

Abstract
Objectives
AXL‐mediated activation of aberrant tyrosine kinase drives various oncogenic processes and facilitates an immunosuppressive microenvironment. We evaluated the anti‐tumor and anti‐metastatic activities of SKI‐G‐801, a small‐molecule inhibitor of AXL, alone and in combination with anti‐PD‐1 therapy.
Methods
In vitro pAXL inhibition by SKI‐G‐801 was performed in both human and mouse cancer cell lines. Immunocompetent mouse models of tumor were established to measure anti‐metastatic potential of SKI‐G‐801. Furthermore, SKI‐G‐801, anti‐PD‐1 or their combination was administered as an adjuvant or neoadjuvant in the 4T1 tumor model to assess their potential for clinical application.
Results
SKI‐G‐801 robustly inhibited pAXL expression in various cell lines. SKI‐G‐801 alone or in combination with anti‐PD‐1 potently inhibited metastasis in B16F10 melanoma, CT26 colon and 4T1 breast models. SKI‐G‐801 inhibited the growth of B16F10 and 4T1 tumor‐bearing mice but not immune‐deficient mice. An antibody depletion assay revealed that CD8+ T cells significantly contributed to SKI‐G‐801‐mediated survival. Anti‐PD‐1 and combination group were observed the increased CD8+Ki67+ and effector T cells and M1 macrophage and decreased M2 macrophage, and granulocytic myeloid‐derived suppressor cell (G‐MDSC) compared to the control group. The neoadjuvant combination of SKI‐G‐801 and anti‐PD‐1 therapy achieved superior survival benefits by inducing more profound T‐cell responses in the 4T1 syngeneic mouse model.
Conclusion
SKI‐G‐801 significantly suppressed tumor metastasis and growth by enhancing anti‐tumor immune responses. Our results suggest that SKI‐G‐801 has the potential to overcome anti‐PD‐1 therapy resistance and allow more patients to benefit from anti‐PD‐1 therapy.
Keywords: AXL, immunotherapy, kinase inhibitor, metastasis, small molecule
SKI‐G‐801, a novel AXL inhibitor, significantly suppressed tumor metastasis and growth by enhancing anti‐tumor immune responses. Our results suggest that SKI‐G‐801 has the potential to overcome resistance to anti‐PD‐1 therapy and expand the number of patients who benefit from anti‐PD‐1 therapy.

Introduction
AXL participates in many signal transduction cascades after being stimulated by growth arrest‐specific gene 6 (GAS6). Overexpression and activation of AXL are associated with cell growth, 1 survival 2 , 3 and differentiation. 4 Notably, AXL is frequently overexpressed in human cancers, including lung cancer, 5 , 6 breast cancer 7 , 8 and renal cell carcinoma, 9 , 10 and aberrant AXL expression predicts poor prognosis. 11 , 12
Distant metastasis is a major cause of death in cancer patients despite recent advances in adjuvant and neoadjuvant strategies to constrain tumors to their primary site. 13 , 14 AXL promotes metastasis via inducing epithelial–mesenchymal transition (EMT), 15 , 16 upregulating the expression of metalloproteinases and cell motility proteins 17 and downregulating E‐cadherin expression. 18 Accordingly, genetic ablation and chemical inhibition of AXL significantly decrease tumor metastasis in vivo, suggesting that AXL is a promising therapeutic target for inhibiting cancer metastasis and dissemination. 19
AXL is abundantly expressed in various immune cells, including natural killer (NK) cells, dendritic cells (DCs) and macrophages. 1 , 20 , 21 Recent studies have suggested multifaceted roles of AXL in anti‐tumor immune responses. AXL performs essential functions in the removal of apoptotic cells by antigen‐presenting cells (APCs), 22 as depicted by autoimmune phenotypes with defected removal of apoptotic cells in AXL knockout mice. 7 AXL inhibition may enhance anti‐tumor immune responses via promoting antigen presentation by macrophages, decreasing anti‐inflammatory cytokine secretion and enforcing T‐cell recruitment and killing activity. AXL activation also induces programmed death‐ligand 1 (PD‐L1) expression in conjunction with the PI3K pathway, 23 and a previous analysis underscored AXL as one of the top‐ranked genes upregulated in anti‐programmed cell death protein 1 (PD‐1) therapy‐resistant tumors. 24 Collectively, this evidence suggests that AXL inhibition is a promising strategy to enhance anti‐tumor immune responses and the efficacy of immune checkpoint blockades.
Here, we comprehensively examined the therapeutic potential of SKI‐G‐801, a small‐molecule AXL inhibitor, as a mono‐ and combination therapy with an anti‐PD‐1 agent in suppressing tumor growth and metastasis, as well as in promoting effective anti‐tumor T‐cell responses in various syngeneic mouse models.
Results
SKI‐G‐801 is a novel, highly selective, small‐molecule AXL inhibitor
A novel small‐molecule AXL kinase inhibitor, SKI‐G‐801 (G‐749 HCl salt, Figure 1a), was synthesised using a structure‐based drug design. The pyridopyrimidine moiety of SKI‐G‐801 binds to the hinge residues Phe 622 (F622) and Met 623 (M623) through hydrogen bonding. The tertiary amine nitrogen of the methylpiperidin ring forms a hydrogen bond with Asp 627 (D627) carbonyl oxygen (Figure 1a). Computational modelling predicted that SKI‐G‐801 would bind to the ATP‐binding pocket of AXL kinase. To investigate the kinase profile of SKI‐G‐801, we performed a kinome profiling assay against 282 kinases using the Millipore kinase profiler (Supplementary figure 1a). The levels of kinase inhibition at 100 nm SKI‐G‐801, tested in duplicate, were compared with those of the DMSO control (0% inhibition). The average values were plotted to represent the human kinome phylogenetic tree using TREEspot from DiscoveRx. SKI‐G‐801 displayed a high kinase selectivity profile, based on the inhibition of 10 of the 282 kinases tested with > 85% cut‐off value (Supplementary figure 1a). Notably, SKI‐G‐801 showed potent activity against AXL kinase, with 91% inhibition. However, SKI‐G‐801 did not show an inhibitory effect on other kinase in vitro (Supplementary figure 1b).
Figure 1.
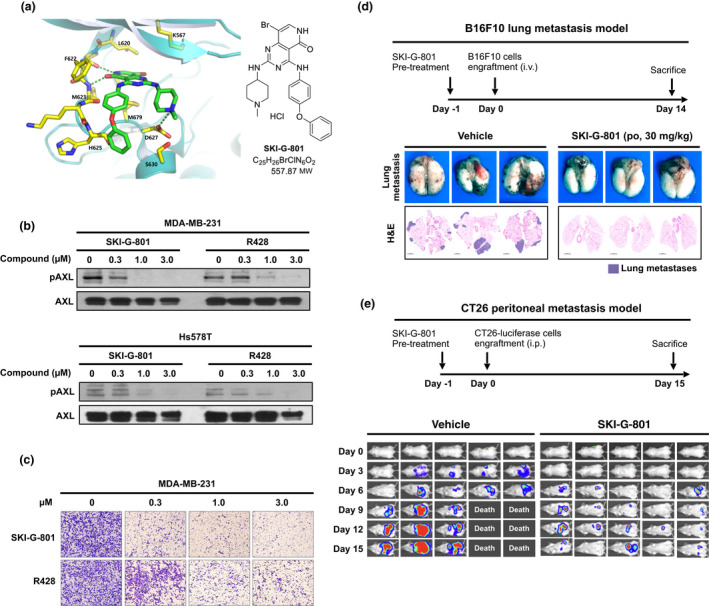
SKI‐G‐801 is a novel AXL kinase inhibitor. (a) Crystal structure of SKI‐G‐801 in AXL kinase domain (left). Chemical structure of SKI‐G‐801 (right). (b) Western blot analysis of pAXL inhibition by SKI‐G‐801 and R428 in MDA‐MB‐231 and Hs578T cells. The experiment was repeated twice. (c) MDA‐MB‐231 cells were incubated with either SKI‐G‐801 or R428 at the indicated concentrations for 48 h, followed by an invasion assay. The experiment was repeated twice. (d) B16F10 cells (1 × 106 cells per mouse, n = 3 per group, derived from two independent experiments) were injected intravenously. SKI‐G‐801 (30 mg kg−1 day−1) was administered from day −1 to day +14. The mice were sacrificed to evaluate the metastasis. (e) CT26‐luciferase cells (1 × 104 cells per mouse, n = 5 per group, derived from two independent experiments) were implanted into the intraperitoneal cavity. SKI‐G‐801 (30 mg kg−1, once a day) was administered from day −1 to day +15. Luciferase expression was determined on days 0, 3, 6, 9, 12 and 15 using the Xenogen in vivo imaging system.
BALB/c mice were orally administered SKI‐G‐801 (10 mg kg−1), and the plasma concentration of the drug was analysed to evaluate the pharmacokinetics. SKI‐G‐801 was rapidly absorbed with a maximum plasma concentration (C max) of 251.0 ng mL−1 and time to C max (T max) of 2 h (Supplementary table 1). Next, we tested the tissue distribution of SKI‐G‐801 using [14C] labelled SKI‐G‐801. SKI‐G‐801 was broadly distributed in all tissues tested in male and female SD rats. Based on the tissue/plasma ratio, the tissue distribution was particularly enriched in the lungs (61.0‐fold to 93.6‐fold), small intestine (25.8‐fold to 177.1‐fold) and liver (113.3‐fold to 142.5‐fold) (Supplementary table 2).
Two human breast cancer cell lines (MDA‐MB‐231 and Hs578T) were treated with increasing concentrations of SKI‐G‐801 (0.3, 1.0 and 3.0 μm), and immunoblotting was performed. Notably, SKI‐G‐801 decreased the phosphorylation of AXL in a concentration‐dependent manner (Figure 1b). As expected, SKI‐G‐801 treatment attenuated the migration and invasion of both cell lines (Figure 1c). Similar results were observed in two AXL‐overexpressing human lung cancer cells (Supplementary figure 2). SKI‐G‐801 also showed more prolonged AXL inhibition (Supplementary figure 3) and less ATP dependency (Supplementary table 3) than R428, another selective small‐molecule AXL inhibitor.
Collectively, these results suggest that SKI‐G‐801 is a potent and selective AXL kinase inhibitor with a favorable preclinical pharmacokinetic profile and broad tissue distribution, which makes it suitable for treating solid tumors.
SKI‐G‐801 inhibits lung and peritoneal metastasis in murine models
We hypothesised that SKI‐G‐801 inhibits metastasis by suppressing AXL signalling. To test our hypothesis, we used two metastatic murine models generating by utilising B16F10 melanoma and luciferase‐transfected CT26 colon carcinoma cells.
C57BL/6 and BALB/c mice were engrafted with B16F10 cells (intravenous) and luciferase‐expressing CT26 cells (intraperitoneal), respectively, and then administered SKI‐G‐801 (30 mg kg−1) after 24 h. Lung metastases of B16F10 melanoma cells, shown as black dots in the lung parenchyma, were significantly reduced by SKI‐G‐801 treatment (Figure 1d). Metastases to other organs, including the mesenteric lymph nodes, liver and kidneys, were also completely ameliorated by SKI‐G‐801, compared to the vehicle (data not shown). In addition, SKI‐G‐801 treatment drastically reduced luminescence intensity in the peritoneal cavity, compared to the vehicle (Figure 1e). The mice treated with the vehicle began to die on day 9 after CT26‐luciferase cell engraftment, whereas a significantly higher proportion of SKI‐G‐801‐treated mice survived during the treatment period (Figure 1e).
Combination of SKI‐G‐801 with anti‐PD‐1 synergistically inhibits spontaneous 4T1 lung metastasis
Immune checkpoint inhibitors (ICIs) targeting the programmed cell death protein 1 (PD‐1)/programmed cell death‐ligand 1 (PD‐L1) have shown impressive results in multiple cancer types. However, only a minority of patients respond to ICIs. 25 Therefore, we next examined whether the combination treatment of SKI‐G‐801 and anti‐PD1 cooperatively inhibited tumor growth and metastasis compared to either monotherapy. The 4T1 tumor is highly metastatic in vivo and is frequently used as a spontaneous metastasis model. 26
Previously, 4T1 cell lines were confirmed to exhibit a basal level of AXL expression and suppressive pAXL signalling by SKI‐G‐801 (Figure 2a). In addition, pAXL expression in 4T1 tumors was measured at 2, 8 and 24 h after SKI‐G‐801 treatment (Figure 2b). The 4T1 breast cancer cells (1 × 106 cells) were engrafted subcutaneously into BALB/c female mice (7 weeks of age). 27 When the tumor sizes reached approximately 500 mm3, the mice were treated with either the vehicle, anti‐PD‐1 (10 mg kg−1, twice a week), SKI‐G‐801 (30 mg kg−1, once a day) or a combination of anti‐PD‐1 and SKI‐G‐801. Mice were sacrificed on day 27, and lung metastasis was evaluated by counting metastatic nodules. Representative images of lung sections from the four treatment arms are shown in Figure 2c. Compared to the vehicle, anti‐PD‐1 alone did not reduce lung metastasis (Figure 2d, left). Remarkably, SKI‐G‐801 alone or in combination with anti‐PD‐1 significantly decreased lung metastases compared to the vehicle or anti‐PD‐1 alone (Figure 2d, left). Combination treatment ameliorated lung metastasis compared to SKI‐G‐801 single treatment. Interestingly, the inhibitory effects of the combination treatment on lung metastasis correlated with the proportion of immune cells in the tumor microenvironment (TME) of primary tumors. Flow cytometry results showed that the proportion of IFN‐γ‐expressing CD8+ T cells, an important immune cell type that influences the anti‐tumor effect, was significantly higher in the combination group than in the other groups (Figure 2e). Furthermore, the proportion of CCR7‐expressing central memory T cells was also significantly more in the combination group (Figure 2e). Central memory T cells play a vital anti‐cancer role by differentiating into effector T cells when challenged with an antigen and have increased self‐renewal capacity. 28
Figure 2.
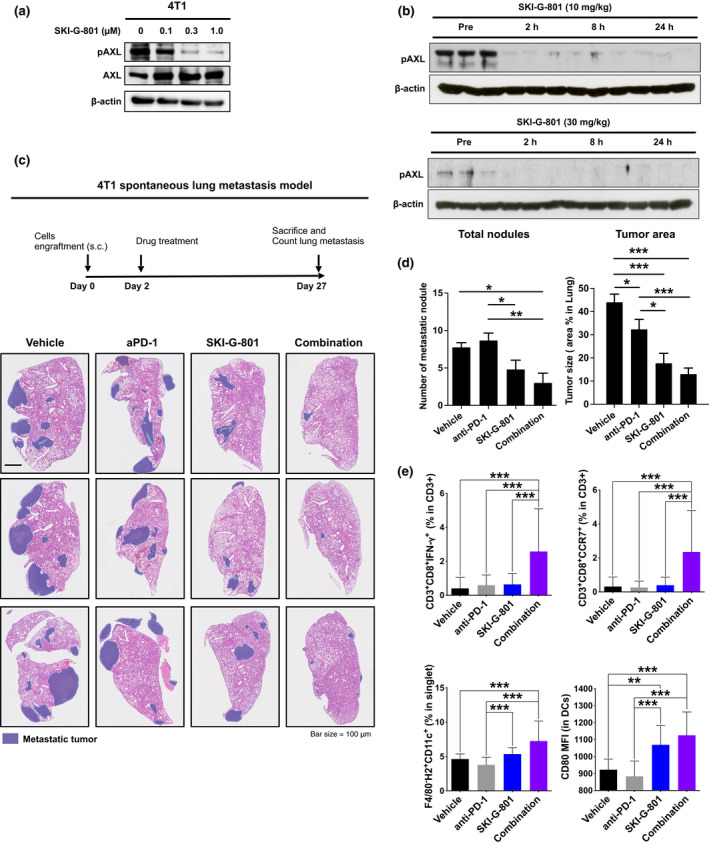
Combination of SKI‐G‐801 and anti‐PD‐1 synergistically inhibits spontaneous lung metastasis in the 4T1 model. (a) Western blot analysis for pAXL inhibition by SKI‐G‐801 in 4T1 cells. Experiments were repeated twice. (b) pAXL were measured in 4T1 tumor‐bearing syngeneic mice. Mice were treated with SKI‐G‐801 10 mg kg−1 (upper) and 30 mg kg−1 (lower) for up to 24 h. (c) Representative images of H&E‐stained lung tissue demonstrating lung metastases are shown. (d) Total number of metastatic nodules was counted, and tumor area in lung tissue was measured. Lung and percentage of tumor area were calculated with ImageJ (n = 4 or 6 per group, derived from two independent experiments). (e) The proportions of IFN‐γ‐expressing cytotoxic T cells (CD3+CD8+IFN‐γ+) and central memory CD8+ T cells (CD3+CD8+CCR7+), measured by flow cytometry, were significantly increased in the combination group compared to other treatment groups. The dendritic cells (DCs) population (F4/80−CD11c+H2+) and CD80 expression on DCs are shown. Values are indicated as means ± SEM, and P‐values were calculated using the Student’s t‐test; *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Flow cytometry analysis was done once.
In addition, the proportion of dendritic cells (DCs) in the TME, and expression of CD80/CD86, the costimulatory signal for T‐cell activation expressed in DCs, were significantly increased in the combination group compared to the vehicle or anti‐PD‐1 alone group (Figure 2e). The combination therapy exerted stronger effects on DCs than those by SKI‐G‐801 alone. SKI‐G‐801 alone and its combination with anti‐PD‐1 yielded similar growth inhibitory effects on the primary tumors (Supplementary figure 4). However, the proportion of infiltrating NK cells, which were reported to have an important role in AXL‐mediated anti‐metastasis, 20 to lung was significantly increased in combination group (Supplementary figure 5). These results suggested that AXL inhibition had more significant therapeutic impact and immunity can alter metastatic response, but may not directly inhibit the primary tumor. This prompted us to evaluate the influence of immunity in anti‐metastatic response.
Anti‐tumor effects of SKI‐G‐801 require CD8+ T cells
Despite the lack of synergy effects of combination therapy on primary tumor growth (Supplementary figure 4), its inhibitory effects of SKI‐G‐801 on metastasis in vivo were evident (Figures 1d and e and 2c). These findings led us to hypothesise that the anti‐metastatic effects of SKI‐G‐801 were immune‐mediated.
Compared to the significant inhibitory effects of SKI‐G‐801 on the growth of B16F10 and 4T1 tumors in the immunocompetent mice, no such effect was observed in the nude mice (Figure 3a). Next, we performed depletion experiments to determine the phenotype of effector cells mediating the anti‐tumor activity of SKI‐G‐801 by injecting anti‐CD8, anti‐CD4 and NK‐depleting anti‐NK 1.1 antibodies in SKI‐G‐801‐treated 4T1 tumor‐bearing immunocompetent mice (Figure 3b). Survival was not compromised following treatment with anti‐CD4 or anti‐NK 1.1 antibodies, compared to the IgG control. However, treatment with anti‐CD8 antibody significantly decreased survival compared to other groups (P < 0.01), which strongly supports our hypothesis that CD8+ T cells contribute significantly to the anti‐tumor activity of SKI‐G‐801.
Figure 3.

CD8+ T cells mediate the therapeutic effects of SKI‐G‐801. (a) B16F10 and 4T1 cells were separately implanted in immunocompetent or immunodeficient (nude) mice, and subsequently, the mice were treated with 30 mg kg−1 SKI‐G‐801 for the indicated number of days (n = 5 per group, derived from one independent experiment). Vehicle‐treated mice are marked in black (■), and SKI‐G‐801‐treated mice are marked red ( ). Significance was determined using the log‐rank test; *P ≤ 0.05. (b) Overall survival rate was monitored in SKI‐G‐801‐treated 4T1 tumor‐bearing mice following injection of anti‐CD8, anti‐NK and anti‐CD4 antibodies, and IgG control (n = 5 per group, derived from one independent experiment). Survival was determined by either death or high tumor volume (= 2500 mm3). Significance was determined by the log‐rank test; **P ≤ 0.01.
). Significance was determined using the log‐rank test; *P ≤ 0.05. (b) Overall survival rate was monitored in SKI‐G‐801‐treated 4T1 tumor‐bearing mice following injection of anti‐CD8, anti‐NK and anti‐CD4 antibodies, and IgG control (n = 5 per group, derived from one independent experiment). Survival was determined by either death or high tumor volume (= 2500 mm3). Significance was determined by the log‐rank test; **P ≤ 0.01.
For further testing, we selected a model of non‐immunogenic tumor, syngeneic CT26 lung cancer, to test the direct anti‐tumor effect of SKI‐G‐801 alone or in combination with anti‐PD‐1. After subcutaneous inoculation with CT26 tumor cells, mice were treated with anti‐PD‐1, SKI‐G‐801 (15 mg kg−1, once a day) and combination treatment for 18 days. Both anti‐PD‐1‐ and SKI‐G‐801‐treated groups exhibited similar anti‐tumor effects (Figure 4a). Compared to the 4T1 metastatic breast cancer model (Supplementary figure 4), the anti‐cancer efficacy to inhibit primary tumor growth was significantly improved. Combination treatment showed significant efficacy on tumor growth, tumor growth inhibition (TGI) (Figure 4b, 4 of 9 in 90% TGI) and survival (Figure 4c, 33% animals survived till the end of experiment).
Figure 4.
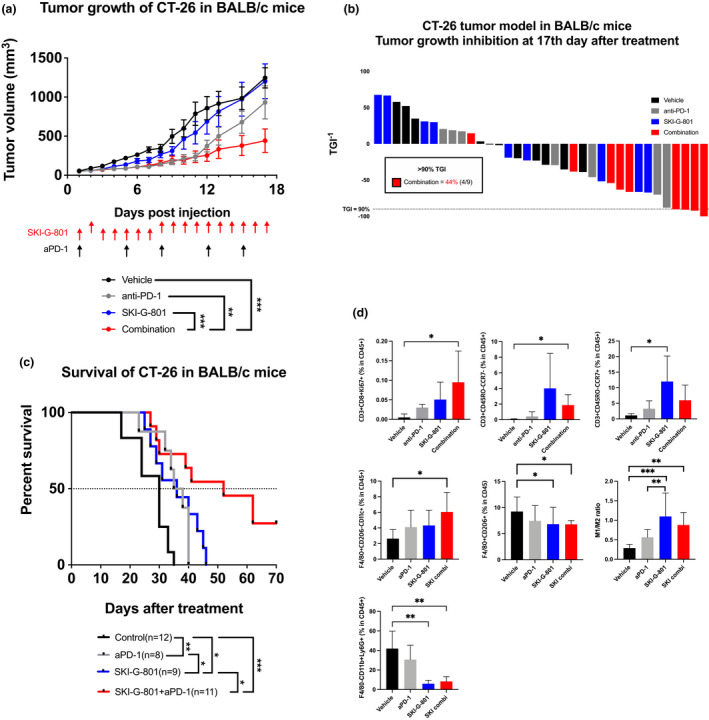
SKI‐G‐801 inhibits CT26 tumor growth by promoting anti‐tumor immunity. (a) Tumor growth was measured after mono‐ or combination therapy [anti‐PD‐1 (10 mg kg−1, intraperitoneal injection, biweekly), SKI‐G‐801 (30 mg kg−1, orally, once a day), SKI‐G‐801 + anti‐PD‐1] (n = 9 per group, derived from one independent experiment). Values are indicated as means ± SEM, and P‐values were calculated using the Student’s t‐test; *P ≤ 0.05 and ***P ≤ 0.001. (b) Tumor growth inhibition (TGI) was measured at day 17; in combination group, 4 of 9 mice showed > 90% of TGI. (c) Survival rate was monitored in each group till the end of experiment. Survival was determined by either death or high tumor volume (= 2500 mm3). Significance was determined by the log‐rank test; *P ≤ 0.05 and **P ≤ 0.01. (d) Immune cell population was analysed by flow cytometry. The gating strategy is shown (Supplementary figure 7). Significantly increased number of proliferating CD8+ Tc cells, effector Tc cells and M1 macrophages were observed in combination group compared to vehicle group. Effector memory Tc cells in SKI‐G‐801 group showed significant increase compared to vehicle group. G‐MDSCs in both SKI‐G‐801 single and combination group decreased significantly compared to the vehicle group. Values are indicated as means ± SEM, and P‐values were calculated using the Student’s t‐test; *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Flow cytometry analysis was done once.
Next, we investigated the changes in the intratumoral leukocyte population following each treatment. Compared to the vehicle, SKI‐G‐801 or anti‐PD‐1 treatment, significant increases were observed in the populations of CD8+KI67+ cytotoxic T (Tc) cells and CD3+CD45RO−CCR7− effector Tc cells following combination treatment with SKI‐G‐801 and anti‐PD‐1. The proportion of CD3+CD45RO−CCR7+ effector memory cells increased in SKI‐G‐801‐treated group (Figure 4d, upper panel), although no substantial change was observed in the CD4+CD25+FoxP3+ T‐cell population (data not shown). Combination treatment also correlated with a significant increase in M1 macrophages and a decrease in M2 macrophages (Figure 4d, lower). Interestingly, granulocyte‐like myeloid‐derived suppressor cells, which inhibit immune reactions and stimulate tumor metastasis, were also significantly decreased in both SKI‐G‐801 alone and combination treatment (Figure 4d, lower). Moreover, tumor infiltered NK cells were observed in the necrotic region of the tumor‐harbouring animals subjected to SKI‐G‐801 alone or combination treatment (Supplementary figure 6).
Neoadjuvant combination of anti‐PD‐1 and SKI‐G‐801 treatment significantly increased survival in 4T1 tumors
Neoadjuvant and adjuvant therapies are often used to treat various solid tumors, including breast cancer. Recently, the use of ICIs for the neoadjuvant or adjuvant treatment of patients with surgically resected solid tumors has demonstrated efficacy and improved relapse‐free survival or overall survival. 29
Based on SKI‐G‐801‐mediated enhancement of anti‐tumor immunity (Figures 3 and 4), we hypothesised a potential synergy between SKI‐G‐801 and anti‐PD‐1 in the neoadjuvant or adjuvant setting. To test this hypothesis, 4T1 tumor‐harbouring animals were treated for 6 days with a vehicle, anti‐PD‐1, SKI‐G‐801 or a combination (n = 4 or 5 per group) either before or after surgical resection, and survival was monitored for 50 days (Figure 5a and b). Notably, among all treatments, the neoadjuvant combination of SKI‐G‐801 and anti‐PD‐1 treatment significantly increased animal survival, compared to the vehicle (P = 0.0017), with 2 of 5 mice cured in this group (Figure 5b, neoadjuvant group). No other treatment completely prevented relapse. These results also indicated that neoadjuvant anti‐PD‐1 alone, but not adjuvant anti‐PD‐1 alone, also significantly increased survival compared to the vehicle, which is consistent with a previous report. 28 Furthermore, mice treated with the neoadjuvant combination tended to survive longer than those treated with adjuvant combination (P = 0.0694).
Figure 5.
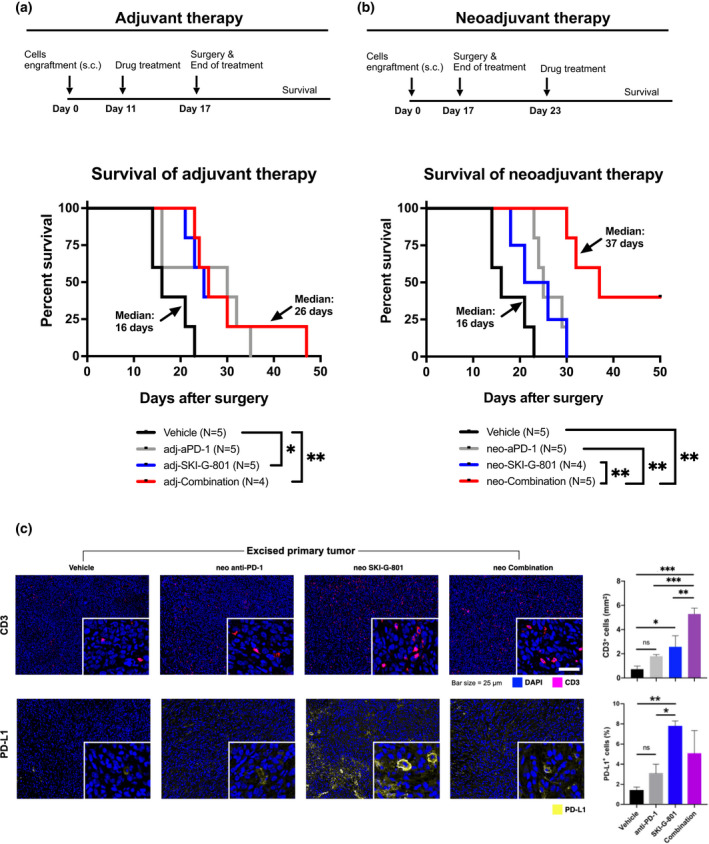
Neoadjuvant combination of anti‐PD‐1 and SKI‐G‐801 treatment most significantly increased survival of mice‐harbouring 4T1 tumors. (a, b) Scheme of neoadjuvant and adjuvant treatment, respectively. Neoadjuvant and adjuvant experiments were performed together, but survivals are displayed separately for readability (n = 4 or 5 per group, derived from one independent experiment). Mice were inoculated with 5 × 104 4T1 cells subcutaneously on day 0, and tumors were removed surgically on day 17. Drugs were administered either before (neoadjuvant) or after surgery (adjuvant) for 6 days. Survival was determined by death. Mice that died during or within 1 week of surgery (considered as death from surgery) were excluded. There were no deaths because of developed primary tumor regrowth (considered as death from tumor burden). Statistical significance was determined by the log‐rank test; *P ≤ 0.05, **P ≤ 0.01. (c) Three tumors per group were stained, and CD3+ and PD‐L1+ cells per unit area (mm−1) or percentage (%) were analysed using Vectra Polaris (left). Nuclei are marked in blue ( ), CD3+ cells are marked in pink (
), CD3+ cells are marked in pink ( ), and PD‐L1+ cells are marked in yellow (
), and PD‐L1+ cells are marked in yellow ( ). IHC was done once and CD3+ cells (per mm2), and percentage of PD‐L1+ cells (%) were calculated (right). Values are indicated as means ± SEM, and P‐values were calculated using the Student’s t‐test; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
). IHC was done once and CD3+ cells (per mm2), and percentage of PD‐L1+ cells (%) were calculated (right). Values are indicated as means ± SEM, and P‐values were calculated using the Student’s t‐test; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Next, in neoadjuvant treatment groups, we performed multispectral immunohistochemistry (IHC) using Vectra Polaris for comprehensive immune profiling of surgically resected 4T1 tumors (n = 3 per group). Vectra Polaris imaging showed increased infiltration of CD3+ T cells upon treatment with the neoadjuvant combination (Figure 5c). An increase in PD‐L1+ cells in tumors was also observed following treatment with SKI‐G‐801 alone or in combination with anti‐PD‐1 (Figure 5c), providing evidence for combination with anti‐PD‐1 therapy.
Taken together, these results suggest that combined treatment with anti‐PD‐1 and SKI‐G‐801 suppresses tumor relapse following surgery, especially when the regimen is administered as a neoadjuvant instead of as an adjuvant treatment, and these effects are achieved via enhancing CD8+ T cell‐mediated anti‐tumor immunity.
Discussion
This study reports the robust anti‐tumor and anti‐metastatic activity of a novel AXL inhibitor, SKI‐G‐801, in multiple syngeneic tumor models. Notably, the suppression of metastasis by SKI‐G‐801 appears to depend on the anti‐tumor immune response, which can be enhanced in combination with anti‐PD‐1 therapy. The therapeutic potential of SKI‐G‐801 was more enhanced as neoadjuvant therapy in a 4T1 triple‐negative breast cancer mouse model, which has been garnering increasing interest in clinical oncology.
Previous studies have consistently reported the suppression of metastasis and chemoresistance in tumor mouse models because of AXL depletion by shRNA or CRISPR knockout. 30 Accordingly, multiple AXL inhibitors have been active for their use in clinical settings. 20 In a biochemical assay, SKI‐G‐801 demonstrated potent inhibitory activity against AXL. SKI‐G‐801 effectively suppressed AXL phosphorylation with concomitant downregulation of vimentin expression and upregulation of E‐cadherin expression in drug‐resistant lung adenocarcinoma cell lines. Consistent with earlier reports, 15 the SKI‐G‐801‐mediated reversal of EMT suppressed tumor cell migration and invasion.
Consistent with these biochemical and in vitro results, we observed significant anti‐metastatic effects in murine models of B16F10 lung metastasis and CT26 peritoneal metastasis. Notably, compared to the vehicle treatment, SKI‐G‐801 treatment ameliorated metastasis to the lungs and other organs including the mesenteric lymph nodes, liver and kidneys in the B16F10 murine model. The effective suppression of micrometastasis to multiple organs could be partly explained by the broad tissue distribution of SKI‐G‐801 as observed in the pharmacokinetic analysis.
In addition to regulating metastasis, AXL signalling plays crucial roles in the immunosuppressive TME by suppressing toll‐like receptor‐mediated pro‐inflammatory responses of APCs and decreasing the anti‐tumor activity of NK cells. 21 , 30 Intriguingly, the significant anti‐tumor activity of SKI‐G‐801 was noted in immunocompetent mice, but not in immunodeficient nude mice, which was abolished by treatment with the anti‐CD8 antibody. These results strongly indicate that the therapeutic effects of SKI‐G‐801 are mediated by CD8+ T cells. SKI‐G‐801 treatment significantly increased cytotoxic and memory CD8+ T cells and activated antigen‐presenting DCs and macrophages as well as T‐cell costimulatory functions in 4T1 and CT26 murine models, further validating the SKI‐G‐801‐mediated enhancement of anti‐tumor immune response. Although an increase in NK cell count was detected post‐treatment, depletion of NK cells did not impact therapeutic efficacy in the 4T1 murine models. Combined AXL and PD‐1 inhibition revealed anti‐tumor synergy in the suppression of metastases, but not in suppressing primary tumor growth in 4T1 breast cancer model. 31 Although the lack of synergy in delaying primary tumor growth is disappointing, we believe that potent inhibition of metastases with combined SKI‐G‐801 and anti‐PD‐1 treatment is still important because most patients with solid tumors die of metastatic disease rather than from the primary tumor. Indeed, the combination treatment of SKI‐G‐801 and anti‐PD‐1 tended to be more effective in reducing lung metastases than monotreatment. Although these findings require further validation, we hypothesise that the anti‐tumor immune responses observed in the primary tumors may contribute to the disruption of the premetastatic microenvironment required for disseminating tumor cells to distant sites. 32
Recent landmark clinical trials have underscored the potential clinical efficacy of adjuvant or neoadjuvant ICIs in patients with operable solid tumors. 33 , 34 , 35 In these studies, neoadjuvant ICIs induced substantial pathological responses and expansion of tumor‐resident T cells, thereby contributing to improved survival in high‐risk operable patients. Consistent with these studies, we also demonstrated that treatment with neoadjuvant anti‐PD‐1, but not adjuvant anti‐PD‐1, significantly increased survival compared to the vehicle treatment in the 4T1 triple‐negative breast cancer model. Importantly, neoadjuvant therapeutic efficacy, compared with adjuvant therapeutic efficacy, showed that anti‐PD‐1 agent was potentiated by SKI‐G‐801 addition, which was reflected in CD8+ T cell‐mediated anti‐tumor immunity. This suggests that the combined inhibition of AXL and PD‐1 pathways may improve the survival of patients following surgical resection of the primary tumor.
In conclusion, our results demonstrated that SKI‐G‐801 is a novel AXL inhibitor that potently suppresses tumor metastasis via a CD8+ T cell‐mediated immunological mechanism. The combination of SKI‐G‐801 with anti‐PD‐1 therapy, particularly as neoadjuvant treatment, exhibited robust synergy to eradicate distant metastases following primary tumor resection in a mouse model of spontaneously metastatic breast cancer. Based on these results, further clinical evaluation of SKI‐G‐801 in combination with anti‐PD‐1 is strongly warranted in locoregionally advanced cancers that are surgically resected but have a high risk of recurrence.
Methods
Kinase assay
A biochemical assay for AXL was performed, and a kinase profile was established according to the half‐maximal inhibitory concentration (IC50) profiler assay (Millipore, UK) and the kinase profiler protocol (Millipore, UK), respectively. The IC50 was calculated using nonlinear regression analysis using GraphPad Prism 6.0 (GraphPad Software, CA, USA).
Pharmacokinetics (PK)
SKI‐G‐801 was administered orally to male ICR mice at 10 mg kg−1. Blood was collected from the abdominal vein into a heparin‐containing collection tube at 0.25, 0.5, 1, 2, 4, 8, 11 and 24 h after administration. Plasma was stored at −80°C until further analysis. The plasma concentration of SKI‐G‐801 was measured using liquid chromatography–mass spectrometry (LC‐MS).
Human cell lines
MDA‐MB‐231 (ATCC HTB‐26, RRID: CVCL_0062) and Hs578T (ATCC HTB‐126, RRID: CVCL_0332) cells were purchased from the American Type Culture Collection (ATCC). Two drug‐resistant lung adenocarcinoma cell lines, HCC78‐CR (crizotinib resistant) and PC9‐GR (gefitinib resistant), were generated by consistent treatment with 1 μm crizotinib and gefitinib.
Western blot
Cells were treated with the vehicle control or SKI‐G‐801 for 4 h and then lysed in radioimmunoprecipitation assay buffer (25 mm Tris–HCl, 150 mm NaCl, 1% NP‐40, 1% sodium deoxycholate and 0.1% SDS) supplemented with a protease and phosphatase inhibitor cocktail (Thermo Scientific, MA, USA). Equivalent amounts of protein were separated using the NuPAGE 4–12% Bis–Tris Gel System (Invitrogen) and then transferred to polyvinylidene difluoride membranes. Membranes were probed with anti‐phospho‐AXL (Y702) (1:1000, CTS, #5724; Cell Signaling Technology, MA, USA), anti‐phospho‐AXL (Y779) (1:1000; R&D, AF2228; R&D Systems, NE, USA), anti‐phospho‐FLT3 (Y842) (1:1000, CTS, #4577; Cell Signaling Technology), anti‐phospho‐H3 (S28) (1:1000, CTS, #9713; Cell Signaling Technology) or anti‐phospho‐KDR (Y1175) (1:1000, CTS, #2478; Cell Signaling Technology) antibodies and then stripped with Restore Western Blot Stripping Buffer (Thermo Scientific). The membranes were then probed again with anti‐AXL (1:1000, CTS, #8661; Cell Signaling Technology), anti‐H3 (1:1000, CTS, #3638; Cell Signaling Technology), anti‐FLT3 (1:1000, CTS, #3462; Cell Signaling Technology) or anti‐actin (1:1000, #MAB1501; Sigma‐Aldrich) antibodies. The reactive bands were visualised using enhanced chemiluminescence. Band intensities were quantified using ImageJ software (US National Institutes of Health, MD, USA).
Invasion and cell proliferation assays
Cells were grown in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% foetal bovine serum. The extracellular matrix (ECM) gel was thawed overnight at 4°C and kept on ice. A Millicell® insert plate (Millicell® Cell Culture Inserts, Millipore, Germany) was chilled to 4°C and kept on ice. Next, 100 µL diluted ECM gel was added to the upper compartment of the Millicell® insert. The insert and ECM gel were incubated at 37°C for 2 h until the ECM gel solidified. 1 × 105 cells were loaded onto the insert and treated with SKI‐G‐801 or the vehicle for 18 h. After treatment, cells on the upper side were obtained using a cotton swab and fixed with 5% glutaraldehyde for 10 min. Then, 1% crystal violet in 2% ethanol was used to stain the insert. Images of the stained cells were captured under a microscope.
Experimental animal and metastasis models
Female BALB/c and C57BL/6 mice were purchased from Orient Bio (Seongnam, Korea). The mice were maintained under pathogen‐free conditions (room temperature, 40–60% humidity) at the Avison BioMedical Research Centre at Yonsei University, according to specific pathogen‐free guidelines of The Institutional Animal Care and Use Committee (IACUC) (ethics number: 2018‐0063). Four cell lines, 4T1 (KCB 200967YJ, RRID: CVCL_0125), B16F10 (KCLB 80008, RRID: CVCL_0159), LLC2 (ATCC CRL‐1642, RRID: CVCL_4358) and CT26 (KCB 201207YJ, RRID: CVCL_7254) were used for inducing tumorigenesis in the experimental animals. A 4T1 spontaneous metastasis model was generated in BALB/c mice. Briefly, the mice were injected with 1 × 106 4T1 cells by subcutaneous injection. After 14 days, lung and tumor tissues were collected, and lung metastasis was observed by microscopic observation. A B16F10 lung metastasis model was generated in C57BL/6 mice by intravenous injection, and the CT26 peritoneal metastasis model was produced in BALB/c mice by intraperitoneal injection. The mice were sacrificed after 14 days. The CT26 tumor model was generated by subcutaneous injection of 1 × 106 CT26 cells. The CT26 and 4T1 solid tumors were measured using a 3D tumor scanner, TM900 (Peria, Belgium). Lung metastasis nodules were measured and counted by whole‐slide image analysis. In vivo experiments included anti‐PD‐1 antibody (Cat # BE0146; Bioxcell, NH, USA).
Haematoxylin and eosin staining and immunohistochemistry analysis
Metastatic organs and primary tumors were fixed overnight in 10% formalin solution. Fixed samples were directly dehydrated and embedded in paraffin, and 4‐μm‐thick sections were cut, mounted on slides, stained with haematoxylin and eosin (H&E) and observed using an electronic microscope. Data were captured, processed and analysed using LEICA Bond RX (Leica Biosystems, Leica Microsystems Inc., IL, USA).
For IHC, sections were stained using anti‐CD3 (ab16669; Abcam, Cambridge, UK), anti‐PD‐L1 (AF1019; R&D Systems, Minneapolis, MN, USA), anti‐NCR1 (ab214468; Abcam, Cambridge, MA, USA) and streptavidin–HRP system (Dako, Carpinteria, CA, USA). CD3+ TIL and PD‐L1+ cells were analysed using the Vectra Polaris system (PerkinElmer, Akron, OH, USA).
Flow cytometry
Experimental mice were sacrificed, and tumor tissues were collected for single‐cell dissociation. The tumor‐infiltrating immune cell population was measured by flow cytometry. The T‐cell population was identified using anti‐CD45 (304032; BioLegend, CA, USA), anti‐CD3 (100320; BioLegend), anti‐CD4 (100526; BioLegend), anti‐CD8 (100750; BioLegend), anti‐FoxP3 (12‐5773‐82; eBioscience, MA, USA), anti‐CD25 (102049; BioLegend) and anti‐PD‐1 (135218; BioLegend) antibodies. Dendritic cells were identified using anti‐H2 (33A15443; Invitrogen), anti‐CD11c (117343; BioLegend), anti‐CD80 (47‐4801‐82; eBioscience) and anti‐CD86 (104729; BioLegend) antibodies. M1 and M2 macrophages were identified using anti‐F4/80 (47‐4801‐82; eBioscience), anti‐CD206 (17‐2061‐80; eBioscience), anti‐CD11c, anti‐CD80 and anti‐CD86 antibodies. MDSCs were identified using anti‐CD11b (101216; BioLegend) and anti‐Ly‐6G (127606; BioLegend) antibodies, whereas NK cells were identified using anti‐CD49b antibody (108906; BioLegend). For staining, the standard intra‐nucleus staining protocol provided with the BioLegend kit was used (424401; BioLegend). All antibody‐stained tumor‐dissociated cells were detected on an LSR2 flow cytometer (BD Bioscience, NJ, USA) and analysed using FlowJo software (FlowJo LLC, OR, USA).
Statistical analysis
Data were analysed statistically using GraphPad Prism 9 (GraphPad Software). The significance in independent groups was calculated by the Student’s t‐test. Data were expressed as means ± SEM, respectively. Overall survivals were analysed by the log‐rank test (Kaplan–Meier), and endpoint of survival was determined as either death or euthanised at high tumor volume (= 2500 mm3). In Figure 5, mice that died during or within 1 week of surgery were excluded. *P < 0.05, **P < 0.01 or ***P < 0.001 was considered statistically significant.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Chun‐Bong Synn: Data curation; Formal analysis; Investigation; Methodology; Visualization; Writing – original draft. Sung Eun Kim: Project administration. Hee Kyu Lee: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Visualization. Min‐Hwan Kim: Conceptualization; Methodology; Project administration; Writing – original draft. Jae Hwan Kim: Investigation. Ji Min Lee: Project administration. Ha Ni Jo: Data curation; Investigation. Wongeun Lee: Data curation; Investigation. Dong Kwon Kim: Data curation; Methodology. Youngseon Byeon: Project administration. Young Seob Kim: Project administration. Mi Ran Yun: Project administration. Chae‐Won Park: Project administration. Jiyeon Yun: Project administration. Sangbin Lim: Project administration. Seong Gu Heo: Project administration. San‐Duk Yang: Project administration. Eun Ji Lee: Project administration. Seul Lee: Project administration. Hunmi Choi: Project administration. You‐Won Lee: Project administration. Jae Seok Cho: Project administration. Do Hee Kim: Project administration. Sungho Park: Project administration. Jung‐Ho Kim: Project administration. Yewon Choi: Project administration. Sung Sook Lee: Project administration. Beung‐Chul Ahn: Project administration. Chang Gon Kim: Project administration. Sun Min Lim: Project administration. Min Hee Hong: Project administration. Hye Ryun Kim: Project administration. Kyoung‐Ho Pyo: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft. Byoung Chul Cho: Data curation; Formal analysis; Funding acquisition; Investigation; Project administration; Supervision; Writing – original draft; Writing – review & editing.
Supporting information
Supplementary figure 1
Supplementary figure 2
Supplementary figure 3
Supplementary figure 4
Supplementary figure 5
Supplementary figure 6
Supplementary figure 7
Supplementary table 1
Supplementary table 2
Supplementary table 3
Acknowledgments
We acknowledge the members of the animal facility and the technicians for their technical assistance. This study was supported by the Deawoong Foundation (DF‐201911‐0000001), Dongin Sports Research Grant of Yonsei University College of Medicine (6‐2019‐0128), Ministry for Health & Welfare Affairs (HI19C0744010020), National Research Foundation of Korea (2017R1D1A1B0303211014) and Oscotec Inc. (2019‐31‐0802).
Contributor Information
Kyoung‐Ho Pyo, Email: pkhpsh@gmail.com.
Byoung Chul Cho, Email: cbc1971@yuhs.ac.
References
- 1. Lee HJ, Jeng YM, Chen YL, Chung L, Yuan RH. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014; 35: 769–775. [DOI] [PubMed] [Google Scholar]
- 2. Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6‐Axl‐mediated survival of endothelial cells. Am J Physiol Heart Circ Physiol 2004; 287: H1207–H1213. [DOI] [PubMed] [Google Scholar]
- 3. Lee WP, Wen Y, Varnum B, Hung MC. Akt is required for Axl‐Gas6 signaling to protect cells from E1A‐mediated apoptosis. Oncogene 2002; 21: 329–336. [DOI] [PubMed] [Google Scholar]
- 4. Collett G, Wood A, Alexander MY et al. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ Res 2003; 92: 1123–1129. [DOI] [PubMed] [Google Scholar]
- 5. Choi YJ, Kim JH, Rho JK et al. AXL and MET receptor tyrosine kinases are essential for lung cancer metastasis. Oncol Rep 2017; 37: 2201–2208. [DOI] [PubMed] [Google Scholar]
- 6. Zhang G, Wang M, Zhao H, Cui W. Function of AXL receptor tyrosine kinase in non‐small cell lung cancer. Oncol Lett 2018; 15: 2726–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Alfonso TM, Hannah J, Chen Z, Liu Y, Zhou P, Shin SJ. AXL receptor tyrosine kinase expression in breast cancer. J Clin Pathol 2014; 67: 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin G, Wang Z, Wang J et al. Expression of Axl and its prognostic significance in human breast cancer. Oncol Lett 2017; 13: 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu H, Liu R, Ma B et al. AXL receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br J Cancer 2015; 113: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou L, Liu X‐D, Sun M et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016; 35: 2687–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu X, Ma W, Zhou Q et al. AXL‐GAS6 expression can predict for adverse prognosis in non‐small cell lung cancer with brain metastases. J Cancer Res Clin Oncol 2017; 143: 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zucca LE, Morini Matushita MA, da Silva Oliveira RJ et al. Expression of tyrosine kinase receptor AXL is associated with worse outcome of metastatic renal cell carcinomas treated with sunitinib. Urol Oncol 2018; 36: 11.e13–e21. [DOI] [PubMed] [Google Scholar]
- 13. Brand TM, Iida M, Stein AP et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin Cancer Res 2015; 21: 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheele CLGJ, Maynard C, van Rheenen J. Intravital insights into heterogeneity, metastasis, and therapy responses. Trends Cancer 2016; 2: 205–216. [DOI] [PubMed] [Google Scholar]
- 15. Antony J, Huang RY. AXL‐driven EMT state as a targetable conduit in cancer. Cancer Res 2017; 77: 3725–3732. [DOI] [PubMed] [Google Scholar]
- 16. Wu F, Li J, Jang C, Wang J, Xiong J. The role of Axl in drug resistance and epithelial‐to‐mesenchymal transition of non‐small cell lung carcinoma. Int J Clin Exp Pathol 2014; 7: 6653–6661. [PMC free article] [PubMed] [Google Scholar]
- 17. Tai KY, Shieh YS, Lee CS, Shiah SG, Wu CW. Axl promotes cell invasion by inducing MMP‐9 activity through activation of NF‐kappaB and Brg‐1. Oncogene 2008; 27: 4044–4055. [DOI] [PubMed] [Google Scholar]
- 18. Aguilera TA, Rafat M, Castellini L et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun 2016; 7: 13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elvin P, Garner AP. Tumor invasion and metastasis: challenges facing drug discovery. Curr Opin Pharmacol 2005; 5: 374–381. [DOI] [PubMed] [Google Scholar]
- 20. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anticancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 2019; 18: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peeters MJW, Rahbech A, Thor SP. TAM‐ing T cells in the tumor microenvironment: implications for TAM receptor targeting. Cancer Immunol Immunother 2020; 69: 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramanian M, Hayes CD, Thome JJ et al. An AXL/LRP‐1/RANBP9 complex mediates DC efferocytosis and antigen cross‐presentation in vivo . J Clin Invest 2014; 124: 1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Skinner HD, Giri U, Yang LP et al. Integrative analysis identifies a novel AXL‐PI3 kinase‐PD‐L1 signaling axis associated with radiation resistance in head and neck cancer. Clin Cancer Res 2017; 23: 2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hugo W, Zaretsky JM, Sun L et al. Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2016; 165: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Syn NL, Teng MWL, Mok TSK, Soo RA. De‐novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 2017; 18: e731–e741. [DOI] [PubMed] [Google Scholar]
- 26. Yang S, Zhang JJ, Huang XY. Mouse models for tumor metastasis. Methods Mol Biol 2012; 928: 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yi Z, Gan‐Lin Z, Xu S et al. Establishment of a murine breast tumor model by subcutaneous or orthotopic implantation. Oncol Lett 2018; 15: 6233–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reiser J, Banerjee A. Effector, memory, and dysfunctional CD8+ T cell fates in the antitumor immune response. J Immunol Res 2016; 2016: 8941260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu J, Blake SJ, Yong MCR et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov 2016; 6: 1382–1399. [DOI] [PubMed] [Google Scholar]
- 30. Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer 2014; 14: 769–785. [DOI] [PubMed] [Google Scholar]
- 31. Gao L, Zhang C, Gao D et al. Enhanced anti‐tumor efficacy through a combination of integrin αvβ6‐targeted photodynamic therapy and immune checkpoint inhibition. Theranostics 2016; 6: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peinado H, Zhang H, Matei IR et al. Pre‐metastatic niches: organ‐specific homes for metastases. Nat Rev Cancer 2017; 17: 302–317. [DOI] [PubMed] [Google Scholar]
- 33. Schmid P, Salgado R, Park YH et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high‐risk, early‐stage triple‐negative breast cancer: results from the phase 1b open‐label, multicohort KEYNOTE‐173 study. Ann Oncol 2020; 31: 569–581. [DOI] [PubMed] [Google Scholar]
- 34. Forde PM, Chaft JE, Smith KN et al. Neoadjuvant PD‐1 blockade in resectable lung cancer. N Engl J Med 2018; 378: 1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blank CU, Rozeman EA, Fanchi LF et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med 2018; 24: 1655–1661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1
Supplementary figure 2
Supplementary figure 3
Supplementary figure 4
Supplementary figure 5
Supplementary figure 6
Supplementary figure 7
Supplementary table 1
Supplementary table 2
Supplementary table 3