

Abstract
Philadelphia chromosome-like acute lymphoblastic leukemia (Ph-like ALL) is a high-risk subtype of B-ALL often associated with genetic variants that alter cytokine receptor signaling, including mutations in the interleukin-7 receptor (IL7R). To investigate whether IL7R variants are leukemia-initiating, we built mouse models expressing activated Il7r (aIL7R). B-cell intrinsic aIL7R mice developed spontaneous B-ALL, demonstrating sufficiency of Il7r activating mutations in leukemogenesis. Concomitant introduction of a knock-out allele in the associated adapter protein Lnk (encoded by Sh2b3) or a dominant-negative variant of the transcription factor Ikaros (Ikzf1) increased disease penetrance. The resulting murine leukemias displayed monoclonality and recurrent somatic Kras mutations and efficiently engrafted into immunocompetent mice. Phosphoproteomic analyses of aIL7R leukemic cells revealed constitutive Stat5 signaling and B cell receptor (BCR)-like signaling despite the absence of surface pre-BCR. Finally, in vitro treatment of aIL7R leukemic B-cells with Jak, mTOR, or Syk inhibitors blocked growth, confirming that each pathway is active in this mouse model of IL7R-driven B-ALL.
INTRODUCTION
Philadelphia chromosome-like acute lymphoblastic leukemia (Ph-like ALL) comprises 15–30% of National Cancer Institute high-risk B-ALL occurring in children, adolescents, and adults [1]. Although Ph-like ALLs share a similar transcriptional profile to Philadelphia chromosome-positive ALL [2], these leukemias exhibit diverse mutations predicted to augment cytokine signaling, promote kinase activation, and limit cell differentiation [1–5]. Activating insertion/deletion (indel) mutations in the IL7R gene have been identified in approximately 12% of Ph-like ALL cases, as well as in 9% of T-ALL [3, 6, 7]. A common IL7R mutation introduces a cysteine residue in the transmembrane domain that promotes receptor homodimerization and cytokine-independent activation [7]. IL-7R plays a critical role in early B progenitor growth and differentiation, where it signals via the JAK-STAT and PI3-kinase (PI3K) pathways to promote cell proliferation and survival prior to BCR rearrangement [8–10]. IL7R mutations are presumed to increase STAT5 signaling, leading to dysregulation of B cell development through inhibition of the pre-BCR pathway and thereby potentially promoting leukemic transformation [6–8, 11–14]. However, this concept has not been definitively tested, and whether malignant B-ALL cells mimic canonical B cell development signals or engage a unique oncogenic signaling program remains unknown.
IL7R activating mutations co-occur in B-ALL with loss-of-function mutations in the negative signaling regulator SH2B3 (encoding LNK) or alterations in the transcription factor IKZF1 (encoding IKAROS) at rates of ~55% or 32%, respectively [3]. B lymphopoiesis is altered in Sh2b3−/− mice, with pro-B cell overproduction due to enhanced sensitivity to IL-7, resulting in increased Stat5 activation [15, 16]. By contrast, IKAROS controls multiple stages of B lymphopoiesis. Introduction of germline homozygous null or DNA-binding domain Ikzf1 mutations results in a complete block in B lineage development [17–19]. In contrast, the introduction of these homozygous changes post B-lineage specification results in a block in the transition from the highly proliferative large pre-B cell stage to the quiescent small pre-B cell stage [20, 21]. Importantly, STAT5 antagonizes IKAROS activity in murine pre-B cells and primary B-ALLs by reciprocally regulating many of the same target genes, including IL7R [22, 23]. Collectively, these data suggest that mutations in IL7R may collaborate with those in SH2B3 and IKZF1 to alter B cell development in ways that drive leukemic transformation.
Here, we present a genetically engineered mouse model (GEMM) of B-ALL that is driven by B-cell intrinsic expression of an activating mutation in Il7r. This model reproduces many of the hallmarks of Ph-like B-ALL, including monoclonal growth, B-precursor immunophenotype, de novo co-mutation of common tumor suppressors and oncogenes, and activation of canonical cytokine and BCR-like signaling. We carefully characterized the biology, leukemic transition, and drug responses in this aIL7R GEMM and propose that it will be broadly useful in future studies to characterize factors regulating Ph-like ALL onset and potential treatments.
RESULTS
B-intrinsic expression of an insertion mutation in Il7r drives B cell precursor ALL
Common IL7R mutations in B-ALL include cysteine insertions within the transmembrane domain [7]. Expression of murine Il7r containing the B-ALL-associated activating L243insPPCL (Il7r p. P243insPPCL) mutation, hereafter referred to as activated “aIL7R”, in Ba/F3 cells permitted IL-3-independent growth and constitutive Stat5 phosphorylation (Supplementary Fig. 1). To generate knock-in mice, we introduced aIL7R with a cis-linked GFP reporter downstream of the murine Rosa26 promoter and a floxed stop cassette (Fig. 1a). Rosa26-aIL7R+/wt mice were crossed to Mb1-cre or CD19-cre strains (referred to as “aIL7R-Mb1” or “aIL7R-CD19,” respectively) to turn on one copy of the mutant allele at the bone marrow (BM) pro-B stage (B220+ surface (s)μHC− IgD− CD24hi BP-1−; Supplementary Fig. 2a). In aIL7R-Mb1 mice, pro-B and all subsequent developmental BM and splenic B cell populations expressed GFP. Mature B cells (B220+ sμHC+ IgD+) maintained surface IL-7R expression and co-expressed GFP, whereas IL-7R expression was absent in mature B cells from controls, demonstrating aIL7R is expressed on the cell surface (Fig. 1b, c). Total surface IL-7R in aIL7R-mb1 late pro-B cells (B220+ sμHC− IgD− CD24hi BP-1+) was not significantly changed relative to controls, while median IL-7R fluorescence in aIL7R-Mb1 mature B cells was approximately one-fifth of that in Mb1-cre late pro-B cells, suggesting that surface aIL7R expression is roughly 20% of endogenous IL-7R (Fig. 1b, c).
Fig. 1. B-intrinsic aIL7R and heterozygous loss of Sh2b3 or Ikaros rapidly promote B cell precursor leukemias.
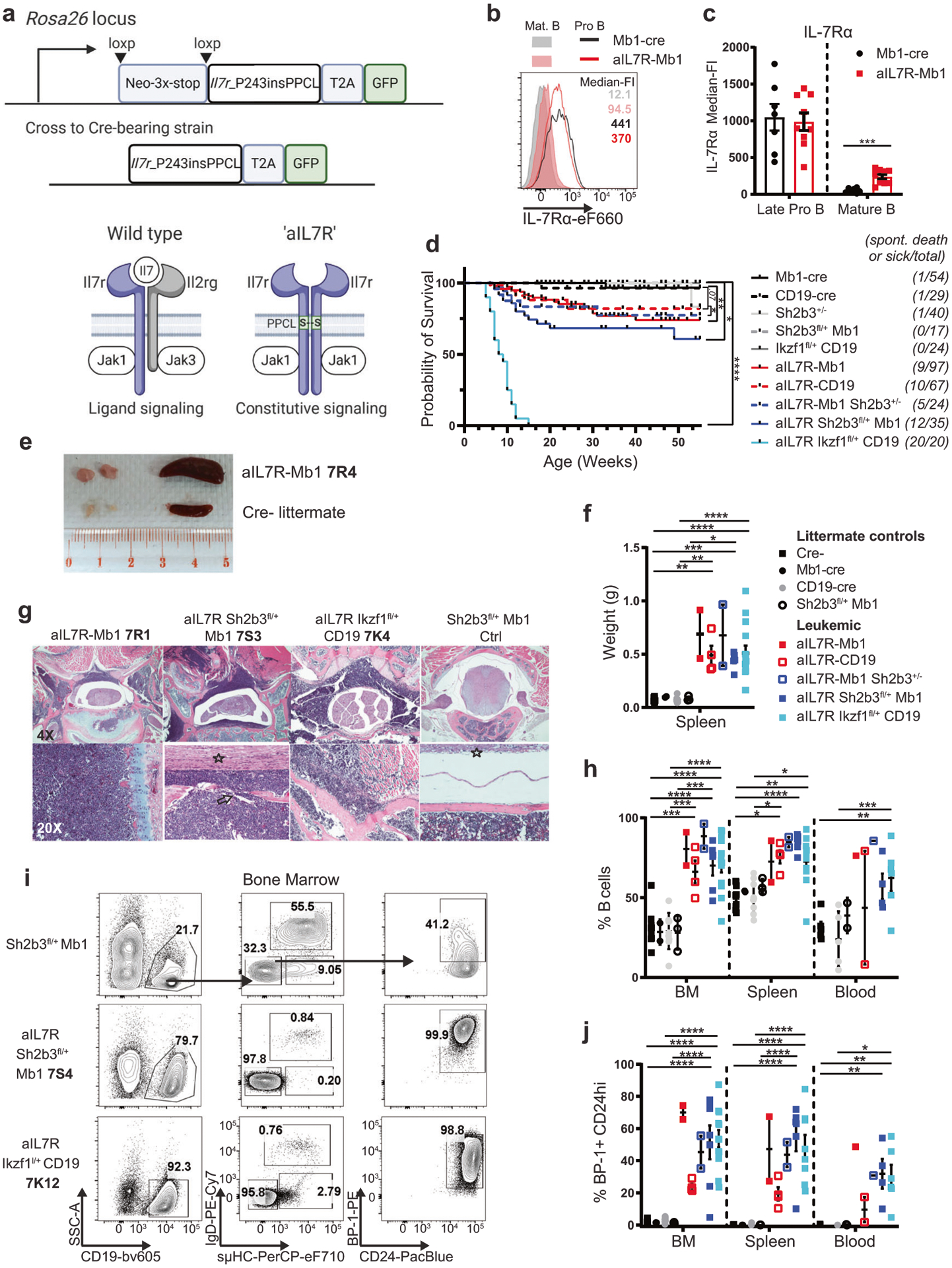
a (Top) Schematic of strategy for generation of knock-in mice expressing B-intrinsic activated mouse aIL7R (Il7r_P243insPPCL). (Bottom) Schematic of protein dimers illustrating ligand-induced wild-type IL-7R (Il7ra-Il2rg) versus constitutive aIL7R homodimer. Other ligand-induced protein dimers could include Il7ra-aIL7R and Il2rg-aIL7R (not pictured). Images created with BioRender.com. b Surface IL-7Rα expression in late pro-B (B220+ IgD− sμHC− CD24hi BP-1+; open histograms) and mature B (B220+ sμHC+ IgD+; filled histograms) cells from Mb1-cre (black) and aIL7R+/− Mb1-cre+/− (aIL7R-Mb1; red) mice. c Quantification of IL-7Rα surface expression in late pro-B and mature B cells. Bar graphs depict mean ± SEM of data from four independent experiments. N = 7 Mb1-cre and 9 aIL7R-Mb1. Significance defined as *** = P ≤ 0.001 by two-tailed Student’s t test. d Survival curves for aIL7R-expressing mice relative to controls (Mb1-cre, CD19-cre, Sh2b3+/−, Sh2b3fl/+ Mb1 or Ikzf1fl/+ CD19). Animals used experimentally prior to 15 weeks of age were excluded. Numbers on left side indicate the number of spontaneous death or disease events per total animals. Total N = 54 Mb1-Cre controls, 29 CD19-cre controls, 40 Sh2b3+/− (global), 17 Sh2b3fl/+ Mb1 (B-intrinsic), 24 Ikzf1fl/+ CD19, 97 aIL7R-Mb1, 67 aIL7R-CD19, 24 aIL7R-Mb1 Sh2b3+/−, 35 aIL7R Sh2b3fl/+ Mb1, and 20 aIL7R Ikzf1fl/+ CD19 mice. Significance defined by Mantel-Cox test. ** = P ≤ 0.005 and **** = P ≤ 0.0001. e Representative spleen and inguinal lymph nodes from a leukemic aIL7R-Mb1 animal and a Cre− littermate control. 7R4 (and similar nomenclature in subsequent panels) represents identifier for specific leukemic animal as detailed in Supplementary Table 1. f Quantification of spleen weights for leukemic animals and littermate controls. Significance defined as **** = P ≤ 0.0001 by one-way ANOVA. g Representative H&E staining of vertebral bone sections from leukemic mice with indicated genotype and an Sh2b3fl/+ Mb1 littermate control. N = 1 Sh2b3fl/+ Mb1 littermate control, 1 Ikzf1fl/+ CD19 littermate control, 1 aIL7R-Mb1, 1 aIL7R-CD19, 3 aIL7R Sh2b3fl/+ Mb1, and 3 aIL7R Ikzf1fl/+ CD19 mice analyzed. h Frequency of B cells (CD19+) in the bone marrow (BM), spleen, and peripheral blood from littermate control and leukemic mice with indicated genotypes. i Representative flow plots of B cell developmental subsets in the BM. j Frequency of CD19+ BP-1+ CD24hi progenitor B cells in the BM, SPL, and peripheral blood. f, h, j Data points represent individual mice and bars represent mean ± SEM. N = 12 Cre− controls (black squares), 2 Mb1-cre controls (black circles), 10 CD19-cre controls (gray circles), 3 Sh2b3fl/+ Mb1 (open black circles), 2 aIL7R-Mb1 (red squares) 4 aIL7R-CD19 (open red squares), 2 aIL7R-Mb1 Sh2b3+/− (open blue squares), 6 aIL7R Sh2b3fl/+ Mb1 (blue squares) and 13 aIL7R Ikzf1fl/+ CD19 (teal squares). h, j Significance defined as ** = P ≤ 0.005, *** = P ≤ 0.001 and **** = P ≤ 0.0001 by two-way ANOVA. aIL7R-Mb1 Sh2b3+/− = B-intrinsic aIL7R & global Sh2b3 het; aIL7R Sh2b3fl/+ Mb1 = B-intrinsic aIL7R & Sh2b3 het; Ikzf1fl/+ CD19 = B-intrinsic mutant Ikzf1 het; aIL7R Ikzf1fl/+ CD19 = B-intrinsic aIL7R & mutant Ikzf1 het.
Longitudinal cohorts revealed that aIL7R-Mb1 and aIL7R-CD19 mice developed spontaneous leukemic disease with low penetrance (~20%) with a median time to euthanasia criteria of 16 or 12 weeks, respectively (Fig. 1d). Leukemic animals displayed lymphadenopathy, splenomegaly, and high levels of neoplastic B cells in peripheral blood (Fig. 1e–h). Disease manifestations included hind limb paralysis with leukemic infiltration of vertebral bone marrow, bone degeneration, and local nerve impingement (Fig. 1g, Supplementary Tables 1, 2). Leukemia infiltration into liver and central nervous system meninges were also observed (Supplementary Fig. 2b and Supplementary Table 2). Leukemia cells (leukemias referred to as “aIL7R alone”) expressed markers consistent with late pro-/large pre-B cells (CD19+ sμHC− IgD− CD24hi BP-1+) (Fig. 1h–j, Supplementary Fig. 2c). Further characterization revealed surface pre-BCR (sμHC+ VpreB1+ or sμHC+) expression in primary leukemia cells from 5 of 6 mice, consistent with a pre-B cell immunophenotype (Supplementary Table 1). Finally, Ig heavy chain sequencing showed clonal leukemia with the expression of one or two predominant VH segments per animal (Supplementary Fig. 2d, Supplementary Table 1). Together, these data demonstrate that aIL7R can promote de novo leukemic transformation.
Loss of Sh2b3 or Ikzf1 exacerbates and accelerates aIL7R-driven leukemia
Loss-of-function mutations in SH2B3 and IKZF1 are also associated with Ph-like ALL, including those with IL7R mutations [3, 16, 22, 23]. To model the genetic interaction between Sh2b3 and aIL7R, we introduced an inducible knock-out allele of Sh2b3 [24] onto the aIL7R background. Alternative breeding strategies produced compound heterozygous animals with either global or B-lineage-restricted loss of Sh2b3 (strains referred to as aIL7R-Mb1 Sh2b3+/− or aIL7R Sh2b3fl/+ Mb1, respectively). To model leukemia-associated IKZF1 deletions, we crossed aIL7R mice to animals expressing an exon 5 floxed dominant-negative isoform of Ikaros in B cells [20] similar to the Ik6 isoform in human B-ALL [25] (strain hereafter referred to as “aIL7R Ikzf1fl/+ CD19”).
As in aIL7R-Mb1/-CD19 mice, aIL7R-Mb1 Sh2b3+/−, aIL7R Sh2b3fl/+ Mb1, and aIL7R Ikzf1fl/+ CD19 animals also developed spontaneous B-ALL with features that closely resembled aIL7R alone leukemias (referred to as “aIL7R/Sh2b3” and “aIL7R/Ikzf1” leukemias) (Fig. 1d–j, Supplementary Tables 1, 2, Supplementary Fig. 2a, b). B-intrinsic Sh2b3 deletion increased leukemia penetrance to ~40% of aIL7R Sh2b3fl/+ Mb1 mice, while co-expression of mutant Ikzf1 resulted in complete penetrance of aIL7R Ikzf1fl/+ CD19 animals by week 15 (Fig. 1d). In the absence of aIL7R, heterozygous Sh2b3 deletion or Ikzf1 mutant expression did not lead to disease (Fig. 1d). Disease presentation, tissue pathology, CD24hi BP-1+ precursor phenotype, and leukemia clonality mirrored the features of aIL7R alone leukemias (Fig. 1e–j, Supplementary Tables 1, 2, Supplementary Fig. 2a, b). Interestingly, hepatic infiltration with leukemia was more pronounced in aIL7R/Ikzf1 leukemias, suggesting potential genotype differences in leukemia localization (Supplementary Fig. 2b, Supplementary Table 2). Unlike the aIL7R alone leukemias, surface pre-BCR expression was not detected in most compound heterozygous leukemias (aIL7R/Sh2b3: only 1 of 8; aIL7R/Ikzf1: 3 of 14) (Supplementary Table 1, Supplementary Fig. 2e). However, some leukemias expressed c-kit and/or surrogate light chain in the absence of surface heavy chain (sμHC− VpreB1+), which was similar to previously described studies of mouse and human “pro-BCR+” pro-B ALL (Supplementary Fig. 2e, Supplementary Table 1) [26–28].
Next, we assessed the capacity to generate secondary leukemias via serial transplantation. Splenocytes from 12 independent aIL7R alone, aIL7R/Sh2b3, and aIL7R/Ikzf1 primary leukemias were transplanted into unirradiated immunocompetent C57BL/6 (B6) recipient mice (Fig. 2a). Regardless of leukemia genotype, all recipient animals rapidly succumbed to disease, although across all starting genotypes the time to required euthanasia varied depending on the specific murine donor (~2–6 weeks; Fig. 2b–d). Transplanted (GFP+) cells were present at high proportions in BM, spleen, and peripheral blood and maintained their CD24hi BP-1+ phenotype (Fig. 2e–h). Collectively, these findings are consistent with reproducible malignant transformation in animals with B-cell intrinsic expression of aIL7R.
Fig. 2. aIL7R leukemias are transplantable into immunocompetent hosts and acquire mutations in clinically relevant genes.
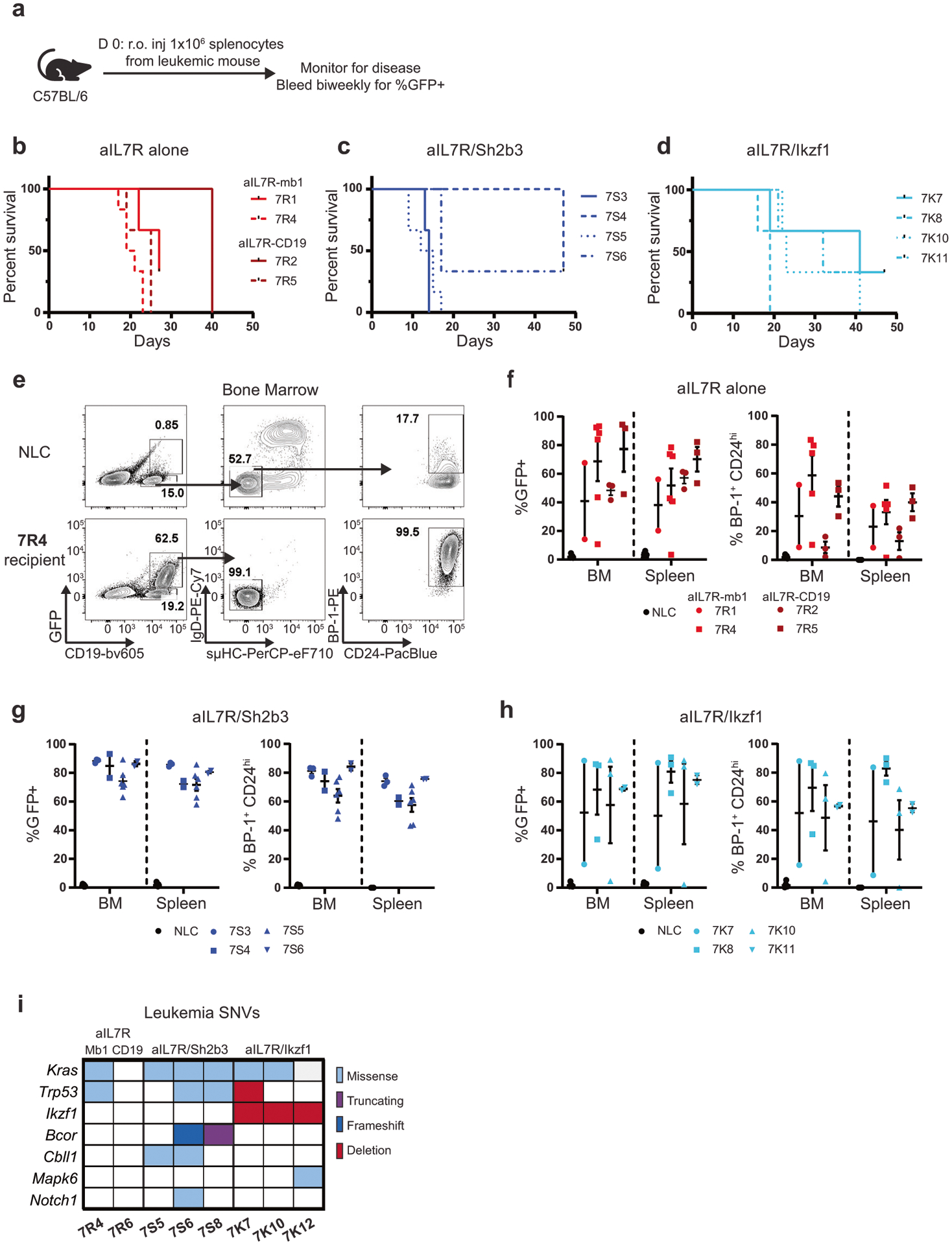
a–h 1e06 primary leukemia cells were transferred retro-orbitally (r.o.) into wild-type C57BL/6 recipient mice. a Schematic of leukemia serial transfer experiments. b–d Survival curves for aIL7R alone (b), aIL7R/Sh2b3 (c), and aIL7R/Ikzf1 (d) leukemia recipient animals. (e) Representative flow plots of B cell subsets in the BM of an aIL7R-Mb1 leukemia (7R4) recipient and a no leukemia control (NLC) that did not undergo adoptive transfer. f–h Frequency of GFP+ (left) and BP-1+CD24hi (right) leukemia cells in the BM and spleen of aIL7R alone (f), aIL7R/Sh2b3 (g), and aIL7R/Ikzf1 (h) leukemia recipients. b–h Data points represent individual recipient mice and bars represent mean + SEM. N = 3 recipients for 7R1, 7R2, 7R5, 7S3, 7S4, 7S6, 7K7, 7K8, 7K10, and 7K11, 6 recipients for 7R4 and 7S5 (2 independent experiments), and 18 no leukemia controls (6 per graph). i Summary of somatic nucleotide variants (SNVs) identified by whole-exome sequencing and validated in IGV in a panel of 8 aIL7R leukemias.
aIL7R leukemias harbor somatic mutations in Kras, Trp53, and Bcor
To determine which secondary mutations were associated with aIL7R-driven leukemia in these models, we performed whole-exome sequencing on eight primary leukemias (2 aIL7R alone, 3 aIL7R/Sh2b3, 3 aIL7R/Ikzf1) and four paired tail DNA samples as controls. We used two somatic variant callers, Pisces and Mutect2 (Supplementary Fig. 3a), to identify somatic nucleotide variants (SNVs) present in leukemia and not tail DNA. Kras variants were highly prevalent irrespective of starting genotype (6 of 8 leukemias) with most located at codons 12, 13, 61, and 146, previously identified “hotspots” mutated in many cancers [29] (Fig. 2i, Supplementary Fig. 3b, Supplementary Table 3). Additional variants included mutations in the tumor suppressor, Trp53 (encoding Tp53), and in the Bcl6 interacting corepressor (Bcor) (Fig. 2i, Supplementary Table 3). Bcor mutations (stop-gain variant or a 1 bp frameshift variant in exon 8) were similar to mutations described in pro-B ALLs from NUP98-PHF23 transgenic mice, a model that mirrors features of cytokine receptor-like factor 2 (CRLF2)-rearranged Ph-like ALL [30]. Mutations in the E3 ubiquitin ligase Cbll1 and the NOTCH1 receptor Notch1 were also present in aIL7R/Sh2b3 leukemias (2 of 3 and 1 of 3, respectively; Fig. 2i, Supplementary Table 3). By contrast, all aIL7R/Ikzf1 leukemias displayed an ostensible loss of the remaining wild-type Ikzf1 allele, as evidenced by absence of exon 5 sequence coverage (Fig. 2i, Supplementary Fig. 3c, Supplementary Table 3). Loss-of-heterozygosity analysis between paired tail and leukemia DNA indicated one aIL7R/Ikzf1 leukemia (7K7) possessed a large deletion in chromosome 11 encompassing both Ikzf1 and Trp53 (Fig. 2i). These data show that aIL7R-driven leukemias acquire somatic mutations in multiple genes previously associated with B-ALL, events that may contribute directly to leukemogenesis.
aIL7R expression preferentially drives expansion of BM pro B cells
To ascertain the effect of aIL7R on B cell development, we performed detailed immunophenotyping of bone marrow and splenic B cells prior to leukemogenesis. aIL7R-Mb1 mice displayed increased marrow cellularity with the expansion of B220+ B cells (Fig. 3a–c). The percentage and the absolute number of late pro B cells (B220+ sμHC− IgD− CD24hi BP-1+; Fig. 3d) was specifically increased. However, mature B cells (B220+ sμHC+ IgD+) were present in numbers equivalent to control mice, consistent with no significant impairment in overall B cell development (Fig. 3d). In the context of aIL7R, global loss of one Sh2b3 allele led to an increase in early pro B and late pro B cells (Fig. 3b, d). Additionally, aIL7R-Mb1 and aIL7R-Mb1 Sh2b3+/− mice displayed a skewing towards increased splenic transitional B cells and reduced marginal zone B cells (Supplementary Fig. 4a–d). While aIL7R-CD19 and aIL7R Ikzf1fl/+ CD19 animals did not exhibit an increase in total B cell numbers (Fig. 3e, Supplemental Fig. 4e–g), aIL7R Ikzf1fl/+ CD19 mice displayed an increased proportion and the absolute number of late pro-B cell progenitor cells (Fig. 3f). These studies show that B-intrinsic aIL7R expression specifically promotes a modest increase in the late pro-B cell compartment and that this phenotype is enhanced by either Sh2b3 loss or the expression of mutant Ikzf1.
Fig. 3. B-lineage aIL7R cooperates with heterozygous loss of Sh2b3 or Ikzf1 to drive a specific expansion of late pro B cells.
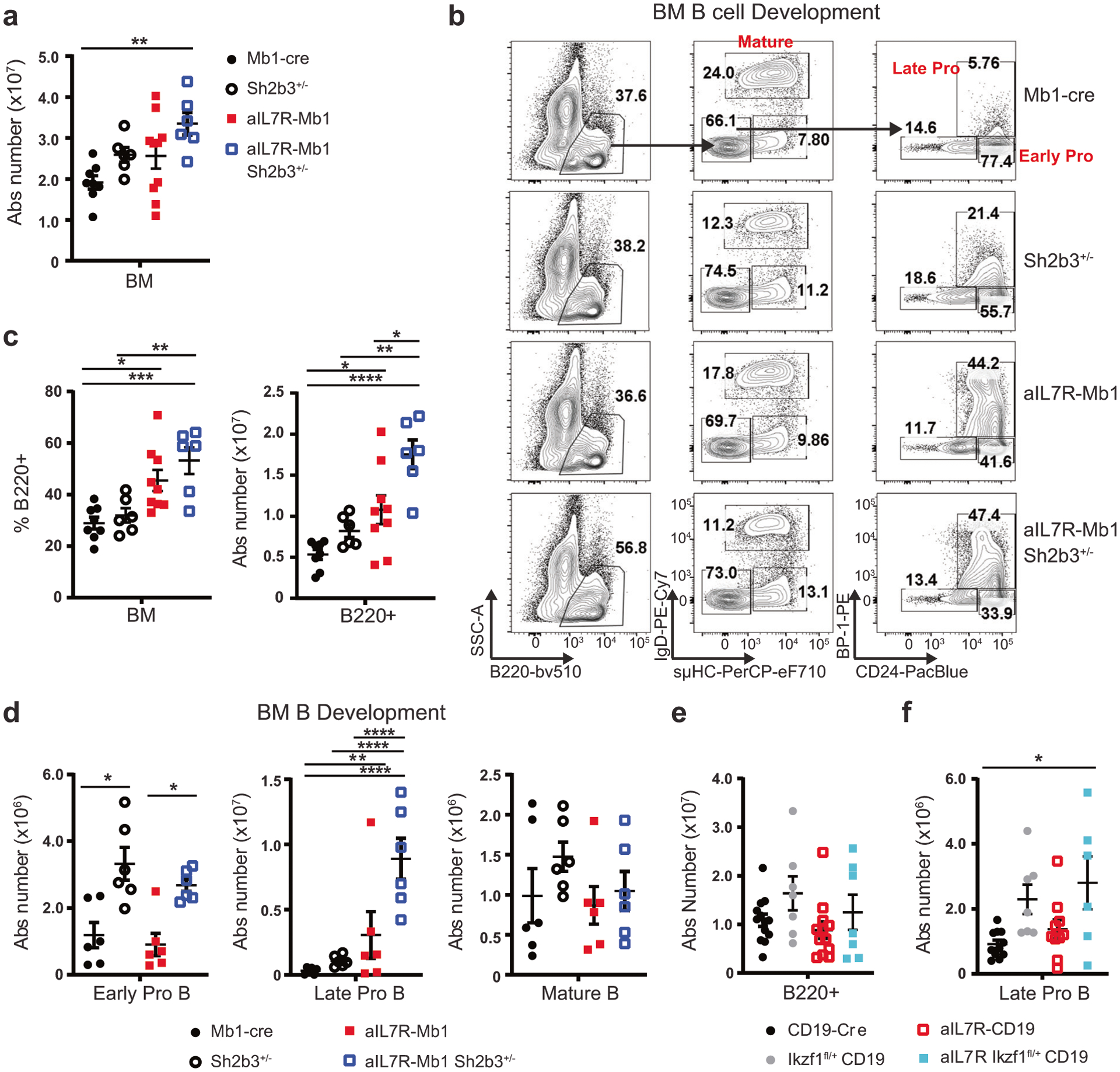
a Absolute number of total BM cells recovered from 1 tibia + femur for animals with indicated genotypes. b Representative flow plots of BM B cell developmental subsets: early pro-B (B220+ sμHC− IgD− CD24hi BP-1−), late pro-B (B220+ sμHC− IgD− CD24hi BP-1+), and mature B (B220+ sμHC+ IgD+) highlighted. c Frequency (left) and absolute number (right) of B220+ B cells in the BM. a, c Data from 4 independent experiments. N = 8 Mb1-cre (black circles), 6 Sh2b3+/− (black open circles), 9 aIL7R-Mb1 (red squares) and 6 aIL7R-Mb1 Sh2b3+/− (open blue squares) mice. d Absolute number of early pro-B (left), late pro-B (center), and mature B (right) cells in the BM. N = 6 replicate mice per genotype. e Absolute number of B220+ B cells in the BM in aIL7R animals with or without B-intrinsic heterozygous mutant Ikzf1 expression (aIL7R Ikzf1fl/+ CD19). f Absolute number of late pro-B cells in the BM. e, f Data from 4 independent experiments. N = 13 CD19-cre (black circles), 7 Ikzf1fl/+ CD19 (gray circles), 12 aIL7R-CD19 (open red squares), and 6 aIL7R Ikzf1fl/+ CD19 (teal squares) mice. a, c–f Data points represent individual mice and bars represent mean ± SEM. Significance defined as * = P ≤ 0.05, ** = P ≤ 0.005, *** = P ≤ 0.001, **** = P ≤ 0.0001 by one-way ANOVA.
aIL7R promotes enhanced survival in normal and leukemic B cell progenitors
IL-7R signaling promotes survival of normal B cell progenitors by upregulating anti-apoptotic BCL-2-like family members [31, 32]. As Bcl2l1 (encoding the Bcl-xl protein) transgenic animals similarly develop a large expansion of pro-B cells and Bcl-xl is most highly expressed in pro-B and pre-B cells [33], we assessed intranuclear Bcl-xl protein expression by flow cytometry in non-leukemic (i.e. lacking in disease symptoms as defined by our IACUC protocol) mice. aIL7R-Mb1 Sh2b3+/− mice displayed elevated Bcl-xl levels in bone marrow B cells (B220+) and pro/pre-B cells (B220+ sμHC− IgD−) (Fig. 4a, b). Similarly, leukemic aIL7R/Sh2b3 B cells exhibited increased Bcl-xl protein expression relative to control pro/pre-B cells (Fig. 4c, d).
Fig. 4. aIL7R with Sh2b3 loss promotes enhanced survival in normal and leukemic B cell progenitors.
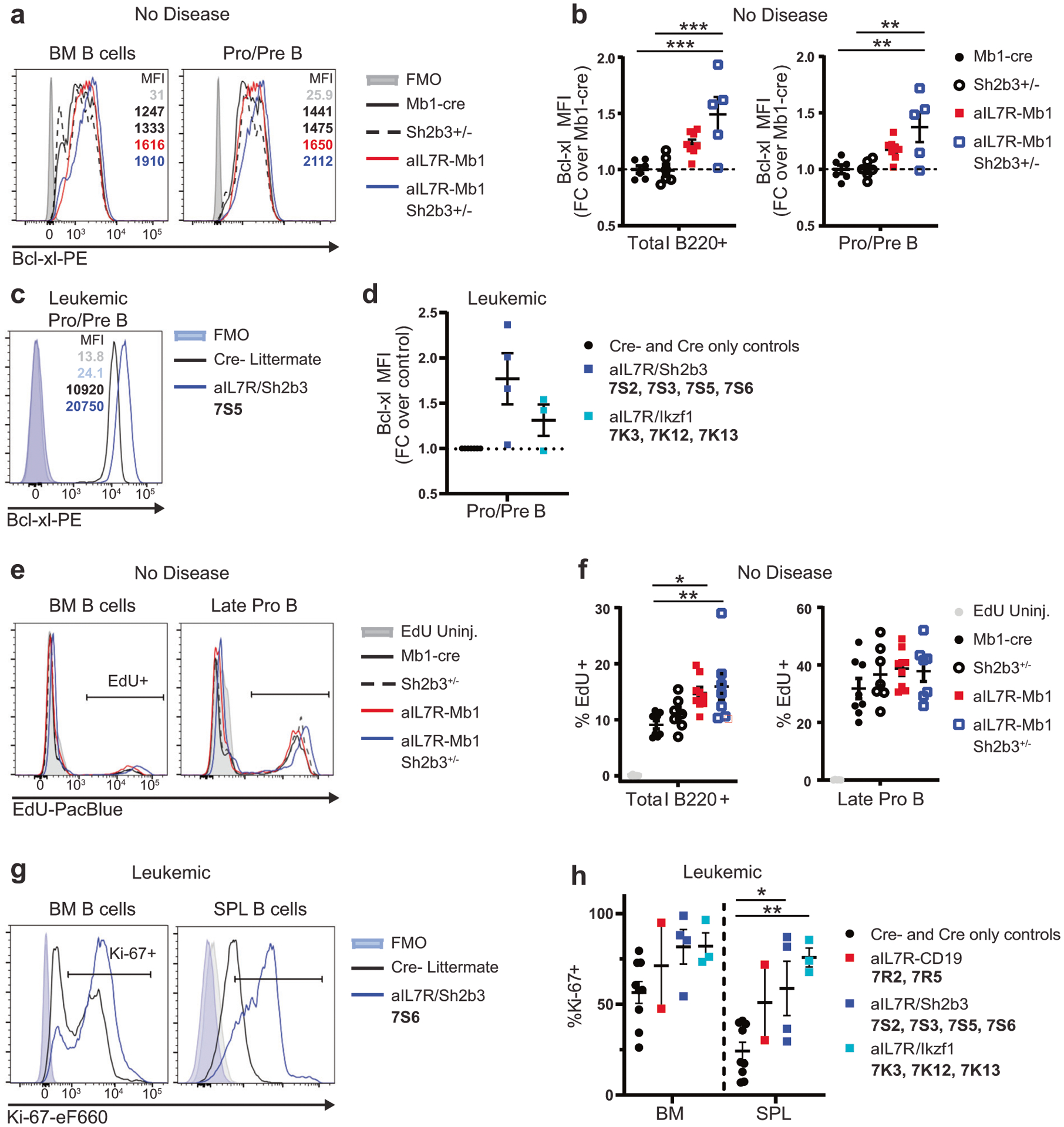
a Representative histograms of Bcl-xl expression in total B220+ (left) and pro/pre-B cells (B220+ sμHC− IgD−; right) in the BM of non-leukemic animals with the indicated genotypes. b Bcl-xl MFI normalized to the average fluorescence in Mb1-cre animals for total B220+ (left) and pro/pre-B cells (right). Data from 3 independent experiments. N = 6 Mb1-cre, 6 Sh2b3+/−, 8 aIL7R-Mb1, and 5 aIL7R-Mb1 Sh2b3+/− mice. FC = fold-change. c Representative histogram of Bcl-xl expression in pro/pre-B cells in the BM from an aIL7R Sh2b3fl/+ Mb1 leukemia (7S5) and a Cre− littermate control. d Relative fold-change in Bcl-xl MFI in leukemic cells normalized to MFI in pro/pre-B cells from Cre− and Cre only control animals. N = 8 controls (black circles), 4 aIL7R Sh2b3fl/+ Mb1 (blue squares), and 3 aIL7R/Ikzf1 (teal squares) mice. e, f 1 mg EdU was injected i.p. 1 h prior to sacrifice. e Representative histograms showing the relative proportions of EdU+ cells in total BM B and late pro-B cells. f Percentage of EdU+ cells in total BM B cells (left) and late pro B cells (right). Data from 3 independent experiments. N = 5 uninjected controls, 8 Mb1-cre, 7 Sh2b3+/−, 9 aIL7R-Mb1, and 7 aIL7R-Mb1 Sh2b3+/− mice. b, f Significance defined as * = P ≤ 0.05, ** = P ≤ 0.005 and *** = P ≤ 0.001 by one-way ANOVA. g Representative histograms of Ki-67 expression in BM B (CD19+; left) and SPL B (B220+; right) cells from an aIL7R/Sh2b3 leukemia (7S6) and a Cre− littermate control. h Percentage of Ki-67+ cells in BM and SPL B cells in leukemic animals versus healthy Cre− and Cre only littermate controls. N = 11 controls (black circles), 2 aIL7R-CD19 (red squares), 4 aIL7R Sh2b3fl/+ Mb1 (blue squares), and 3 aIL7R/Ikzf1 (teal squares). Significance defined as * = P ≤ 0.05 and ** = P ≤ 0.005 by one-way ANOVA. b, d, f, h Data points represent individual mice and bars represent mean ± SEM.
To assess the role of aIL7R-driven cell proliferation, we performed in vivo EdU labeling studies. While aIL7R-Mb1 and aIL7R-Mb1 Sh2b3+/− mice had a greater frequency of EdU+ BM B cells, the rate of incorporation within the late pro-B cell compartment was not different from that of control mice (Fig. 4e, f). In contrast, ex vivo staining of B cells from leukemic aIL7R mice demonstrated increased Ki-67+ cells in both bone marrow and spleen (Fig. 4g, h). Taken together, these experiments suggested that aIL7R with loss of Sh2b3 may promote enhanced survival of normally proliferating marrow progenitors in non-leukemic animals via upregulation of Bcl-xl. Further, aIL7R/Sh2b3 leukemic cells exhibit both upregulation in Bcl-xl, possibly due to conserved signaling events, and increased cell proliferation mediated via acquired leukemia-enhancing genetic lesions. However, as changes in Bcl-xl expression were not evident in non-leukemic or leukemic aIL7R Ikzf1fl/+ CD19 mice, this pathway is not uniformly active across aIL7R-associated leukemias.
aIL7R pro-B cells lack constitutive Stat5 activation yet are less sensitive to IL-7 depletion in vivo
We next assessed whether phosphorylated (p)-Stat5 was increased in flow cytometry-sorted late pro-B cells from littermate control, aIL7R-Mb1, and aIL7R Sh2b3fl/+ Mb1 mice. Surprisingly, we observed no genotype-specific differences in basal or IL-7-induced pStat5 levels via intracellular phosphoflow cytometry (Fig. 5a, b). Alternatively, when analyzing splenic B cells, we found that IL-7 induced higher levels of pStat5 in aIL7R-expressing mature B cells relative to controls, although basal levels remained similar across genotypes (Fig. 5c, d). Overall, these data suggest that aIL7R is capable of transducing pStat5 signals in primary B cells in the presence of ligand but lacks evidence for constitutive Stat5 signaling as assessed by flow cytometry. The lack of measurable constitutive Stat5 signaling likely reflects the relatively low level of aIL7R expression in our Rosa-promoter based model.
Fig. 5. aIL7R-expressing B cell progenitors are less sensitive to IL-7 depletion in vivo.

a, b Late pro-B cells were sorted, rested, then stimulated with or without 10 ng/mL recombinant murine IL-7 (mIL-7). a Representative histograms of phosphorylated (p)Stat5 in unstimulated and mIL-7-stimulated late pro-B cells. b pStat5 Median-FI normalized to the average fluorescence of unstimulated pro-B cells from Cre− littermate control animals. Data from 3 independent experiments. N = 5 Cre− littermate, 6 aIL7R-Mb1, and 7 aIL7R Sh2b3fl/+ Mb1 mice. c, d Naïve B cells were enriched from total splenocytes and stimulated with 25 ng/mL mIL-7. c Representative histograms of pStat5 in unstimulated and IL-7 stimulated cells. d pStat5 Median-FI normalized to the average fluorescence of unstimulated B cells from Mb1-cre animals. Data from 3 independent experiments. N = 6 Mb1-cre, 6 Sh2b3+/−, 8 aIL7R-Mb1, and 6 aIL7R-Mb1 Sh2b3+/− mice. Significance defined by **** = P ≤ 0.0001 by two-way ANOVA. e–h aIL7R Sh2b3fl/+ Mb1 and Cre− littermate control mice were treated i.p. with 0.5 mg/mouse isotype control or neutralizing anti-IL-7 mAb every third day for 16 days (6 injections) and sacrificed on day 18. e Schematic of in vivo blocking antibody treatment. f Absolute number of B220+ B cells in the BM from 1 tibia + femur in isotype- (black circles) and anti-IL-7-treated (open squares) animals. g Representative flow cytometry plots showing the frequency of late pro-B cells in isotype- and anti-IL-7-treated animals. h Absolute number of late pro-B cells in the BM from isotype- and anti-IL-7-treated animals. f, h Data from 2 independent experiments. N = 5 Cre− controls per treatment group and 5 (isotype) or 6 (anti-IL-7) aIL7R Sh2b3fl/+ Mb1 mice. Significance defined by multiple t tests using the Holm-Sidak method. b, d, f, h) Data points represent individual mice and bars represent mean ± SEM.
We next tested whether the expansion of the pro-B cell compartment in aIL7R animals requires endogenous IL-7. In these experiments, we treated aIL7R Sh2b3fl/+ Mb1 and control animals in vivo with IL-7 blocking or isotype negative control antibodies (Fig. 5e). Strikingly, treatment with the blocking antibody depleted both total BM B (B220+) and late pro-B cells in control animals, but had no effect upon aIL7R progenitors (Fig. 5f–h). These data imply that aIL7R provides an IL-7-independent signal that promotes pro-B cell growth in a non-leukemic setting.
To test whether aIL7R-driven leukemias require IL-7 for survival/maintenance, two aIL7R/Sh2b3 primary leukemias (7S4 and 7S5) were transplanted into B6 recipient animals that were subsequently treated with anti-IL-7 or isotype control antibodies and monitored for signs of disease (Supplementary Fig. 5d). Anti-IL-7- and isotype-treated animals developed disease with similar kinetics, and IL-7 blockade had no impact on survival (Supplementary Fig. 5e), indicating that IL-7 is not required for growth of aIL7R-derived leukemias in vivo. We performed parallel studies in human Ph-like ALL PDX models, including one with IL7R mutation and SH2B3 deletion (PALJDL) [34], and assessed in vitro and in vivo dependencies upon IL-7 (n = 11 PDX models; Supplementary Table 4). While cells from all PDX models showed increased pSTAT5 levels following in vitro stimulation with IL-7, in vivo treatment of the PALJDL PDX model with the IL-7 blocking antibody had no effect upon leukemia proliferation, as measured by longitudinal bioluminescent imaging (Supplementary Fig. 5f–i). Taken together, these data suggest that aIL7R promotes IL-7-independent growth of B cell progenitors and that IL-7 blockade is unlikely to control leukemic burden in B-ALL expressing aIL7R.
aIL7R murine leukemias are sensitive in vitro and in vivo to rapamycin
Ph-like ALL cells have previously been shown to be sensitive to in vitro and in vivo treatment with JAK and/or mTOR pathway inhibitors, implying that survival may be, in part, dependent upon both signaling pathways [34–36]. To assess Jak/Stat and mTOR signaling activation in our aIL7R-driven mouse leukemias, we measured ex vivo phosphorylation of Stat5, as well as the PI3-K/Akt/mTORC1 targets ribosomal protein S6 and the translational repressor 4E-BP1. aIL7R alone and aIL7R/Ikzf1 leukemias showed a 1.5-fold increase in pStat5 levels in BM B (CD19+) cells and no significant differences in basal pS6 relative to control B cells from the BM and/or spleen (Supplementary Fig. 5a, b). Similar to our pStat5 findings, aIL7R/Ikzf1 bone marrow leukemia cells exhibited a ~2-fold increase in p4E-BP1 levels relative to control BM B cells and splenic cells from all types of aIL7R leukemias increased p4E-BP1 compared to control cells (~2–6-fold; Fig. 6a). Taken together, these data suggest that mTOR and likely also Stat5 signaling are activated in aIL7R leukemias and that our GEMMs recapitulate human Ph-like ALL biology.
Fig. 6. aIL7R leukemias are responsive to rapamycin therapy in vivo.
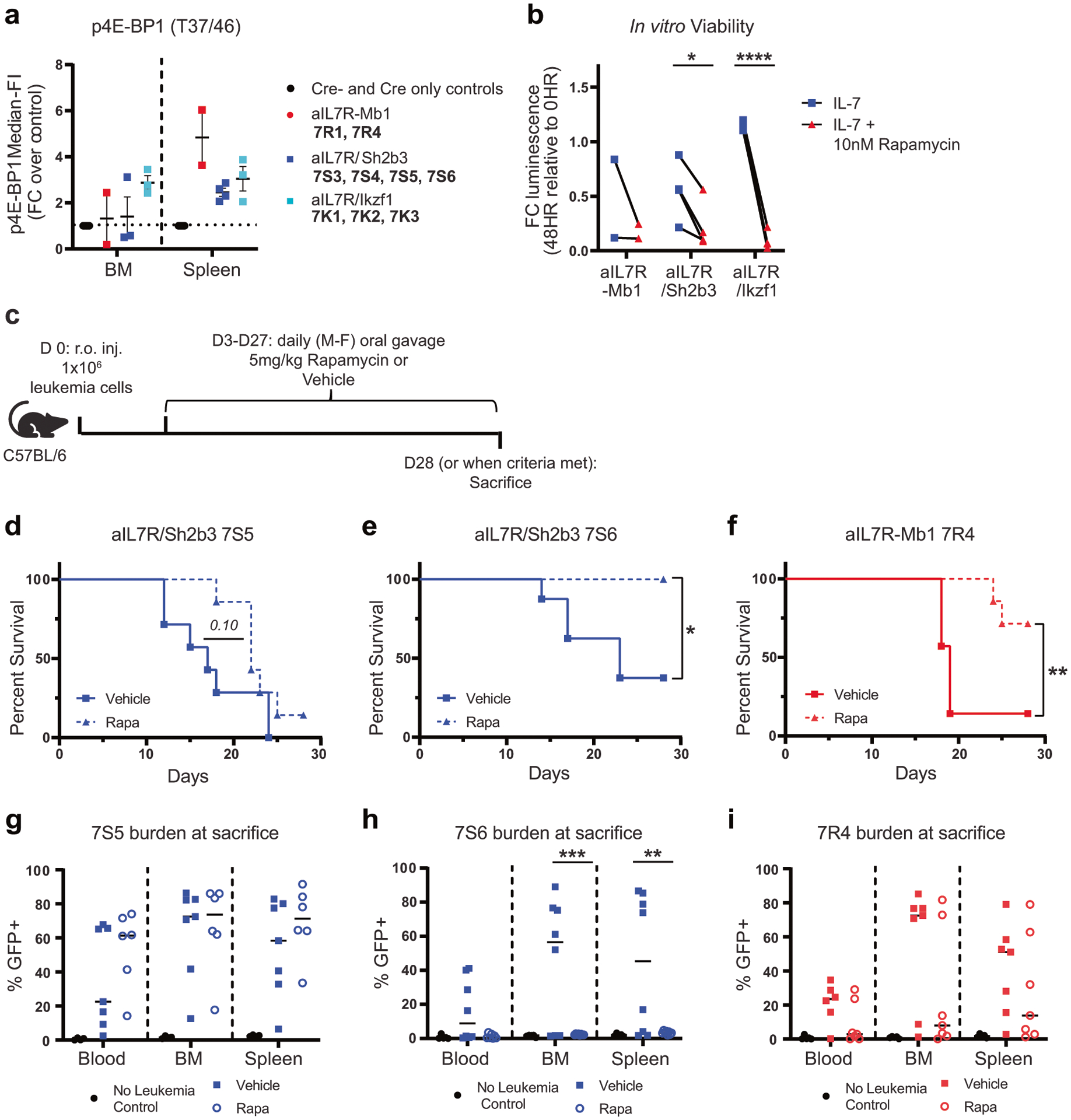
a Fold-change median-FI of p4E-BP1 in BM and spleen B cells (CD19+) ex vivo from aIL7R leukemic animals relative to Cre− or Cre only littermate controls. N = 8 controls, 2 aIL7R-Mb1, 4 aIL7R Sh2b3fl/+ Mb1, and 3 aIL7R/Ikzf1. b CellTiter-Glo luminescent cell viability of aIL7R leukemias cultured for 48 h with mIL-7 with (red triangle) or without (blue square) rapamycin (Rapa). Luminescence reported as fold-change relative to ex vivo (0 h) levels. Significance defined as *: P = 0.024 and ****: P < 0.0001 by two-tailed paired t tests. N = 2 aIL7R-Mb1, 4 aIL7R Sh2b3fl/+ Mb1 and 4 aIL7R/Ikzf1. c–i Primary aIL7R leukemia cells were transferred retro-orbitally (r.o.) into C57BL/6 recipient mice and then given rapamycin or vehicle by oral gavage daily for 4 weeks. c Schematic of in vivo rapamycin treatment experiments. d–f Survival curves for recipients of murine leukemias 7S5 (d), 7S6 (e), and 7R4 (f) treated with vehicle (solid line) or rapamycin (dashed line). Significance defined by Mantel-Cox test. *: P = 0.023 and **: P = 0.0084. Data from 2 independent experiments. g–i Frequency of GFP + leukemic 7S5 (g), 7S6 (h), and 7R4 (i) cells in peripheral blood, BM, and spleen of recipient animals at sacrifice. Closed squares = vehicle and open circles = rapamycin. Data points represent individual mice and lines represent mean. N = 7 7S5, 8 7S6, and 7 7R4 recipients per treatment group. N = 5 no leukemia controls per graph.
Consistent with prior reports [34], we confirmed that rapamycin treatment of PALJDL PDX mice effectively inhibited in vivo leukemia proliferation (Supplementary Fig. 5i). To assess whether murine aIL7R leukemias were responsive to mTOR inhibition, we cultured leukemia cells with the mTOR inhibitor rapamycin and found it significantly reduced viability of aIL7R/Sh2b3 and aIL7R/Ikzf1 leukemias (Fig. 6b). To test the effect of mTOR inhibition in vivo, we transplanted an aIL7R alone leukemia (7R4) or aIL7R/Sh2b3 leukemias (7S5 and 7S6) into B6 mice and treated animals with rapamycin or vehicle for 4 weeks (Fig. 6c). Rapamycin treatment extended recipient survival for leukemias 7R4 and 7S6, but exerted minimal impact on 7S5 recipients (Fig. 6d–i). Five of 8 (62.5%) 7S6 leukemia recipients treated with vehicle succumbed to disease (median survival of 23 days), while all rapamycin-treated mice survived and showed no signs of leukemia at sacrifice (Fig. 6e, h). Similarly, 6 of 7 (85.7%) vehicle-treated 7R4 leukemia recipients met euthanasia criteria (median survival = 19 days), compared to only 2 of 7 (28.6%) receiving rapamycin (Fig. 6f, i). Together, these data demonstrate that the mTOR pathway is active in aIL7R leukemias and that rapamycin is partially effective at controlling leukemia burden in vivo. The sensitivity of aIL7R leukemias to rapamycin further validates the response previously observed in our human Ph-like ALL PDX model (Supplementary Fig. 5i).
IL7R-mutant leukemias display activation of “BCR-like” effector molecules
To identify signaling programs that might mediate IL-7 independence in aIL7R leukemia cells, we performed phospho-tyrosine immunoprecipitation coupled with mass spectrometry from cell lysates derived from surface pre-BCR− secondary murine leukemias (1 aIL7R alone - 7R4, 2 aIL7R/Sh2b3 - 7S5 and 7S6, and 2 aIL7R/Ikzf1 – 7K7 and 7K10). We identified 122 unique phosphosites from 109 proteins (Supplementary Table 5). As anticipated, tyrosine phosphorylation of Stat5a/b and Stat3 were increased in leukemia cells isolated from secondary hosts (Fig. 7a, Supplementary Tables 5, 6, Supplementary Fig. 6). Despite the absence of surface pre-BCR expression, we surprisingly observed phosphorylation of multiple canonical “BCR-like” effectors, including Syk, Cbl, Btk, and others (Fig. 7a, Supplementary Tables 5, 6, Supplementary Fig. 6). We next assessed whether this tyrosine phosphorylation signature was conserved in human Ph-like ALL PDX models. We performed phospho-tyrosine immunoprecipitation of cells harvested from 6 discrete Ph-like ALL PDX models (Supplementary Table 4) and quantified tyrosine phosphorylation by mass spectrometry (Supplementary Table 7). Similar to the aIL7R murine leukemias, we identified enrichment of pBTK and pSYK residues, as well as pSTAT5A and pJAK3, in the PALJDL model (Fig. 7b). Notably, a similar BCR-like signaling signature was detected in additional Ph-like ALL PDX models with other JAK pathway genetic alterations. These findings suggest that a common adaptation in IL7R-driven leukemia is the activation of BCR-like signaling independent of surface pre-BCR expression that was recently reported in other Ph-like ALL cases [37].
Fig. 7. IL-7R-mutant leukemias display activation of a “BCR-like” signaling program.

a Diagram of phosphorylated “BCR-like” peptides (gold) and phosphorylated Stat peptides (blue) identified by phospho-tyrosine immunoprecipitation and mass spectrometry analysis of six secondary aIL7R murine leukemias (1 aIL7R-Mb1, 7R4—2 recipients; 2 aIL7R/Sh2b3—7S5 and 7S6; 2 aIL7R/Ikzf1—7K7 and 7K10). Created with BioRender.com. b Quantification of peptides identified by phospho-tyrosine immunoprecipitation in six Ph-like ALL patient-derived xenograft models directly harvested from spleens of NSG mice. PDX models included an activating IL7R/SH2B3-null (IL-7R/SH2B3; PALJDL), three independent CRLF2-overexpressing/JAK2 gain-of-function ALLs (CRLF2-R/JAK2; ALL121, PAMDKS, and PAWAKV), a NUP214-ABL1 fusion (PAKVKK), and an EBF1-PDGFRB fusion (PAKKCA). c–g The murine aIL7R leukemias from (a) were cultured with or without 1 uM of entospletinib (Ento) or ruxolitinib (Rux) for 30 min and then stimulated with or without 10 ng/mL mIL-7 for an additional 15 mins. Phospho-tyrosine residues were immunoprecipitated and analyzed by mass spectrometry. c Scatter plot of all pY peptides for the IL-7 alone vs entospletinib + IL-7 conditions. Data are presented as fold-change relative to the average of untreated conditions for all peptides within each respective genotype. pStat3/Stat5 (red), pSyk (blue), pPik3ap1 (green), and pCbl (orange) peptides are highlighted. d Bar graph of pY699 Stat5b levels (in response to the indicated treatments) relative to the untreated conditions across genotypes. e Bar graph of pY342 Syk levels relative to untreated. f Bar graph of pY694 Pik3ap1 levels relative to untreated. g Bar graph of pY672 Cbl levels relative to untreated. d–g White bars = untreated; gray bars = IL-7 only; red bars = IL-7 + Ento; blue bars = IL-7 + Rux. Data points represent individual mice and bars represent mean with range. h–m Splenocytes harvested ex vivo from primary or secondary murine aIL7R leukemias were cultured with ruxolitinib (h, i), bortezomib (j, k), or entospletinib (l, m) for 24 h in the presence of 10 ng/mL mIL-7 and cell viability was assessed via CellTiter-Glo luminescence. Representative dose curves for leukemias 7R4 (red), 7S6 (blue) and 7K8 (teal) treated with ruxolitinib (h), bortezomib (j), or entospletinib (l). Data points represent luminescence values at specific inhibitor doses normalized to the luminescence at the lowest dose (100 pM or 10 pM (bortezomib)). Summary of EC50s for aIL7R leukemias at 24 h treated with ruxolitinib (i), bortezomib (k), or entospletinib (m). Data points represent individual leukemias and lines represent means. N = 1 aIL7R-Mb1, 2 aIL7R-CD19, 4 aIL7R Sh2b3fl/+ Mb1, and 5 aIL7R/Ikzf1 leukemias.
aIL7R leukemias are sensitive to Syk inhibition in vitro
To determine whether the BCR-like signaling program present in aIL7R murine leukemias and Ph-like ALL PDX models was directly associated with IL-7R activation, we used a targeted proteomic approach to monitor tyrosine phosphorylation of these substrates in the presence of acute stimulation with IL-7. We also used entospletinib and ruxolitinib to assess whether phosphorylation of these peptides was associated with SYK- or JAK-dependent signaling, respectively. As expected, Stat5 phosphorylation was enhanced by IL-7 and diminished with exposure to the Jak1/2 inhibitor, ruxolitinib, but was minimally affected by treatment with the Syk inhibitor, entospletinib (Fig. 7c–d). Additionally, we found IL-7-independent phosphorylation of Syk that was abrogated by entospletinib, but minimally affected by ruxolitinib (Fig. 7c–e). Finally, Cbl and Pik3ap1 (BCAP) peptides were also inhibited by entospletinib, suggesting that these proteins are downstream of Syk in this BCR-like pathway (Fig. 7b, f, g). These results suggest that aIL7R leukemias manifest a unique IL-7-independent BCR-like program in addition to an IL-7-inducible Jak/Stat program.
We subsequently tested whether this BCR-like signaling program is essential for survival of aIL7R leukemias in vitro. We cultured aIL7R leukemia cells with ruxolitinib, entospletinib, the proteasome inhibitor bortezomib as a positive control, and compounds that target proteins downstream of Syk, including the PI3Kδ inhibitor idelalisib, the Btk inhibitor ibrutinib, or the MEK inhibitor trametinib. As predicted in prior reports [28], viability of aIL7R leukemic cells was sensitive to ruxolitinib (Fig. 7h, i), idelalisib (Supplementary Fig. 7a, b), and bortezomib (Fig. 7j, k). While individual leukemias showed varying degrees of sensitivity to these inhibitors, aIL7R leukemias were also sensitive to entospletinib in vitro (Fig. 7l, m). Finally, aIL7R/Ikzf1 leukemias displayed specific sensitivity to ibrutinib (Supplementary Fig. 7c, d) and trametinib (Supplemental Fig. 7e, f), which suggest that this genotype may rely on different essential pathways compared with aIL7R alone or aIL7R/Sh2b3. These data further validate that aIL7R leukemias exhibit active BCR-like signaling, especially via the Syk and PI3K pathways, and suggest that these compounds could be effective as part of combinations with Jak and/or mTOR inhibitors to target aIL7R-driven leukemias.
DISCUSSION
To date, preclinical modeling of Ph-like ALL has been performed predominantly using cell lines or patient-derived xenografts implanted into immunodeficient animals. While these models have contributed substantively to the understanding of Ph-like ALL, each has limitations (i.e. cell passage limits, lack of an immune-competent tumor microenvironment, etc.) that may impact translation. Here, we report a new activated IL7R GEMM as a model of human Ph-like ALL and other leukemia subtypes and demonstrate that B cell-intrinsic expression of aIL7R is sufficient to initiate B-ALL in mice. Importantly, the resultant leukemias reproducibly acquire mutations in oncogenes and tumor suppressors commonly associated with Ph-like ALL. Furthermore, concomitant heterozygous loss of Sh2b3 or co-expression of a dominant-negative Ikzf1 variant that frequently occur in human ALL markedly increased leukemia penetrance. Notably, our GEMM effectively mirrors key signaling programs in human IL7R-mutant ALL, including constitutive STAT5 and mTOR signaling, as well as a unique BCR-like signaling program despite the absence of surface pre-BCR, which resulted in rapid, penetrant, and spontaneous disease in immune-competent animals. Finally, based on our initial in vitro and in vivo studies, we predict that this preclinical aIL7R GEMM can effectively model candidate therapeutic interventions in an immune-competent setting and is representative of this high-risk human B-ALL subtype.
Leukemogenic potential has previously been described for primary murine hematopoietic cells or lymphoid progenitors overexpressing aIL7R mutant proteins following adoptive transfer into immunocompromised mice [10, 14, 38]. In these studies, mutant IL7R, alone or in combination with those in NRAS or NOTCH1, led to T-cell ALLs, myeloproliferative disorders, or IgM+ B cell lymphomas [14, 38]. More recently, Geron et al. has demonstrated a similar leukemogenic potential for human hematopoietic progenitors overexpressing the PPCL mutant version of IL-7R [39]. In contrast, our study provides the first demonstration that B lineage-restricted expression of aIL7R at or below endogenous IL-7R levels is sufficient to initiate B-ALL in primary B cell progenitor cells. We speculate that leukemic transformation in the aIL7R model requires aIL7R-induced signaling and stereotypical secondary genetic hits. While additional work is needed to determine how SH2B3 or IKZF1 deletions specifically contribute to disease, our genetic studies demonstrate that some acquired lesions in aIL7R leukemias appear genotype-specific with aIL7R/Ikzf1 leukemias consistently exhibiting total loss of functional Ikzf1. Our findings showing co-occurrence of mutant Trp53 in primary aIL7R/Sh2b3 leukemias also provide direct demonstration of previously modeled synergistic impacts of Sh2b3 and Tp53 in mouse cancer mortality [16]. These observed genomic landscapes suggest that although aIL7R-Mb1/-CD19, aIL7R Sh2b3fl/+ Mb1, and aIL7R Ikzf1fl/+ CD19 animals all develop B-ALL, their journey to leukemia transformation may indeed be quite distinct.
In addition to associated genetic alterations, we predict that active IL-7R signaling may additionally play a direct role in ALL progression. Prior to disease, the pro-B cell expansion observed in aIL7R mice mirrors the effects of in vivo exogenous IL-7 administration to wildtype animals [40–42]. IL-7 hypersensitivity of pre-leukemic B cells and preferential expansion at the expense of normal progenitor cells has been reported by several groups [16, 43–45]. IL-7R has also been shown to be required for the leukemogenesis of BCR-ABL1-transduced murine pre-B cells in vivo, while IL-7 can diminish the inhibitory effect of dasatanib treatment on similarly derived BCR-ABL1+ leukemogenic cells in vitro [46, 47]. Indeed, our in vitro cytokine stimulation studies using both aIL7R mouse leukemias and Ph-like ALL PDX cells suggest that endogenous IL-7 may provide an important survival signal for Ph-like leukemogenesis and maintenance. However, our limited studies of in vivo IL-7 antibody blockade did not appreciably affect leukemia proliferation in either our aIL7R mouse or human PALJDL models. Notably, recent work by Abdelrasoul et al. demonstrated a suppressive effect of in vivo IL-7 receptor blockade on BCR-ABL1+ xenograft models [47]. These findings suggest that combinatorial therapy using anti-IL-7/IL-7R in association with an additional target pathway may be a valuable next step in preclinical studies.
Ph-like ALL is characterized by a kinase-activated gene expression profile driven by JAK/STAT, PI3K pathway, and other signaling networks. Strikingly, our quantitative phosphoproteomic analyses independently identified multiple canonical “BCR-like” effectors in all aIL7R mouse leukemias and in our human IL7R-mutant/SH2B3-deleted and several other primary Ph-like ALL PDX models. Our findings are concordant with recent studies by Hurtz and colleagues, who reported BCR-like activation in CRLF2-rearranged Ph-like ALL cell lines and PDX cells lacking μHC surface expression via orthogonal transcriptional analysis and targeted immunoblotting to support the concept that BCR-like signaling was orchestrated via CD79A/B and SRC family kinases [37]. Upstream events mediating the BCR-like signaling program in aIL7R Ph-like and other leukemias remain to be determined; however, alternative non-BCR pathway proteins potentially capable of scaffolding Syk activation include integrins, C-type lectins, Fc or complement receptors (reviewed in [48]), and the surrogate light chain-only pro-BCR complex [26, 28]. Integrins comprise an important candidate, as large pre-B cells from Ikzf1fl/fl CD19 mice have enhanced expression of integrins α5, α6, and activated β1, and show an increase in FAK activation [20]. While technically challenging, it will also be important to determine whether this BCR-like program is active in aIL7R-expressing progenitors prior to overt disease development, or whether this phenomenon is a feature of transformation and/or secondary mutation acquisition. Regardless, consistent with recent data in CRLF2-rearranged Ph-like ALL [37] and KMT2A-rearranged infant B-ALL [49], our results suggest that targeting this BCR-like program with Syk inhibitors (in parallel with other pathway agents) could potentially provide therapeutic benefit for aIL7R leukemias in vivo in preclinical models and in patients.
In summary, our aIL7R GEMM provides the first definitive demonstration of the leukemia-initiating role of aIL7R, a robust new platform for detailed and iterative investigation of combinatorial therapies to treat primary aIL7R B-ALL, and a system for identifying additional biochemical and genetic events that facilitate B cell leukemic transformation, growth, and survival. Our inducible strain should further permit analogous lineage-restricted modeling of other aIL7R-driven leukemias, including T-ALL.
MATERIALS AND METHODS
Cell lines
Nalm6 (human pre-B; ATCC) and Ba/F3 (murine pro-B; Immunex, Seattle, WA, USA) cells were maintained in RPMI-1640 (HyClone) supplemented with 10% fetal calf serum (Omega Scientific), 1X GlutaMAX (Invitrogen), 20 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), 55 μm 2-mercaptoethanol (2ME; GIBCO), sodium pyruvate, and 10 ng/mL recombinant murine IL-3 (mIL-3 – Ba/F3 only; R&D Systems). Nalm6 cells were validated by surface immunophenotyping. Ba/F3 cells were authenticated by their inability to grow in the absence of murine IL-3.
Ba/F3 assays
Parental Ba/F3 cells were transduced with pRRL-MND-2A-mCherry lentiviral vectors containing WT or mutant murine Il7r (Il7r_P243insPPCL) followed by a self-cleaving T2A-linked mCherry reporter. Transduction efficiency (% mCherry+) was measured after 4 days. For assessment of cytokine independence, parental, Il7r_WT, and Il7r_PPCL Ba/F3 cells were subjected to mIL-3 withdrawal for 6 days. Cell counts, viability, and %mCherry+ were quantified at days 3 and 6.
Patient-derived xenografts
Secondary Ph-like ALL xenograft samples were provided by S. Tasian (Children’s Hospital of Philadelphia, Philadelphia, PA). 1e06 splenocytes were injected retro-orbitally (r.o.) into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) animals. Leukemia burden was assessed biweekly by flow cytometric analysis of peripheral blood. Recipient animals were euthanized when ill according to standard parameters set on the IACUC protocol.
Animals
Murine Il7r_P243insPPCL coding region was cloned into the pRosa26-DEST vector (Gateway) with an upstream loxp-flanked 3×-stop codon and a downstream self-cleaving T2A-linked GFP reporter. This targeting vector was then inserted into the Rosa26 locus via homologous recombination after electroporation into C57BL6/J ES cells (Biocytogen). These activated Il7r “aIL7R” mice were crossed to Mb1-Cre [50] and CD19-Cre [51] mice. aIL7R-Mb1 mice were also crossed to Sh2b3 knockout and floxed mice [24] and aIL7R mice were crossed to B-intrinsic Ikzf1 exon 5 floxed animals [20]. NSG (005557), Mb1-Cre (020505), CD19-Cre (006785), and C57BL/6 J (000664) mice were purchased from Jackson Laboratories. All mice were bred and maintained in a specific pathogen-free animal facility, and studies were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of Seattle Children’s Research Institute. Mice were sorted into experimental groups based on genotype and matched for age and sex. Experimental groups were co-housed whenever possible. aIL7R and littermate mice used for immunophenotyping, cell cycle analysis, phospho-Stat5 assays, and in vivo IL-7 blockade were 6–11 weeks old. aIL7R mice were monitored for signs of spontaneous disease and were euthanized when ill according to standard parameters set on the IACUC protocol.
Reagents and antibodies
The LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Invitrogen) was used according to the manufacturer’s instructions. Anti-mouse and anti-human antibodies used in the study are described in detail in Supplementary Table 8.
Flow cytometric and cell cycle analysis
Single-cell suspensions and antibody staining of peripheral blood, spleen, and BM were obtained as previously described [52]. Transcription factor labeling was performed following surface staining for 20 min at 4 °C by using the True-Nuclear Transcription Factor Buffer Set (BioLegend) according to the manufacturer’s instructions. For Ki-67 staining, cells were fixed for 30 min at 4 °C with fixation/permeabilization solution (BD Biosciences), followed by staining with Ki-67 under the same conditions. In vivo EdU labeling 1 h prior to sacrifice and detection for cell cycle analysis was performed as previously described [53]. All flow cytometric data were collected on an LSR II (BD) and analyzed by using FlowJo software (TreeStar).
In vitro cytokine stimulations and Phosphoflow analysis
Mouse splenic B cells were enriched using CD43 depletion (Miltenyi) and purified B cells were cultured in supplemented RPMI-1640 at 37 °C. B cells were seeded at 5e05 cells/well in a 96-well plate with or without recombinant murine IL-7 (mIL-7) [54] (25 ng/mL) for 25 mins and fixed with 2% PFA in PBS for 10 min at 37 °C. Cells were resuspended in cold Perm Buffer III (BD Biosciences) and stained with surface and phospho-site antibodies for 35 min at 4 °C in the dark. For ex vivo aIL7R leukemia analysis, 1e06 cells harvested from BM and spleen were directly fixed, permeabilized, and stained as above for phosphoflow analysis. Mouse bone marrow late pro-B cells were FACS-sorted into 96-well plates at a density of 1.5e04 cells/well in supplemented RPMI-1640 medium, rested for 2 h at 37 °C and stimulated with mIL-7 (10 ng/mL) for 25 min. Cells were then fixed, permeabilized, and stained for phosphoflow analysis. Il7r_WT and Il7r_PPCL expressing Ba/F3 cells were sorted on mCherry expression and maintained in supplemented RPMI-1640 with 10 ng/mL mIL-3. Cells were washed to remove cytokine, starved for 5 h, stimulated with 10 ng/mL mIL-7 or 10 ng/mL mIL-3 for 20 mins, fixed and stained for phosphoflow analysis.
For human xenografts, splenocytes from NSG recipients were plated at a density of 5e05 cells/well in a 96-well plate in supplemented RPMI-1640 with or without recombinant human IL-7 (PeproTech) or human TSLP (R&D Systems) (10 ng/mL) for 25 min at 37 °C. Cells were then fixed, permeabilized, and stained for phosphoflow analysis.
Histopathology
Tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, and tissue sections stained with Hematoxylin and eosin (H&E) according to standard practices. Histology images were acquired using a Nikon OptiPhot-2 microscope and a Canon Eos 5D Mark II camera. Tissue sections were examined by a board-certified veterinary pathologist (H.D.L.) who was blinded to the study design.
Igh sequencing
Murine Igh gene transcripts were amplified from leukemic splenocyte cDNA as described previously, using only the first round PCR of the nested PCR strategy [55]. Amplified sequences were purified using the NucleoSpin Gel & PCR Cleanup Kit (Macherey-Nagel), cloned into pJET1.2 plasmid DNA using the CloneJET PCR Cloning Kit (Thermo Fisher Scientific) and Sanger sequencing of individual transformed bacterial colonies was performed using the pJET1.2 forward and reverse sequencing primers. 96 colonies were sequenced per leukemia sample.
Exome sequencing and analysis
Genomic DNA (gDNA) from sorted primary aIL7R leukemia samples (spleen) and paired tail gDNA was submitted for whole-exome sequencing using Twist Mouse Exome Panel with paired end 100 bp NGS reads on NovaSeq (Illumina). FASTQ files were aligned using BWA to mm10 reference mouse genome. Unique leukemia somatic variants were identified by either Mutect2 [56] or Pisces [57]. Mutect2 variants were subsequently filtered using FilterMutectCalls and annotated using ANNOVAR [58]. Pisces variants in the tumor sample were annotated using ANNOVAR and then filtered for coverage and if present in more than one read in the normal sample. Variants were visualized using Integrated Genome Viewer (IGV; Broad Institute).
aIL7R leukemia serial transfer studies
1e06 splenocytes from aIL7R animals with spontaneous disease were injected r.o. into C57BL/6 mice. Leukemic burden was assessed biweekly by flow cytometric analysis of peripheral blood. Recipient animals were euthanized when ill according to IACUC protocol.
In vivo IL-7 blockade and rapamycin treatment
aIL7R Sh2b3fl/+ Mb1 and cre-negative littermate control mice were treated i.p. with 0.5 mg/mouse isotype control or neutralizing anti-IL-7 mAb (Bio X Cell) every third day for 16 days (6 injections) and sacrificed on day 18. For murine leukemias, B6 recipient mice were pre-treated i.p. with isotype or anti-IL-7 every other day for 6 days (3 injections), then injected r.o. with 1e06 splenocytes from primary aIL7R/Sh2b3 leukemias. Recipient mice were treated every other day with isotype or anti-IL-7 until they met euthanasia criteria.
NSG mice were injected r.o. with 1e06 IL-7Rins/SH2B3null (PALJDL) PDX cells transduced to express a firefly luciferase-eGFP fusion protein (ffluc-GFP; gift from Michael Jensen). In vivo bioluminescent imaging of PALJDL-ffluc-GFP cells was performed as previously described [59] using an IVIS Spectrum Imager (PerkinElmer). 8 days post-injection (after leukemic cells were detectable by bioluminescent imaging), mice were injected i.p. with 0.5 mg/mouse isotype or anti-IL-7, or with 0.1 mg/kg rapamycin (Selleck Chemicals) as a positive control. Mice were treated every other day for 8 weeks and in vivo bioluminescent imaging was used ~2x weekly to track leukemia burden over time. All animals were sacrificed on day 58.
1e06 aIL7R primary leukemia cells were injected r.o. into C57BL/6 recipient mice. Three days later, recipients were treated by gavage with 5 mg/kg rapamycin or vehicle (5% dextrose in water) daily (Mon-Fri) for up to 4 weeks or until euthanasia. Leukemic burden was assessed weekly by flow cytometric analysis of peripheral blood.
In vitro drug sensitivity assay
Splenocytes from primary or secondary leukemic animals were plated in supplemented RPMI-1640 with 10 ng/mL mIL-7 in 96 well plates at a density of 5e04 cells/well. Cells were cultured for 24 h with or without ruxolitinib, rapamycin, bortezomib, entospletinib, ibrutinib, idelalisib (Selleck Chemicals), or trametinib (Cayman Chemical). Inhibitors were tested at a 10-fold dilution curve ranging from 10 uM to 100 pM or 1 uM to 10 pM (Bortezomib). Viability of cells was measured at 24 and 48 h by adding 10 uL of CellTiter-Glo reagent (Promega) per well and incubating for 10 mins with shaking at room temperature (RT) before luminescence is recorded. Luminescence was read by using a SpectraMax i3x microplate reader (Molecular Devices).
Mass spectrometry-based phosphoproteomic analysis
Secondary murine aIL7R leukemias were harvested ex vivo from spleen and plated into 10 cm dishes at a density of 1e07 cells/mL. Cells were incubated at 37 °C with or without 1 uM entospletinib or ruxolitinib (Selleck Chemicals) for 30 mins, and then with or without 10 ng/mL mIL-7 for an additional 15 min. Cells were harvested, washed with 1X PBS (HyClone), and lysed. For Ph-like PDX models, cells were harvested ex vivo from spleen and 1e08 cells/sample were directly lysed.
Cell pellets were lysed in 8 M Urea containing phosphatase inhibitors and sonicated. Protein concentration was then determined by a BCA assay (Pierce). 1–2 mg of each sample was reduced with 5 mM DTT at 56 °C for 45 min, then alkylated with 15 mM iodoacetamide for 30 min at RT in the dark. Samples were diluted 4-fold with 100 mM ammonium acetate, pH 8.9, and digested with Lysyl endopeptidase (Lys-C, from Wako) for 4 h at RT at a ratio of 1:100 (Lys-C to total protein), followed by sequencing grade modified trypsin (Promega) at a ratio of 1:100 (trypsin to total protein), overnight at room temperature. Following digestion, peptides were desalted and concentrated using Sep-Pak Plus C18 cartridges (Waters) according to the manufacturer’s recommendations. Samples were then dried by vacuum centrifugation, lyophilized, and stored at −80 °C until further processing.
Protein-G agarose (Sigma) was reconstituted in water for 1 h at room temperature and washed three times in IP buffer (100 mM Tris pH 7.4, 0.3% NP-40). Beads were resuspended in IP buffer containing three antibodies (4G10 from Sigma, pY100 and pY1000 from Cell Signaling Technologies) and placed on a rotator for 6–8 h at 4 °C. Beads were washed with IP buffer and added to lyophilized peptides in IP buffer, overnight at 4 °C. Beads were washed with IP buffer, wash buffer (100 mM Tris pH 7.4), and with water. Peptides were eluted with 15% Acetonitrile containing 0.15% trifluoroacetic acid. For human PDX samples, heavy labeled peptides were added, and in all cases the acetonitrile was removed using vacuum centrifugation.
Online peptide separation coupled to MS/MS was performed with a nanoLC system (nanoAcquity UPLC system, Waters) and a Q-exactive mass spectrometer (Thermo Fisher Scientific). Peptide samples were loaded onto a trap column (Bruker C18 MAGIC beads, 200 A pore size, 4 cm packing length, 100 μm column inner diameter) connected to an analytical column (Bruker C18 MAGIC beads, 100 A pore size, 20 cm packing length, 75 μm column inner diameter) with an incorporated electrospray emitter. Peptide separation was achieved using a gradient from 3 to 40% (V/V) of ACN in 0.1% FA over 40 or 115 min at a flow rate of 250 nL/min.
A top 10 method was used for data-dependent/discovery acquisitions. Full MS scans were acquired at a resolution of 70,000, a target automatic gain control (AGC) value of 1e6, with a maximum fill time of 100 ms. MS2 scans were acquired at a resolution of 17,500 with an AGC target of 5e4 and a maximum fill time of 200 ms, and a normalized collision energy of 28.
Parallel Reaction Monitoring (PRM) methods were used to target specific phosphorylated peptides. The acquisition method combined 5 sequential PRM events followed by a full scan event. Precursor ions of the peptides were targeted in preliminary experiments without time-scheduling and confirmed precursors were targeted ±5 min of the observed elution times in the actual experimental runs. PRM events were acquired with an orbitrap resolution of 17,500, an AGC value of 2e5, and maximum fill times of 100 ms, and a normalized energy of 28. The precursor ion of each targeted peptide was isolated using a 2 m/z window. Full MS scans were acquired at a resolution of 70,000, an AGC value of 3e6, and maximum fill times of 240 ms.
Data processing and analysis
For DDA analysis, MS data files were searched using the COMET algorithm [60] and the output was imported into the Trans-Proteomic Pipeline [61] with the following parameters: variable oxidation of methionine (15.99 Da), variable phosphorylation (79.96 Da) of serine, threonine, or tyrosine, up to 4 variable modifications per peptide, fixed carbamidomethylation of cysteine (57.02 Da), two missed cleavages, maximum charge of 7. Peptide false discovery rate (FDR) was set to 1%.
Quantitative analysis of pY peptides was performed with Skyline software [62]. Quantifiable fragment ions for each peptide were confirmed and an intensity value was determined by summing the area under the curve gleaned from a subset of fragment ion and/or precursor ion isotopic peaks (minimum peak number = 3). For mouse pY quantification, the intensity value for each peptide was normalized to that for a phosphorylated peptide from Fgr within the same sample prior to assessing changes in relative peptide abundance. For human PDX models, the intensity values for endogenous peptides were first normalized to that of the corresponding spiked-in heavy peptide and then to that of GSK3B pY216.
Statistical analysis
All statistical analyses were performed using graphPad Prism version 8.4.2 unless specified above. All specific statistical tests and P-values are indicated in the relevant figures. In all summary figures, a single data point represents an individual mouse, and bars indicate the mean ± SEM. One-way and two-way ANOVA tests were adjusted for multiple comparisons using Tukey’s (one-way) or Sidak’s (two-way) multiple comparisons tests. No sample size calculations were performed. Experiments were generally repeated 2 or more times with at least two mice or leukemias per genotype/group. An exception is the characterization of the mice with spontaneous leukemia - those experiments typically contained just the sick mouse and a healthy littermate control, as it was infrequent that multiple animals with spontaneous disease would meet euthanasia criteria at the same time. Data were only excluded when there were a technical issue with administering the EdU i.p. injections and no labeling was detected upon flow cytometry analysis (Fig. 4e–f).
Materials availability
The Rosa26-aIL7R mouse strain and primary murine aIL7R leukemia samples are available from the corresponding author upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Karen Sommer and Andrea Repele for laboratory management, Jennifer Haddock for administrative support, Beth Lawlor for helpful comments and critical review of the manuscript, the University of Washington Histology and Imaging Core, and the Northwest Genomics Center at the University of Washington. This work was supported by the National Institutes of Health under award numbers: TL1-TR000422 (to KRT), DP3-DK111802 (to DJR), NCI-R01CA201135-A1 (to RGJ), 1K08CA184418 (to SKT), 1U01CA232486 (to SKT), 1U01CA243072 (SKT), 1K08DK114568-01 (to EJA), Department of Defense Translational Team Science Award CA180683P1 (to SKT), the V Foundation for Cancer Research (to SKT), the Seattle Children’s Research Institute (SCRI) Center for Immunity and Immunotherapies (CIIT) Program for Cell and Gene Therapy (PCGT), the Children’s Guild Association Endowed Chair in Pediatric Immunology (to DJR), and the Hansen Investigator in Pediatric Innovation Endowment (to DJR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
COMPETING INTERESTS
SKT receives research funding from Incyte Corporation, Gilead Sciences, and MacroGenics, Inc. for unrelated studies and is a member of the scientific advisory board of Aleta Biotherapeutics for unrelated studies.
Footnotes
DATA AVAILABILITY
The mapped reads for the whole-exome sequencing have been deposited in the Sequence Read Archive database (BioProject ID: PRJNA735253). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [63] partner repository with the dataset identifiers PXD026438 and PXD026322.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-021-01326-x.
REFERENCES
- 1.Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood. 2017;130:2064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines J, Peters S, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts KG, Morin RD, Zhang JH, Hirst M, Zhao YJ, Su XP, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology-Am Soc Hematol Educ Program. 2014;2014:174–80. [DOI] [PubMed] [Google Scholar]
- 6.Zenatti PP, Ribeiro D, Li WQ, Zuurbier L, Silva MC, Paganin M, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43:932–U931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, Kronnie GT, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208:901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. 2014;14:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corfe SA, Paige CJ. The many roles of IL-7 in B cell development; Mediator of survival, proliferation and differentiation. Semin Immunol. 2012;24:198–208. [DOI] [PubMed] [Google Scholar]
- 10.Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in health and disease. Nat Immunol. 2019;20:1584–93. [DOI] [PubMed] [Google Scholar]
- 11.Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol. 2012;13:300–U124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchner M, Swaminathan S, Chen ZS, Muschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol Rev. 2015;263:192–209. [DOI] [PubMed] [Google Scholar]
- 13.Treanor LM, Zhou S, Janke L, Churchman ML, Ma ZJ, Lu TH, et al. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J Exp Med. 2014;211:701–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoyama K, Yokoyama N, Izawa K, Kotani A, Harashima A, Hozumi K, et al. In vivo leukemogenic potential of an interleukin 7 receptor alpha chain mutant in hematopoietic stem and progenitor cells. Blood. 2013;122:4259–63. [DOI] [PubMed] [Google Scholar]
- 15.Takaki S, Sauer K, Iritani BM, Chien S, Ebihara Y, Tsuji K, et al. Control of B cell production by the adaptor protein Lnk: definition of a conserved family of signal-modulating proteins. Immunity. 2000;13:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Y, Chikwava K, Wu C, Zhang HB, Bhagat A, Pei DH, et al. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. J Clin Investig. 2016;126:1267–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang JH, Nichogiannopoulou A, Wu L, Sun L, Sharpe AH, Bigby M, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5:537–49. [DOI] [PubMed] [Google Scholar]
- 18.Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, et al. The IKAROS gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–56. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida T, Ng SYM, Zuniga-Pflucker JC, Georgopoulos K. Early hematopoietic lineage restrictions directed by Ikaros. Nat Immunol. 2006;7:382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi I, Yoshida T, Jena N, Qi XQ, Zhang JW, Van Etten RA, et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014;15:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwickert TA, Tagoh H, Gultekin S, Dakic A, Axelsson E, Minnich M, et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat Immunol. 2014;15:283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katerndahl CDS, Heltemes-Harris LM, Willette MJL, Henzler CM, Frietze S, Yang RD. et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat Immunol. 2017;18:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge Z, Gu Y, Xiao LC, Han Q, Li JY, Chen BA, et al. Co-existence of IL7R high and SH2B3 low expression distinguishes a novel high-risk acute lymphoblastic leukemia with Ikaros dysfunction. Oncotarget. 2016;7:46014–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allenspach E, Shubin N, Cerosaletti K, Mikacenic C, Gorman J, MacQuivey M, et al. The autoimmune risk R262W variant of the adaptor SH2B3 improves survival in sepsis. J Immunol [In preparation/revision] 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullighan CG, Su XP, Zhang JH, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N Engl J Med. 2009;360:470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meffre E, Fougereau M, Argenson JN, Aubaniac JM, Schiff C. Cell surface expression of surrogate light chain (Psi L) in the absence of mu on human pro-B cell lines and normal pro-B cells. Eur J Immunol. 1996;26:2172–80. September [DOI] [PubMed] [Google Scholar]
- 27.Lemmers B, Arnoulet C, Fossat C, Chambost H, Sainty D, Gabert J, et al. Fine characterization of childhood and adult acute lymphoblastic leukemia (ALL) by a proB and preB surrogate light chain-specific mAb and a proposal for a new B cell ALL classification. Leukemia. 2000;14:2103–11. [DOI] [PubMed] [Google Scholar]
- 28.Lemmers B, Gauthier L, Guelpa-Fonlupt V, Fougereau M, Schiff C. The human (Psi L+mu(-)) proB complex: Cell surface expression and biochemical structure of a putative transducing receptor. Blood. 1999;93:4336–46. [PubMed] [Google Scholar]
- 29.Goldberg L, Gough SM, Lee F, Dang C, Walker RL, Zhu YLJ, et al. Somatic mutations in murine models of leukemia and lymphoma: Disease specificity and clinical relevance. Genes Chromosomes Cancer. 2017;56:472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gough SM, Goldberg L, Pineda M, Walker RL, Zhu YJ, Bilke S, et al. Progenitor B-1 B-cell acute lymphoblastic leukemia is associated with collaborative mutations in 3 critical pathways. Blood Adv. 2017;1:1749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu LW, Chaudhury P, Osmond DG. Regulation of cell survival during B lymphopoiesis: Apoptosis and Bcl-2/Bax content of precursor B cells in bone marrow of mice with altered expression of IL-7 and recombinase-activating gene-2. J Immunol. 1999;162:1931–40. [PubMed] [Google Scholar]
- 32.Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, et al. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol. 2010;11:171–U197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grillot DAM, Merino R, Pena JC, Fanslow WC, Finkelman FD, Thompson CB, et al. bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J Exp Med. 1996;183:381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120:3510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120:833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129:177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hurtz C, Wertheim GB, Loftus JP, Blumenthal D, Lehman A, Li Y, et al. Oncogene-independent BCR-like signaling adaptation confers drug resistance in Ph-like ALL. J Clin Investig. 2020;130:3637–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cramer SD, Hixon JA, Andrews C, Porter RJ, Rodrigues GOL, Wu XL, et al. Mutant IL-7R alpha and mutant NRas are sufficient to induce murine T cell acute lymphoblastic leukemia. Leukemia. 2018;32:1795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geron I, Savino AMS, Tal N, Brown J, Turati V, James C, et al. An instructive role for IL7RA in the development of human B-cell precursor leukemia. BioRxiv; 2020. 10.1101/2020.01.27.919951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Damia G, Komschlies KL, Faltynek CR, Ruscetti FW, Wiltrout RH. Administration of recombinant human interleukin-7 alters the frequency and number of myeloid progenitor cells in the bone-marrow and spleen of mice. Blood. 1992;79:1121–9. [PubMed] [Google Scholar]
- 41.Komschlies KL, Gregorio TA, Gruys ME, Back TC, Faltynek CR, Wiltrout RH. Administration of recombinant human IL-7 to mice alters the composition of b-lineage cells and t-cell subsets, enhances t-cell function, and induces regression of established metastases. J Immunol. 1994;152:5776–84. [PubMed] [Google Scholar]
- 42.Morrissey PJ, Conlon P, Braddy S, Williams DE, Namen AE, Mochizuki DY. Administration of IL-7 to mice with cyclophosphamide-induced lymphopenia accelerates lymphocyte repopulation. J Immunol. 1991;146:1547–52. [PubMed] [Google Scholar]
- 43.Digel W, Schmid M, Heil G, Conrad P, Gillis S, Porzsolt F. Human interleukin-7 induces proliferation of neoplastic-cells from chronic lymphocytic-leukemia and acute leukemias. Blood. 1991;78:753–9. [PubMed] [Google Scholar]
- 44.Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of Pax5-inherited susceptibility. Cancer Discov. 2015;5:1328–43. [DOI] [PubMed] [Google Scholar]
- 45.Nakayama J, Yamamoto M, Hayashi K, Satoh H, Bundo K, Kubo M, et al. BLNK suppresses pre-B-cell leukemogenesis through inhibition of JAK3. Blood. 2009;113:1483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Appelmann I, Rillahan CD, de Stanchina E, Carbonetti G, Chen C, Lowe SW, et al. Janus kinase inhibition by ruxolitinib extends dasatinib- and dexamethasone-induced remissions in a mouse model of Ph plus ALL. Blood. 2015;125:1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdelrasoul H, Vadakumchery A, Werner M, Lenk L, Khadour A, Young M, et al. Synergism between IL7R and CXCR4 drives BCR-ABL induced transformation in Philadelphia chromosome-positive acute lymphoblastic leukemia. Nature. Communications 2020;11:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mocsai A, Ruland J, Tybulewicz VLJ. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loftus JP, Yahiaoui A, Brown PA, Niswander LM, Bagashev A, Wang M, et al. Combinatorial efficacy of entospletinib and chemotherapy in patient-derived xenograft models of infant acute lymphoblastic leukemia. Haematologica. 2021;106:1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA. 2006;103:13789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu CH, et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med. 2011;208:2033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wray-Dutra MN, Chawla R, Thomas KR, Seymour BJ, Arkatkar T, Sommer KM, et al. Activated CARD11 accelerates germinal center kinetics, promoting mTORC1 and terminal differentiation. J Exp Med. 2018;215:2445–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bandaranayake AD, Correnti C, Ryu BY, Brault M, Strong RK, Rawlings DJ. Dae-dalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res. 2011;39:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwartz MA, Kolhatkar NS, Thouvenel C, Khim S, Rawlings DJ. CD4(+) T cells and CD40 participate in selection and homeostasis of peripheral b cells. J Immunol. 2014;193:3492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dunn T, Berry G, Emig-Agius D, Jiang Y, Lei S, Iyer A, et al. Pisces: an accurate and versatile variant caller for somatic and germline next-generation sequencing data. Bioinformatics. 2019;35:1579–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hale M, Lee B, Honaker Y, Leung WH, Grier AE, Jacobs HM, et al. Homology-directed recombination for enhanced engineering of chimeric antigen receptor T Cells. Mol Ther Methods Clin Dev. 2017;4:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eng JK, Jahan TA, Hoopmann MR. Comet: An open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. [DOI] [PubMed] [Google Scholar]
- 61.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, et al. A guided tour of the trans-proteomic pipeline. Proteomics. 2010;10:1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez-Riverol Y, Csordas A, Bai JW, Bernal-Llinares M, Hewapathirana S, Kundu DJ. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.