

Abstract
In solution, the charge of a protein is intricately linked to its stability, but electrospray ionization distorts this connection, potentially limiting the ability of native mass spectrometry to inform about protein structure and dynamics. How the behavior of intact proteins in the gas phase depends on the presence and distribution of ionizable surface residues has been difficult to answer because multiple chargeable sites are present in virtually all proteins. Turning to protein engineering, we show that ionizable side chains are completely dispensable for charging under native conditions, but if present, they are preferential protonation sites. The absence of ionizable side chains results in identical charge state distributions under native-like and denaturing conditions, while coexisting conformers can be distinguished using ion mobility separation. An excess of ionizable side chains, on the other hand, effectively modulates protein ion stability. In fact, moving a single ionizable group can dramatically alter the gas-phase conformation of a protein ion. We conclude that although the sum of the charges is governed solely by Coulombic terms, their locations affect the stability of the protein in the gas phase.
Keywords: protein folding, gas-phase conformations, ion mobility mass spectrometry
Characterizing the interactions, stabilities, and conformations of proteins is of central importance in biochemical and pharmaceutical sciences. Native mass spectrometry (MS) informs about the molecular weight of protein complexes to reveal oligomeric states and ligand binding.1−3 Combined with ion mobility (IM), it can be used to determine collision cross sections (CCS) and identify conformational changes.4−6 If protein complexes are additionally subjected to collisional activation inside the mass spectrometer, the resulting collision-induced unfolding (CIU) can be followed by IM-MS, informing about their gas-phase stabilities.7−10
IM-MS has been successfully applied to characterize the lipid preferences of membrane proteins,11,12 define structural heterogeneity associated with glycosylation,13 follow structural transitions in disordered proteins,14 and determine the impact of disease-associated mutations on protein stability.15 By combining alanine scanning with CIU, it is possible to quantify the contributions of individual residues on the conformational stabilities of protein ions.16 IM-MS is furthermore employed in the pharmaceutical industry,17 for example to characterize drug conjugates.18
Common to all of these applications is a reliance on the generation of native-like protein ions through electrospray ionization (ESI), where the protein is desolvated and charges are added to facilitate its detection in the mass spectrometer. A significant body of work over many decades has been dedicated to understanding the mechanism of protein charging in ESI and protein structures in the gas phase.19−23 However, while the three-dimensional structures of protein complexes can be preserved for the time frame of an MS experiment, their surface charges inevitably differ between solution and gas phase.20,24,25 In positive-mode ESI, the number of charges acquired by native proteins scales with their solvent-accessible surface area (SASA), as predicted by the charge residue model (CRM), and is in virtually all cases lower than the number of basic surface residues.26−28 Protein stability in solution depends on a delicate balance of surface electrostatics.29 Therefore, the charging process can distort protein stability by adding Coulombic repulsion between ESI charges.30−32 This discrepancy raises the question to what extent the surface properties of a protein can affect its behavior in the gas phase and thus in IM-MS. For example, drug conjugation of antibodies can alter their surface charges, and membrane proteins have a lower percentage and a more uneven distribution of ionizable residues on their surface than soluble proteins.33 Here, we clarify the role of ionizable residues in native MS using engineered proteins where the number of ionizable side chains can be altered without affecting their native structures.
Results
Ionizable Groups Connect Charging and Folding in Native MS
First, to fully uncouple solution- and gas-phase charge, we turned to a cellulose-binding domain derived from an exoglycanase from Cellulomonas fimi. The wild-type protein has a low charge density, with three basic and one acidic residue in its 106-residue sequence (EXGWT) and includes one disulfide bridge. Replacement with uncharged residues (K28Q, D36Q, R68Q, and H90W), plus an N-terminal acetylation (EXGQQQW), has yielded a protein in which the C-terminus is the only ionizable site, while maintaining a near-identical structure as the wild-type (Figure 1A, Figure S1).34,35 ESI-MS of EXGQQQW revealed two populations with masses of 11 246 and 11 290 Da instead of the expected 11 158 Da. Enzymatic digestion and LC-MS/MS analysis did not show any unexpected sequence variations or modifications. Collisional activation of the protein in the ion trap of the mass spectrometer resulted in a reduction of ion mass, revealing the expected molecular weight with 0–8 sodium adducts (Figure S2A). ESI-MS in negative mode similarly showed up to eight chloride adducts (Figure S2B). EXGWT, on the other hand, yielded the expected mass of 11 080 Da, with only minor adduct formation (Figure S2C). The presence of 0–8 sodium adducts for the EXGQQQW 6+ ion suggests that they are not the sole charge carriers and could bind to the backbone or side chain carbonyls.36,37 EXGQQQW may also retain NH4+, as shown previously for proteins with disproportionately few basic residues,38 which can be dissociated by collisional activation to reveal the more stable Na adducts. Such a “mixed charging” scenario can also involve protonation of side chains such as P and Q.39
Figure 1.
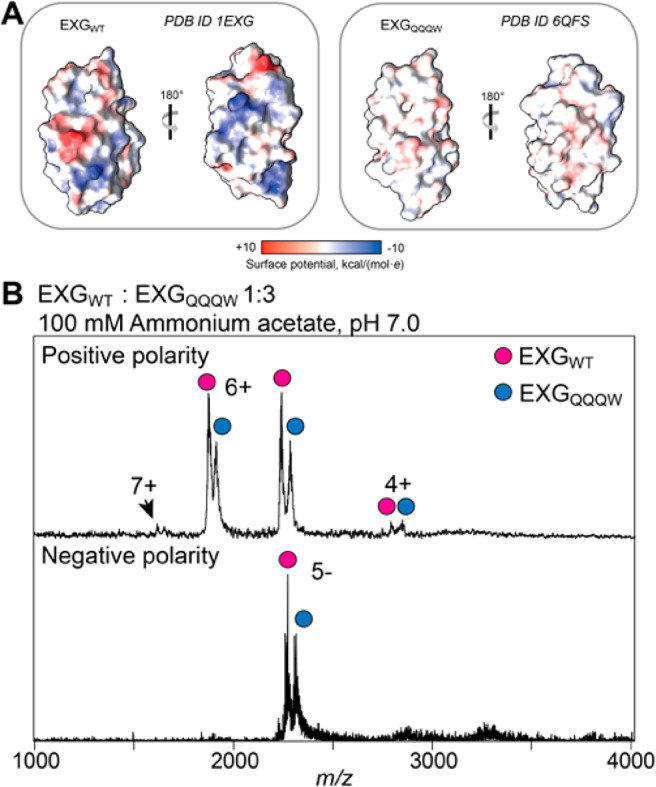
Charging of EXG variants in ESI-MS. (A) Wt EXGWT and the chargeless variant EXGQQQW have highly similar three-dimensional structures. Protein surfaces are rendered according to Coulombic potential at pH 7.0, showing the effect of four replacements (K28Q, D36Q, R68Q, and H90W) on the surface charge of EXGQQQW. (B) ESI-Mass spectra of a 1:3 mixture of EXGWT and EXGQQQW show identical charge state envelopes for both variants in positive (top) and negative (bottom) ionization modes.
We recorded ESI-mass spectra of a mixture of EXGWT and EXGQQQW in 100 mM ammonium acetate (AmAc), pH 7.0, in positive and negative ionization modes. We found that we required ∼3 times the concentration of EXGQQQW to obtain a signal intensity comparable to that of EXGWT (Figure 1B). Both proteins exhibited virtually identical narrow charge state distributions (CSDs). In positive mode, we found a maximum charge (zmax) of 7+, just below the 8+ charge predicted by the SASA. Furthermore, the average charge of 5.4+ is in good agreement with the value of 5.5+ predicted based on the SASA of the crystal structure (Table 1).38 These findings clearly illustrate that ESI charging is independent of surface properties, but rather reflects key features of protein solution structure. This interpretation is in good agreement with reports that chemical modifications of side-chain charges do not affect ESI charges of compactly folded protein ions.28,40 To understand further the role of ionizable residues, we tested whether we could reduce the charge of EXGWT and EXGQQQW ions in the gas phase. Trimethylamine-N-oxide (TMAO) is a basic osmolyte that strips protons off ionized proteins through collisions in the gas phase.41,42 After addition of 50 mM TMAO to the ESI solution, both proteins shift to lower charge states (Figure S3). However, we also observed that the intensity of the EXGQQQW peaks was significantly reduced. To explore the origin of this phenomenon, we increased the charge-reducing effect of TMAO through increased collisional activation in the ion source. At a cone voltage of 300 V, the average charge of the EXGWT was reduced to 2.7, while the EXGQQQW signal was lost. We conclude that a subset of charges on EXGWT are bound to high-affinity sites that render them resistant to stripping by TMAO, whereas EXGQQQW can be decharged completely and is lost as a neutral species. In summary, although both variants acquire the same number of charges, the presence of basic residues in EXGWT results in a higher gas phase basicity (GB).
Table 1. SASAs, Predicted and Experimental Maximum Charge (zmax), and Predicted and Experimental Average Charge (zavg) for All Protein Variants.
| protein | SASA (Å2) | pred. zmax | exp. zmax | pred. zavg | exp. zavg |
|---|---|---|---|---|---|
| EXGWT | 5080 | 8.2 | 7 | 5.5 | 5.5 |
| EXGQQQW | 4594 | 8.3 | 7 | 5.4 | 5.4 |
| GFPWT | 11 170 | 13.1 | 12 | 9.7 | 9.7 |
| GFPAc | 9941 | 13.1 | 11 | 9.2 | 9.2 |
| GFPBas | 11 601 | 13.2 | 12 | 10.1 | 10.1 |
| TTHA | 4273 | 6.5 | 5 | 5.2 | 4.8 |
Protein Folded States Can Be Distinguished in the Absence of Ionizable Residues
Because both proteins exhibited identical charge states under all conditions, we asked whether we could still distinguish coexisting conformational states by MS. Using circular dichroism (CD) spectroscopy, we found that the addition of the common denaturant acetonitrile to the buffer (50% v/v) converted EXGQQQW to random coil, whereas EXGWT appears to retain more of its native secondary structure (Figure 2A). MS analysis of the proteins in 50% acetonitrile showed identical CSDs with only a minor shift to higher charge states compared to native conditions (Figure 2B). Despite unfolding in solution, the charge states of EXGWT and EXGQQQW again remain close to the limit imposed by the SASA of the native protein.
Figure 2.
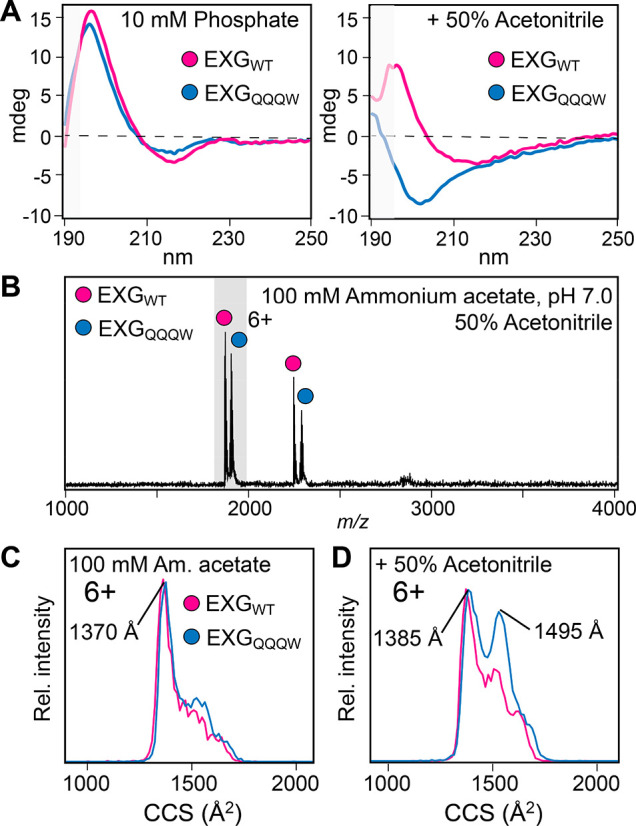
Folded states of EXGWT and EXGQQQW in solution and in the gas phase. (A) CD spectroscopy in the far-UV region shows near-identical secondary structures in 10 mM sodium phosphate buffer pH 7. In 50% acetonitrile, EXGQQQW, but not EXGWT, converts to random coil. Greyed-out parts of the curves exhibited HT voltages above 700 V, indicating increased spectral noise. (B) ESI-MS shows a slight increase of the average charge to 5.7 for both proteins sprayed from 50% acetonitrile. (C) In 100 mM AmAc, the 6+ charge states of EXGWT and EXGQQQW have similar CCS distributions. (D) In 50% acetonitrile, the CCS distribution for EXGQQQW shows an extra peak around 1550 Å2, indicating a second, more unfolded population.
Next, we employed IM-MS, where protein ions are separated according to mobility, which depends on their charges and their CCSs.5 In this manner, we can distinguish conformational ensembles with identical mass-to-charge ratios. Analysis of the main 6+ charge state under native conditions revealed identical CCS distributions for EXGWT and EXGQQQW, with a narrow peak at 1370 Å2 and a shoulder around 1500 Å2 (Figure 2C). Comparison to the expected value of 1240 Å2 theoretically calculated from the crystal structure suggests a compact conformation in the gas phase. The small increase over the predicted CCS may be due to the disordered N-terminus, as well as some Coulombic stretching due to the high charge. Consistent with this interpretation, the CCS determined for the 5+ charge state is centered at a peak of 1280 Å2 and exhibits a good agreement with the theoretical value (Figure S4). We then investigated the effect of 50% acetonitrile on the CCS distributions of both proteins. For the 6+ charge state of EXGWT, we found a small increase in the unfolded fraction with a CCS of around 1500 Å2, while most of the protein retained the same CCS as under native conditions. For the 6+ ions of the chargeless variant, the intensity of the CCS distribution around 1500 Å2 increased notably, indicating a nearly 1:1 ratio of compact to unfolded protein (Figure 2D). It appears therefore that despite their identical CSDs, an increased population of EXGQQQW variant occupies an unfolded state, reflecting the solution behavior observed by CD spectroscopy.
CIU was then employed to test whether ionizable residues affect the folding landscape of EXG in the gas phase. Briefly, protein ions are subjected to thermal activation in the mass spectrometer by raising the acceleration voltage of ions entering the collision cell, leading to an increase in high-energy collisions with the neutral buffer gas. The resulting unfolding events can be monitored by following the change in CCS as a function of collision energy.7 Surprisingly, the CIU traces obtained for EXGWT and EXGQQQW showed no significant differences in stability, with both reaching an unfolded state at 15 V (Figure S5). We reason that for the same charge state, the gas-phase stabilities of both variants are governed mainly by the energy required to disrupt their identical or highly similar internal hydrogen-bonding networks.
Surface Charge Distributions Control the Stability of Protein Ions
Having established that charge depletion does not necessarily change gas-phase unfolding, we asked whether we could modulate the stability of protein ions by instead adding ionizable sites. For this analysis, we utilized two green fluorescent protein (GFP) variants that maintain fluorescence and have near-identical secondary and tertiary structure to the wild-type (GFPWT) protein (28 basic and 34 acidic residues), in which a large number of surface-exposed residues have been mutated to have either acidic (GFPAc, 19 basic and 49 acidic residues) or basic (GFPBas, 41 basic and 26 acidic residues) side chains (Figure 3A).43 Varying only in the charge of the surface-exposed amino acids, this system is therefore ideal to study how an excess of ionizable side chains affects protein ion stability. As expected, mass spectra of GFPWT, GFPAc, and GFPBas exhibit near-identical CSDs with average charge states of 9.7+, 9.2+, and 10.1+, respectively (Figure 3B, Table 1), reflecting their highly similar SASAs. IM-MS analysis yield CCSs of approximately 2000–2100 Å2 for all three variants, in good agreement with the 2118 Å2 predicted from the GFPWT structure. However, the CIU profiles of the 9+ and 8+ charge states of GFPWT and the basic and acidic variants reveal different gas-phase stabilities, as well as a smaller CCS for the unfolded state of GFPAc (Figure 3C, Figure S6). To assess the difference in stability, we performed CIU of all GFP variants. By analyzing the proteins pairwise, we could quantify the stabilities of the most compact state of each variant relative to the GFPWT.10 The wild type and acidic variant unfold at low collision voltages, whereas the basic variant is more stable (Figure S7). Interestingly, solution unfolding analysis has not revealed significant differences in stability of supercharged GFP variants.43 Because all three variants share the same three-dimensional fold and hydrophobic core, the data show that despite making no notable contribution to ion charge, increasing the number of basic residues on the protein surface can have a significant impact on the gas phase stabilities of positively charged protein ions.
Figure 3.
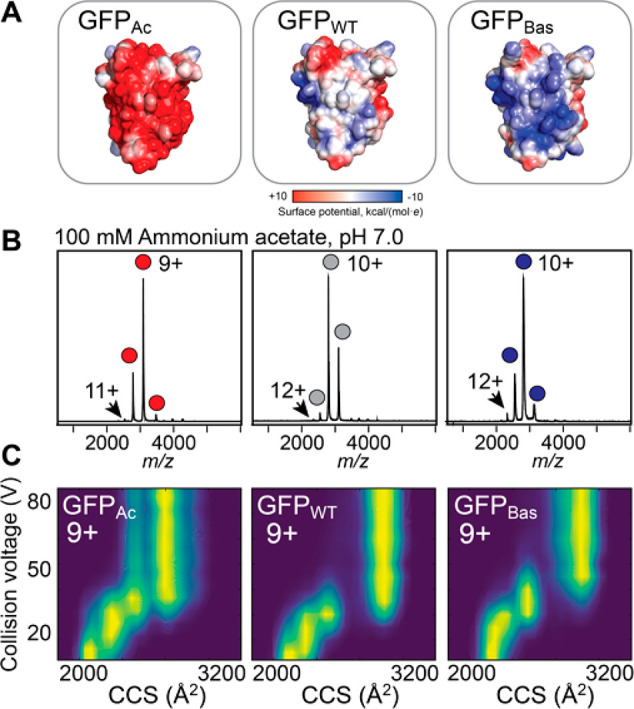
Charging and gas-phase stabilities of WT and supercharged GFP variants. (A) Coulombic surface potentials predicted for the three GFP variants. Structural models were generated using Rosetta; the His6 tag is omitted for clarity. (B) Mass spectra of GFPAc, GFPWT, and GFPBas show near-identical CSDs dominated by the 9+ and 10+ charge states. (C) CIU profiles of the 9+ charge state of each variant indicate that GFPBas has a higher resistance to unfolding than GFPWT and GFPAc.
We thus asked if the increased stability of GFPBas could be related to the distribution of the ESI charges. We devised a Monte Carlo (MC) scheme for transferring protons (See Supporting Information) where we omit the dynamics of the overall structure and simplify the otherwise slow rearrangement of side chains in vacuum,44,45 thus getting a more exhaustive sampling compared to hybrid MC-MD approaches46,47 while also removing any bias from the surface details in the atomistic structure used as input. Briefly, we used the GFPWT crystal structure and Rosetta structures for GFPAc and GFPBas on which we randomly placed protons on ionizable sites to obtain a 9+ charge state. Because surface side chains may rearrange in response to both the gas-phase conditions and to changes in the charge locations, we discarded all atomic coordinates apart from the Cα and Cβ atoms. At each MC step a proton was moved at random to another residue, after which a short energy minimization was run to let the charges adjust their positions in response to the local potential, comprised of Coulombic interactions, a mean-field solvation at the protein surface, and a short-range repulsion from the backbone defined by the Cα atoms. MC moves were accepted or rejected based on a Metropolis criterion based on the GB’s of protonated sites (Table S1), Coulombic energy, and solvation energy (Figure S8, S9). For each system a total of 107 iterations were performed in 50 replicates. Notably, the lowest energy states were not found in all replicates, but in several of them, indicating that the combined simulation data are sufficient, but that millions of iterations are required, in line with what has been observed for MC schemes with static structures without side chain rearrangements,48 underlining the challenge to achieve sufficient sampling for pure and hybrid MC. In all cases the lowest energy states were zwitterionic, with salt bridges forming at the protein surface. We find that the GFPBas has the lowest Coulombic energy, followed by GFPWT (Table S2). Conversely, the GB contribution is most negative for GFPAc and least negative for GFPBas, which can be attributed to their different number of D and E residues, as their GBs are the highest among the amino acids. GFPBas evidently minimizes its total energy by optimizing its charge locations, which is less important for GFPAc where the low-energy states are dominated by the GBs of protonated side chains. Protonation of high-GB sites does not stabilize the structure per se, whereas lowering of the Coulombic energy does, which explains why GFPBas is more resistant to gas-phase unfolding than the other variants, and why GFPAc is the most unstable.
Moving Individual Charged Groups Alters Protein Conformations in the Gas Phase
The observations from GFP variants raise the question of whether the gas-phase structure of a protein can be affected not just by the number, but also the location of the charges. We thus turned to a third protein system, the small metal-binding protein TTHA1718 from Thermus thermophilus, which contains 9 lysines and 1 histidine residue. By exchanging surface lysines at positions 5, 20, 30, or 61 for glutamate, we generated four variants with near-identical structures that differ in the location of one basic and one acidic residue each (TTHAK5E, TTHAK20E, TTHAK30E, and TTHAK61E).49 Because acidic residues are neutralized during positive-mode ESI, this system enables us to observe the effect of moving a single protonation site on the gas-phase conformation of the protein. To be able to probe a broad range of charge states, we performed MS analysis in ammonium acetate and in dH2O which resulted in CSDs ranging from 9+ to 4+ (Figure 4A, S10). The 4+ charge states of the TTHA variants exhibited CCSs of 940–960 Å2, in good agreement with the 920 Å2 predicted from the NMR structure of the WT protein (Figure S10). The 4+ charge ions were found to unfold readily as the trap voltage was raised above 5 V, and therefore, no CIU traces could be recorded. However, the 5+ charge state showed significant differences between variants: for TTHAK20E and TTHAK30E, a small population with a native-like CCS could be detected, as well as more extended states with CCSs of 1100 Å2 and 1200 Å2, indicating partial unfolding even under the gentlest ESI conditions. TTHAK5E and TTHAK61E, on the other hand, showed only an unfolded state with a CCS of approximately 1200 Å2 (Figure 4B, C). The lysines are the likely protonation sites due to their high GB. We thus speculated that changing the location of a single lysine residue could change the local Coulombic strain experienced by the protein. However, for the 5+ ion, the number of lysines is greater than the number of ESI charges, meaning multiple charge configurations are still possible. We therefore also considered the higher charge states observed in dH2O, the 8+ ion, which should have fully protonated lysines, also exhibited different CCSs (Figure 4C). The CCS values of 1300–1600 Å2 are substantially lower than the 2000 Å2 expected for a fully extended conformation, indicating that local structural differences can persist even in highly charged ions. We conclude that changing the location of the ionizable sites on the protein surface, and thus the location of charges, affects the gas-phase conformation of protein ions.
Figure 4.
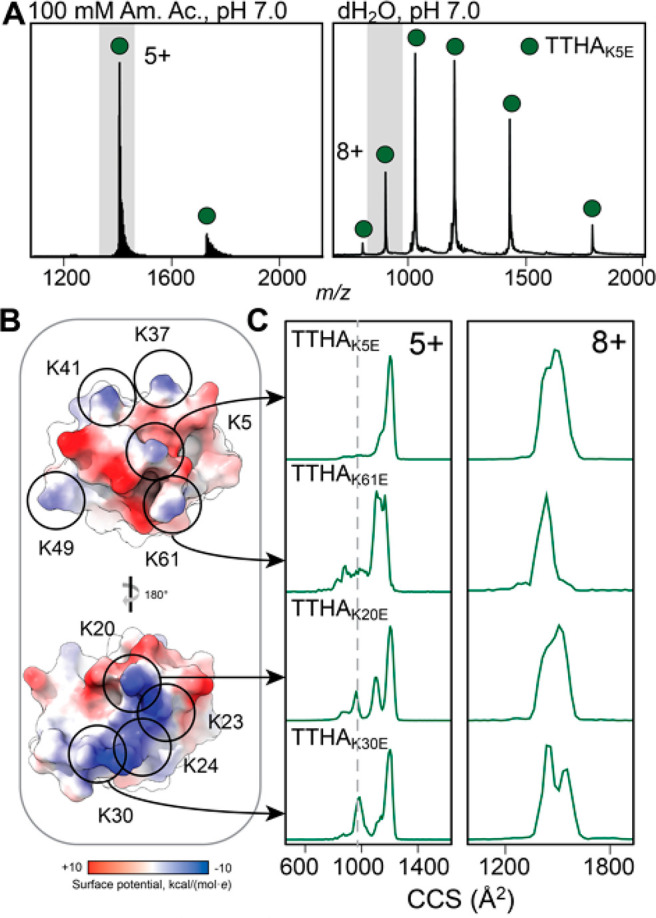
Changing the location of individual lysine residues affects the gas-phase conformation of TTHA1718 variants. (A) ESI mass spectra of TTHAK5E in ammonium acetate show a narrow CSD of 4+ and 5+ ions and in dH2O show a broad CSD ranging from 4+ to 9+. (B) The locations of the 9 lysine residues are indicated on the Coulombic surface representation of WT TTHA1718 (PDB ID 2ROE). (C) The CCS distributions of the 5+ charge states of TTHAK5E, TTHAK20E, TTHAK30E, and TTHAK61E sprayed from AmAc at a trap voltage of 5 V show a small native-like population for TTHAK20E and TTHAK30E, whereas TTHAK5E and TTHAK61E are mostly unfolded. The 8+ charge states from dH2O, which can be considered mostly unfolded, reveal variant-specific differences in their CCS distributions. The dashed line indicates the theoretical CCS of TTHA1718.
Discussion
Protein charging in positive ESI can be dependent or independent of basic residues: native-like proteins charge according to their SASA regardless of surface charge, whereas the maximum charge for unfolded proteins depends on the number of basic residues.38,39 Our results show that droplet charge at the final step of desolvation, which is governed by the Rayleigh limit of droplet stability,50 determines the final charge of compact protein ions, not their proton affinity. In the absence of ionizable residues, this charging limit applies even to unfolded proteins: presumably, denatured EXG remains trapped in the ESI droplet and is eventually released in a compact state, giving rise to a native-like CSD, but then easily unfolds in the gas phase due to the absence of a folded protein core. The generation of high charge states commonly associated with unfolded proteins in ESI-MS thus requires ionizable residues which likely drive the expulsion of the protein from the ESI droplet. This interpretation is supported by recent observations from chemically modified proteins.28 For native-like proteins, however, a lack of ionizable sites does not appear to alter the conformational landscape. An excess of ionizable side chains, on the other hand, has a direct impact on the conformational stability, as shown for three GFP variants. In solution, GFPAc and GFPBas are less stable than GFPWT due to charge repulsion.43 In the gas phase, GFPBas, which contains 41 potential protonation sites, displays the highest stability, whereas GFPWT and GFPAc, with 28 and 19 basic residues, respectively, are more susceptible to CIU. Our simulations suggest this stability stems from the scarcity of carboxylate side chains in GFPBas, making the protons distribute over the high number of basic residues for a favorable Coulombic energy. It seems likely that the larger number of basic residues in this variant also allows for a more optimal distribution of charges that minimizes repulsion and creates more opportunities for salt bridges that stabilize compact states in the gas phase.16,51 The importance of charge locations is emphasized by the different conformational preferences of TTHA variants in the gas phase. Furthermore, the combination of basic and acidic residues raises the possibility of salt bridge formation on the protein surface, which can also alter gas phase stability.52
Together, our findings show that the charge states of native protein ions are governed solely by the Rayleigh limit, whereas the spatial distribution of the charges affects the conformational landscape of the ion. The conclusions have several implications for the use of native MS to study protein folding and interactions:
-
(1)
Ionizable residues are not prerequisite for ESI of intact protein complexes. This finding implies that native MS is suitable for the analysis of integral membrane proteins, despite their uneven surface charge in solution.33
-
(2)
The fact that proteins lacking basic residues favor the CRM over CEM reveals the limitations of using charge state signatures to assess the distribution of folded states.53 As a result, proteins with few ionizable groups may display diverging charging patterns upon unfolding.
-
(3)
Previous studies have shown that point mutations can affect the CIU pattern of native-like protein ions by modulating their structure in solution.9,15,54,55 Our findings reveal that even identically folded proteins can exhibit different CIU fingerprints due to altered surface electrostatics during ESI. Because these differences arise from the locations of ESI charges, not from solution folding, care must be taken when using CIU data to probe solution structures.
-
(4)
Gas-phase unfolding is routinely used to identify compounds that can stabilize protein complexes by occupying specific binding pockets.11,56−58 We find that moving charged sites can have a dramatic effect on a protein’s conformation in the gas phase. We speculate that for small proteins with few ionizable sites, such as TTHA, masking a charge site, for example with an ionic ligand, could potentially affect its gas-phase stability. This possibility should be taken into consideration when studying ligand-mediated stabilization of protein complexes in the gas phase.
Methods
Protein Preparation
EXGWT and the chargeless mutant EXGQQQW were expressed and purified as described previously, and the chargeless variant subjected to N-terminal acetylation.34,35 GFPWT, GFPAc, GFPBas, TTHAK5E, TTHAK20E, TTHAK30E, and TTHAK61E were expressed and purified as described.43,49 The EXG variants were stored at −80 °C at a concentration of 90 μM and exchanged into 100 mM AmAc, pH 6.9, prior to MS analysis using Biospin 6 columns (BioRad). The TTHA variants were stored at 120 μM in dH2O and diluted to 12 μM in dH2O for MS analysis. Mass spectra were recorded at concentrations of 15 μM for wild-type and 45 μM for chargeless EXG, and 5 μM for all GFP variants. Horse heart cytochrome C for TWIMS calibration was purchased from Sigma and prepared as described above.
Native Mass Spectrometry
nESI capillaries were purchased from Thermo. Positive ionization mode mass spectra were acquired on a Micromass LCT ToF modified for analysis of intact protein complexes (MS Vision, The Netherlands) equipped with an offline nanospray source. For the LCT, the capillary voltage was 1.5 kV and the RF lens 1.5 kV. The cone voltage was set to 100 V for normal acquisition and ramped between 50 and 300 V for collisional activation. The pressure in the ion source was maintained at 9.0 mbar. Data were analyzed using MassLynx V4.1 (Waters, UK). Negative ionization mode mass spectra were acquired on an Orbitrap Fusion (Thermo Fisher Scientific, Waltham, MA) equipped with an offline nanoelectrospray source. The instrument was operated in intact protein mode. The capillary voltage was −1.8 kV, the transfer tube temperature was maintained at 40 °C and the pressure in the ion-routing multipole was 0.011 Torr. Collisional activation was performed by increasing the HCD energy in the ion-routing multipole. High-purity nitrogen was used as collision gas. Spectra were recorded using the Orbitrap mass analyzer at a resolution of 60 000 with a high mass mode acquisition window of 1000–5000 m/z and a scan time of 1 ms. Data were analyzed using Xcalibur 3.0 (Thermo Scientific, Waltham, MA).
Ion Mobility Mass Spectrometry of EXG and TTHA1718
IMMS data for EXG and TTHA1718 were recorded on a Waters Synapt G1 TWIMS MS modified for analysis of intact protein complexes (MS Vision, The Netherlands) and equipped with an offline nanospray source. The capillary voltage was 1.5 kV. The cone voltage was set to 10 V, and the source temperature was maintained at 20 °C. The source pressure was adjusted to 8 mbar. The ion trap voltage was ramped from 5 to 25 V for collisional activation, transfer voltage was 10 V. IM settings were: Wave height 12 V, wave velocity 450 m/s for EXG, and wave height 10 V, wave velocity 250 m/s for TTHA1718. IMS gas was nitrogen with a flow of 15 mL/min and collision gas argon with a flow of 4 mL/min. Horse heart cytochrome C and human insulin were used for TWIMS calibration for EXG and TTHA1718, respectively.
Ion Mobility Mass Spectrometry of GFP
IMMS measurements of equimolar mixtures of GFPWT with either GFPAc or GFPBas were performed on a Waters Synapt G1 ion mobility mass spectrometer equipped with a linear field drift tube to facilitate direct CCS determination, and an offline nanospray source. Protein samples were introduced using in-house produced gold-coated borosilicate capillaries. The pressure in the source region was maintained at 5.0 mbar. Mass spectra were recorded at drift voltages between 40 and 120 V at an ion trap voltage of 20 V for CCS determination. For collisional unfolding measurements, the ion trap voltage was ramped from 10 to 100 V in 5 V increments. The drift gas was helium and collision gas was argon. Stabilities of the native-like states of states GFPBas or GFPAc were calculated relative to GFPWT, measuring either GFPBas or GFPWT with GFPAc as an internal standard in the same MS experiment, from three independent repeats. Importantly, we observed the same CSDs on all four MS platforms when using the same solution conditions. Theoretical CCS values of EXG (PDB 6QFS) and GFP (PDB 2B3P as well as Rosetta structures of the GFPAc and GFPBas variants) were calculated using IMPACT using the PA method and an empirical correction factor of 1.14.59,60 All IMMS data were analyzed using the PULSAR software package.10
Acknowledgments
Prof. Astrid Gräslund, Stockholm University, is gratefully acknowledged for providing the CD spectrometer, and Prof. David R. Liu, Harvard University, for the kind gift of the GFP plasmids.
Glossary
Abbreviations
- ESI-MS
electrospray ionization-mass spectrometry
- SASA
solvent-accessible surface area
- CRM
charged residue model
- CEM
chain ejection model
- AmAc
ammonium acetate
- CSD
charge state distribution
- CCS
collision cross section
- CIU
collision-induced unfolding
- TMAO
trimethylammonia-N-oxide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00458.
Experimental details for computational simulations; protein sequences; supplementary tables summarizing energy terms for MC (ST1) and total energies (ST2); and supplementary figures showing the structure of EXG (S1), salt adduction to EXG (S2), charge reduction of EXG (S3), CCS distributions of EXG (S4), CIU profiles of EXG (S5) and GFP (S6), quantification of GFP stability (S7), a schematic of the MD strategy (S8), solvation potentials (S9), and mass spectra of TTHA variants (S10) (PDF)
Author Present Address
▲ OMass Therapeutics, The Schrödinger Building, Oxford Science Park, Oxford OX4 4GE, UK
Author Contributions
⬡ M.L.A., C.S., and J.T.S.H. contributed equally.
M.L. is supported by a KI faculty-funded Career Position, a Cancerfonden Project grant (19 0480), and a VR Starting Grant (2019-01961). C.S. is supported by a Novo Nordisk Foundation Postdoctoral Fellowship (NNF19OC0055700). J.C.P. and E.G.M. acknowledge funding from MS SPIDOC by the European Union’s Horizon 2020 research and innovation program (801406) and thank the MS SPIDOC consortium for valuable discussions. S.A.C. and J.L.P.B. thank BBSRC BB/L017067/1 and Waters Corp for an iCASE studentship.
The authors declare no competing financial interest.
Supplementary Material
References
- Benesch J. L.; Robinson C. V. Mass Spectrometry of Macromolecular Assemblies: Preservation and Dissociation. Curr. Opin. Struct. Biol. 2006, 16, 245–251. 10.1016/j.sbi.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Lössl P.; van de Waterbeemd M.; Heck A. J. The Diverse and Expanding Role of Mass Spectrometry in Structural and Molecular Biology. EMBO J. 2016, 35 (24), 2634–2657. 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J.; Robinson C. V. Twenty Years of Gas Phase Structural Biology. Structure 2013, 21 (9), 1541–1550. 10.1016/j.str.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Allison T. M.; Landreh M. Ion Mobility in Structural Biology. Compr. Anal. Chem. 2019, 83, 161–195. 10.1016/bs.coac.2018.10.001. [DOI] [Google Scholar]
- Jurneczko E.; Barran P. E. How Useful Is Ion Mobility Mass Spectrometry for Structural Biology? The Relationship between Protein Crystal Structures and Their Collision Cross Sections in the Gas Phase. Analyst 2011, 136 (1), 20–28. 10.1039/C0AN00373E. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Rose R. J.; Van Duijn E.; Lorenzen K.; Heck A. J. R. Ion Mobility Mass Spectrometry of Proteins and Protein Assemblies. Chem. Soc. Rev. 2010, 39 (5), 1633–1655. 10.1039/B914002F. [DOI] [PubMed] [Google Scholar]
- Hopper J. T. S.; Oldham N. J. Collision Induced Unfolding of Protein Ions in the Gas Phase Studied by Ion Mobility-Mass Spectrometry: The Effect of Ligand Binding on Conformational Stability. J. Am. Soc. Mass Spectrom. 2009, 20 (10), 1851–1858. 10.1016/j.jasms.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Dixit S. M.; Polasky D. A.; Ruotolo B. T. Collision Induced Unfolding of Isolated Proteins in the Gas Phase: Past, Present, and Future. Curr. Opin. Chem. Biol. 2018, 42, 93–100. 10.1016/j.cbpa.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-jabiry A.; Palmer M.; Langridge J.; Bellamy-Carter J.; Robinson D.; Oldham N. J. Combined Chemical Modification and Collision Induced Unfolding Using Native Ion Mobility-Mass Spectrometry Provides Insights into Protein Gas-Phase Structure. Chem. - Eur. J. 2021, 27 (55), 13783. 10.1002/chem.202101857. [DOI] [PubMed] [Google Scholar]
- Allison T. M.; Reading E.; Liko I.; Baldwin A. J.; Laganowsky A.; Robinson C. V. Quantifying the Stabilizing Effects of Protein–Ligand Interactions in the Gas Phase. Nat. Commun. 2015, 6, 8551. 10.1038/ncomms9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Allison T. M.; Ulmschneider M. B.; Degiacomi M. T.; Baldwin A. J.; Robinson C. V. Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510 (7503), 172–175. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla J. R.; Agasid M. T.; Mehmood S.; Robinson C. V. Membrane Protein–Lipid Interactions Probed Using Mass Spectrometry. Annu. Rev. Biochem. 2019, 88 (1), 85–111. 10.1146/annurev-biochem-013118-111508. [DOI] [PubMed] [Google Scholar]
- Roberts D. S.; Mann M.; Melby J. A.; Larson E. J.; Zhu Y.; Brasier A. R.; Jin S.; Ge Y. Structural O-Glycoform Heterogeneity of the SARS-CoV-2 Spike Protein Receptor-Binding Domain Revealed by Top-Down Mass Spectrometry. J. Am. Chem. Soc. 2021, 143, 12014–12024. 10.1021/jacs.1c02713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinen B. A.; Gadkari V. V.; Stull F.; Ruotolo B. T.; Bardwell J. C. A. SERF Engages in a Fuzzy Complex That Accelerates Primary Nucleation of Amyloid Proteins. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 23040–23049. 10.1073/pnas.1913316116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin S. M.; Parson K. F.; Yadav P.; Juliano B.; Li G. C.; Sanders C. R.; Ohi M. D.; Ruotolo B. T. Ion Mobility–Mass Spectrometry Reveals the Role of Peripheral Myelin Protein Dimers in Peripheral Neuropathy. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2015331118. 10.1073/pnas.2015331118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy-Carter J.; O’Grady L.; Passmore M.; Jenner M.; Oldham N. J. Decoding Protein Gas-Phase Stability with Alanine Scanning and Collision-Induced Unfolding Ion Mobility Mass Spectrometry. Anal. Sens. 2021, 1, 63–69. 10.1002/anse.202000019. [DOI] [Google Scholar]
- Campuzano I. D. G.; Sandoval W. Denaturing and Native Mass Spectrometric Analytics for Biotherapeutic Drug Discovery Research: Historical, Current, and Future Personal Perspectives. J. Am. Soc. Mass Spectrom. 2021, 32, 1861–1885. 10.1021/jasms.1c00036. [DOI] [PubMed] [Google Scholar]
- Deslignière E.; Ehkirch A.; Duivelshof B. L.; Toftevall H.; Sjögren J.; Guillarme D.; D’atri V.; Beck A.; Hernandez-Alba O.; Cianférani S. State-of-the-Art Native Mass Spectrometry and Ion Mobility Methods to Monitor Homogeneous Site-Specific Antibody-Drug Conjugates Synthesis. Pharmaceuticals 2021, 14, 498. 10.3390/ph14060498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Ahadi E.; Rodriguez A. D.; Vahidi S. Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85 (1), 2–9. 10.1021/ac302789c. [DOI] [PubMed] [Google Scholar]
- Susa A. C.; Xia Z.; Tang H. Y. H.; Tainer J. A.; Williams E. R. Charging of Proteins in Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 332–340. 10.1007/s13361-016-1517-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone A. T.; Williams E. R. Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J. Am. Chem. Soc. 2003, 125 (8), 2319–2327. 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebarle P.; Verkerk U. H. Electrospray: From Ions in Solution to Ions in the Gas Phase, What We Know Now. Mass Spectrom. Rev. 2009, 28 (6), 898–917. 10.1002/mas.20247. [DOI] [PubMed] [Google Scholar]
- Testa L.; Brocca S.; Grandori R. Charge-Surface Correlation in Electrospray Ionization of Folded and Unfolded Proteins. Anal. Chem. 2011, 83 (17), 6459–6463. 10.1021/ac201740z. [DOI] [PubMed] [Google Scholar]
- Verkerk U. H.; Kebarle P. Ion-Ion and Ion–Molecule Reactions at the Surface of Proteins Produced by Nanospray. Information on the Number of Acidic Residues and Control of the Number of Ionized Acidic and Basic Residues. J. Am. Soc. Mass Spectrom. 2005, 16 (8), 1325–1341. 10.1016/j.jasms.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Breuker K.; McLafferty F. W. Stepwise Evolution of Protein Native Structure with Electrospray into the Gas Phase, 10–12 to 102 S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (47), 18145–18152. 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltashov I. A.; Mohimen A. Estimates of Protein Surface Areas in Solution by Electrospray Ionization Mass Spectrometry. Anal. Chem. 2005, 77 (16), 5370–5379. 10.1021/ac050511+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Mora J. F. Electrospray Ionization of Large Multiply Charged Species Proceeds via Dole’s Charged Residue Mechanism. Anal. Chim. Acta 2000, 406 (1), 93–104. 10.1016/S0003-2670(99)00601-7. [DOI] [Google Scholar]
- Pimlott D. J. D.; Konermann L. Using Covalent Modifications to Distinguish Protein Electrospray Mechanisms: Charged Residue Model (CRM) vs. Chain Ejection Model (CEM). Int. J. Mass Spectrom. 2021, 469, 116678. 10.1016/j.ijms.2021.116678. [DOI] [Google Scholar]
- Strickler S. S.; Gribenko A. V.; Gribenko A. V.; Keiffer T. R.; Tomlinson J.; Reihle T.; Loladze V. V.; Makhatadze G. I. Protein Stability and Surface Electrostatics: A Charged Relationship. Biochemistry 2006, 45, 2761–2766. 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- Schnier P. D.; Gross D. S.; Williams E. R. Electrostatic Forces and Dielectric Polarizability of Multiply Protonated Gas-Phase Cytochrome c Ions Probed by Ion/Molecule Chemistry. J. Am. Chem. Soc. 1995, 117, 6747–6757. 10.1021/ja00130a015. [DOI] [Google Scholar]
- Mao Y.; Woenckhaus J.; Kolafa J.; Ratner M. A.; Jarrold M. F. Thermal Unfolding of Unsolvated Cytochrome c: Experiment and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1999, 121, 2712–2721. 10.1021/ja980324b. [DOI] [Google Scholar]
- Zhong Y.; Han L.; Ruotolo B. T. Collisional and Coulombic Unfolding of Gas-Phase Proteins: High Correlation to Their Domain Structures in Solution. Angew. Chem., Int. Ed. 2014, 53 (35), 9209–9212. 10.1002/anie.201403784. [DOI] [PubMed] [Google Scholar]
- Mbaye M. N.; Hou Q.; Basu S.; Teheux F.; Pucci F.; Rooman M. A Comprehensive Computational Study of Amino Acid Interactions in Membrane Proteins. Sci. Rep. 2019, 9, 12043. 10.1038/s41598-019-48541-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Højgaard C.; Kofoed C.; Espersen R.; Johansson K. E.; Villa M.; Willemoës M.; Lindorff-Larsen K.; Teilum K.; Winther J. R. A Soluble, Folded Protein without Charged Amino Acid Residues. Biochemistry 2016, 55, 3949–3956. 10.1021/acs.biochem.6b00269. [DOI] [PubMed] [Google Scholar]
- Hervø-Hansen S.; Højgaard C.; Johansson K. E.; Wang Y.; Wahni K.; Young D.; Messens J.; Teilum K.; Lindorff-Larsen K.; Winther J. R. Charge Interactions in a Highly Charge-Depleted Protein. J. Am. Chem. Soc. 2021, 143, 2500. 10.1021/jacs.0c10789. [DOI] [PubMed] [Google Scholar]
- Timofeev O.; Zhu M. M.; Gross M. L. Information for Proteomics: ESI-MS Titration by Sodium Ions Gives the Number of Carboxylate Groups in Peptides. Int. J. Mass Spectrom. 2004, 231, 113–117. 10.1016/j.ijms.2003.09.015. [DOI] [Google Scholar]
- Rodriquez C. F.; Guo X.; Shoeib T.; Hopkinson A. C.; Siu K. W. M. Formation of [M - NH + MNa](m-n)+ and [M - NH + MK](m-n)+ Ions in Electrospray Mass Spectrometry of Peptides and Proteins. J. Am. Soc. Mass Spectrom. 2000, 11, 967–975. 10.1016/S1044-0305(00)00162-8. [DOI] [PubMed] [Google Scholar]
- Kaltashov I.; Mohimen A. Estimates of Protein Surface Areas in Solution by Electrospray Ionization Mass Spectrometry. Anal. Chem. 2005, 77 (16), 5370–5379. 10.1021/ac050511+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnier P. D.; Gross D. S.; Williams E. R. On the Maximum Charge State and Proton Transfer Reactivity of Peptide and Protein Ions Formed by Electrospray Ionization. J. Am. Soc. Mass Spectrom. 1995, 6 (11), 1086–1097. 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- Carbeck J. D.; Severs J. C.; Gao J.; Wu Q.; Smith R. D.; Whitesides G. M. Correlation between the Charge of Proteins in Solution and in the Gas Phase Investigated by Protein Charge Ladders, Capillary Electrophoresis, and Electrospray Ionization Mass Spectrometry. J. Phys. Chem. B 1998, 102, 10596–10601. 10.1021/jp980768u. [DOI] [Google Scholar]
- Gault J.; Lianoudaki D.; Kaldmäe M.; Kronqvist N.; Rising A.; Johansson J.; Lohkamp B.; Laín S.; Allison T. M.; Lane D. P.; Marklund E. G.; Landreh M. Mass Spectrometry Reveals the Direct Action of a Chemical Chaperone. J. Phys. Chem. Lett. 2018, 9 (14), 4082–4086. 10.1021/acs.jpclett.8b01817. [DOI] [PubMed] [Google Scholar]
- Kaldmäe M.; Österlund N.; Lianoudaki D.; Sahin C.; Bergman P.; Nyman T.; Kronqvist N.; Ilag L. L.; Allison T. M.; Marklund E. G.; Landreh M. Gas-Phase Collisions with Trimethylamine-N-Oxide Enable Activation-Controlled Protein Ion Charge Reduction. J. Am. Soc. Mass Spectrom. 2019, 30 (8), 1385–1388. 10.1007/s13361-019-02177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. S.; Phillips K. J.; Liu D. R. Supercharging Proteins Can Impart Unusual Resilience. J. Am. Chem. Soc. 2007, 129 (33), 10110–10112. 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl T.; Östlin C.; Dawod I. E.; Brodmerkel M. N.; Marklund E. G.; Martin A. V.; Timneanu N.; Caleman C. Structural Heterogeneity in Single Particle Imaging Using X-Ray Lasers. J. Phys. Chem. Lett. 2020, 11, 6077–6083. 10.1021/acs.jpclett.0c01144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D.; Marklund E. G.; Larsson D. S. D.; Caleman C. Proteins, Lipids, and Water in the Gas Phase. Macromol. Biosci. 2011, 11, 50–59. 10.1002/mabi.201000291. [DOI] [PubMed] [Google Scholar]
- Konermann L. Molecular Dynamics Simulations on Gas-Phase Proteins with Mobile Protons: Inclusion of All-Atom Charge Solvation. J. Phys. Chem. B 2017, 121 (34), 8102–8112. 10.1021/acs.jpcb.7b05703. [DOI] [PubMed] [Google Scholar]
- Bakhtiari M.; Konermann L. Protein Ions Generated by Native Electrospray Ionization: Comparison of Gas Phase, Solution, and Crystal Structures. J. Phys. Chem. B 2019, 123, 1784–1796. 10.1021/acs.jpcb.8b12173. [DOI] [PubMed] [Google Scholar]
- Ewing S. A.; Donor M. T.; Wilson J. W.; Prell J. S. Collidoscope: An Improved Tool for Computing Collisional Cross-Sections with the Trajectory Method. J. Am. Soc. Mass Spectrom. 2017, 28, 587–596. 10.1007/s13361-017-1594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X.; Choi S.; Lang L.; Mowray D.; Dokholyan N. V.; Danielsson J.; Oliveberg M. Physicochemical Code for Quinary Protein Interactions in Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (23), E4556–E4563. 10.1073/pnas.1621227114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. N.; Flagan R. C.; Beauchamp J. L. Droplet Evaporation and Discharge Dynamics in Electrospray Ionization. J. Phys. Chem. A 2002, 106, 9957–9967. 10.1021/jp025723e. [DOI] [Google Scholar]
- Li J.; Lyu W.; Rossetti G.; Konijnenberg A.; Natalello A.; Ippoliti E.; Orozco M.; Sobott F.; Grandori R.; Carloni P. Proton Dynamics in Protein Mass Spectrometry. J. Phys. Chem. Lett. 2017, 8 (6), 1105–1112. 10.1021/acs.jpclett.7b00127. [DOI] [PubMed] [Google Scholar]
- Steinberg M. Z.; Elber R.; McLafferty F. W.; Gerber R. B.; Breuker K. Early Structural Evolution of Native Cytochrome c after Solvent Removal. ChemBioChem 2008, 9 (15), 2417–2423. 10.1002/cbic.200800167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Robinson C. V. Do Charge State Signatures Guarantee Protein Conformations?. J. Am. Soc. Mass Spectrom. 2012, 23 (7), 1161–1168. 10.1007/s13361-012-0393-z. [DOI] [PubMed] [Google Scholar]
- McAlary L.; Harrison J. A.; Aquilina J. A.; Fitzgerald S. P.; Kelso C.; Benesch J. L. P.; Yerbury J. J. Trajectory Taken by Dimeric Cu/Zn Superoxide Dismutase through the Protein Unfolding and Dissociation Landscape Is Modulated by Salt Bridge Formation. Anal. Chem. 2020, 92, 1702–1711. 10.1021/acs.analchem.9b01699. [DOI] [PubMed] [Google Scholar]
- Liuni P.; Deng B.; Wilson D. J. Comparing Equilibrium and Kinetic Protein Unfolding Using Time-Resolved Electrospray-Coupled Ion Mobility Mass Spectrometry. Analyst 2015, 140, 6973–6979. 10.1039/C5AN00843C. [DOI] [PubMed] [Google Scholar]
- Tomlinson L. J.; Eyers C. E. Ion Mobility-Mass Spectrometry to Evaluate the Effects of Protein Modification or Small Molecule Binding on Protein Dynamics. Methods Mol. Biol. 2020, 2084, 179–190. 10.1007/978-1-0716-0030-6_11. [DOI] [PubMed] [Google Scholar]
- Fantin S. M.; Huang H.; Sanders C. R.; Ruotolo B. T. Collision-Induced Unfolding Differentiates Functional Variants of the KCNQ1 Voltage Sensor Domain. J. Am. Soc. Mass Spectrom. 2020, 31, 2348–2355. 10.1021/jasms.0c00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Zhuang X.; Pi Z.; Liu S.; Liu Z.; Song F. Determining the Effect of Catechins on SOD1 Conformation and Aggregation by Ion Mobility Mass Spectrometry Combined with Optical Spectroscopy. J. Am. Soc. Mass Spectrom. 2018, 29, 734–741. 10.1007/s13361-017-1864-z. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Hall Z.; Giles K.; Hoyes J.; Robinson C. V.; Ruotolo B. T. Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem. 2010, 82 (22), 9557–9565. 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- Marklund E. G.; Degiacomi M. T.; Robinson C. V.; Baldwin A. J.; Benesch J. L. P. Collision Cross Sections for Structural Proteomics. Structure 2015, 23 (4), 791–799. 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.