

Abstract
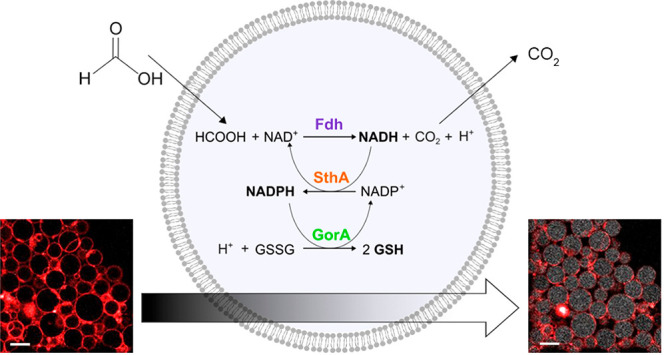
Effective metabolic pathways are essential for the construction of in vitro systems mimicking the biochemical complexity of living cells. Such pathways require the inclusion of a metabolic branch that ensures the availability of reducing equivalents. Here, we built a minimal enzymatic pathway confinable in the lumen of liposomes, in which the redox status of the nicotinamide cofactors NADH and NADPH is controlled by an externally provided formate. Formic acid permeates the membrane where a luminal formate dehydrogenase uses NAD+ to form NADH and carbon dioxide. Carbon dioxide diffuses out of the liposomes, leaving only the reducing equivalents in the lumen. A soluble transhydrogenase subsequently utilizes NADH for reduction of NADP+ thereby making NAD+ available again for the first reaction. The pathway is functional in liposomes ranging from a few hundred nanometers in diameter (large unilamellar vesicles) up to several tens of micrometers (giant unilamellar vesicles) and remains active over a period of 7 days. We demonstrate that the downstream biochemical process of reduction of glutathione disulfide can be driven by the transfer of reducing equivalents from formate via NAD(P)H, thereby providing a versatile set of electron donors for reductive metabolism.
Keywords: Redox cofactors, metabolic pathways, synthetic biology, liposome confinement, formic acid
Introduction
Metabolism is an intricate network of reactions, catalyzed by enzymes, that enables living systems to sustain themselves autonomously and to adapt to different environmental conditions. Although there are numerous metabolic pathways, leading to thousands of different compounds, only a limited number of molecules have been conserved under evolutionary pressure in each domain of life.1 Among these hub metabolites2 are the nicotinamide adenine dinucleotides NAD(H) and NADP(H). The central role of these nucleotides in metabolism is illustrated by the fact that, in bacteria, more than 1000 different redox reactions require NAD+ or NADPH as cofactors.3 Their primary role is to transfer energy in the form of reducing equivalents in catabolic and anabolic processes. In addition to cellular redox homeostasis, NAD(P) cofactors fulfill several other functions in both prokaryotes and eukaryotes, including those of second messengers,4 regulators of gene expression,5,6 and signaling molecules for cell division and growth.7
An imbalance in the concentration or redox status of these cofactors in vivo, as happens for instance when NAD(P)-dependent enzymes NADH oxidase8,9 and nucleotide pyridine transhydrogenase are overexpressed,10 affects large parts of the transcriptome and many metabolic fluxes. From a biotechnological perspective, the exhaustion of reducing equivalents constitutes one of the main limiting factors in the microbial conversion of natural gas11 and biomass12 into high-value chemicals and biofuels. The ability to maintain the NAD+/NADH and NADP+/NADPH ratios within threshold values is made possible through sophisticated regulation mechanisms, i.e., from gene expression to the activation or inactivation of proteins.
The regeneration of redox cofactors in vitro has been explored for potential applications both in the biotechnological production of valuable chemicals13−15 and in the field of synthetic biology.16 Strategies have been developed to optimize the yield of metabolic pathways by avoiding depletion of the cofactor in the desired oxidative state. A remarkable example of cofactor metabolic engineering is the work of Opgenorth and co-workers.17 They constructed an artificial pathway that leads to the formation of polyhydroxybutyrate, regardless of which nicotinamide-nucleotide cofactor is present in the reduced form. Although this “molecular purge valve” has not been tested in a cell-like system, it offers advantages for bypassing the redox balance through self-regulation. Beneyton et al.18 designed a microfluidic platform of water–oil droplets as vessels in which, following pico-injection of glucose-6-phosphate, NAD+ can be reduced into NADH. The oxidized form is obtained via NADH dehydrogenase, which is present in inverted E. coli membrane vesicles and part of the droplet compartment. The microfluidic device allows the control of a large population of vesicles in minimal volumes. Nonetheless, this work is limited to NAD(H) and requires the use of bacterial membranes with undefined composition and possible side-reactions.
Cell free systems provide a suitable platform to investigate and optimize a metabolic pathway for redox homeostasis. First, they can exclude coexistent fluxes of other pathways that would disturb the electron transfer.19,20 Second, the use of purified enzymes guarantees a high level of control of the reactants and products, which facilitates the design and optimization of metabolic pathways according to fundamental principles, such as thermodynamic spontaneity (negative ΔG) and favorable equilibrium constants.16 An example of how these concepts can be used is the multienzymatic pathway crotonyl–coenzyme A (CoA)/ethylmalonyl–CoA/hydroxybutyryl–CoA cycle for the fixation of CO2,21 in which a series of optimization rounds of the initial pathway design increased the final yield of fixed CO2 20-fold, by first identifying the rate-limiting steps and the dead-end reactions and then troubleshooting them by replacing or engineering the “faulty” enzymes.
Here, we present a minimal enzymatic pathway for the control of the redox state of both NAD+/NADH and NADP+/NADPH in phospholipid vesicles via the supply of formate. The pathway is functional in biomimicking compartments of different sizes, ranging from a few hundred nanometers (large unilamellar vesicles) up to several tens of micrometers (giant unilamellar vesicles). We also demonstrate how downstream biochemical processes can take place through the transfer of reducing equivalents from formate via NAD(P)H to glutathione disulfide.
Results
Thermodynamically Feasible Pathway for Cofactor Regeneration
The design of a metabolic pathway for the generation of the redox cofactors NADH and NADPH in lipid vesicles requires a reduced substrate that can pass the membrane and luminal enzymes that catalyze the transfer of reducing equivalents to NAD+ and NADP+. Considering the NAD(P)-dependent biochemical reactions reported in the KEGG (Kyoto Encyclopedia of Genes and Genomes) database and the enzymes annotated in the BRENDA (The Comprehensive Enzyme Information System) collection, we identified formate as a potential source of reducing power. This C1 compound exhibits a lower standard reduction potential (E0′ = −0.43 V22) than the cofactors (E0 = −0.32 V), is highly membrane permeable,23 and can be oxidized to carbon dioxide by specific NAD+-oxidoreductases. The product carbon dioxide is also membrane permeable, and therefore the reaction will not be limited by protein-mediated transport rates or reconstitution efficiency.
In the framework of cellular metabolism, many redox reactions in catabolism require NADP(H) instead of NAD(H). The two redox pairs NAD+/NADH + H+ and NADP+/NADPH + H+ have essentially the same E0′ values.24 Thus, we designed the pathway to include a transhydrogenase to catalyze transfer of electrons between the two cofactors. Finally, we chose glutathione disulfide (GSSG) as a suitable electron sink for transfer of the reducing equivalents from NADPH, forming reduced glutathione (GSH) and simultaneously regenerating NADP+. The redox potential of the couple GSSG/GSH is estimated to be −0.24 V.25 The complete reaction scheme of our designed cofactor regeneration pathway is shown in Figure 1a.
Figure 1.

Design and feasibility of the redox regeneration pathway. (A) Scheme of the reactions and their coupling for generation of NADH, NADPH, and GSH. (B) Purity of the enzymes after Ni-Sepharose and size-exclusion chromatography (SEC). The SEC profile was monitored at 280 nm. On the top right corner of each frame, we show the sodium dodecyl sulfate (SDS)-polyacrylamide gel of the corresponding protein peak.
Liposomal NADH Formation
We expressed the gene for the NAD+-dependent formate dehydrogenase fromStarkeya novella(Fdh–EC 1.17.1.9)29 in E. coliand purified the protein to homogeneity (Figures 1b and S1 for the full SDS-polyacrylamide gel). The conversion of formate in solution was followed by monitoring the production of NADH, which, unlike NAD+, is autofluorescent. The kinetic analysis of the enzyme (Figure S2a and Table 1) showed that Fdh has a relatively high affinity for formate (KM = 2.15 mM),30 which allows maximal rates even at low millimolar concentrations of substrate.
Table 1. Overview of the Pathway Enzymes and Their Properties.
| systematic name | EC number | organism | molecular weight (kDa) (native mass) | oligomeric state | substrates | KM (mM) | kCAT (s–1) | refs | |
|---|---|---|---|---|---|---|---|---|---|
| Fdh | formate:NAD+ oxidoreductase | 1.17.1.9 | S. novella | 93 | dimer | NAD+ | 0.11 | 1.08 | this study |
| formate | 2.15 | 0.87 | |||||||
| SthA | NADPH:NAD+ oxidoreductase | 1.6.1.1 | E. coli | 432 | octamer | NADH | 2.63 | 9.7 | this study |
| (thio)NADP+ | 0.03 | 19.9 | |||||||
| GorA | glutathione:NADP+ oxidoreductase | 1.8.1.7 | E. coli | 102 | dimer | GSSG | 0.07 | 733.3 | (27, 28) |
| NADPH | 0.02 | 661.8 |
To explore the functionality of formate dehydrogenase inside the lumen of phospholipid vesicles, we tested the Fdh activity in 400 nm-extruded large unilamellar vesicles (LUVs). As shown in Figure 2a, NADH was formed upon addition of external formate. At fixed concentrations of protein and cofactor in the lumen of the liposomes, the rate at which NAD+ was reduced was tunable by variation of the concentration of formate. Furthermore, by varying the internal cofactor concentration (Figure 2b), we could tune the maximal achievable NADH concentration (Figure 2c; see Figure S3 for the calibration with NADH containing vesicles). Finally, we found that the Fdh inhibitor thiocyanate,29 which is membrane permeable,31 inhibits the luminal formate dehydrogenase (Figure 2a).
Figure 2.

Intraluminal NADH formation. (A) Dependence of the formate dehydrogenase reaction on externally added formate. The signal from the NADH autofluorescence was followed in large unilamellar vesicles (LUVs) containing 2.0 μM Fdh and 1.0 mM NAD+ upon addition of formate at the indicated concentrations w/wo thiocyanate (pink, SCN–). The relative fluorescence intensity units (RFUs) are normalized to the full reduction of 1.0 mM NAD+. The vesicles are diluted in buffer D. Error bars are represented as s.e.m. (n = 4). (B,C) NAD+ dependence. Different concentrations of NAD+ together with 2.0 μM Fdh were encapsulated in LUVs and then diluted in buffer D for activity assays. The reduction of NAD+ upon addition of 5 mM external formate was measured. The linearity between the concentration of NADH and fluorescence intensity in the vesicles is shown in panel C. Data points from independent quadruplicates (n = 4) are shown; error bars illustrate s.e.m. (D–F) NADH formation in GUVs. In panel D, 5.0 mM sodium formate (buffer I) was flown into a microfluidic device containing trapped giant vesicles. The autofluorescence of NADH allowed us to follow the reduction of NAD+ over time. The GUV membranes are labeled with a fluorescent lipid dye, 0.1 mol % Atto 633 DPPE. Scale bar: 20 μm. (E) Schematic of a portion of one of the microfluidic channels, showing the bucket-like structures, in which the GUVs are trapped [Reproduced with permission from the work of Yandrapalli and Robinson.26 Copyright 2021 Royal Society of Chemistry]. (F) Averaged fluorescence of multiple vesicles (n = 61). The dark gray region denotes the time it takes for the exchanged solution to reach the vesicles in the microfluidic device. In panel G, the external solutions were alternated between buffers without and with 0.5 mM formate (white and gray regions respectively, corresponding to buffers H and J) and the fluorescence of multiple vesicles (n = 138) was averaged.
Malate Dehydrogenase as an External Scavenger System
Next, we tested if the observed activity of Fdh occurred exclusively within the LUVs. In fact, it is well-known that the encapsulation procedure of enzymes and small molecules in the lumen of LUVs usually also leads to a small fraction that remains bound to the outer surface of membrane vesicles (extensively discussed by Walde and Ichikawa32). We prepared vesicles containing only Fdh and added the cofactor NAD+ to the external medium. Upon formate addition, we detected a slow and steady reduction to NADH, likely due to a small amount of formate dehydrogenase available on the exterior of the liposomes (Figure 3a). Similarly, when we encapsulated only NAD+ and added Fdh only externally (Figure 3b), we observed some NADH formation, suggesting that not only the enzyme but also a small amount of cofactor remains attached to the outer surface of the vesicles. In the latter case, the small amount of external NAD+ is immediately reduced by the externally provided Fdh, while in the setup of Figure 3a the much larger pool of 0.5 mM external NAD+ is steadily reduced by the Fdh molecules still attached externally to the membranes, explaining the difference in absolute fluorescent values. In the attempt to eliminate the stickiness of the reactants to the external vesicular leaflets, we included sodium chloride in the buffers to prevent possible electrostatic interactions. A higher ionic strength (addition of 100 mM NaCl) significantly reduced the extraluminal reaction (filled black circles, Figure 3a,b) but not completely.
Figure 3.
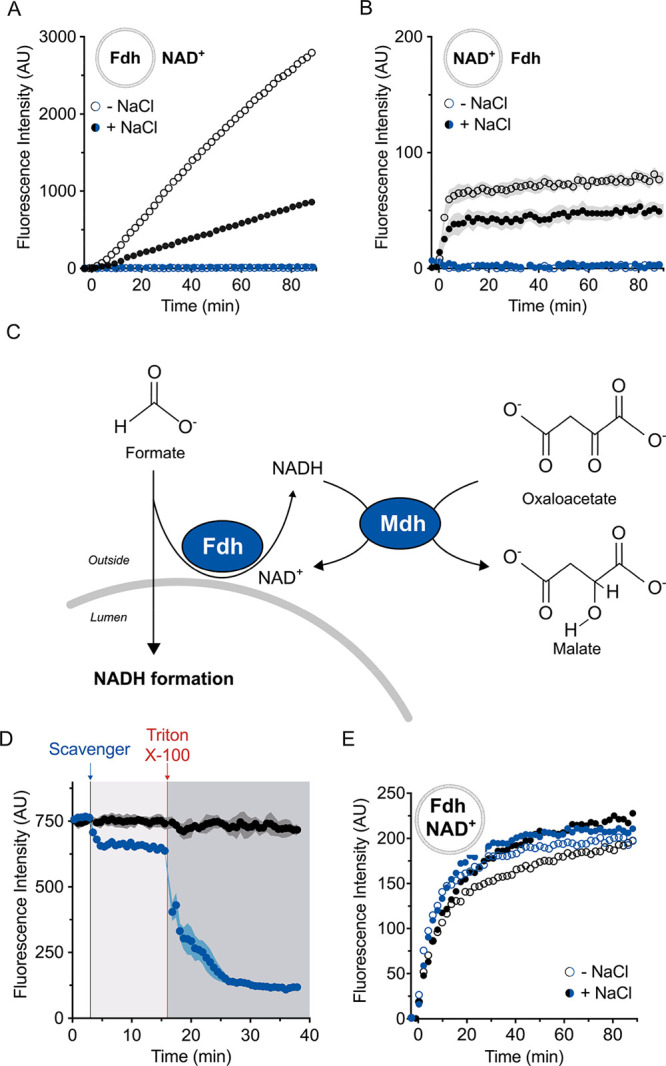
External scavenger system. (A,B) Enzyme and cofactor “stickiness” to synthetic vesicles. In panel A, 2.0 μM Fdh was encapsulated and 0.5 mM NAD+ was present externally. In panel B, the reverse configuration was used (internal 0.5 mM NAD+, 1.0 μM Fdh supplemented to the external buffer). Although the compartmentalization should separate the cofactor from the enzyme and prevent NADH formation, we observed an increase in fluorescence intensity (empty black circles). The presence of 100 mM NaCl dampened the formation of NADH (filled black circles). The external scavenger system composed of 0.2 μM malate dehydrogenase (Mdh) and 0.5 mM oxaloacetate eliminated the external signal w/wo 100 mM NaCl (respectively filled and empty blue circles). In both the panels, error bars correspond to s.e.m. (n = 4). (C) Schematic of the scavenger system that prevents the generation of extraluminal NADH. The presence of external Mdh and oxaloacetate (both membrane-impermeable) ensured the reoxidation of any possible noncompartmentalized reduced cofactor molecule. (D) Confirmation of compartmentalization by vesicle solubilization. After incubation in buffer B (white region), the scavenger system was added outside the LUVs with encapsulated 1.0 mM NADH (blue line, light gray region). Upon the addition of Triton X-100 (dark gray region), the compartmentalization was lost leading to complete oxidation of the cofactor by the scavenger system. In the absence of the scavenger system (black line), there was no change in fluorescence of NADH upon solubilization. Data from six independent measurements (n = 6) are shown, and error bars indicate s.e.m. (E) Formation of NADH inside LUVs which is not affected by supplement of NaCl. The reaction was triggered (t = 0 min) by 5.0 mM formate. The error bars are not reported for clarity (n = 4). The employed buffers in panels A, B, and E are described in detail in the Materials and Methods section.
We therefore developed a scavenger system (Figure 3c) to remove all external NADH. The purified malate dehydrogenase (Mdh) fromE. coli (Figure S1a,b) converts oxaloacetate into malate with the concomitant oxidation of NADH to NAD+. The equilibrium of this reaction lies toward malate and NAD+ by almost 300 000 times.33 In addition, malate dehydrogenase has a high kCAT of 930 s–1.34 Therefore, each molecule of external cofactor, that is reduced into NADH, is rapidly reoxidized by Mdh along with the production of malate. The effectiveness of the external scavenger system was demonstrated in vesicles with NADH (Figure 3d). The scavenger system decreased the NADH fluorescence by ∼10%, which reflects the residual NADH on the outside. Subsequent detergent-mediated solubilization showed that the rest of the fluorescence (∼90%) arose from the compartmentalized reduced cofactor. When Fdh was incorporated in the lumen and the substrate NAD+ was added only on the outside (or vice versa) (Figure 3a,b), the external scavenger system prevented the generation of extraluminal NADH, even after the addition of formate. In contrast, the vesicles with enzyme and cofactor (Figure 3e) catalyzed the formation of NADH from formate in the presence of the external scavenger system. No significant difference in the luminal reaction rate was observed in the presence or absence of the scavenger (respectively blue or black symbols) or at different ionic strengths (full circles), showing that the large majority of the activity takes place inside the lumen. Nonetheless, to avoid any possible misinterpretations, we decided to use the scavenger system in all subsequent experiments.
NADH Formation Inside GUVs
Giant unilamellar vesicles (GUVs) have an increased volume relative to LUVs, allowing both an easier mechanical manipulation, and their direct and individual observation by optical microscopy. We prepared GUVs by the PVA gel-assisted swelling method,35 with the enzyme(s) and cofactor(s) present in the swelling solution (see Materials and Methods). We utilized a microfluidic device developed by Yandrapalli and Robinson26 to trap populations of GUVs and observe changes in fluorescence over time (Figure 2d–f); a schematic of the traps in this device is shown in Figure 2e (the entire device design is in Figure S4). The device allows us to alternate external solutions while still observing the same GUVs. Therefore, we used the microfluidic setup to wash away external Fdh and NAD+ before the substrate formate was introduced. In Figure 2d, selected timelapse confocal images show the changes in the fluorescence of internal NADH (white). The NADH levels plateau after approximately 3 h (Figure 2f), which is evident from the population averaged analysis of individual vesicles. The position of a GUV within the trap did not have an effect on the kinetics of NADH formation (Figure S5a), but the size of the GUV had some effect. Larger vesicles reached higher levels of NADH, and the smallest vesicles displayed a slower initial rate (Figure S5b). The density of vesicles surrounding the measured GUV had some effect on the final concentration of NADH (Figure S5c). Nonetheless, the significance of these observations (vesicle size and vesicle environment) is relatively low as the measurements had overlapping error bars (Figure S5d–f).
The property of the microfluidic device to completely exchange the external solutions provided a further level of control of the compartmentalized reaction, as we could sequentially trigger and attenuate the enzymatic reduction of NAD+ (Figure 2g). When sodium formate was flowed through the device (gray shaded regions), the rate of NADH formation increased; the NADH formation declined when buffer devoid of substrate was used. This switching between different activity regimes could be repeated many times, highlighting the possibility of tuning the flow of electrons when the system is encapsulated in GUVs.
Transhydrogenation between NADH and NADPH
For the transfer of reducing equivalents from NADH to NADP+, we selected the soluble pyridine nucleotide transhydrogenase SthA (EC 1.6.1.1) fromE. coli. In vivo, the primary role of this enzyme is to prevent the formation of excessive amounts of NADPH,10 but we used SthA in vitro for NADPH formation, exploiting the reversible nature of the reaction. The detection of NADPH formation is challenging, because NADH and NADPH are spectroscopically virtually indistinguishable from each other. For this reason, transhydrogenation assays are usually carried out with one of the cofactors replaced with a thio-analogue.36 Following the purification of SthA to homogeneity (Figure 1b), we determined the kinetic parameters for transhydrogenation (Figure S2b), using NADH and thioNADP+ as substrates. Both NADH and thioNADP+ inhibited the enzyme at high concentrations. Once we determined the ideal substrate concentration range, we reverted from thioNADP+ to the native cofactor NADP+. For this, we used the high-affinity NADPH-specific sensor, iNap1,37 which allowed monitoring of the transhydrogenation without the need of using a thio-analogue (Figure 4a,b). With iNap1, we detected NADPH formation as a change in the ratio of fluorescence excitation at 420 and 485 nm, using the fixed emission wavelength of 530 nm (Figure 4a). Starting from 1.0 mM NADH, the full reduction of 200 μM NADP+ was performed in bulk solution in less than 20 min by 0.08 μM SthA (empty circles, Figure 4b). In the case of coupling the reaction with Fdh, the formate oxidation caused a NADH build-up, which then led to NADP+ reduction catalyzed by SthA; the Fdh-catalyzed reaction was almost completely inhibited by 30 mM thiocyanate. As expected, the ratiometric readout of the sensor iNap1 remained unchanged when we monitored exclusively the formate oxidation with NAD+, confirming the NADPH specificity of iNap1.
Figure 4.

SthA-mediated transhydrogenation. (A) Fluorescent excitation spectrum of the NADPH sensor iNap1 before (dashed line) and after NADPH production (solid line) by the Fdh-SthA coupled reaction, with wavelengths used for excitation highlighted. The formed NADPH was quantified using the ratio in fluorescence at the excitation wavelengths of 420 and 485 nm. (B) Bulk solution NADPH formation. Empty circles depict the formation of NADPH from NADH mediated by SthA (0.08 μM SthA, 1.0 mM NADH, 0.2 mM NADP+, and 0.2 μM iNap1). Filled green circles represent NADPH formation using formic acid as an electron donor to reduce NAD+ to NADH which is subsequently used in the transhydrogenation reaction (0.25 μM Fdh, 1.0 mM NAD+, 0.08 μM SthA, 0.2 mM NADP+, and 0.2 μM iNap1). In the latter mixture, thiocyanate (SCN–) inhibits electron flow at the Fdh stage (pink symbols, 0.25 μM Fdh, 1.0 mM NAD+, 0.08 μM SthA, 0.2 mM NADP+, 0.2 μM iNap1, and 30 mM SCN–). Black circles: The conversion of NAD+ into NADH by Fdh is not detected by iNap1 (1.0 μM Fdh, 0.2 mM NAD+, and 0.2 μM iNap1). Each condition was repeated in biological quadruplicate (n = 4) and tested in buffer B. Error bars are reported as s.e.m. (C) Reduced cofactor detection in LUVs equipped with the iNap1 sensor (2.0 μM Fdh, 0.21 μM SthA, 1.0 mM NAD+, 0.2 mM NADP+, 1.0 μM iNap1, buffer C). At the excitation wavelength of 370 nm (left graph), the reduction of both nicotinamide cofactors can be observed without distinguishing NADPH from NADH. The ratio of the excitation wavelengths 420/485 permits the quantification of exclusively NADPH in the right-hand graph. The data sets from four independent experiments (n = 4) are displayed, and the error bars indicate the s.e.m. (D) Ratiometric time series of GUVs in the microfluidic traps with the encapsulated Fdh and SthA reactions and the sensor iNap1. The reactions were started by flowing in 5 mM external formate. Scale bar: 20 μm. (E) Ability to specifically sense NADPH formation in GUVs containing Fdh and SthA. The coupled reaction (2.0 μM Fdh, 1.0 mM NAD+, 0.21 μM SthA, 1.0 μM iNap1) can take place only in the presence of 0.5 mM NADP+ (green circles), when buffer I with 5 mM formate is flowed in the microfluidic chip (n = 114). Only a relatively small increase in the 420/485 ratio (green crosses) is visible in the absence of NADP+ (n = 109).
Next, we investigated the functionality of the Fdh-SthA network in the lumen of LUVs (Figure 4c). Figure 4c displays the combined formation of the reduced cofactors NADH and NADPH (left graph) or exclusively NADPH generation (right graph). At the excitation wavelength of 370 nm, vesicles containing Fdh only exhibited a lower increase in fluorescence than vesicles also containing SthA and NADP+. This difference in fluorescence reflects the total pool of reduced cofactors within the compartment. We ruled out the possible direct reduction of NADP+ by Fdh by incorporating the enzyme in the presence of only NAD+ or NADP+ (Figure S6). Using the 420/485 ratio, the Fdh-SthA liposomes showed NADPH generation upon formate addition, whereas only a small change in 420/485 was observed in Fdh-LUVs. We also analyzed this coupled reaction system inside GUVs. Indeed, we observed the anticipated changes in fluorescence of the iNap1 sensor upon addition of formate (Figure S7).
Following this, we monitored the formation of NADPH over time by trapping GUVs (with relevant encapsulated components for NADP+ reduction) in the microfluidic device and flowed in buffer containing sodium formate (buffer I). The fluorescence emission of GUVs excited at 405 and 488 nm was followed for approximately 3.5 h (Figure 4d,e). The reduction of NADP+ reached a plateau after approximately 2.5 h, and we confirmed that the iNap1 is highly specific for NADPH; the signal is negligibly affected by high concentrations of NADH (Figure 4e, green crosses).
Engineering of an Electron Sink
To induce flow through the pathway, we included an electron sink that takes reducing equivalents from NADPH. We picked glutathione disulfide (GSSG), which is reduced by NADPH to 2 molecules of glutathione (GSH), a reaction catalyzed by the flavoprotein glutathione reductase GorA (EC 1.8.1.7—see Figure 1b for the purified protein). We monitored the formation of GSH by using the Ellman’s reagent38,39 (DTNB). In the presence of 400 μM NADPH, GorA alone catalyzed the complete reduction of 200 μM GSSG within 3 min (Figure 5a). With Fdh, SthA, and GorA, and formate as the electron donor, the full conversion of GSSG took 15–60 min, depending on the transhydrogenase concentration. We used a concentration of NADP+/NADPH that was less than the amount of NADPH required for the complete reduction of GSSG (Figure S8), and thus multiple cycles of NADPH formation (from NADH via formate oxidation) and NADP+ regeneration (by GorA) had taken place. In the absence of SthA and NADP+, only less than 4% of GSH was formed, presumably due to the nonspecific NADH-dependent activity of GorA27 (Figure 5b, empty squares). The exclusion of NAD+ from the reaction mixture completely abolished GSH formation (Figure 5b, empty triangles). As final evidence of the pathway being dependent on the formation of NADH, we tested the three-enzyme system in the presence of thiocyanate, the inhibitor29 of Fdh (Figure 5b, pink circles). Indeed, less than 2% of reduced glutathione was formed compared to the reduction of the GSSG pool in the absence of thiocyanate. Nonetheless, the residual slow formation of NADH still supported the SthA-mediated catalysis, yielding some NADPH formation of GSH by GorA, albeit at a very slow rate.
Figure 5.

Addition of an electron sink to the redox pathway. (A,B) Pathway activity in bulk solution. (A) GSH formation. Empty circles: GorA alone (0.05 μM) catalyzes reduction of 200 μM GSSG by oxidizing 400 μM NADPH. Filled symbols: In the presence of Fdh, SthA, and GorA, 5.0 mM formate triggers the complete pathway activity (1.0 μM Fdh, 1.0 mM NAD+, 0.2 mM NADP+, 200 μM GSSG, and 0.05 μM GorA), leading to a GSH accumulation rate depending on the SthA concentration (0.21 μM SthA squares, 0.08 μM SthA triangles, 0.04 μM SthA diamonds). (B) Exclusion of the transhydrogenase and NADP+ (empty squares) inhibited the GSH formation by 4% comparing it at 15′ with the full pathway including 0.21 μM SthA. Exclusion of SthA and NAD+ (empty triangles) fully prevented GSH formation. The pathway is also inhibited by 30 mM thiocyanate (SCN–, pink). Data points and error bars (s.e.m.) of both panels A and B result from six independent measurements (n = 6) in buffer B. (C) Kinetics of NADPH formation in the presence of an electron drain to glutathione in LUVs. The luminal inclusion of 0.5 μM GorA allows the electrons to from NADPH to reduce GSSG. All samples include 0.38 μM Fdh, 1.0 mM NAD+, 0.06 μM SthA, 0.05 mM NADP+, and 1.0 μM iNap1 sensor, while 5.0 mM formate was externally added (n = 6). LUVs were prepared in buffer C and diluted in buffer D for activity assays. (D) Time-dependence of NADPH formation of the full pathway (2.0 μM Fdh, 1.0 mM NAD+, 0.08 μM SthA, 0.2 mM NADP+, 0.05 μM GorA and 0.2 μM iNap1, 5.0 mM ammonium formate, buffer B) in bulk with different concentrations of GSSG as an electron sink. Steady state levels of NADPH are detected until the exhaustion of the electron sink. Independent replicates (n = 2) are reported, while the error bars are not shown for clarity. (E) Stability of the cofactor regeneration pathway inside vesicles. After storage at 4 °C, the activity of LUVs containing 0.38 μM Fdh, 1.0 mM NAD+, 0.06 μM SthA, 0.05 mM NADP+, 0.5 μM GorA, 2.5 mM GSSG, 1.0 μM iNap1, and buffer C was monitored at different time intervals, upon addition of 5.0 mM sodium formate (left main graph). The activity is defined as the difference in the 420/485 ratio measured in the first 10 min after the provision of formate, converted into the percentage of the estimated activity on day 1 (n = 6). On the right, the inset shows the size distribution profile of the 400 nm extruded vesicles on days 1 (upper graph) and 21 (bottom graph).
We then equipped LUVs with the complete pathway, the two cofactors and GSSG (see Figure 1a), and the NADPH sensor iNap1. Upon addition of 5.0 mM formate, we examined the NADPH formation at different concentrations of encapsulated GSSG (Figure 5c). We reasoned that by increasing the amount of glutathione disulfide as an electron drain, it should be possible to prolong the flux through the pathway and observe a phase with an approximately constant steady state concentration of NADH and NADPH. Compared to vesicles without GSSG, the accumulation of NADPH slowed down in the presence 0.5 and 2.5 mM GSSG but did not lead to a period with constant steady state concentration of NADPH (Figure 5c, dark green circles and black triangles, respectively). At 5.0 mM GSSG (black circles), a prolonged phase was found in which the NADPH concentration remained ∼10 μM NADPH, even after 10 h (Figure S9). This result indicates that the 100-fold excess of GSSG over NADP+ delays the accumulation of NADPH and leads to a long steady state phase. This behavior can also be seen in solution experiments (Figure 5d), where the full reduction of the NADPH pool is delayed at higher GSSG concentrations.
Similar experiments can also be conducted in GUVs where the NADPH concentration is monitored by observing the fluorescent readout from NADPH sensor iNap1. As before, the GUVs were trapped in the microfluidic device (Figure S4) and the external solution exchanged for a buffer supplemented with 5 mM formate. The fluorescence emission of GUVs excited at 405 and 488 nm was followed for approximately 5 h for vesicles containing additionally 0.25 μM GorA and 2.5 mM GSSG (Figure S10, black circles). Also here, we saw that the presence of the electron drain resulted in a long steady state phase with a partially reduced NAD+/NADPH pool.
Since enzymatic deactivation in aqueous solutions40,41 is one of the main limiting steps for cell-free systems lacking an efficient proteostasis mechanism, we tested the stability of the complete redox regeneration pathway in LUVs. After storage at 4 °C, we measured the activity of the whole pathway in terms of NADPH formation for a period of 2 weeks (Figure 5e). The structural integrity of the liposomes over storage was confirmed by dynamic light scattering (Figure 5e, inset). Promisingly, more than 95% of the metabolic activity was retained after 3 days, and, even after 1 week, the vesicles still conserved about 60% of the original activity. Only at day 14 could we assess a significant drop in the pathway functionality, corresponding to <20%. A systematic analysis of the individual enzymes allowed us to identify SthA as the critical component for the long-term stability of the redox cofactor regeneration pathway (Figure S11a–c).
Discussion
We designed a minimal enzymatic pathway for the regeneration of the redox cofactors NAD(H) and NAD(P)H in a thermodynamically and kinetically feasible fashion. The Fdh-mediated oxidation of formate leads to the formation of NADH, which was used to form NADPH via a transhydrogenase reaction, which in turn was used to reduce glutathione disulfide. The pathway was characterized in solution and in large and giant unilamellar vesicles (LUVs and GUVs), indicating the flexibility and functionality of the pathway in different cell-free systems. In GUVs the redox state could be controlled via the feed of formate in the microfluidic device.
Historically, formate dehydrogenases have been utilized for NADH regeneration in vivo and in vitro.29 With the aim of using the minimum number of enzymes to interconvert both cofactors, we opted to couple the NADH buildup with transhydrogenation. Transhydrogenases have previously been used in cell-free systems42,43 for the production of pharmaceutical chemicals such as hydromorphone and fatty acid surrogates; however, all previous studies required the use of analogues of the NAD(P)H cofactors to monitor the reactions. By employing the sensor iNap1,37 we were able to quantify the enzymatically formed NADPH itself. To date, this is the first applicative use of the sensor to discriminate the reduced cofactors NADH and NADPH from each other. GorA was selected to showcase the applicability of our synthetic redox system with glutathione -disulfide as an electron sink, due to its essential function of scavenger of reactive oxygen and nitrogen species in living cells.44,45 Alternatively, a different NADPH-dependent dehydrogenase could be used in place of GorA, such as a stereoselective reductase or oxidase.46 Overall, our synthetic pathway in vesicles is remarkably stable, but SthA is the Achilles’ heel for long-term usage, which may need to be replaced by a more stable variant in the future.
Our synthetic redox pathway is fed by formate, which is an ideal electron donor47 as most biological membranes are highly permeable for formic acid.23 Since the pKa of formate/formic acid is ≈3.75, at pH 7.0 most of the compound is present in the anionic form (A–). Yet, the acid–base equilibrium between formic acid and formate is fast, and we find that the diffusion across the membrane does not limit the supply of the electron donor. Importantly, the product of the reaction, CO2, is also membrane permeable and diffuses out of the vesicles and contributes to pulling the flux48 of the whole pathway in the desired direction.
We also developed a method to ensure that the observed reactions take place exclusively within the vesicle lumen. Methods for the preparation of phospholipid vesicles can lead to unwanted activities when enzymes or small molecules stick to the surface of the vesicles,32 which could result in the overestimation of luminal activity. Electrostatic interactions between phospholipids and both Fdh and NAD+ contribute to the stickiness (Figure 2a,b). While addition of NaCl decreased the generation of extraluminal NADH, it did not lead to complete elimination, suggesting that other types of interactions also take place. Although the stickiness of nicotinamide cofactors is not commonly observed (however, see ref (49)), our work shows that it is very difficult to remove all external NAD+. Our “external scavenger system” makes use of the thermodynamic and kinetic properties of the reaction catalyzed by malate dehydrogenase, as well as the impermeable nature of oxaloacetate and malate. We could convert any externally formed NADH immediately back into NAD+, allowing the quantitative analysis of luminal NADH formation.
An important discussion point in the field of bottom-up synthetic biology or synthetic biochemistry relates to the optimal compartment size,50,51 as several parameters such as surface-to-volume ratio, transport capacity in relation pathway fluxes, space for all macromolecules, and excluded volume effects come into play. As such, we explored the feasibility of our biochemical reactions within differently sized phospholipid vesicles (from about 0.2 to 10–50 μm in diameter). While we could measure the enzymatic activity in ensembles of 400 nm-extruded vesicles (effective diameter range from 50 ± 5 to 215 ± 25 nm, Figures 5e and S12), the micrometer-size GUVs allowed us to observe the reactions by microscopy and gain control over the pathway on a single vesicle level through the feeding of formate. This comparison also highlights some differences in the kinetics of the regeneration pathway, which may partly be explained by the differences in assay temperature affecting the enzymatic activities52 (30 °C in LUVs, 19 °C in GUVs) and partly by the differences in the effective concentration of enzymes and reactants in the vesicles. Kuchler et al. have extensively discussed the point of spatially confined enzymatic reactions,50 explaining how the volume of entrapment can affect the concentration of the components inside vesicles. In addition, we observe a difference in the stability of LUVs and GUVs, which is in line with previous observations.53,54 The hydrodynamic radius of LUVs did not change significantly after 3 weeks storage of the vesicles at 4 °C (see Figures 5e and S12), while the GUVs maintained structural integrity for about 1 week.
A major advantage of using formate as a feed (and having CO2 as reaction product) is that no membrane-embedded transporter proteins are required for pathway functionality. Not only is membrane reconstitution of transporters challenging, it may also lead to a fraction of proteins in the nonpreferred orientation, e.g. as encountered by Kleineberg et al.55 Our redox regeneration system bypasses the limitations of membrane reconstitution and the requirement of transporters for specific lipids highlighted in several bottom-up metabolic modules.56,57 The permeation of formic acid through the phospholipid membrane is a crucial aspect of our pathway design, which can also be used in biomimicking systems with low permeability such as polymerosomes wherein transporters are not functional.58
A potential next application of our redox system is its integration with synthetic metabolism in vesicles or droplet systems,51,59,60 thereby generating a higher level of complexity combining ATP as fuel57,61 and nicotinamide cofactors for redox homeostasis. Recently, water-in-oil droplets containing a pathway for CO2 fixation62 (cycle CETCH version 7.0) have yielded the production of glycolate in a light-driven manner by coupling the compartmentalization of thylakoid membranes and formation of ATP and NADPH in the aqueous lumen. For instance, the encapsulation of our redox enzymes would ensure the availability of reducing equivalents in the absence of light even after the long-term inactivation of the thylakoid modules. Besides, the reducing power of our pathway can readily be integrated with any larger metabolic network, in which the flow of electrons is directed toward other components for the breakdown of complex molecules.
The in vivo synergic action of a formate dehydrogenase and a membrane-bound transhydrogenase has recently been presented63 as advantageous for aerobic C1-assimilation. Kim and colleagues63 developed an E. coli strain capable of formatotrophic growth by a reductive glycine pathway. Potentially, our pathway might be transplanted in engineered strains of this kind to increase the yield of manufactured valuable chemicals.
In conclusion, we devised a system that ensures the availability of reducing power in the form of the two main biological redox cofactors. That this can be performed efficiently for long periods of time with a minimal number of enzymes that can be incorporated in vesicles of varying sizes, i.e., the range of the smallest bacteria such as Pelagibacter to that of large mammalian cells. The low metabolic burden that our pathway demands, together with no need for membrane proteins, makes this system attractive for both in vitro and in vivo applications. Since it is based on the permeation of formic acid into the liposomal lumen and diffusion out of its reaction product (CO2), our pathway would not unbalance the carbon stoichiometry of other metabolic reactions with which it could work synergistically. This means that the reduction of the upstream cofactor (NAD+) does not involve the simultaneous formation and consequent accumulation of a dead end metabolite, which should otherwise be metabolically recycled with another enzymatic module, adding an unnecessary layer of complexity to the system.64 Besides, as a very cheap and optimal substrate to donate reducing equivalents to nicotinamide cofactors (which in the reduced forms are significantly expensive65), formic acid has also been advocated as a promising feedstock47,66 for the manufacture of high value chemicals. Our minimal system could be tailored or enhanced to support targeted biosynthesis pathways in industrial or food biomanufacturing,16 boosting the half-life of production at the expense of a low-cost trigger.
Materials and Methods
Materials
LB-Broth Miller (Formedium, LMM0102), d(+)-sucrose (Formedium, SUC01), Difco granulated agar (Thermo Fisher Scientific, DF0145-17-0), ampicillin sodium salt (Carl Roth, K029.4), kanamycin sulfate (Carl Roth, T832.2), sodium chloride (Merck KGaA, 106404), l-(+)-arabinose (Sigma, A3256), potassium dihydrogen phosphate (Merck KGaA, 104873), potassium hydrogen phosphate-trihydrate (Merck KGaA, 105099), deoxyribonuclease I from bovine pancreas (Sigma, DN25), magnesium sulfate heptahydrate (Merck KGaA, 105886), PMSF (Carl Roth, 6367.2), EDTA dipotassium salt-dihydrate (Sigma, ED2P), imidazole (Carl Roth, X998.4), Tris ultrapure (AppliChem GmbH, A1086), glycerol (Boom B.V., 76050772), β-NAD+ hydrate (Sigma, N1636), β-NADP+ disodium salt (Sigma, NADP-RO), Thio-NADP+ monopotassium salt, oxidized form (Oriental Yeast Co., Ltd.), β-NADH disodium salt hydrate (Sigma, N8129), β-NADPH tetra-sodium salt (Sigma, NADPH-RO), l-glutathione oxidized disodium salt (Sigma, G4626), l-glutathione reduced (Sigma, G4251), sodium formate (Sigma, 71539), ammonium formate (Sigma, 156264), sodium thiocyanate (Merck KGaA, 106627), oxaloacetic acid (Sigma, O4126), 5,5′-dithiobis(2-nitrobenzoic acid) (Sigma, D8130). The synthetic lipids 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, 850725C), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, 850375C), and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG, 840475C) were purchased from Avanti Polar Lipids, Inc. in chloroform solutions with a purity >99%. No unexpected or unusually high safety risks were found using the above-mentioned chemicals or performing the experimental procedures described below.
Construction of the Expression Plasmids
The used genes (fdh, sthA, gorA, and iNap1) were amplified using the respective primers (Table S1) and subsequently cloned in various E. coli expression vectors from the FX cloning kit.67 The synthetic gene fdh was codon-optimized for E. coli expression, obtained from Thermo Fisher Scientific, Inc. and inserted in the pMA-RQ vector (available in suppl. material). pMA-RQ_fdh was the template for fdh, pRDNA3.1-hygro-cyto-iNap1 was that for iNap1, and the E. coli K-12 genome was that for sthA and gorA. pRDNA3.1-hygro-cyto-iNap1 was a gift from Dr. Yi Yang (Laboratory Synthetic Biology and Biotechnology, East China University of Science and Technology). The inserts were first subcloned into the pINIT_kan vector by cutting with SapI and ligation using the FX cloning procedure. The presence of the genes within the plasmids was checked by DNA sequencing. After this, the genes of interest were transferred from the initial plasmid to specific expression vectors by FX cloning protocol. Chemically competent E. coli MC1061 cells were transformed with 5 μL of the ligation mixture containing the pINIT_gene and the expression vector of choice (pBXNH3 or pBXC3H). The selection of cells containing pBXC3H_ FdH, pBXC3H_SthA, pBXNH3_GorA, or pBXNH3_iNap1 was carried out on LB plates with ampicillin 100 μg/mL +7% sucrose.
The mdh gene was PCR-amplified from Escherichia coli K-12 genomic DNA with primers Mdh8hisNcoI-Fw and MdhXbaI-Rv (see Table S1) by use of Phusion HF DNA polymerase (Thermo Fisher Scientific, Inc.). The insert and the plasmid pBAD2468 were digested with the restriction enzymes NcoI and XbaI, purified from the presence of salt and restriction enzymes by using the PCR Clean-up kit (Macherey-Nagel), then ligated with T4 DNA Ligase (Thermo Fisher Scientific, Inc.) to form the expression plasmid pBAD24_MdH8XNhis. E. coli MC1061 cells were transformed as described above. Also in this case, the presence of the correct gene sequence in the vector was checked by sequencing.
Gene Expression
We used E. coli MC1061 as expression host for all genes. In a typical experiment, a 200 mL culture was grown in LB + ampicillin 100 μg/mL until 0.5 ≥ OD ≥ 0.8 at 37 °C, 200 rpm and, then, transferred to 20 °C and allowed to cool prior to the addition of inducer. The induction was started with the addition of l-arabinose at final concentrations of 0.01% w/v for fdh, sthA, and iNap1 and 0.05% w/v for gorA, and the culture was left overnight, with the exception of the pBXNH3_iNap1 strain, which needed 48 h of induction time for appreciable production of iNap1. Instead, the mdh overexpression was kept at a temperature of 37 °C and, then, induced by 0.1% (w/v) l-arabinose for 3 h. Following overexpression, the cells were harvested by centrifugation (15 min, 6000g, 4 °C), resuspended in buffer A (50 mM KPi pH 7.5, 150 mM NaCl), and washed in the same medium. Eventually, the pellet was resuspended in a final volume of 30–35 mL buffer A and flash-frozen in liquid nitrogen and stored at −80 °C. All the buffers used are listed in Table S2.
Protein Purification
The purification procedure was the same for all the overproduced proteins. The frozen cells were slowly thawed on ice, after which 0.1 mg/mL DNAase, 1 mM MgSO4, and 1 mM PMSF were added. Cells were disrupted by sonication for 10 min (3 s on, 6 s off cycle, 70% amplitude) at 4 °C using a VCX130 Vibra-Cell sonicator (Sonics & Materials, Inc., Newton, CT), followed by DNAase inactivation by the addition of 1 mM K-EDTA pH 7.0. Next, the resulting samples were centrifuged at 48 254g for 30 min, at 4 °C, and the supernatant was diluted one to one with buffer A (final volume 60–70 mL), mixed, and split in two tubes. One of the two tubes was flash-frozen and stored at −80 °C, while the rest of the diluted soluble fraction was shaken by nutation for half an hour at 4 °C with the addition of 10 mM imidazole pH 7.5 and 1 mL Ni2+-Sepharose resin (Ni Sepharose 6 Fast Flow Cytiva), already prewashed with 20 column volumes (CV) Milli-Q and then equilibrated with 20 CV of buffer A. After binding, the mixture was poured into an Econo-Pac chromatographic column (Bio-Rad Laboratories, Inc.) and allowed to flow through by gravity. The resin material was washed with 20 CV of Buffer A and 50 mM imidazole, followed by elution with a first fraction of 700 μL and subsequent ones of 500 μL with Buffer A and 500 mM imidazole. The protein purity was checked by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and the concentration was determined from the absorbance spectrum (NanoDrop Technologies, Inc.) and the extinction coefficient at 280 nm (calculated by the Protparam tool, available at https://web.expasy.org/protparam/). For the extinction coefficient of the transhydrogenase, the presence of FAD was considered with the assumption of one molecule of FAD per protomer. The samples containing the highest concentrations were centrifuged in 1.0 mM EDTA at 21 230g for 5 min at 4 °C and then loaded on a size-exclusion chromatography (SEC) column Superdex 200 Increase 10/300 GL (GE Healthcare) for further purification and removal of imidazole. All the proteins were eluted with buffer A, with the exception of SthA, which required Tris 50 mM pH 7.5, 150 mM NaCl, and 10% (v/v) glycerol. The SEC column was connected to a NGC Quest 10 Chromatography System (Bio-Rad Laboratories, Inc.), whose UV detector provided the protein elution profile at the wavelength of 280 nm. Following the application of the protein fractions to an Amicon Ultra 0.5 mL (Merck Millipore Ltd.) centrifugal filter device with 10 kDa cutoff, samples were concentrated to 2–6 mg mL–1 and glycerol was added to 10% (v/v), flash frozen in 80 μL aliquots, and stored at −80 °C. The protein purity was assessed on a SDS-PAGE, prepared with a 15% separating gel and a 5% stacking gel.
Enzymatic Assays for SthA and Fdh
The activity of Fdh was tested by recording the reduction of NAD+ at 340 nm (εNADH, 340 nm = 6.22 mM–1 cm–1) at 30 °C in a SPARK 10 M plate reader (Tecan). The length of the light path for the plate reader wells was estimated by calculating the difference in absorption of 200 μL aqueous solutions at 977 and 970 nm. The reaction mixtures were prepared in buffer B, employing 20 μL of 2.0 μM Fdh and 20 μL NAD+ at the chosen concentration, to be subsequently loaded into the wells of a 96-well flat-bottom transparent polystyrene plate (Greiner Bio-One International GmbH) and incubated at 30 °C for 3 min. The addition of 10 μL ammonium formate in a total volume of 200 μL started the reaction. A final concentration of 20 mM formic acid was used to determine the kinetic parameters for NAD+, which concentration was tested in the range of 10–1000 μM. Setting the amount of cofactor to 2.0 mM, we estimated KM and kCAT for formic acid by varying its concentration (0.3–18.0 mM). For both the substrates, the obtained initial velocities were analyzed with the Michaelis–Menten equation (eq 1) and the kinetic parameters were estimated:
| 1 |
where v stands for velocity of the reaction, VMAX is the maximal velocity, [S] is the used substrate concentration, and KM corresponds to the Michaelis–Menten constant; kCAT is Vmax/[enzyme].
In the case of SthA, we monitored the formation of thioNADPH at 400 nm (εthioNADPH, 400 nm = 11.7 mM–1 cm–1). To avoid any possible overlap with the absorption at 400 nm caused by high concentrations of added NADH, 10 μL of 0.04 μM SthA was added to the reaction mixture, already containing the cofactors thioNADP+ and NADH at the concentrations of interest in buffer B. Fixing thioNADP+ at 150 μM, we varied the NADH concentrations from 0.25 to 18.0 mM. In the same way, keeping NADH constant at 15.0 mM, we performed the transhydrogenation reaction in the presence of thioNADP+ (10–300 μM). Both the native and the substrate analogue displayed substrate inhibition, a phenomenon taken into account in the analysis (eq 2) used for fitting of the data:
| 2 |
where KI is the inhibitory constant, while v, VMAX, [S] and KM have the same meanings as in eq 1. Four different purification batches were used for the determination of the kinetic parameters for both SthA and Fdh.
The Fdh stability in solution (Figure S11c) was monitored by absorbance in a similar manner as described for the kinetic measurements. The protein stock at −80 °C was thawed, diluted to 5.0 μM in buffer B, and then stored in the fridge for a period of 3 weeks. At different time points, 0.5 μM Fdh was tested for its capacity to reduce 0.5 mM NAD+ upon the addition of 5 mM ammonium formate. The initial velocity of day 1 was set to 100% to estimate the loss of activity during the storage period.
Glutathione Measurements
The capacity of GorA to produce reduced glutathione from glutathione disulfide was measured by TNB2– formation using Ellman reagent (DTNB).38,39 DTNB was freshly prepared in buffer B and wrapped in aluminum foil to prevent photodamage. According to Eyer’s work69 on the effect of temperature and pH on the absorption of TNB2–, we estimated its molar extinction coefficient at 412 nm to be 13.8 × 103 M–1 cm–1. All the reactants were solubilized in buffer B, which was used for all measurements. The reaction mixtures were loaded into the wells of a 96-well plate and then incubated with 10 μL 4 mM DTNB (final 0.2 mM) at 30 °C for 5 min after mixing. The final volume for both the negative controls (no substrate addition) and the other samples was 200 μL. For these last ones, the addition of final 5 mM ammonium formate, or 0.2 mM β-NADPH for the only glutathione reductase reaction, started the assays. The measurements were carried out at 30 °C with shaking at 270 rpm. The glutathione formation was followed by the increase in absorption at 412 nm, performed in the SPARK 10 M plate reader (Tecan). All measurements were repeated in biological triplicates.
The common components of all reaction mixtures were 1.0 μM Fdh, 0.2 mM GSSG, and 0.05 μM GorA. The final cofactor concentrations were 1.0 mM for NAD+ and 0.2 mM for NADP+, while the inhibitor thiocyanate was used at a concentration of 30 mM.
NADPH Measurements in Bulk Solution
The formation of NADPH by SthA, both alone and coupled with other enzymes, was followed by the variation of the excitation ratio 420/485 of the fluorescent sensor iNap1. An aliquot of the sensor purified in buffer A and previously stored in −80 °C was thawed and added in 105.250-QS microcuvettes (Hellma Analytics) at the final concentration of 0.2 μM. Each cuvette had already been filled with buffer B, the nucleotide cofactors and enzymes prepared in buffer B, and the total volume was 120 μL (114 μL and 6 μL of substrate to start the reaction). The samples were incubated at 30 °C for 5 min in a FP-8300 spectrofluorometer (Jasco, Inc.), and the fluorescent excitation spectrum was recorded from 350 ± 1 to 500 ± 1 nm at an emission wavelength of 530 ± 1 nm. Each reaction (at 30 °C) was started through the addition of the substrate. In the case of the transhydrogenase reaction, 6 μL of 20 mM β-NADH was added to the mixture containing 0.08 μM SthA and 0.2 mM NADP+. In the case of coupling with the formate dehydrogenase, 6 μL 100 mM of ammonium formate was added to start the formation of NADPH in mixtures containing 0.25 μM Fdh, 1.0 mM NAD+, and 0.2 mM NADP+. A 1.00 μM aliquot of Fdh was employed together with 0.2 mM NAD+ for the control without NADP+ and SthA, as well for the 30 mM thiocyanate inhibition; in the latter case the NAD+ concentration was 1.0 mM. The same setup with the addition of different GSSG concentrations and the presence of 0.05 μM GorA was used to monitor the full pathway in bulk solution (Figure 5d).
The quantification of NADPH formation was carried out using the normalizing equation (R – Rmin)/(Rmax – Rmin) used for iNap1 measurements by Tao et al.,37 with the addition of a normalization factor (a) to convert the concentration into micromolar units:
| 3 |
where [NADPH] is the concentration NADPH in μM, R is the measured ratio 420/485 of excitation wavelengths, Rmin is the ratio 420/485 before the addition of the substrate, Rmax is the ratiometric readout corresponding to the full NADP+ reduction into NADPH.
Large Unilamellar Vesicles and Enzyme Encapsulation
The liposomes were prepared as previously described,57 although with a different lipid ratio enclosing the vesicles and the lack of the reconstitution step for membrane proteins. The synthetic lipids in chloroform were 50 mol % DOPC, 25 mol % DOPG, and 25 mol % DOPE. The lipid mixture was dried in a rotary evaporator (Rotavapor R3̅, Büchi Labortechnik AG), then, resuspended in diethyl ether, and dried again to remove all traces of organic solvents. The dried lipids were rehydrated with 50 mM KPi pH 7.0 to a final concentration of 20 mg mL–1. The lipid suspension was sonicated with a VCX130 Vibra-Cell sonicator (Sonics & Materials, Inc., Newton, CT) at 0 °C, setting the amplitude to 70% and using 16 cycles (5 s on/45 s off). Subsequently, three freeze–thaw cycles were carried out between liquid nitrogen and a water-bath at room temperature. Then, the liposomes were divided into 1 mL aliquots contained in tubes with pierced lids and stored in liquid nitrogen to avoid oxidation. Following thawing of aliquot(s), LUVs were formed by extrusion, 13 times, through a polycarbonate filter with 400 nm pores (Avestin Europe GmbH). After dilution to 4 mg mL–1 with 50 mM KPi pH 7.5 and 100 mM NaCl (buffer C), the vesicles were collected by centrifugation (30 min, 325 000g, 4 °C) and suspended in 200 μL (20 mg mL–1) of the same buffer. The encapsulation of protein(s) and substrate(s) within the LUVs was always carried out with five freeze–thaw cycles using 200 μL as the total volume of the encapsulation mixture, including 66 μL LUVs (6.6 mg), while the concentration of the components to be incorporated varied according to the specific experiment. Then, the liposomes were extruded as described in the previous step, diluted to 5 mL of buffer C and centrifuged for 30 min at 325 000g, at 4 °C. Finally, the vesicles were resuspended in 50 μL buffer C per 6.6 mg of lipid, yielding a final concentration of 133.2 mg of lipid mL–1, and kept in the fridge or on ice for activity measurements.
Large Unilamellar Vesicles Activity
All assays involving enzymatically active LUVs were carried out in a FP-8300 spectrofluorometer, by measuring the fluorescence excitation spectrum (350 ± 1 to 500 ± 1 nm) at the emission wavelength of 530 ± 1 nm. Regardless of the encapsulation content, 2.9 μL of 400 nm-diameter vesicles (133.2 mg lipid mL–1) were typically resuspended to a final concentration of 3.2 mg of lipid mL–1 in buffer D (50 mM KPi pH 7.5, 100 mM NaCl, 0.2 μM malate dehydrogenase, and 0.5 mM oxaloacetate) for a total volume of 114 μL, unless otherwise indicated. After incubating the samples at 30 °C for 5 min in the fluorescence cuvette, the measurement was started by adding 6 μL of 20× stock substrate solution (frequently 100 mM ammonium formate). For activity measurements over time, the vesicles were stored at 4 °C, without any additional extrusion step (Figures 5e and S11c).
In the case of the assessment of the substrate/enzyme stickiness (Figure 3a,b,e), the vesicles were resuspended in buffer D or its variations (without sodium chloride, without scavenger systems, or without both), depending on the experimental need. Regardless of the specific buffer, 0.5 mM NAD+ was always included in the resuspension solution for the experiments depicted in Figure 3a; 0.5 μM of Fdh was used in the assays as shown in Figure 3b. Buffer B was chosen to prepare and resuspend the NADH-liposomes used for Figures 3d and S3. Their structural integrity was destabilized by the addition of Triton X-100 to a final concentration of 0.5%.
To estimate the amount of NADPH formed, we equipped the LUVs with 1.0 μM of the fluorescence-based sensor iNap1. The protocols for the encapsulation of reactants and enzymes (freeze–thawing cycles), and the preparation of iNap1-vesicles by centrifugation are the same as described under Large Unilamellar Vesicles and Enzyme Encapsulation. The concentrations of the encapsulated reactants and enzymes were 2.0 μM Fdh, 0.21 μM SthA, 1.0 mM NAD+, 0.2 mM NADP+, 1.0 μM iNap1 for Figure 4c and the same composition without SthA and NADP+ as a control for the Fdh-reaction. As above, the excitation spectrum was recorded from 350 ± 1 to 500 ± 1 nm, employing the emission wavelength of 530 ± 1 nm. The 420/485 excitation ratio was used to quantify the luminal concentration of NADPH, after fitting the data with eq 3. The intensity of the excitation wavelength at 370 nm was employed to monitor the reduction of NAD+ and NADP+, for which the cofactors and the SthA transhydrogenase were encapsulated in the vesicles.
The stability of the LUVs over time was monitored over a period of storage of the liposomes for 3 weeks at 4 °C (Figure 5e for the full pathway and Figure S11c for the Fdh-reaction). At the chosen time point, 2.9 μL of vesicle sample (133.2 mg lipid mL–1) were diluted in buffer D to the final concentration of 3.2 mg of lipid mL–1, and 5.0 mM formic acid was used to start the assay. The initial velocity of NAD(P)H formation was used to quantify the activity relative to that of freshly prepared vesicles. The redox pathway vesicles contained 0.38 μM Fdh, 0.06 μM SthA, 0.5 μM GorA, 1.0 mM NAD+, 0.05 mM NADP+, 2.5 mM GSSG, and 1.0 μM iNap1. The Fdh-vesicles contained 0.5 μM Fdh and 1.0 mM NAD+.
Size Distribution of Large Unilamellar Vesicles
The size distribution of the 400 nm-extruded LUVs was determined as previously described23 by dynamic light scattering (DLS) with the use of a DynaPro NanoStar Detector (Wyatt Technology, Santa Barbara, CA). In order to follow the liposomal size distribution over the storage period of 3 weeks at 4 °C, we freshly diluted the vesicles (133.2 mg of lipid mL–1) for each time point. The final lipid concentration of ∼2 μg/mL permitted us to reach the optimal number of 2 million counts at a constant temperature of 20 °C. The resulting DLS profiles were obtained by averaging 10 acquisitions of 20 s each, whose corresponding correlation curves showed an optimal level of overlap between each other.
Preparation of Giant Unilamellar Vesicles
GUVs were prepared according to the gel-assisted swelling method, as previously described by Weinberger et al.35 Briefly, a solution of poly(vinyl alcohol) (PVA) at 5% (w/w) with 50 mM sucrose was prepared by continuous stirring and heating at 90 °C until dissolved. A 5 μL drop was deposited on each of two clean glass slides and spread smoothly (region of gel approximately 1.5 cm × 1.5 cm) and dried in an oven at 50 °C for 30 min. Lipids were dissolved in chloroform at a final concentration of 4 mM with a composition DOPC/DOPG/DOPE of 49.9/25/25 mol % and 0.1 mol % Atto 633-DPPE. A 2 μL drop of the lipid suspension was deposited and spread over each dried PVA film. The lipid film was then dried under vacuum for 1 h to remove any traces of residual solvent. A chamber was formed using a Teflon spacer (height approximately 8 mm) between the two glass slides and secured with clips.
The swelling buffer (buffer E, F, or G; see below) was then injected via one of two small holes in the spacer using a needle. We found the yield and quality of GUVs to be significantly improved with the inclusion of sodium chloride in the swelling buffers (100 mM). The chambers were kept at 4 °C during formation, which took approximately 60–90 min. The GUV formation was followed by phase contrast microscopy. After the GUV formation, the chambers were tapped against a surface to mechanically detach the GUVs from the PVA. The vesicle suspensions were then removed using a needle and kept on ice until use. The osmolarity of the GUV swelling buffer was measured using an Osmomat 3000 (Gonotec, Berlin), and the osmolarities of any dilution or exchange buffers were matched to this value. The buffers used for GUV preparation and activity measurements are listed in Table S2. In case of buffers H–L, the solutions were supplemented with NaCl to osmotically balance the luminal medium.
Imaging and Image Analysis
A confocal laser scanning microscopy (LSM 710, Carl Zeiss AG Jena, Germany) equipped with a C-Apochromat 40×/1.2 NA objective was used for all imaging data. Three lasers (405, 488, and 633 nm) were employed for fluorescence excitation. The pinhole was set to 1 Airy unit. The experiments were performed at ambient room temperature (controlled to 19 °C). For images of GUVs in bulk, we collected 8-bit images of GUVs of 512 pixels × 512 pixels (212.55 μm × 212.55 μm). For time lapse series of GUVs in microfluidic devices (see the section Microfluidic Device Preparation and Operation), 8-bit images were collected at different predetermined trap positions (using the “Positions” feature of the microscope setup) of 512 pixels × 512 pixels (303.64 μm × 303.64 μm). For all comparable and replicate experiments, identical laser intensity, gain, and detector wavelength settings were used.
For image intensity analysis, a ROI was selected inside each GUV (a circular region from the center to approximately 1/3 of the radius from the membrane); see Figure S13 for a typical ROI selection example. The intensity value for each vesicle was determined using the Measure feature in Fiji (ImageJ). For measurements of the bulk solutions surrounding the vesicles, ROIs were taken from random regions in the image, sufficiently far from GUVs (so as not to have any fluorescence contribution from the vesicles). For bulk measurements, absolute intensity values were compared. For imaging analysis for microfluidic measurements, ROIs within GUVs were selected with the same parameters as described above, with the additional constraint that the ROI does not encompass the vesicle membrane throughout the time-lapse in case the GUV moves. For intensity analysis over the different frames, the plugin Time Series Analyzer V3 in Fiji was used. When GUVs did move throughout the image series, the ROI was adjusted to new positions. For NADH formation, the absolute intensity values were used. For NADPH formation, the normalized ratio between the 405 and 488 nm channels was calculated. Briefly, the signal for each frame at 405 nm was divided by the signal at 488 nm. The value for frame 1 was then used to normalize all subsequent frames. Each vesicle is normalized by its own initial value. For the images displayed in Figure 4d, the processing was done in Fiji. Briefly, background values were first subtracted from each image and then the 405 nm image was divided by the 488 nm image. Values of infinity (x/0) were set to 0. Pixel values exceeding 5 AU were removed using the “Remove Outliers” function. The minimum and maximum pixel values were set to 0 and 5, respectively.
Microfluidic Device Preparation and Operation
Microfluidic devices (scheme in Figure S4) were prepared using the method and design described by Yandrapalli et al.26 In short, PDMS oligomer and curing agent were mixed at a 10:1 ratio and poured over a silicon master wafer (feature height 40 μm), for a final PDMS thickness of approximately 8 mm. The PDMS was then cured at 80 °C for 3 h. The PDMS was cut to size, and holes of 1.5 mm were punched using a Biopsy puncher (Integra Miltex, Kai Medical). The PDMS was bonded to a cleaned glass coverslip by exposing the lower side of the PDMS and the glass slide to air plasma (1 min, 0.5 mbar, Plasma Etch, NV USA). Devices were then left for 30 min at 60 °C.
For use, the microfluidic devices were first filled with β-casein (2 mg mL–1 in Milli-Q) via centrifugation (5 min at 900g) and incubated for 30 min. The device was then connected to a syringe pump (Harvard Apparatus) with a 1 mL Hamilton syringe in withdrawal mode, and Buffer H was added to the inlet reservoir. In this way, the β-casein is washed away, and subsequent solutions/samples were added to the microfluidic trapping device. The following flow rates were used: exchanging casein for Buffer H, 10 μL min–1, 80 μL; adding GUVs, 0.5 μL min–1, 40 μL; exchanging external solution for Buffer H, 1 μL min–1, 40 μL; exchanging Buffer H for Buffer I or J, 1 μL min–1. For the results shown in Figure 2g, Buffers H and J were sequentially exchanged for each other. The time duration for the final exchange varied between experiments and is indicated where relevant.
Acknowledgments
We thank Yi Yang for the provision of the template vector pRDNA3.1-hygro-cyto-iNap1 and Marco Fraaije for assistance and discussion on SthA properties. M.P. and D.J.S. are supported by the NWO Gravitation program (Building a Synthetic Cell) grant 024.003.019. B.P. is supported by ERC Advanced Grant (ABCvolume) grant no. 670578. E.J.E. and D.J.S. are supported by the Dutch Ministry for Education, Culture and Science (OCW), Bonus Incentive Scheme.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00406.
SthA absorbance spectrum over storage (supplementary methods), purity of the protein components employed in our cell-free system (Figure S1), kinetic data for purified Fdh and SthA (Figure S2), linear correlation between encapsulated NADH and fluorescence of LUVs (Figure S3), design of microfluidic chips used for monitoring changes in GUV fluorescence over time (Figure S4), influence of different experimental parameters and environments on NADH formation in GUVs (Figure S5), Fdh cofactor specificity within 400 nm large unilamellar vesicles (Figure S6), luminal intensity of GUVs with and without the iNap1 sensor encapsulated along with NAD+, NADP+, Fdh, and SthA (Figure S7), glutathione reduction in bulk solution by GorA dependent on the NADPH concentration (Figure S8), long-term NADPH dynamics in the presence of the glutathione drain in LUVs (Figure S9), the formation of NADPH in GUVs in the presence of the glutathione drain (Figure S10), stability of the cofactor regeneration pathway inside vesicles (Figure S11), structural integrity of large unilamellar vesicles containing Fdh and NAD+ over time (Figure S12), measuring luminal GUV NADH intensity (Figure S13), primers for cloning (Table S1), buffers used in this work (Table S2), and plasmid maps of Fdh and iNap1 template vectors (PDF)
The authors declare no competing financial interest.
Notes
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supporting Information. All data, code, and materials used in the analysis are available upon request to the lead author.
Supplementary Material
References
- Schmidt S.; Sunyaev S.; Bork P.; Dandekar T. Metabolites: A helping hand for pathway evolution?. Trends Biochem. Sci. 2003, 28 (6), 336–41. 10.1016/S0968-0004(03)00114-2. [DOI] [PubMed] [Google Scholar]
- Pfeiffer T.; Soyer O. S.; Bonhoeffer S. The evolution of connectivity in metabolic networks. PLoS Biol. 2005, 3 (7), 1269–75. 10.1371/journal.pbio.0030228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Li S.; Liu L. Engineering redox balance through cofactor systems. Trends Biotechnol. 2014, 32 (6), 337–43. 10.1016/j.tibtech.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Pollak N.; Dölle C.; Ziegler M. The power to reduce: Pyridine nucleotides - Small molecules with a multitude of functions. Biochem. J. 2007, 402 (2), 205–18. 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Colavitti R.; Rovira I. I.; Finkel T. Redox-dependent transcriptional regulation. Circ. Res. 2005, 97 (10), 967–74. 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- Wang E.; Bauer M. C.; Rogstam A.; Linse S.; Logan D. T.; Von Wachenfeldt C. Structure and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol. Microbiol. 2008, 69 (2), 466–78. 10.1111/j.1365-2958.2008.06295.x. [DOI] [PubMed] [Google Scholar]
- Beaufay F.; De Bolle X.; Hallez R. Metabolic control of cell division in α-proteobacteria by a NAD-dependent glutamate dehydrogenase. Commun. Integr. Biol. 2016, 9 (1), 1–5. 10.1080/19420889.2015.1125052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm A. K.; Blank L. M.; Oldiges M.; Schmid A.; Solem C.; Jensen P. R.; et al. Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J. Biol. Chem. 2010, 285 (23), 17498–506. 10.1074/jbc.M109.095570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heux S.; Cachon R.; Dequin S. Cofactor engineering in Saccharomyces cerevisiae: Expression of a H2O-forming NADH oxidase and impact on redox metabolism. Metab. Eng. 2006, 8 (4), 303–14. 10.1016/j.ymben.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sauer U.; Canonaco F.; Heri S.; Perrenoud A.; Fischer E. The Soluble and Membrane-bound Transhydrogenases UdhA and PntAB Have Divergent Functions in NADPH Metabolism of Escherichia coli. J. Biol. Chem. 2004, 279 (8), 6613–9. 10.1074/jbc.M311657200. [DOI] [PubMed] [Google Scholar]
- Conrado R. J.; Gonzalez R. Envisioning the bioconversion of methane to liquid fuels. Science (Washington, DC, U. S.) 2014, 343 (6171), 621–3. 10.1126/science.1246929. [DOI] [PubMed] [Google Scholar]
- Jeffries T. W. Engineering yeasts for xylose metabolism. Curr. Opin. Biotechnol. 2006, 17 (3), 320–326. 10.1016/j.copbio.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Weckbecker A.; Gröger H.; Hummel W. Regeneration of Nicotinamide Coenzymes: Principles and Applications for the Synthesis of Chiral Compounds. Adv. Biochem Eng. Biotechnol. 2010, 120 (July), 195–242. 10.1007/10_2009_55. [DOI] [PubMed] [Google Scholar]
- Hummel W.; Gröger H. Strategies for regeneration of nicotinamide coenzymes emphasizing self-sufficient closed-loop recycling systems. J. Biotechnol. 2014, 191, 22–31. 10.1016/j.jbiotec.2014.07.449. [DOI] [PubMed] [Google Scholar]
- Wang M.; Chen B.; Fang Y.; Tan T. Cofactor engineering for more efficient production of chemicals and biofuels. Biotechnol. Adv. 2017, 35 (8), 1032–9. 10.1016/j.biotechadv.2017.09.008. [DOI] [PubMed] [Google Scholar]
- Shi T.; Han P.; You C.; Zhang Y. H. P. J. An in vitro synthetic biology platform for emerging industrial biomanufacturing: Bottom-up pathway design. Synth Syst. Biotechnol [Internet] 2018, 3 (3), 186–95. 10.1016/j.synbio.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opgenorth P. H.; Korman T. P.; Bowie J. U. A synthetic biochemistry molecular purge valve module that maintains redox balance. Nat. Commun. 2014, 5 (May), 1–8. 10.1038/ncomms5113. [DOI] [PubMed] [Google Scholar]
- Beneyton T.; Krafft D.; Bednarz C.; Kleineberg C.; Woelfer C.; Ivanov I.; et al. Out-of-equilibrium microcompartments for the bottom-up integration of metabolic functions. Nat. Commun. 2018, 9 (1), 1–10. 10.1038/s41467-018-04825-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley Q. M.; Karim A. S.; Jewett M. C. Cell-free metabolic engineering: Biomanufacturing beyond the cell. Biotechnol. J. 2015, 10 (1), 69–82. 10.1002/biot.201400330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassens N. J.; Burgener S.; Vögeli B.; Erb T. J.; Bar-Even A. A critical comparison of cellular and cell-free bioproduction systems. Curr. Opin. Biotechnol. 2019, 60, 221–9. 10.1016/j.copbio.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander T.; Schada von Borzyskowski L.; Burgener S.; Cortina N. S.; Erb T. J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 2016, 354 (6314), 900–4. 10.1126/science.aah5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer R K; Jungermann K; Decker K Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 1977, 41 (1), 100–180. 10.1128/br.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabba M.; Frallicciardi J.; van ’t Klooster J.; Henderson R.; Syga Ł; Mans R.; et al. Weak Acid Permeation in Synthetic Lipid Vesicles and Across the Yeast Plasma Membrane. Biophys. J. 2020, 118 (2), 422–34. 10.1016/j.bpj.2019.11.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton K.; Wilson T. H. The free-energy changes for the reduction of diphosphopyridine nucleotide and the dehydrogenation of L-malate and L-glycerol 1-phosphate. Biochem. J. 1953, 54 (1), 86–94. 10.1042/bj0540086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer F. Q.; Buettner G. R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol. Med. 2001, 30 (11), 1191–212. 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Yandrapalli N.; Robinson T. Ultra-high capacity microfluidic trapping of giant vesicles for high-throughput membrane studies. Lab Chip 2019, 19 (4), 626–33. 10.1039/C8LC01275J. [DOI] [PubMed] [Google Scholar]
- Mata A. M.; Carmen M.; Lopez-Barea J. Purification by affinity chromatography of glutathione reductase (EC 1.6. 4.2) from Escherichia coli and characterization of such enzyme. Z. Naturforsch., C: J. Biosci. 1984, 39 (9–10), 908–915. 10.1515/znc-1984-9-1009. [DOI] [PubMed] [Google Scholar]
- Henderson G. B.; Murgolo N. J.; Kuriyan J.; Osapay K.; Kominos D.; Berry A.; et al. Engineering the substrate specificity of glutathione reductase toward that of trypanothione reduction. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (19), 8769–73. 10.1073/pnas.88.19.8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov V O; Lamzin V S NAD+-dependent formate dehydrogenase. Biochem. J. 1994, 301 (3), 625–43. 10.1042/bj3010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkov V. I.; Popov V. O. Catalytic mechanism and application of formate dehydrogenase. Biochemistry 2004, 69 (11), 1252–67. 10.1007/s10541-005-0071-x. [DOI] [PubMed] [Google Scholar]
- Gutknecht J.; Walter A. SCN– and HSCN transport through lipid bilayer membranes. Biochim. Biophys. Acta, Biomembr. 1982, 685 (3), 233–40. 10.1016/0005-2736(82)90062-1. [DOI] [PubMed] [Google Scholar]
- Walde P.; Ichikawa S. Enzymes inside lipid vesicles: Preparation, reactivity and applications. Biomol. Eng. 2001, 18 (4), 143–77. 10.1016/S1389-0344(01)00088-0. [DOI] [PubMed] [Google Scholar]
- Guynn R. W.; Gelberg H. J.; Veech R. L. Equilibrium constants of the malate dehydrogenase, citrate synthase, citrate lyase, and acetyl coenzyme A hydrolysis reactions under physiological conditions. J. Biol. Chem. 1973, 248 (20), 6957–65. 10.1016/S0021-9258(19)43346-2. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Kirsch J. F. Identification of functional paralog shift mutations: Conversion of Escherichia coli malate dehydrogenase to a lactate dehydrogenase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (44), 17353–7. 10.1073/pnas.0708265104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger A.; Tsai F. C.; Koenderink G. H.; Schmidt T. F.; Itri R.; Meier W.; et al. Gel-assisted formation of giant unilamellar vesicles. Biophys. J. 2013, 105 (1), 154–64. 10.1016/j.bpj.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RydstroM J.; Hoek J.B.; Ernster L. 2 Nicotinamide Nucleotide Transhydrogenases. Enzym. 1976, 13, 51–88. 10.1016/S1874-6047(08)60240-1. [DOI] [Google Scholar]
- Tao R.; Zhao Y.; Chu H.; Wang A.; Zhu J.; Chen X.; et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 2017, 14 (7), 720–8. 10.1038/nmeth.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman G. L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82 (1), 70–7. 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Rahman I.; Kode A.; Biswas S. K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1 (6), 3159–65. 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- Yoshioka S.; Izutsu K.; Aso Y.; Takeda Y. Inactivation Kinetics of Enzyme Pharmaceuticals in Aqueous Solution. Pharm. Res. 1991, 8, 480–4. 10.1023/A:1015899011324. [DOI] [PubMed] [Google Scholar]
- Polizzi K. M.; Bommarius A. S.; Broering J. M.; Chaparro-Riggers J. F. Stability of biocatalysts. Curr. Opin. Chem. Biol. 2007, 11 (2), 220–5. 10.1016/j.cbpa.2007.01.685. [DOI] [PubMed] [Google Scholar]
- Boonstra B.; Rathbone D. A.; French C. E.; Walker E. H.; Bruce N. C. Cofactor regeneration by a soluble pyridine nucleotide transhydrogenase for biological production of hydromorphone. Appl. Environ. Microbiol. 2000, 66 (12), 5161–6. 10.1128/AEM.66.12.5161-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouri T.; Shimizu T.; Kamiya N.; Goto M.; Ichinose H. Design of a cytochrome P450BM3 reaction system linked by two-step cofactor regeneration catalyzed by a soluble transhydrogenase and glycerol dehydrogenase. Biotechnol. Prog. 2009, 25 (5), 1372–8. 10.1002/btpr.231. [DOI] [PubMed] [Google Scholar]
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; Van Dien S. J.; Rabinowitz J. D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5 (8), 593–9. 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masip L.; Veeravalli K.; Georgiou G. The many faces of glutathione in bacteria. Antioxid. Redox Signaling 2006, 8 (5–6), 753–62. 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- Matsuda T.; Yamanaka R.; Nakamura K. Recent progress in biocatalysis for asymmetric oxidation and reduction. Tetrahedron: Asymmetry 2009, 20 (5), 513–57. 10.1016/j.tetasy.2008.12.035. [DOI] [Google Scholar]
- Claassens N. J.; Sánchez-Andrea I.; Sousa D. Z.; Bar-Even A. Towards sustainable feedstocks: A guide to electron donors for microbial carbon fixation. Curr. Opin. Biotechnol. 2018, 50 (ii), 195–205. 10.1016/j.copbio.2018.01.019. [DOI] [PubMed] [Google Scholar]
- Abu R.; Woodley J. M. Application of Enzyme Coupling Reactions to Shift Thermodynamically Limited Biocatalytic Reactions. ChemCatChem 2015, 7 (19), 3094–105. 10.1002/cctc.201500603. [DOI] [Google Scholar]
- Borba A.; Lairion F.; Disalvo A.; Fausto R. Interaction of nicotinamide and picolinamide with phosphatidylcholine and phosphatidylethanolamine membranes: A combined approach using dipole potential measurements and quantum chemical calculations. Biochim. Biophys. Acta, Biomembr. 2009, 1788 (12), 2553–62. 10.1016/j.bbamem.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Küchler A.; Yoshimoto M.; Luginbühl S.; Mavelli F.; Walde P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11 (5), 409–20. 10.1038/nnano.2016.54. [DOI] [PubMed] [Google Scholar]
- Śmigiel W. M.; Lefrançois P.; Poolman B. Physicochemical considerations for bottom-up synthetic biology. Emerg Top Life Sci. 2019, 3, 445 10.1042/ETLS20190017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcus V. L.; Prentice E. J.; Hobbs J. K.; Mulholland A. J.; Van Der Kamp M. W.; Pudney C. R.; et al. On the Temperature Dependence of Enzyme-Catalyzed Rates. Biochemistry 2016, 55 (12), 1681–8. 10.1021/acs.biochem.5b01094. [DOI] [PubMed] [Google Scholar]
- Szoka F.; Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. 10.1146/annurev.bb.09.060180.002343. [DOI] [PubMed] [Google Scholar]
- Moscho A.; Orwar O. W. E.; Chiu D. T.; Modi B. P.; Zare R. N. Rapid preparation of GUVs. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (October), 11443–7. 10.1073/pnas.93.21.11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleineberg C.; Wölfer C.; Abbasnia A.; Pischel D.; Bednarz C.; Ivanov I.; et al. Light-Driven ATP Regeneration in Diblock/Grafted Hybrid Vesicles. ChemBioChem 2020, 21 (15), 2149–60. 10.1002/cbic.201900774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biner O.; Fedor J. G.; Yin Z.; Hirst J. Bottom-up construction of a minimal system for cellular respiration and energy regeneration. ACS Synth. Biol. 2020, 9 (6), 1450–9. 10.1021/acssynbio.0c00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols T.; Sikkema H. R.; Gaastra B. F.; Frallicciardi J.; Smigiel W. M.; Singh S.; Poolman B. A synthetic metabolic network for physicochemical homeostasis. Nat. Commun. 2019, 10 (1), 1–13. 10.1038/s41467-019-12287-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Meins J. F.; Schatz C.; Lecommandoux S.; Sandre O. Hybrid polymer/lipid vesicles: State of the art and future perspectives. Mater. Today 2013, 16 (10), 397–402. 10.1016/j.mattod.2013.09.002. [DOI] [Google Scholar]
- Luisi P. L.; Ferri F.; Stano P. Approaches to semi-synthetic minimal cells: A review. Naturwissenschaften 2006, 93 (1), 1–13. 10.1007/s00114-005-0056-z. [DOI] [PubMed] [Google Scholar]
- Blain J. C.; Szostak J. W. Progress toward synthetic cells. Annu. Rev. Biochem. 2014, 83, 615–40. 10.1146/annurev-biochem-080411-124036. [DOI] [PubMed] [Google Scholar]
- Sikkema H. R.; Gaastra B. F.; Pols T.; Poolman B. Cell Fuelling and Metabolic Energy Conservation in Synthetic Cells. ChemBioChem 2019, 20 (20), 2581–92. 10.1002/cbic.201900398. [DOI] [PubMed] [Google Scholar]
- Miller T. E.; Beneyton T.; Schwander T.; Diehl C.; Girault M.; Mclean R.; et al. Light-powered CO2 fixation in a chloroplast mimic with natural and synthetic parts. Science 2020, 368 (May), 649–54. 10.1126/science.aaz6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Lindner S. N.; Aslan S.; Yishai O.; Wenk S.; Schann K.; Bar-Even A. Growth of E. coli on formate and methanol via the reductive glycine pathway. Nat. Chem. Biol. 2020, 16 (May), 538. 10.1038/s41589-020-0473-5. [DOI] [PubMed] [Google Scholar]
- Chowdhury A.; Maranas C. D. Designing overall stoichiometric conversions and intervening metabolic reactions. Sci. Rep. 2015, 5 (1), 1–20. 10.1038/srep16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Saba T.; Yiu H. H. P.; Howe R. F.; Anderson J. A.; Shi J. Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways. Chem. [Internet] 2017, 2 (5), 621–54. 10.1016/j.chempr.2017.04.009. [DOI] [Google Scholar]
- Yishai O.; Lindner S. N.; Gonzalez de la Cruz J.; Tenenboim H.; Bar-Even A. The formate bio-economy. Curr. Opin. Chem. Biol. 2016, 35, 1–9. 10.1016/j.cbpa.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Geertsma E. R.; Dutzler R. A versatile and efficient high-throughput cloning tool for structural biology. Biochemistry 2011, 50 (15), 3272–8. 10.1021/bi200178z. [DOI] [PubMed] [Google Scholar]
- Guzman L. M.; Belin D.; Carson M. J.; Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. J. Bacteriol. 1995, 177 (14), 4121–30. 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer P.; Worek F.; Kiderlen D.; Sinko G.; Stuglin A.; Simeon-Rudolf V.; Reiner E. Molar absorption coefficients for the reduced Ellman reagent: reassessment. Anal. Biochem. 2003, 312 (2), 224–7. 10.1016/S0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.