

Abstract
Traumatic brain injury (TBI) is a critical public health concern and major contributor to death and long-term disability. After the initial trauma, a sustained secondary injury involving a complex continuum of pathophysiology unfolds, ultimately leading to the destruction of nervous tissue. One disease hallmark of TBI is ectopic protease activity, which can mediate cell death, extracellular matrix breakdown, and inflammation. We previously engineered a fluorogenic activity-based nanosensor for TBI (TBI-ABN) that passively accumulates in the injured brain across the disrupted vasculature and generates fluorescent signal in response to calpain-1 cleavage, thus enabling in situ visualization of TBI-associated calpain-1 protease activity. In this work, we hypothesized that actively targeting the extracellular matrix (ECM) of the injured brain would improve nanosensor accumulation in the injured brain beyond passive delivery alone and lead to increased nanosensor activation. We evaluated several peptides that bind exposed/enriched ECM constituents in the brain and discovered that nanomaterials modified with peptides that target hyaluronic acid (HA) displayed widespread distribution across the injury lesion, in particular colocalizing with perilesional and hippocampal neurons. Modifying TBI-ABN with HA-targeting peptide led to increases in activation in a ligand-valency-dependent manner, up to 6.6-fold in the injured cortex compared to a nontargeted nanosensor. This robust nanosensor activation enabled 3D visualization of injury-specific protease activity in a cleared and intact brain. In our work, we establish that targeting brain ECM with peptide ligands can be leveraged to improve the distribution and function of a bioresponsive imaging nanomaterial.
Keywords: hyaluronic acid, controlled cortical impact, calpain-1, peptides, protease activity, light sheet microscopy, CUBIC
Traumatic brain injury (TBI) affects over 1.5 million Americans per year, and an estimated 3.17 million patients live with chronic neurodisability due to TBI.1,2 After the primary injury, a progressive secondary injury unfolds within the brain over the course of hours to months caused by a complex continuum of pathophysiology characterized by hallmarks such as neuronal apoptosis, excitotoxicity, inflammation, and blood–brain barrier (BBB) dysfunction.3 One signature of secondary injury pathophysiology is ectopic protease activity. For instance, the calcium-dependent cysteine protease calpain-1 cleaves cytoskeletal proteins and contributes to apoptotic and necrotic cell death,4 and its activity is correlated with worsened TBI outcome.5−7 Protease activity measurements have the potential to improve the understanding of TBI disease biology and can serve as clinical biomarkers for disease progression,8,9 but there are few approaches to measure protease activity in the living brain. In order to measure TBI-associated calpain-1 activity, we previously engineered a fluorogenic activity-based nanosensor for TBI (TBI-ABN) comprised of a Förster resonance energy transfer (FRET)-based peptide substrate of calpain-1 conjugated to a polymeric nanomaterial scaffold.10 When administered intravenously in a TBI mouse model, we demonstrated that TBI-ABN could accumulate in the injured brain tissue and activate in the context of injury. To sample ectopic protease activity in the injured tissue, this technology relied on size-dependent accumulation in brain tissue via transient BBB permeability, a hallmark of TBI pathophysiology. This passive accumulation of nanomaterials localized to diseased tissue has been called an “enhanced permeation and retention” (EPR)-like effect,11,12 and a similar phenomenon is described in other diseases such as cancer,13,14 arthritis,15 and myocardial infarction.16,17 However, in TBI, reliance on passive targeting alone is generally limited because unlike chronic conditions such as cancer and arthritis, BBB disruption after TBI is transient, restricting the time window for nanomaterial delivery to hours after injury.11,18 Furthermore, increased intracranial pressure from ischemia-associated edema creates a pressure gradient that likely limits nanomaterial diffusion across the extracellular space (ECS).19 Thus, strategies to control the tissue-level distribution of activity-based nanosensors beyond passive targeting have the potential to increase protease-specific signal generation and therefore increase the sensitivity of nanosensors, as was previously demonstrated when the addition of tumor-targeting ligands to an MMP-9-sensitive ABN enabled ultrasensitive detection of low tumor burdens in a urinary readout.20
As a complementary approach in addition to passive delivery, active targeting via peptide or protein affinity ligands (e.g., transferrin,21,22 HER2 antibodies,23,24 RVG,18,25 Lyp-1)26,27 can improve the transport and/or tissue-level distribution of payloads. The extracellular matrix (ECM) represents an attractive biological target for nanomaterials due to its abundance and high density of binding moieties28 and therefore has the potential to act as a binding reservoir for exogenously delivered nanomaterials. Active targeting to ECM has been successfully applied to nanoparticle and protein therapeutics within the contexts of cancer29−32 and arthritis-associated inflammation.33,34 The dysregulated vasculature associated with cancer and inflammation exposes tissue ECM to materials in systemic circulation. In addition, aberrantly deposited ECM constituents can serve as disease-specific “neo-antigens” to improve distribution and efficacy of therapeutics. For example, the fusion of a collagen binding domain to chemokine CCL4 increased the accumulation of systemically administered CCL4 within collagen-rich tumors, leading to an enhancement of intratumor infiltration of immune cells when used in combination with checkpoint inhibitor immunotherapy.32 In the brain, tissue is largely inaccessible to systemically administered synthetic nanomaterials under physiological conditions due to the selective and tightly regulated BBB. After TBI, transient BBB disruption initiated by the injury enables access to the brain parenchyma,10,11,18,35 including the ECM. In addition, the composition of ECM in the brain is distinct from that of peripheral organs: The brain is enriched with hyaluronic acid (HA) and sulfate proteoglycans, while fibrillar collagens and elastin are less represented.36,37 This composition is furthermore changed in disease, with tenascins, laminin, chondroitin sulfate proteoglycans, and heparin sulfate proteoglycans focally upregulated in the TBI lesion.36,37 Local levels of HA are also perturbed due to changes in HA metabolism; hyaluronidases can degrade HA into bioactive fragments,36,38 and expression of hyaluronic acid synthases are increased after experimental brain injury.38 In addition to the tissue ECM present in the brain parenchyma, vascular injury leads to fibrin deposition during natural clotting and transient exposure of collagen IV in the basement membrane.36 Each of these brain ECM components are potential targets for nanomaterials in the context of TBI. The approach of targeting the ECM after brain injury is highlighted by the recent identification of TBI-specific peptides via in vivo phage display.39 This unbiased screening approach against whole brain tissue after TBI identified a peptide, CAQK, whose receptors were discovered to be upregulated tenascin and versican proteoglycans in the ECM. CAQK modification enhanced the accumulation, acute retention, and activity of nanomaterials carrying siRNA payloads in a penetrating brain injury model.39 Beyond this demonstration, active targeting to the ECM after TBI has been largely unexplored.
In this manuscript, we hypothesized that actively targeting nanomaterials to the brain ECM can increase the bioavailability and therefore activity of a diagnostic payload after systemic delivery in a TBI mouse model. We first performed an in vivo evaluation of nanomaterials modified with a selection of peptides that target major brain ECM constituents: proteoglycans, collagen IV, fibrin, and HA. In a controlled cortical impact (CCI) model of TBI in mice, nanomaterials modified with peptides that targeted HA led to widespread distribution across cortical and hippocampal perilesional tissue after intravenous administration, and tissue localization was distinct from nanomaterials modified with peptides that targeted other ECM components. To determine whether this ECM-targeting-mediated change in tissue distribution could be harnessed into a functional outcome, we added HA-targeting to TBI-ABN, a nanosensor that activates in response to calpain-1 protease activity. We modified TBI-ABNs with HA-targeting peptide at various levels of substitutions, quantified calpain-1 nanosensor activation in injured brain tissue, and observed that targeting could increase activation up to 6.6-fold over that of a nontargeted nanosensor, which suggests that increasing nanomaterial avidity to the ECM translates into enhanced nanosensor sensitivity. At the tissue level, we observed that the activation of HA-targeting TBI-ABN throughout the perilesional brain tissue colocalized with both neuronal and endothelial cell populations. Finally, light sheet imaging of TBI-ABN in cleared brain revealed nanosensor activation in proximity to the impact lesion in both the hippocampus and cortex. In summary, we establish that targeting an activity-based nanosensor to the brain ECM can significantly increase signal generation when applied to an animal model of TBI after systemic administration, enabling tissue-level visualization of aberrant calpain-1 protease activity in the injured brain.
Results & Discussion
Nanomaterials Targeted to Hyaluronic Acid Have Widespread Distribution in the Injured Brain
In order to generate nanomaterials that interact with the brain ECM after intravenous delivery, we first identified peptide ligands from literature that bind to ECM constituents that are either exposed or enriched in the injured brain.36 The selected ECM targets and ligand sequences were proteoglycans (CAQK),39 collagen IV (KLWVLPK),40 fibrin (CREKA),41 and hyaluronic acid (STMMSRSHKTRSHHV).42,43 Throughout the manuscript, these ECM-targeting peptides are referred to as PGpep, CIVpep, FIBpep, and HApep, respectively. These peptides are low-molecular-weight, linear sequences with comparable physicochemical properties (Table 1). In previous work, we have established that the physiochemical properties of peptides influence nanoparticle pharmacokinetics in a mouse model of TBI.44 Our nanomaterial scaffold was a 40 kDa 8-arm polyethylene glycol (PEG) which was selected for several reasons. First, each arm can participate in chemical conjugation, thus allowing for multivalent ligand presentation. Second, we previously established that its ∼10 nm hydrodynamic diameter facilitates accumulation in the injured brain after intravenous administration10 since it is larger than the ∼5.5 nm renal filtration limit which prevents rapid kidney excretion,45 yet smaller than the pores in the ECS which allows for diffusion into brain tissue.19,46 Third, PEG is a component used to extend in vivo circulation half-life in multiple FDA-approved formulations.47 The ECM-targeting peptides were synthesized with the fluorescent molecule fluorescein (FAM) for quantification and a cysteine for reaction with maleimides on the 8-arm PEG. The PEG scaffold was reacted with 1 mol equiv of fluorescent molecule VivoTag S-750 (VivoTag 750), and the remaining moieties were fully reacted with ECM-targeting peptides. The FAM on the peptide was utilized for quantitative biodistribution and histology and VivoTag 750 on the PEG scaffold was utilized for near-infrared surface imaging of organs. A nontargeted nanomaterial control was synthesized by modifying PEG with cysteine (Cys) instead of ECM-targeting peptide. In addition, the linear form of the well-studied peptide RGD48 was included as a control due to the known elevation of integrin expression in inflammation.49−51
Table 1. Brain ECM Targeting Peptides and Their Properties.
| peptide name | ECM target | peptide sequence | pIa | GRAVYb |
|---|---|---|---|---|
| PGpep | proteoglycans | CAQK39 | 9.13 | –0.78 |
| CIVpep | collagen IV | KLWVLPK40 | 10.69 | 0.21 |
| FIBpep | fibrin | CREKA41 | 9.12 | –1.52 |
| HApep | hyaluronic acid | STMMSRSHKTRSHHV42 | 12.13 | –1.27 |
pI = isoelectric point.
GRAVY = grand average of hydropathy.
We evaluated the biodistribution of nanomaterials modified with each ECM-targeting peptide after intravenous delivery in a CCI mouse model of TBI (Figure 1A). CCI is a well-established model of TBI that has reproducible molecular phenotypes,52 including elevation of calpain-1 activity.10,53 Injuries were created by performing a 5 mm craniotomy over the right hemisphere of the brain and impacting the exposed dura with an electromagnetically driven probe 2 mm in diameter at a speed of 3 m/s and depth of 2 mm. ECM-targeting peptide-modified PEG nanomaterial (25 nmoles of peptide per injection based on FAM absorbance, corresponding to a dosage range of 1136–1389 nmol/kg) was injected intravenously at 6 h post-CCI. Mice were perfused, and the organs were harvested at 1 h postinjection, an experimental timeline established by previous work.39 Surface imaging of the VivoTag 750 label in intact organs shows that nanomaterial accumulation in the brain was localized to the injured brain hemisphere (Figure 1B). This accumulation of intravenously delivered nanomaterials into the injured hemisphere due to local, transient permeability of the dysregulated BBB is consistent with previous work from our group10,18,44 and others.11,12 Nanomaterials modified with CIVpep had high liver accumulation over all other materials. One potential hypothesis for this observation is the abundance of collagens I, III, IV, and V in the basement membrane of the liver.54 We also observed significant nanomaterial accumulation in the kidneys, in particular for FIBpep- and PGpep-modified nanomaterials.
Figure 1.

Nanomaterial modification with HA-targeting peptide leads to widespread distribution in the injured brain after systemic administration. (A) Schematic of ECM-targeting nanomaterials and overview of experimental design. At 6 hours post-CCI, ECM-targeting nanomaterials were intravenously administered. After 1 h, organs were harvested for analysis of nanomaterial biodistribution and histology. (B) Surface imaging of VivoTag 750 from major organs of one representative mouse per nanomaterial (n = 3, white line indicates outline of organ). (C) Bulk quantification of percent injected dose of nanomaterial per gram of tissue (% ID/g) based on FAM fluorescence (n = 3, mean ± SEM; ****, p ≤ 0.0001, two-way ANOVA and Tukey’s multiple comparisons post hoc test within each organ group). (D) Representative images of the injured cortex in coronal brain slices (n = 3; blue, nuclei; green, FAM-labeled ECM-targeting peptide on nanomaterial; scale bar = 500 μm).
While surface imaging gives spatial distribution within an organ, it is limited by imaging depth, and we therefore performed a bulk analysis of homogenized organs to quantify nanomaterial accumulation. Bulk quantitative biodistribution analysis was based on fluorescence signal from the FAM label on the peptide which was used to calculate the percent injected dose per gram tissue (%ID/g tissue) from a standard of known peptide concentrations. We observed that bulk quantitative analysis was largely consistent with surface imaging (Figure 1C). CIVpep-modified nanomaterials significantly accumulated in the liver over all nanomaterials modified with other ECM peptides (between 8.9- and 24.8-fold). Moreover, nanomaterials modified with PGpep and FIBpep had the highest overall kidney accumulation, followed by RGD. The ECM-targeting peptides did not appear to have significant impacts on total accumulation in the injured brain (%ID/g between 0.80 and 1.68), consistent with previous observations that targeting has modest effects on total tissue accumulation.21,22,24,26 Considering that the targeting peptides we investigated bind to the ECM, we posit that nanomaterials must first encounter the ECM through passive targeting; therefore, ECM-targeting peptides would be unlikely to contribute to changes in total brain accumulation. This is further corroborated by similar blood half-lives measured for each ECM-targeting nanomaterial (Figure S1); previous work has established that passive accumulation after systemic administration correlates with blood half-life due to a maintenance of a concentration gradient of nanomaterials between the blood and target tissue.55
To investigate the spatial distribution of nanomaterials in the injured brain at greater depth and resolution than could be achieved with surface imaging, we completed a qualitative comparison of nanomaterial distribution in brain sections following immunostaining with an anti-FAM antibody (Figure 1D, Table S1). We observed a marked increase in the distribution of HApep-modified nanomaterials in the injured area of the brain compared to the other peptide-modified nanomaterials. This observation was consistent across triplicate brains (Figure S2). To verify that the FAM-immunostaining was specific, we used an alternate anti-FAM antibody and observed the same outcome (Figure S3). The nontargeted nanomaterial served as a negative control for nonspecific staining since no FAM was present on that nanomaterial. The widespread distribution of HApep-modified nanomaterial in the tissue proximal to the injury was observed despite the lack of appreciable differences measured in bulk analysis of FAM signal. This may be due to the reduced sensitivity of measurement in the bulk analysis since antibody staining was used to image nanomaterial in brain sections. In addition, analysis of brain sections revealed that nanomaterial was largely restricted to the cortical and hippocampal tissue in close proximity to the injury, whereas in bulk quantification the whole hemisphere was homogenized potentially diluting total nanomaterial signal. Our results were also consistent with previous studies that establish active targeting has greater impacts on intratissue distribution of nanoparticles over bulk accumulation.22
HA-Targeted Nanomaterial Accumulates within Injured Brain Tissue and Colocalizes with Hippocampal and Cortical Neurons
Having demonstrated that nanomaterials modified with peptides that target HA have improved distribution within the injured brain over that of peptides that target other ECM components, we sought to further characterize the tissue-level distribution of this nanomaterial. We stained and imaged both the uninjured contralateral and injured ipsilateral hemispheres in coronal brain sections and observed that nanomaterial localization is specific to the injured hemisphere with minimal signal in the uninjured hemisphere (Figure 2A), consistent with our previous observation (Figure 1B). Nanomaterial accumulation coincided with areas of tissue trauma in the right hemisphere, where the CCI was applied. Within the injury, there was significant nanomaterial signal in the perilesional cortical and hippocampal regions.
Figure 2.

HA-targeting nanomaterial distributes across perilesional brain tissue in CCI-injured brains. (A) Coronal brain sections from mice i.v.-administered HApep-modified nanomaterial at 6 h post-CCI after 1 h of circulation (blue, nuclei; green, HApep on nanomaterial; scale bar = 500 μm). (B) HApep-modified nanomaterial in the injured cortex stained for neurons (NeuN, red; scale bar = 500 μm). Insets show (i) perilesional cortex and (ii) hippocampus (scale bar = 100 μm). (C) Injured cortex labeled with biotinylated hyaluronic acid binding protein (bHABP, magenta; scale bar = 500 μm). Insets show (i) perilesional cortex and (ii) hippocampus (scale bar = 50 μm). Arrows note instances of nanomaterial colocalization with HABP staining.
Next, to identify nanomaterial localization around brain cells of interest, we performed immunostaining for neurons, a major source of calpain-156,57 and the cellular target for neuroprotective therapeutics.58 HApep-modified nanomaterial was found to colocalize with subsets of both cortical and hippocampal neurons (Figure 2B). In the perilesional cortex (Figure 2B-i), nanomaterial localized to neurons adjacent to tissue trauma, and similarly, nanomaterial localized to neurons in the hippocampal region (Figure 2B-ii).
We next compared the localization of our HA-targeting nanomaterial to the spatial distribution of endogenous HA. A biotinylated hyaluronic acid binding protein (bHABP) was used to visualize native HA, as described previously.59−62 Consistent with the published literature,60−62 regions with HABP binding were enriched in the hippocampus and cerebral cortex (Figure 2C). The staining of bHABP in both the cortex (Figure 2C-i) and hippocampus (Figure 2C-ii) was mainly diffuse, although enrichment was observed around the cell bodies of hippocampal neurons and select neurons in the cortex. This observation is consistent with literature, as HA is a known component of perineuronal nets (PNNs) that surround neuronal cell bodies.60 In some instances, our HA-targeting nanomaterial colocalized with cells with HABP perineuronal staining in both the cortex and hippocampus (Figure 2C-i,ii, arrows). Differences observed between the distribution of HABP staining of brain slices and systemic administration of HApep-modified nanomaterial are likely due to limited access of intravenously administered nanomaterial across the damaged vasculature and extracellular space.19,46 Additionally, there are likely differences between the specificity of HABP and the short peptide ligand HApep since the modes of discovery and composition are divergent.42,59
Targeting HA Enhances the In Vivo Sensitivity of an Activity-Based Nanosensor for TBI (TBI-ABN)
We previously developed an activity based nanosensor for TBI (TBI-ABN) for detection of calpain-1 protease activity in a mouse model of TBI.10 The TBI-ABN consists of a FRET pair (fluorophore: Cy5, quencher: QSY21) separated by the calpain-1-cleavable peptide sequence QEVYGAMP, which is derived from a native calpain-1 substrate, αII-spectrin,63 attached to the 40 kDa 8-arm PEG, the same polymeric scaffold that we used to evaluate brain ECM-targeting peptides (Figure 1). When active calpain-1 protease is present, the FRET substrate on TBI-ABN is cleaved, leading to dequenching of Cy5 fluorescence. We previously demonstrated that TBI-ABN could passively accumulate in the injured brain and generate fluorescent signal in response to locally activated calpain-1 after systemic administration in CCI-injured mice.10 As a demonstration that spatial localization in the TBI microenvironment can increase the activity of nanomaterials, we proposed that the addition of active targeting ligands would improve signal generation from TBI-ABN. We previously established that the addition of tumor targeting increased the sensitivity of an activity-based protease nanosensor for cancer via in silico and experimental analyses.20 In the context of TBI, we hypothesized that the incorporation of HA targeting in our TBI-ABN design would increase nanosensor activation since HA-targeting nanomaterial colocalized within perilesional cortical and hippocampal neurons (Figure 2), and neurons are a major cellular source of calpain-1.56,57
We synthesized TBI-ABNs with varying degrees of HA targeting in order to investigate the relationship between ligand valency and TBI-ABN activation in vivo. This was motivated by the phenomenon that multivalent molecular interactions confer enhanced overall binding strength (i.e., avidity) and increase the likelihood for binding events to occur.64 Previous studies have established the importance of tuning binding avidity for nanomaterial targeting; for example, increasing densities of transferrin targeting ligand on gold nanoparticles led to an increase in nanoparticle localization in tumor cells, although a higher ligand density also increased the off-target uptake by hepatocytes.22 In order to create TBI-ABNs with a matched amount of calpain-1 FRET substrate peptide and varying levels of targeting for comparison studies, a 1:1 stoichiometry of calpain FRET substrate and the 8-arm PEG was reacted and split into three parts, and HApep was added at stoichiometric ratios of 0, 4, and 7 to yield non-, moderate-, and high-targeting TBI-ABNs respectively (Figure 3A). Absorbance measurements verified that stoichiometric ratios of HApep to calpain FRET peptide on the TBI-ABN were 0, ∼3.2, ∼8.5 for non-, moderate-, and high-targeting TBI-ABNs respectively (Table S2). In order to verify that HApep modification of TBI-ABN did not significantly impact nanosensor performance, non-, moderate-, and high-targeting TBI-ABNs were incubated with human calpain-1 enzyme at various concentrations to construct Michaelis–Menten kinetics curves. Maximum cleavage velocities for non-, moderate-, and high-targeting TBI-ABNs were 120.0, 73.72, and 76.13 RFU/min, respectively, indicating that the incorporation of HApep moderately decreased calpain-1 cleavage kinetics of TBI-ABN (Figure 3B). A similar decrease in cleavage kinetics was observed when nontargeted TBI-ABNs were mixed with unconjugated HApep in molar ratios matching those of the moderate- and high-targeting TBI-ABNs, indicating that the presence of HApep independent of conjugation led to modest decreases in calpain-1 cleavage kinetics (Figure S4).
Figure 3.
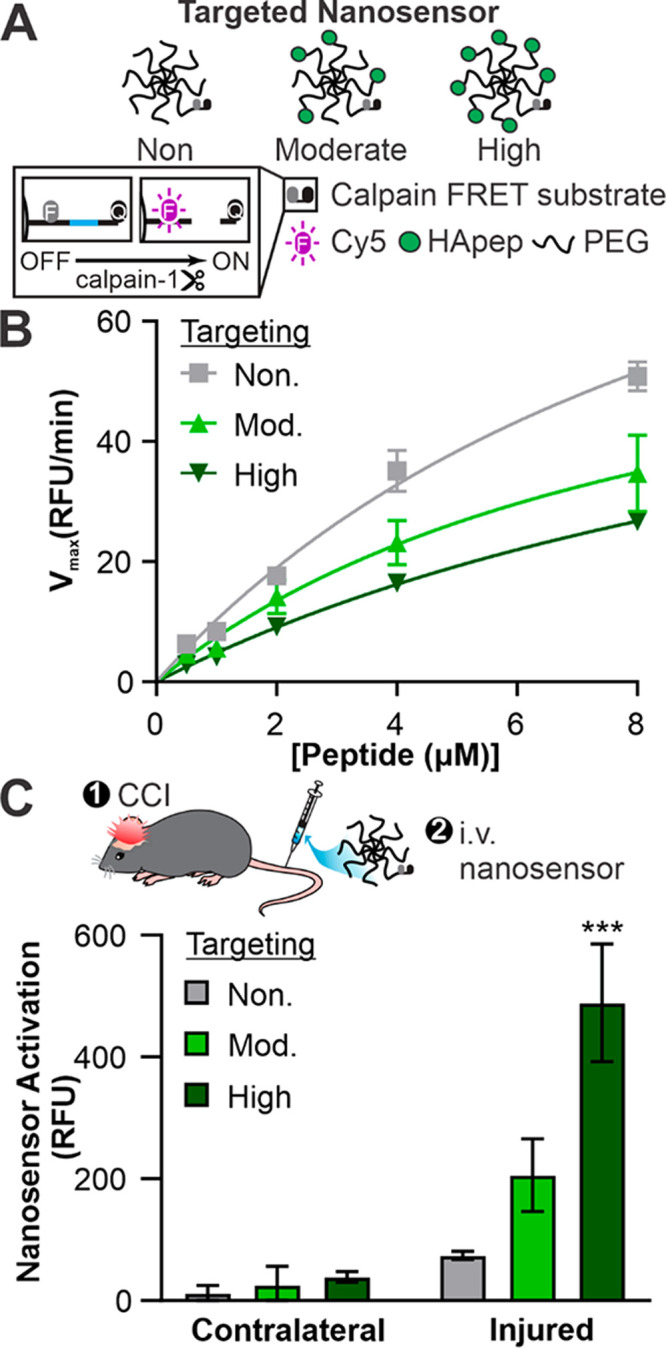
Hyaluronic acid targeting improves nanosensor signal generation in a CCI mouse model of TBI. (A) Schematic of HApep-modified nanosensors with no, moderate, and high targeting. (B) Michaelis–Menten cleavage kinetics of nanosensors incubated with human calpain-1 (n = 3, mean ± SD). (C) Activated nanosensor signal measured in cortical brain tissue lysate collected from the contralateral and injured hemispheres (n = 3, mean ± SEM; ***, p ≤ 0.001, two-way ANOVA with Sidak’s multiple comparisons post hoc test compared to nontargeted groups).
Next, we quantified TBI-ABN activation after intravenous administration in a mouse model of TBI. Non-, moderate-, and high-targeting nanosensors were administered via tail vein at matched calpain-1 FRET substrate concentrations 3 h after CCI, a time point when calpain-1 activation was measured to be locally increased in injured brain tissue.10 One hour after nanosensor administration, fresh perilesional tissue and corresponding uninjured tissue from the contralateral hemisphere were collected and homogenized for bulk fluorescence analysis of dequenched FRET substrate from the TBI-ABN. Consistent with our previous observations, nanosensor activation was significantly greater in homogenate isolated from the injured hemisphere over the uninjured contralateral hemisphere by 6.2-, 8.1-, and 12.5-fold for non-, moderate-, and high-targeting TBI-ABNs, respectively (Figure 3C). Comparing signal generation in the injured hemisphere across degrees of targeting, moderate and high targeting increased the levels of activated nanosensor signal by approximately 2.8- and 6.6-fold, respectively, over the nontargeted control. This increase in signal generation was observed despite the moderately decreased cleavage kinetics of HApep-modified TBI-ABNs measured in vitro with human calpain-1 enzyme (Figure 3B). Furthermore, the Cy5 signal from activated nanosensor was greater than vehicle control and consistent across triplicate mice within each group (Figure S5). These results demonstrate that targeting TBI-ABN to the ECM increases activation within the injured brain in a valency-dependent manner. Although the high-targeting TBI-ABNs resulted in the highest overall signal in the brain, we also observed a concomitant increase in sensor activation in off-target organs compared to moderate- and nontargeted TBI-ABNs (Figure S6), suggesting that a moderate level of targeting may be desirable to achieve higher specificity of brain-to-off-target organ signal generation.
Targeting Hyaluronic Acid Enables Widespread TBI-ABN Activation in Injured Brain Tissue
Having established that HApep modification of TBI-ABNs leads to an increase in bulk nanosensor activation in homogenized tissue lysate, we then set out to establish spatial activation of TBI-ABN in the injured brain tissue. The same experimental paradigm was repeated in triplicate, and coronal brain sections were imaged for activated nanosensor via dequenched Cy5 signal and nanosensor localization via the FAM label on HApep. We observed distinct nanosensor activation in all three HApep modification levels (non-, moderate-, and high-targeting) compared to the vehicle control (Figure 4), consistent with previous work.10 In mice administered TBI-ABN with moderate or high HApep valency, there was an observable increase in both nanosensor distribution within the injured cortex and nanosensor activation. The nanosensor tissue distribution resembled that of HApep nanomaterial (Figure 2) with accumulation in both the perilesional cortex and hippocampus, indicating the FRET substrate did not have a large impact on distribution, as expected with the low stoichiometry modification (Figure 4). The pattern of nanosensor activation in the perilesional cortex and hippocampus were consistent across triplicate brains within each group, although one brain with high-targeting had significantly increased signal generation compared to replicates in the same group (Figure S7). We note that the pattern of nanosensor distribution was distinct from the pattern of nanosensor activation, indicating some level of specificity of nanosensor activation. Overall, the brain sections reflected quantitative measurements made in bulk homogenates (Figure 3) and further supported our conclusion that incorporation of HA-targeting ligands increased nanosensor activation in the brain compared to nontargeted control. Since the high-targeting TBI-ABN yielded the maximal fold increase of nanosensor activation in the brain, subsequent studies were performed with this nanomaterial.
Figure 4.

Hyaluronic acid peptide targeting increases overall TBI-ABN activation and distribution within coronal sections of the injured hemisphere. Injured hemispheres from CCI-injured mice (representative brains from triplicate) after intravenous administration of vehicle or nanosensors with non-, moderate-, or high-targeting modification (blue, nuclei; magenta, activated nanosensor; green, HApep on nanosensor; scale bar = 500 μm).
Next, we investigated the distribution of activated TBI-ABN within the brain’s cellular milieu because understanding the cellular localization of proteolytic activity could offer insight into TBI pathophysiology. Neurons are a major source of calpain-1 in the brain,56,57 and we have established that HA-targeting nanomaterial predominantly accumulates within cortical and hippocampal neurons proximal to the injury (Figure 2). Furthermore, endothelial cells can also contribute to abnormal calpain activation,65,66 and we expected nanosensor to have access to endothelial cells due to the vascular administration route.10 We therefore investigated the activation of HA-targeting TBI-ABN in the context of neuronal and endothelial cell staining (Figure 5). In the perilesional cortex (Figure 5A), activated nanosensor signal colocalized with NeuN+ neurons and CD31+ endothelial cells. In the hippocampus (Figure 5B), activated nanosensor colocalized mainly with neurons, consistent with HA-targeting nanomaterial accumulation. Importantly, activation of nanosensor was distinct from the presence of nanosensor imaged by the FAM label on HApep in both NeuN+ neurons and CD31+ endothelial cells. Interestingly, the pattern of TBI-ABN activation in perilesional hippocampal and cortical neurons that we observed in our study was in accordance with the pattern of neurodegeneration observed in previous studies that stained for degenerating neurons up to ∼1 week after injury.53,67
Figure 5.

HA-targeting nanosensor activates within neuronal and endothelial cells in the perilesional cortex and hippocampus. (A) Perilesional cortex and (B) hippocampus imaged for nanosensor distribution and activated nanosensor (box in schematic indicates imaging location). Immunostaining was performed for HApep (green, FAM), neurons (yellow, NeuN), and endothelial cells (cyan, CD31) (blue, nuclei; magenta, activated nanosensor; scale bar = 200 μm for larger images and 25 μm for insets). Colocalization of nanosensor activation with neurons or endothelial cells is denoted with arrows or stars, respectively.
The robust activation of HA-targeting TBI-ABN enabled the capture of macroscopic 3D spatial heterogeneity of protease activation across the TBI lesion in intact brains. We performed whole brain tissue clearing after TBI-ABN administration in CCI-injured mice with Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis (CUBIC),68 which enabled removal of light-scattering components while matching the brain refraction index to the imaging media (Figures 6A and S8). Cleared brains were imaged with light sheet fluorescence microscopy (LSFM) for TBI-ABN localization and activation (Figure 6B,C). The imaged volume was a 9.7 × 4.5 × 3.3 mm3 rectangular prism that encompassed the injured and contralateral cortices. A 3D rendering was generated of both TBI-ABN localization and activation based on the fluorescence of FAM on HApep and Cy5 on the dequenched FRET substrate, respectively (Figure 6B, Video S1). Robust nanosensor activation was observed in the perilesional cortex in all directions from the injury lesion, approximately 2 mm away from the injury epicenter, and in the hippocampal formation directly below the injury. A cylindrical pattern of nanosensor activation around the impact epicenter was observed, as demonstrated by horizontal cross sections at three optical sectioning depths (Figure S9). Furthermore, cross-sectional views of the injury (Figure 6C), generated using sagittal, horizontal, and coronal clipping planes (Figure S10), demonstrated that robust sensor activation was confined to the area proximal to the injury with minimal nanosensor activation detected in the contralateral hemisphere. Our 3D imaging corroborated the injury-specific activation observed in 2D imaging from brain sections (Figures 4 and 5). Overall, volumetric optical sectioning of cleared brains enabled spatial visualization of activated nanosensor, showing the extent of calpain-1 protease activity in relation to the injury.
Figure 6.

Light sheet fluorescence microscopy (LSFM) of cleared tissues enables 3D reconstruction of nanosensor activation within the injured brain. (A) Image of injured brain before and after whole brain CUBIC clearing. (B) 3D rendered view. (C) Sagittal, horizontal, and coronal cross sections (magenta, activated nanosensor; green, HApep on nanosensor; scale bar = 1 mm; Contra. = contralateral hemisphere). See Video S1 and Figure S10 for the clipping planes used to generate the cross sections.
Conclusions
Aberrant protease activity is linked to a worsening of TBI pathophysiology and prognosis5−7 and tools to measure protease activity in the injured brain would allow for greater understanding of TBI progression. In this manuscript, the addition of HA-targeting peptides to a previously developed activity-based nanosensor for TBI10 led to a robust signal that enabled visualization of protease activity in an intact brain. The HA-targeting peptide,42 which is reported to have a dissociation constant between ∼0.1–10 μM depending on HA length,42,43 was selected following an in vivo screen of ECM-binding peptides. We observed that the addition of HA-targeting peptide increased TBI-ABN activation in a valency-dependent manner, consistent with observations that avidity strengthens with increasing peptide valency.64 Beyond nanosensors, our results provide impetus to apply ECM targeting as a generalizable strategy to improve nanomaterial distribution and function within the injured brain after systemic administration.
Activity-based nanosensors are useful tools to probe protease activity in various disease states and can thus serve as imaging tools or biomarkers for diagnosis and prognosis.8,9 We have shown that active targeting to brain ECM is a viable method to improve bioavailability of our nanosensor in the brain post-TBI and thus increase both protease-dependent signal generation and sensitivity. Beyond improvements in sensitivity, the TBI-ABN design can incorporate the measurement of multiple proteases to further specify TBI-associated protease activity. Multiplexed detection of several disease-specific proteases has enhanced diagnostic specificity of urinary nanoreporters for lung69 and prostate cancer.70 In TBI, MMP-9 is a possible candidate for multiplexing as MMP-9 levels are elevated in patients post-TBI, and MMP-9 is implicated in BBB permeability.71 Tethering substrates for multiple proteases onto a single nanomaterial scaffold and visualization in 3D would allow for the study of spatial activation of proteases in relation to each other; for example, calpain-1 has been shown to activate MMP-9 in TBI72 and stroke.73 In the future, the TBI-ABN platform could be redesigned for in vivo measurements with quenchable quantum dots74 and tissue-penetrating near-infrared optical imaging through cranial windows, or with superparamagnetic iron oxide nanoparticles75 and magnetic resonance imaging (MRI). Furthermore, HA-targeting has the potential to be applied to a greater range of nanomaterials, including therapeutic nanomaterials. Our observation that HA-targeting nanomaterial colocalized with hippocampal and perilesional neurons provides motivation for its use in the delivery of neuroprotective agents, since neuronal apoptosis is a hallmark of secondary injury and hippocampal neurodegeneration is associated with memory deficits after TBI.76
Methods
Synthesis of ECM-Targeted and TBI-ABN Poly(ethylene glycol) (PEG) Conjugates
The following targeting peptides were synthesized by Lifetein (Somerset, NJ): PGpep (X(FAM)-CAQK), CIVpep (KLWVLPKGG-K(FAM)-GGC), FIBpep (X(FAM)-CREKA), HApep (X(FAM)-CSTMMSRSHKTRSHHV), and RGD (X(FAM)-CRGD). Calpain substrate FRET peptide (QSY21-QEVYGAMP-K(Cy5)-PEG2-GC-NH2) was synthesized by CPC Scientific, Inc. (Sunnyvale, CA). X stands for 6-aminocaproic acid, and PEG2 stands for poly(ethylene glycol). The 40 kDa 8-arm PEG maleimide (tripentaerythritol) was purchased from Jenkem Technology (Beijing, China). For ECM-targeting peptide screening, PEG maleimide was batch-reacted with 1 mol of l-cysteine-functionalized VivoTag S-750 (PerkinElmer, Boston, MA). The reaction was then split to ensure matched VivoTag 750 modification for each conjugate and reacted with 8.4 mol equiv of each ECM-targeting peptide followed by quenching with an excess of l-cysteine. For HA-targeting TBI-ABNs: The 8-arm PEG maleimide was batch reacted with 1 mol equiv of calpain substrate FRET peptide, split, and reacted with 0 mol equiv (nontargeting), 4 mol equiv (moderate-targeting), or 8.4 mol equiv (high-targeting) of HApep in the presence of 50 mM triethylamine (TEA), then quenched with an excess of l-cysteine. All conjugates were dialyzed with water, and final concentrations were determined by absorbance of VivoTag 750, FAM, or Cy5 using a Spark multimode microplate reader (Tecan Trading AG, Switzerland).
Controlled Cortical Impact (CCI) TBI Mouse Model
All mouse procedures were approved by the University of California San Diego’s Institutional Animal Care and Use Committee (IACUC). A total of 61 female C57BL/6J mice (8–13 weeks old, Jackson Laboratories) weighing between 18 and 22 g were used for all in vivo studies. Following anesthetization with 2.5% isoflurane, buprenorphine analgesia was administered. A 5 mm craniotomy was performed over the right hemisphere between bregma and lambda, and controlled cortical impact was performed using the ImpactOne (Leica Biosystems) with a 2 mm diameter stainless-steel probe at a velocity of 3 m/s, depth of 2 mm, and dwell time of 300 ms. The center of the injury impact was centered around −2.0 mm (±0.5 mm) lateral from the midline and −2.0 mm (±0.5 mm) caudal from bregma. Group sizes were n = 3 for each experiment unless noted, based on a type I error rate of 5% and a power of 80% with an anticipated effect size greater than 2 with a 50% variance.
Extracellular Matrix (ECM) Peptide In Vivo Screen
Six hours after CCI, 25 nmol of ECM-targeting peptide-PEG nanomaterial (quantified via FAM absorbance) in 150 μL of 5% dextrose was intravenously administered via the tail vein. Nanomaterial doses were administered to mice in a weight range of 18–22 g, corresponding to a dosage range of 1136–1389 nmol/kg. Control cysteine mice received a matching dose as quantified by VivoTag 750 absorbance. Triplicate mice were obtained per ECM-targeting peptide. One hour after injection, mice were transcardially perfused with USP saline followed by 10% formalin. Surface fluorescence of organs (brain, heart, lung, liver, spleen, and kidneys) was measured with an Odyssey scanner (Li-Cor Biosciences) within 6 h of collection. For generation of tissue for quantitative biodistribution of homogenized tissue, triplicate mice were perfused with 10 mL of ice-cold PBS. For quantification of nanomaterial blood half-life, 5–10 μL of blood was collected into heparinized tubes (n = 3). VivoTag 750 signal was detected on a Li-Cor scanner, and the %ID of nanomaterial in the blood was calculated based on a standard of known concentrations and estimated blood volume.
HA-Targeted TBI-ABN In Vivo Study
Three hours after CCI, 6 nmol (concentration based on substrate peptide) of non-, moderate-, and high-targeting TBI-ABN in 150 μL of 5% dextrose was injected intravenously via the tail vein. Nanomaterial doses were administered to mice with a weight range of 18–22 g, corresponding to a dosage range of 273–333 nmol/kg. Vehicle control received equivolume of 5% dextrose. Following a 1 h circulation time, mice were sacrificed by transcardial perfusion of either USP saline followed by 10% formalin (for histology) or 10 mL of ice-cold PBS (for whole-organ homogenization). Each condition was repeated in triplicate for histology. The study was independently repeated again in triplicate for homogenization tissue generation. A third independent study was performed for CUBIC tissue clearing in one mouse.
Quantitative Biodistribution of Homogenized Tissue
Organ tissue was flash frozen at −80 °C and minced, and lysis buffer (6% w/v sodium dodecyl sulfate (SDS), 150 mM Tris-HCl pH 7.4, 100 mM dithiothreitol (DTT), and 2 mM ethylenediaminetetraacetic acid (EDTA)) was added to achieve a concentration of 250 mg tissue/mL. Tissue was further processed with a Tissue-Tearor with 4.5 mm probe (Fisher) at medium-high speed for 20–30 s until lysate was homogenized. Samples were heated at 90 °C for 10 min with agitation at 800 rpm and vortexed to mix, and the homogenate was analyzed for FAM fluorescence (ECM-targeting peptide screen) or Cy5 fluorescence (TBI-ABN activation). The percent injected dose per gram of tissue (% ID/g) was calculated based on a known nanomaterial concentration standard.
Immunostaining of Brain Tissue Slices
Following transcardial perfusion with 10% formalin, necropsied organs were further fixed in 10% formalin at 4 °C overnight. Organs were washed in PBS, transferred to 30% w/v sucrose–PBS overnight, then frozen in OCT (Tissue-Tek). Coronal tissue slices (10 μm thick) were obtained within the 2 mm diameter injury region, then stained using conventional protocols. See Table S1 for a list of antibodies and staining reagents. Briefly, tissues were blocked for 1 h in 2% bovine serum albumin, 5% serum of secondary antibody, and 0.1% Triton X-100. For NeuN staining, blocking buffer included 2 μg/mL donkey anti-mouse Fab. Primary antibody incubations were done in blocking buffer overnight at 4 °C. Secondary antibodies were applied for 1 h at room temperature, and the sample was washed in PBS and mounted with Fluoromount-G (Southern Biotech). To probe for endogenous hyaluronic acid, tissues were blocked for 1 h, incubated overnight with bHABP, incubated for 1 h with streptavidin AF647 conjugate, washed, and mounted. Images were collected on a Nikon Eclipse Ti2 microscope fitted with a Hamamatsu Orca-Flash 4.0 digital camera. Activated TBI-ABN was visualized with imaging settings for Cy5. Images for direct comparison were collected using the same exposure and LED intensity settings.
In Vitro Reaction Kinetics Assay
The calpain-1 reaction kinetics assay was run as previously described.10 Briefly, conjugates were incubated with 26.6 nM human calpain-1 (Sigma-Aldrich, C6108) in 50 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 50 mM NaCl, 2 mM EDTA, 5 mM CaCl2, and 5 mM β-mercaptoethanol. Fluorescence readings were taken every 90 s at 37 °C for 1 h. Reaction curves were normalized to controls, and their initial velocities were fitted to a Michaelis–Menten curve in GraphPad Prism (9.2.0).
CUBIC Tissue Clearing
Brains were cleared following a published protocol (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational Analysis, or CUBIC).68 Following transcardial perfusion with 10% formalin, the simple immersion protocol for dissected whole brain tissue was followed without modifications. Tissues were kept static in fresh reagent-2 at room temperature between clearing and imaging, with no longer than 1–1.5 weeks between the last day of clearing and imaging.
3D Light Sheet Fluorescence Microscopy (LSFM)
LSFM was accomplished using a Zeiss Lightsheet Z.1. The microscope was fitted with a 2.5× objective, solid-state lasers (excitation wavelengths 488 nm for FAM and 640 nm for sensor), and a PCO.edge 16 bit sCMOS camera for detection. The cleared brain was embedded in low-melt 2% agarose (ThermoFisher) then submerged in CUBIC reagent-2 overnight. The next day, magnetic staples were superglued to the agarose-embedded tissue, and the sample was hung vertically in front of the LSFM objective (2.5×) using a custom magnetic fixture. The brain was slowly lowered into the imaging chamber filled with reagent-2 for at least 20 min prior to image collection. A z-stack (range of 3.258 mm with a 3 μm step size) was obtained. The 3D area, encompassing the injured and contralateral hemispheres, was approximately 9.7 mm (x-axis) by 4.5 mm (y-axis) by 3.3 mm (z-axis). 3D images, clipping planes, and videos were generated in Arivis Vision4D.
Software and Statistics
GraphPad Prism (9.2.0) was used to perform statistics. All images were processed in ImageJ (1.53g).
Acknowledgments
R.M.K. acknowledges support from the NHLBI training program Integrative Bioengineering of Heart Vessels Blood (T32 HL105373). J.A.K. acknowledges support from the National Science Foundation (NSF) Graduate Research Fellowship Program under Grant No. DGE-1650112. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF. We also thank the University of California–San Diego School of Medicine microscopy core for use of their light sheet microscopy imaging facilities, supported under NINDS P30NS047101.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c09064.
Immunostaining reagents, concentration measurements for calpain substrate peptide, blood half-life of ECM-targeting nanomaterials, triplicate histology of ECM-targeting nanomaterials, representative brains with alternative anti-FAM antibody, maximal cleavage velocities of TBI-ABNs, raw signal from TBI-ABN activation in brain lysates for each mouse in the triplicate, TBI-ABN fold-change in activation for off-target organs, triplicate histology of nanosensor activation, CUBIC clearing progress for brains, LSFM horizontal cross sections across imaging depths, 3D clipping planes (PDF)
Video S1: 3D nanosensor localization and activation in the CUBIC-cleared injured brain (MP4)
Author Contributions
† R.M.K. and J.A.K. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Institutes of Health Director’s New Innovator Award (1DP2NS111507) and a National Science Foundation (NSF) CAREER Award (CBET 2046926).
The authors declare no competing financial interest.
Supplementary Material
References
- Peterson A. B.; Zhou H.; Thomas K. E.; Daugherty J.. Surveillance Report of Traumatic Brain Injury-Related Hospitalizations and Deaths by Age Group, Sex, and Mechanism of Injury. United States, 2016/2017. Centers for Disease Control and Prevention, 2021. https://www.cdc.gov/traumaticbraininjury/pdf/TBI-surveillance-report-2016-2017-508.pdf.
- Maas A. I. R.; Menon D. K.; Adelson P. D.; Andelic N.; Bell M. J.; Belli A.; Bragge P.; Brazinova A.; Büki A.; Chesnut R. M.; et al. Traumatic Brain Injury : Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol. 2017, 16, 987. 10.1016/S1474-4422(17)30371-X. [DOI] [PubMed] [Google Scholar]
- Kandell R. M.; Waggoner L. E.; Kwon E. J. Nanomedicine for Acute Brain Injuries: Insight from Decades of Cancer Nanomedicine. Mol. Pharmaceutics 2021, 18 (2), 522–538. 10.1021/acs.molpharmaceut.0c00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Larner S. F.; Liu M. C.; Zheng W.; Hayes R. L.; Wang K. K. W. Multiple AlphaII-Spectrin Breakdown Products Distinguish Calpain and Caspase Dominated Necrotic and Apoptotic Cell Death Pathways. Apoptosis 2009, 14 (11), 1289–1298. 10.1007/s10495-009-0405-z. [DOI] [PubMed] [Google Scholar]
- Gan Z. S.; Stein S. C.; Swanson R.; Guan S.; Garcia L.; Mehta D.; Smith D. H. Blood Biomarkers for Traumatic Brain Injury: A Quantitative Assessment of Diagnostic and Prognostic Accuracy. Front. Neurol. 2019, 10, 1–14. 10.3389/fneur.2019.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R.; Shahim P.; Tegner Y.; Blennow K.; Zetterberg H.; Smith D. H. Serum SNTF Increases in Concussed Professional Ice Hockey Players and Relates to the Severity of Postconcussion Symptoms. J. Neurotrauma 2015, 32 (17), 1294–1300. 10.1089/neu.2014.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R.; Giovannone N.; Hanten G.; Wilde E. A.; McCauley S. R.; Hunter J. V.; Li X.; Levin H. S.; Smith D. H. Evidence That the Blood Biomarker SNTF Predicts Brain Imaging Changes and Persistent Cognitive Dysfunction in Mild TBI Patients. Front. Neurol. 2013, 4, 190. 10.3389/fneur.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudani J. S.; Warren A. D.; Bhatia S. N. Harnessing Protease Activity to Improve Cancer Care. Annu. Rev. Cancer Biol. 2018, 2, 353–376. 10.1146/annurev-cancerbio-030617-050549. [DOI] [Google Scholar]
- Sanman L. E.; Bogyo M. Activity-Based Profiling of Proteases. Annu. Rev. Biochem. 2014, 83, 249–273. 10.1146/annurev-biochem-060713-035352. [DOI] [PubMed] [Google Scholar]
- Kudryashev J. A.; Waggoner L. E.; Leng H. T.; Mininni N. H.; Kwon E. J. An Activity-Based Nanosensor for Traumatic Brain Injury. ACS Sensors 2020, 5, 686–692. 10.1021/acssensors.9b01812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj V. N.; Lifshitz J.; Adelson P. D.; Kodibagkar V. D.; Stabenfeldt S. E. Temporal Assessment of Nanoparticle Accumulation after Experimental Brain Injury : Effect of Particle Size. Sci. Rep. 2016, 6, 1–12. 10.1038/srep29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd B. J.; Galle A.; Daglas M.; Rosenfeld J. V.; Medcalf R. Traumatic Brain Injury Opens Blood-Brain Barrier to Stealth Liposomes via an Enhanced Permeability and Retention (EPR)-like Effect. J. Drug Target. 2015, 23 (9), 847–853. 10.3109/1061186X.2015.1034280. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46 (8), 6387–6392. [PubMed] [Google Scholar]
- Fang J.; Nakamura H.; Maeda H. The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect. Adv. Drug Delivery Rev. 2011, 63 (3), 136–151. 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Prasad L. K.; O’Mary H.; Cui Z. Nanomedicine Delivers Promising Treatments for Rheumatoid Arthritis. Nanomedicine (London, U. K.) 2015, 10 (13), 2063–2074. 10.2217/nnm.15.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz L. D.; Kaufman D.; Keller M. W.; Kong Y. Time Course of Coronary Endothelial Healing after Injury Due to Ischemia and Reperfusion. Circulation 1994, 90 (5), 2439–2447. 10.1161/01.CIR.90.5.2439. [DOI] [PubMed] [Google Scholar]
- Prajnamitra R. P.; Chen H. C.; Lin C. J.; Chen L. L.; Hsieh P. C. H. Nanotechnology Approaches in Tackling Cardiovascular Diseases. Molecules 2019, 24 (10), 2017. 10.3390/molecules24102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E. J.; Skalak M.; Lo Bu R.; Bhatia S. N. Neuron-Targeted Nanoparticle for SiRNA Delivery to Traumatic Brain Injuries. ACS Nano 2016, 10, 7926–7933. 10.1021/acsnano.6b03858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne R. G.; Nicholson C. In Vivo Diffusion Analysis with Quantum Dots and Dextrans Predicts the Width of Brain Extracellular Space. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (14), 5567–5572. 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E. J.; Dudani J. S.; Bhatia S. N. Ultrasensitive Tumour-Penetrating Nanosensors of Protease Activity. Nat. Biomed. Eng. 2017, 1 (4), 0054 10.1038/s41551-017-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett D. W.; Su H.; Hildebrandt I. J.; Weber W. A.; Davis M. E. Impact of Tumor-Specific Targeting on the Biodistribution and Efficacy of SiRNA Nanoparticles Measured by Multimodality in Vivo Imaging. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (39), 15549–15554. 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C. H. J.; Alabi C. A.; Webster P.; Davis M. E. Mechanism of Active Targeting in Solid Tumors with Transferrin-Containing Gold Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (3), 1235–1240. 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. W.; Hong K.; Kirpotin D. B.; Colbern G.; Shalaby R.; Baselga J.; Shao Y.; Nielsen U. B.; Marks J. D.; Moore D.; et al. Anti-HER2 Immunoliposomes: Enhanced Efficacy Attributable to Targeted Delivery. Clin. Cancer Res. 2002, 8 (4), 1172–1181. [PubMed] [Google Scholar]
- Kirpotin D. B.; Drummond D. C.; Shao Y.; Shalaby M. R.; Hong K.; Nielsen U. B.; Marks J. D.; Benz C. C.; Park J. W. Antibody Targeting of Long-Circulating Lipidic Nanoparticles Does Not Increase Tumor Localization but Does Increase Internalization in Animal Models. Cancer Res. 2006, 66 (13), 6732–6740. 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- Joo J.; Kwon E. J.; Kang J.; Skalak M.; Anglin E. J.; Mann A. P.; Ruoslahti E.; Bhatia S. N.; Sailor M. J. Porous Silicon – Graphene Oxide Core-Shell Nanoparticles for Targeted Delivery of SiRNA to the Injured Brain. R. Soc. Chem. 2016, 1, 407–414. 10.1039/C6NH00082G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo J. H.; Kwon E. J.; Zhang A. Q.; Singhal P.; Bhatia S. N. Comparison of Modular PEG Incorporation Strategies for Stabilization of Peptide-SiRNA Nanocomplexes. Bioconjugate Chem. 2016, 27 (10), 2323–2331. 10.1021/acs.bioconjchem.6b00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K. Y.; Kwon E. J.; Lo J. H.; Bhatia S. N. Drug-Induced Amplification of Nanoparticle Targeting to Tumors. Nano Today 2014, 9 (5), 550–559. 10.1016/j.nantod.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz C.; Stewart K. M.; Weaver V. M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123 (24), 4195–4200. 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Shen S.; Liao Z.; Shi W.; Wang Y.; Zhao J.; Hu Y.; Yang J.; Chen J.; Mei H.; Hu Y.; et al. Targeting Fibronectins of Glioma Extracellular Matrix by CLT1 Peptide-Conjugated Nanoparticles. Biomaterials 2014, 35 (13), 4088–4098. 10.1016/j.biomaterials.2014.01.046. [DOI] [PubMed] [Google Scholar]
- Briquez P. S.; Hauert S.; de Titta A.; Gray L. T.; Alpar A. T.; Swartz M. A.; Hubbell J. A. Engineering Targeting Materials for Therapeutic Cancer Vaccines. Front. Bioeng. Biotechnol. 2020, 8, 19. 10.3389/fbioe.2020.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara J.; Ishihara A.; Sasaki K.; Lee S. S.-Y.; Williford J.-M.; Yasui M.; Abe H.; Potin L.; Hosseinchi P.; Fukunaga K.; et al. Targeted Antibody and Cytokine Cancer Immunotherapies through Collagen Binding. Sci. Transl. Med. 2019, 11 (487), eaau3259. 10.1126/scitranslmed.aau3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williford J. M.; Ishihara J.; Ishihara A.; Mansurov A.; Hosseinchi P.; Marchell T. M.; Potin L.; Swartz M. A.; Hubbell J. A. Recruitment of CD103+ Dendritic Cells via Tumor-Targeted Chemokine Delivery Enhances Efficacy of Checkpoint Inhibitor Immunotherapy. Sci. Adv. 2019, 5 (12), 0–16. 10.1126/sciadv.aay1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumata K.; Ishihara J.; Mansurov A.; Ishihara A.; Raczy M. M.; Yuba E.; Hubbell J. A. Targeting Inflammatory Sites through Collagen Affinity Enhances the Therapeutic Efficacy of Anti-Inflammatory Antibodies. Sci. Adv. 2019, 5, 1–9. 10.1126/sciadv.aay1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumata K.; Ishihara J.; Fukunaga K.; Ishihara A.; Yuba E.; Budina E.; Hubbell J. A. Conferring Extracellular Matrix Affinity Enhances Local Therapeutic Efficacy of Anti-TNF-α Antibody in a Murine Model of Rheumatoid Arthritis. Arthritis Res. Ther. 2019, 21 (1), 1–10. 10.1186/s13075-019-2075-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price L.; Wilson C.; Grant G.. Chapter 4: Blood–Brain Barrier Pathophysiology Following Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz D., Grant G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, 2016; pp 1–10. [PubMed] [Google Scholar]
- George N.; Geller H. Extracellular Matrix and Traumatic Brain Injury. J. Neurosci. Res. 2018, 96 (4), 573–588. 10.1002/jnr.24151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L. W.; Cua R.; Keough M. B.; Haylock-Jacobs S.; Yong V. W. Pathophysiology of the Brain Extracellular Matrix: A New Target for Remyelination. Nat. Rev. Neurosci. 2013, 14 (10), 722–729. 10.1038/nrn3550. [DOI] [PubMed] [Google Scholar]
- Xing G.; Ren M.; Verma A. Divergent Temporal Expression of Hyaluronan Metabolizing Enzymes and Receptors with Craniotomy vs. Controlled-Cortical Impact Injury in Rat Brain: A Pilot Study. Front. Neurol. 2014, 5 (SEP), 1–9. 10.3389/fneur.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann A. P.; Scodeller P.; Hussain S.; Joo J.; Kwon E.; Braun G. B.; Mölder T.; She Z.; Kotamraju V. R.; Ranscht B.; Krajewski S.; et al. A Peptide for Targeted, Systemic Delivery of Imaging and Therapeutic Compounds into Acute Brain Injuries. Nat. Commun. 2016, 7, 1–11. 10.1038/ncomms11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J. M.; Zhang L.; Tong R.; Ghosh D.; Gao W.; Liao G.; Yuet K. P.; Gray D.; Rhee J. W.; Cheng J. Spatiotemporal Controlled Delivery of Nanoparticles to Injured Vasculature. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (5), 2213–2218. 10.1073/pnas.0914585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simberg D.; Duza T.; Park J. H.; Essler M.; Pilch J.; Zhang L.; Derfus A. M.; Yang M.; Hoffman R. M.; Bhatia S.; et al. Biomimetic Amplification of Nanoparticle Homing to Tumors. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (3), 932–936. 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolg C.; Hamilton S. R.; Zalinska E.; McCulloch L.; Amin R.; Akentieva N.; Winnik F.; Savani R.; Bagli D. J.; Luyt L. G.; et al. A RHAMM Mimetic Peptide Blocks Hyaluronan Signaling and Reduces Inflammation and Fibrogenesis in Excisional Skin Wounds. Am. J. Pathol. 2012, 181 (4), 1250–1270. 10.1016/j.ajpath.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust H. J.; Sommerfeld S. D.; Rathod S.; Rittenbach A.; Ray Banerjee S.; Tsui B. M. W.; Pomper M.; Amzel M. L.; Singh A.; Elisseeff J. H. A Hyaluronic Acid Binding Peptide-Polymer System for Treating Osteoarthritis. Biomaterials 2018, 183, 93–101. 10.1016/j.biomaterials.2018.08.045. [DOI] [PubMed] [Google Scholar]
- Waggoner L. E.; Madias M. I.; Hurtado A. A.; Kwon E. J. Pharmacokinetic Analysis of Peptide-Modified Nanoparticles with Engineered Physicochemical Properties in a Mouse Model of Traumatic Brain Injury. AAPS J. 2021, 23 (100), 1–12. 10.1208/s12248-021-00626-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo Choi H.; Liu W.; Misra P.; Tanaka E.; Zimmer J. P; Itty Ipe B.; Bawendi M. G; Frangioni J. V Renal Clearance of Nanoparticles. Nat. Biotechnol. 2007, 25 (10), 1165–1170. 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance E. A.; Woodworth G. F.; Sailor K. A.; Shih T. Y.; Xu Q.; Swaminathan G.; Xiang D.; Eberhart C.; Hanes J. A Dense Poly(ethylene Glycol) Coating Improves Penetration of Large Polymeric Nanoparticles within Brain Tissue. Sci. Transl. Med. 2012, 4 (149), 149ra119 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk J. S.; Xu Q.; Kim N.; Hanes J.; Ensign L. PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Delivery Rev. 2016, 99, 28–51. 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher M. D.; Ruoslahti E. Cell Attachment Activity of Fibronectin Can Be Duplicated By Small Synthetic Fragments of the Molecule. Nature 1984, 309, 30–33. 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- Mezu-Ndubuisi O. J.; Maheshwari A. The Role of Integrins in Inflammation and Angiogenesis. Pediatr. Res. 2021, 89 (7), 1619–1626. 10.1038/s41390-020-01177-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abumiya T.; Lucero J.; Heo J. H.; Tagaya M.; Koziol J. A.; Copeland B. R.; Del Zoppo G. J. Activated Microvessels Express Vascular Endothelial Growth Factor and Integrin α(v)B3 during Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 1999, 19 (9), 1038–1050. 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Ward P. A. Inflammation and α vβ 3 Integrin. Am. J. Respir. Crit. Care Med. 2012, 185 (1), 5–6. 10.1164/rccm.201110-1859ED. [DOI] [PubMed] [Google Scholar]
- Xiong Y.; Mahmood A.; Chopp M. Animal Models of Traumatic Brain Injury. Nat. Rev. Neurosci. 2013, 14 (2), 128–142. 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall E. D.; Sullivan P. G.; Gibson T. R.; Pavel K. M.; Thompson B. M.; Scheff S. W. Spatial and Temporal Characteristics of Neurodegeneration after Controlled Cortical Impact in Mice: More Than a Focal Brain Injury. J. Neurotrauma 2005, 22 (2), 252–265. 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- Natarajan V.; Harris E. N.; Kidambi S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. BioMed Res. Int. 2017, 4097205. 10.1155/2017/4097205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault S. D.; Walkey C.; Jennings T.; Fischer H. C.; Chan W. C. W. Mediating Tumor Targeting Efficiency of Nanoparticles through Design. Nano Lett. 2009, 9 (5), 1909–1915. 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- Hamakubo T.; Kannagi R.; Murachi T.; Matus A. Distribution of Calpains I and II in Rat Brain. J. Neurosci. 1986, 6 (11), 3103–3111. 10.1523/JNEUROSCI.06-11-03103.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czogalla A.; Sikorski A. F. Spectrin and Calpain: A “Target” and a “Sniper” in the Pathology of Neuronal Cells. Cell. Mol. Life Sci. 2005, 62 (17), 1913–1924. 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Mahmood A.; Chopp M. Emerging Treatments for Traumatic Brain Injury. Expert Opin. Emerging Drugs 2009, 14 (1), 67–84. 10.1517/14728210902769601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascall V. C.; Heinegård D. Aggregation of Cartilage Proteoglycans III. Characteristics of the Proteins Isolated from Trypsin Digests of Aggregates. J. Biol. Chem. 1974, 249 (13), 4232–4241. 10.1016/S0021-9258(19)42507-6. [DOI] [PubMed] [Google Scholar]
- Ueno H.; Suemitsu S.; Murakami S.; Kitamura N.; Wani K.; Matsumoto Y.; Aoki S.; Okamoto M.; Ishihara T. Hyaluronic Acid Is Present on Specific Perineuronal Nets in the Mouse Cerebral Cortex. Brain Res. 2018, 1698, 139–150. 10.1016/j.brainres.2018.08.011. [DOI] [PubMed] [Google Scholar]
- Arranz A. M.; Perkins K. L.; Irie F.; Lewis D. P.; Hrabe J.; Xiao F.; Itano N.; Kimata K.; Hrabetova S.; Yamaguchi Y. Hyaluronan Deficiency Due to Has3 Knock-Out Causes Altered Neuronal Activity and Seizures via Reduction in Brain Extracellular Space. J. Neurosci. 2014, 34 (18), 6164–6176. 10.1523/JNEUROSCI.3458-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochlamazashvili G.; Henneberger C.; Bukalo O.; Dvoretskova E.; Senkov O.; Lievens P. M.-J.; Westenbroek R.; Engel A. K.; Catterall W. A.; Rusakov D. A.; et al. The Extracellular Matrix Molecule Hyaluronic Acid Regulates Hippocampal Synaptic Plasticity by Modulating Postsynaptic L- Type Ca 2 + Channels. Neuron 2010, 67 (1), 116–128. 10.1016/j.neuron.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockholm D.; Bartoli M.; Sillon G.; Bourg N.; Davoust J.; Richard I. Imaging Calpain Protease Activity by Multiphoton FRET in Living Mice. J. Mol. Biol. 2005, 346 (1), 215–222. 10.1016/j.jmb.2004.11.039. [DOI] [PubMed] [Google Scholar]
- Jeong W.-j.; Bu J.; Kubiatowicz L. J.; Chen S. S.; Kim Y. S.; Hong S. Peptide–Nanoparticle Conjugates: A Next Generation of Diagnostic and Therapeutic Platforms?. Nano Converg. 2018, 5 (1), 1–18. 10.1186/s40580-018-0170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindis C.; Elbaz M.; Escargueil-Blanc I.; Augé N.; Heniquez A.; Thiers J. C.; Nègre-Salvayre A.; Salvayre R. Two Distinct Calcium-Dependent Mitochondrial Pathways Are Involved in Oxidized LDL-Induced Apoptosis. Arterioscler., Thromb., Vasc. Biol. 2005, 25 (3), 639–645. 10.1161/01.ATV.0000154359.60886.33. [DOI] [PubMed] [Google Scholar]
- Smith M. A.; Schnellmann R. G. Calpains, Mitochondria, and Apoptosis. Cardiovasc. Res. 2012, 96 (1), 32–37. 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. C.; Mao H.; Yang K. H.; Abel T.; Meaney D. F. A Modified Controlled Cortical Impact Technique to Model Mild Traumatic Brain Injury Mechanics in Mice. Front. Neurol. 2014, 5, 100. 10.3389/fneur.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susaki E. A.; Tainaka K.; Perrin D.; Yukinaga H.; Kuno A.; Ueda H. R. Advanced CUBIC Protocols for Whole-Brain and Whole-Body Clearing and Imaging. Nat. Protoc. 2015, 10 (11), 1709–1727. 10.1038/nprot.2015.085. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick J. D.; Warren A. D.; Soleimany A. P.; Westcott P. M. K.; Voog J. C.; Martin-Alonso C.; Fleming H. E.; Tammela T.; Jacks T.; Bhatia S. N. Urinary Detection of Lung Cancer in Mice via Noninvasive Pulmonary Protease Profiling. Sci. Transl. Med. 2020, 12, 1–10. 10.1126/scitranslmed.aaw0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudani J. S.; Ibrahim M.; Kirkpatrick J.; Warren A. D.; Bhatia S. N. Classification of Prostate Cancer Using a Protease Activity Nanosensor Library. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (36), 8954–8959. 10.1073/pnas.1805337115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilfoyle M. R.; Carpenter K. L. H.; Helmy A.; Pickard J. D.; Menon D. K.; Hutchinson P. J. A. Matrix Metalloproteinase Expression in Contusional Traumatic Brain Injury: A Paired Microdialysis Study. J. Neurotrauma 2015, 32 (20), 1553–1559. 10.1089/neu.2014.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp R. C.; Jastaniah A.; Dubrovskyi O.; Gaisina I.; Tai L.; Thatcher G. R. J. Extending the Calpain-Cathepsin Hypothesis to the Neurovasculature: Protection of Brain Endothelial Cells and Mice from Neurotrauma. ACS Pharmacol. Transl. Sci. 2021, 4 (1), 372–385. 10.1021/acsptsci.0c00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubokawa T.; Solaroglu I.; Yatsushige H.; Cahill J.; Yata K.; Zhang J. H. Cathepsin and Calpain Inhibitor E64d Attenuates Matrix Metalloproteinase-9 Activity after Focal Cerebral Ischemia in Rats. Stroke 2006, 37 (7), 1888–1894. 10.1161/01.STR.0000227259.15506.24. [DOI] [PubMed] [Google Scholar]
- Samanta A.; Zhou Y.; Zou S.; Yan H.; Liu Y. Fluorescence Quenching of Quantum Dots by Gold Nanoparticles: A Potential Long Range Spectroscopic Ruler. Nano Lett. 2014, 14 (9), 5052–5057. 10.1021/nl501709s. [DOI] [PubMed] [Google Scholar]
- Shin T. H.; Kang S.; Park S.; Choi J.-s.; Kim P. K.; Cheon J. A Magnetic Resonance Tuning Sensor for the MRI Detection of Biological Targets. Nat. Protoc. 2018, 13 (11), 2664–2684. 10.1038/s41596-018-0057-y. [DOI] [PubMed] [Google Scholar]
- Girgis F.; Pace J.; Sweet J.; Miller J. P. Hippocampal Neurophysiologic Changes after Mild Traumatic Brain Injury and Potential Neuromodulation Treatment Approaches. Front. Syst. Neurosci. 2016, 10 (FEB), 1–10. 10.3389/fnsys.2016.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.