

Summary
We demonstrate early progress toward constructing a high-throughput, single-molecule protein sequencing technology utilizing barcoded DNA aptamers (binders) to recognize terminal amino acids of peptides (targets) tethered on a next-generation sequencing chip. DNA binders deposit unique, amino acid-identifying barcodes on the chip. The end goal is that, over multiple binding cycles, a sequential chain of DNA barcodes will identify the amino acid sequence of a peptide. Toward this, we demonstrate successful target identification with two sets of target-binder pairs: DNA-DNA and Peptide-Protein. For DNA-DNA binding, we show assembly and sequencing of DNA barcodes over six consecutive binding cycles. Intriguingly, our computational simulation predicts that a small set of semi-selective DNA binders offers significant coverage of the human proteome. Toward this end, we introduce a binder discovery pipeline that ultimately could merge with the chip assay into a technology called ProtSeq, for future high-throughput, single-molecule protein sequencing.
Subject areas: Biochemistry, Biochemistry applications, Sequence analysis, Proteomics, Transcriptomics
Graphical abstract
Highlights
-
•
Designed ProtSeq protein sequencing method compatible with widely used NGS technology
-
•
Built Target-Switch SELEX to isolate aptamers specific to N-terminal amino acids (AAs)
-
•
Showed binding, ligation, cleavage, and NGS of six DNA binders in ordered barcode chain
-
•
Developed pipeline to deconvolve AAs from DNA barcodes to identify putative proteins
Biochemistry; Biochemistry applications; Sequence analysis; Proteomics; Transcriptomics
Introduction
Rapid improvements in DNA and RNA sequencing technology over the last decade have resulted in a wealth of molecular information. Although DNA sequencing captures a cellular blueprint, genomic data cannot capture the layers of information transmitted from DNA through transcription and translation. Similarly, RNA sequencing yields information on transcriptional activity and mRNA production, but mRNA levels are not strictly correlated to protein levels. Instead, protein levels are regulated by a multitude of post-transcriptional and post-translational mechanisms (Payne, 2015; Haider and Pal, 2013). Current DNA and RNA sequencing technologies therefore do not provide concrete, high-throughput information on cellular protein composition. High-throughput, whole-proteome protein sequencing may allow identification of proteoforms associated with different cellular states and produce insight into processes like translational fidelity, post-translational modifications, and proteoform dynamics in cells and subcellular compartments.
Existing methods of identifying amino acid (AA) residues are limited by instrument resolution and sample size in the case of mass spectrometry (MS) (Sheynkman et al., 2016), throughput in the case of high-pressure liquid chromatography (Pham et al., 2003), and the inability to account for large-scale mutations that create gene structures unique to an individual in the case of template proteogenomics (Castellana et al., 2010).
Several approaches toward single-molecule proteomics are currently being explored (Alfaro et al., 2021), including nanopore technologies that rely on variations in ionic current during passage of a peptide through a pore, although complexities due to the diversity of AA mass, charge, and configuration limit current usage of these approaches (Nicolaï et al., 2020, Hu et al., 2021). Near single-cell techniques such as nanoPOT have been successful with characterizing nanoliter volumes containing as few as 10 cells using MS (Zhu et al., 2018; Williams et al., 2020) but are limited by MS detection for single-molecule readouts. Another approach, termed “fluorosequencing,” utilizes single-molecule imaging of arrayed peptides with fluorescently labeled N-terminal AAs followed by Edman degradation to cleave off terminal AAs (Swaminathan et al., 2015). This approach has recently been shown to work on cysteine and lysine residues (Swaminathan et al., 2018) and would enable identification of many proteins (Yao et al., 2015). The proposed technology of Swaminathan et al. would overcome many existing hurdles to large-scale protein sequencing, although this imaging-based approach currently relies on expert chemists to develop enough unique binders to unmodified and modified AAs for protein identification and enough multi-cycle Edman resistant fluorescent dyes to label those binders. Although there are other technologies in development, including improvements in MS detection, current experimental methods are not yet capable of true single-molecule sequencing of proteins or complexes that are unknown or expressed at low levels (Slavov, 2021; Timp and Timp, 2020).
To address current experimental limitations to exploring the proteomic landscape, computational models can be used to predict protein structures and therefore putative protein functions (Senior et al., 2020). Although potentially extremely impactful, current robust modeling methods require either protein sequence or crystal structure as an input, thus limiting the space of application across the human proteome. Both currently available experimental and computational methods fall short at capturing protein function and cell state across the human proteome with the same ease as DNA and RNA sequencing.
We developed components of the aspirational protein sequencing platform called ProtSeq to address the limitations to existing technologies, including the discovery of binders to AAs for protein identification. Ultimately, the ProtSeq platform may be particularly useful for single cells or small blood volumes, proteins with low expression, and single AA mutations, where the goal is to understand complex disease phenotypes. In addition, the envisioned ProtSeq approach allows for sequencing of many samples simultaneously, since samples can be barcoded and proteins with high expression can be filtered to enhance the signal from peptides with low levels of expression.
ProtSeq DNA-peptide binding platform: design and construction
Barcoded binders identified DNA targets and peptide targets through generation of a barcode chain
In this section we first describe the design and conceptualization of technologies required for ProtSeq. Below, we present the design of “Barcode Cycle Sequencing” (BCS), a cyclic method for converting an AA sequence into a DNA sequence using binders to unique AAs (Table 1). In the results we detail (1) single-molecule imaging to assess the signal-to-noise ratio during each stage of AA identity capture, (2) a binder discovery pipeline called “Target-Switch SELEX″ to facilitate discovery of binders to unique AAs for use in BCS, and (3) the sequencing output produced by BCS processed through a computational pipeline to infer likely target matches based on the binder-associated DNA barcode. ProtSeq was built to scale, so that in the future, proteome database matching and sequencing of multiple samples may be possible applications.
Table 1.
Glossary of terms
| Term | Definition |
|---|---|
| AA | Amino acid |
| Backbone | Short AA sequence composed of AAs from a defined group of residues, used in Target Switch SELEX |
| Barcode | DNA sequence conferring unique identity of component (e.g., aptamer, peptide, foundation) |
| Barcode chain | Foundation ligated to multiple binder barcodes |
| Barcoded binder | Binder containing DNA elements for BCS (ligation spacer, binder barcode, restriction site spacer, 5-T nucleotide spacer) |
| BCS | Barcode Cycle Sequencing, assay introduced in this paper for capturing, recording, and sequencing binding events on an NGS chip |
| Binder | Entity that binds the target, may refer to a variety of molecules, including aptamers, proteins, peptides, DNA, nanobodies, small molecules |
| Bridge | ssDNA oligonucleotide that facilitates ligation between the binder and foundation |
| CLR target | ssDNA target attached to a P7 with no complementary binder; utilized as a false-positive binding control |
| Colocalization linkers | Pair of ssDNA oligonucleotides (forward and reverse) used in target-foundation deposition on the NGS chip |
| Empty target | No modification to P7 adapter on chip |
| Foundation | ssDNA oligonucleotide (containing a target-specific barcode) onto which binder barcodes are ligated |
| MS | Mass spectrometry |
| NGS | Next-generation sequencing |
| P5 & P7 | ssDNA sequences that allow DNA sequences to bind and generate clusters on Illumina DNA sequencing flow cells |
| POC | Peptide-oligonucleotide conjugate, peptide with C-terminal linkage to a short oligonucleotide |
| PP-C | PP dipeptide followed by the C backbone |
| PP-CD | Alternating target between PP-C and PP-D backbone |
| ProtSeq | Protein sequencing method introduced in this paper that utilizes BCS and Target Switch SELEX |
| SELEX | Systematic Evolution of Ligands by EXponential enrichment |
| ssDNA | Single-stranded DNA |
| Switch and non-switch protocols | Target Switch SELEX protocols associated with different stringency gradients |
| Target | Entity to which the binder binds, may refer to a variety of molecules, including peptides, proteins, DNA, small molecules |
| Target Switch SELEX | Aptamer-discovery method introduced in this paper for discovering N-terminal dipeptides |
| 5Phos target | 5′ Phosphorylated foundation attached to a P7; utilized as a false-positive DNA ligation/encoding control |
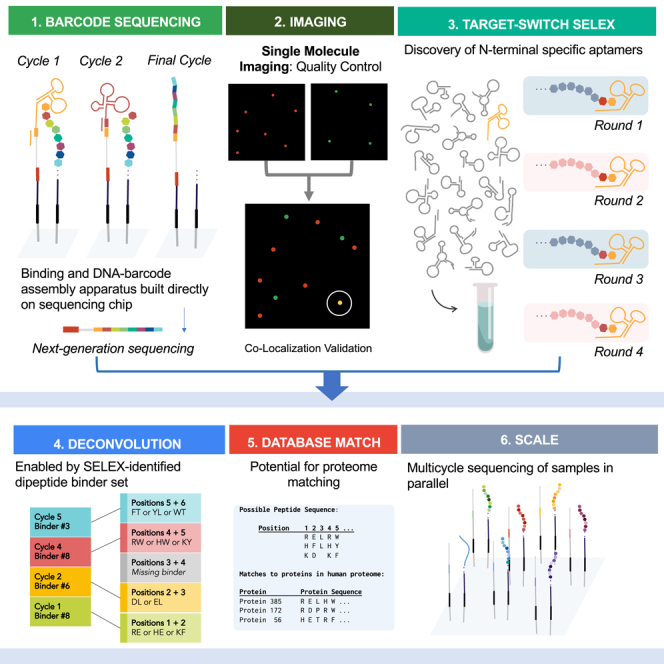
The BCS assay was a binding platform that recorded interactions between DNA-barcoded targets displayed on the NGS chip and DNA-barcoded binders flowed onto the chip. “Targets” and “binders” were illustrated as peptides and aptamers but may refer to any combination of target-binder pairs, including aptamers, proteins, peptides, cDNA, and nanobodies. Aptamers are short, single-stranded DNA (ssDNA) molecules that fold into unique conformations to allow for binding specificity to biological targets such as proteins and peptides. Each target was displayed near a short ssDNA sequence referred to as a “foundation” (Figures 1.0 and 1.1), onto which a chain of sequential DNA barcodes, or a “barcode chain” (Figure 1.7), would be constructed. MiSeq chips contain two different ssDNA sequencing adapters, “P5” and “P7” (Illumina). Foundation barcodes were deposited to a P7 sequence in close proximity to the target via ligation (Figure 1.1B), and two assisting DNA “cololinkers” were washed away (Figure 1.1C). “Barcoded binders,” binders with DNA barcodes attached, were flowed onto the chip to bind to displayed targets (Figure 1.2). Upon binding, the binder's DNA barcode was transferred from the binder onto a nearby foundation, first by ligation of the barcode to the foundation (Figure 1.3), followed by restriction enzyme cleavage to release the binder (Figure 1.4). Enzymatic cleavage created a new ligation site onto which the unique barcode of the next binder could be ligated. Although not included in BCS experiments discussed below, a degradation step (Figure 1.5) would be included to reveal sequential N-terminal amino acids in the finalized version of ProtSeq. In subsequent binding cycles, barcoded binders continued to transfer their barcodes by ligation to the cleavage site of the previous binder (Figure 1.6) to generate a barcode chain (Figure 1.7). After completion of all binding cycles, the barcode chain was sequenced. Binder barcodes and their positions in the chain were used to reconstruct the target based on the binding profile associated with each barcode. Buffer solutions used in each step are listed in Table S1.
Figure 1.

Barcode Cycle Sequencing (BCS): A strategy for converting amino acids into DNA barcodes directly on a next-generation sequencing chip
This schematic depicts the seeding of foundations and subsequent per round barcode capture. Step 0 depicts the off-chip construction of a target-foundation complex to ensure colocalization between the foundation and target, as described in Figure 2. Step 1 includes the tethering of the peptide-foundation complex onto solid substrate on the flow cell. Step 2 includes incubating the bound proteins or peptides with a barcoded binder library under conditions that allow the appropriate aptamer to bind specifically to the appropriate N-terminal amino acid. Step 3 includes ligating the aptamer tail to a second oligonucleotide bound to the substrate. Step 4 includes cleaving off the binder, leaving the DNA barcode associated with that particular amino acid bound to the second oligonucleotide. For a full-fledged ProtSeq technology, after or at the same time with binder removal, Step 5 would consist of a degradation step in which the terminal amino acid is cleaved. After a washing cycle, Steps 2–5 are repeated, generating a chain of DNA barcodes that reflect binding events to the colocalized target. Refer to Figure S8 for alternative methods.
Chip-based binding assay required multiple DNA components
The BCS platform was built directly onto the Illumina Miseq v2 Nano and MiSeq v3 NGS sequencing chips. Each target-foundation pair was displayed through ligation to two nearby P7 adapters.
To achieve colocalization between a target and its foundation on the chip, the target and foundation were first linked together in solution using a pair of ssDNA “colocalization linkers,” where the “forward cololinker (FC)” had complementarity to the foundation, the “reverse cololinker (RC)” had complementarity to the target, and both cololinkers had a region of complementarity to each other (Figure 2A). This pre-formed target-foundation complex was then flowed onto the chip at 30 mL of 120 pM solution and guided to dock onto P7 adapters via regions of complementarity within the cololinkers (Figure 2B). The 5′ ends of the foundation and target were then ligated to two P7 adapters, and the cololinkers were washed twice with pure formamide, leaving behind the target and foundation tethered to two spatially associated P7 adapters (Figure 1.1C).
Figure 2.

Barcode Cycle Sequencing (BCS) components: a strategy for converting amino acids into DNA barcodes directly on a next-generation sequencing chip
(A) Foundations are assembled off-chip. Two cololinkers that are partially complementary to each other and complementary to P7 adapters are used to link a barcoded foundation with the oligo-tethered target to be sequenced. Experimentally, this target may consist of DNA, a Spot-Tag with residues, or a 10-mer peptide.
(B) Targets and foundation barcodes are deposited in close proximity on the sequencing chip by ligating the target and foundation barcode to proximal P7 adapters on the sequencing chip. The cololinkers allow the foundation barcode and target to localize to adapters in close proximity. Cololinkers are washed away prior to binding events and no longer present.
(C) Depicts the gray region of B in detail. The 5′ end of the oligo portion of each binder contains a restriction site spacer, which is hybridized to a complementary universal bridge. The bridge provides a double-stranded substrate that the restriction enzyme can act upon. Full sequences can be found in Table S2 and molecular details in Figure S2.
After target and foundation deposition, barcoded binders were flowed onto the chip (Figure 1.2). Upon binding, the 5′ end of the barcoded binder and the 3′ end of the foundation were ligated together with assistance from an ssDNA “bridge” sequence designed to bring the barcoded region of the binder into close proximity with the foundation (Figure 2B). After ligation, the binder was cleaved with restriction enzyme EcoR1, leaving behind the assigned DNA barcode attached to the foundation (Figure 1.4), and the next binder set was introduced to repeat the cycle (Figure 1.6).
Building the BCS assay directly on an NGS chip required consideration of several elements, including (1) reducing spatial separation between a target and its foundation to increase signal, (2) maximizing separation between different targets to reduce noise, (3) loading as many targets as possible to optimize readouts per run, and (4) avoiding overclustering of DNA barcodes during NGS sequencing to prevent sequencing failure. Numerous unit tests of the foundation, cololinkers, bridge, and barcoded binders contributed to the final design of BCS components described in the subsections below. Sequences for foundation, cololinkers, and bridge sequences are provided in Table S2.
Building the BCS assay on Illumina MiSeq chips allowed for compatibility with NGS
We elected to build the assay directly on a single Illumina sequencing chip to ensure compatibility with widely available industry standard NGS technology. MiSeq chips were selected for purposes of development due to relatively affordable kit and sequencer costs and small loading volume, although the assay has been designed to generalize to other chip types (NextSeq, etc.). P7 was chosen as the DNA adapter for tethering both the target and the barcode chain due to single-molecule imaging findings that showed that the P5 adapter was removed upon exposure to TFA, a chemical required for one approach to single-AA cleavage of a peptide target (Figure S1).
Foundation barcode sequences provided a base for DNA barcode chain deposition
We built barcode chains onto a foundation sequence, as opposed to non-specific nearby P7 adapters, to (1) ensure compatibility with NGS technology by avoiding multiple species in a single sequencing cluster, (2) tune the separation between target and barcode to maximize probability of barcode deposition, and (3) design the capability to transfer a target barcode to the base of the barcode chain to debug the system and ultimately scale for multiplexing of different samples. Binder barcodes were ligated onto a 31-nt foundation containing (from 5′ to 3′) a 16-nt foundation base, an 8-nt foundation barcode, and a 7-nt bridge-binding sequence. Foundation barcodes were designed in various lengths. Barcode sequences were designed to have similar GC content, avoid four nucleotide repeats, and possess a hamming distance of two or greater. We observed that different 8-nt foundation barcodes had differing efficiencies of target deposition and binder barcode capture. Ultimately, four foundations that demonstrated consistency in target deposition and rate of binder barcode capture were selected.
Cololinkers assisted in the formation of a spatially localized foundation-target complex
The forward and reverse cololinkers and their regions of complementarity are shown in Figure 2A. Cololinkers were optimized for length, sequence, T-spacers, and ratio of forward to reverse cololinker. The forward and reverse cololinkers were both 100 nt long. From 5′ to 3′, the forward cololinker contained a 16-nt foundation base (complement), 20-nt P7 complement, 44-T nucleotide spacer, and 20-nt region for hybridization with the reverse cololinker. From 5′ to 3′, the reverse cololinker contained a 20-nt target base (complement), 20-nt P7 complement, 40-T nucleotide spacer, and 20-nt region for hybridization with the forward cololinker.
Unique barcodes on targets and binders were used to identify the target-binder pair
Targets (shown in Figure 2A as a DNA target, Spot-Tag target, or peptide-oligonucleotide conjugate target) contained a 40-nt DNA region consisting of from 5′ to 3′ a 20-nt target base, 21- or 25-nt DNA spacer, and the target. Barcoded binders contained a series of elements allowing for identification of the binding sequence, binding cycle, and position on the chip. A “barcoded binder” refers to a binder attached to a DNA sequence containing the following elements, from 5′ to 3’: a 9-nt ligation spacer, an 8- or 12-nt binder barcode unique to a particular binder sequence and binding cycle, a 24-nt restriction site spacer containing an EcoR1 cleavage site, a 5-T nucleotide spacer, and the binding region.
Bridge sequence facilitated proper encoding between binder barcode and foundation
The ssDNA “bridge” sequence was designed to bring the binder barcode and foundation into close proximity through complementarity to the 5′ end of every barcoded binder and the 3′ end of every foundation. From 5′ to 3′, the bridge contained a 24-nt restriction site spacer (complement), an 8- or 12-nt universal bridge, a 9-nt ligation spacer (complement), and a 7-nt overhang. The universal base was designed with 5-Nitroindole, a universal base analogue that exhibits high duplex stability and hybridizes indiscriminately with each of the four natural bases (Loakes and Brown, 1994) to allow for permissive binding to any binder barcode.
Direct DNA sequencing on the BCS chip required customized steps
To prepare for sequencing, we ligated a custom NGS adapter with 5′ P5 complementarity directly onto barcode chains. To facilitate preferential ligation of the NGS adapter to barcode chains, we incorporated a 16-nt NGS ligation bridge containing a 7-nt complementary region to the binder ligation spacer and a 9-nt complementary region to the NGS sequencing adapter. In order to run a DNA sequencing assay on a chip with a pre-loaded library, we reprogrammed the sequencer to skip initial chip washing steps to prevent the library from being removed from the chip prior to sequencing.
Results
Here we demonstrated the ability to capture and record binding events on two types of binder pairs, DNA-DNA binders and peptide-nanobody binders. For DNA-DNA binders, we demonstrated the ability to record six consecutive binding events. In parallel, we conducted simulations to determine the binder characteristics (e.g., binding specificity) that would provide optimal coverage of the human proteome and built a BCS-compatible aptamer discovery pipeline called Target-Switch SELEX to find binders with those ideal characteristics.
Ideal binder-target system using a DNA-DNA binding pair showed success in stages of BCS assay
We ran the BCS assay with a set of DNA sequences (binders) and their complementary sequences (targets). Our purpose was to use an ideal binder-target system to characterize binding kinetics and binder-target specificity, as well as develop an internal binding-affinity ladder utilizing DNA-DNA binding pairs for future experiments. The binding assay had three main steps: (1) deposition of the target colocalized with its uniquely labeled foundation on the chip, (2) capture of target-binder interaction via barcode ligation to the foundation, and (3) restriction enzyme cleavage of the DNA binder.
Single-molecule imaging confirmed target-foundation deposition on the chip
Single-molecule imaging allowed us to visualize foundation-target colocalization on the sequencing chip and the impact of varying ratios of forward to reverse cololinker on in-solution assembly of the foundation-target complex. To characterize the effect of forward:reverse cololinker ratio on colocalization, we labeled foundations and targets with fluorescent Alexa 488 and ATTO 647, respectively (sequences in Table S3). We then assembled foundation-target complexes in solution using forward:reverse cololinker ratios of 1:0.5, 1:1, 1:3, and 1:5 and visualized the complexes under TIRF microscopy averaged across five imaged areas. In two separate experiments, we observed peak colocalization between foundations and targets using the 1:1 forward to reverse cololinker ratio (64.2% ± 7.9% and 61.77% ± 7.01%), compared with 1:0.5 (24.7% ± 6.8%), 1:3 (61.2% ± 5.9% and 57.7% ± 4.0%), and 1:5 (61.2% ± 5.9% and 55.5% ± 4.4%) (Figure 3).
Figure 3.

Single-molecule imaging of target-foundation colocalization
(A) Single-molecule images of RC coupled with Atto 647 for three different FC:RC ratios 1:0.5, 1:1, and 1:3. Imaging exposure time was 500 ms with a laser output of 7 mW. Scale bars are 10 μm.
(B) Single-molecule imaging demonstrating colocalization of FC:RC at ratio 1:1. Scale bars are 10 μm.
(C) Microscopy reveals efficient co-localization of barcode foundations with peptide targets on the BCS chip. Colocalization efficiency at the different cololinker ratios where 1:1 possessed the highest efficiency. Two experiments were performed and five tiles each were analyzed. Error bars represent standard error. A ratio of 1:1 (FC:RC) was selected for BCS experiments to ensure the highest possible number of ligated targets had an associated foundation.
Binder barcodes ligated with high fidelity to their associated foundations
We demonstrated over a single cycle that binders ligated to the foundations associated with their respective targets. The DNA binder-DNA target validation assay demonstrated a significant difference between binder barcodes captured correctly at their target sites compared with binder barcodes detected at non-target and incorrect target sites. Oligonucleotide sequences for DNA binders and DNA targets used in all binding experiments are listed in Table S4. For DNA Target 6, we discovered a log2 fold change of 6.02 and 8.25, respectively, for non-target and incorrect target sites (Figure 4). For DNA Target 9, we discovered a log2 fold change of 5.55 and 6.71, respectively, for non-target and incorrect target sites (Figure 4). Negative controls were barcoded binders with no complementary targets. “Non-targets” tested were targets containing only the target base and DNA spacer (no binding region), with one foundation-only replicate. Binders and negative controls were tested against targets and non-targets in quadruplicate. The DNA Binder 6-DNA Target 6 and DNA Binder 9-DNA Target 9 pairs produced 19,663 ± 7,394 counts and 27,211 ± 25,621 counts averaged across four replicates, respectively, which exceeded counts for incorrect and non-targets (all counts listed in Table S5). Despite <10 total capture events for both negative controls across all foundations, we did observe a small degree of off-target capture between incorrect and non-targets, and DNA Binder 6 and DNA Binder 9. This indicates that, upon binding, barcodes may occasionally ligate to an incorrect nearby foundation or that some exchange of target-foundation pairs takes place during incubation prior to deposition on the chip. Overall, results indicated that binders ligate with high fidelity to the foundations associated with their bound targets over a single BCS cycle.
Figure 4.

BCS performance for single cycle DNA target-binder pairs
This dot plot shows that a DNA control binder is specific for its cDNA target, a region of 16 (DNA Target 6) or 24 nt (DNA Target 9), and successfully barcodes correctly for a single cycle of BCS. Two negative controls were used, where a DNA binder was applied with no target (DNA Binder 4.1 and DNA Binder 4.2). Two DNA binder-target pairs were used (DNA Target 6 to DNA Binder 6 and DNA Target 9 to DNA Binder 9) demonstrating robust binding and DNA-barcode encoding in a single cycle with multiple replicates on a single chip. Table of counts included in Table S5.
Cleavage and ligation over multiple cycles demonstrated capture of six sequential binder barcodes
Using a DNA-DNA binding pair system, we demonstrated successful deposition of barcoded binders alongside their corresponding foundations, ligation of binder barcodes to those foundations, sequential ligation of binder barcodes onto a growing foundation over multiple cycles of binding, and reconstruction of the original binders and their targets through computational analysis.
Over multiple cycles, we demonstrated successful sequential capture of six barcodes on the foundation (Figure 5A). See Figure S2 for schematic of barcode chain construction and Table S6 for exact sequencing counts. As a measure of the overall ability of binders to distinguish their intended target from all other targets and null controls, we calculated the distribution of all binders across all foundations and found that the proportion of correct barcoding events ranged from 62% to 78% (Figure S3). When we calculated the per-cycle dropout of exact matches for the highest performing binder-target pairs, we observed an exact-match barcoding efficiency of 58%, as determined by the exponential decay rate of perfect matches observed in DNA Binder 10 and DNA Binder 11 (Figure 5B). Two of the binder-target pairs showed a strong drop in performance at cycle 3. To confirm that the perfect match decay rate was an accurate predictor of cycle efficiency, we also inspected the per-cycle signal and noise and found that, although the signal is relatively constant (with the exception of DNA Binder 12 and DNA Binder 13 in cycle 3) across cycles, the noise increases in a cycle-wise fashion.
Figure 5.

BCS performance for multiple cycle DNA target-binder pairs
(A) Histogram reporting the counts of reads of barcodes added in an experiment with six cycles of barcode ligation for four DNA-DNA binder pairs. Each DNA barcode within the chain encoded for an expected position based on cycle number. “False” is defined as any binder other than the corresponding binder-target pair appearing with the listed foundation. Cycle consistency of the DNA-DNA binder experiment shows roughly uniform matching counts across all cycles of DNA Target 10 and DNA Target 11. Results confirm it is possible to achieve serial ligation of six barcodes in the expected positions. Table of counts is included in Table S6 and analysis of barcode identification after six cycles in Figure S3.
(B) When constrained to perfect matches (i.e., the expected target at each cycle up to a certain cycle), there is an exponential drop-off in binding for DNA Binder 10 and DNA Binder 11 that inversely correlates to cycle number.
Transition from ideal DNA hybridization pair to peptide-nanobody pair revealed successful binding capture
We then transitioned to use of a peptide-nanobody binding system, where peptides were displayed as “peptide-oligonucleotide conjugates” (POCs) via C-terminal linkage to a short oligonucleotide tethered to the chip. We used a high-affinity Spot-Tag binder system composed of a 12-AA Spot-Tag and anti-Spot-Tag nanobody engineered to recognize the Spot-Tag specifically and with high affinity (Kd 6 nM) (Virant et al., 2018). We chose this nanobody for small size (15 kDa), stability, and commercial availability. The C-terminal recognition motif LPETG is intended for site-specific sortase-mediated conjugation to small molecules, and conjugation of nanobodies to oligonucleotides has been demonstrated without affecting nanobody functionality (Fabricius et al., 2018).
In order to control for the potential effects of foundation barcode sequences, each of the targets was associated with multiple different foundations (six replicates for the Spot-Tag, three for all others). Target sequences and their associated foundations are listed in Table S7. Using a barcoded Spot Nanobody as the binder, we demonstrated specific barcoding of its corresponding peptide target log2 fold change of 4.29 above that of non-Spot-Tag associated foundations (Figure 6). In addition, no binder ligation was observed in the absence of Spot-Tag conjugation indicating that nanobody ligation specificity was independent of the foundation sequences. However, the overall binding rate was lower for the barcoded nanobody compared with the DNA-DNA control (sequencing counts listed in Table S8).
Figure 6.

BCS performance for protein-peptide binder system spot-tag
Spot-Tag binding performance counts demonstrated enrichment of nanobody-peptide binding on BCS. Experiments are run in replicates with different barcodes associated for each replicate. Difference in sequencing counts between experimental replicates is thought to be due to the difference in barcode used for each replicate. The impact of barcode sequence was screened and analyzed to derive a set of barcodes used for downstream experimentation. No known variables (GC content, sequential base pairs, etc.) were found to be related to a barcode's impact on sequencing noise outside of target type (DNA versus nanobody, etc.). Experiments were repeated and validated, confirming the protocol utilization for a DNA-DNA binding system and peptide-nanobody binding system. Table of counts is included in Table S8.
Our nanobody experiments demonstrated appropriate deposition of POCs and foundations onto the chip, binding of DNA-barcoded nanobodies to their peptide targets, and capture and sequencing of nanobody binder barcodes.
Aptamer binder set presents a potential avenue for protein sequencing
After separate successes with DNA and protein binders, our focus shifted toward developing an initial aptamer binder set suitable for protein identification, with the intention of applying the same techniques toward the development of a larger and more specific binder set for protein sequencing. A single experiment testing a published aptamer (Kd = 500 pM) (Tasset et al., 1997) for thrombin (sequences in Table S4) demonstrated putative enrichment of aptamer binding over controls on the BCS platform (Table S9). The thrombin protein had seven POC-binding sites, so without additional controls, enrichment could not be fully attributed solely to aptamer-protein binding. Nevertheless, this preliminary finding suggested that aptamers could be a viable avenue for creating a binder set compatible with the BCS platform.
Although modified aptamers called SOMAmers have been used in protein profiling, aptamers have not yet been discovered specifically for protein sequencing (Kim et al., 2014). Based on the work of several research groups that have isolated aptamers against free AAs (arginine [Geiger et al., 1996], phenylalanine [Cheung et al., 2019], tryptophan [Yang et al., 2011]), we believe that aptamers could be used to create a set of binders for protein identification, and eventually protein sequencing.
Simulation of a small set of semi-selective binders provided significant coverage of the human proteome
We built a theoretical model to determine the properties required of the aptamer binders to assess different levels of proteomic coverage. In our simulation, each binder had a determined binding profile that included information on the specificity of each binder to N-terminal dipeptide AA targets (represented graphically in Figure S4). After each binding event, a DNA barcode remained and an AA was removed, resulting in the construction of a DNA barcode chain. The possible AA sequence of the peptide was determined from an algorithmic review of the barcodes in sequence. The probable full-length protein was derived by identifying a barcode sequence corresponding to a distinctive amino acid sequence. The scaffolded sequences were then aligned against a proteome map to identify known proteins.
The results demonstrated that different binder specificities could provide vital information for a range of resolutions spanning the proteome. The strategy for ProtSeq was to increase the signal-to-noise of each binding event by designing aptamers to a dipeptide, rather than to a single AA. In our simulation, each dipeptide aptamer binding event provided a set of guesses for the identity of the two N-terminal AAs while each round of degradation only removed one AA. This allowed each AA, except the original N-terminal and C-terminal AAs (which were only read once), to be read by two rounds of aptamer binding (Figure 7A). When selecting aptamers to bind to the N-terminal dipeptide, our simulation showed that extremely specific aptamers were not necessary to match and rank peptides to sequences in comprehensive protein sequence databases.
Figure 7.

A binder set for the algorithmic identification and coverage of the human proteome
(A) A dipeptide aptamer binder provides putative identities for the two N-terminal amino acids. As each round of Edman degradation removes only one amino acid, each amino acid (except the original N-terminal amino acid) is exposed to two rounds of aptamer binding, enabling algorithmic identification of individual residues based on overlap between likely candidates identified across two rounds.
(B) A simulation predicts that a small set of semi-selective binders offers significant coverage of the human proteome. Binder sets of various sizes and selectivity were evaluated to see what percent of the proteome could be identified. In the simulation, each binder in a set binds to a sample of the 400 possible dipeptides (20 possibilities for two N-terminal amino acids). A protein is identified if the barcode series for a sequenced fragment is unique. See the text for details of the simulation. For each actual binding set, the real-world performance would be contingent on the set-specific binding characteristics (or parameters).
The simulation was grounded in the following tenets: (1) each given protein was digested and cleaved into fragments at each lysine, (2) each protein was considered identified when one of its fragments had a distinct barcode match in the proteome, (3) dipeptides recognized by a given binder set (composed of between 1 and 100 binders) were randomly chosen out of 400 combinations, (4) 20 randomly sampled sets of binders were selected for each combination of dipeptides bound and number of binders (i.e., 250 dipeptides recognized by 50 binders) and the percentage of the proteome identified was averaged across those 20 scenarios, and (5) 11 cycles of terminal AA degradation were performed. The simulation did not model noise (e.g., binders failing to bind or binding incorrectly). In the experimental system, some noise would be mitigated by the redundancy in dipeptide reads and by reading multiple copies of the same protein.
We simulated the estimated percentage of the human proteome potentially identifiable for binder sets consisting of 1–100 binders, where each binder bound up to 400 different dipeptides (Figure 7B). Post-translational modifications and protein isoforms were ignored. Results showed the estimated percentage of the human proteome potentially identified by a given binder set. For 20 AAs, there were 400 possible dipeptide combinations. For the purpose of this simulation, aptamers within the same binder set had the same specificity, where specificity was defined by the number of dipeptides bound by a single binder. A binder that bound to only one dipeptide was “perfectly specific,” and a binder that bound to all 400 dipeptides was “perfectly non-specific.”
This simulation showed that a set of ten binders, each with specificity for nine dipeptides, could potentially identify 90% of proteins in the human proteome defined by UniProtKB/Swiss-Prot (What is UniProt's human proteome?, 2019), where there is one canonical protein per protein-encoding gene. Relative quantification of protein/peptide concentrations in the sample can be calculated from the number of derived peptide sequences associated with those proteins or peptides. If binders had perfect affinity for one dipeptide each, approximately 75 binders would be needed to achieve the same percentage of proteome coverage. Conversely, if all binders bound non-specifically to 300 dipeptides, even a binder set with 100 aptamers would be unable to reach the same percentage of proteome coverage. The simulation showed that even a small set of approximately ten binders could identify most proteins if each binder had specificity for a small group of dipeptides (<10). These results suggested that aptamers with moderate binding specificity and selectivity would enable us to accurately quantify mixtures of known proteins with relative ease. In the event of AA identification errors, downstream computation algorithms would be used to correct or detect inaccurate readbit results with a certain level of confidence. Real-world performance would depend on the actual binding characteristics of a real binding set, where measurements of the affinity and specificity would be used as inputs to the simulation. The simulation described above applies to protein identification. However, the same process could be utilized for protein sequencing with a larger set of binders possessing greater specificity.
Target-Switch SELEX: an approach to developing N-terminal dipeptidase-specific aptamers
Initially motivated to develop a small binder set with the potential for a high proteomic readout yield, we designed a binder discovery pipeline for creating specialized aptamers that bind N-terminal dipeptides. Dipeptides were chosen as binding targets, as opposed to single AAs, to provide built-in redundancy during the reading process to allow each peptide (except for the terminal peptide) to be read twice over rounds of single AA degradation. Aptamers are generated through an in vitro process of directed evolution, termed systematic evolution of ligands by exponential enrichment (SELEX) (Ellington and Szostak, 1990; Tuerk and Gold, 1990), in which a diverse, random DNA library is incubated with a target molecule and screened for binding to the target over multiple rounds of selection. SELEX is the primary method for discovering aptamers and generated aptamers for cells (Shangguan et al., 2006), proteins (Liu et al., 2011), and small molecules (McKeague and DeRosa, 2012). AAs are complex targets, with similarities in both size and chemical structure. Thus, developing site-specific aptamers capable of recognizing only N-terminal AAs within the context of a protein or peptide remains a significant challenge (Ruscito et al., 2017).
Many SELEX variations have sought to increase target specificity (White, R., Rusconi, C., Scardino, E., Wolberg, A., Lawson, J., Hoffman, M., Sullenger, B., 2001; Jenison et al., 1994) through counter or subtractive selection (Jenison et al., 1994), where the pool of aptamers is challenged against a structurally similar alternate target and depleted. One example, the “toggle”-SELEX method, alternated between two targets in different rounds of selection to identify aptamers to conserved motifs on both human and porcine thrombin or a unique motif on only human thrombin (White et al., 2001).
To address the lack of binders to N-terminal dipeptides, we developed a SELEX method, Target-Switch SELEX, designed to isolate aptamers specific to two consecutive N-terminal AAs. Target-Switch SELEX differs from other SELEX methods in two key ways: (1) a target “switch,” which contains the same N-terminal dipeptide and differing remainders of the sequence, and (2) multiple semi-automated selections run in parallel, including both multiple targets and multiple independent selections per target. The Target-Switch methodology incorporated aspects of toggle-SELEX and counter-SELEX in order to isolate aptamers to a specific portion of a target regardless of the surrounding environment. Furthermore, analyzing the sequenced output of each selection round across multiple parallel selections allowed comparison of enrichment across and within selections. These differences in our SELEX workflow allowed us to identify multiple aptamers enriched for targets with the N-terminal dipeptide Proline-Proline with a measured Kd (Jarmoskaite et al., 2020) of 3.65 μM (Figure 9C).
Figure 9.

Aptamer 4 Kd and backbone dependence
(A) Top ten sequences for each selection for each target. Two selections each were performed for Backbone, Beads, and Bradykinin. Three selections were performed for PPC and PPCD. High enrichment (>3, equivalent to 1,000-fold) was seen for 4 sequences for Backbone, 1 sequence for beads, all of the top 10 sequences (total 20) for bradykinin, 18/30 sequences for PPC, and 11/30 sequences for PP-CD.
(B) Results of a single point binding assay for 10 potential aptamer candidates. Binding, indicated by fluorescent signal (y axis), was measured for 10 aptamers at 100 nM. Apt 4 shows higher binding than the controls (non-aptamer [straight line] and buffer [dotted line]) for target PP-C. Apt 1,2,3,4,7,8,9 show higher binding than controls for PP-D. Data were normalized to the positive control (FAM conjugated directly to beads).
(C) Binding curve for Apt 4 binding to 100 nM PP-D plotted here. Data were fitted via the “fit_hyperbola” function in the biofits library (https://github.com/jimrybarski/biofits). Apt 4's Kd was found to be 3.65 μM (±1.99).
(D) Binding curve for Apt 4 for targets PP-C and PP-D for 100 nM to 2.5 mM concentrations. Apt 4 shows saturation binding against PP-D and no binding against PP-C. Data were normalized to the positive control (FAM conjugated directly to beads).
“Target-Switch” peptide design divided the potential binding space into four groups
For peptide target design, we divided the binding space into four groups (A through D) of five AAs each. During creation of these groups, we attempted to represent diversity in basicity, hydrophobicity, and AA charge. We also created four eight-residue “backbones” (A through D), where each backbone was composed of seven residues from a single group plus a C-terminal lysine (Table S10). Arrangement into sets allowed for coverage of desired permutations of N′-AA-AA-backbone-C’ (e.g., constant-constant-variable, constant-variable-variable). As a representative example, for group B we designed a set of peptides containing bibi-1-X, bibi-X, bibi+1-X, where bi represented one of five peptides in group B and X represented backbones composed of A–D group residues. In this example, the possible combinations for a proline-proline dipeptide (bibi-X, where bi is proline (P)) are PP-A, PP-B, PP-C, and PP-D. A similar schematic for target design can be used to generalize this approach to finding binders for other terminal AA targets.
Target-Switch protocol isolated aptamers with low micromolar affinity to proline-proline dipeptide targets
As the entire design matrix represented 960 testable targets, we simplified our initial experiments by focusing our efforts on a single dipeptide PP. PP was chosen as the N-terminal dipeptide of interest because the bulky cyclic side chain created multiple potential binding sites. Although ideally four versions of the PP dipeptide should be tested (PP-A through PP-D), we chose to perform five initial rounds of selection using only PP-C and PP-D to first characterize the enrichment patterns and performance of Target-Switch aptamers before attempting to scale (Figure 8A).
Figure 8.

Isolation of N-terminal amino acid-binding aptamers using a semi-automated and parallel replicates-and-switch selection strategy
(A) Schematic diagram of replicates-and-switch selection strategy. Twelve selections comprising replicates of each target mixtures were run for five rounds in parallel. The workflow begins with a negative selection against streptavidin beads on an initial pool of ssDNA and split across 12 random pools. Two parallel selections were performed on each control reference target and three parallel selections on the target (Proline-Proline) with and without the switching of backbones (C and D backbones) in alternating rounds. A representative pool of ssDNA from every round of every selection was sequenced and analyzed for round-to-round enrichment of sequences. Refer to Figures S5, S6, and S7 for digestion quality control assay, enrichment profile of top 10 aptamers, and contamination analysis, respectively.
(B) Target compositions and amino acid sequences in Non-Switch and Switch SELEX.
All targets were 10mer peptides conjugated to a magnetic bead (Figure 8B). Twelve selections were run in parallel, against five total targets: two targets of interest and three control targets. Three selections were run against each target of interest and two selections against each control target. All rounds of positive selection were sequenced and used for analysis of enrichment across rounds and targets. In addition, automation was used in several steps to ensure minimization of potential errors across samples and to facilitate running parallel selections.
In order to isolate aptamers to only the N-terminal dipeptide, regardless of the surrounding environment, we developed a target “switch” protocol where rounds of selection alternated between two targets with the same N-terminal dipeptide sequence and differing backbone sequences. Switching between two different backbones should decrease enrichment for aptamers bound non-specifically to other portions of the peptide. To increase our chances of isolating an aptamer specific to the N-terminal dipeptide PP, both “switch” and “non-switch” protocols were utilized, with multiple selections for each. The targets of interest included two versions of the PP dipeptide. The non-switch protocol used PP-C for every round. The “PP-CD” switch protocol alternated between PP-C and PP-D in each round of selection.
The control targets consisted of a beads-only negative control, as well as a specificity filter target and an enrichment reference target. The specificity filter target was created by conjugating beads to a 50/50 mixture of C and D backbones. This was utilized as a method to track binding to both backbones for use in analysis of top hits in switch and non-switch protocols. The enrichment reference target was used as a comparison against which we measured round-to-round enrichment. Our enrichment reference target was lysine-tagged bradykinin, a naturally occurring, “sticky” 9mer peptide that was chosen for its small size and ability to enrich aptamers quickly, as observed in our previous experiments. Sequences for SELEX aptamers, peptide targets, and NGS for SELEX experiments are listed in Tables S11 and S12.
Starting with a random library of 1015 unique oligonucleotides, we performed a single round of negative selection against streptavidin beads in order to reduce the likelihood of enriching promiscuous and randomly binding candidates, followed by five rounds of positive selection against the targets. Between rounds, libraries were amplified by PCR and converted back into ssDNA by enzymatic digestion (Figure S5). To gradually increase selection pressure, binding stringency was increased over each round of positive selection by reducing the ratio of target to aptamer. Switch and non-switch protocols followed two different stringency patterns. For the non-switch protocol, stringency was increased in every round. In the switch protocol, stringency was increased in every other round, since switching the backbone was a stringency change in itself. All targets started at a concentration of 842.7 nM and exact stringency gradients (Table S13). Replicates of all targets across all rounds (R2-R5) were sequenced via high-throughput sequencing (Illumina NextSeq).
Following five rounds of selection, we used the sequencing data collected after every round to identify the best binders. Several groups have demonstrated that the best binders cannot be identified solely by the highest copy number in the final pool (Cho et al., 2013). Thus, binders were identified by an enrichment term, “growth”, defined by the log ratio of counts of putative binders between round 5 and round 2 of selection. To reduce the likelihood of selecting low-quality binders for which counts increased by chance, we introduced a penalization term that penalized low-count binders. Details of this analysis can be found in supplemental information.
We observed that enrichment for all targets increased rapidly from rounds 2 to 3 and plateaued over rounds 3 to 5 (Figure S6). In addition, we observed that bradykinin, PP-C, and PP-CD targets had log enrichment values of 3.5, 3.2, and 3.0 indicating that these targets had putative binders (Figure 9A). To examine these binders further, we pulled out the top 10 binders by enrichment per replicate for each target. Enrichment for each target clustered among replicates, indicating that selections for these targets were isolating binders of interest (Figure 9A). Further analysis of target replicates indicated that overall there was little overlap between binders across replicates (analysis for top 10 in Figure S7B). Owing to the size of the initial random pools there is a low likelihood that identical sequences would be found in different replicates or targets, suggesting that these were instead contaminant sequences that may have been due to use of automation, aerosolized DNA, or other sources. This analysis allowed us to filter these likely contaminant sequences out when we selected a short list of candidates to test binding characteristics in vitro.
To identify the final aptamer sequences to fully characterize, we performed two filtering steps. We selected candidate aptamers from PP-CD binders that had high enrichment (greater than 2, which correlates to at least a 100-fold increase from R2 to R5) and demonstrated selective binding to PP-CD. Filtering of candidate sequences produced 26 candidates of which 10 were selected for final testing (sequences listed in Table S11). These final 10 candidates were chosen based on a variety of factors: highest enrichment ratio, total sequencing counts, representation within each selection replicate, and zero sequence contamination in SELEX replicates. Aptamer binding performance was assessed via fluorescence in plate reader assay described in STAR Methods.
At a single concentration (100 nM), seven aptamers showed higher fluorescent signal than non-aptamer and buffer-only controls toward the target PP-D. One aptamer showed higher fluorescent signal than controls toward the target PP-C (Figure 9B). Two aptamers were chosen for further testing, Apt 1 and 4. Apt 1 showed potential saturation binding toward PP-C but non-specific binding toward PP-D (Figure S7A), whereas Apt 4 showed saturation binding toward PP-D, with a Kd of 3.4 μM (Figure 9C) but no binding toward PP-C (Figure 9D). Although we were hoping to not see backbone preference for Apt 4, our preliminary results do not demonstrate aptamers completely agnostic to a backbone influence. However, further testing and optimization is still required to decipher how much of the binding is dependent on the backbone.
Target-Switch SELEX results demonstrate a scalable, semi-automated aptamer discovery pipeline that produced multiple binders to custom designed 10mer targets sharing the same N-terminal dipeptide. These results demonstrated progress toward aptamer seed sequences for proline-proline N-terminal aptamers. Promising aptamer candidates from Target-Switch SELEX displayed affinity for their dipeptide targets in the micromolar range, which is comparable with several discovered aptamers to AAs (Ames and Breaker, 2011; Cheung, K.M., Yang, K.-A., Nakatsuka, N., Zhao, C., Ye, M., Jung, M.E., Yang, H., Weiss, P.S., Stojanović, M.N., Andrews, A.M., 2019; Majerfeld and Yarus, 1994) and small-molecule binders (McKeague and DeRosa, 2012). BCS unit test experiments demonstrated an ideal operating range for binders with nanomolar affinities, so additional affinity optimization of seed sequences would be required. Future developments toward building a robust, single-molecule protein sequencing technology with aptamers would include exploring the vast space of chemical and physical modifications possible to DNA. In the long term, this will be necessary to increase aptamer binding affinity before testing and merging Target-Switch SELEX with BCS.
Discussion
Here we introduced a path toward creating ProtSeq, a proposed protein sequencing method for converting AA residues into DNA barcode sequences directly on an NGS chip. Although we did not yet demonstrate the capability to sequence full proteins, we were able to demonstrate progress toward several key components, including (1) a barcode-cycle binding assay on which we were able to bind, ligate, cleave, and sequence DNA-barcoded binders; 2) a computational pipeline to identify DNA barcodes from sequencing reads, connect to binder, deconvolve AA sequence, and identify putative proteins; and (3) a method called Target-Switch SELEX, which may be used to discover aptamer binders to N-terminal dipeptides. Below, we describe modified approaches (Figure S8A) and lessons learned.
We established a semi-automated SELEX pipeline capable of identifying aptamers to proteins and peptides, toward the goal of discovering aptamers to N-terminal dipeptides. Preliminary experiments identified aptamers with micromolar affinity toward 10mer peptides sharing the same N-terminal dipeptide. The next step toward finding backbone-agnostic binders with this design will be to incorporate all four backbones into rounds of selection and extend the experiment to additional rounds of selection. Unlike traditional SELEX in which top hits are tested for affinity to a single target, “top hits” from Target-Switch SELEX must be affinity tested against multiple targets. Although we only tested the affinity for two enriched candidates, a large pool will need to be screened to identify seed sequences for dipeptide binders. Top hits for Target-Switch SELEX may not be discovered with traditional aptamer enrichment analysis since selection against multiple targets with the same experiment may produce a heterogeneous mix of binders with different PCR amplification rates. To expand to additional N-terminal amino acid dipeptide targets, additional targets, designed with the Target-Switch methodology and four backbones, can be generated and utilized (Table S10).
Improved signal via increased aptamer affinity and specificity would benefit Protseq. Experimental sequences identified through Target-Switch SELEX may be used as a computational starting point for designing a seed pool with modified aptamer sequences to improve binding affinities or target specificities to explore a much larger input pool size than experimentally possible (Tolle et al., 2015; Bashir et al., 2021). Also, the experimental option space for aptamer modifications is large and rapidly expanding. Modifications can include both physical (such as sequence length, cross-linking to maintain secondary structure) and chemical (i.e., introduction of non-natural base pairs or amino acids) alterations to the aptamer sequence. Modified DNA aptamers, such as locked nucleic acids (Hernandez et al., 2009), base-modified aptamers (Gordon et al., 2019), and SOMAmers (Gawande et al., 2017; Gold et al., 2010), have been shown to increase aptamer affinity, including toward small-molecule targets. Modifications could be made on discovered DNA aptamers, or modified pools could be used as initial input pools. Target-Switch SELEX on modified aptamer pools may ultimately be able to isolate aptamers with nanomolar affinities and high enough specificity to produce a sufficient signal on the BCS platform.
In addition to enhancing aptamer or barcode properties, another approach to reduce noise would be to modify the target display platform by either switching to an in-solution binding assay or using a customized BCS chip. We tested using an in-solution binding assay as an alternative to a solid substrate, where aptamers were ligated to POC targets post binding using a bridge-based proximity ligation strategy (Pawlosky et al., 2019). In preliminary unit tests using DNA-DNA binding as a proxy for peptide-aptamer binding, we demonstrated that ligation kinetics can be optimized to favor spatially associated oligos in solution (Figures S8B–S8D, alternative ligation, STAR Methods) (sequences are reported in Table S14). However, the in-solution assay encountered a similar issue to the solid substrate of inadequate spatial separation between targets. Even with strong binders in a maximally diluted binding solution, stochastic interaction between non-binders is more challenging to minimize in a liquid substrate than on a solid substrate where one can control the distance between fixed substrates without additional barriers utilized for future isolation.
A solid substrate binding platform consisting of a custom BCS chip with targets distributed sparsely over a wide area would allow for tight spatial control between target and ligation foundation while maintaining the ability to remove non-binding elements off the chip. In place of Illumina chips, custom-built flow cells could have nucleic acids printed on the glass, including individual molecules with unique foundation barcodes, in a known pattern and an automated fluidics system to allow for POC deposition and barcode building without the constraints of pre-existing adapters on a DNA sequencing chip. The barcoded foundation representing a protein or peptide sequence may then be amplified and transferred to any existing DNA sequencing platform.
In summary, single-molecule protein sequencing technologies hold several interesting avenues of future exploration, including use of aptamers with modified AAs and use of a custom chip for BCS. However, in the process of building these technologies, we have discovered that implementing solutions in an integrated system is a balancing act between the advantages they confer and the changes they produce. In our experience, modification to one part of the process may require major changes to the rest of the pipeline. For example, changing one component of the SELEX buffer would entail changing the BCS buffer and recharacterizing all BCS components to fully understand the effects of the change. We have learned that, in order to create a functioning, integrated, end-to-end binder discovery and protein sequencing platform, every element must be built and tested in parallel to achieve compatibility between the elements.
Limitations of the study
As is, what is presented here is only the early foundation of a single-molecule protein sequencing technology. In order to be taken to completion the following steps would also need to be accomplished: (1) discovery of enough N-terminal amino acid aptamer binders with appropriate affinities and specificities, (2) establishment of compatible degradation techniques, and (3) sequential binding, barcoding, degradation, and identification of peptide/protein targets. Modifications to aptamers for improved affinity and specificity are covered in the discussion section. The number of aptamers needed would be dependent on their binding characteristics, which can be used as inputs to our simulation.
An eventual consideration for end-to-end protein sequencing will be the need for a ProtSeq-compatible technique to cleave N-terminal peptides. Preliminary testing showed that using Edman degradation on the current version of the BCS platform would be challenging, as the primary reagent trifluoroacetic acid (TFA) depurinated DNA components of the BCS machinery. We even discovered with single-molecule imaging the cleavage of the P5 adapter occurs after a single round of TFA exposure and degradation of the P7 adapter after four rounds. Additional work demonstrated that the degradation of the P7 adapter could be mitigated by using either polypurine or 2′-O-methylated modified RNA oligo adapters, since they appeared to be partially resistant to TFA degradation. However, utilizing custom adapters would require an expanded BCS protocol, where the barcodes would be assembled on one chip, amplified, and then transferred to a sequencing cartridge, and may result in the loss of reads. We propose the consideration of alternative strategies for terminal peptide degradation, such as modified exopeptidases or C-terminal chemical digestion (Casagranda and Wilshire, 1994; Bergman et al., 2001).
Finally, end-to-end sequencing of protein and peptide molecules is still required. We developed a protocol to attach DNA oligos to peptides and proteins at a specific terminus of select amino acids, where proteins are tagged prior to cleavage to allow for unique molecule barcoding and assistance of protein reassembly in analysis. Once enough suitable aptamer binders are discovered, tagged molecules could then follow the steps of the BCS protocol for single-molecule sequencing.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Bradykinin (Rabbit) Antibody | Abcam | ab14391, RRID:AB_2133420 |
| Donkey anti-Rabbit Secondary Antibody (555) | Thermo Scientific | A-31572, RRID:AB_162543 |
| Chemicals, peptides, and recombinant proteins | ||
| Spot-Tag Experiment Spot-Tag∗ (peptide target) target called Spot-Tag.O1 (N-terminus)-PDRVRAVSHWSSGGG-Cys (C-terminus)-3'ATCCCTTCTCTTCCTGTATACTAATAGGTG CACGTAGATTC/5Phos/ |
this paper | |
| Spot-Tag Experiment Bradykinin∗ (peptide target control for non-specific binding) target called Brady.O1 (N-terminus)-RPPGFSPFR-Cys (C-terminus)-3’ATCCCTTCTCTTCCTGTATA CTAATAGGTGCACGTAGATTC/5Phos/ |
this paper | |
| SELEX peptide PP-C PPNHFENEIK bt | this paper | |
| SELEX peptide PP-D PPTKYVGTGK bt | this paper | |
| SELEX peptide Bradykinin RPPGFSPFRK bt | PubChem | 439201 |
| In-Solution Experiment Peptide NC1 KQNTSQNTSC | this paper | |
| In-Solution Experiment Peptide NC2 KQNTYQNTSC | this paper | |
| In-Solution Experiment Peptide NC3 QNTSYQNTSC | this paper | |
| EcoRI | NEB | Cat#R0101S |
| Cutsmart buffer | NEB | Cat#B7204S |
| Hybridization buffer (0.025% TWEEN20 in 1x PBS) | this paper | |
| Blocking buffer (0.025% TWEEN20 in 1x PBS + 10mg/mL BSA) | this paper | |
| Chip-blocking buffer (10 uM of P5 Complementary oligo (5’-TCTCGGTGGTCGCCGTATCATT-3’)/P7 Complementary oligo (5’-ATCTCGTATGCCGTCTTCTGCTTG-3’) sequences + 10 uM POC Tail blocking sequence (5’-TAGGGAAGAGAAGGACATA TGATTATCCACGTGCATCTAAG-3’ ) in 60 uL of Blocking Buffer) |
this paper | |
| Aptamer incubation buffer (0.025% TWEEN20 in 1x PBS + 0.1 mg/mL BSA) | this paper | |
| Bovine Serum Albumin | Thermo Scientific | A2153-50G |
| Phosphate Buffered Saline (PBS) | Thermo Scientific | 11205D |
| Tween-20 | Sigma-Aldrich | P9416 |
| FluoSpheres™ Streptavidin-Labeled Microspheres, 0.04 μm, yellow-green fluorescent (505/515) | Thermo Scientific | F8780 |
| TransFluoSpheres™ Streptavidin-Labeled Microspheres, 0.04 μm (488/645) | Thermo Scientific | T10711 |
| Phenyl isothiocyanate (PITC) | Sigma Aldrich | 317861-5G |
| Dimethyl allylamine | Sigma Aldrich | 05937-25ML |
| Pyridine | Sigma Aldrich | 270970-4X25ML |
| Trifluoroacetic acid (TFA) | Fisher Scientific | O4901-500 |
| SimplyBlue SafeStain | ThermoFisher | LC6060 |
| Glycerol | Sigma Aldrich | G5516-500ML |
| Human Thrombin | haemtech | HCT-0020 |
| Formamide | Sigma Aldrich | 11814320001 |
| T4 DNA ligase | NEB | M0202S |
| HT1 buffer | Illumina | 20015892 |
| Lambda exonuclease | NEB | M0262L |
| Mag-Bind Total Pure NGS beads | Omega-Biotek | M1378-02 |
| Herculase II Phusion polymerase | Agilent | 600679 |
| 70% ethanol | Fisher Scientific | BP8201500 |
| 200 proof ethanol | Fisher Scientific | BP2818500 |
| MgCl2 | ThermoFisher | AM9530G |
| dNTP mix | ThermoFisher | R1122 |
| fluorescein biotin | Biotium | #80019 |
| DMSO, Anhydrous | Thermo Scientific | D12345 |
| Critical commercial assays | ||
| MiSeq Reagent Nano Kit v2 (300-cycles) | Illumina | MS-103-1001 |
| MiSeq Reagent Nano Kit v2 (500 cycles) | Illumina | MS-103-1003 |
| Miseq Reagents Kits v2 (50 Cycles) | Illumina | MS-102-2001 |
| MiSeq® Reagent Kit v3 (150 cycle) | Illumina | MS-102-3001 |
| PhiX Control v3 | Illumina | FC-110-3001 |
| Blunt/TA Ligase Master Mix | NEB | Cat#M0367L |
| SoluLINK Protein-Oligonucleotide Conjugation Kit | Vector Laboratories | S-9011-1 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher | 23227 |
| Luna qPCR Master Mix | NEB | M3003X |
| Qubit ssDNA kit | ThermoFisher | Q10212 |
| Qubit™ dsDNA HS Assay Kit | ThermoFisher | Q32854 |
| 2100 Bioanalyzer | Agilent | G2939BA |
| Tapestation | Agilent | G2991AA |
| Pippin Prep system | Sage Science | |
| Deposited data | ||
| Raw sequencing data for BCS | Mendeley | https://data.mendeley.com/datasets/f9hdn5xc3v/1 |
| Raw sequencing data for Target-Switch SELEX | Harvard Dataverse | https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/W903IJ |
| Oligonucleotides | ||
| All oligonucleotide sequences listed in supplemental information | ||
| Software and algorithms | ||
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| MiSeq Control Software | Illumina | https://www.illumina.com/systems/sequencing-platforms/miseq/products-services/miseq-control-software.html |
| Colab | https://colab.research.google.com/ | |
| Kd analysis hyperbola formula | Jarmoskaite et al. (2020) | https://github.com/jimrybarski/biofits/blob/master/biofits/function.py |
| Custom analysis code | Github | https://github.com/google-research/google-research/tree/master/protseq |
| Other | ||
| H-8 DNA & RNA Synthesizer | K&A LABORGERÄTE | N/A |
| 1290 Infinity II HPLC | Agilent | N/A |
| MiSeq 500 | Illumina | SY-410-1003 |
| Ti2-E microscope | Nikon | N/A |
| Ti2-LAPP TIRF module | Nikon | N/A |
| Andor iXon Ultra 897 EMCCD camera | Oxford Instruments | N/A |
| SPECTRA X LED light engine | Lumencor | N/A |
| 520/35 bandpass emission filter | Semrock | FF01-520/35-25 |
| 676/29 nm bandpass emission filter | Semrock | FF01-676/29-25 |
| Bravo liquid handler | Agilent | G5574AA |
| 1.5ml microfuge tubes, DNA LoBind | Eppendorf | cat#022431021 |
| 96-well plates, DNA Lo-Bind | Eppendorf | 30129512 |
| Nunc plates | VWR | 73520-120 |
| Mastercycler® nexus gradient, 115 V/50 – 60 Hz (US) | Eppendorf | 6331000025 |
| Mastercycler® nexus eco, 115 V/50 – 60 Hz (US) | Eppendorf | 6332000029 |
| Mastercycler® nexus flat eco, 110 V/50 – 60 Hz (JP/South America/TW/US) | Eppendorf | 1010015267 |
| Adhesive PCR Plate Seals | Thermo Fisher | AB0558 |
| Plate reader | Biotek Synergy HTX | S1LFA |
Resource availability
Lead contact
Further information and requests for reagents should be directed to Lead Contact Annalisa Pawlosky (apawlosky@google.com).
Materials availability
Sequences of aptamers, oligos, peptides, and peptide-oligo constructs generated in this study are listed in the key resources table.
Method details
General information for Barcode Cycle Sequencing (BCS)
The following protocol was used in the developmental experiments and was adapted for the following barcode ligation, Spot-Tag, and thrombin experiments. Aptamers and foundation oligos were either purchased from IDT, or synthesized in-house by K&A LABORGERÄTE H-8 DNA & RNA Synthesizer and purified via HPLC (Agilent 1290 Infinity II). Peptide-oligonucleotide constructs were either coupled in-house (protocol below) with purchased peptide sequences or commercially obtained from Genscript. Aptamer incubation and later DNA barcode sequencing was performed on MiSeq Reagent Kits, supplemented with PhiX Control v3, and sequenced on a MiSeq500 (Illumina). Bound aptamers were ligated to the barcode foundations using T4 ligase (blunt/TA Master mix formulation) and cleaved with EcoRI in Cutsmart Buffer, all purchased from New England Biolabs. Excess aptamers and the hybridization buffer were washed away with Cutsmart® buffer. All buffers were diluted with Ambion™ Nuclease-Free water. Analysis of NGS-data DNA barcode alignment was accomplished with a custom analysis pipeline running on a Colaboratory notebook environment. Specialized buffer solutions are listed in Table S1.
DNA BCS methods (1, 6 cycle) methods
Foundation hybridization
First step of BCS experiments is to hybridize cololinkers, foundations, and targets to form colocalized constructs. Sequencing unit components (FC, RC, barcoded foundations, and barcoded targets) were thawed on ice and hybridized with a 10 nM FC concentration (foundation, target, reverse cololinker in excess). In a 96 well plate, sequencing unit components were combined (1 target per well), and annealed using the following cycling parameters on a thermocycler 5 minutes at 95°C, 1 minutes at 85°C, 2 minutes at 75°C, 3 minutes at 65°C, 5 minutes at 55°C, 5 minutes at 45°C, 5 minutes at 35°C, and 40 minutes at 25°C.
Foundation ligation and blocking
After sequencing units of each target are hybridized, colocalized constructs are ligated to the flow cell to ensure targets and foundations are available for aptamer incubation. Hybridized colocalized constructs were diluted to get a 500 pM working solution in Hybridization Buffer of each target foundation. In single tube, equal amounts of each target foundation was combined with a final concentration of all foundations at 120 pM and 10 μL 2xBlunt/TA MM (T4) Ligase, then diluted in Hybridization Buffer for total volume of 100 μL solution hereafter referred to as the Foundation Mix. 30 μL Foundation Mix was added to the sequencing chip twice in succession and incubated for 15 minutes at 28°C. After incubation, the sequencing chip was washed with 100 μL of 100% formamide and incubated for 90 seconds at 40°C.
To reduce availability of flow cell surfaces and ssDNA ligated to the flow cell for non-specific binding of aptamers during aptamer incubation, we blocked the chip surface and the exposed ssDNA. First the chip was washed with the Blocking Buffer, then with Chip Blocking Solution. Chip Blocking Buffer was added to the chip twice and incubated for 15 minutes at 37°C.
Barcoding cycles and barcode sequencing
The sequencing process begins by incubating the first BCS Compatible aptamer pool, followed by washout of unbound aptamers and addition of a ligase to covalently connect the aptamer to the BF. This cycle of incubation and ligation is performed multiple times, where ligation is performed after each incubation or after all aptamer pools have been introduced. Aptamers (or DNA binders) were combined with bridge oligo at 1:2 ratio in Hybridization Buffer, heated to 95°C for 5 minutes in PCR tube and cooled at RT on benchtop for 1 hour. Immediately prior to incubation of aptamers and bridges on chip, 10 mg/mL BSA was added to achieve final BSA concentration of 100 μg/mL. The final aptamer solution was loaded onto the sequencing chip and incubated at RT for 30 minutes.
Aptamers (or DNA binders) bound to targets were ligated to the colocalized foundations using 2x T4 ligase. Following ligation, a restriction enzyme (NEB, EcoRI) was introduced (along with an excess of the complementary sequence to the restriction site and spacers) to cleave the peptide-binding sequence of the aptamer from the aptamer barcode on the 5’ end, leaving only the aptamer barcode and the short consensus sequence for subsequent ligation attached to the BF. The process of binder incubation, barcode ligation, and restriction digest can be repeated for multiple rounds. The aptamers in each round contain unique barcodes (even when the peptide binding sequences are the same), such that missed incorporation events (e.g., apparent deletions) may be easily identified and accounted for in subsequent data analysis steps.
The final step in the sequencing process is the addition of a next-generation sequencing (NGS) adapter complementary to P5 adapter sequence to facilitate direct amplification and sequencing of the DNA barcode chain on the chip. Using a similar bridge ligation strategy, the adapter is ligated to the 3’ end of the sequence of aptamer barcodes that represent the series of aptamer binding events. Excess adapter is removed via a washing step and the chip is loaded in the MiSeq for sequencing. MiSeq instruments run instructions are adapted to remove initial washing and library loading steps from the cartridge to the sequencing chip.
Target-foundation deposition colocalization validation methods
First, we built and validated a widefield fluorescence microscope capable of imaging single molecules on the ProtSeq platform. All imaging was undertaken on a Nikon Ti2-E microscope equipped with the Nikon Ti2-LAPP TIRF system and Andor iXon Ultra 897 EMCCD camera. For single molecule imaging a 60x 1.49 NA TIRF objective was used. As proof-of-concept that single molecule imaging can be achieved, fluorescence-tagged oligonucleotides and single-molecule glass slide controls displayed similar bead size and fluorescence intensity (Figure S1). Figure S1B shows the intensity distribution of all the fluorescent spots in an image snapshot. The local maxima of every 10,000 grayscale count (in the case of channel one : 488 nm excitation and 645 nm emission, Figure S1B) can be used to distinguish spots with various peak intensities. For example, the first interval (grayscale count from 0 - 10,000 grayscale count) in Figure S1C indicates only one streptavidin bead bound to one biotinylated oligo. The second or third interval suggests a cluster of (two or three) streptavidin beads were binding to one biotinylated oligo. Data from size comparison analysis and intensity distribution suggests that single oligo molecules were detected.
After we achieved single molecule imaging, we used the system to investigate ground truth of ProtSeq platform’s components such as colocalization efficiency. Forward and reverse colocalization linkers (FC and RC) were tagged with fluorescent Streptavidin beads and imaged on a flow cell. The FC consisted of the barcode foundation-complementary region at the 5’ end, followed by a sequence complementary to the glass-bound oligo, followed by a flexible T-spacer, with a short, high GC-content sequence at the 3’ end complementary to the RC. In turn, the 3’ end of the RC was complementary to the 3’ end of the FC, followed by a long T-spacer, followed by a sequence complementary to the glass-bound oligo, followed by a sequence complementary to another oligo. The FC and RC was biotinylated at the 5’ end. The FC, LC, and Streptavidin beads, and flow cell surface were blocked separately with a BSA buffer (1x PBS, .05% Tween, 10 mg/ml BSA) for 1 hour at RT. In two separate reactions, the FC was incubated with FluoSpheres™ Streptavidin-Labeled Microspheres, 0.04 μm, yellow-green fluorescent (505/515), and the RC with TransFluoSpheres™ Streptavidin-Labeled Microspheres, 0.04 μm (488/645) in a 1:4 oligo to beads ratio such that each biotinylated oligo likely binding to at least one bead for 30 minutes at RT. The FC and RC were combined in a 1:2 ratio for 1 hour at RT. The solution was loaded onto a Illumina MiSeq v2 chip and incubated for 30 minutes at 37°C to allow for the FC and RCs to hybridize to the P7 adapters in the chip and then washed prior to imaging. The imaging system is a wide-field upright fluorescence microscope with a 60X Nikon objective (NA = 1.49). Glass piece of the chip was taken out from the MiSeq cassette and imaging was performed on the external top surface of the chip. The beads inside the chip were excited at 488 nm with SPECTRA X LED light engine and the emitted fluorescence signal was collected at 515 nm (with a 520/35 bandpass emission filter) and 645 nm (with a 676/29 nm bandpass emission filter). Images were acquired with an Andor EMCCD camera with 16 micron pixel size and 2 second exposure time.
It is desirable to ensure as many POC’s have DNA foundations nearby as possible thus we mixed the dye labeled POC: forward linker: reverse linker: DNA foundations at ratios of 1:1:1:0.5, 1:1:1:1, 1:1:1:3 and 1:1:1:5 to try ascertain a peak efficiency. The mixtures were ligated onto chips that were subsequently imaged with a Nikon TE2 TIRF microscope. Peak colocalization efficiency of (64.17%,+/- 7.88%) was achieved with 1:1:1:1 ratio compared to (24.66%, +/- 6.83%), (61.22%, +/- 5.87%) and (57.69%, +/- 3.95%) for 1:1:1:0.5, 1:1:1:3 and 1:1:1:5 concentrations respectively.
Edman degradation
For Edman degradation, attached peptides were coupled with phenyl isothiocyanate (PITC) in coupling buffer (0.4 M dimethyl allylamine in 3:2 (v/v) pyridine:water, pH 9.5), cleaved in trifluoroacetic acid (TFA), and washed prior to imaging. All reagents for Edman degradation were purchased from Sigma-Aldrich.
Conjugate Spot-Tag nanobody and thrombin to DNA tail methods
Spot-tag peptide (sequence PDRVRAVSHWSS) was purchased from Genscript as peptide-oligo-conjugate. Spot-tag peptide-oligo target included a spacer of three glycine residues at the C-terminus.
Spot-tag nanobodies (Spot VHH) were obtained from Chromotek, and conjugated to the 3’ amino-modified end of a 5’ phosphorylated oligo (sequence /5Phos/GC CGT GTC CTT TGT TAA CCG GGA TAA CGA ATT CCT ATA GGC GCA GTT TTT TTT TTT TTT T/3AmMO/) in a non-site directed manner using the SoluLINK Protein-Oligonucleotide Conjugation Kit according to manufacturer instructions. Site-directed conjugation can also be achieved using the sortase-enzyme method, with which we were able to obtain ∼30% labeling efficiency (data not shown).
Success of Spot-tag nanobody-oligo conjugation was confirmed by denaturing PAGE electrophoresis and protein staining by SimplyBlue SafeStain (ThermoFisher). Nearly no unconjugated protein was observed on the gel, while multiple higher molecular weight bands were present presumably corresponding to multiple oligos conjugated to a single nanobody. Conjugation reactions were not purified any further, and were stored in 1x PBS with 20% glycerol at -20°C prior to use. Importantly, for BCS experiments nanobodies with multiple oligos are less of a concern because they will either 1) be non-functional, in which case they will not bind Spot-tag and be washed away, or 2) will bind to the Spot-tag, following which either of the multiple tails can then become ligated to the nearby foundation. Final protein concentration was determined by Pierce™ BCA Protein Assay Kit (ThermoFisher) and conjugates were used at ∼ 200 nM final concentration on the chip.
Thrombin protein (ThermoFisher) conjugation followed the same procedure, however each protein has 7 binding sites, so there will be a range from 0-7 DNA oligos conjugated to each thrombin protein.
Spot-tag inding validation methods
As a proof-of-concept experiment to validate the ability of the BCS platform to record specific binding events in a complex environment, the Spot-Tag-oligo conjugates (Spot-Tag.O1) and 6 other control targets were seeded onto a MiSeq Nano v2 sequencing chip. The other peptide target was Bradykinin conjugated to a 5’ phosphorylated DNA tail (Brady.O1). Two null targets (oligo tails without target) comprised a 5’ phosphorylated oligo (5’Phos.O1), and an oligo lacking a 5’ phosphate, which therefore can not be attached to the chip (CLR.Null.Block). Two DNA controls (DNA Target 6.O1 and DNA Target 4.O1), continuous oligo sequences that contained both a 5’ phosphorylated linking region to tether to the P7 primers and a binding region to hybridize to a complementary strand, served as positive controls. The binding region and DNA tail sequences of each target is reported in Table S7.
Each control target was tested in triplicates and Spot-Tag in sextuplicate. Their respective FC, RC, and BF were thawed on ice before each set of sequencing units were combined in 91 μL of Hybridization Buffer (0.025% TWEEN20 in 1x PBS) in separate wells to generate solutions of 10 nM FC, with RCs, BFs and targets in excess. FCs and RCs were kept in a stock solution with a ratio of 3:1 FC:RC in Hybridization Buffer. The components were added in the order of Hybridization Buffer, FC and RC stock, and BFs. Targets were added to the mixtures immediately prior to hybridization. Sequences of each set of targets are reported in Table S7. The final ratios of individual pieces are 5:1 BF:FC, 3:1 FC:RC, and 10:1 Target:RC.
To assemble the sequencing units, the complete mixtures were mixed thoroughly, spun down for 30 seconds, sealed, and heated in a thermocycler with the following conditions: 5 minutes at 95°C, 1 minute at 85°C, 2 minutes at 75°C, 3 minutes at 65°C, 5 minutes at 55°C, 5 minutes at 45°C, 5 minutes at 35°C, and 40 minutes at 25°C.
Prior to seeding the colocalized constructs, the sequencing chip was washed with 100 μL Hybridization Buffer twice. Each mixture of colocalized constructs were diluted to 0.5 nM and and 1.14 μL of each mixture was combined with 10 μL of 2x Blunt/TA MM Ligase Master Mix and 44 μL of Hybridization Buffer, and gently mixed for a final concentration of 120 pM of colocalized constructs. To ligate the colocalized constructs onto the chip, the sequencing chip was washed with 30 μL of Foundation Mix twice and heated at 28°C for 15 minutes on a hotplate. Then it was washed once with 100 μL of 100% formamide to remove unligated colocalized constructs. The chip was heated again at 40°C for 90 seconds on a hotplate, washed with 500 μL of Blocking Buffer (0.025% TWEEN20 in 1x PBS + 10 mg/ml BSA) once, washed with 30 μL of Chip Blocking Solution twice (10 μM of P5 Complementary oligo (5’-TCTCGGTGGTCGCCGTATCATT-3’)/P7 Complementary oligo (5’-ATCTCGTATGCCGTCTTCTGCTTG-3’) sequences + 10 μM POC Tail blocking sequence (5’-TAGGGAAGAGAAGGACATATGATTATCCACGTGCATCTAAG-3’)), incubated for 37°C for 15 minutes on a hotplate, and washed with 100 μL Hybridization Buffer twice for 60 seconds one immediately before loading the prepared binder library (see barcoded-binder library preparation section below).
Barcoded-binder library preparation
Four DNA barcoded “binders” were incubated with the targets, each consisting of a binder region, a DNA spacer region, a restriction site, DNA barcode indicative of the binder region identity, and ligation site. Two DNA binders, DNA Binder 4.2 and DNA Binder 6, contained a binder region consisting of DNA that were complementary to DNA Target 4 and DNA Target 6 respectively. These binders were positive controls that should bind to DNA Target 4 and DNA Target 6 with high affinity and specificity. Another DNA binder, DNA Binder 9, contained a binder region consisting of a scramble DNA sequence that should bind to none of the targets present, serving as a negative control to measure noise. The last binder was the Spot-tag nanobody-oligo conjugate.
Prior to incubation each binder was hybridized to a universal bridge (5’-CTGCGCCTATAGGAATTCGTTATC/i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd//i5NitInd/GGACACGGCCGTTATC-3’), an oligo that was partially complementary to the restriction site spacer and partially complementary to the consensus sequence (Figure 2B–C). Each /i5NitInd/ is a 5-Nitroindole, a universal base analogue that exhibits high duplex stability and hybridizes indiscriminately with each of the four natural bases (Loakes and Brown, 1994). The DNA binders and the Spot-tag nanobody target were hybridized with their respective bridges in separate reactions. The DNA binders were added to 2x excess bridge oligos per DNA binder in Hybridization Buffer to generate a 50 μL solution with an end concentration of 200 nM of each DNA binder (600 nM of all DNA binders combined). The solution was heated to 95°C for 5 minutes at room temperature (RT) (22-24°C) for an hour.
To hybridize the Spot-tag nanobody target to the universal bridge, it was added to 5x excess bridge per Spot-tag nanobody target in Hybridization Buffer to generate a 49 μL solution with an end concentration of approximately 400 nM Spot-tag nanobody target. In the preparations of nanobody-oligo conjugates, the DNA tails are added in excess and are not purified away. It is possible that the excess of unconjugated DNA tails present in the solution hybridize to the Spot-tag-oligo conjugates, preventing hybridization of the universal bridge needed for the subsequent ligation of the Spot-tag nanobody barcode to the nearby foundation. A ratio of 5:1 bridge:Spot-tag nanobody target was used such that any excess DNA tail that were in the solution but not conjugated to Spot-tag nanobody target from the protein-oligo conjugation reaction were hybridized to a bridge, promoting bridge hybridization with all oligo tails conjugated to Spot-tag nanobody targets. This solution was heated to 37°C for 30 minutes and cooled at RT for 30 minutes. After cooling, the solutions containing the DNA binders and Spot-tag nanobody targets, both hybridized to universal bridges were combined and 1 μL of Blocking Buffer (0.025% TWEEN20 in 1x PBS + 10 mg/mL BSA) was added. The final binder library solution had a concentration of 100 nM of each DNA binder (300 nM of all DNA binders combined) and 200 nM of Spot-tag nanobody target.
Barcoded-binder library incubation, binder barcode ligation, and restriction digest
After the step of washing the sequencing chip with 100 μL Hybridization Buffer twice for 60 seconds (see Build and Tether Foundations to Solid Substrate section above), the chip was washed with Aptamer Incubation Buffer (0.025% TWEEN20 in 1x PBS + 0.1 mg/ml BSA) for 60 seconds. The binder library was gently mixed and the sequencing chip was slowly loaded with 30 μL binder library solution twice. The sequencing chip was incubated with the binder library solution on a hotplate at 25°C for 30 minutes. After incubation, the chip was washed with 100 μL of Aptamer Incubation Buffer for 90 seconds three times to wash away unbound and weakly bound binders.
To prepare the ligation reaction, 7 μL of 2x Blunt/TA MM Ligase solution was diluted in 63 μL of Hybridization buffer and gently mixed. 30 μL of the diluted ligase solution was loaded onto the chip twice before the chip was incubated for 5 minutes in a hotplate at 28°C to ligate the DNA tail of the binders to its bound target’s respective foundation oligo. The ligation reaction was terminated by washing the plate with 100 μL of 1x CutSmart solution for 60 seconds three times.
The rest of the binder besides the consensus region and binder barcode was removed from the barcode-foundation construct with a restriction digestion reaction. The restriction enzyme mix was prepared by adding 10 μL of 20 units/μL EcoRI to 30 μL 10 μM Restriction bridge (5’-CTGCGCCTATACGAATTCGTTATC-3’), 10 μL of 10x CutSmart solution, and 77 μL of Nuclease-Free H2O before the contents were gently mixed. 30 μL of the restriction enzyme mix was loaded onto the chip twice and incubated at 40°C on a hotplate for 30 minutes. To terminate the ligation reaction and wash off any hybridized DNA, the chip was loaded with 100 μL of 100% formamide, incubated at 40°C on a hotplate for 90 seconds, and washed with 500 μL of Hybridization Buffer.
Alternative ligation methods
Ligation reactions were performed in 10 microliters total volume consisting of 5 μL Blunt/TA ligation master mix (NEB) and 5 μL DNA in 1X cutsmart buffer (Figure S7B) on “0A”-“0B” pairs and “50A”-“50B” dsDNA pairs. DNA concentration varied between 490 pM and 1 nM, achieved by serial dilution of 1 nM DNA across 12 wells. Reactions were incubated on the benchtop for 5 minutes and inactivated in a PCR thermal cycler for 10 minutes at 95C. Each qPCR reaction was set up using 22.5 zmol ligation product, Trilink forward primer, and Trilink reverse primer (Table S14), with NEB Luna qPCR Master Mix according to manufacturer recommendations.
Ligation reactions were performed in 20 μL reactions using T4 DNA ligase. Each ligation reaction contained 2 μL T4 ligase DNA buffer, 1 μL T4 DNA ligase diluted 1:50, 0.5 mM ATP, and DNA at desired concentration (Figure S7C). Reactions were incubated on the benchtop for desired amount of time and inactivated in a PCR thermal cycler for 10 minutes at 95°C. Each qPCR reaction was set up using 9 microliters of ligation product, Trilink forward primer, and Trilink reverse primer, with NEB Luna qPCR Master Mix according to manufacturer recommendations.
Ligation reactions were performed using T4 DNA ligase with 2 μL T4 ligase DNA buffer, 1 μL T4 DNA ligase diluted 1:50, 0.5 mM ATP, and 1.25 nM DNA at desired ratios of binders to non-binders (Figure S7D). Reactions were incubated on the benchtop for one minute and inactivated in a PCR thermal cycler for 10 minutes at 95°C. The qPCR reaction was set up as described above.
Thrombin-HD22 BCS methods
The HD22 aptamer discovered to the thrombin protein was used with our oligo-tagged thrombin on BCS (Tasset et al., 1997). The thrombin aptamer was ordered from IDT (100 nmole DNA Oligo HPLC) with the 5’ BCS binding components attached (Table S4). Thrombin protein (Thermofisher) was modified as mentioned above to be compatible for ligation on BCS chips. Aptamers were refolded at 95C for 5 minutes, cooled to RT on the bench for an hour before applied to the BCS chip at a concentration of 200 nM. All other steps of the procedure follow the Spot-tag protocol described above with 3 target replicates for thrombin, 3 replicates for a control DNA binding pair (DNA Binder 4.2), 3 replicates of a control ssDNA binder CLR and 3 replicates of a ligation control 5’Phos.
NGS sequencing
The final step in the sequencing process was the addition of Next Generation Sequencing (NGS) adapters. 1.5 μL of 2:1 1μM Universal NGS Adapter (/5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT ) + Universal NGS Adapter Bridge 9/5 (5’-TTCCGATCTCGTTA-3’ ) was added to 10 μL of 10x CutSmart, 25 μL of 2x Blunt/TA MM Ligase, and diluted in 63.5 μL of Nuclease-Free H2O. 30 μL of the NGS ligation mix was loaded onto the sequencing chip twice and the chip was incubated at 40°C on a hotplate for 2 minutes and 45 seconds. The chip was washed with 500 μL of Nuclease-Free H2O twice with 90 seconds in between the washes. 20 μL of 20 pM denatured PhiX (Illumina) was diluted in 580 μL of HT1 buffer (Illumina) and loaded into the sample well of the sequencing cartridge. A 45 to 600 cycle read was conducted using MiSeq V2 chemistry.
Target-Switch SELEX general information
DNA libraries were purchased from TriLink Biotechnologies and all DNA primers were purchased from Integrated DNA Technologies with HPLC purification. All peptides were purchased from Genscript. 10X PBS and Tween-20 were purchased from Sigma-Aldrich. Lambda Exonuclease and buffer were purchased from New England Biolabs. Mag-Bind Total Pure NGS beads were purchased from Omega-Biotek. The bioanalyzer and all reagents, the Bravo liquid handler, and Herculase II Phusion polymerase and buffer were purchased from Agilent. Tubes, plates, and thermocyclers were purchased from Eppendorf. Nunc plates were purchased from VWR. Both 70% and 200 proof ethanol was purchased from Fisher Scientific. Nuclease-free water, MgCl2, Bovine Serum Albumin, dNTP mix, Dynabeads M280 Streptavidin, and QuBit reagents were purchased from Thermo Scientific.
Target-bead conjugation
Target-bead conjugations were performed fresh before each round of incubation. Biotinylated peptide targets were conjugated to M280 streptavidin beads using the Agilent Bravo liquid handling platform. Beads were vortexed to homogeneity before 25 μL beads were added to the appropriate volume for 75 ng peptide target for each conjugation reaction. The beads and target incubated on a chilled plate for 2 minutes to allow the biotin and streptavidin to interact and form a tight bond before the beads were washed several times with SELEX buffer (1x PBS, 0.025% Tween-20, 0.1 mg/mL BSA, 1 mM MgCl2). The final product of the bead conjugation reaction was resuspended in 50 μL of SELEX buffer.
Negative SELEX
DNA aptamer generation was carried out with a protocol involving aptamers in solution and biotinylated targets conjugated to streptavidin beads. The initial library of 1015 aptamers was pulled from the library stock and underwent 30 minutes of negative selection against streptavidin beads in SELEX buffer. The supernatant was kept and put directly into a positive selection against the peptide targets. This positive selection was the first step of 5 rounds of SELEX with the following workflow: selection, amplification (small-scale PCR + large-scale PCR), and single strand generation.
Positive SELEX
Prior to every selection step, aptamers were annealed in Refold Buffer (1x PBS, 0.025% Tween-20, 1 mM MgCl2) for 5 minutes at 95°C and at least 30 minutes at RT.
Selections were carried out in SELEX Buffer for 30 minutes (negative selection) or 1 hour (positive selections) with rotation. Amplification was performed in two steps- small scale PCR and large scale PCR
After washing off non-binders, the remaining target-aptamer conjugates were put directly into a small-scale PCR reaction of 1 reaction (50 μL) per sample. PCR reaction conditions consist of all of the DNA retained from the wash steps, 3 μM forward primer, 3 μM reverse primer, Herculase buffer, 0.2 mM DNTP, 0.0.5 units/μL Herculase polymerase in a final volume of 50 μL.
After this PCR reaction was cleaned, an aliquot of the products was placed into a large-scale PCR with 24 reactions of 50 μL each. The purpose of this large-scale PCR was to amplify the DNA as much as possible without introducing excess PCR bias. PCR reaction conditions consist of 0.17 ng DNA , 6 μM forward primer, 6 μM reverse primer, 1X Herculase buffer, 0.2 mM DNTP, 0.5 units/μL Herculase polymerase in a final volume of 50 μL.
Both small scale and large scale PCR was performed using a Mastercycler Nexus with conditions as follows: 5 minutes at 95°C, 13 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 30 seconds, and 72°C for 5 minutes. PCR reactions were purified using Mag-Bind® TotalPure NGS beads from Omega Bio-Tek and were performed using the Agilent Bravo liquid handling platform. SsDNA and Mag-Bind® TotalPure NGS beads were incubated at a 3:5 ratio and washed with 70% ethanol.
To generate single stranded DNA from the large scale PCR products, digestion with lambda exonuclease was performed at optimized times. Digestion was tracked qualitatively using a bioanalyzer. Cleaned digestions were quantified and used as input into the next selection.
NGS preparation and sequencing
Samples after the SELEX rounds were prepared for sequencing. The samples were normalized to a concentration of 10 ng/μl. A 50 μL PCR reaction (2 μL of 6.25 μM forward and reverse primers, 10 μL of 10 ng/μL DNA sample, 36 μL Master Mix) was set up for each sample to amplify the DNA and the reaction was performed using the Mastercycler Nexus (PCR condition: 98°C for 5 minutes, 10 cycles of 98°C for 30 seconds, 65°C for 30 seconds, 72°C for 30 seconds and 72°C for 5 minutes). After the reaction, the PCR product was cleaned on the Agilent Bravo liquid handling platform. The Tapestation was then used to quantify the size of the PCR product to determine if the PCR reaction was successful. The samples should have DNA size of 170-190 bp. The concentration of the PCR product was determined using the qubit dsDNA assay. The PCR products were then pooled in a tube according to the concentrations of each product. The concentration of the pooled products were determined using the qubit dsDNA assay. PCR product was purified by selecting DNA size 177 bp (Pippin Prep system, Sage Science). The concentration of the purified product was determined using the qubit dsDNA assay. After purification, 10 μL of the purified product was finally sent for NGS sequencing.
Quantification and statistical analysis
DNA barcode chain alignment analysis
NGS reads are processed post-NGS DNA barcode chain sequencing. Each sequence is truncated at the first occurrence of a P7 sequence, which suggests the end of the DNA-barcode chain. For initial clustering of sequences from fastq files, and to determine total sequence count, all similar sequences are grouped together with a corresponding count representing their frequency. For the purposes of this manuscript all analyses required 100% identity (perfect-match). After grouping, BCS components (e.g. foundation and barcode sequences), are identified in the DNA-barcode chain sequence. A named list consisting of each component name and unique barcode sequence used in the experiment is passed as input, where both binder identity and cycle number are encoded in the binder barcode. The foundation binder barcode is used to generate a mapping of the foundation name to a list of all possible binder names. Using this foundation dictionary and the sequencing counts matrix as input, a table is created with each unique sequence from the fastq annotated with the order of components (if any) found in the component dictionary. Only exact matches to the components were permitted.
If the experiment contained multiple barcoding cycles, then a set of additional tables were created to summarize the expected vs. observed cycle positions of barcodes across the experiment. Subsequent tables were generated by barcode position, with counts of binder IDs in rows and foundations as columns, in order to determine the on- vs off-target binding rates for each target-binder pair and to determine the per-cycle efficiency of the BCS machinery.
Formulas defining growth and pen_growth
The number of times a given aptamer sequence appeared in the sequencing data set is the aptamer count. We defined two rounds of SELEX, before and after, as the subset of sequencing data to track the unique aptamer sequences. Before is the subset from round 2 and after is the subset from round 5. We applied a logarithmic scaling factor to each aptamer count to accommodate the wide range of aptamer counts, from 0 to 105
| before = log10 (beforect + 1) |
| after = log10 (afterct + 1) |
Growth is defined as the enrichment of a given aptamer between the before round, round 2, and the after round, round 5.
| growth = after − before = log10 [(beforect + 1)/(afterct + 1)] |
We calculated a raw_penalty value that penalizes sequences that have low count numbers in both round 2 and round 5, multiplied it by a factor y and applied it to the growth factor by subtracting the product of y and raw_penalty.
Technicality:
If before < c, we can use c in the formulas instead, where:
Kd measurement
200 pmol peptide (PPC, PPD) was conjugated to 100 μL Dynabeads™ M-280 Streptavidin
(Thermo Fisher Scientific) following the manufacturer's protocol and resuspended to original concentration in the SELEX buffer. 5 mg fluorescein biotin (Biotinium, #80019) was resuspended in DMSO. 650 pmol fluorescein biotin was conjugated to 100 μL Dynabeads™ M-280 Streptavidin (Thermo Fisher Scientific) following manufacturer's protocol, as a positive control, and resuspended to original concentration. 5' end FAM labeled aptamer candidates #1-10 were purchased from IDT.
Peptide-conjugated beads were diluted to 0.03 mg/mL, or 1:320 of original concentration for the binding assay. 100 μL diluted peptide-conjugated beads or fluorescein conjugated beads were aliquoted into individual wells of a 96 well plate. Plate was placed on a magnetic rack for 2 minutes and the supernatant was removed. FAM labeled aptamers synthesized with attached dye molecules were annealed at 95C and allowed to cool to temperature. 100 μL of 5' end FAM labeled aptamer candidates at varying concentrations (blank control, 100 nM, 250 nM, 500 nM, 750 nM, 1 μM, 2.5 μM, 5 μM, 10 μM, 20 μM), diluted in SELEX buffer, was annealed at 95°C, cooled to RT, and added to appropriate wells. Plate was sealed with plate seal (AB 0558 Adhesive PCR film, ThermoFisher) and rotated in the dark at RT for 1 hour. After incubation, seal was removed and beads were washed 3 times with 100 μL SELEX buffer and resuspended in 100 μL SELEX buffer. Beads were transferred to black plate and single endpoint fluorescent readout was measured using plate reader (Biotek Synergy HTX).
Kd analysis
Computations for Kd analysis were run with python scripts for c following the outline published by Jarmoskaite et al. (2020). The Hyperbola formula is from https://github.com/jimrybarski/biofits/blob/master/biofits/function.py.
Acknowledgments
The authors would like to acknowledge Philip Nelson, John Platt, Erica Brand, Jason Miller, and Patrick Riley for project support and management advice and Tiffany Ly, John Hazard, and Amy Chung-Yu Chou for administrative support. In addition, the authors would like to thank Joshua Cutts, Orion Pritchard, and Samuel Yang for protocol feedback, automation support, and microscopy assistance, as well as Marytheresa Ifediba and Maureen Mckeague for technical discussions. The work presented here was funded through Google Research.
Author contributions
Conceptualization, A.P.; methodology, Z.C., J.M.H., D.W., P.J., V.A.C., M. Chavarha, M.G., M.B., B.W, A.P.; software, M.B., B.W., A.B., M. Coram; validation, M.G., D.W., S.S., Z.C., J.M.H., M.F., A.H.-T.L., A.T., P.J., A.P.; formal analysis, B.W., A.B., P.J., D.W., M.G., M.B., A.P.; investigation, M. Chavarha, S.S., Y.C., D.W., J.M.H., M.G., Z.C., M.F., A.H.-T.L., L.S., A.T., P.J., A.P.; writing - original draft, J.M.H., A.B., D.W., S.S., M. Chavarha, Y.C., M.F., V.A.C., L.C., S.A., M.D., B.W., P.J., A.P.; writing - review & editing, J.M.H., V.A.C., L.C., B.W., P.J., A.P.; visualization, S.S., A.B., B.W., D.W., Y.C., J.M.H., L.C., M. Coram, P.J., A.P.; supervision, AP; project administration, AP.
Declaration of interests
A.B., M.D., M.C., B.W., P.J., M.B., and A.P. are employees and shareholders of Alphabet. Google has filed patent applications related to this work, including PCT/US2020/050574, PCT/US2020/053716, PCT/US2020/040130, PCT/US2020/053715. US20210079398A1, US20210079557A1, US20210102248A1, and WO2021051011A1. M.C. is an inventor of US10546650B2.
All work was completed while authors were affiliated with Alphabet. Current affiliations for authors who have moved on from Alphabet are the following:
J.H., S.S., Z.C., M.F., L.C., University of California, San Francisco (UCSF); M.G., 10x Genomics; D.W., Genentech, employee and shareholder; Y.C., University of Washington, Seattle; S.A., Alkahest, employee; L.S., Insitro, employee; A.H.-T.L., University of California Los Angeles (UCLA); V.A.C., Washington University School of Medicine, St. Louis.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103586.
Supplemental information
Data and code availability
Raw sequencing has been deposited at Mendeley (https://data.mendeley.com/datasets/f9hdn5xc3v/1) and Harvard Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi%3A10.7910%2FDVN%2FW903IJ) and all original code has been deposited on Github (https://github.com/google-research/google-research/tree/master/protseq). They are publicly available as of the date of this article's publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Alfaro J.A., Bohländer P., Dai M., Filius M., Howard C.J., van Kooten X.F., Ohayon S., Pomorski A., Schmid S., Aksimentiev A., et al. The emerging landscape of single-molecule protein sequencing technologies. Nat. Methods. 2021;18:604–617. doi: 10.1038/s41592-021-01143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames T.D., Breaker R.R. Bacterial aptamers that selectively bind glutamine. RNA Biol. 2011;8:82–89. doi: 10.4161/rna.8.1.13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir A., Yang Q., Wang J., Hoyer S., Chou W., McLean C., Davis G., Gong Q., Armstrong Z., Jang J., et al. Machine learning guided aptamer refinement and discovery. Nat. Commun. 2021;12:2366. doi: 10.1038/s41467-021-22555-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman T., Cederlund E., Jörnvall H. Chemical C-terminal protein sequence analysis: improved sensitivity, length of degradation, proline passage, and combination with edman degradation. Anal. Biochem. 2001;290:74–82. doi: 10.1006/abio.2000.4922. [DOI] [PubMed] [Google Scholar]
- Casagranda F., Wilshire J.F. C-terminal sequencing of peptides. The thiocyanate degradation method. Methods Mol Biol. 1994;32:335–349. doi: 10.1385/0-89603-268-X:335. [DOI] [PubMed] [Google Scholar]
- Castellana N., Pham V., Arnott D., Lill J., Bafna V. Template proteogenomics: sequencing whole proteins using an imperfect database. Mol. Cell Proteomics. 2010;9:1260–1270. doi: 10.1074/mcp.M900504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K.M., Yang K.-A., Nakatsuka N., Zhao C., Ye M., Jung M.E., Yang H., Weiss P.S., Stojanović M.N., Andrews A.M. Phenylalanine monitoring via aptamer-field-effect transistor sensors. ACS Sens. 2019;4:3308–3317. doi: 10.1021/acssensors.9b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M., Oh S.S., Nie J., Stewart R., Eisenstein M., Chambers J., Marth J.D., Walker F., Thomson J.A., Soh H.T. Quantitative selection and parallel characterization of aptamers. Proc. Natl. Acad. Sci. U S A. 2013;110:18460–18465. doi: 10.1073/pnas.1315866110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Fabricius V., Lefèbre J., Geertsema H., Marino S.F., Ewers H. Rapid and efficient C-terminal labeling of nanobodies for DNA-PAINT. J. Phys. D: Appl. Phys. 2018;51:474005. doi: 10.1088/1361-6463/aae0e2. [DOI] [Google Scholar]
- Gawande B.N., Rohloff J.C., Carter J.D., Carlowitz I.von, Zhang C., Schneider D.J., Janjic N. Selection of DNA aptamers with two modified bases. Proc. Natl. Acad. Sci. U S A. 2017;114:2898–2903. doi: 10.1073/pnas.1615475114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger A., Burgstaller P., von der Eltz H., Roeder A., Famulok M. RNA aptamers that bind L-arginine with sub-micromolar dissociation constants and high enantioselectivity. Nucleic Acids Res. 1996;24:1029–1036. doi: 10.1093/nar/24.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold L., Ayers D., Bertino J., Bock C., Bock A., Brody E.N., Carter J., Dalby A.B., Eaton B.E., Fitzwater T., et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C.K.L., Wu D., Pusuluri A., Feagin T.A., Csordas A.T., Eisenstein M.S., Hawker C.J., Niu J., Soh H.T. Click-particle display for base-modified aptamer discovery. ACS Chem. Biol. 2019;14:2652–2662. doi: 10.1021/acschembio.9b00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider S., Pal R. Integrated analysis of transcriptomic and proteomic data. Curr. Genomics. 2013;14:91–110. doi: 10.2174/1389202911314020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez F.J., Kalra N., Wengel J., Vester B. Aptamers as a model for functional evaluation of LNA and 2’-amino LNA. Bioorg. Med. Chem. Lett. 2009;19:6585–6587. doi: 10.1016/j.bmcl.2009.10.039. [DOI] [PubMed] [Google Scholar]
- Hu Z.-L., Huo M.-Z., Ying Y.-L., Long Y.-T. Biological nanopore approach for single-molecule protein sequencing. Angew. Chem. Int. Ed. Engl. 2021;60:14738–14749. doi: 10.1002/anie.202013462. [DOI] [PubMed] [Google Scholar]
- Jarmoskaite I., AlSadhan I., Vaidyanathan P.P., Herschlag D. How to measure and evaluate binding affinities. eLife. 2020;9 doi: 10.7554/eLife.57264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenison R., Gill S., Pardi A., Polisky B. High-resolution molecular discrimination by RNA. Science. 1994;263:1425–1429. doi: 10.1126/science.7510417. [DOI] [PubMed] [Google Scholar]
- Kim M.-S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S., et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., You M., Pu Y., Liu H., Ye M., Tan W. Recent developments in protein and cell-targeted aptamer selection and applications. Curr. Med. Chem. 2011;18:4117–4125. doi: 10.2174/092986711797189619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loakes D., Brown D.M. 5-Nitroindole as an universal base analogue. Nucl. Acids Res. 1994;22:4039–4043. doi: 10.1093/nar/22.20.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerfeld I., Yarus M. An RNA pocket for an aliphatic hydrophobe. Nat. Struct. Biol. 1994;1:287–292. doi: 10.1038/nsb0594-287. [DOI] [PubMed] [Google Scholar]
- McKeague M., DeRosa M.C. Challenges and opportunities for small molecule aptamer development. J. Nucleic Acids. 2012;2012:1–20. doi: 10.1155/2012/748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaï A., Rath A., Delarue P., Senet P. Nanopore sensing of single-biomolecules: a new procedure to identify protein sequence motifs from molecular dynamics. Nanoscale. 2020;12:22743–22753. doi: 10.1039/d0nr05185c. [DOI] [PubMed] [Google Scholar]
- Pawlosky A., Hong J., Shao S., Church V., Dimon M., Berndl M. U.S. Patent No. 17/019109. U.S. Patent and Trademark Office; 2019. Methods and Compositions for Protein and Peptide Sequencing. [Google Scholar]
- Payne S.H. The utility of protein and mRNA correlation. Trends Biochem. Sci. 2015;40:1–3. doi: 10.1016/j.tibs.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham V., Tropea J., Wong S., Quach J., Henzel W.J. High-throughput protein sequencing. Anal. Chem. 2003;75:875–882. doi: 10.1021/ac0206317. [DOI] [PubMed] [Google Scholar]
- Ruscito A., McConnell E.M., Koudrina A., Velu R., Mattice C., Hunt V., McKeague M., DeRosa M.C. In vitro selection and characterization of DNA aptamers to a small molecule target: DNA aptamers for small molecule targets. Curr. Protoc. Chem. Biol. 2017;9:233–268. doi: 10.1002/cpch.28. [DOI] [PubMed] [Google Scholar]
- Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A., et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Shangguan D., Li Y., Tang Z., Cao Z.C., Chen H.W., Mallikaratchy P., Sefah K., Yang C.J., Tan W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. 2006;103:11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheynkman G.M., Shortreed M.R., Cesnik A.J., Smith L.M. Proteogenomics: integrating next-generation sequencing and mass spectrometry to characterize human proteomic variation. Annu. Rev. Anal. Chem. 2016;9:521–545. doi: 10.1146/annurev-anchem-071015-041722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2021;60:1–9. doi: 10.1016/j.cbpa.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan J., Boulgakov A.A., Hernandez E.T., Bardo A.M., Bachman J.L., Marotta J., Johnson A.M., Anslyn E.V., Marcotte E.M. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol. 2018;36:1076–1082. doi: 10.1038/nbt.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan J., Boulgakov A.A., Marcotte E.M. A theoretical justification for single molecule peptide sequencing. Plos Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasset D.M., Kubik M.F., Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J. Mol. Biol. 1997;272:688–698. doi: 10.1006/jmbi.1997.1275. [DOI] [PubMed] [Google Scholar]
- Timp W., Timp G. Beyond mass spectrometry, the next step in proteomics. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aax8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolle F., Brändle G.M., Matzner D., Mayer G. A versatile approach towards nucleobase-modified aptamers. Angew. Chem. Int. Ed. Engl. 2015;54:10971–10974. doi: 10.1002/anie.201503652. [DOI] [PubMed] [Google Scholar]
- Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Virant D., Traenkle B., Maier J., Kaiser P.D., Bodenhöfer M., Schmees C., Vojnovic I., Pisak-Lukáts B., Endesfelder U., Rothbauer U. A peptide tag-specific nanobody enables high-quality labeling for dSTORM imaging. Nat. Commun. 2018;9:930. doi: 10.1038/s41467-018-03191-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- What is UniProt’s Human Proteome? [WWW Document], 2019. UniProt. https://www.uniprot.org/help/human_proteome
- White R., Rusconi C., Scardino E., Wolberg A., Lawson J., Hoffman M., Sullenger B. Generation of species cross-reactive aptamers using “toggle” SELEX. Mol. Ther. 2001;4:567–573. doi: 10.1006/mthe.2001.0495. [DOI] [PubMed] [Google Scholar]
- Williams S.M., Liyu A.V., Tsai C.-F., Moore R.J., Orton D.J., Chrisler W.B., Gaffrey M.J., Liu T., Smith R.D., Kelly R.T., et al. Automated coupling of nanodroplet sample preparation with liquid chromatography-mass spectrometry for high-throughput single-cell proteomics. Anal. Chem. 2020;92:10588–10596. doi: 10.1021/acs.analchem.0c01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Bing T., Mei H., Fang C., Cao Z., Shangguan D. Characterization and application of a DNA aptamer binding to l-tryptophan. Analyst. 2011;136:577–585. doi: 10.1039/C0AN00550A. [DOI] [PubMed] [Google Scholar]
- Yao Y., Docter M., van Ginkel J., de Ridder D., Joo C. Single-molecule protein sequencing through fingerprinting: computational assessment. Phys. Biol. 2015;12 doi: 10.1088/1478-3975/12/5/055003. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Piehowski P.D., Zhao R., Chen J., Shen Y., Moore R.J., Shukla A.K., Petyuk V.A., Campbell-Thompson M., Mathews C.E., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat. Commun. 2018;9:882. doi: 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing has been deposited at Mendeley (https://data.mendeley.com/datasets/f9hdn5xc3v/1) and Harvard Dataverse (https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi%3A10.7910%2FDVN%2FW903IJ) and all original code has been deposited on Github (https://github.com/google-research/google-research/tree/master/protseq). They are publicly available as of the date of this article's publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.