

Abstract
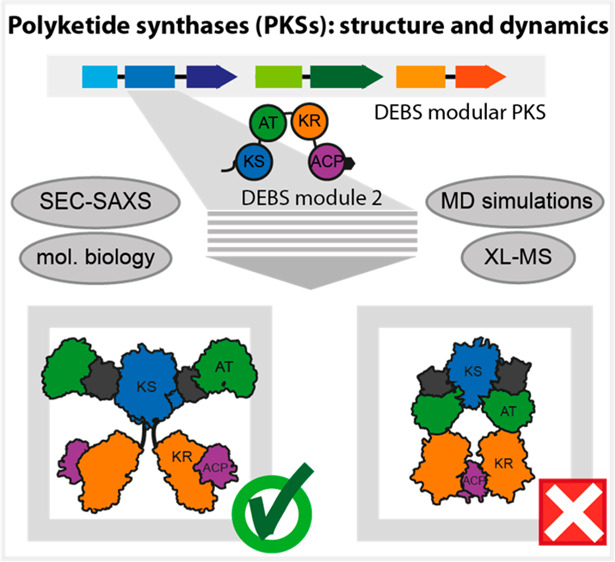
Polyketide synthases (PKSs) are versatile C–C bond-forming enzymes that are broadly distributed in bacteria and fungi. The polyketide compound family includes many clinically useful drugs such as the antibiotic erythromycin, the antineoplastic epothilone, and the cholesterol-lowering lovastatin. Harnessing PKSs for custom compound synthesis remains an open challenge, largely because of the lack of knowledge about key structural properties. Particularly, the domains—well characterized on their own—are poorly understood in their arrangement, conformational dynamics, and interplay in the intricate quaternary structure of modular PKSs. Here, we characterize module 2 from the 6-deoxyerythronolide B synthase by small-angle X-ray scattering and cross-linking mass spectrometry with coarse-grained structural modeling. The results of this hybrid approach shed light on the solution structure of a cis-AT type PKS module as well as its inherent conformational dynamics. Supported by a directed evolution approach, we also find that acyl carrier protein (ACP)-mediated substrate shuttling appears to be steered by a nonspecific electrostatic interaction network.
Keywords: natural compound synthesis, assembly lines, multidomain proteins, hybrid approach, directed evolution
Introduction
Modular polyketide synthases (PKSs) are a class of megasynthases that produce compounds of high pharmaceutical importance.1 In modular PKSs, modules are linked covalently or noncovalently,2−4 and are hypothesized to self-organize into linear scaffolds that feature product synthesis in a vectorial manner (Figure 1A).5,6 In cis-AT PKSs, the catalytic core of each PKS module comprises the domains needed for the condensation of small acyl precursors, the ketosynthase (KS), the acyl transferase (AT), and the acyl carrier protein (ACP). In addition, the domains ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) can be present to modify the condensation product at the β-position. In most cases, a thioesterase (TE) domain at the C-terminus of the modular PKSs is responsible for product release.7,8
Figure 1.

Schematic architecture of DEBS, structural models, and constructs used in this study. (A) Overview of DEBS. The three polypeptides (DEBS1–3), the encoded modules (M1–M6), the loading module (LM), the TE, and the final product are shown. Black tabs depict docking domains. The KR domain of M3 is redox-inactive and termed kr.33 (B) Models for the partially reducing PKS modules; DEBS M3 is based on SAXS analysis that implemented high-resolution structural information on KS-AT models and FAS,9 and PIKS M5 is based on cryoEM analysis.14 DEBS M3 model: Domains KS and AT form a rigid dimeric unit of an overall extended shape, and are separated by a noncatalytic linker-domain (LD). AT is followed by a structured post-AT linker10,11,34 that interacts with both LD and KS, and connects AT to KR. PIK M5 model: In the arch-shaped KS-AT dimer, the AT is oriented downward. It interacts with the respective upward-facing LD and the KS of the other protomer of the dimer. The post-AT linker is not resolved within the PIKS M5 structure. Schemes of the domain structures including domain boundaries are attached. Note that for DEBS M3, domain borders are also given in construct A numbering with labeling in italics, which has been used in this study (Table S1). Post-AT linker sequence as defined in refs (21 and 22). (C) DEBS M2-derived constructs MBP-M2-DD2 (construct A) and KR1-ACP1-M2-TE (construct B).
Over the last years, structural evidence has been obtained for two distinct structural arrangements of PKS modules that differ most substantially in the relative arrangement of the condensation domains KS and AT. Small-angle X-ray scattering (SAXS) data obtained on the 6-deoxyerythronolide B synthase module 3 (DEBS M3) revealed an extended conformation of the KS-AT dimer9 in support of previous X-ray crystallographic data collected on the didomain subunit.10−12 In contrast, a cryo-electron microscopy (cryoEM) study on the related pikromycin synthase module 5 (PIKS M5), reporting the first structure of a complete PKS module received at moderate resolution, suggested an arch-shaped KS-AT dimer conformation (Figure 1B).13,14 Whereas the DEBS M3 structural model agrees with the structure of the evolutionarily related mammalian fatty acid synthase (FAS),15 with the cryoEM structure of the lovastatin synthase iterative module LovB,16 and with 3D-models generated from partial X-ray structures (mycocerosic acid synthase (MAS)-like PKS17 and spinosyn synthase module 2 (SPNS M2)18), the structural model derived from the EM reconstruction of PKS M5 differs substantially from the others. The two structural models lead to substantial divergence in the molecular mechanism, notably concerning both the intramodular elongation and intermodular translocation steps.19,20 The model derived from PIKS M5 structural data suggested loading of the KS active site by the upstream ACP (translocation step) via a side entrance, whereas, for elongation, the extender substrate reaches the binding site via a newly observed bottom entrance. Understanding the structural and mechanistic key features of PKSs is essential to inform their engineering for producing novel compounds with potentially new bioactivities.21,22
To address the question of the structural and conformational properties of a PKS module, we have investigated DEBS module 2 (DEBS M2) by integrating SAXS and cross-linking mass spectrometry (XL-MS) with coarse-grained (CG) structural modeling and simulation. We considered the combination of noninvasive solution structure methods with robust structure calculation by Bayesian inference as particularly suited for addressing the question of the overall structural appearance of a PKS module. The hybrid approach enables the detailed study of the ensemble of conformations of a PKS module in solution, providing insight beyond the currently available structural information. DEBS is the ideal modular PKS for this approach, because it is a prototypical PKS and it has served as a model system before to examine the mechanism23 and engineering potential24−27 as well as the structure of the enzymatic domains or larger assemblies, e.g., KR1, DH4, KS3-AT3, KS5-AT5, ACP2 (numbers indicate module), docking domains (DDs) between DEBS2 and DEBS3, and TE.2,7,9−11,28−30 Further, DEBS is phylogenetically situated in the same clade with other actinobacterial modular PKSs, so that insight gained likely applies to PKS modules in general.31
We applied our approach to two DEBS M2 constructs (Figure 1C). Construct A contains the native M2 (KS-AT-KR-ACP-DD) with an N-terminally fused maltose binding protein (MBP). It represents a stand-alone PKS module with a domain organization comparable to the previously studied DEBS M3 and PIKS M5 (essentially just differing in the N-terminal MBP fusion). In construct B, M2 is decorated with the natural upstream domains KR1 and ACP1 (“1” for M1), constituting the processing part of DEBS M1, and a C-terminal TE domain (Figure 1C). The two constructs were chosen to provide complementary information on domain–domain interfaces in PKS modules (Table S1).
Although our data show that extended (“DEBS M3 model”) and arch-shaped (“PIKS M5 model”) conformations of module DEBS M2 are possible in solution, they likewise reveal the propensity of DEBS M2 to adopt the extended conformation in solution. In this conformation, domains KR and ACP are positionally highly variable, representative of an overall conformationally dynamic processing part of a PKS module in accordance with previous data.32 In a focus on the domain–domain interplay in modular PKSs, we complemented our hybrid approach with a directed evolution experiment based on phage display. Enhanced CG simulations, XL-MS analysis, and phage display suggest that interactions of ACP with catalytic domains are steered by networks of weak electrostatic interactions.
Our findings shed light on the structure and dynamics of a PKS module, and offer new insight into the interplay between domains in PKSs. Collectively, these results provide guidance in the engineering of PKSs for the custom synthesis of novel polyketides.
Experimental Section
Plasmids, Bacterial Cell Culture, and Protein Purification
Plasmids harboring genes encoding individual PKS modules were generated via In-Fusion Cloning (Takara) and restriction and ligation-based techniques. Proteins were expressed and purified using similar protocols. For holo-proteins (where the ACP domain is post-translationally modified with a phosphopantetheine arm), E. coli BL21 cells were cotransformed with a plasmid encoding for the phosphopantetheine transferase Sfp from B. subtilis (pAR35735). Proteins either contained a C-terminal His6-tag or an N-terminal MBP-tag for purification. Twin-Strep-tagged proteins (KS3-AT3 for phage display) were purified using 5 mL of StrepTactin in a column.36 The column was washed with 5 CV lysis buffer to elution with 3 column volumes of lysis buffer containing 2.5 mM destiobiotin. Eluates from His6-tag, Strep-tag, and MBP-tag purifications were further purified by anion exchange chromatography and size exclusion chromatography (SEC). Samples were stored as aliquots at −80 °C until further use. For more information, see the Supporting Information.
Enzymatic Assays and Triketide Analysis
PKS enzymatic assay of KR1-ACP1-M2-TE was performed according to published procedures.34 The resulting product was extracted twice with 450 μL dried in vacuo and submitted for LC-MS analysis. For more information, see the Supporting Information.
SAXS Analysis and CG Structural Modeling
SEC-SAXS analysis was performed on the Bio-SAXS beamline BM29 at the European Synchrotron Radiation Facility (ESRF).37 We computed the SAXS intensities for each of the individual CG conformations using the package FoXS.38,39 We used the Bayesian Inference of Ensembles (BioEn) method to determine the statistical weights of Aext, Aarch, and Bext ensembles of conformations underlining the measured SAXS intensities.40−42 BioEn computations were performed considering the SAXS intensities in the range of 0.02 < q < 0.3 Å–1. From the scattering curves, the radius of gyration Rg was calculated using the Guinier approximation.43 We reduced the sampled ensembles to the conformations that together fulfill more than 99% of the statistical cumulative weight for evaluation of the measured cross-links. Systematic deviations between the measured and simulated scattering curves at low q range suggested moderate aggregation of the systems in solution. To quantify and account for the fraction of aggregation, we combined each simulated curve with the intensities of a globular model at a statistical weight that matched the experiment. We built initial structural arrangements for Aext and Bext as informed by previous SAXS investigations of DEBS.9 The initial structural arrangement of Aarch was constructed by docking the modeled protein domains within the cryoEM density of the related PikAIII complex (EMD-5664).14 We generated a canonical ensemble of models Aext, Aarch, and Bext using Monte Carlo (MC) simulations based on coarse-grained (CG) potential energies.44 Folded protein domains were treated as rigid bodies described by one interaction site per residue located at their Cα atoms. The protein domains were linked by flexible linkers represented as Gaussian chain polymers and placed in a cubic box of 50 nm size. The solvent dielectric constant was set to ε = 80. The post-AT regions of Aarch were modeled as flexible linkers. We fixed the position and orientation of KS-AT2 and used step sizes of 3 Å and 3.14 rad for the MC translational and rotational movements of all other domains. For all models, conformations were sampled in a temperature replica-exchange simulation scheme covering the range of 298 to 598 K in steps of 20 K. For Aarch, we also performed replica-exchange simulations collecting replicas between 298 and 398 K every 20 K to preserve the arch-shaped conformation that were otherwise disrupted at higher temperatures. For each model, after an initial equilibration, a total of 9.0 × 105 MC conformations were produced to exhaustively sample relevant conformations. To augment model flexibility and the sampled conformational space of the models, we considered only the conformations produced at highest temperature for further analysis. Aarch conformations sampled at 598 K are denoted as Aarch598. For more information, see the Supporting Information.
XL-MS Analysis
Amounts of 150–175 μg of constructs A and B at a final protein concentration of 1 μg/μL or less were incubated with 2 mM SDA (100 mM stock in DMSO) for 30 min at room temperature. The cross-linking reactions were quenched with 50 mM Tris-HCl. Proteins were digested by trypsin, and peptides were acidified with trifluoroacetic acid (TFA) to a final concentration of 0.5% (v/v), desalted on MicroSpin Columns (Harvard Apparatus) following manufacturer’s instructions, and vacuum-dried. To enrich cross-linked peptide species by peptide size exclusion chromatography, we subjected fractions that eluted first and contained the cross-linked peptide pairs to LC-MS/MS analysis. Cross-linked peptides were measured in technical duplicates on an Orbitrap Fusion Tribrid mass spectrometer or on a Q Exactive HF-X coupled to a Dionex UltiMate 3000 UHPLC system (Thermo Fisher Scientific) equipped with an in house-packed C18 column (ReproSil-Pur 120 C18-AQ, 1.9 μm pore size, 75 μm inner diameter, 30 cm length, Dr. Maisch GmbH). ProteomeDiscoverer 1.4 (Thermo Fisher Scientific) was used for converting raw files into.mgf format (signal-to-noise ratio 1.5, 1000–10000 Da precursor mass). The generated.mgf files were subjected to pLink v. 1.23 (pFind group)45 to identify cross-linked peptides. All spectra were evaluated manually. For more information, see the Supporting Information.
Structural Mapping of the XL-MS Data
We mapped the measured cross-links of construct A over the 1000 Aext and Aarch conformations ranked by weight that fulfill 99.9% of the BioEn scattering curves. The same analysis was performed to map the measured XL-MS data of construct B over the 5000 conformations of model Bext that fulfill 99% of the BioEn scattering curve. For all selected conformations, we calculated the Cα pair-distances across the reported cross-linked residues. For more information, see the Supporting Information.
Phage Display Methodology
Phage display experiments were carried out according to the protocol described by Tonikian et al.46 A library of ACP1(2) mutants fused to the N-terminus of the minor coat protein P3 of the M13 bacteriophage was generated as described before.46 A primer for randomization of five amino acids in the chain translocation epitope of ACP1 was designed using NNK at the target positions (P-MK162:5′ CTG GCG TCG CTG CCC GCG NNK GAG CGC NNK NNK GCG CTG TTC NNK CTC GTG CGC NNK CAC GCG GCC GCC GTC CTC 3′ (N: A/C/G/T K: G/T). Panning was performed in a StrepTactinXT-coated microtiter plate with wells coated with (3)KS3-AT3--Strep. The blocked phage library was incubated for 1 h on the coated and blocked wells prior to washing. Wash steps were increased each round over the course of the panning protocol (10×, 12×, 14×, 16×, 18×). Elution of bound phage was done with 100 μL of 100 mM hydrochloric acid for 20 min. The elution was propagated and the titer was determined. For propagation, 2 mL of log phase E. coli SS320 (OD600 0.8) was added to 200 μL of eluted phage and infection was allowed for 30 min at 37 °C, 200 rpm, whereupon 20 μL of M13K07 helper phage (1.0 × 1012 PFU/mL) was added. Phages were harvested and pellets containing the phage were resuspended in 1 mL of blocking buffer. To test the eluted phage for increased and specific binding, a specificity ELISA was performed. To further confirm the relative binding intensity of the newly enriched mutants, we used a titration ELISA using purified phage. The advantage of this assay is that defined phage amounts can be used and the signal can be normalized across different phage preparations. For more information, see the Supporting Information.
Results
SAXS Analysis and XL-MS Data Collection
Constructs A and B were purified as both apo- and phosphopantetheinylated holo-proteins using established protocols,47 yet codon harmonization of the M2 gene was used to increase expression yields for both constructs (Figure S1, Table S2). We note that the body of data has been collected on the apoprotein (CG structural modeling, SAXS, XL-MS, and phage display) unless otherwise stated. The proteins purified to homogeneity were subjected to tandem size exclusion chromatography small-angle X-ray scattering (SEC-SAXS) and eluted as dimeric species (Figure S2A). Scattering data from the region of stable Rg values were analyzed, assuming that higher oligomer and aggregates are essentially eliminated (Figures S2B–D, Figures S3A–D, and SI Note 1). The scattering derived parameters, Rg values of 77.2 ± 0.2 Å for construct A and 76.6 ± 0.2 Å for construct B (Guinier approximation), respectively (Figure S3B and Table S3), were significantly larger than those previously obtained for DEBS M3 (M3-TE: 61.3 ± 0.5 Å).9 Although the Rg value of constructs A and B are essentially similar, molecular weight (MW) estimates by the volume of correlation (Vc) well reflect calculated MWs; i.e., 408 (exp.) vs 403 kDa (calc.) for construct A, and 484 vs 488 kDa for construct B (SI Note 1, Figure S3B and Tables S3 and S4).48 In addition to SAXS analysis, XL-MS was employed to gain distance information for specific residues in solution. We used the heterobifunctional cross-linker succinimidyl 4,4-azipentanoate to map interactions of lysine residues with other residues within and across domain boundaries (Cα-Cα-distance ∼20 Å (ref49) (Tables S5–S7). Focusing on construct A, we observed a number of XLs between the flexible ACP domain and catalytic domains (from ACPK1775 and ACPK1784), yet also across interfaces of less flexible domains and units, e.g., KS:AT and KS:KR (from KSK727), AT:LD and AT:KR (from ATK1071), and the post-AT linker (from K1273) and domains LD, AT, and KR (Table S6). Overall constructs A and B showed a strong overlap in mapped interactions (Table S6 and S7). The biological relevance of the M2-TE region of construct B was established by measuring triketide lactone product formation using a synthetic substrate (Figure S4).
Coarse-Grained Modeling of Two PKS Module Conformations
In integrating SAXS and XL-MS into a complex computational model building approach, we decided to employ structural models of the extended and the arch-shaped KS-AT dimer (KS-AT2) conformation for CG modeling of construct A (Aext and Aarch, Figure 2A, B, Figure S5, and SI Material and Methods). Model Aext was based on available X-ray crystallographic data10,11 and exhibits an extended KS-AT2 conformation with rigid post-AT linkers folding back to the KS domain. Model Aarch was informed by the PIKS M5 cryoEM density14 and carries an arch-shaped KS-AT2 with flexible post-AT loops (Figure S6). We generated an ensemble of structural models in CG simulations44 and performed Bayesian ensemble refinement.40−42 An entropic penalty ensured that the weights of the individual conformations were minimally adjusted to match the measured SAXS intensities (Figure S7).41,42 We note that for CG structural modeling, SEC-SAXS frames over the entire dimeric elution peak were averaged for constructs A and B, respectively (Figure S2A). The Bayesian inference highlighted a small degree of protein aggregation in solution, also visible for both constructs in the SEC profiles (Figure S2A), for which we accounted in silico by aggregate correction (SI Note 2). Computed Rg values from the BioEn ensembles of Aext and Aarch were equal to 67.6 and 64.0 Å based on the Guinier approximation (Figures S8–10, SI Notes 2 and 3), which is in good agreement with the previous data collected on DEBS M3-TE.9 The in silico intensities computed for Aext are in excellent agreement with the experimental SAXS data (Figure 2A). By contrast, the calculated intensities for Aarch show a clear divergence from measured data (Figure 2B and Figure S11). All complex arrangements were sorted based on their statistical weight, and we analyzed the positional variability of the domains of those that cumulatively explain 90% of the scattering intensities (Figure 2A, B). In Aext, the KR and thereby the attached ACP domain are highly mobile (Figure 2A and Figure S12). In Aarch, the flexibility of the post-AT linker enables occasional migration of KR toward the condensing domains; however, these conformations contribute only at a marginal statistical weight (Figure 2B, SI Note 4). In equilibrium with an extended conformation, an arch-shaped conformation thus appears to be feasible both topologically and energetically, in an arrangement resembling the cryoEM structure of PikAIII (SI Note 2).13,14 However, SAXS data identify the extended conformation as the prevalent structural conformation of DEBS M2 in solution.
Figure 2.
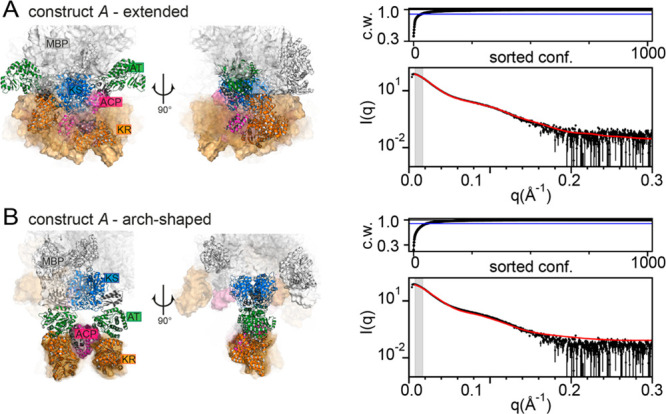
Structural models of DEBS M2 in solution. (A) Extended and (B) arch-shaped conformation. (A and B, left) Model conformations with high statistical weight in the ensembles of Aext and Aarch, respectively. Conformations at the highest weight are shown as cartoons. The conformations at lower cumulative weights, reflecting less populated conformations in solution, are depicted in surface representation (transparency correlated to statistical weights). Domain coloring: MBP, white; KS, blue; LD, gray; AT, green; KR, orange; ACP, magenta; DD, gray. (A and B, top right) Cumulative weights (c. w.) of the weight-sorted conformations fulfilling 90% (blue line) and 100% (black line) of the fittings. (A and B, bottom right) Experimental (black) and computed (red) scattering intensities as I(q) versus q.
XL-MS Distance Information within PKS Modules
In a next step, we mapped the measured XLs of construct A over the extended and arch-shaped configurations to locate candidate regions of domain–domain interactions in M2 (SI Note 5). Overall, the Aext model is in very good agreement with the measured cross-linking data (Tables S6 and S7, SI Note 6). High numbers of cross-linked peptide spectral matches (CSMs), supporting a specific interaction, are observed across the post-AT linker and KR, particularly between K1273 (post-AT linker) and E1474, Y1475, and A1476 (KR, Figure 3A). These residues emerge as a hot-spot of interaction that constrain an otherwise undirected random motion of KR around the post-AT linker and KS, supporting the high flexibility of M2 in solution (Figure 3B and Movie S1). The attachment of the post-AT linker to the KS-AT2 core and the treatment of the KS-AT2 core as rigid body generally prevented interactions between KR and the peripheral regions of AT. As such, one observed cross-link in this region (XL7, AT2K1071-KRQ1623), which cannot be explained by conformational variability of the Aext model, indicates that dissociation of the post-AT linker from the KS-AT2 core, providing a higher degree of flexibility to KR, or conformational variability within the KS-AT2 core may be possible at low frequency. Alternatively, XL7 may result from formation of higher oligomers, as observed in SEC (Figure S2A), in which the peripheral stretch of the AT could interact with the mobile KR of another module (Figure S13, Table S6). Another set of XLs was observed between MBP and KS-AT2, in which the MBP domain interacts ubiquitously, yet at low frequencies with the KS-AT dimer (Figure S14, Table S6). This can be rationalized by the high mobility of the MBP domain, which is connected by long linkers to the condensing domain (Table S9). In contrast, Aarch satisfies only a small set of the XLs measured mainly across KR and ACP (Table S6). Overall, XL-MS data thus support an extended conformation of M2 in solution.
Figure 3.

XL-MS distance information within PKS modules. (A) High-frequency XLs between KR (E1474, Y1475, and A1476) and the post-AT linker (K1273). (B) Schematic illustration of KR flexibility from multiple generated conformations. Post-AT linker in light green. (C–E) Candidate regions of domain–domain interaction of ACP with KS and KR based on the Aext configurations at high statistical weights that satisfy the measured XLs with residue-pair distance lower than 21 Å. (C) Interactions between ACPK1775 (yellow sphere) and D538, G539, and Y540 of KS (red spheres). The active serine of ACP (S1790, cyan sphere) is oriented far from the entrance of the KS binding pocket with the catalytic KSC571 highlighted in green. We note that only a subset of observed ACP-KS interactions place the ACP active serine within proximity to the KS active site (Figure S15). (D) Interactions between ACPK1784 (yellow sphere) and G539, and Y540 of KS (red spheres). The active serine of ACP (S1790, cyan sphere) is oriented in proximity of the entrance of the KS binding pocket with the catalytic KSC571 highlighted in green. (E) Interaction of ACPK1775 (yellow sphere) and V1295, S1303, and E1304 of KR (red spheres).
Positional Variability of ACP
The attachment of ACP via a 14-amino acid linker to the mobile KR implies a high conformational variability. By computational modeling and XL-MS, we isolated candidate regions of domain–domain interaction of ACP with KS-AT2 and KR (Figure 3C–E and Figure S15). We observed high frequencies of interactions across K1775 and K1784 of ACP and D538, G539, and Y540 of KS and across K1775 of ACP and V1295, S1303, and E1304 of KR. The candidate regions are populated by charged residues: D1172, D1773, R1778, E1785 and D1789 of ACP; D535, R553, Y556, R612, R711, and D714 of KS; and D1296, E1299, R1302, E1449, D1450, R1716, R1718, D1722, and R1723 of KR (Table S6). These data indicate that domain–domain interactions are based on networks of electrostatic interactions, similarly as found before in type I and type II FASs.50−52 In our simulations, ACP extensively samples the entrance of the KS binding pocket (Figure S15), and we isolated a candidate orientation with ACPS1790 pointing toward the active site and the catalytic C571 of KS (Figure 3D). Note that, because of its small size, the ACP domain is represented in computational models with low weight, thereby impeding a more detailed description of interfaces. However, XL-MS confirms modeling data and highlights interaction hot spots of ACP distributed over KS-AT2 and KR that are steered by networks of electrostatic interactions (Table S6).
Structural Properties of Construct B
Analogously to construct A, the SAXS intensities measured for construct B in solution are explained by a structural model (Bext) that carries the extended KS-AT2 conformation (Figure S16, SI Note 7). In Bext, KR1 (KR of M1) is highly mobile and occupies a large conformational space, whereas ACP1 displays low mobility and samples specific regions of KS-AT2 (Figure S17, SI Note 8). In addition, the lowered flexibility of KR (of M2) in Bext confirms the structural constraints imposed by the TE (Figure S18, SI Note 8). We note that XL-MS was performed with phosphopantetheinylated construct B. Overall fewer CSMs were observed compared to (apo-)construct A (Table S7), which was also found in a single experiment with phosphopantetheinylated construct A (yielding fewer CSMs compared to (apo-)construct A; Table S6). This observation indicates that the phosphopantetheine moiety constrains ACP docking during substrate shuttling, as suggested previously in structural13,53,54 and functional studies55−57 on type I PKSs and the related FASs (SI Note 9). In construct B, no CSMs were observed between ACP1 and KS, which may be due to the suppression of this interaction by the competitive ACP1-KR1 interactions or by the high dynamics of ACP1 due to the unconstrained KR1 (no N-terminal KS1-AT1 dimer in construct B). In conclusion, Bext is in good agreement with the measured CSMs (Table S5, SI Note 10). Interestingly, the interaction pattern of Aext is also captured in Bext, highlighting the transferability of the XL-MS data to a different construct of DEBS (Table S6, SI Note 11).
Characterization of an Interface via Phage Display
Having observed that the ACP-covering cross-links do not converge on specific interfaces on either catalytic domain of the KS-AT2 core (Tables S6 and S7), we sought to further our understanding of catalytic domain recognition by ACP in an additional experiment based on directed evolution through phage display. We decided to work with the intermodular translocation interface between ACP and KS, i.e., ACP interacting with KS of the downstream module for translocating the growing acyl chain, because this interface is the only one that has been described so far in modular PKSs in more detail.58 Specifically, we set up a phage display approach with the aim of increasing the affinity of the non-native interface between ACP of module 1 (ACP1) and KS of module 3 (KS3). The sequence identity between KS of M2 (KS2) and KS3 is 58% on the protein level (EMBOSS pairwise assembly, www.ebi.ac.uk).59 For phage display, ACP1 was expressed on phage, and its helix 1 (H1), previously determined to play a crucial role during intermodular chain translocation,58 was mutagenized (Figure S19). We note that phage display was performed with apo-ACP phage library due to the difficulty in performing and monitoring phosphopantetheinylation of the ACP displayed at the phage surface.
In our phage display experiment, ACP1H1 mutants (mutants of ACP1 at helix 1) with higher affinity toward KS3-AT3 were enriched. The didomain construct KS3-AT3 was used instead of standalone KS3, because it can be produced as a stable protein by recombinant production.10 For ACP1, three interfaces are available in principle for docking, i.e., two interfaces on KS3 (one for intermodular translocation and one for intramodular condensation), and one on AT3 for transacylation. As H1 plays a major role in the intermodular translocation reaction,24,58 while only marginally contributing to domain–domain interactions during condensation58 and transacylation,60−62 a putative consensus sequence on H1 should evolve from selection for the translocation interface. From the initial library of 3.1 × 106 possible ACP1H1 mutants, 300 enriched mutants were selected for ELISA screening, from which we finally identified six mutants (ACP1-Lib1-Mut“X”) exhibiting an increased binding compared to wild-type ACP1, albeit only marginally, and BSA as a control (Figure 4A). The BSA control was considered as a mean to eliminate ACP1H1 mutants that did presumably not evolve specificity to KS3-AT3, because of also nonspecifically interacting with BSA. We did not observe a consensus in mutations between enriched and ELISA-selected ACP1H1 mutants, denoted ACP1-Lib1-Mut“X” (Figure 4B). The integrity of the selected mutants was eventually confirmed by the activity of bimodular LM+M1+M3-TE assembly lines in which the full-length M1 construct carried mutated ACPs (SI Note 12) (Figure 4C)26,27,47
Figure 4.

Directed evolution of a chimeric ACP:KS interface. Analysis of enriched mutants from ACP1-Library1. (A) ELISA of ACPs presented on the phage surface. Results for selected mutants (Lib1-MutX) and wild-type ACPs are shown. Signal was obtained in KS3-AT3-coated wells (black bars) and the degree of unspecific binding was assessed by comparing it to the signal in BSA coated wells (gray bars). For each ACP, four individually grown phage cultures were tested (except three cultures for ACP1 and ACP2), and the individual data points and data mean are indicated. Data range of ACP2 is indicated by a gray bar for reference. (B) Sequence alignment of obtained mutants compared to wild-type ACP1 and ACP2. Randomized positions are indicated with a yellow asterisk. (C) Turnover rates of wild-type and chimeric bimodular PKSs harboring the mutations enriched in the ACP1-Lib1 biopanning experiments. All bimodular PKS consisted of LM, M1, and Module“X”-TE. Either wild-type DEBS M1 was used as the first module (M1) or one of the six mutants obtained through the first directed evolution experiments (M1-Lib1-Mut“X”). Initial rate data were obtained at individual PKS protein concentrations of 4 μM and nonlimiting concentrations of propionyl-CoA, methylmalonyl-CoA, and NADPH. Measurements were performed in triplicate from one batch per construct M1-Lib1-Mut“X”, and mean and standard deviation are given.
For the two most active mutations, Lib1-Mut3 and Lib1-Mut5, new positions at H1 were chosen for further randomization. Both libraries were treated with the same protocol and analyzed with the same methods as the original library, except for an additional ELISA titration experiment that was conducted to further evaluate binding of ACP1H1 mutants compared to wild-type. The new libraries did neither produce a consensus sequence nor result in high affinity or high turnover bimodular PKSs (Figure S20, Table S8, and SI Note 13). Collectively, these data argue against the occurrence of a specific ACP:KS translocation interface, and, thus, agree with the positional variability of ACP in docking at a catalytic domain suggested from cross-linking and modeling data. We note that several studies propose that the spatial distribution of ACP is sensitive to the acyl-phosphopantetheine moiety,13,53,54 and future studies with phosphopantetheinylated ACP and the native substrate will be needed to further understand the specificity in substrate shuttling as well as the impact of acyl-phosphopantetheinylation.
Discussion
Structural analysis of the evolutionarily related iterative PKSs and FASs suggests a high conformational variability of modular PKSs,17,53,63 which makes the high-resolution structure determination of PKS modules challenging. Structural characterization of flexible proteins is possible by the synergistic application of SAXS, XL-MS, and integrative modeling that enables the quantitative assessment and modeling of structural heterogeneity and conformational variability. In applying the combined approach, we were able to substantiate the relevance of the two strikingly different arch-shaped and extended conformations that have both been proposed as prevalent structures of a PKS module. Overall, our experimental data and modeling results on DEBS M2 show that both conformations are possible in principle; however, the extended structure appears to be dominant in solution.
The extended structure of modular PKSs in solution is itself dynamic, and we present direct insight into the conformational variability of a PKS module. Whereas the KS-AT dimeric unit is relatively rigid, as reported by previous structural studies on type I PKS10−12,16,32 and treated as rigid core in this study, computational modeling constrained by SAXS data suggests a high conformational variability of KR. Combined with XL-MS data highlighting the interaction of KR with the post-AT linker, a picture of KR moving undirected and randomly just constrained by the post-AT linker as an anchoring point emerges. A PKS module can also feature dimerizing units in the processing part; i.e., the dimeric domain DH, and a dimerization element present in about half of the modules with only KR as modifying domain.64 Both units will constrain conformational dynamics, and KR will no longer be a separately moving domain (Movie S1), but part of a larger assembly with decreased mobility. In conclusion, this study argues that in PKS modules, a compact KS-AT dimeric unit carries a processing part with conformational dynamics depending on its module organization.17
The substrate-shuttling ACP domain is of key importance for any PKS module. In our study, performed on the nonphosphopantetheinylated domain, we observe a lack of high-frequency XLs from ACP to KS, AT and KR, which indicates that ACP does not form highly specific interfaces with those domains. To further our understanding of the specificity of ACP-domain interactions, we employed a directed evolution approach for evolving the ACP:KS translocation interface toward higher affinities. Directed evolution approaches are widely used to narrow down specific interactions as a result of an increase in affinity. Our results show that despite intensive screening only few higher affinity hits emerged from ELISA analysis, among which no consensus sequence could be found. We interpret this observation as arguing against the existence of specific ACP:KS translocation interfaces, in line with our solution structure data. Overall, our data suggest that substrate shuttling by ACP of type I PKS is facilitated by weak interactions, similarly as described earlier for type I and type II FAS.51,52 We conclude that ACP moves freely, and rather randomly collides with domains of the type I assembly, where electrostatic networks influence the approach of ACP to the individual domains. As such our findings essentially support a previously suggested model in which ACP is unrestrained, but restricted in its trajectory by the other domains of the multienzyme assembly.20
We note that the body of data for this study was collected with nonphosphopantetheinylated proteins, mainly due to technical reasons. For example, domains were represented as rigid bodies in CG structural modeling, and the phosphopantetheine moiety was not represented in the model. Since ACPs of type I systems do not accommodate phosphopantetheine and acyl-chains, leading just to local changes around the active serine in apo-, holo-, and acylated-ACP, but leaving the ACP fold overall unchanged,65−67 our data are representative of the domain–domain portion of the interactions. However, several structural studies revealed that the spatial distribution of ACP is sensitive to the acyl moiety in type I PKSs and the related FASs.13,53,54 Further, recent enzyme kinetic studies on ATs from type I PKSs and FASs reported the variation in KM with the identity of the acyl unit, suggesting the impact of acyl-phosphopantetheine moiety on ACP docking.55−57 Accordingly, studies are required on phosphopantetheinylated and acylated ACPs to understand substrate shuttling. In particular, the impact of the phosphopantetheine and the identity of the acyl-moiety on this process is just poorly characterized for type I PKSs and FASs until to date.65−67
Besides giving insight into the structural and conformational properties of PKSs, our study provides guidelines for PKS engineering. Taking into account recently published data,34,68 the extended conformation is likely self-sufficient for synthetic progress in PKS assembly lines. We therefore suggest to work with the extended model in PKS engineering approaches, as well as to base active site remodeling on the biomolecular processes connected to this structural arrangement. For example, our data support the KS functional mode to be “conventional”, i.e., using the generic KS binding tunnel identical to type I FASs and iterative PKSs, and approaches on active site design can be informed by data available also on those systems. Our findings can further explain why engineering approaches involving exchanges of the KR domain or the entire processing part have been successful in the past,69−72 as well as encourage consideration of such approaches in PKS engineering projects. XL-MS data indicate just few cross-links between KR and other domains (to the AT domain, Tables S6 and S7), so that KR exchanges may be well accessible in modules with only KR as modifying domain. We assume that also exchanges of the entire processing part (in larger PKS modules) are amenable, because condensing and processing parts appear separated in structural and conformational properties; as also implied by several successful engineering approaches (e.g., refs (72 and 73)). Data for ACP should be interpreted in a similar light. As our direct insight in ACP-mediated domain–domain interactions argues against well-defined docking events (between apo-ACP and catalytic domains), productive substrate shuttling appears possible in chimeric PKS with non-native ACP as long as conflicting electrostatic interactions are avoided.
Acknowledgments
We thank Mirko Joppe for providing the diketide substrate, Alicia D’Souza for assistance in cloning pADD01, and Lynn Buyachuihan for help with the protein purification. We are grateful for Florian Buhr’s and Harald Schwalbe’s support in generating the harmonized gene sequences, Gregor Witte for initial support in analyzing the SAXS data, and Andreas Ernst and Mateusz Putyrski for support in setting up and running the phage display experiments.
Glossary
Abbreviations
- PKS
polyketide synthase
- DEBS
deoxyerythronolide B synthase
- PIKS
pikromycin synthase
- KS
ketosynthase
- AT
acyl transferase
- ACP
acyl carrier protein
- KR
ketoreductase
- DH
dehydratase
- ER
enoylreductase
- TE
thioesterase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00043.
Description of plasmids and chemicals, enzymatic assays, and compound analysis; protocols for data collection and analysis in XL-MS, CG structural modeling, SAXS, and phage display; this includes Supporting Notes 1–13, Figures S1–S20, and Tables S1–S10 for details and additional data on all the methods and material used in this study (PDF)
Movie S1, prevalent conformations of construct A in solution (MP4)
Author Contributions
† M.K. and E.R. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Support was received from the Volkswagen Foundation (Lichtenberg Professorship to M.G.; grants 85701 and 91809), the LOEWE program (Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz) of the state of Hesse conducted within the framework of the MegaSyn Research Cluster (to M.G. and G.H.), the Max Planck Society (to G.H.), the Deutsche Forschungsgemeinschaft (to H.U., SFB860), and the NIH (to C.K., grant number R01 GM087934).
The authors declare no competing financial interest.
Supplementary Material
References
- Staunton J.; Weissman K. J. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 2001, 18, 380–416. 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- Broadhurst R. W.; Nietlispach D.; Wheatcroft M. P.; Leadlay P. F.; Weissman K. J. The structure of docking domains in modular polyketide synthases. Chem. Biol. 2003, 10, 723–731. 10.1016/S1074-5521(03)00156-X. [DOI] [PubMed] [Google Scholar]
- Buchholz T. J.; Geders T. W.; Bartley F. E. 3rd.; Reynolds K. A.; Smith J. L.; Sherman D. H. Structural basis for binding specificity between subclasses of modular polyketide synthase docking domains. ACS Chem. Biol. 2009, 4, 41–52. 10.1021/cb8002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risser F.; Collin S.; Dos Santos-Morais R.; Gruez A.; Chagot B.; Weissman K. J. Towards improved understanding of intersubunit interactions in modular polyketide biosynthesis: Docking in the enacyloxin IIa polyketide synthase. J. Struct. Biol. 2020, 212, 107581. 10.1016/j.jsb.2020.107581. [DOI] [PubMed] [Google Scholar]
- Cortes J.; Haydock S. F.; Roberts G. A.; Bevitt D. J.; Leadlay P. F. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 1990, 348, 176–178. 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- McDaniel R.; Ebert-Khosla S.; Hopwood D. A.; Khosla C. Engineered biosynthesis of novel polyketides. Science 1993, 262, 1546–1550. 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]
- Tsai S. C.; Miercke L. J.; Krucinski J.; Gokhale R.; Chen J. C.; Foster P. G.; Cane D. E.; Khosla C.; Stroud R. M. Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: versatility from a unique substrate channel. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 14808–14813. 10.1073/pnas.011399198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldes J. W.; Akey D. L.; Kittendorf J. D.; Sherman D. H.; Smith J. L.; Fecik R. A. Structural and mechanistic insights into polyketide macrolactonization from polyketide-based affinity labels. Nat. Chem. Biol. 2006, 2, 531–536. 10.1038/nchembio822. [DOI] [PubMed] [Google Scholar]
- Edwards A. L.; Matsui T.; Weiss T. M.; Khosla C. Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide B synthase. J. Mol. Biol. 2014, 426, 2229–2245. 10.1016/j.jmb.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Chen A. Y.; Kim C. Y.; Cane D. E.; Khosla C. Structural and mechanistic analysis of protein interactions in module 3 of the 6-deoxyerythronolide B synthase. Chem. Biol. 2007, 14, 931–943. 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Kim C.-Y.; Mathews I. I.; Cane D. E.; Khosla C. The 2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11124–11129. 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher J. R.; Smaga S. S.; Hansen D. A.; Brown W. C.; Gerwick W. H.; Sherman D. H.; Smith J. L. Cyanobacterial polyketide synthase docking domains: a tool for engineering natural product biosynthesis. Chem. Biol. 2013, 20, 1340–1351. 10.1016/j.chembiol.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher J. R.; Dutta S.; Hansen D. A.; Hale W. A.; Chemler J. A.; Dosey A. M.; Narayan A. R.; Håkansson K.; Sherman D. H.; Smith J. L.; Skiniotis G. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature 2014, 510, 560–564. 10.1038/nature13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S.; Whicher J. R.; Hansen D. A.; Hale W. A.; Chemler J. A.; Congdon G. R.; Narayan A. R.; Håkansson K.; Sherman D. H.; Smith J. L.; Skiniotis G. Structure of a modular polyketide synthase. Nature 2014, 510, 512–517. 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T.; Leibundgut M.; Ban N. The crystal structure of a mammalian fatty acid synthase. Science 2008, 321, 1315–1322. 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liang J.; Chen L.; Zhang W.; Kong L.; Peng C.; Su C.; Tang Y.; Deng Z.; Wang Z. Structural basis for the biosynthesis of lovastatin. Nat. Commun. 2021, 12, 867. 10.1038/s41467-021-21174-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst D. A.; Townsend C. A.; Maier T. The architectures of iterative type I PKS and FAS. Nat. Prod. Rep. 2018, 35, 1046–1069. 10.1039/C8NP00039E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Gay D. C.; Demeler B.; White M. A.; Keatinge-Clay A. T. Divergence of multimodular polyketide synthases revealed by a didomain structure. Nat. Chem. Biol. 2012, 8, 615–621. 10.1038/nchembio.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittner A.; Grininger M. Modular Polyketide Synthases (PKSs): A New Model Fits All?. ChemBioChem 2014, 15, 2489–2493. 10.1002/cbic.201402432. [DOI] [PubMed] [Google Scholar]
- Weissman K. J. Uncovering the structures of modular polyketide synthases. Nat. Prod. Rep. 2015, 32, 436–453. 10.1039/C4NP00098F. [DOI] [PubMed] [Google Scholar]
- Klaus M.; Grininger M. Engineering strategies for rational polyketide synthase design. Nat. Prod. Rep. 2018, 35, 1070–1081. 10.1039/C8NP00030A. [DOI] [PubMed] [Google Scholar]
- Malico A. A.; Nichols L.; Williams G. J. Synthetic biology enabling access to designer polyketides. Curr. Opin. Chem. Biol. 2020, 58, 45–53. 10.1016/j.cbpa.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry B.; Li X.; Robbins T.; Cane D. E.; Khosla C. A Turnstile Mechanism for the Controlled Growth of Biosynthetic Intermediates on Assembly Line Polyketide Synthases. ACS Cent. Sci. 2016, 2, 14–20. 10.1021/acscentsci.5b00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S.; Lowry B.; Yuzawa S.; Kenthirapalan S.; Chen A. Y.; Cane D. E.; Khosla C. Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 4110–4115. 10.1073/pnas.1118734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa S.; Backman T. W. H.; Keasling J. D.; Katz L. Synthetic biology of polyketide synthases. J. Ind. Microbiol. Biotechnol. 2018, 45, 621–633. 10.1007/s10295-018-2021-9. [DOI] [PubMed] [Google Scholar]
- Klaus M.; D’Souza A. D.; Nivina A.; Khosla C.; Grininger M. Engineering of Chimeric Polyketide Synthases Using SYNZIP Docking Domains. ACS Chem. Biol. 2019, 14, 426–433. 10.1021/acschembio.8b01060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus M.; Buyachuihan L.; Grininger M. Ketosynthase Domain Constrains the Design of Polyketide Synthases. ACS Chem. Biol. 2020, 15, 2422–2432. 10.1021/acschembio.0c00405. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay A. T. Crystal structure of the erythromycin polyketide synthase dehydratase. J. Mol. Biol. 2008, 384, 941–953. 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay A. T.; Stroud R. M. The structure of a ketoreductase determines the organization of the β-carbon processing enzymes of modular polyketide synthases. Structure 2006, 14, 737–748. 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Alekseyev V. Y.; Liu C. W.; Cane D. E.; Puglisi J. D.; Khosla C. Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 2007, 16, 2093–2107. 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivina A.; Yuet K. P.; Hsu J.; Khosla C. Evolution and Diversity of Assembly-Line Polyketide Synthases. Chem. Rev. 2019, 119, 12524–12547. 10.1021/acs.chemrev.9b00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst D. A.; Jakob R. P.; Zahringer F.; Maier T. Mycocerosic acid synthase exemplifies the architecture of reducing polyketide synthases. Nature 2016, 531, 533–537. 10.1038/nature16993. [DOI] [PubMed] [Google Scholar]
- Garg A.; Xie X.; Keatinge-Clay A.; Khosla C.; Cane D. E. Elucidation of the cryptic epimerase activity of redox-inactive ketoreductase domains from modular polyketide synthases by tandem equilibrium isotope exchange. J. Am. Chem. Soc. 2014, 136, 10190–10193. 10.1021/ja5056998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Sevillano N.; La Greca F.; Deis L.; Liu Y. C.; Deller M. C.; Mathews I. I.; Matsui T.; Cane D. E.; Craik C. S.; Khosla C. Structure-Function Analysis of the Extended Conformation of a Polyketide Synthase Module. J. Am. Chem. Soc. 2018, 140, 6518–6521. 10.1021/jacs.8b02100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittner A.; Paithankar K. S.; Drexler D. J.; Himmler A.; Grininger M. Probing the modularity of megasynthases by rational engineering of a fatty acid synthase Type I. Protein Sci. 2019, 28, 414–428. 10.1002/pro.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T.; Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528–1535. 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- Pernot P.; Round A.; Barrett R.; De Maria Antolinos A.; Gobbo A.; Gordon E.; Huet J.; Kieffer J.; Lentini M.; Mattenet M.; Morawe C.; Mueller-Dieckmann C.; Ohlsson S.; Schmid W.; Surr J.; Theveneau P.; Zerrad L.; McSweeney S. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 2013, 20, 660–664. 10.1107/S0909049513010431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D.; Hammel M.; Tainer J. A.; Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013, 105, 962–974. 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D.; Hammel M.; Tainer J. A.; Sali A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44, W424–429. 10.1093/nar/gkw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Różycki B.; Kim Y. C.; Hummer G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure 2011, 19, 109–116. 10.1016/j.str.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köfinger J.; Stelzl L. S.; Reuter K.; Allande C.; Reichel K.; Hummer G. Efficient Ensemble Refinement by Reweighting. J. Chem. Theory Comput. 2019, 15, 3390–3401. 10.1021/acs.jctc.8b01231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummer G.; Köfinger J. Bayesian ensemble refinement by replica simulations and reweighting. J. Chem. Phys. 2015, 143, 243150. 10.1063/1.4937786. [DOI] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. 10.1107/S0021889892001663. [DOI] [Google Scholar]
- Kim Y. C.; Hummer G. Coarse-grained models for simulations of multiprotein complexes: application to ubiquitin binding. J. Mol. Biol. 2008, 375, 1416–1433. 10.1016/j.jmb.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Wu Y. J.; Zhu M.; Fan S. B.; Lin J.; Zhang K.; Li S.; Chi H.; Li Y. X.; Chen H. F.; Luo S. K.; Ding Y. H.; Wang L. H.; Hao Z.; Xiu L. Y.; Chen S.; Ye K.; He S. M.; Dong M. Q. Identification of cross-linked peptides from complex samples. Nat. Methods 2012, 9, 904–906. 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- Tonikian R.; Zhang Y.; Boone C.; Sidhu S. S. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat. Protoc. 2007, 2, 1368–1386. 10.1038/nprot.2007.151. [DOI] [PubMed] [Google Scholar]
- Lowry B.; Robbins T.; Weng C. H.; O’Brien R. V.; Cane D. E.; Khosla C. In vitro reconstitution and analysis of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 2013, 135, 16809–16812. 10.1021/ja409048k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo R. P.; Tainer J. A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsom A.; Schneider M.; Fischer L.; Brock O.; Rappsilber J. Serum Albumin Domain Structures in Human Blood Serum by Mass Spectrometry and Computational Biology. Mol. Cell Proteomics 2016, 15, 1105–1116. 10.1074/mcp.M115.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C.; Haushalter R. W.; Lee D. J.; Markwick P. R.; Bruegger J.; Caldara-Festin G.; Finzel K.; Jackson D. R.; Ishikawa F.; O’Dowd B.; McCammon J. A.; Opella S. J.; Tsai S. C.; Burkart M. D. Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 2014, 505, 427–431. 10.1038/nature12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini E.; Gajewski J.; Klaus M.; Hummer G.; Grininger M. Analysis and engineering of substrate shuttling by the acyl carrier protein (ACP) in fatty acid synthases (FASs). Chem. Commun. (Cambridge, U. K.) 2018, 54, 11606–11609. 10.1039/C8CC06838K. [DOI] [PubMed] [Google Scholar]
- Misson L. E.; Mindrebo J. T.; Davis T. D.; Patel A.; McCammon J. A.; Noel J. P.; Burkart M. D. Interfacial plasticity facilitates high reaction rate of E. coli FAS malonyl-CoA:ACP transacylase, FabD. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 24224–24233. 10.1073/pnas.2009805117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignole E. J.; Smith S.; Asturias F. J. Conformational Flexibility of Metazoan Fatty Acid Synthase Enables Catalysis. Nat. Struct. Mol. Biol. 2009, 16, 190–197. 10.1038/nsmb.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J. W.; Iyer K. R.; Hasan S. M. N.; Cowen L. E.; Mazhab-Jafari M. T. Electron cryomicroscopy observation of acyl carrier protein translocation in type I fungal fatty acid synthase. Sci. Rep. 2019, 9, 12987. 10.1038/s41598-019-49261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittner A.; Paithankar K. S.; Huu K. V.; Grininger M. Characterization of the Polyspecific Transferase of Murine Type I Fatty Acid Synthase (FAS) and Implications for Polyketide Synthase (PKS) Engineering. ACS Chem. Biol. 2018, 13, 723–732. 10.1021/acschembio.7b00718. [DOI] [PubMed] [Google Scholar]
- Geyer K.; Sundaram S.; Susnik P.; Koert U.; Erb T. J. Understanding Substrate Selectivity of Phoslactomycin Polyketide Synthase by Using Reconstituted in Vitro Systems. ChemBioChem 2020, 21, 2080–2085. 10.1002/cbic.202000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegemann F.; Grininger M. Transacylation Kinetics in Fatty Acid and Polyketide Synthases and its Sensitivity to Point Mutations**. ChemCatChem 2021, 13, 2771–2782. 10.1002/cctc.202002077. [DOI] [Google Scholar]
- Kapur S.; Chen A. Y.; Cane D. E.; Khosla C. Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22066–22071. 10.1073/pnas.1014081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman S. B.; Wunsch C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Wong F. T.; Jin X.; Mathews I. I.; Cane D. E.; Khosla C. Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry 2011, 50, 6539–6548. 10.1021/bi200632j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F. T.; Chen A. Y.; Cane D. E.; Khosla C. Protein-protein recognition between acyltransferases and acyl carrier proteins in multimodular polyketide synthases. Biochemistry 2010, 49, 95–102. 10.1021/bi901826g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanaga A.; Iwasawa S.; Shinohara Y.; Kudo F.; Eguchi T. Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 1802–1807. 10.1073/pnas.1520042113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benning F. M. C.; Sakiyama Y.; Mazur A.; Bukhari H. S. T.; Lim R. Y. H.; Maier T. High-Speed Atomic Force Microscopy Visualization of the Dynamics of the Multienzyme Fatty Acid Synthase. ACS Nano 2017, 11, 10852–10859. 10.1021/acsnano.7b04216. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Piasecki S. K.; Keatinge-Clay A. T. Structural studies of an A2-type modular polyketide synthase ketoreductase reveal features controlling alpha-substituent stereochemistry. ACS Chem. Biol. 2013, 8, 1964–1971. 10.1021/cb400161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran L.; Broadhurst R. W.; Tosin M.; Cavalli A.; Weissman K. J. Insights into Protein-Protein and Enzyme-Substrate Interactions in Modular Polyketide Synthases. Chem. Biol. 2010, 17, 705–716. 10.1016/j.chembiol.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Perez D. R.; Leibundgut M.; Wider G. Interactions of the Acyl Chain with the Saccharomyces cerevisiae Acyl Carrier Protein. Biochemistry 2015, 54, 2205–2213. 10.1021/bi5014563. [DOI] [PubMed] [Google Scholar]
- Ploskon E.; Arthur C.; Evans S.; Williams C.; Crosby J.; Simpson T.; Crump M. A mammalian type I fatty acid synthase acyl carrier protein domain does not sequester acyl chains. J. Biol. Chem. 2008, 283, 518–528. 10.1074/jbc.M703454200. [DOI] [PubMed] [Google Scholar]
- Cogan D. P.; Li X.; Sevillano N.; Mathews I. I.; Matsui T.; Craik C. S.; Khosla C. Antibody Probes of Module 1 of the 6-Deoxyerythronolide B Synthase Reveal an Extended Conformation During Ketoreduction. J. Am. Chem. Soc. 2020, 142, 14933–14939. 10.1021/jacs.0c05133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford D.; Jacobsen J. R.; Luo G.; Cane D. E.; Khosla C. A functional chimeric modular polyketide synthase generated via domain replacement. Chem. Biol. 1996, 3, 827–831. 10.1016/S1074-5521(96)90068-X. [DOI] [PubMed] [Google Scholar]
- Kellenberger L.; Galloway I. S.; Sauter G.; Bohm G.; Hanefeld U.; Cortes J.; Staunton J.; Leadlay P. F. A polylinker approach to reductive loop swaps in modular polyketide synthases. ChemBioChem 2008, 9, 2740–2749. 10.1002/cbic.200800332. [DOI] [PubMed] [Google Scholar]
- Annaval T.; Paris C.; Leadlay P. F.; Jacob C.; Weissman K. J. Evaluating Ketoreductase Exchanges as a Means of Rationally Altering Polyketide Stereochemistry. ChemBioChem 2015, 16, 1357–1364. 10.1002/cbic.201500113. [DOI] [PubMed] [Google Scholar]
- Hagen A.; Poust S.; Rond T.; Fortman J. L.; Katz L.; Petzold C. J.; Keasling J. D. Engineering a Polyketide Synthase for In Vitro Production of Adipic Acid. ACS Synth. Biol. 2016, 5, 21–27. 10.1021/acssynbio.5b00153. [DOI] [PubMed] [Google Scholar]
- Eng C. H.; Yuzawa S.; Wang G.; Baidoo E. E.; Katz L.; Keasling J. D. Alteration of Polyketide Stereochemistry from anti to syn by a Ketoreductase Domain Exchange in a Type I Modular Polyketide Synthase Subunit. Biochemistry 2016, 55, 1677–1680. 10.1021/acs.biochem.6b00129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.