

Abstract
Peptides were designed to inhibit the protein-protein interaction of CD2 and CD58 to modulate the immune response. This work involved the design and synthesis of eight different peptides by replacing each amino acid residue in peptide 6 with alanine as well as grafting the peptide to the sunflower trypsin-inhibitor framework. From the alanine scanning studies, mutation at position 2 of the peptide was shown to result in increased potency to inhibit cell adhesion interactions. The most potent peptide from the alanine scanning was further studied for its detailed 3D structure and binding to CD58 protein using surface plasmon resonance and flow-cytometry. This peptide was used to graft to the sunflower trypsin inhibitor to improve the stability of the peptide. The grafted peptide, SFTI-a1, was further studied for its potency as well as its thermal, chemical, and enzymatic stability. The grafted peptide exhibited improved activity compared to our previously grafted peptide and was stable against thermal and enzymatic degradation.
Keywords: alanine scanning, protein-protein interaction, CD2, CD58, cell adhesion, immunomodulation
Graphical Abstract
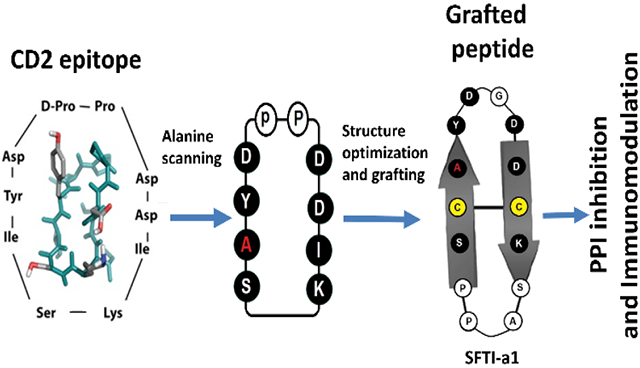
Peptides from CD2 protein epitope were designed to inhibit CD2-CD58 interactions. The most potent peptide was grafted on to sunflower trypsin inhibitor (SFTI) framework. The grafted peptide was shown to suppress T cell immune response in vitro and was shown to be stable against thermal and enzymatic degradation.
1. INTRODUCTION
Immune response in humans is mediated by signaling from protein molecules in a highly orchestrated process. T-cell receptor (TCR) and major histocompatibility complex (MHC) molecules interact with one another in a highly specific manner between T cells and antigen-presenting cells (APC) to generate the immune response (Bretscher, 1999). Apart from TCR and MHC, co-stimulatory molecules play a major role in holding the T cells and APC in close contact to generate signals for other cytokines (L. Chen & Flies, 2013; Davis et al., 2003; van der Merwe & Davis, 2003). Among the costimulatory molecules, CD58 (also called lymphocyte function-associated antigen-3 (LFA-3)) on APC binds to CD2 on T cells in the early stages of the immune response. CD2 and CD58 molecules have been shown to be upregulated in autoimmune diseases such as rheumatoid arthritis (Balanescu et al., 2002; Chamian et al., 2005; Raychaudhuri et al., 2009). Signaling from CD2-CD58 generates cytokine production, T cell responsiveness to IL-12, and induction of TNF-α (Chamian et al., 2005; L. Chen & Flies, 2013; Davis et al., 2003; Espagnolle et al., 2007; Hutchings, Clarkson, Chalkley, Barclay, & Brown, 2003). Modulation of CD2-CD58 interactions by antibodies and fusion proteins has been shown to reduce the inflammatory response (Chamian et al., 2005; Mrowietz, 2002; Shao et al., 2018; Webber, Hirose, & Vincenti, 2011).
The involvement of co-stimulatory molecules in inflammatory response make CD2 and CD58 molecules attractive targets for understanding the mechanism(s) of protein-protein interaction (PPI) inhibition in cell adhesion in autoimmune and inflammatory diseases. We have designed peptides from CD2 that can bind to CD58 and inhibit the PPI of CD2-CD58 (A. Gokhale, Kanthala, Latendresse, Taneja, & Satyanarayanajois, 2013; A. Gokhale, Weldeghiorghis, Taneja, & Satyanarayanajois, 2011; A. S. Gokhale et al., 2015; Sable et al., 2016). Among several designed peptides, peptide 6 demonstrated potential activity to inhibit the adhesion between CD2 protein-expressing T cells and CD58 protein overexpressing Caco-2 cells. Peptide 6 (Table 1) exhibited the most potent activity with an IC50 value of 6.9 nM in cell adhesion assay and was able to suppress the T cell immune response in cells derived from transgenic mice that develop arthritis similar to human arthritis (A. Gokhale et al., 2013; A. S. Gokhale et al., 2015). Considering these results, peptide 6 was selected as a starting compound for further studies. To improve the stability of this active peptide (peptide 6), we adopted a novel strategy of multicyclic peptide grafting. When this active peptide sequence was grafted onto the sunflower trypsin inhibitor (SFTI) framework, a slight decrease in vitro activity with IC50 value, 43 nM was observed (Sable et al., 2016). There could be many possibilities for this observed decrease in the activity such as a) grafting onto SFTI framework that results in rigidity to the structure, b) steric hindrance created by side chains present in amino acid residues in a sequence, or c) change in conformation of the peptide and hence orientation of important amino acid residue in the grafted peptide due to extra amino acid residues present in SFTI-a compared to peptide 6.
TABLE 1.
The sequence of peptide 6 and alanine scanning peptides along with cell adhesion inhibition activity of peptides using two models, namely, Jurkat T cells and OVACR-3 cells and Jurkat T cells and HFLS-RA cells. Substitution /replacement of amino acid by alanine is shown in bold.
| Code | Sequence | Cell adhesion inhibition IC50 (μM) |
|
|---|---|---|---|
| Jurkat- OVCAR-3 |
Jurkat- HFLS-RA |
||
| Peptide 6 | Cyclo (1,10) S1I2Y3D4p5P6D7D8I9K10 | 0.0069 ±0.0004 | 0.036±0.020 |
| AS1 | Cyclo (1,10)A1I2Y3D4p5P6D7D8I9K10 | >50 | ND |
| AS2 | Cyclo(1,10)S1A2Y3D4p5P6D7D8I9K10 | 0.0011 ± 0.0009 | 0.0031 ± 0.0012 |
| AS3 | Cyclo(1,10)S1I2A3D4p5P6D7D8I9K10 | >50 | ND |
| AS4 | Cyclo(1,10)S1I2Y3A4p5P6D7D8I9K10 | >50 | ND |
| AS5 | Cyclo(1,10)S1I2Y3D4p5P6A7D8I9K10 | >50 | ND |
| AS6 | Cyclo(1,10)S1I2Y3D4p5P6D7A8I9K10 | >50 | ND |
| AS7 | Cyclo(1,10)S1I2Y3D4p5P6D7D8A9K10 | 0.143 ± 0.096 | ND |
| AS8 | Cyclo(1,10)S1I2Y3D4p5P6D7D8I9A10 | >50 | ND |
L amino acids are represented by the capital letter with single letter code for amino acids. D amino acids are indicated by small letters. Peptide 6 was reported in our earlier publication, Ameya et al., J. Medicinal Chem. 54 (2011) 5307-5319. Amino acids are represented by single-letter code.
In the grafted peptide, amino acids in the loop, disulfide bond, and prolines are important in retaining the stability of the peptide, and we decided to investigate the importance of amino acids in peptide 6 by alanine scanning (Figure 1/Table 1). Alanine scanning is widely used for the evaluation of the importance or functional role of amino acid residues present in peptides/proteins. Alanine is the best choice for substitution for several reasons, such as a) alanine eliminates the side-chain beyond the β carbon, b) alanine will not contribute to major changes in the backbone conformation (as can occur in the case of glycine or proline), and c) alanine does not enforce extreme electrostatic or steric effects (Kristensen et al., 1997; Lefevre, Remy, & Masson, 1997; Weiss, Watanabe, Zhong, Goddard, & Sidhu, 2000).
FIGURE 1.

Design of peptides from CD2 adhesion domain and grafting of peptides to sunflower trypsin inhibitor using cyclic peptide 6 and alanine scanning. A) F & C strands of CD2 adhesion domain that binds to CD58 protein. Amino acid residues that are important in binding to CD58 are indicated with green circles. Amino acids from the adhesion domain used for the design of peptide is shown in a box. B) Peptide 6 was designed from the adhesion domain of CD2 that binds to CD58. D-Pro-L-Pro was used to induce β-turn and to stabilize the β-strand structure of the peptide with cyclization. C) Alanine scanning of peptide 6 resulted in peptide AS2. Ala replacement is shown in red. D) Sunflower trypsin inhibitor (SFTI-wt) peptide used as a template to graft the CD2 peptide onto SFTI. E) SFTI-a peptide designed based on SFTI and CD2 peptide 6. F) SFTI-a1 was designed based on peptides AS2 and SFTI-a. Cys residues involved in the disulfide bond from SFTI template are shown in yellow. Amino acids grafted from peptide 6 are shown in filled circles (dark). Amino acids that are present in peptide 6, as well as SFTI-wt, are shown in dark shaded circles and highlighted with a red line. Amino acids introduced in the loop region for grafting are shown in the light shaded circle.
The work in the current study involved the design and synthesis of eight different peptides by replacing each amino acid residue in peptide 6 with alanine (Table 1) as well as grafting the peptide to the sunflower trypsin-inhibitor framework (Table 2). Our purpose was to evaluate the importance of amino acid residues in peptide 6 (a lead molecule) using alanine scanning and to determine the ability of peptides to inhibit CD2-CD58 PPI using lymphocyte-epithelial cell adhesion assay. From the alanine scanning studies, mutation at position Ile2 of the peptide resulted in an increase in potency to inhibit cell adhesion interaction. The most potent peptide from alanine scanning was further studied for its detailed three-dimensional (3D) structure using NMR and binding to CD58 protein using surface plasmon resonance and flow-cytometry. This peptide was used to graft to the sunflower trypsin inhibitor to improve the stability of the peptide (Figure 1). The grafted peptide SFTI-a1 was further studied for its potency and its thermal, chemical, and enzymatic stability. The grafted peptide exhibited improved activity compared to our previous grafted peptide and was stable against thermal, chemical, and enzymatic degradation. Furthermore, the grafted peptide was able to inhibit the PPI of CD2 and CD58 and modulate the CD2-generated signaling for T-cell proliferation.
TABLE 2.
The sequence of grafted peptides and cell adhesion inhibition activity of peptides using two models, namely, Jurkat T cells and OVACR-3 cells and Jurkat T cells and HFLS-RA cells.
| Peptide Code |
Peptide Sequence | Lymphocyte Cell Adhesion Inhibition Assay IC50 (μM) |
|
|---|---|---|---|
| Jurkat - OVCA R-3 |
Jurkat- HFLS- RA |
||
| SFTI-a* | Cyclo[C1K2A3S4A5P6P7S8C9Y10D11G12D13D14] | 0.043 ± 0.025 | 0.051 ± 0.025 |
| SFTI-a1 | Cyclo[C1K2S3A4P5P6S7C8A9Y10D11G12D13D14] | 0.023 ± 0.046 | 0.037 ± 0.001 |
| SFTI-a2 | Cyclo[C1K2A3S4A5P6P7S8C9(homotyrosine)10D11G12D13D14] | >50 | ND |
| SFTI-a3 | Cyclo[C1K2A3S4A5P6P7S8C9(3-Amino-3-(1-Napthyl)Propanoic Acid)10D11G12D13D14] | >50 | ND |
| SFTI-a4 | Cyclo[c1k2a3s4a5p6p7s8c9y10d11g12d13d14] | >50 | ND |
| SFTI-a5 | Cyclo[d1d2g3d4y5c6s7p8p9a10s11a12k13c14] | >50 | ND |
Disulfide bonds are indicated by underline.
L amino acids are represented by capital letter with single letter code for amino acids. D amino acids are indicated by small letters.
Sable et al., ACS Chem. Biol. 2016 11(8):2366-2374. Amino acids are represented by single letter code
2. EXPERIMENTAL SECTION
2.1. Cell Lines
Human ovarian carcinoma cells (OVCAR-3) and human T-lymphocyte cells (Jurkat, E6-1) were purchased from the American Type Culture Collection (ATCC). OVCAR-3 cells were cultured in RPMI-1640 medium from ATCC, with 20% FBS, 0.01 mg/mL bovine insulin, and 0.1 mg/mL penicillin/streptomycin. Jurkat cells were cultured in RPMI-1640 medium (ATCC) with 10% FBS, 0.01 mg/mL bovine insulin and 0.1 mg/mL penicillin/streptomycin. Human fibroblast-like synoviocytes rheumatoid arthritis (HFLS-RA) cells were purchased from Cell Applications, Inc., San Diego, CA, USA, and maintained in human synoviocyte growth medium purchased from Cell Applications, Inc. OVCAR-3 cells and HFLS-RA cells were used as a model for cells expressing CD58 protein, and Jurkat cells were used as a model for human T cells.
2.2. Chemicals
Fmoc-protected amino acids, piperidine, trifluoroacetic acid (TFA), 2-chlorotrityl chloride resin, 2 - (6 - chloro - 1H - benzotriazole - 1 - yl) - 1,1,3,3 - tetramethylaminium hexafluorophosphate (HCTU), and 1, 1-[bis(dimethylamino) methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) were purchased from Advanced ChemTech (Louisville, KY). Diisopropylethylamine (DIEA), triisopropylsilane (TIPS), and N-methylmorpholine (NMM) were purchased from Sigma Aldrich (St. Louis, MO). Dimethylformamide (DMF) was purchased from Protein Technologies (Tucson, AZ). Dichloromethane (DCM), methanol (MeOH), and acetonitrile (CH3CN) were purchased from VWR International, LLC (Radnor, PA).
2.3. Design and Synthesis of Alanine Scanning Peptides
Eight different sequences were designed by replacing each amino acid residue at a time. F-moc-based solid phase synthesis method was used for the synthesis of all sequences (Ieronymaki et al., 2015). Linear peptides were synthesized by automatic Fmoc-based solid phase-peptide synthesis and peptide was cleaved from the resin without removal of side-chain protecting group. In the case of SFTI-a1, after cyclization of the backbone, disulfide bond cyclization was performed.
2.3.1. Preloaded H-Pro-CTC resin
One gram of 2-chlorotrityl chloride resin (1.2 mmol/g) was swelled with DCM (10 mL) for 30 min and drained. A solution of Fmoc-proline (405 mg) and DIEA (1.05 mL) in DCM (10 mL) was added to the resin and shaken for 30 min. The resin was then drained and washed with DCM (10 mL, 5 x 2 min). A solution of DCM:MeOH:DIEA (10 mL, 85:15:5) was added to the resin and shaken for 15 min then drained. This step was repeated twice for a total of 3 times. The resin was then washed with DMF (10 mL, 5 x 30 sec) and DCM (10 mL, 5 x 30 sec).
A small amount of resin (2-3 mg) was weighed into a 3 ml quartz cuvette to which was added 20% piperidine in NMP (3 mL). The solution was allowed to react for 20 min with occasional stirring. After the resin fully settled to the bottom of the cuvette, the UV absorption was measured at 290 nm with a UV spectrometer, and the resin loading level was determined.
2.3.2. Synthesis of Linear Sequence
The title peptides were synthesized with the previously loaded H-Pro-CTC resin on a Tribute Peptide Synthesizer (Protein Technologies, Tucson, AZ) utilizing a standard Fmoc peptide chemistry protocol on a 100 μmol scale. Fivefold excess of Fmoc-amino acids (Fmoc-Ala-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-d-Pro-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Ser (tBu)-OH) and HCTU, in the presence of 10 equivalents of NMM were used for each of the amino acid coupling steps. After the synthesis was completed, the resin was deprotected with 20% piperidine in NMP then washed with DMF (10 mL, 5 x 30 sec) and DCM (10 mL, 5 x 30 sec). The peptide was cleaved from the resin with a solution of 1% TFA in DCM (3 mL) for 5 min. The solution was drained and collected in a round bottom flask. This procedure was repeated for a total of 10 times. The solutions were combined and evaporated, yielding the crude linear peptide as an off white solid.
2.3.3. Cyclization of Synthesized Linear Peptide
The crude linear peptide was dissolved in DMF (60 mL) to which was added HATU (114 mg) and DIEA (170 uL). The solution was allowed to mix for 2 hrs. After completion, the solvent was removed obtain the cyclized peptide as an off white solid. The crude cyclized peptide was deprotected using TFA/water/TIPS (4 mL, 95:2.5:2.5) for 2 hours in a 50 mL centrifuge tube. Cold diethyl ether was then added to the peptide solution to precipitate the crude peptide. The peptide was centrifuged for 10 minutes (4,000 rpm), and the ether layers decanted. Fresh cold diethyl ether was added and the pelleted peptide was re-suspended. The peptide was centrifuged again and the procedure was repeated 5 times in total. After the final ether wash, the peptide pellet was dissolved in 5 mL water containing 0.1% TFA, frozen and lyophilized.
2.3.4. Design and Synthesis of Grafted Peptides
Sunflower trypsin inhibitor (SFTI-wt) framework was chosen for grafting of the peptide. Peptides were synthesized using the previously reported method (Sable et al., 2016). Cyclic peptides were synthesized, as explained above. After cyclization and deprotection of side chains, the disulfide bonds were introduced to the peptide by oxidation reaction in the presence of iodine.
2.3.5. Analysis
Peptides were analyzed by analytical HPLC and mass spectrometry for the percentage of purity and correct molecular ion for the peptide. Analytical HPLC was performed in a C18 column [(Agilent Zorbax 300 SB-C18 (5 μm, 4.6 × 250 mm, SN USHH007525)] with Agilent guard column Zorbax 300 SB-C18 (5 μm, 4.6 × 12.5 mm). The analysis was performed using mobile phases A (0.1% TFA in water) and B (0.1% TFA in acetonitrile) and detected at a wavelength of 215 nm. The gradient started from 5% B to 55% B in 50 min, with 1 mL/min flow rate. Supporting information Table 1 provides analytical data for peptides designed for alanine scanning. Analytical data for peptides designed for grafted peptides are shown in Table 2 and in Supporting Information.
2.4. Lymphocyte Cell Adhesion Assay
Approximately 1 × 104 OVCAR-3 cells were coated per well in 96-well plates, and cell adhesion assay was carried out as described in our previous publication inhibition (Liu, Chow, & Jois, 2004). All stock solutions of peptides (2 mg/mL) were prepared in PBS. Different concentrations of these cyclic peptides were prepared by diluting the above stock solution in serum-free RPMI1640 medium and the cells were treated with 100 μl of peptides with different concentration. The Jurkat cells were fluorescently tagged with 2',7'-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein, Acetoxymethyl Ester (BCECF AM) dye and incubated with peptide-pretreated OVCAR-3 cells. For the analysis of the inhibition assay, the fluorescence of the obtained by bound Jurkat cells after washing was determined with a microplate fluorescence analyzer (Biotek Synergy, Winooski, VT, USA) with excitation wavelength at 485 nm and emission wavelength at 528 (± 20) nm. For AS2 and SFTI-a1 grafted peptides, similar experiments were repeated with HFLS-RA cells and Jurkat cells.
2.5. Nuclear Magnetic Resonance (NMR)
The samples were prepared in 90% H2O/10% D2O at a concentration of 1 mM. A Bruker Avance 500 MHz spectrometer was used to record spectra at 298 K. Different 2D NMR experiments were carried out, including TOCSY (MLEV-17 spin lock with an 80 ms mixing time) and ROESY and NOESY (200 ms mixing time). Chemical shifts were processed with Top Spin (Bruker) and referenced to the water resonance at 4.77 ppm. NMR spectra were processed using Bruker processing software, and files were converted to SPARKY format. Analysis of NMR data was done using SPARKY software (Lee, Tonelli, & Markley, 2015).
2.6. Circular Dichroism (CD)
CD spectroscopic studies were performed on a JASCO J-815 CD spectrometer. Peptides were dissolved in water (40 μM), and data were collected using a 1 mm path-length quartz cuvette. Data were collected from 185-–380 nm wavelengths with four accumulations per sample. The wavelength vs. CD ellipticity spectra obtained were analyzed using spectral analysis software. The baseline obtained with solvent was subtracted from each spectrum.
2.7. Surface Plasmon Resonance (SPR) Analysis
The human recombinant adhesion domain of CD58 protein (22 KD) was purchased from Abcam (Cambridge, UK). The CM5 type of sensor chip (GE Health Care Life Sciences) was used to immobilize CD58 protein, and analysis was done using SPR (GE Health Care Life Sciences, Biacore X100). The protein was immobilized in pH 4 acetate buffer condition on flow cell 2 with a flow rate of 5 μL/min, and flow cell 1 was used as a reference. The HBS-EP buffer was used as a run buffer for the SPR study. Different concentrations of peptide ranging from 0.25 to 100 μM were used to titrate with immobilized CD58 protein, and the binding response was measured based on a change in the angle of reflected polarized light with changes in sensor chip surface (because of binding). The binding curve results were fitted into 1:1 Langmuir equation to obtain the Kd values.
For the grafted peptides, surface plasmon resonance experiments were done using Reichert 2SPR (2-Channel) from Reichert Technologies, Life Sciences, Depew, NY, USA. CD58 protein (Abcam) was immobilized over the carboxymethyl dextran hydrogel surface sensor chip (CM-5) over the left channel with pH 4.0, 10 mM sodium acetate buffer; the right channel was used as a reference. HBS-EP buffer from GE Healthcare was used as a run buffer. Different concentrations of SFTI-a1 peptide was titrated and the binding response was measured. Binding curves were fitted to 1:1 binding model for kinetic evaluation in Tracedrawer software to evaluate the Kd value.
2.8. Competitive Binding Experiment
Competitive binding experiments were performed to confirm that the designed peptide binds to CD58 protein on the cell surface. 1 million OVCAR-3 cells were taken for each sample. The cells were fixed with formaldehyde and incubated for 15 min with different concentrations of SFTI-a1 peptide. After 15 min, 20 μL of FITC-conjugated CD58 antibody (Santa Cruz) was added to each Eppendorf tube and incubated for 45 additional min. Negative control was prepared without antibody treatment (cells only). After 45 min, the mixture was centrifuged at 2500 rpm for 10 min and pellets were resuspended in 2 mL of cold PBS. Cell suspensions were subjected to flow cytometry analysis. Cell count vs. fluorescence unit histograms were plotted after analysis of 10,000 cells from each sample.
2.9. Docking
Autodock Tools and Autodock version 4.2 (Morris et al., 2009) were used to dock the peptides onto CD58 protein adhesion domain from the crystal structure of CD58 (J. H. Wang et al., 1999). The peptide structures were docked to the protein using the Lamarckian genetic algorithm method, in which 10 million energy evaluations are done by starting with 150 starting confirmations. The top 50 low-energy docked structures were analyzed. For visualization of the lowest energy structures, PyMol software was used. Computational studies were carried out on a local workstation as well as on a high-performance computer (HPC) at LSU, Baton Rouge via the Louisiana Optical Network Initiative (LONI).
2.10. Thermal Stability
Circular dichroism was used to determine the stability of the peptide backbone at various temperatures. JASCO J-815 CD spectrometer (Jasco, Easton MD) was used for temperature interval measurement. Experiments were done with gradual 5 °C increases in temperature from 25 to 85 °C; 4 scans were taken for each temperature, and graphs were plotted against wavelength from 185 nm to 320 nm. Samples were taken from 25, 55, and 85 °C and mass of the sample was analyzed using mass spectrometry. Peptide samples were also incubated at 25, 55, and 85 °C for 30 min and analyzed for stability by HPLC method using a C-18 column.
2.11. Disulfide Bond Stability
The stability of the disulfide bonds was analyzed by circular dichroism (CD) spectrometer. Peptides were incubated for 30 min in the presence of 0.1 and 1 mM dithiothreitol. Changes in addition of dithiothreitol (DTT) to the peptide were also analyzed by mass spectrometry.
2.12. Serum Stability
The stability of the peptides in the presence of human serum was analyzed for 24 h. Stock solutions of the peptide (2 mM) were prepared in PBS. Peptides were incubated with pooled human serum (Innovative Research, Inc, Novi MI) in a ratio of 1:9 (peptide:100% human serum/150:1350 μL). At each time point, 150 μL of the solution was taken out and ice-cold acetonitrile was added to it. The mixture was centrifuged at 10,000 rpm for 10 min and the supernatant was passed through a Sep-pak column (Waters) and lyophilized. Lyophilized samples were reconstituted with 300 μL of ACN: water (50:50) and analyzed by HPLC (Shimadzu) for change in peptide concentration.
2.13. Liver Microsome Stability
For liver microsome stability assay, pools of liver microsome enzymes were purchased from Corning (Corning, NY, USA). 20 μL peptide, 200 μL of 10 mM NADPH, 200 μL of 40 mM CaCl2, and 50 μL of human liver microsome solution (20 mg/mL) were mixed and incubated for 2 h at 37 °C with constant shaking (Knights, Stresser, Miners, & Crespi, 2016). At each time point, 250 μL of the mixture was taken out, and ice-cold acetonitrile was added to it. The mixture was kept on ice for 2 min and centrifuged for 10 min at 10,000 rpm. The supernatant was passed through a Sep-pak column (Waters) and lyophilized. The dry sample was reconstituted with acetonitrile/water mixture for HPLC (Shimadzu) analysis. Verapamil was used as control to validate the microsomal assay.
2.14. Chemical Stability
For chemical stability, peptide was incubated in pH 1, pH 7.4 and pH 9.6 buffers for 30 days and samples were analyzed at different time intervals using HPLC (C-18 column HCl (0.1 M) pH 1, 0.1 M PBS pH 7.4 buffer, and carbonate/bicarbonate buffer was used as pH 9.6 buffer).
2.15. Calcium Flux Assay
Calcium concentrations inside Jurkat cells were compared using Fluo-8 calcium dye (Abcam). 10,000 HFLS-RA cells in each well were seeded in 96-well plates. After 72 h, HFLS-RA cells were treated with different concentrations of peptide and incubated for 1 h. Jurkat cells were washed three times with PBS and incubated with Fluo-8 calcium dye for 1 h. After 1 h, dye-loaded Jurkat cells were washed three times with PBS and activated with the CD3 antibody. Jurkat cells were resuspended on PBS supplemented with calcium chloride and added over the peptide-treated HFLS-RA cells. Immediately, samples were analyzed using a fluorescence plate reader for measurement of change on the fluorescence of Fluo-8 calcium dye. Fluorescence change was measured for 2 h on a Synergy H1 Hybrid plate reader (Biotek), and a graph of relative fluorescence change vs. time was plotted.
2.16. Western Blot
One million HFLS-RA cells were seeded in T-75 cell culture flasks and incubated for 72 h. After 72 h, the medium was removed, and cells were washed with PBS. Peptide concentrations were prepared in serum-free RPMI 1640 medium, added over the HFLS-RA cells, and incubated for 45 min. After 45 min, 3 million CD3-activated Jurkat cells were added to each flask. The co-culture flasks were incubated at 37 °C for 1 h. After 1 h, Jurkat cells were taken out by gentle pipetting and lysed. The lysate was quantified for total protein analysis. 25 μg of protein was loaded into each well for analysis. Western blot experiments were performed, and images were obtained using the Biorad Imaging System and analyzed using paired T-test in GraphPad Software (San Diego, CA).
2.17. T-Cell Suppression Assay in Transgenic Mice
HLA-DQ8 Transgenic mice used in this study lack endogenous class II molecules (DQ8.AEo) and were described previously (Taneja & David, 2010). Mice were bred and maintained by the Department of Comparative Medicine at the Mayo Clinic, and all experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committee. T-cell suppression assays were performed as described in our previous studies (Sable et al., 2016). Briefly, HLA-DQ8 mice were immunized with 100 μL of type II collagen (CII, 2 mg mL−1; Chondrex) emulsified 1:1 with complete Freund’s adjuvant (CFA; Difco), by intradermal injection at the base of the tail. Ten days post-immunization, mice were sacrificed and lymph nodes were harvested and made into single-cell suspension. LNCs (1 × 106) were then challenged with 100 μL of RPMI containing concanavalin A (20 μg/mL, positive control), native collagen (50 μg/mL), and medium alone (negative control). To the cells challenged in vitro with CII at 50 μg/mL, peptide SFTI-a1 was added at a concentration of 100 μg/mL, and cells were incubated for 48 h at 37 °C. 3H thymidine was added to the wells 18h prior to harvesting the plate as described (Sable et al., 2016). Results are depicted as stimulation index (S.I.).
2.18. Immunogenicity Studies
DQ8 mice were immunized with 200 μL of the peptide at 50 μg/mL. Ten days after immunization, T cell proliferation was carried out as described (Sable et al., 2016). Concanavalin (ConA) (1 μg/mL) was used as a positive control. Lymph node cells were challenged with the peptide in a dose-dependent manner. The stimulation index of more than 2 was considered positive and results are depicted as stimulation index with respect to peptide concentration.
3. RESULTS AND DISCUSSION
3.1. Design, Synthesis Cell Adhesion-Inhibition Activity of Peptides
In our previous report, we described the design and structure-activity relationships of peptide 6 (Figure 1, Table 1) from the adhesion domain of CD2 protein (Figure 1A) (A. Gokhale et al., 2011). As shown by cell adhesion inhibition assay, the peptide exhibited inhibition of PPI between CD2 and CD58 and between Jurkat cells and epithelial cells such as Caco-2 and OVCAR-3 with an IC50 value for inhibition of 6.9 nM. In peptide 6, the amino acid sequences Ser-Ile-Tyr-Asp and Asp-Asp-Ile-Lys are derived from F and C strands of CD2 protein (J. H. Wang et al., 1999). To stabilize this sequence to acquire β–turn/strand conformation and to cyclize the peptide, we introduced the D-Pro-L-Pro sequence into the peptide (Figure 1B). To evaluate the importance of amino acid residues in the peptide to bind to CD58, we replaced each amino acid in peptide 6 with alanine. Eight cyclic peptides were designed with the systematic replacement of alanine with amino acids that were from CD2 protein (Table 1). Since the D-Pro-L-Pro sequence is known to maintain a stable β-strand structure (Chatterjee et al., 2008; Kantharaju et al., 2009), the D-Pro-L-Pro sequence was not replaced with alanine. All peptides were synthesized using Fmoc-based solid-state automatic peptide synthesis and cyclized after being removed from the resin, as described in our previous reports (Sable et al., 2016). The peptides were purified by preparative HPLC and analyzed by analytical HPLC to evaluate the purity of the peptide. All the peptides exhibited >95% purity (except AS8) as indicated by HPLC. The peptides were analyzed for correct molecular ion using MALDI-TOF mass spectrometry. Analytical data for the peptides are provided in Supporting Information (Tables S1 -S5 and Figures S1-S20).
In this study, we wanted to investigate the amino acid residues in peptide 6 (Figure 1B) that are important for the inhibition of CD2-CD58 protein interactions by alanine scanning. The ability of these peptides to inhibit CD2-CD58 PPI was evaluated by lymphocyte-epithelial cell adhesion assay, where CD2-expressing Jurkat cells (T cells) and CD58-overexpressing OVCAR-3 cells were used (Liu et al., 2004). Among the alanine scanning peptides, AS2 and AS7 showed significant cell adhesion inhibitory activity (Table 1). The IC50 value for AS2 was found to be 1.1 ± 0.9 nM and AS7 exhibited inhibition activity with an IC50 value of 143 ± 96 nM in Jurkat cell-OVCAR-3 cell adhesion assay. Despite the minimal structural change in the peptide backbone, lymphocyte cell adhesion assay revealed that the side chains of amino acids have a vital role in binding to CD58 protein and, ultimately, inhibition of CD2-CD58 interaction (Table 1). Since AS2 peptide exhibited improved activity compared to peptide 6, we chose AS2 peptide (Figure 1C) for further study.
The stability of peptides could be enhanced for in vivo administration using different strategies, including substitution of D-amino acids or unnatural amino acids, or grafting the peptide to a multicyclic peptide framework such as sunflower trypsin inhibitor framework (Craik & Du, 2017; Gentilucci, De Marco, & Cerisoli, 2010; Lesner, Legowska, Wysocka, & Rolka, 2011). We used a sunflower trypsin-inhibitor template (SFTI-wt) (Figure 1D) because the peptide has 14 amino acids and exhibits β-strand structure, as in the case of peptide 6 or the CD58 adhesion domain of CD2 that has F and C strands. We used the AS2 peptide sequence (Figure 1C) for grafting on to sunflower trypsin-inhibitor template (Figure 1D). The SFTI-wt peptide is a bicyclic peptide with disulfide bridge cyclization in addition to backbone cyclization and known to have exceptional thermal and enzymatic stability. SFTI template has been used for attaining oral bioavailability (Qiu et al., 2017). Except for the disulfide bridge, other amino acids at crucial positions can be replaced without altering the stability of the SFTI peptide. Previously we reported SFTI grafting of compound 6 (Sable et al., 2016). Structures of F and C strands from CD2, and peptide 6 and SFTI-wt template were overlapped and replaced the amino acids of SFTI-wt with peptide 6 resulting in SFTI-a (Figure 1E). One of the β-strands of peptide 6 was compared with SFTI-wt structure β–strand and replaced the amino acids Pro-Phe-Cys-Ile with Asp-Tyr-Cys-Ser (Asp, Tyr and Ser are from peptide 6). The right side of strand structure Arg-Cys-Thr-Lys in SFTI-wt was replaced with Asp-Asp-Cys-Lys-Ala (Asp-Asp and Lys are from peptide 6). Since AS2 peptide was derived from peptide 6, we used similar approach to design grafted peptide SFTI-a1. In this case, Asp-Tyr-Ala-Ser sequence from AS2 left side of β-strand was replaced in SFTI-wt by Asp-Tyr-Ala-Cys-Ser resulting in SFTI-a1. To retain a total of 14 amino acids in the peptide sequence, we removed some amino acids or replaced amino acids in the loop region of the peptide resulting in peptide SFTI-a1 (Figure 1F).
SFTI-a and SFTI-a1 were further modified to evaluate the structure-activity relationships of the composition of amino acids in the peptide. From the analysis of the crystal structure of CD2-CD58 complex and mutation of amino acids in CD2 and CD58 protein structures, it was reported that amino acids Tyr86, Lys34, Asp32 from CD2 protein were important in binding to CD58 protein (Figure 1A). Replacement of Tyr86 by Ala in CD2 protein was known to decrease the binding of CD2 to CD58 by thousandfold (Kim et al., 2001). Tyr86 from CD2 interacts with Phe46 and Lys34 side chain of CD58 forming a hot-spot of protein-protein interaction. Tyr10 in the SFTI-a peptide corresponds to Tyr86 in the CD2 protein structure. Since Tyr10 in the peptide is an important residue for binding to CD58, we wanted to investigate whether changing the aromatic side chain such as Tyr will affect the binding of the peptide to protein CD58. In peptide SFTI-a2 (Table 2), tyrosine was replaced with homotyrosine. In peptide SFTI-a3, tyrosine was replaced with a bulky functional non-natural amino acid 3-amino-3 (1-napthyl propanoic acid). SFTI-a was also subjected to modification in its sequence. In SFTI-a structure chirality of the amino acids was changed (L to D amino acids) resulting in SFTI-a4. It is known that changing the chirality results in orientation of side chains and hence in many peptides sequence is reversed when chirality is changed. In SFTI-a5, the chirality was changed and the sequence was reversed compared to peptide SFTI-a. Cell adhesion inhibition activity indicated that among the grafted peptides, SFTI-a1 inhibited the adhesion of OVCAR-3 cells and Jurkat cells with an IC50 value of 23.34 ± 4.62 nM (Figure 2A), while peptides SFTI-a2 to a4 exhibited inhibition activity with IC50 value >50 μM (Table 2).
FIGURE 2.
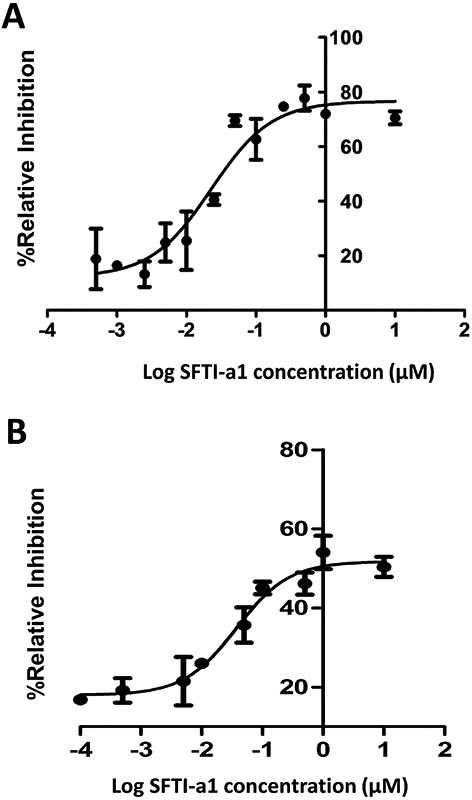
Cell adhesion inhibition activity of peptide SFTI-a1. A) Dose response curve for inhibition of adhesion between Jurkat cells (T cells, CD2 expression) and OVCAR-3 cells (express CD58) by SFTI-a1. B) Dose-response curve for inhibition of cell adhesion between Jurkat cells (express CD2) and HFLS-RA cells (express CD58) by SFTI-a1.
Peptides that exhibited IC50 in the lower nanomolar concentration in Jurkat cell-OVCAR-3 cell adhesion assay were also evaluated for their inhibition activity using Jurkat cells, and human fibroblast-like synoviocytes-rheumatoid arthritis (HFLS-RA) cells adhesion assays. HFLS-RA cells are derived from joints of rheumatoid arthritis patients and, hence, are relevant as a model for immunomodulation activity of peptides (D. P. Chen, Wong, Tam, Li, & Lam, 2011; Sable et al., 2016). Peptide AS2 inhibited cell adhesion between Jurkat cells (CD2 protein) and HFLS-RA cells (CD58 protein) with an IC50 value of 3.1 ± 1.2 nM, while, SFTI-a1 inhibited the adhesion of HFLS-RA cells and Jurkat cells with an IC50 value of 37.23 ± 1.02 nM (Figures 2B). In the same cellular assay, peptide 6 has an IC50 value of 36.99 ± 20.02 nM. As in the case of OVACR-3 cell and Jurkat cell adhesion assay, AS2 peptide exhibited improved cell adhesion inhibition activity compared to peptide 6. Among the grafted peptides, SFTI-a1 showed improved adhesion inhibition activity compared to SFTI-a. Cell adhesion inhibition data for grafted peptides clearly indicate that substitution of unnatural amino acids such as amino napthyl propanoic acid or modified tyrosine, did not produce cell adhesion inhibition activity. Hence, bulky functional groups, the chirality of amino acids in the peptide, determine the binding to CD58.
3.2. NMR Studies
To investigate the structure-activity relationship of peptides, we performed detailed NMR studies of one potent peptide that exhibited inhibitory activity (peptide AS2) and one peptide that exhibited inhibitory activity >50 μM in cell adhesion assays (peptide AS3) from alanine scanning peptides. Among the grafted peptides, an NMR study was conducted on SFTI-a1 peptide (Tables 1 and 2).
3.2.1. Peptide AS2
The TOCSY and ROESY NMR data obtained for the peptide AS2 indicated a well-defined structure. The spectra obtained were very clear with amide resonances spread over a range of 1 ppm (Supporting Information, Figure S21). The amide region of TOCSY spectra exhibited eight resonances corresponding to all the amino acids in the peptide except prolines. The sequence-specific assignment was performed to resolve amino acid resonances in the peptide that were similar. The NOE connectivity between Lys10 NH-Ser1 NH and Ser1 NH -Ala 2 NH is indicative of a possible β-turn around Lys10 and Ser1 (Supporting Information, Figure S22-23). The NH-NH connectivities in the amide region found in ROESY spectra together with TOCSY spectra, suggested that the peptide exhibited turn structure in solution. Detailed analysis of amide ROEs, the NH-NH connectivities, coupling constant information, and chemical shifts of resonances are provided in the Supporting Information (Tables S3).
3.2.2. Peptide AS3
The TOCSY and ROESY NMR data obtained for peptide AS3 suggested it has a well-defined structure. The amide resonances were spread over a range of 1.3 ppm, suggesting well-defined conformation for the peptide. The ROE connectivity between Lys10 NH-Ser1 NH and Ser1 NH Ile 2 NH is indicative of a possible β-turn around Lys10 and Ser1 (Supporting Information, Figures S24-26, Table S4). The NH-NH connectivities in the amide region found in ROESY spectra together with TOCSY spectra, suggested that the peptide acquires a well-defined cyclic structure. Results obtained after the assignment of NMR spectra suggested that there was no significant difference found in either peptide AS2 or AS3. From the NMR results, we can infer that the replacement of one amino acid residue from the cyclic sequence with alanine did not cause a significant difference in the overall structure of the peptides.
3.2.3. Peptide SFTI-a1
SFTI-a1 peptide exhibited a large number of resonances in the fingerprint region of TOCSY spectra. TOCSY data were analyzed based on characteristic resonances. For example, alanine exhibits a peak due to a methyl group at 1.2 ppm that is very distinct. TOCSY NMR spectra of SFTI-a1 have six sets of resonances for alanine, suggesting that there is a possibility of multiple conformations in the solution for the peptide (Supporting Information, Figure S27). ROESY spectra were complicated by connectivity between different protons and multiple resonances due to conformational heterogeneity. Hence, a detailed 3D structure of the peptide could not be elucidated from NMR spectra.
3.2.4. Circular Dichroism Study
The peptides that exhibited potent lymphocyte cell adhesion inhibition activity in in vitro assay, AS2, AS7, and a control peptide AS3 without activity, were selected for secondary structure analysis by circular dichroism spectra. All three peptides exhibited a negative band around 217 nm and a positive band around 190, suggesting possible β-hairpin/β-turn structure of the peptide in solution (Perczel, Hollosi, Foxman, & Fasman, 1991). The CD spectra of AS2 and AS7, along with that of control peptide AS3, were also compared with that of peptide 6, which was a lead compound for this project. CD spectra of peptide 6 had a positive band around 200 nm and a negative band around 210-215 nm suggesting a β-hairpin structure of the peptide. When peptide 6 and AS2 spectra were compared, there was a change in the amplitude of the negative band around 215 nm indicating a slight change in the structure of AS2 compared to peptide 6 (Supporting Information, Figure S28). All peptides showed negative and positive peaks around similar wavelengths, indicating a minimal change in the secondary structure (β- hairpin) as compared to compound 6 after replacing each of the amino acid residues with alanine one at a time.
The CD spectra of SFTI-a1 showed a positive band at 190 nm and a negative band at 203 nm, which suggests the presence of β-turn/β-strand in the peptide (Supporting Information, Figure S28) (Perczel et al., 1991). CD spectra of SFTI-a1 were not similar to those of peptide 6 or other alanine scanning peptides because of the presence of disulfide bonds, β-turn of SFTI framework, and added amino acid residues. The CD spectra were similar to that of the previously published grafted peptide that inhibits cell adhesion (Sable et al., 2016).
3.3. Molecular Modeling of AS2 and AS3 Peptides
Based on NMR data of peptides, distance restraints were generated for AS2 and AS3 peptides. NMR-restrained MD simulations were carried out on these peptides using InsightII software. The final structures that were consistent with NMR data were represented as possible 3D structures of these peptides in solution (Figure 3)(Dolenc, Missimer, Steinmetz, & van Gunsteren, 2010).
FIGURE 3.

Proposed model for the structures of A) AS2 and B) AS3. Structures were generated using NMR-restrained molecular dynamics simulations followed by energy minimization. Amino acid residues that are in β-turns in the structure are shown by sticks and are labeled with three-letter codes for amino acids. C) Overlay of structures of peptide 6 (backbone blue) and AS2 (backbone red). Ile2 and Ile9 from peptide 6 are shown as sticks in green. Ala2 and Ile9 from AS2 are shown in magenta. There is a slight change in the backbone structure of AS2 compared to peptide 6. D) Side view of overlay of peptide 6 and AS2. E) Overlay of structures of peptides AS2 (red) and AS3 (green). Replacement of Tyr 3 by Ala changes the overall backbone structure slightly, but the bulky side chain of Tyr is important in interaction with CD58 protein. Hydrogen bonds are shown by dashed lines. pro5 represents the D amino acid proline.
3.3.1. Structure of AS2
Fifty intra- and inter-residue ROE/NOE-based distances were used to generate the structure of AS2. The backbone rmsd of 23 structures that satisfy the NMR constraints was 1.661 ± 0.491. A representative structure from a family of structures is shown in Figure 3A. The structure of AS2 exhibited a β-turn structure at Asp4-pro5-Pro6-Asp7 (Rose, Gierasch, & Smith, 1985). The dihedral angles at pro5 (i+1) and Pro6 (i+2) were ϕ = 66, ψ = 153, and ϕ = −80, ψ = 26. The dihedral angle, along with chirality of pro5 being D suggested that the β-turn is type II′ (Bean, Kopple, & Peishoff, 1992). At least four intramolecular hydrogen bonds were observed in the peptide structure. The β-turn at Asp4-pro5-Pro6-Asp7 was stabilized by a hydrogen bond between C=O of Asp4 and the NH of Asp7. Other hydrogen bonds observed were between the NH of Ala2 to C=O of Asp8, A2 C=O to the NH of Asp4, Asp7 C=O to the NH of Ile9, and Tyr3 C=O to Lys10 side chain. There was a second β-turn in the peptide at Asp8-Ile9-Lys10-Ser1; however, this turn was not stabilized by a hydrogen bond between I and i+4; rather, it was stabilized by a hydrogen bond between NH of Ala2 and C=O of Asp8 as indicated above. The phi, psi angles at Ile9 (i+1) and Lys10 (i+2) were ϕ = −60, ψ = −20, and ϕ = −87, ψ = 39. These dihedral angles indicate a type I β-turn structure at Asp8-Ile9-Lys10-Ser1. The possibility of β-turn conformation at Asp8-Ile9-Lys10-Ser1 was supported by the NOE observed between Lys10 NH-Ser1 NH (NOE between i+2 and i+3). Temperature dependence of NMR data also suggested the possibility of intramoleculary hydrogen-bonded or solvent-shielded amides of Tyr3, Asp4, Asp8 and Lys10, as indicated by low-temperature coefficient (Δ δ/ΔT < 4.5 ppb/K). Overall, the peptide exhibited two β turns with a twist in the backbone between the two β turns (Wüthrich, 1986).
3.3.2. Structure of AS3
Fifty-nine intra- and inter-residue ROE/NOEs (Jacobsen, 2007; Wüthrich, 1986)were used to generate the structure of AS3 using a simulated annealing procedure with NMR restraints. The backbone rmsd of 26 structures that satisfy the NMR constraints was 0.794 ± 0.206. A representative structure from a family of structures is shown in Figure 3B. The overall structure of AS3 was similar to that of AS2 with β-turns. The β-turn was at Asp4-pro5-Pro6-Asp7 with phi and psi around pro5 and Pro6, ϕ = 71, ψ = −138, and ϕ = −67, ψ = 12 was considered type II′. This turn was stabilized by hydrogen bonding between C=O of Asp4 and the NH of Asp7. There were γ-turns at Ala2-Tyr3-Asp4 and Asp7-Asp8-Ile9 with hydrogen bonding between C=O of Pro6 and the NH of Asp8, C=O of Ile2 and the NH of Asp4, the NH of Ala3 and Lys10 C=O, and the NH of Ile2 and C=O of Asp8. The temperature coefficients of chemical shifts of amide resonances of Ser1, Ala3, Asp8, and Lys10 were < 4.5 ppb/K, supporting the hydrogen-bonding pattern observed in the structure of the peptide(Jacobsen, 2007).
Based on the NMR and molecular modeling activity of Ala scan peptides and peptide 6, we can correlate the improved activity of peptide AS2 compared to peptide 6 to the importance of amino acids that are involved in CD58 protein binding. When Ile2 in peptide 6 was replaced by Ala, the cell adhesion inhibition activity of the peptide was improved. Ile2 is placed next to an important amino acid Tyr3 that is known to be a “hot-spot” in CD2-CD58 interaction(Kim et al., 2001). By introducing flexible residue next to hot spot, residue may be advantageous to bind to the protein. This alanine amino acid may provide more flexibility or less steric hindrance to neighboring amino acids Ser and Tyr side chains as compared to Ile for hydrogen bonding (Ser) or hydrophobic interaction (Tyr). The replacement of Ala at positions, 1, 3-6 resulted in significant loss of cell adhesion inhibition activity of peptides (AS1, AS3-6, IC50 >50 μM), but when position 9 Ile was replaced with Ala (AS7) the inhibition activity was 0.143 μM. Compared to the IC50 value of peptide 6 and AS2 peptides, the activity of AS7 was significantly different but not complete loss of activity. The Ile9 in the peptide is next to Lys10. In CD2-CD58 interaction, amino acid corresponding to Lys10 is also a hot-spot residue. Furthermore, Lys10 replacement by Ala resulted in the loss of inhibition activity of the peptide (as seen for AS8). Thus, amino acid residues next to hot-spot residues seem to play a major role in stearic hindrance to bind to CD58 protein or effect the conformation of side chains to bind to CD58 protein. Replacement of amino acid residues that are capable of producing ionic interaction or hydrogen bonding, including Ser, Tyr, Asp, and Lys, leads to complete loss of activity.
To understand the nature of the interaction of designed CD2 peptides with CD58 protein, we compared the structures of peptide 6 with AS2 as well as with F and C strands of CD58 protein (Kim et al., 2001; J. H. Wang et al., 1999). When peptides are designed from protein epitopes, they may not acquire the same three-dimensional structure as in native protein as proteins have tertiary structure and side-chain interactions stabilize the globular structure of the protein. When peptide 6 and AS2 peptide were compared (Figures 3C & D), there was a slight change in the backbone structure of AS2 compared to peptide 6. Ile9 orientation was similar in peptide 6 and peptide AS2. Because Ala side chain is relatively small compared to Ile, there was a change in the orientation of side chain of Ile2 as well as Lys10 in peptide 6 compared to AS2. To compare the structure of active peptide with an inactive peptide, we overlapped the structures of AS2 and AS3. The overall backbone structure of both peptides was similar (Figure 3E). However, in peptide AS3, important amino acid Tyr3 was replaced by Ala3 and hence there was a significant loss of inhibition activity in peptide AS3 compared to AS2.
Side chain conformation is flexible in solution, and the structure of the peptides derived from NMR data will have side-chain flexibility depending on the number of constraints that define the side-chain orientation. We looked at the side chain flexibility of peptide AS2 in our NMR restrained MD simulated low energy 20 structures (Supporting information Figure S29). Based on the MD simulated structure, it is very clear that there are few possible orientations of side chains for AS2 peptide. In peptide 6 and AS2 side chains, Ile9 and Ile2/Ala2 were one side of the peptide surface whereas Tyr3 was on the opposite surface of the plane formed by the peptide backbone (Figures 3C and D).
To elucidate the nature of the interaction of the peptide with CD58 protein, we compared the F and C strands of CD2 protein in the complex of CD2-CD58 protein structure (Figures 4A and B). In the crystal structure, Tyr86 and Lys34 hot –spot residues were orientated towards CD58 while Ile residues that are next to the hot-spot residues, Ile85 and Ile33, were facing away from the surface of CD2-CD58 interaction (J. H. Wang et al., 1999). We found a similar orientation of Ile and Tyr residues in peptide 6 and AS2 structures. These Ile residues may not directly involve in the interaction of peptide-protein binding; however, they help to orient the hot-spot residues towards the receptor protein. In the case of peptides 6 and AS2, replacing the Ile residue with Ala may help to remove steric hindrance with receptor binding and hence improved the activity of the peptide.
FIGURE 4.

A) F and C strands of CD2 peptide showing side chains of amino acids orientation with respect to the plane of the backbone surface. Hot-spot residues Tyr86 and Lys34 side chains orient on one side of the sheet surface, whereas Ile33 and Ile85 side chains orient towards the other surface. B) Crystal structure of the complex of CD2-CD58 with F and C strands of CD2 highlighted. Tyr86 and Ile34 are involved in an interaction with CD58, whereas Ile33 and Ile85 are oriented away from the interface of PPI.
3.3.3. SPR Analysis
We presume that peptides designed from CD2 bind to CD58 and inhibit CD2-CD58 interaction. Using cellular assays, we have shown that peptide SFTI-a1 exhibit cell adhesion inhibition activity. To show that cell adhesion inhibition is due to the binding of CD2 peptides to CD58, we carried out surface plasmon resonance studies. The CD58 protein adhesion domain (Abcam) was immobilized on the SPR chip surface and peptides were used as analyte (de Mol & Fischer, 2010). Grafted peptide SFTI-a1 showed concentration-dependent binding with the immobilized recombinant CD58 protein. Figure 5A shows that the binding response increases with an increase in the concentration of peptide. The Kd value of the binding of peptide SFTI-a1 to protein CD58 in SPR analysis was found to be 0.32 μM. It is reported that CD2-CD58 protein-protein interactions are specific but weak, with a Kd value in the range of 1–10 μM, depending on the method used to determine Kd (Merwe & Davis, 2003; J. H. Wang et al., 1999). Thus, weak interaction between CD2 and CD58 can be inhibited by alanine scanning peptide or grafted peptide SFTI-a1 at nanomolar concentrations. The Kd value of SFTI-a1 peptide to bind to CD58 protein at nanomolar concentrations indicates that SFTI-a1 peptide is specific in binding to CD58 protein. These results are consistent with the IC50 value of inhibition of adhesion of SFTI-a1 at a nanomolar concentration (Table 2).
FIGURE 5.

Binding of SFTI-a1 to CD58 protein. A) SPR analysis of SFTI-a1 binding to the CD58 adhesion domain. The adhesion domain of CD58 was immobilized on a CM-5 chip and SFTI-a1 was used as analyte. Kinetics of association and dissociation were monitored at different concentrations of SFTI-a1. Chip surface without protein was used as control. Inset:expanded region of sensorgrams at 0, 0.5 and 1 μM of SFTI-a1. B) Binding of SFTI-a1 to cells overexpressing CD58 (OVCAR cells) monitored by flow cytometry. The cell population without antibody labeling is at the left. With FTIC-Ab CD58, the cell population shifted to the right. When SFTI-a1 and SFTI-Ab CD58 were incubated with OVCAR-3 cells, there was a concentration-dependent shift of the OVCAR-3 cell population to the left, indicating the binding of SFTI-a1 to CD58 on cells.
3.3.4. Competitive Binding Experiment
As indicated previously, peptide SFTI-a1 binds to CD58 protein on the cell surface and inhibits Jurkat cell epithelial cell interaction by inhibiting CD2-CD58 interaction. To evaluate the binding of SFTI-a1 peptide to the CD58 protein adhesion domain on the epithelial cell surface, we performed competitive binding of FITC-Ab CD58 to CD58 with SFTI-a1 peptide. FITC-Ab CD58 is known to bind to the adhesion domain of CD58. Thus, the replacement of FITC-Ab CD58 from the cell surface by peptides indicates the binding of the peptide to the adhesion domain of CD58. Different concentrations of SFTI-a1 were added with a constant concentration of FITC-Ab CD58 (20 μL, Santa Cruz Biotechnology, Dallas, TX, USA) and incubated with OVCAR-3 cells that express CD58. Cells with FTIC-Ab CD58 without peptide shifted to the right side in the histogram compared to cells without FITC-Ab CD58. Upon the addition of SFTI-a1, the cell population was shifted to the left side, as shown in the histogram (Figure 5B) in a concentration-dependent manner. These results indicate that SFTI-a1 competes with FITC-Ab CD58 to bind with CD58 on the cell surface. The results also indicate that the peptide binds to the adhesion domain of CD58 protein.
3.4. Docking
To model the interaction of peptides with CD58 protein, docking calculations were performed using Autodock (Morris et al., 2009). 3D structures of AS2 and AS3 were generated from molecular modeling based on NMR data. The SFTI-a1 peptide structure was generated using InsightII molecular modeling software (BIOVIA San Diego, CA, USA)
Peptides AS2 and SFTI-a1 were docked to CD58 domain I by creating a grid around the adhesion domain of CD58. Lowest energy docked structures were analyzed for the binding modes of peptides on the CD58 protein surface. In the case of peptide AS2, the lowest energy docked structure had a docking energy of −3.78 kcal/mol with a cluster of three structures. AS2 peptide was bound near the CD58 binding surface with hydrogen-bonding interactions between Ser1 of the peptide to Glu37 of protein, Lys10 of the peptide to Glu39, Asp7 to Lys34, and Asp4 to Lys29. Tyr3 of the peptide formed hydrophobic interaction with Phe46 and Lys34 of protein with Lys34 side chain sandwiched between two hydrophobic groups. In the CD2-CD58 crystal structure (J. H. Wang et al., 1999), this interaction (Phe46 and Lys34 from CD58 to Tyr of CD2) was considered a hot spot. This peptide AS2 mimics the adhesion domain of CD2 in binding to CD58 and binds to CD58 (Figure 6A). All-important amino acid residues that are reported to be present on CD2-CD58 protein-protein interaction interface, including Glu25, Lys29, Asp33, Lys34, Glu37, and Glu39 (Kim et al., 2001; J. H. Wang et al., 1999), were seen to form interactions with the AS2 peptide. Ile9 side chain was oriented away from CD58 protein indicating that Ile9 may not be important in binding to CD58 protein.
FIGURE 6.

Proposed model for binding of AS2 and SFTI-a1 to CD58 protein by docking. Low energy docked structure of A) AS2, B) SFTI-a1 to CD58 adhesion domain. Important amino acid residues in the adhesion domain of CD58 are labeled with single-letter codes for amino acids. Peptides are labeled with three-letter codes for amino acids and are shown as sticks. Protein is shown in secondary structures with amino acids that participate in the interaction with SFTI-a1 shown as thin lines. Amino acids from protein are shown using a single letter code to distinguish from amino acids from the peptide.
Docking of peptide SFTI-a1 to CD58 protein resulted in a low energy docked structure of SFTI-a with docking energy of −2.99 kcal/mol with three structures in the docking cluster. Docking studies showed that peptide SFTI-a1 binds with CD58 protein in the CD2 binding region. Hydrogen bonding was observed between Ser5 of peptide and Asp33 of protein, Lys4 side chain with Lys32 backbone C=O, Asp1 with Lys32, Asp2 and Asp13 with Lys29, and Tyr12 side chain with Lys34 side chain. Tyr12 from peptide and Phe46 side chain from protein sandwiched Lys34 of CD58 protein. Lys34 and Phe46 formed pi-cation interaction (Figure 6B). Overall, SFTI-a1 was bound near the important amino acids that are defined in the CD2-CD58 crystal structure (Kim et al., 2001; J. H. Wang et al., 1999). The decrease in binding affinity of SFTI-a1 compared to that of AS2 peptide could be explained in terms of docking. The AS2-docked structure covered a major portion of the CD2 binding region of the adhesion domain of CD58, from Glu39 to Lys32 (Figure 6A), while SFTI-a1 covered part of adhesion domain (Figure 6B). We believe this difference in binding leads to a decrease in affinity toward CD58 by SFTI-a1 compared to AS2 peptide. Furthermore, we believe that the existence of multiple conformations of the peptide is also responsible for the observed decrease in cell adhesion activity of SFTI-a1.
3.5. Thermal Stability of Peptides
Sunflower trypsin inhibitor peptides and grafted peptides are known to exhibit thermal stability compared to non-grafted peptides (Colgrave, Korsinczky, Clark, Foley, & Craik, 2010; Lesner et al., 2011). To assess the thermal stability of peptides, we used circular dichroism and monitored the changes in CD spectra of peptides at different temperatures. SFTI-a1 was dissolved in water, and CD spectra of the peptide were acquired from 25 to 85 °C. CD spectra of the peptide were not significantly changed with increased temperature, indicating stable conformation of the peptide in solution (Figure 7A). Although CD spectra are sensitive to conformational changes, degradation of peptides with temperature or disulfide bond changes may not be shown in CD spectra. To confirm the stability of the peptide, HPLC analysis of the peptide was conducted after incubating at 55 and 85 °C. There was no significant change in HPLC peak area for the peptide, suggesting that the peptide was stable at the high temperatures studied (Figure 7B). Furthermore, mass spectrometric analysis of a peptide sample incubated at different temperatures in solution state suggested that the peptide was intact as there was no change in its mass (Supporting Information, Figure S30). These results suggest that SFTI-a1 was resistant to thermal degradation.
FIGURE 7.
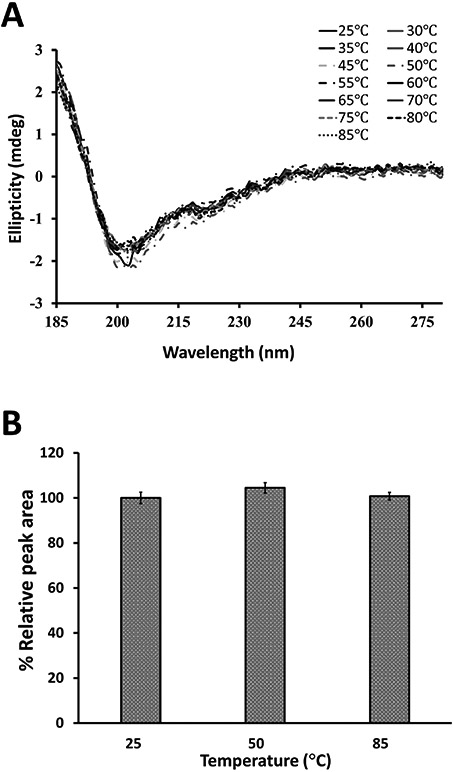
Thermal stability of SFTI-a1. A) CD spectra of SFTI-a1 in the water at different temperatures. There was no significant difference in CD spectra of SFTI-a1 from 25 to 85 °C. B) Thermal stability analysis by HPLC. Peptides were incubated at different temperatures, and samples were analyzed by HPLC. Relative peak area was plotted at different temperatures. Results from triplicate experiments are presented.
Disulfide bonds are known to provide structural rigidity and increase the stability of grafted peptides, an approach used for rational drug design of stable and potent peptides (Poth, Chan, & Craik, 2013). We determined the stability of the peptide in the presence of 0.1 and 1 mM of dithiothreitol. CD and mass spectroscopy data showed that the peptide was stable with 0.1 and 1 mM of dithiothreitol (Supporting Information, Figures S31, 32). Dithiothreitol (1 mM) could break disulfide bonds, as suggested by a change in CD spectra and the presence of an extra peak of almost equivalent intensity with an increase of mass of the peptide equivalent to 2 hydrogens.
3.6. Serum Stability of Peptides
Peptides are known to be cleaved by proteases in the blood, depending on the sequence of the peptide (Jenssen & Aspmo, 2008). Hence, the stability of peptide-based drugs in serum is important to know before in vivo studies are carried out. To evaluate the in vitro stability of SFTI-a1 in serum, pooled human serum was purchased from Innovative Research (Innovative Research, Inc, Novi, MI) and peptide SFTI-a1 were incubated in serum as described in the Experimental Section. Using HPLC analysis, the amount of intact peptide was determined. After incubation with human serum for 24 h, we did not find a significant decrease in the peak area of the peptide HPLC peak (Figure 8A). The molecular mass of the intact peptide was also confirmed by mass spectrometry (Figure 8B). In SFTI peptide, the binding loop contains amino acids Lys-Ser that are resistant to enzymatic degradation (Lesner et al., 2011). SFTI-a1 has Lys-Ser (Figure 1) in the sequence. Retaining this Lys-Ser-containing loop is known to maintain enzymatic stability (Qiu et al., 2017). Thus, stability studies clearly indicate the importance of amino acid residues of the SFTI framework for imparting stability to grafted peptides.
FIGURE 8.
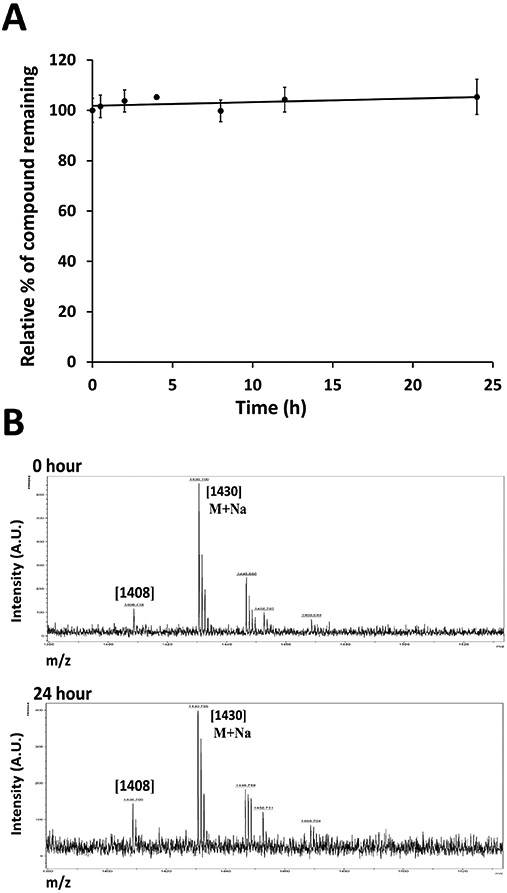
A) In vitro serum stability of peptide studied by HPLC. SFTI-a1 was incubated in human serum, and aliquots were obtained at different time points and analyzed by HPLC. Zero-point peak area was taken as 100%. B) Mass spectrometry analysis of samples of SFTI-a1 incubated at different time points. MALDI-TOF mass spectrometry was used for analysis. m/z corresponding to the peptide ion and sodium adduct were seen.
3.7. Liver Microsome Stability
The liver is the major site for metabolism of drugs, and almost 60% of the marketed drugs are metabolized by CYP mediated biotransformation (McGinnity, Soars, Urbanowicz, & Riley, 2004). Our ultimate goal is to make the peptide suitable for oral administration. To evaluate the hepatic metabolism and possibility of first-pass effect during oral administration, SFTI-a1 stability was assessed in liver microsomes (Knights et al., 2016) (Corning, NY, USA). SFTI-a1 was incubated in liver microsomes 0.5 mg/mL. At different time points, 250 μL of the sample was taken, ice-cold acetonitrile was added, and the sample was centrifuged. The supernatant was lyophilized, and the reconstituted solution was analyzed by HPLC for intact peptide. In the presence of liver microsome enzymes, degradation of the peptide was observed (Figure 9A). After 2 h of incubation with liver microsome enzymes, 46% of the peptide remained. We also confirmed the presence of peptide after 2 h by using mass spectrometry. The exact mechanism of peptide degradation could not be identified from mass spectrometry (Figure 9B) as a majority of liver enzymes are cytochromes and carry out oxidation-reduction and hydroxylation mechanisms (Jia & Liu, 2007). However, deamidating enzymes are found in liver S9 fractions (Zvereva, Semenistaya, Krotov, & Rodchenkov, 2016). The presence of s significant amount of peptide up to 2 h indicates that the peptide is not rapidly metabolized by the liver by phase I metabolism. This result also suggests the possibility of limited first-pass metabolism for the peptide. Although this study provides information about possible metabolism of the peptide in the liver, it does not provide information about oral bioavailability of the peptide SFTI-a1. Orally administration of the drugs reach the systmetic circulation depending on the absorption at the intestinal barrier and the absorption depends on lipophilicity and overall structure of the molecule (Renukuntla, Vadlapudi, Patel, Boddu, & Mitra, 2013). As a control verapamil was used and half-life calculated was around 30 min as reported in the literature (Ong et al., 2016) (Supporting Information S34&S35).
FIGURE 9.
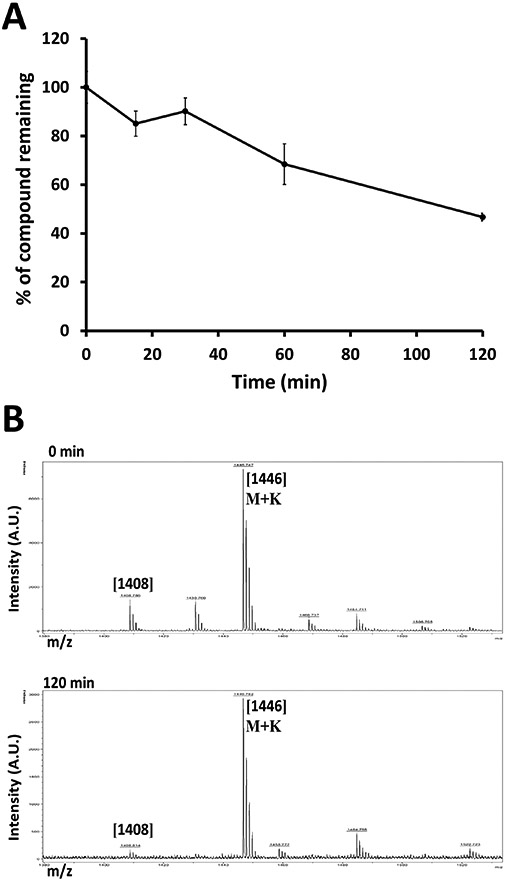
Stability of SFTI-a1 in liver microsomes. SFTI-a1 was incubated in liver microsomes, and samples were aliquoted at different time points and analyzed by A) HPLC, B) Mass spectrometry analyses for 0 and 120-min samples are represented in the figure.
3.8. Chemical Stability of Peptides
If a peptide has to be administrated via the oral route, its stability under different pH conditions is important as the pH of the digestive system varies from 2 to 7.4 (Fallingborg, 1999). The stability of SFTI-a1 was evaluated in three buffer conditions corresponding to pH 1.5, 7.4, and 9.6. Samples were analyzed by HPLC for intact peptide. Results indicated that the peptide was not degraded significantly under the incubation conditions during 30 days of incubation (Figure 10ABC). Although the medium does not have a pepsin enzyme to simulate the gastrointestinal fluids, one can conclude that SFTI-a1 peptide is stable at different pH conditions.
FIGURE 10.
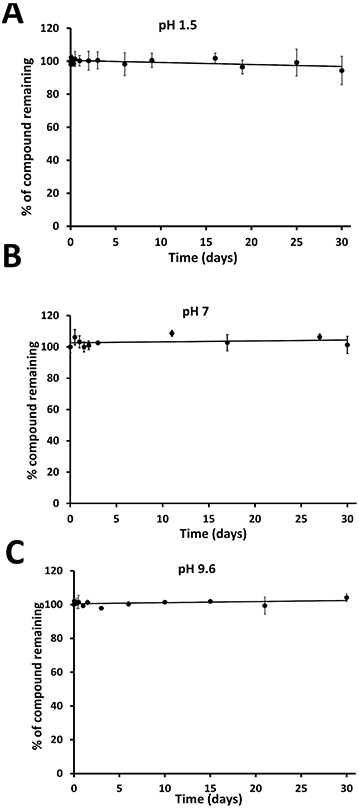
Stability of SFTI-a1 at different pH in buffers. SFTI-a1 was incubated in different buffers at pH A) 1.5, B) 7, and C) 9.6, and samples were aliquoted at certain time intervals and analyzed by HPLC. Zero time point peak area was taken as 100%, and other time point data were represented as a percentage of zero time point data.
3.9. Calcium Flux Assay
To understand the molecular mechanism of peptide SFTI-a1, we carried out studies related to CD2 signaling that is mediated by the cytoplasmic tail of CD2 protein via Zap-70 protein and calcium flux. An increase in calcium concentration in the cytoplasm of T cells is an indicator of activation of T cells and helps to generate the immune response (Joseph, Reicher, & Barda-Saad, 2014; Trebak & Kinet, 2019). T-cell engagement with APC produces cytoskeletal rearrangement and a dramatic increase in intracellular concentration. TCR triggering by APC results in the intracellular release of calcium from endoplasmic reticulum within seconds. We wanted to evaluate whether there is any change in the intracellular concentration of calcium and its signaling in the presence of peptide SFTI-a1 bound to CD58 compared to the absence of SFTI-a1. The co-culture of Jurkat cells with HFLS-RA cells could significantly increase the calcium concentration in Jurkat cells. After treating with 20 nM and 200 nM of the peptide, calcium flux in Jurkat cells was inhibited in a concentration-dependent manner compared to without peptide (Figure 11AB). This result suggests that peptide treatment inhibits the calcium signaling and, hence, the activation of T cells in an in vitro model.
FIGURE 11.

Molecular mechanism of inhibition of protein-protein interaction and its effect on CD2 signaling by inhibition of SFTI-a1. A) Calcium flux measurement by Jurkat cells in the presence and absence of HFLS-RA cells and in the presence of SFTI-a1. B) Quantitative data from calcium flux measurements. In the presence of CD3 and HFLS-RA cells, there was a drastic increase in calcium flux due to the CD2 signaling mechanism. In the presence of SFTI-a1 at 10 and 200 nM, calcium flux was decreased significantly compared to controls. C) Phosphorylation of Zap-70 and effect of SFTI-a1 on Zap-70 phosphorylation at 20 and 200 nM. There was a decrease in phosphorylation of Zap-70 in the presence of SFTI-a1 compared to control. D) Quantitative analysis of phosphorylation of Zap-70. GAPDH was used as control.
3.10. Western Blot Analysis
CD2-CD58 interactions are known to generate signaling for immune response. Upon binding to CD58, the CD2 cytoplasmic tail initiates signaling that leads to downstream signaling to generate cytokines. One of the main proteins involved in this signaling is Zap-70, which is known to phosphorylate. Zap-70 phosphorylation produces an immediate downstream signaling of TCR- and T-cell activation (Wang et al., 2010). To evaluate whether inhibition of CD2-CD58 interaction by the designed peptide from CD2 will inhibit Zap-70 phosphorylation and signaling for immune response, Western blot analysis was carried out on Jurkat cells in the presence and absence of peptides. Western blot of Jurkat cell lysate revealed that Zap-70 phosphorylation is increased in Jurkat cells after culturing them in the presence of HFLS-RA cells. After treating the HFLS-RA cells with SFTI-a1 peptide, the peptide inhibited the phosphorylation of Zap-70 significantly (Figure 11CD). This result shows that blocking the CD2-CD58 interaction could inhibit the phosphorylation of Zap-70 and the activation of T cells. This decrease in signaling might be a result of insufficient contact and decreased strength of TCR-MHC binding. These results are consistent with calcium flux studies.
3.11. T-Cell Suppression Activity of SFTI-a1
The HLA-DQ8.AEo transgenic mouse model exhibits autoimmune disease RA similar to that in humans. CD2 is involved in T-cell co-stimulation during an immune response and in RA, whereas, in APC, CD58 is known to be upregulated. We wanted to evaluate whether inhibition of CD2-CD58 interaction by SFTI-a1 will suppress the T-cell immune response in transgenic mice. HLA-DQ8 mice injected with type II collagen (CII) produce antigen-specific cellular and humoral response, resulting in these mice developing arthritis (Taneja et al., 2007; Taneja et al., 2005). Lymph node cells from those mice were harvested and cultured with and without CII and peptide SFTI-a1 in vitro. T-Cell proliferation was measured by incorporation of 3H-thymidine. SFTI-a1 suppressed antigen-specific T-cell immune response, whereas controls containing CII without the peptide SFT1-a1 showed robust CII-induced T-cell immune response, indicating that SFTI-a1 suppresses T-cell immune response from cells derived from the RA model (Figure 12 A & B). Thus, SFTI-a1 can be used for antigen-specific immune response in autoimmune diseases such as RA.
FIGURE 12.
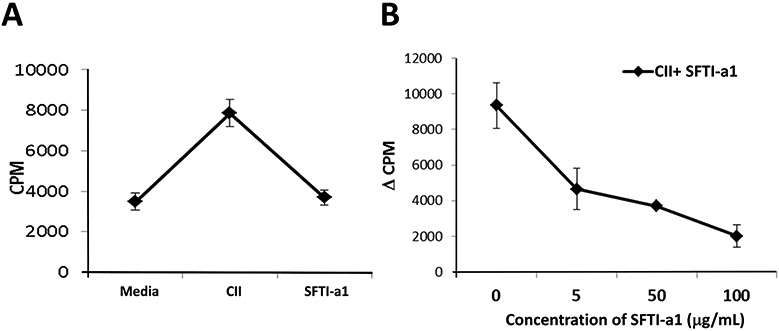
T-cell suppression assay. Lymph node cells (LNCs) harvested from type II collagen (CII)-immunized DQ8 mice were cultured in vitro in the presence or absence of CII and SFTI-a1 (n = 3/group), and T-cell proliferation was measured. A) SFT1-a1 suppressed antigen-specific T-cell response compared to CII control at 100 μg/mL. B) Dose-dependent T-cell suppression by SFTI-a1 peptide.
3.12. Immunogenicity of SFTI-a1
Sunflower trypsin inhibitor and grafted peptides are not known to be immunogenic (Craik & Du, 2017; Poth et al., 2013). However, the SFTI-wt peptide sequence was grafted with amino acids from CD2 protein, an immunoglobulin family member protein. To evaluate whether SFTI-a1 peptide itself generated the immune response in DQ8 mice, mice primed with SFTI-a1 were sacrificed 10 days later, and lymph node cells (LNC) were isolated from these mice. A single-cell suspension was made from the isolated LNCs which were challenged with SFTI-a1 in an in vitro assay and T-cell proliferation was measured. SFTI-a1 showed low immune response than the stimulation index (S.I) of 2 in T cells, indicating that SFTI-a1 is not immunogenic. As a control, concanavalin was used which generated a significant immune response compared to SFTI-a1 (Supporting informationFigure S33).
4. CONCLUSIONS
To investigate the structure-activity relationships of CD2-CD58 PPI inhibitory peptide 6 (a lead compound from previous studies), the alanine scanning technique was used. This method evaluated the importance of each amino acid residue present in peptide 6. Eight different peptides were designed and synthesized by replacing each amino acid present in peptide 6 one at a time with alanine amino acid, which is considered to be the least reactive and causes the fewest changes in the backbone structure of the peptide. Based on NMR study results, we concluded that the replacement of amino acid residues by alanine does not cause any significant change in the overall structure of the peptide. The most potent peptide resulting from alanine scanning was grafted onto the SFTI frame to achieve enhanced inhibition activity of the grafted peptide previously reported from our lab. Grafted peptide SFTI-a1 could inhibit the cell-cell adhesion with a nanomolar IC50 value. Grafting of peptides to a stable bicyclic framework has shown improved stability and possible oral bioavailability (Qiu et al., 2017). Grafting of peptides to the SFTI framework retained the potency of CD2-CD58 protein-protein interaction inhibition. Binding studies using SPR and flow cytometry indicated that SFTI-a1 binds to CD58 and inhibits CD2-CD58 interaction. Stability studies suggest that peptide SFTI-a1 is thermally stable and exhibits significant stability under different pH conditions.
Furthermore, a microsomal stability assay indicated that the peptide is stable for at least 2 h in liver microsomes. To understand the molecular mechanism of the peptide SFTI-a1, we carried out studies related to CD2 signaling that is mediated by the cytoplasmic tail of CD2 protein via Zap-70 protein and calcium flux. The signaling due to this pathway ultimately results in T-cell activation, cytokine production, and proliferation of T cells. Our Western blot analysis and calcium flux assay of Jurkat cells, in the presence of HFLS-RA, demonstrated a novel finding that the inhibition of CD2-CD58 protein-protein in a co-culture condition inhibits the activation of Jurkat cells in vitro. Zap-70 is an instantaneous signaling pathway for T cells (Wang et al., 2010).
Supplementary Material
ACKNOWLEDGMENTS
Part of the funding for this project was supported by the National Institute of General Medical Sciences of the National Institute of Health under grant number P20GM12345. R. Sable was supported by the Louisiana Biomedical Research Network an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 GM103424 and the LBRN Summer Research Program supported by the Louisiana of Board of Regents under grant number LEQSF(2015-20)-INBRE-Match. The authors would like to thank the NMR and mass spectrometry facility at Louisiana State University, Baton Rouge. We would also like to thank the BBC core facility and HPC, LONI, for the computational facility.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interest
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Data Sharing
Research data are not shared without permission from the University. Most of the data is presented in the supporting information.
REFERENCES
- Balanescu A, Radu E, Nat R, Regalia T, Bojinca V, Predescu V, & Predeteanu D (2002). Co-stimulatory and adhesion molecules of dendritic cells in rheumatoid arthritis. Journal of cellular and molecular medicine, 6(3), 415–425. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12417058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean JW, Kopple KD, & Peishoff CE (1992). Conformational analysis of cyclic hexapeptides containing the D-Pro-L-Pro sequence to fix .beta.-turn positions. Journal of the American Chemical Society, 114(13), 5328–5334. doi: 10.1021/ja00039a050 [DOI] [Google Scholar]
- Bretscher PA (1999). A two-step, two-signal model for the primary activation of precursor helper T cells. Proceedings of the National Academy of Sciences of the United States of America, 96(1), 185–190. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9874793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamian F, Lowes MA, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, … Krueger JG (2005). Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proceedings of the National Academy of Sciences of the United States of America, 102(6), 2075–2080. doi: 10.1073/pnas.0409569102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee B, Saha I, Raghothama S, Aravinda S, Rai R, Shamala N, & Balaram P (2008). Designed peptides with homochiral and heterochiral diproline templates as conformational constraints. Chemistry, 14(20), 6192–6204. doi: 10.1002/chem.200702029 [DOI] [PubMed] [Google Scholar]
- Chen DP, Wong CK, Tam LS, Li EK, & Lam CW (2011). Activation of human fibroblast-like synoviocytes by uric acid crystals in rheumatoid arthritis. Cell Mol Immunol, 8(6), 469–478. doi: 10.1038/cmi.2011.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, & Flies DB (2013). Molecular mechanisms of T cell co-stimulation and co-inhibition. Nature reviews. Immunology, 13(4), 227–242. doi: 10.1038/nri3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrave ML, Korsinczky MJ, Clark RJ, Foley F, & Craik DJ (2010). Sunflower trypsin inhibitor-1, proteolytic studies on a trypsin inhibitor peptide and its analogs. Biopolymers, 94(5), 665–672. doi: 10.1002/bip.21415 [DOI] [PubMed] [Google Scholar]
- Craik DJ, & Du J (2017). Cyclotides as drug design scaffolds. Curr Opin Chem Biol, 38, 8–16. doi: 10.1016/j.cbpa.2017.01.018 [DOI] [PubMed] [Google Scholar]
- Davis SJ, Ikemizu S, Evans EJ, Fugger L, Bakker TR, & van der Merwe PA (2003). The nature of molecular recognition by T cells. Nature immunology, 4(3), 217–224. doi: 10.1038/ni0303-217 [DOI] [PubMed] [Google Scholar]
- de Mol NJ, & Fischer MJ (2010). Surface plasmon resonance: a general introduction. Methods in molecular biology, 627, 1–14. doi: 10.1007/978-1-60761-670-2_1 [DOI] [PubMed] [Google Scholar]
- Dolenc J, Missimer JH, Steinmetz MO, & van Gunsteren WF (2010). Methods of NMR structure refinement: molecular dynamics simulations improve the agreement with measured NMR data of a C-terminal peptide of GCN4-p1. J Biomol NMR, 47(3), 221–235. doi: 10.1007/s10858-010-9425-9 [DOI] [PubMed] [Google Scholar]
- Espagnolle N, Depoil D, Zaru R, Demeur C, Champagne E, Guiraud M, & Valitutti S (2007). CD2 and TCR synergize for the activation of phospholipase Cgamma1/calcium pathway at the immunological synapse. Int Immunol, 19(3), 239–248. doi: 10.1093/intimm/dxl141 [DOI] [PubMed] [Google Scholar]
- Fallingborg J (1999). Intraluminal pH of the human gastrointestinal tract. Dan Med Bull, 46(3), 183–196. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/10421978 [PubMed] [Google Scholar]
- Gentilucci L, De Marco R, & Cerisoli L (2010). Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Current pharmaceutical design, 16(28), 3185–3203. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/20687878 [DOI] [PubMed] [Google Scholar]
- Gokhale A, Kanthala S, Latendresse J, Taneja V, & Satyanarayanajois S (2013). Immunosuppression by co-stimulatory molecules: inhibition of CD2-CD48/CD58 interaction by peptides from CD2 to suppress progression of collagen-induced arthritis in mice. Chemical biology & drug design, 82(1), 106–118. doi: 10.1111/cbdd.12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A, Weldeghiorghis TK, Taneja V, & Satyanarayanajois SD (2011). Conformationally constrained peptides from CD2 to modulate protein-protein interactions between CD2 and CD58. Journal of medicinal chemistry, 54(15), 5307–5319. doi: 10.1021/jm200004e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale AS, Sable R, Walker JD, McLaughlin L, Kousoulas KG, & Jois SD (2015). Inhibition of cell adhesion and immune responses in the mouse model of collagen-induced arthritis with a peptidomimetic that blocks CD2-CD58 interface interactions. Biopolymers. doi: 10.1002/bip.22692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchings NJ, Clarkson N, Chalkley R, Barclay AN, & Brown MH (2003). Linking the T cell surface protein CD2 to the actin-capping protein CAPZ via CMS and CIN85. The Journal of biological chemistry, 278(25), 22396–22403. doi: 10.1074/jbc.M302540200 [DOI] [PubMed] [Google Scholar]
- Ieronymaki M, Androutsou ME, Pantelia A, Friligou I, Crisp M, High K, … Tselios T (2015). Use of the 2-chlorotrityl chloride resin for microwave-assisted solid phase peptide synthesis. Biopolymers, 104(5), 506–514. doi: 10.1002/bip.22710 [DOI] [PubMed] [Google Scholar]
- Jacobsen NE (2007). NMR Spectroscopy Explained: Simplified Theory, Applications and Examples for Organic Chemistry and Structural Biology: Wiley. [Google Scholar]
- Jenssen H, & Aspmo SI (2008). Serum stability of peptides. Methods in molecular biology, 494, 177–186. doi: 10.1007/978-1-59745-419-3_10 [DOI] [PubMed] [Google Scholar]
- Jia L, & Liu X (2007). The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr Drug Metab, 8(8), 822–829. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/18220563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph N, Reicher B, & Barda-Saad M (2014). The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1838(2), 557–568. doi: 10.1016/j.bbamem.2013.07.009 [DOI] [PubMed] [Google Scholar]
- Kantharaju, Raghothama S, Raghavender US, Aravinda S, Shamala N, & Balaram P (2009). Conformations of heterochiral and homochiral proline-pseudoproline segments in peptides: context dependent cis-trans peptide bond isomerization. Biopolymers, 92(5), 405–416. doi: 10.1002/bip.21207 [DOI] [PubMed] [Google Scholar]
- Kim M, Sun ZY, Byron O, Campbell G, Wagner G, Wang J, & Reinherz EL (2001). Molecular dissection of the CD2-CD58 counter-receptor interface identifies CD2 Tyr86 and CD58 Lys34 residues as the functional "hot spot". Journal of molecular biology, 312(4), 711–720. doi: 10.1006/jmbi.2001.4980 [DOI] [PubMed] [Google Scholar]
- Knights KM, Stresser DM, Miners JO, & Crespi CL (2016). In vitro drug metabolism using liver microsomes. Current protocols in pharmacology, 7.8. 1–7.8. 24. [DOI] [PubMed] [Google Scholar]
- Kristensen C, Kjeldsen T, Wiberg FC, Schaffer L, Hach M, Havelund S, … Andersen AS (1997). Alanine scanning mutagenesis of insulin. The Journal of biological chemistry, 272(20), 12978–12983. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9148904 [DOI] [PubMed] [Google Scholar]
- Lee W, Tonelli M, & Markley JL (2015). NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics, 31(8), 1325–1327. doi: 10.1093/bioinformatics/btu830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre F, Remy MH, & Masson JM (1997). Alanine-stretch scanning mutagenesis: a simple and efficient method to probe protein structure and function. Nucleic Acids Res, 25(2), 447–448. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9016577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesner A, Legowska A, Wysocka M, & Rolka K (2011). Sunflower trypsin inhibitor 1 as a molecular scaffold for drug discovery. Current pharmaceutical design, 17(38), 4308–4317. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/22204429 [DOI] [PubMed] [Google Scholar]
- Liu J, Chow VT, & Jois SD (2004). A novel, rapid and sensitive heterotypic cell adhesion assay for CD2-CD58 interaction, and its application for testing inhibitory peptides. Journal of immunological methods, 291(1-2), 39–49. doi: 10.1016/j.jim.2004.04.026 [DOI] [PubMed] [Google Scholar]
- McGinnity DF, Soars MG, Urbanowicz RA, & Riley RJ (2004). Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab Dispos, 32(11), 1247–1253. doi: 10.1124/dmd.104.000026 [DOI] [PubMed] [Google Scholar]
- Merwe P. A. v. d., & Davis SJ (2003). Molecular Interactions Mediating T Cell Antigen Recognition. Annual review of immunology, 21(1), 659–684. doi: 10.1146/annurev.immunol.21.120601.141036 [DOI] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, & Olson AJ (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem, 30(16), 2785–2791. doi: 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowietz U (2002). Treatment targeted to cell surface epitopes. Clinical and experimental dermatology, 27(7), 591–596. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12464155 [DOI] [PubMed] [Google Scholar]
- Ong V, Hough G, Schlosser M, Bartizal K, Balkovec JM, James KD, & Krishnan BR (2016). Preclinical Evaluation of the Stability, Safety, and Efficacy of CD101, a Novel Echinocandin. Antimicrob Agents Chemother, 60(11), 6872–6879. doi: 10.1128/AAC.00701-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perczel A, Hollosi M, Foxman BM, & Fasman GD (1991). Conformational analysis of pseudocyclic hexapeptides based on quantitative circular dichroism (CD), NOE, and x-ray data. The pure CD spectra of type I and type II .beta.-turns. Journal of the American Chemical Society, 113(26), 9772–9784. doi: 10.1021/ja00026a010 [DOI] [Google Scholar]
- Poth AG, Chan LY, & Craik DJ (2013). Cyclotides as grafting frameworks for protein engineering and drug design applications. Biopolymers, 100(5), 480–491. doi: 10.1002/bip.22284 [DOI] [PubMed] [Google Scholar]
- Qiu Y, Taichi M, Wei N, Yang H, Luo KQ, & Tam JP (2017). An Orally Active Bradykinin B1 Receptor Antagonist Engineered as a Bifunctional Chimera of Sunflower Trypsin Inhibitor. Journal of medicinal chemistry, 60(1), 504–510. doi: 10.1021/acs.jmedchem.6b01011 [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, … Plenge RM (2009). Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet, 41(12), 1313–1318. doi: 10.1038/ng.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renukuntla J, Vadlapudi AD, Patel A, Boddu SH, & Mitra AK (2013). Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm, 447(1-2), 75–93. doi: 10.1016/j.ijpharm.2013.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GD, Gierasch LM, & Smith JA (1985). Turns in peptides and proteins. Adv Protein Chem, 37, 1–109. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2865874 [DOI] [PubMed] [Google Scholar]
- Sable R, Durek T, Taneja V, Craik DJ, Pallerla S, Gauthier T, & Jois S (2016). Constrained Cyclic Peptides as Immunomodulatory Inhibitors of the CD2:CD58 Protein-Protein Interaction. ACS chemical biology, 11(8), 2366–2374. doi: 10.1021/acschembio.6b00486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao T, Shi W, Zheng JY, Xu XX, Lin AF, Xiang LX, & Shao JZ (2018). Costimulatory Function of Cd58/Cd2 Interaction in Adaptive Humoral Immunity in a Zebrafish Model. Frontiers in immunology, 9, 1204. doi: 10.3389/fimmu.2018.01204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja V, & David CS (2010). Role of HLA class II genes in susceptibility/resistance to inflammatory arthritis: studies with humanized mice. Immunol Rev, 233(1), 62–78. doi: 10.1111/j.0105-2896.2009.00858.x [DOI] [PubMed] [Google Scholar]
- Taneja V, Krco CJ, Behrens MD, Luthra HS, Griffiths MM, & David CS (2007). B cells are important as antigen presenting cells for induction of MHC-restricted arthritis in transgenic mice. Mol Immunol, 44(11), 2988–2996. doi: 10.1016/j.molimm.2006.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja V, Taneja N, Behrens M, Griffiths MM, Luthra HS, & David CS (2005). Requirement for CD28 may not be absolute for collagen-induced arthritis: study with HLA-DQ8 transgenic mice. Journal of immunology, 174(2), 1118–1125. doi: 10.4049/jimmunol.174.2.1118 [DOI] [PubMed] [Google Scholar]
- Trebak M, & Kinet JP (2019). Calcium signalling in T cells. Nat Rev Immunol, 19(3), 154–169. doi: 10.1038/s41577-018-0110-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, & Davis SJ (2003). Molecular interactions mediating T cell antigen recognition. Annual review of immunology, 21, 659–684. doi: 10.1146/annurev.immunol.21.120601.141036 [DOI] [PubMed] [Google Scholar]
- Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, & Weiss A (2010). ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol, 2(5), a002279. doi: 10.1101/cshperspect.a002279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. h., Smolyar A, Tan K, Liu J. h., Kim M, Sun Z.-y. J., … Reinherz EL (1999). Structure of a Heterophilic Adhesion Complex between the Human CD2 and CD58 (LFA-3) Counterreceptors. Cell, 97(6), 791–803. doi: 10.1016/S0092-8674(00)80790-4 [DOI] [PubMed] [Google Scholar]
- Wang JH, Smolyar A, Tan K, Liu JH, Kim M, Sun ZY, … Reinherz EL (1999). Structure of a heterophilic adhesion complex between the human CD2 and CD58 (LFA-3) counterreceptors. Cell, 97(6), 791–803. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10380930 [DOI] [PubMed] [Google Scholar]
- Webber A, Hirose R, & Vincenti F (2011). Novel strategies in immunosuppression: issues in perspective. Transplantation, 91(10), 1057–1064. doi: 10.1097/TP.0b013e3182145306 [DOI] [PubMed] [Google Scholar]
- Weiss GA, Watanabe CK, Zhong A, Goddard A, & Sidhu SS (2000). Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proceedings of the National Academy of Sciences of the United States of America, 97(16), 8950–8954. doi: 10.1073/pnas.160252097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K (1986). NMR of Proteins and Nucleic Acids: Wiley. [Google Scholar]
- Zvereva I, Semenistaya E, Krotov G, & Rodchenkov G (2016). Comparison of various in vitro model systems of the metabolism of synthetic doping peptides: Proteolytic enzymes, human blood serum, liver and kidney microsomes and liver S9 fraction. J Proteomics, 149, 85–97. doi: 10.1016/j.jprot.2016.08.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.