be used to map molecular structures to their chemical properties, but the data sets for these tasks are relatively small, which can limit accuracy and generalizability. To overcome this limitation, it would be preferable to model these properties on the basis of the underlying quantum chemical characteristics of small molecules. However, it is difficult to learn higher level chemical properties from lower level quantum calculations. To overcome this challenge, we pretrained deep learning models to compute quantum chemical properties and then reused the intermediate representations constructed by the pretrained network. Transfer learning, in this way, substantially outperformed models based on chemical graphs alone or quantum chemical properties alone. This result was robust, observable in five prediction tasks: identifying sites of epoxidation by metabolic enzymes and identifying sites of covalent reactivity with cyanide, glutathione, DNA and protein. We see that this approach may substantially improve the accuracy of deep learning models for specific chemical structures, such as aromatic systems.
Graphical Abstract
INTRODUCTION
Artificial intelligence, driven by a collection of techniques known as deep learning, has made significant strides in solving important problems in a wide range of scientific and technical fields.1–6 In the chemistry domain, deep learning has found applications in drug metabolism,7–9 toxicity,10–12 chemical reactions,13 material design,14,15 catalyst design,16 and drug design.17,18 In quantum chemistry, deep learning has been shown to calculate quantum chemical properties in a fraction of a second and at a level of accuracy previously seen only with time-consuming methods,19–21 such as density functional theory (DFT)22 or coupled cluster analysis,23 which sometimes take hours to compute the properties of a single molecule.
Higher-level chemical properties are practically important but are much more difficult to compute from first principles. Though molecule-level properties, such as toxicity and bioactivity, are commonly studied, we are most interested in site-level (atom- and bond-level) properties, which can give insight into which specific parts of a small molecule are mechanistically important, which in turn can guide chemists in altering a molecule’s properties. Atom- and bond-level properties include whether a site is metabolized by liver enzymes, whether it covalently reacts with biological macromolecules, or if it causes interference with high throughput screening assays. These sorts of properties emerge from underlying quantum phenomena, and predicting these chemical properties across new molecules is practically important. By training on data sets where the properties are known from experiments, deep learning models can be built to predict these properties.9,10 These data sets, however, are relatively small, which can limit accuracy and generalizability of models.
To overcome this limitation, it would be best to model high-level atom and bond properties, where data is sparse, from lower-level and easily computable quantum chemical features. Encouragingly, some relationships between QC and higher-level properties have been reported in prior work. For example, in the case of spontaneous covalent reactivity, electron deficient atoms of a molecule have a propensity to form bonds with electron rich atoms of proteins.24 This spontaneous electrophilic reactivity can lead to severe adverse drug reactions such as drug-induced liver failure.25 Likewise, carbon–hydrogen bonds with exceptionally high energy are easily broken by P450 enzymes, allowing oxygen substitution.26 These oxidation reactions sometimes lead to the formation of reactive, electrophilic species.27 As such, quantum measurements of properties such as bond energy and electrophilic delocalizability often correlate with these behaviors.9,10 These correlations can be used to construct quantitative structure activity relationships (QSAR) to study how chemical modifications may reduce or eliminate these phenomena.28 Unfortunately, in practice, lower-level quantum chemistry and these chemical properties have not yet proven effective in improving the accuracy over learning from chemical structures directly on limited data sets.
In this paper, we investigate using transfer learning to make use of quantum chemistry information to improve models of higher-level chemical properties, such as metabolism and reactivity. We hypothesize that these properties can be better modeled using representations derived from models first trained on quantum chemistry data sets. We hypothesize that deep learning models trained to compute quantum chemical observables will learn an intermediate representation that improves modeling of higher-level chemical properties.
Our approach is understandable as an example of “transfer learning”, where information from one task is used to improve performance on another, related task. This approach has been successfully used to model molecule-level properties in the past,29 and we expect this approach might also work for individual atoms and bonds within a molecule. We also find a close analogy to our specific approach in image recognition. Here, large networks are commonly trained on large databases of images. The trained network’s intermediate representation, sometimes called its “bottlenecks”, is used to model for more specialized imaging tasks. Analogously, we will train a network to model lower-level quantum properties using very large databases of computed results. Then we will use the bottlenecks of this network to model higher-level chemical properties, where data sets are smaller and the task is harder. One major advantage of this approach is that knowledge from one task, where there is a great deal of data, can improve modeling of another related task, where data are more difficult acquire.
Our approach to transfer learning closely matches how transfer learning is leveraged in image analysis, by pretraining networks on large databases of images.30 The hidden layers of the network are then reused on differing imaging tasks, which might have far less data. In this way information learned in the first task is transferred to the second. In our approach, a model is trained to map molecule structures to the output of a QM simulation, where very large data sets can be computationally generated. The network’s trained hidden layers is then reused to build a model of higher-order biochemical properties such as drug metabolism or reactivity, where data sets are much smaller.
Our approach also parallels how quantum operators understood in quantum mechanics to operate on a hidden wave function. Theoretically, the wave function represents the complete information about the chemical properties of a molecule. Observables, such as total energy, dipole moment, or polarizability, are mathematically related to the wave function by “operators”.31 Operators, in this way, are a powerful theoretical connections between the hidden quantum representation of a molecule and its properties. However, operators for predicting higher-level chemical behaviors are not easy to derive. In the case of this study, the network learns a shared hidden representation of the molecule analogous to a wave function, and it also learns operators to reduce this representation into observable properties.
We hypothesize, nonetheless, that site-level chemical properties can be better modeled by a network that transfers information, encoded in an atom- and bond-level latent space, from quantum mechanics data. We test our hypothesis in two ways. First, we show that deep learning representations trained on a subset of quantum chemical properties can be used to predict other quantum chemical properties not observed during training. Second, we show how deep learning representations trained on quantum chemical properties can be used to predict higher-level chemical properties including epoxidation by enzyme-mediated metabolism and covalent reactivity with important biochemical substrates including cyanide, deoxyribonucleic acid (DNA), glutathione (GSH), and protein. These transfer models outperform comparison models using calculated quantum chemical properties as input. These experiments suggest that the quantum chemical representation derived by deep learning is generalizable between lower-level and higher-level chemical properties using straightforward, data-driven methods.
DATA AND METHODS
Epoxidation Data Set.
We collected epoxidation data from previously published work by Hughes et al.9 Briefly, this data set consisted of 523 molecules with labels for each bond indicating whether an epoxide was observed to form as a result of oxidation by drug metabolizing enzymes in in vitro or in vivo experimental data compiled in the Accelrys Metabolite Database (AMD). To construct cross-validation holdout sets, molecules were grouped by metabolic networks (common metabolic descendants of the same parent molecules) and molecules were included in the same cross-validation fold if they shared a metabolic network, as described by Hughes et al. This process resulted in 384 cross-validation folds.
Reactivity Data Set.
We collected reactivity data from previously published work by Hughes et al.32 Briefly, this data set contained a total of 1364 molecules with labels for each atom indicating whether it was observed to form covalent bonds with cyanide, DNA, GSH, or protein. These labels were derived in vitro and in vivo experimental data compiled in the Accelrys Metabolite Database (AMD). To cross-validate their results, we used the same 10-fold cross validation scheme from Hughes et al., wherein molecules were grouped by pairwise similarities so that similar molecules were included in the same cross-validation fold. Models trained on this data set used a multitarget structure, and sites with missing labels for either cyanide, DNA, GSH, or protein were masked during training. We excluded molecules containing atomic numbers greater than 18, due to limitations of the 6–31G basis set used to compute QC properties for our transfer learning training data (see below). This resulted in the exclusion of several molecules containing the larger halogens bromine and iodine.
Computing Quantum Chemical Representations with Deep Learning.
We trained a graph-based deep learning model of quantum chemical properties using training data extracted from PubChemQC.33,a The PubChemQC project aims to perform DFT calculations on all of PubChem. Their calculations were carried out at the B3LYP level using the 6–31G* basis set. For our training set, we selected 500 000 diverse molecules covering a range of atom types and molecule sizes. We constructed and trained a message passing neural network (MPNN)34 to predict these calculated quantum chemical properties. The MPNN took as input simple descriptors of the atom type (one-hot encoding of atomic number) and formal charge (integer from −2 to +2). The MPNN used five steps of message passing, an atom, bond, and global state vector size of 64 for transfer learning on epoxidation and reactivity tasks and 128 for transfer learning on quantum chemical properties. The state update function was a fully connected layer with exponential linear unit (eLU) activation. The atom states output from this network were further processed by additional networks, which were trained to predict up to three molecule-level properties (total energy, highest occupied molecular orbital energy or HOMO, and excitation gap energy), four atom-level properties (Mulliken charge and population, total and bonded valence), and two bond-level properties (Mayer order and bond length). The molecule-level properties were predicted by first applying a set2set encoder to the atom states, as described in previous work.34,35 The resulting molecule-level state was then passed to a fully connected layer with linear output to predict the molecule-level properties. The atom-level properties were predicted directly from atom states by a neural network with one hidden layer of 32 units and exponential linear activation (eLU) and linear output. The bond-level properties were predicted from atom states by first producing a bond-level encoding according the formula B = f(A1,A2) + f(A2,A1), where f is a single hidden layer neural network with 32 units and eLU activation. This bond representation was then used to predict bond-level properties using a fully connected layer with linear output.
The model was constructed and trained using Tensorflow,36 DeepMind GraphNets,37 and the tflon deep learning toolkit.b Models were trained by minibatch stochastic gradient descent on the mean square error using the Adam optimizer38 for 250 000 iterations with a learning rate of 10−3, a continuous learning rate decay of 0.99, and a minibatch size of 64 molecules.
After training on the PubChemQC data set, each model was used to compute graph-based encodings for each atom in the epoxidation and reactivity data sets. These graph-based encodings included just the final atom state of the MPNN, prior to the quantum property decoder networks. In addition, the MPNN trained on the complete set of 15 properties was used to compute quantum chemical properties for each atom, bond and molecule in the epoxidation and reactivity data sets.
Models for Transfer Learning from Quantum Chemistry (TLQC).
We constructed two simple neural network architectures to perform transfer learning from quantum chemistry (TLQC) for both the epoxidation and reactivity data sets.
After computing graph-based encodings of quantum state for each atom, we trained a neural network to predict bond-level epoxidation labels for each bond in the epoxidation data set. This neural network contained three layers. First, a weave layer39,40 computed a bond-level intermediate encoding from atom-level encodings derived from the deep learning quantum chemistry model. Given atom-level encoding vectors A1 and A2, a bond-level encoding B was computed by B = f(A1,A2) + f(A2,A1), where f is a single hidden layer neural network with 16 units and tanh activation. The bond encoding was then passed to an additional hidden layer of 8 units with tanh activation and finally to an output layer with one unit and sigmoid activation. The neural network was trained via gradient descent with the OpenOpt L-BFGS optimizer.41 This architecture and training strategy is similar to the architecture used to predict epoxidation in the original publication analyzing this data set.9
We also trained a neural network to predict atom-level reactivity labels for each atom in the reactivity data set. This neural network had three layers. Atom encodings derived from the deep learning quantum chemistry model were passed to a neural network with two hidden layers of 32 and 16 units and tanh activation and finally to an output layer with 4 units and sigmoid activation. Each of the four outputs correspond to predictions of reactivity with cyanide, DNA, GSH, and protein. This neural network was trained via gradient descent with the OpenOpt L-BFGS optimizer. This architecture and training strategy is similar to the architecture used to predict reactivity in the original publication analyzing this data set.10
Topological and Quantum Chemistry Models.
We compared models with two different inputs, graph-based topological descriptors (Top) or quantum chemical descriptors (QC). These models were used to predict both bond-level epoxidation and atom-level reactivity sites. The architectures used for these models were similar to those used for the TLQC approach, with appropriate changes to the input representation.
For the Top model, the atom-level input descriptors were the same as those used to train the quantum chemical representations, a vector encoding atom type and formal charge. Topological representations of local atom neighborhoods were constructed automatically by the neighborhood network component of the model. This architecture generalizes and optimizes the graph-based descriptors that we used in the past to build the highest performing models in site-of-metabolism and site-of-reactivity modeling.9,10 This approach, however, is distinct from algebraic topological descriptors, which are used widely in chemical informatics to summarize whole molecules.42–46 As effective as are these descriptors, they have not been widely used to model individual atoms or bonds within a small molecule context.
For the QC model, the input atom-level quantum properties were processed similarly to the Top models. For the epoxidation task, bond-level and molecule-level quantum properties were concatenated with the bond-level representation constructed from atom-level properties by the weave network. For the reactivity task, bond-level properties were summed over bonds incident to each atom and concatenated to the atom-level representation along with molecule-level properties. These neural networks were trained via gradient descent with the OpenOpt L-BFGS optimizer.
Unlike our prior work, which included detailed comparisons with every published model,9,10 the goal of this study is not to produce the most performant models. Rather, our goal is to understand, with well-controlled experiments, how deep learning can make use of QC information from very simple descriptors. Though we found that the performance was similar to that of the most performant models, performance was not directly comparable because we did not use exactly the same data because we excluded molecules with element-types where QC parameters were not available.
Representation Optimization.
Input representations for Top and QC models were constructed by neighborhood convolution, a standard approach in machine learning for chemical informatics.40 For topological descriptors, individual atoms are initially described by a vector consisting of the atom type (one-hot encoding) as well as formal charge with total vector size s. Neighborhood convolution appends to each atom vector the sum of these initial vectors over successively larger neighborhoods around an atom, where a neighborhood is defined as all other atoms at a given distance, specified in the number of bonds, from the target atom. This process results in atom descriptions consisting of the total number of atoms of a given type at a given depth for all depths up to a specified depth D, with total vector size sD. For Top and QC models we experimented with D = 2, 3, 4, 5 and chose the models with the best average accuracy on all the tasks.
Model Evaluation.
We chose the top-2 metric as our primary accuracy metric. Top-2 measures the proportion of molecules for which an observed site of metabolism or reactivity is ranked in the top 2 among all sites in the molecule. This metric is commonly used in machine learning applied to metabolism and chemical reactions.8–10,32,47,48 Top-2 is particularly relevant to medicinal chemists, as it measures a model’s ability to identify the most metabolically labile or chemically reactive sites within a given molecule and evaluate metabolism enzyme regioselectivity. To measure each model’s ability to perform intermolecular comparisons, we also evaluated and reported area under the receiver operator curve (AUC) statistics in the Supporting Information.
Statistical Methods.
Comparison statistical tests for top-2 scores, AUCs and Z-scores were performed using the paired McNemar test,49 paired Z-test,50 and Welch’s t test,51 respectively.
RESULTS AND DISCUSSION
In the following sections, we discuss the design and implementation of the TLQC approach. First, we demonstrate that it achieves good accuracy in predicting QC properties. Second, we demonstrate that TLQC computes unobserved QC properties with minimal increase in error and substantially better than an alternative approach not utilizing transfer learning. Third, we demonstrate that TLQC can be used to predict higher-level chemical behaviors including epoxidation of small molecules and covalent reactivity between small molecules and nucleophilic trapping agents (cyanide and GSH) or biological macromolecules (DNA and protein). These tasks are both important in medicinal chemistry. Further, we demonstrate that TLQC is stable under variations of the QC properties used during training and that these variations are all generalizable across tasks. Finally, we examine a case where TLQC provides benefits relative to a standard approach to deep learning in chemistry.
Transfer Learning Computes Unobserved Quantum Chemical Properties.
Representations of quantum chemistry trained on one set of computed QC properties can generalize to compute other QC properties (Figure 1A). We trained a message passing neural network deep learning model (Data and Methods) to predict the following four groups of quantum chemical properties: (1) total energy, (2) highest occupied molecular orbital energy (HOMO) and gap energy, (3) Mulliken population, charge, bonded valence and total valence, (4) Mayer bond order and bond length. We then trained four additional networks, leaving out one group of properties in each case. We computed graph-based molecular representations from the final message passing layer of these four networks and then trained new decoders for each of the left out property groups. Evaluating these transfer models on each of the property groups demonstrates only a small increase in error compared to the model trained on all the properties simultaneously. The increase in error is 1% or less for all the tested properties except Mulliken charges. Furthermore, these transfer learning networks perform significantly better on seven of the nine properties when compared to a network in which topological descriptors of the molecule are used as input to the decoder network instead of a representation learned from quantum chemistry. These results demonstrate that the intermediate representations learned by deep learning on QC properties contain information required to compute other, unobserved QC properties.
Figure 1.
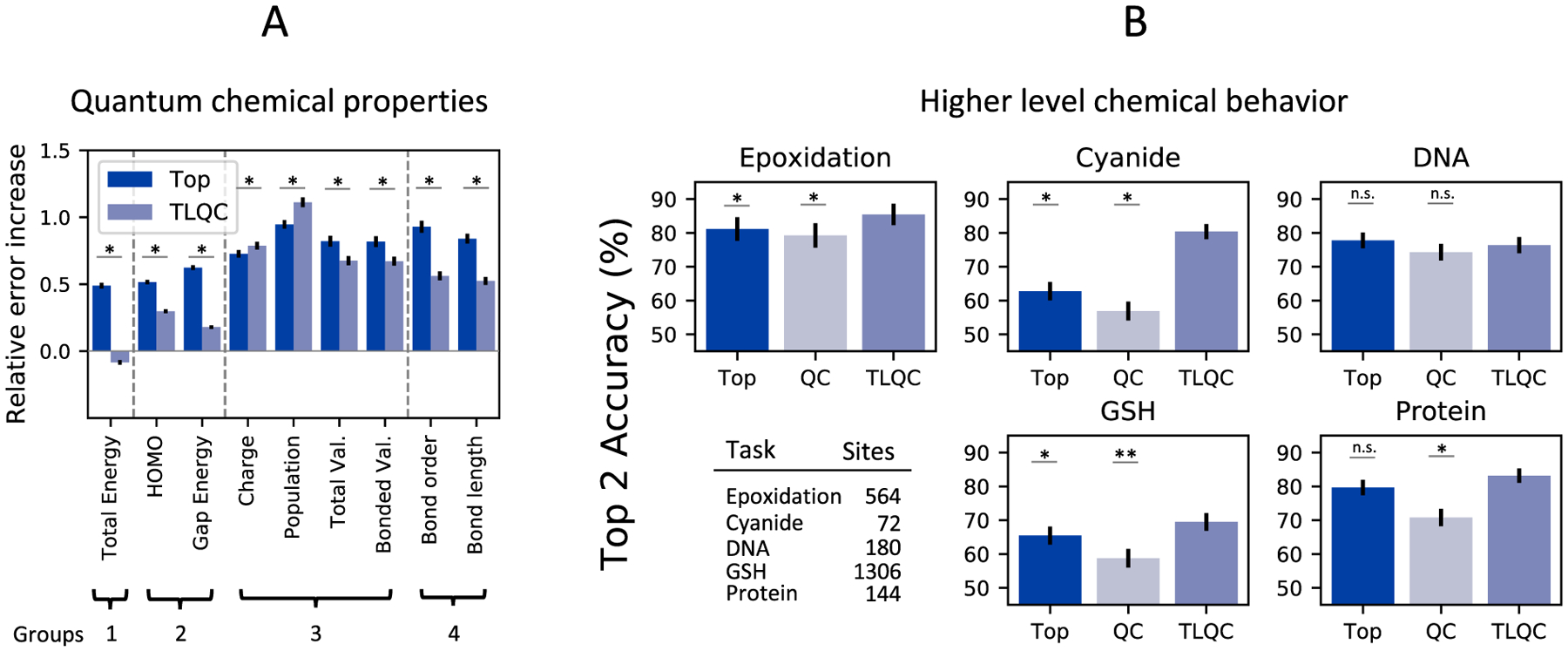
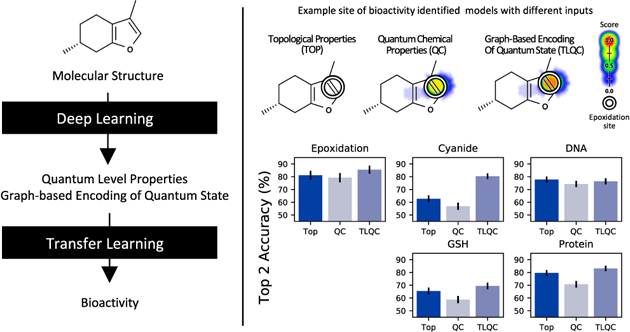
Transfer learning from quantum chemistry can calculate quantum chemical properties and predict higher-level chemical behaviors. (A) Transfer learning using graph representations learned from quantum chemistry (TLQC) can be used to calculate values of unobserved QC properties by training new decoders. TLQC performs better than decoders trained on topology-based graph representations for most QC properties. (B) TLQC predicts the formation of epoxides by P450-mediated metabolism as well as reactivity of small molecules with four important substrates: cyanide, glutathione (GSH), DNA, and protein. This approach improves accuracy on four of the five tasks compared to models using just topological descriptors (TOP) or using quantum chemical descriptors (QC). *p-value <0.05, **p-value <0.001.
Transfer Learning Predicts Sites of Epoxidation and Reactivity.
We used two previously published data sets to test whether TLQC could predict metabolism and chemical reactivity of small molecules. When bioactivated by metabolism, small molecules can form chemically reactive species that covalently bind with biological macromolecules such as proteins and DNA. These complexes can cause clinically evident toxicity through cellular dysfunction or immune-mediated hypersensitivity.25 The first data set comprised 523 small molecules with labels at each bond where an epoxide was observed to form by oxidative metabolism9 (Data and Methods). Epoxides are an important class of chemically reactive metabolites. The second data set comprised 1364 molecules with labels at each atom indicating whether that atom was observed to form a covalent bond with cyanide, GSH, DNA, or protein.10 We trained neural network architectures to predict these five chemical behaviors, with different architectures for the epoxidation models and reactivity models to account for the difference in output prediction (atom-level vs bond-level; see Data and Methods).
For each neural network, we varied the molecular representations used as input between three alternatives: topological descriptors (Top), QC descriptors, and transfer learning from quantum chemistry (Data and Methods). Topology-based descriptors are known to perform well on these and other chemical prediction tasks9,10,47,48 and typically outperform representations based on a limited set of QC descriptors. Topological representations are an important experimental comparison, as bridging the performance gap between topology-based deep learning approaches and approaches based on quantum chemical calculation is an open problem. To select optimal representations, we varied the size of the neighborhood used to construct the atom representation vectors (Data and Methods). We present results from the QC and topological models with the best average performance across all five tasks (Supplementary Table S2, Supplementary Table S3). No optimization of neighborhood depth was performed for the TLQC models. To minimize the number of parameters of TLQC models, we decreased the state size of the quantum representation to 64. This size was chosen by evaluating the accuracy drop with decreasing state sizes (128, 96, 64, 48, 32), for which we observed a large increase in error with state sizes below 64 (Supplementary Table S4). The final models produce well-scaled probabilistic predictions on each of the five tasks (Supplementary Figure S2).
Representations of quantum chemistry derived from deep learning can be used to predict higher-level chemical behaviors (Figure 1B). On the epoxidation task, TLQC outperforms models trained with QC representations (85.4% vs 78.9% top-2 accuracy, p = 0.002 McNemar test) in leave-one-out cross-validated experiments and also outperforms topological representations (85.4% vs 81.1% top-2 accuracy, p = 0.036 McNemar test). Furthermore, TLQC outperforms topological representations on three of the four reactivity tasks: cyanide (top-2 accuracy 80.4% vs 62.7%, p = 0.035 McNemar test), GSH (top-2 accuracy 69.5% vs 65.5%, p = 0.018 McNemar test), and protein (top-2 accuracy 83.2% vs 79.6%, p = 0.541 McNemar test). Importantly, TLQC also improves on the overall accuracy of the topological approach on the task of protein reactivity (AUC 92.9% vs 90.2%, p = 0.038, AUC Z-test, Supplementary Figure S1). Accurate predictions of protein reactivity are key to predicting in vivo toxicity, and there is a relative paucity of data to construct deep learning models for this task (Figure 1). Additionally, TLQC outperforms the QC model at all the reactivity tasks by a substantially wider margin. Together, these data suggest that TLQC can achieve similar or better performance compared to topological representations when predicting higher-level chemical behaviors.
Transfer Learning from Quantum Chemistry Is Stable and Generalizable.
TLQC representations produce stable performance in predicting epoxidation and reactivity under varying choices of QC properties used for representation training (Figure 2). We varied the number of QC properties used to train quantum representations across all 15 combinations of the four categories of QC properties. The quantum representations computed in each of these domains perform comparably, with no relationship between number of included QC properties and top-2 accuracy for four of the five prediction tasks. These data suggest that quantum representations learned from even one QC property are robust enough to decode higher-level chemical behaviors. Some tasks may benefit from increasingly accurate quantum representations afforded by training on more than one QC property.
Figure 2.
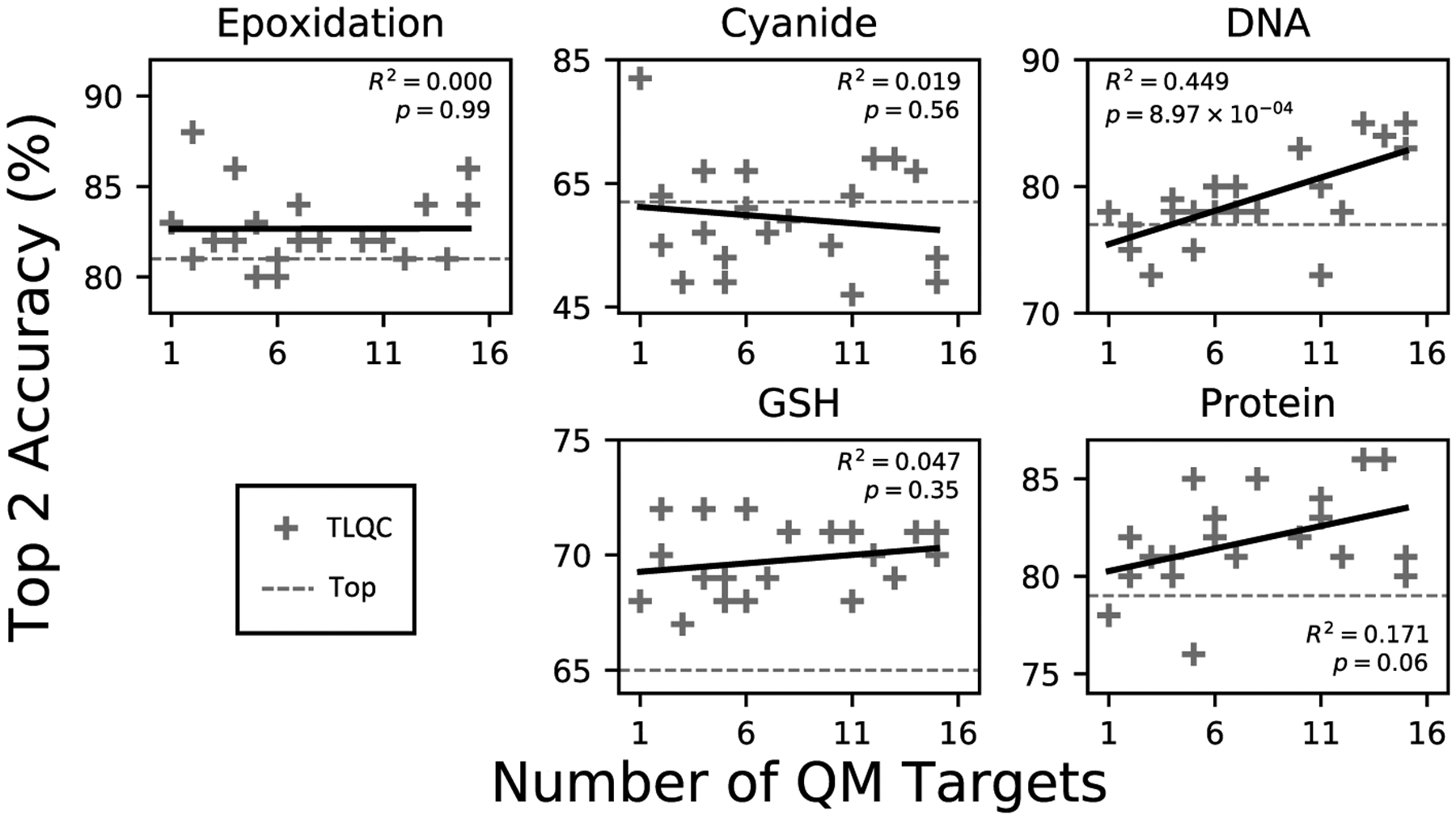
The accuracy of TLQC is stable with respect to the choice of quantum chemical descriptors used during pretraining. Each data point represents transfer learning from one of 20 subsets of the 15 calculated quantum chemical descriptors. For four of the five targets, most TLQC models performed better than TOP (dashed line). The accuracy on most targets is not correlated with the number of quantum chemical descriptors used for transfer learning, suggesting a robust quantum chemical representation can be built even from one molecule-level quantum chemical descriptor.
TLQC representations built from various sets of QC properties do not show clear performance benefits in all tasks (Figure 3). Using the quantum representations constructed in the previous experiment, we plotted the pairwise top-2 accuracy on each of the five prediction tasks. Accuracy on one task does not predict accuracy on the others, as evidenced by the absent or weak correlations between pairs of tasks. These data suggest that quantum representations may be generalizable across tasks regardless of the set of QC properties used to construct these representations.
Figure 3.
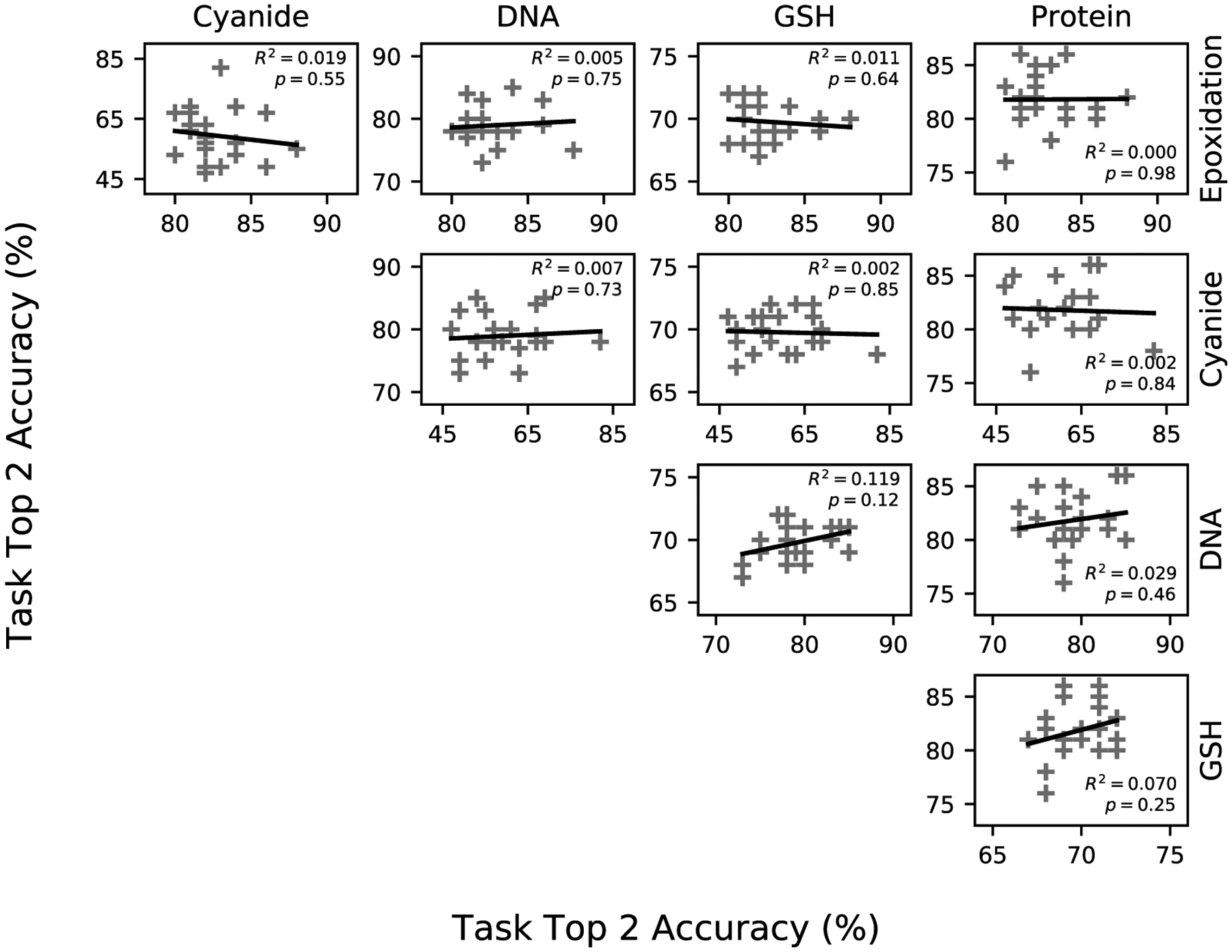
Quantum chemical representations used in transfer learning are generalizable. Correlation between accuracies on all pairs of the five. Each data point represents transfer learning from one of 20 subsets of the 15 calculated quantum chemical descriptors. No pair of tasks is correlated, suggesting that each quantum transfer model is generalizable across different tasks, and no choice of QC properties improves performance on all tasks equally.
Transfer Learning from Quantum Chemistry on Specific Chemical Moieties.
Transfer learning may also yield improvements in our ability to model important chemical moieties with deep learning (Figure 4). For example, aromatic systems can sometimes be identified topologically by graph-based algorithms.40 However, these systems arise due to delocalization of electrons within molecular orbitals, a process explictly modeled by quantum chemistry. Properties of aromatic systems vary and two aromatic systems labeled by graph-based algorithms may exhibit substantially different electronic structures.52 Sites of epoxidation occurring on bonds within topologically identified aromatic systems are better identified by TLQC compared to a topology representation (top-2 score 83.9% vs 78.7%, p = 0.080 McNemar test, Figure 4B). Furthermore, TLQC models assign higher scores to the correct aromatic bonds relative to scores assigned to nonepoxidized bonds (mean Z-score 4.36 vs 3.95, p = 0.169 t test, Figure 4C). Example molecules 1–3 show three compounds for which TLQC provides higher confidence predictions of sites of aromatic epoxidation compared to topological models. These predictions are similarly supported by the QC model, suggesting that transfer of knowledge from the QC domain plays a role in improving the model’s understanding of these structures.
Figure 4.
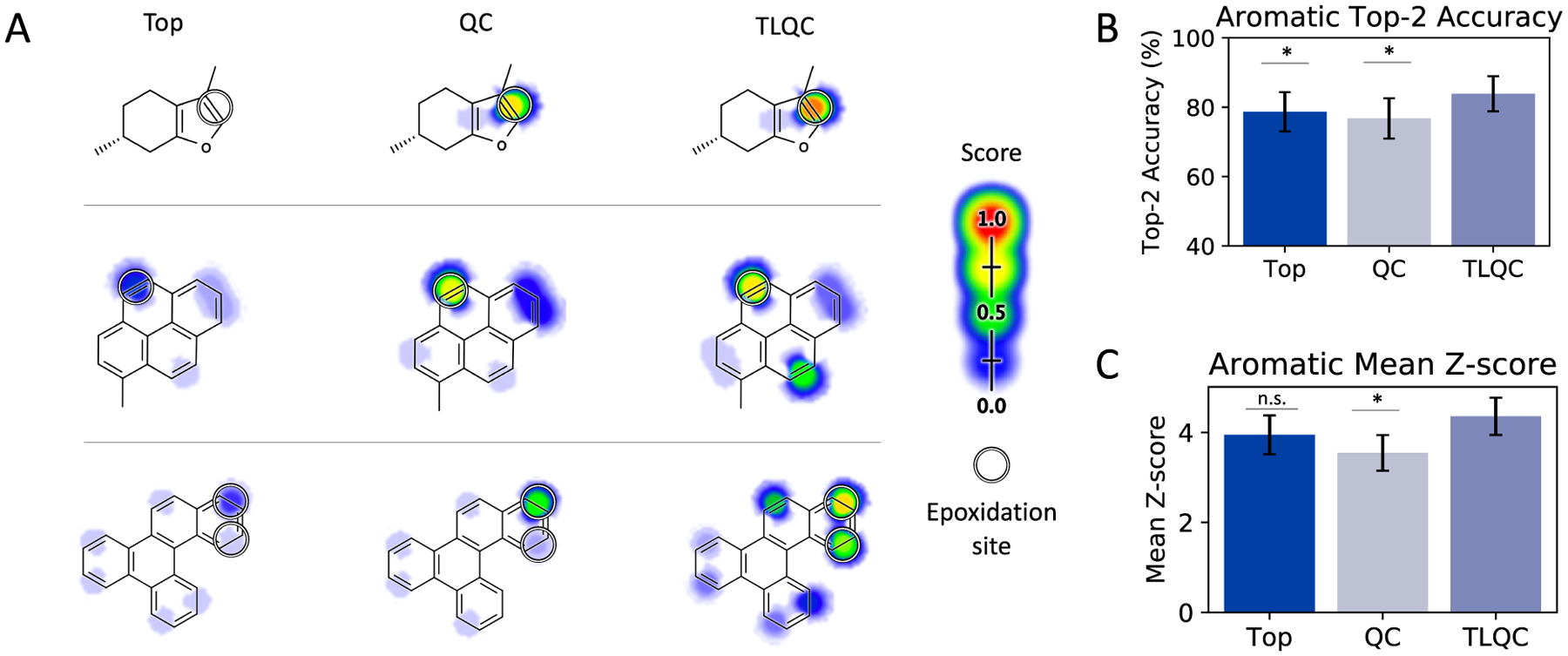
Quantum transfer learning improves identification of epoxidation sites within aromatic systems. (A) Predicted and actual sites of epoxidation for example molecules 1–3 are shown for the Top, QC, and TLQC models. (B) Top-2 accuracy for aromatic sites. (C) Mean Z-score for sites of epoxidation within aromatic systems relative to the distribution of scores assigned to nonsites.
LIMITATIONS
Important limitations on TLQC arise from the choice of the training data when constructing quantum chemical representations. For example, the choice of basis set affects the capabilities of the model. In our work, the 6–31G* basis set was used to compute quantum chemical properties. This basis set only covers atoms up to atomic number 18. In drug design, larger halogens such as bromine and iodine can increase the strength of halogen bonding interactions between proteins and drug ligands.53 Metalloporphyrins (metal coordinating complexes containing porphyrin and either cobalt, copper, and iron) play important enzymatic roles in material design54 and in a variety of biological processes including photosynthesis, xenobiotic metabolism, and oxygen transport.55,56
Another important limitation of TLQC is the volume of data required to train new decoders for other chemical properties or higher-level chemical behaviors. Deep learning methods are notoriously data hungry. The transfer learning approach can significantly decrease the data required to train new models by transferring information between related problems,57 provided the problems are closely related. However, data on hundreds or even thousands of molecules may still be necessary to derive decoders of sufficient accuracy to be useful.
CONCLUSIONS
In this paper, we trained deep learning models to compute site-level quantum chemical properties. Using transfer learning, we reused intermediate representations constructed by deep learning to (1) compute other initially unobserved quantum chemical properties and (2) predict site-level chemical behaviors with high accuracy. In particular, transfer learning from quantum chemistry substantially outperformed models that were derived using the computed QC properties alone in five prediction tasks: identifying sites of epoxidation by metabolic enzymes and identifying sites of covalent reactivity with cyanide, GSH, DNA, and protein. We further investigated the relative strengths of these approaches in predicting epoxidation of aromatic chemical moieties.
This work shows that both lower-level quantum chemical properties and higher-level chemical behaviors, such as metabolism and reactivity, can be computed from deep learning models that only use a very simple input representation. Deep learning models trained to compute quantum mechanical observations learn an intermediate, graph-based latent representation that captures information about the quantum state of a molecular system. Transfer learning allows us to reuse this representation by providing it as input to a second neural network trained to compute other properties. This approach is analogous to the use of wave functions and operators in quantum mechanic.
Constructing quantum representations and operators in a data driven manner allows us to bridge the gap between the theory of quantum mechanics and the chemical behaviors of interest to medicinal chemists. This is a general approach, which might extend to compute chemical synthesis reactions and other chemical properties.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the developers of the open-source cheminformatics tools: Open Babel, and RDKit. Research reported in this publication was supported by the National Library Of Medicine of the National Institutes of Health under Award Number R01LM012222 and R01LM012482 and by the National Institutes of Health under Award Number GM07200. The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by NIH grants nos. 1S10RR022984-01A1 and 1S10OD018091-01. We also thank both the Department of Immunology and Pathology at the Washington University School of Medicine, the Washington University Center for Biological Systems Engineering and the Washington University Medical Scientist Training Program for their generous support of this work.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jpca.0c06231
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.0c06231.
More performance metrics (AUC and reliability), more quantum domains used to train transfer models, top-2 accuracy performance of alternate topological and QC representations, and accuracy related to state size (PDF)
The authors declare no competing financial interest.
Preprocessed version available at https://bitbucket.org/mkmatlock/coordinate_free_quantum_chemistry.
Available at https://bitbucket.org/mkmatlock/tflon/.
Contributor Information
Kathryn Sarullo, Department of Pathology and Immunology, School of Medicine, Washington University in St. Louis, Saint Louis, Missouri 63110, United States.
Matthew K. Matlock, Department of Pathology and Immunology, School of Medicine, Washington University in St. Louis, Saint Louis, Missouri 63110, United States;.
S. Joshua Swamidass, Department of Pathology and Immunology, School of Medicine, Washington University in St. Louis, Saint Louis, Missouri 63110, United States;.
REFERENCES
- (1).Segler MHS; Kogej T; Tyrchan C; Waller MP Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci 2018, 4, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Esteva A; Kuprel B; Novoa RA; Ko J; Swetter SM; Blau HM; Thrun S Dermatologist-level Classification of Skin Cancer with Deep Neural Networks. Nature 2017, 542, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Titano JJ; et al. Automated Deep-Neural-Network Surveillance of Cranial Images for Acute Neurologic Events. Nat. Med 2018, 24, 1337–1341. [DOI] [PubMed] [Google Scholar]
- (4).Wood DE; et al. A Machine Learning Approach for Somatic Mutation Discovery. Sci. Transl. Med 2018, 10, eaar7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kumar RD; Swamidass SJ; Bose R Unsupervised Detection of Cancer Driver Mutations with Parsimony-guided Learning. Nat. Genet 2016, 48, 1288–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Evans DC; Watt AP; Nicoll-Griffith DA; Baillie TA Drug-Protein Adducts: An Industry Perspective on Minimizing the Potential for Drug Bioactivation in Drug Discovery and Development. Chem. Res. Toxicol 2004, 17, 3–16. [DOI] [PubMed] [Google Scholar]
- (7).Dang NL; Matlock MK; Hughes TB; Swamidass SJ The Metabolic Rainbow: Deep Learning Phase I Metabolism in Five Colors. J. Chem. Inf. Model 2020, 60, 1146–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hughes TB; Swamidass SJ Deep Learning to Predict the Formation of Quinone Species in Drug Metabolism. Chem. Res. Toxicol 2017, 30, 642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hughes TB; Miller GP; Swamidass SJ Modeling Epoxidation of Drug-like Molecules with a Deep Machine Learning Network. ACS Cent. Sci 2015, 1, 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hughes TB; Dang NL; Miller GP; Swamidass SJ Modeling Reactivity to Biological Macromolecules with a Deep Multitask Network. ACS Cent. Sci 2016, 2, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mayr A; Klambauer G; Unterthiner T; Hochreiter S DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci 2016, DOI: 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- (12).Dang NL; Hughes TB; Miller GP; Swamidass SJ Computationally Assessing the Bioactivation of Drugs by N-Dealkylation. Chem. Res. Toxicol 2018, 31, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kayala MA; Baldi PF In Advances in Neural Information Processing Systems 24; Shawe-Taylor J, Zemel RS, Bartlett PL, Pereira F, Weinberger KQ, Eds.; Curran Associates, Inc., 2011; pp 747–755. [Google Scholar]
- (14).Sanchez-Lengeling B; Aspuru-Guzik A Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering. Science 2018, 361, 360–365. [DOI] [PubMed] [Google Scholar]
- (15).Ma W; Cheng F; Liu Y Deep-Learning-Enabled On-Demand Design of Chiral Metamaterials. ACS Nano 2018, 12, 6326–6334. [DOI] [PubMed] [Google Scholar]
- (16).Poree C; Schoenebeck F A Holy Grail in Chemistry: Computational Catalyst Design: Feasible or Fiction? Acc. Chem. Res 2017, 50, 605–608. [DOI] [PubMed] [Google Scholar]
- (17).Zhavoronkov A; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol 2019, 37, 1038–1040. [DOI] [PubMed] [Google Scholar]
- (18).Stokes JM; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yao K; Herr JE; Brown SN; Parkhill J Intrinsic Bond Energies from a Bonds-in-Molecules Neural Network. J. Phys. Chem. Lett 2017, 8, 2689–2694. [DOI] [PubMed] [Google Scholar]
- (20).Schütt KT; Arbabzadah F; Chmiela S; Müller KR; Tkatchenko A Quantum-Chemical Insights from Deep Tensor Neural Networks. Nat. Commun 2017, 8, 13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Smith JS; Nebgen BT; Zubatyuk R; Lubbers N; Devereux C; Barros K; Tretiak S; Isayev O; Roitberg AE Approaching Coupled Cluster Accuracy with a General-Purpose Neural Network Potential Through Transfer Learning. Nat. Commun 2019, 10, 2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hohenberg P; Kohn W Inhomogeneous Electron Gas. Phys. Rev 1964, 136, B864–B871. [Google Scholar]
- (23).Bartlett RJ Coupled-Custer Approach to Molecular Structure and Spectra: a Step Toward Predictive Quantum Chemistry. J. Phys. Chem 1989, 93, 1697–1708. [Google Scholar]
- (24).Attia SM Deleterious Effects of Reactive Metabolites. Oxid. Med. Cell. Longevity 2010, 3, 238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Knowles SR; Uetrecht J; Shear NH Idiosyncratic Drug Reactions: The Reactive Metabolite Syndromes. Lancet 2000, 356, 1587–1591. [DOI] [PubMed] [Google Scholar]
- (26).Rydberg P; Gloriam DE; Zaretzki J; Breneman C; Olsen L SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug Metabolism. ACS Med. Chem. Lett 2010, 1, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tarpey MM Methods for Detection of Reactive Metabolites of Oxygen and Nitrogen: In Vitro and In Vivo Considerations. Am. J. Physiol.: Regul., Integr. Comp. Physiol 2004, 286, R431–R444. [DOI] [PubMed] [Google Scholar]
- (28).Cherkasov A; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem 2014, 57, 4977–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wu K; Wei G-W Quantitative toxicity prediction using topology based multitask deep neural networks. J. Chem. Inf. Model 2018, 58, 520–531. [DOI] [PubMed] [Google Scholar]
- (30).Mahajan D; Girshick RB; Ramanathan V; He K; Paluri M; Li Y; Bharambe A; van der Maaten L Exploring the Limits of Weakly Supervised Pretraining. arXiv:1805.00932 2018. [Google Scholar]
- (31).Moyal JE Quantum Mechanics as a Statistical Theory. Math. Proc. Cambridge Philos. Soc 1949, 45, 99–124. [Google Scholar]
- (32).Hughes TB; Miller GP; Swamidass SJ Site of Reactivity Models Predict Molecular Reactivity of Diverse Chemicals with Glutathione. Chem. Res. Toxicol 2015, 28, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nakata M; Shimazaki T PubChemQC Project: A Large-Scale First-Principles Electronic Structure Database for Data-Driven Chemistry. J. Chem. Inf. Model 2017, 57, 1300–1308. [DOI] [PubMed] [Google Scholar]
- (34).Gilmer J; Schoenholz SS; Riley PF; Vinyals O; Dahl GE Neural Message Passing for Quantum Chemistry. arXiv:1704.01212 2017. [Google Scholar]
- (35).Vinyals O; Bengio S; Kudlur M Order Matters: Sequence to Sequence for Sets. arXiv:1511.06391 2015. [Google Scholar]
- (36).Abadi M; Barham P; Chen J; Chen Z; Davis A; Dean J; Devin M; Ghemawat S; Irving G; Isard M TensorFlow: A System for Large-Scale Machine Learning. Proceedings of the 12th USENIX Conference on Operating Systems Design and Implementation 2016, 265–283. [Google Scholar]
- (37).Battaglia PW; et al. Relational Inductive Biases, Deep Learning, and Graph Networks. arXiv:1806.01261 2018. [Google Scholar]
- (38).Kingma D; Ba J Adam: A Method for Stochastic Optimization. arXiv:1412.6980 2014. [Google Scholar]
- (39).Kearnes S; McCloskey K; Berndl M; Pande V; Riley P Molecular Graph Convolutions: Moving Beyond Fingerprints. J. Comput.-Aided Mol. Des 2016, 30, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Matlock MK; Dang NL; Swamidass SJ Learning a Local-Variable Model of Aromatic and Conjugated Systems. ACS Cent. Sci 2018, 4, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ngiam J; Coates A; Lahiri A; Prochnow B; Le QV; Ng AY On Optimization Methods for Deep Learning. Proceedings of the 28th International Conference on Machine Learning (ICML-11) 2011, 265–272. [Google Scholar]
- (42).Cang Z; Wei G-W Analysis and Prediction of Protein Folding Energy Changes Upon Mutation By Element Specific Persistent Homology. Bioinformatics 2017, DOI: 10.1093/bioinformatics/btx460. [DOI] [PubMed] [Google Scholar]
- (43).Cang Z; Wei G-W TopologyNet: Topology Based Deep Convolutional and Multi-task Neural Networks for Biomolecular Property Predictions. PLoS Comput. Biol 2017, DOI: 10.1371/journal.pcbi.1005690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cang Z; Mu L; Wei G-W Representability of Algebraic Topology for Biomolecules in Machine Learning Based Scoring and Virtual Screening. PLoS Comput. Biol 2018, 14, e1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nguyen DD; Cang Z; Wei G-W A Review of Mathematical Representations of Biomolecular Data. Phys. Chem. Chem. Phys 2020, 22, 4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wu K; Wei G-W Quantitative Toxicity Prediction Using Topology Based Multitask Deep Neural Networks. J. Chem. Inf. Model 2018, 58, 520. [DOI] [PubMed] [Google Scholar]
- (47).Zaretzki J; Rydberg P; Bergeron C; Bennett KP; Olsen L; Breneman CM RS-Predictor Models Augmented with SMARTCyp Reactivities: Robust Metabolic Regioselectivity Predictions for Nine CYP Isozymes. J. Chem. Inf. Model 2012, 52, 1637–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zaretzki J; Matlock M; Swamidass SJ XenoSite: Accurately Predicting CYP-Mediated Sites of Metabolism with Neural Networks. J. Chem. Inf. Model 2013, 53, 3373–3383. [DOI] [PubMed] [Google Scholar]
- (49).McNemar Q Note on the Sampling Error of the Difference Between Correlated Proportions or Percentages. Psychometrika 1947, 12, 153–157. [DOI] [PubMed] [Google Scholar]
- (50).Hanley JA; McNeil BJ The Meaning and Use of the Area Under a Receiver Operating Characteristic (ROC) Curve. Radiology 1982, 143, 29–36. [DOI] [PubMed] [Google Scholar]
- (51).Welch BL The Generalization of ‘Student’s’ Problem When Several Different Population Variances are Involved. Biometrika 1947, 34, 28–35. [DOI] [PubMed] [Google Scholar]
- (52).Cyrañski MK; Krygowski TM; Wisiorowski M; van Eikema Hommes NJ; Schleyer P. v. R. Global and Local Aromaticity in Porphyrins: An Analysis Based on Molecular Geometries and Nucleus-Independent Chemical Shifts. Angew. Chem., Int. Ed 1998, 37, 177–180. [Google Scholar]
- (53).Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- (54).Chou J-H; Kosal ME; Nalwa HS; Rakow NA; Suslick KS Applications of Porphyrins and Metalloporphyrins to Materials Chemistry. J. Cheminf 2003, DOI: 10.1002/chin.200317263. [DOI] [Google Scholar]
- (55).Nelson DL; Lehninger AL; Cox MM Lehninger Pinciples of Biochemistry; Macmillan, 2008. [Google Scholar]
- (56).de Montellano PRO; Voss JJD Cytochrome P450; Springer Science + Business Media, 2005; pp 183–245. [Google Scholar]
- (57).Pan SJ; Yang Q A Survey on Transfer Learning. IEEE Trans. Knowl. Data Eng 2010, 22, 1345–1359. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.