

Abstract
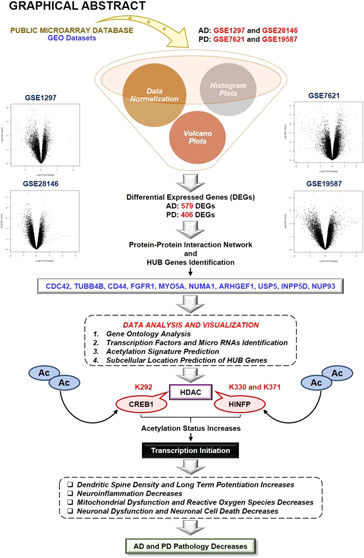
Integration of omics data and deciphering the mechanism of a biological regulatory network could be a promising approach to reveal the molecular mechanism involved in the progression of complex diseases, including Alzheimer’s and Parkinson’s. Despite having an overlapping mechanism in the etiology of Alzheimer’s disease (AD) and Parkinson’s disease (PD), the exact mechanism and signaling molecules behind them are still unknown. Further, the acetylation mechanism and histone deacetylase (HDAC) enzymes provide a positive direction toward studying the shared phenomenon between AD and PD pathogenesis. For instance, increased expression of HDACs causes a decrease in protein acetylation status, resulting in decreased cognitive and memory function. Herein, we employed an integrative approach to analyze the transcriptomics data that established a potential relationship between AD and PD. Data preprocessing and analysis of four publicly available microarray datasets revealed 10 HUB proteins, namely, CDC42, CD44, FGFR1, MYO5A, NUMA1, TUBB4B, ARHGEF9, USP5, INPP5D, and NUP93, that may be involved in the shared mechanism of AD and PD pathogenesis. Further, we identified the relationship between the HUB proteins and transcription factors that could be involved in the overlapping mechanism of AD and PD. CREB1 and HINFP were the crucial regulatory transcription factors that were involved in the AD and PD crosstalk. Further, lysine acetylation sites and HDAC enzyme prediction revealed the involvement of 15 and 27 potential lysine residues of CREB1 and HINFP, respectively. Our results highlighted the importance of HDAC1(K292) and HDAC6(K330) association with CREB1 and HINFP, respectively, in the AD and PD crosstalk. However, different datasets with a large number of samples and wet lab experimentation are required to validate and pinpoint the exact role of CREB1 and HINFP in the AD and PD crosstalk. It is also possible that the different datasets may or may not affect the results due to analysis parameters. In conclusion, our study potentially highlighted the crucial proteins, transcription factors, biological pathways, lysine residues, and HDAC enzymes shared between AD and PD at the molecular level. The findings can be used to study molecular studies to identify the possible relationship in the AD–PD crosstalk.
1. Introduction
Neurodegenerative diseases (NDDs) such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) are forms of dementia characterized by the progressive loss of neuronal cells due to the accumulation of toxic protein aggregates. Further, excessive neuronal cell death due to protein aggregates causes synaptic dysfunction, memory impairment, and cognitive defects.1 AD is the most prevalent form of dementia best characterized by the presence of amyloid plaques and neurofibrillary tangles produced by unsystematic proteolytic processing of amyloid peptide-protein and hyperphosphorylation of the tau protein.2 For example, Kollmer et al., 2019, demonstrated that β-amyloid (Aβ) fibrils from meningeal Alzheimer’s brain tissue are polymorphic but consist of similarly structured protofibrils.3 Similarly, Bu et al., 2017, concluded that blood-derived Aβ protein induces AD pathologies that result in the functional impairment of neurons.4 In contrast, PD, which is the second most common NDDs, is characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta. The pathological feature of PD is the accumulation of the toxic α-synuclein5 protein and the formation of Lewy bodies, which cause neuronal cell death and ultimately lead to synaptic dysfunction and memory loss.6,7 Mounting evidence suggests the common overlapping molecular phenomenon in the pathology of AD and PD. However, the exact molecular pathways and signaling molecules being involved are poorly understood. Moreover, the active treatment of AD and PD is still unknown due to a lack of understanding of the molecular mechanism of disease progression. Accumulating evidence suggests that protein acetylation and deacetylation play a significant role in the pathogenesis of AD and PD.8−10 For instance, Choi et al., 2019, demonstrated that acetylation of tau facilitated the recruitment of Hsp40, Hsp70, and Hsp110, which causes tau association with E3 ligases and results in its degradation through a proteasomal pathway.11 Similarly, Wang et al., 2020, concluded that AMPK reduces tau acetylation and rescues memory impairment by activating sirtuin 1 in APP/PS1 mice.12 Further, Fan et al., 2020, concluded that PGC-1α translocation due to its acetylation promotes neuroprotection from oxidative damage in a PD experimental model.13
In the pathogenesis of NDDs such as AD and PD, HDACs/HATs are involved in the regulation of biological processes such as apoptosis and autophagy, cell-cycle arrest, inflammatory and immune response, oxidative stress, and mitochondrial dysfunction, which cause neuronal cell death and lead to memory impairment and cognitive defects.14−18 Different experimental studies have confirmed the role of HDAC and its inhibitors in the pathogenesis of AD and PD. For instance, the overexpression of HDAC3 in the hippocampus increases spatial memory deficits and amyloid plaque load, whereas HDAC2 dysregulation in the nucleus basalis of Meynert was observed during the progression of AD.19,20 Similarly, the inhibition of HDAC through valproic acid increases histone acetylation levels and decreases the expression of proinflammatory biomarkers in the LRRK2 R1441G mice model of PD.21 Further, the inhibition of HDAC4/5 with the administration of LMK235 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium (MPP+) and α-synuclein-induced neuronal cell death.22 Apart from HDAC and its inhibitors, acetylated lysine residues of histone and non-histone substrates also play a crucial role in AD and PD pathogenesis. For example, Yakhine-Diop et al., 2018, demonstrated the acetylation of H4 at K5, K8, and K12 and its increased expression in IPD cells in PD pathogenesis. The authors also confirmed the acetylation of α-tubulin at K40 and the role of PCAF/p300 in α-tubulin acetylation.15 Similarly, Pilkington et al., 2020, concluded that the acetylation of Aβ at K16 and K28 promotes the extent of aggregation and inhibits fibril formation and oligomerization. However, the authors concluded that the acetylation of Aβ at K16 is preferred over the acetylation at K28.23 A recent study demonstrated the crucial role of lysine residues in the PTM crosstalk, namely, acetylation, ubiquitination, and SUMOylation in AD and PD pathogenesis. The authors concluded that the inhibition of PARP1 acetylation (K249, K331, K337, K528, K600, K637, K700, and K796) and the simultaneous activation of ubiquitination and SUMOylation at identical lysine residues rescue neuronal cell death.24 Further, lysine residues are also crucial for subcellular localization of proteins, where the loss of K304 resulted in CREB nuclear localization and modification of HDAC1 at K444 and K476, resulting in increased biological activity.25,26 In addition, Kirsh et al., 2002, demonstrated that SUMOylation of HDAC4 at K559 takes place at a nuclear pore complex RanBP2 and is coupled to its nuclear import.27 Thus, the identification of crucial lysine acetylating/deacetylating residues in novel pathological biomarkers provide considerable significance in unraveling a novel therapeutic approach for the treatment of AD and PD. Extensive research is ongoing on proteomics, transcriptomics, and epigenetic-based approaches to determine the molecular signatures and pathways involved in disease progression using a network biology approach based on microarray datasets. This will enable us to understand the molecular basis of the disease and the exact mechanism of disease progression.
Further, the acetylation and deacetylation of transcription factors (TFs) play a vital role in regulating cellular and molecular processes, which activate different neuronal signal transduction pathways such as PI3K/Akt and MAPK pathways, cAMP/PKA pathways, and Ca2+/CaMK cascade. For instance, Fusco et al., 2016, concluded that the acetylation of CREB1 at K122 increases Hes-1 expression under low glucose concentrations, facilitating neurogenesis by removing sirtuin 1 on the Hes-1 promoter region.28 Similarly, Paz et al., 2014, demonstrated that the acetylation of CREB at K136 facilitated its interaction with the CBP bromodomain that augmented the recruitment of this coactivator to the promoter.29 Thus, these pieces of evidence concluded the importance of acetylation of TFs in gene regulation.
Herein, we aim to investigate the potential conventional biomarkers and regulatory TFs involved in the pathogenesis of AD and PD simultaneously with the help of microarray datasets and the network biology approach. The identified proteomic and transcriptomic signatures were further analyzed to investigate the potential lysine residue for acetylation and deacetylation activity, along with the determination of the type of HDAC enzyme being involved in the disease progression. Lastly, the study focuses on investigating conserved amino acid residues involved in the lysine acetylation/deacetylation process along with the structural selectivity of molecular signatures, which could be crucial for protein acetylation or deacetylation activity.
2. Results and Discussion
2.1. Transcriptomic Signatures of AD and PD
The obtained datasets were normalized through quantile normalization and log2 transformation. Statistically, in microarray data, the intensity values are relative numbers, and thus log2 transformation is necessary to make variations similar across the order of magnitude. Boxplots of data before normalization and after normalization were created to check the background corrections in the datasets (Supplementary Figure 1). Further, independent histograms of normalized data with a color intensity such as green for control and red for the disease were prepared to check the variation in the required datasets (Supplementary Figure 2B). Our results identified 4736 (GSE7621), 2961 (GSE19587), 1989 (GSE1297), and 3634 (GSE28146) differentially expressed genes (DEGs) (Figure 1) (Table 1). Independent volcano plots of different datasets were used to measure the extent of DEGs in AD and PD (Supplementary Figure 2A). After identifying DEGs, the probe IDs were converted into respective gene symbols, and then Venn analysis of DEGs was performed. Venn analysis results demonstrated 579 DEGs in AD while 406 DEGs in PD.
Figure 1.

Methodology of the study: workflow and steps that were considered along with the datasets collected and processed to identify shared molecular signatures between AD and PD. The figure also highlights the involvement of the acetylation mechanism and HDAC enzymes in the AD and PD crosstalk.
Table 1. Datasets Obtained from the GEO Database for AD and PD.
| GEO accession number | platform | sample source | total samples | control samples | disease samples | total DEGs | upregulated DEGs | downregulated DEGs |
|---|---|---|---|---|---|---|---|---|
| Alzheimer’s Disease | ||||||||
| GSE1297 | Affymetrix Human Genome U133A Array | hippocampal region | 31 | 9 | 22 | 1989 | 949 | 1040 |
| GSE28146 | Affymetrix Human Genome U133 Plus 2.0 Array | hippocampal region | 30 | 8 | 22 | 3634 | 1718 | 1916 |
| Parkinson’s Disease | ||||||||
| GSE7621 | Affymetrix Human Genome U133 Plus 2.0 Array | Substantia nigra | 25 | 9 | 16 | 4736 | 2508 | 2228 |
| GSE19587 | Affymetrix Human Genome U133A 2.0 Array | Substantia nigra | 22 | 10 | 12 | 2961 | 1457 | 1504 |
2.2. PPI Interaction Analysis
PPI interaction analysis confirmed the presence of 492 proteins with 2335 physical interactions and 311 proteins and 1014 physical interactions in the AD and PD network, respectively. The clustering coefficients of AD and PD networks were found to be 0.244 and 0.248, respectively, which implies a higher coexpression of DEGs in AD networking than in PD networking. Further, the characteristic path lengths of AD and PD networks were 3.504 and 3.390, respectively. Herein, the network centralization was found to be 0.107 and 0.200, whereas the network heterogeneity was found to be 1.028 and 1.057 for AD and PD PPI networks, respectively. The analysis found that the network densities of AD and PD networks are 0.019 and 0.021, respectively, which indicates that a particular node in the PD PPI network has more participants compared to the AD PPI network (Figure 2).
Figure 2.

It represents the protein–protein interaction network of the top 15 ranked or HUB genes involved in Alzheimer’s disease, Parkinson’s disease, Alzheimer’s disease–Parkinson’s disease union merged network, and Alzheimer’s disease–Parkinson’s disease intersection merged network. Further, the top 15 proteins of the individual network were mapped against the clusters of AD, PD, AD–PD intersection, and AD–PD union network to extract HUB proteins.
Further, network biology using PPI networking becomes an important tool to establish a relationship between two proteins and identify the interactive pattern of proteomics data.30 In addition, PPI networking provides an in-depth understanding of the biological characteristics of proteins encoded through DEGs and helps in estimating their biological significance.31,32 The PPI network is characterized by the presence of nodes and edges along with other topological features, namely, clustering coefficient, characteristic path length, network density, and network centralization.33 The protein in the networks were represented as nodes marked in a circle, while their biological association with other proteins were represented as edges marked as lines.34 The clustering of the network determines the extent to which genes in the network coexpressed in biological conditions based on distance calculation. Thus, the higher the clustering coefficient, the lower the probability of proteins coexpressing in the biological network.35 The characteristic path length denotes the best possible configuration of the biological network.36 Network homogeneity refers to a nonuniformity in character,37,38 while network centralization or centrality identifies the network’s essential vertices or proteins.39,40 Another essential feature of biological networks is network density, which measures the average number of connections of a particular protein or node divided by the total number of participant proteins in the network.41 Statistically, the topological coefficient is a relative measure for the extent to which a particular protein in the given network shares neighbors with other proteins. The proteins that have one or no neighbors are assigned a topological coefficient of zero.42 The topological analysis of the PPI network provides a way to identify HUB proteins, which pass signaling stimulus to other proteins or nodes in the network. Subsequently, HUB proteins were identified based on topological features of the PPT network, especially node degree (number of proteins interacting with single protein), which may serve as potential biomarkers in AD and PD therapeutics.
2.3. Network Clustering and Proteomic Signatures of AD and PD
The merging of two PPI networks was done in two steps. In the first step, the PPI networks of AD and PD were combined by a union to ensure complete coverage of relevant proteins involved in the study, followed by extraction of common proteins (nodes) of the individual PPI network. The AD–PD union PPI network consists of 784 proteins and 3344 physical/functional interactions, while the AD–PD intersection biological network consists of 19 proteins and five physical/functional interactions (Figure 2). The top 15 highly connected proteins of individual AD, PD, AD–PD (union), and AD–PD (intersection) PPI networks were extracted. The HUB proteins were marked according to their presence in the respective PPI cluster. CDC42, CD44, FGFR1, MYO5A, NUMA1, TUBB4B, ARHGEF9, USP5, INPP5D, and NUP93 were found to be the most prominent proteins found in clusters of AD, PD, and AD–PD (union) PPI networks. Table 2 describes the role of HUB proteins in the pathogenesis of AD and PD (Supplementary Table 1). Here, our network analysis study demonstrates the involvement of CDC42, CD44, FGFR1, MYO5A, NUMA1, TUBB4B, ARHGEF9, USP5, INPP5D, and NUP93 in the onset and progression of AD and PD. Studies demonstrated that these proteins were associated with different biological processes. For instance, the activation of FAK/Rac1/CDC42-GTPase signaling rescued the impaired microglial migration response to Aβ42 in triggering the receptor expressed on myeloid cells 2 loss-of-function.43 Similarly, the inhibition of FGFR1 effectively blocked the GLP-promoted NPC proliferation in the mouse model of AD.44 However, the exact role of FGFR1 and CDC42 in the AD and PD crosstalk is still missing. In addition, Lim et al., 2018, concluded that CD44 activates tau pathology, whereas Neal et al., 2018, concluded that GPNMB attenuates astrocyte inflammatory response through the CD44 receptor.45,46 Further, loss of MYO5A resulted in structural and functional alterations in the rat brain through alterations in dopamine metabolism, whereas TUBB4B may be a part of the signaling cascade involved in the etiology of PD and is related to an inflammatory response.47,48 ARHGEF9 encodes collybistin involved in the postsynaptic clustering of glycine and inhibitor γ-aminobutyric acid receptors.49 Further, Griffin et al., 2020, concluded the upregulation of ARHGEF9 during astroglia response to Aβ oligomers.50 USP5, a stress granule protein, increases TNFα expression through the ubiquitin-proteasome pathway and regulates inflammatory response through Smurf1.51 Recently, Tsai et al., 2021, demonstrated that INPP5D was positively associated with amyloid plaque density in the human brain.52 Thus, these pieces of evidence concluded that the abovementioned HUB proteins are associated with neurological diseases in some manner through the regulation of different biological phenomena, yet their relationship in the AD and PD crosstalk is still missing. Further, HUB proteins, namely, NUMA1 and NUP93, lack the potential involvement in the pathogenesis of either AD and PD.
Table 2. Role of HUB Genes in the Pathogenesis of Alzheimer’s Disease and Parkinson’s Disease Identified with the Help of MalaCards.
| HUB genes | description | involvement in Alzheimer’s disease | involvement in Parkinson’s disease |
|---|---|---|---|
| CDC42 | Cell Division Cycle 42 | establishes neuron polarity, regulates cell morphology and mortality, and regulates cell cycle | inhibits the activating features of microglia |
| TUBB4B | Tubulin β 4B | regulates inflammatory response | serves as a target for PD-associated toxins |
| CD44 | CD44 Molecule (Indian Blood Group) | interacts with mutant p53 activity | causes α-synuclein-induced migration of BV-2 microglial cells |
| FGFR1 | Fibroblast Growth Factor Receptor 1 | involved in axonal projection and inhibits apoptosis | elevates DA levels and protects the specific midbrain neurons |
| MYO5A | Myosin VA (Heavy-Chain, Myoxin) | induces cell motility | mutant MYO5A exhibits alterations in dopamine metabolism |
| NUMA1 | Nuclear Mitotic Apparatus Protein 1 | identifies transported MSC in the brain | helps in mitotic spindle formation |
| ARHGEF9 | CDC42 Guanine Nucleotide Exchange Factor (GEF) 9 | plays a role in integrin signaling and axon guidance signaling | encodes synaptic proteins, and loss of function results in intellectual disability |
| USP5 | Ubiquitin-Specific Peptidase 5 | compromises tau levels | deletion causes increased p53 activity |
| INPP5D | Inositol Polyphosphate-5-Phosphatase, 145 kDa | modulates inflammatory response | involved in immune response |
| NUP93 | Nucleoporin 93 kDa | promotes nuclear accumulation of mRNA | inhibits mRNA transport |
2.4. Gene-Set Enrichment Analysis and Pathway Analysis
To identify the complicated relationship between the highly dense connected components of PPI networks (AD, PD, AD–PD union, and AD–PD intersection), pathway analysis and GO analysis were performed. Moreover, we extracted the top 10 biological pathways, cellular components, and molecular functions of highly interconnected proteins involved in neurodegeneration, as demonstrated in Figure 3. Moreover, after GO analysis, the extracted highly interconnected proteins were subjected to pathway analysis, which enables the identification of the molecular pathway, followed by the interconnected proteins in the progression of AD and PD. Figure 3 demonstrates the top 10 biological pathways in which these proteins were involved. Gap junction (TUBB4B), GnRH signaling pathway (CDC42), and Rap1 signaling pathway (CDC42 and FGFR1) were critical pathways in which HUB proteins were involved and may be potential biological pathway targets for the AD and PD crosstalk. For instance, Esteves et al., 2017, demonstrated that nicotine effectively prevented prefrontal long-term potentiation and memory deficits induced by streptozotocin in AD,53 whereas Carvajal-Oliveros et al., 2021, demonstrated that nicotine suppresses the PD-like phenotype induced by synphilin-1 overexpression through increased dopamine levels.54 Similarly, a study concluded that the balance between dopamine and adenosine signals regulates the PKA/Rap1 pathway in spiny neurons, where D1R and A2AR agonist enhanced PKA-mediated Rap1 phosphorylation in vivo and in vitro.55 Further, studies demonstrated that impaired GnRH production is directly linked to oxidative stress and mitochondrial dysfunction in neurons.56,57 Another significantly enriched pathway is the gap junction that is involved in the pathogenesis of AD and PD.58,59 For instance, Angeli et al., 2020, demonstrated the altered expression of glial gap junction proteins, namely, Cx43, Cx30, and Cx47, in the 5XFAD model of AD,60 whereas Maulik et al., 2020, concluded that Aβ regulates the gap junction protein connexin 43 in cultured primary astrocytes.61 Consistent with this, the results demonstrated the importance of CDC42, TUBB4B, and FGFR1 in the pathogenesis of AD and PD. Further, these three HUB proteins were a potential target for identifying the relationship between AD and PD.
Figure 3.

Represents the bar graph of the top 10 biological processes, molecular functions, and biological pathways of HUB proteins along with their p-value and involved HUB proteins. The axis of the bar represents the p-value. The figures also represent the critical cellular components in which HUB proteins lie with their corresponding p-value. Terms with a P-value ≤ 0.05 were considered significant.
2.5. CREB1 and HINFP: Essential Regulatory Molecules in AD and PD Crosstalk with High Acetylation Marks
The functions of a particular protein depend on its subcellular location. Mounting evidence demonstrated that acetylation was highly observed on nuclear proteins involved in chromatin regulation and transcription. This observation is consistent with the known nuclear function of acetyltransferase, deacetylase, and acetylated lysine-binding bromodomain proteins.62 For instance, a study demonstrated that in response to oxidative stress, TyrRS becomes highly acetylated, which causes its nuclear translocation, where sirtuin 1 and PCAF/p300 regulate its nuclear translocation in an acetylation-dependent manner.63 Further, Zhao et al., demonstrated that silica nanoparticle-mediated sirtuin 1 suppression markedly increased p53 acetylation and cytoplasmic localization.64
Herein, we analyzed the cellular location of HUB proteins with CELLO version 2.5: subCELlular LOcalization predictor. Among the 10 HUB proteins extracted, 40% were cytoplasmic proteins, 50% were nuclear proteins, and 10% were extracellular proteins. CD44 (1.974), FGFR1 (2.078), INPP5D (3.954), MYO5A (3.300), and NUMA1 (1.858) were predicted as nuclear proteins, while CDC42 (2.037) was predicted as an extracellular protein. Similarly, ARHGEF (2.770), NUP93 (2.534), TUBB4B (3.682), and USP5 (2.207) were predicted as cytoplasmic proteins. Studies demonstrated that acetylation activates STAT3 through the nuclear translocation of CD44, whereas the acetylation of histone proteins controls FGFR1 polymorphisms and isoform splicing.65,66 In addition, lysine acetylation of SCF FBXL19 ubiquitin E3 ligase increases its activity and stabilization that targets CDC42 for its ubiquitination and degradation.67
Further, we identified HUB protein–TF interaction and detected central regulatory molecules using topological features. Thus, we extracted seven regulatory TFs, namely, FOXC1 (8), GATA2 (5), CREB1 (4), FOXL1 (3), NFIC (3), HINFP (3), and SREBF1 (3). Subsequently, the cross-validation of TFs in the pathogenesis of AD and PD was identified with the help of MalaCards, as demonstrated in Table 3. TFs are transcriptional regulators that are involved in the pathogenesis of AD and PD.68−71 In this study, we also studied the potential relationship between TFs and HUB proteins to identify mutual transcriptional regulators of the identified HUB proteins. The identified TFs are FOXC1, GATA2, CREB1, FOXL1, NFIC, HINFP, and SREBF1 as a regulator of HUB proteins commonly expressed in AD and PD pathogenesis (Figure 4A). For instance, Xu et al., 2019, concluded that the deletion of CREB1 diminishes the effect of DJ1 on TH regulation through the deregulation of the CaMKKβ/CaMIV/CREB1 pathway.72 Similarly, the deletion of CREB1 promotes proinflammatory changes in the mouse hippocampus.73 Moreover, He et al. concluded that the deacetylation of EZH2 through SIRT6 causes an increased association between EZH2 and FOXC1 that exerts anti-inflammatory response, whereas Emelyanov et al., 2018, concluded the positive correlation between dopamine and GATA2 expression in PD.74,75 FOXL1 is implemented in the pathogenesis of NDDs, while NFIC was identified as novel loci in AD.76−78 Studies demonstrated that HINFP is a coactivator in the sterol-regulated transcription of PCSK9, a target gene of SREBP2 involved in the tau alterations, which contribute to disturbed cholesterol homeostasis in AD.79,80 Lastly, genetic mutation analysis concluded that genetic polymorphism rs11868035 was associated with susceptibility to PD in the Chinese population.81,82 Thus, the evidence mentioned above proves the potential link of identified TFs in the progression and pathogenesis of AD and PD and acts as a specific biomarker for their therapeutics. However, their potential role in the AD and PD crosstalk is still missing.
Table 3. Biological Significance of Top Interacting Transcription Factors in the Progression of Alzheimer’s Disease and Parkinson’s Disease, along with Their Degree of Node and Interacting Partners.
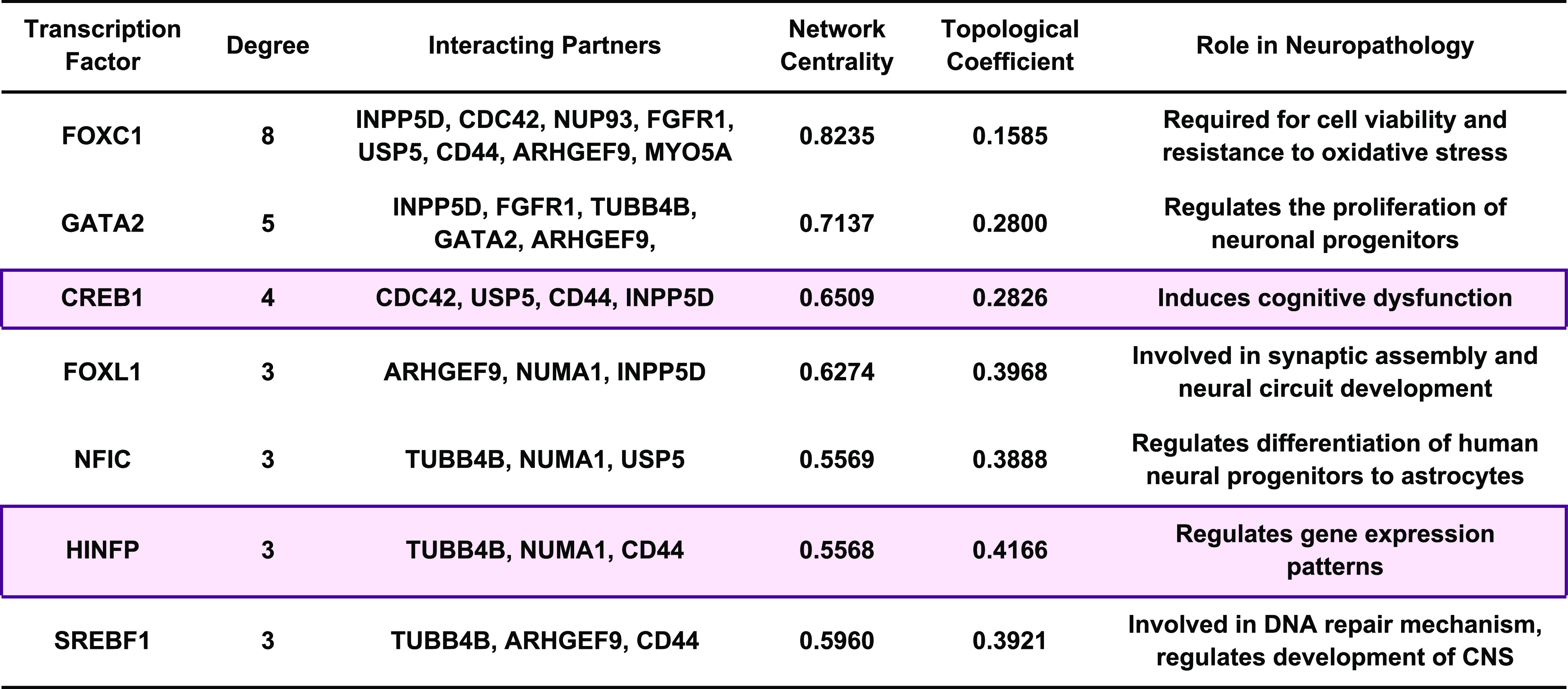
Figure 4.

(A) PPI network of HUB genes with associated regulatory transcription factors. Among the transcription factors, FOXC1 (8) has the highest number of interacting proteins, followed by GATA2 (5), CREB1 (4), FOXL1 (3), NFIC (3), HINFP (3), and SREBF1 (3). The total number of proteins and physical/functional interaction in HUB proteins and transcription factors in protein–protein interaction networks are 52 and 73. (B) Acetylation signatures of non-histone protein substrates, such as HUB genes and transcription factors. CREB1 and HINFP are the most prominent acetylated transcription factors, whereas CDC42, CD44, and TUBB4B are the most crucial non-histone protein acetylating substrates.
Moreover, HUB genes and TFs were analyzed for their acetylation signature to understand the involvement of acetylation and deacetylation processes associated with HUB genes and TFs in the pathogenesis of AD and PD. Herein, CDC42 (10), CD44 (11), FGFR1 (11), MYO5A (13), NUMA1 (14), ARHGEF9 (11), USP5 (14), and NUP93 (15) were predicted as the most non-histone acetylating substrates among HUB proteins, while CREB1 (16) and HINFP (10) were predicted as non-histone acetylating substrates among TFs (Figure 4B) (Supplementary Figure 3). Lately, to study the epigenetic regulation of HUB proteins and TFs, we investigated histone modification sites found in the coding region of HUB proteins and TFs implicated with NDDs and identified a range of sites.83,84 Thus, this raises the possibility that PTMs, namely, acetylation, deacetylation, ubiquitination, SUMOylation, methylation, and others, are the primary means of alteration in these proteins that need further investigation. Further, histone acetylation signatures are primarily related to the markers of activity at regulatory elements, namely, promoters and enhancers.85 Moreover, understanding the specific role of histone acetylation at different genomic elements has the potential to improve disease therapeutics by increasing the target specificity.86 In addition, histone signatures enable us to understand the biological phenomenon, namely, chromosome packaging, transcriptional activation, and DNA packaging. Further, studies demonstrated the correlation between histone acetylation levels and gene expression in vivo and in vitro studies. For instance, curcumin, a CREB HAT activity inhibitor, causes a reduction in acetylation levels of both histone H4 and H3, whereas HDAC inhibitors, namely, butyric acid and valproic acid, inhibit the H4 acetylation and CREB1 activity in vivo.87,88 Similarly, Guo et al., 2011, demonstrated that excessive alcohol exposure decreases CREB-binding protein expression and acetylation status of both H3 and H4 in the cerebellum of ethanol-induced rats.89 Similarly, another study identified that GATA1 displaces GATA2, which is associated with transcriptional repression, and causes a reduction in the histone H3K4 acetylation status.90 In addition, Li and Liu et al. concluded that HINFP forms a complex with NPAT that recruits the HAT cofactor TRRAP to facilitate H4 acetylation at the PCSK9 promoter, whereas Gruber et al. concluded that the requirement for the acetyltransferase activity of HAT1 for proliferation might point to the HAT1-dependent acetylation of non-histone substrates, for example, Hinfp, a factor also shown to bind H4 promoters.91,92 Forma et al., 2018, demonstrated that an increased expression of FOXA1 and FOXC1 was associated with increased acetylation levels of histone H3, whereas He et al. concluded that increased FOXC1 protein levels in RAW246.7 cells were associated with altered levels of H3 acetylation.74,93
2.6. Potential Lysine Residues for Protein Acetylation
The correlation between acetylation and HDAC enzymes has been studied extensively in the past.94−96 For instance, MS-275, a class I HDAC inhibitor, promotes rapid acetylation of the YB-1 RNA-binding protein at K81,97 whereas the HDAC1 complex is able to regulate histone H3 acetylation at K18.98 Further, Topuz et al., 2019, demonstrated that administration of the HDAC inhibitor, namely, sodium butyrate, increases H2B acetylation at K5 that leads to increased spatial learning and long-term memory in the rat hippocampus.99 Similarly, Choi et al., 2017, demonstrated that increased acetylation of peroxiredoxin 1 at K197 through HDAC6 inhibition leads to the recovery of Aβ-induced impaired axonal transport.100 In addition, the role of SIRT1 in regulating pathogenic tau acetylation at K174 and in suppressing the spread of tau pathology has been demonstrated in a mouse model of tauopathy.101 Thus, based on the abovementioned evidence, we identified acetylation sites and HDAC enzymes of CREB1 and HINFP through two online tools, namely, MuSite deep and PSKAcePred. For MuSite Deep, statistically, a high confidence score relates to the high probability of lysine acetylation at a particular lysine amino acid. A score above 0.5 is considered as a high confidence score and a high probability of lysine acetylation, whereas a score below 0.2 is considered as a low confidence score where the probability of lysine acetylation is negligible, and a score between 0.2 and 0.5 is considered as the site with moderate probability. Further, for PSKAcePred, a score above 0.7 is considered a high confidence score and the probability of lysine acetylation is very high, whereas a score between 0.5 and 0.7 is considered a moderate confidence score and the probability of lysine acetylation is relatively less as compared to acetylation at a high confidence score lysine site. The CREB1 peptide sequence (P16220.2) has 15 potential acetylating lysine residues. The respective acetylation-site prediction scores were determined with the help of MuSite deep and PSKAcePred, as shown in Supplementary Table 2. MuSite predicted K330 as an essential lysine acetylation site with a high confidence score of 0.557. Similarly, PSKAcePred predicted K94, K292, K303, K304, and K309 as potential protein acetylation lysine residues with a high confidence score of 0.872, 0.737, 0.856, 0.924, and 0.994, respectively. From the protein acetylation-site prediction of CREB1, it may be concluded that K304, K309, and K330 were essential for acetylating lysine residues. The type of HDAC enzymes involved in the deacetylation of CREB1 was predicted and found that HDAC1, HDAC2, and SIRT7 were important in CREB1 deacetylation, where HDAC1 was involved in K292 (3.35) deacetylation, HDAC2 involved in K330 (10.67), and SIRT7 involved in K94 (13.26), K303 (12.54), and K304 (12.90) deacetylation. The results demonstrated that the binding propensity of SIRT7 and HDAC2 is very low as compared to the binding propensity of HDAC1. Thus, the results show that K292 is a critical lysine residue for CREB1 acetylation and deacetylation processes with HDAC1 as its deacetylating enzyme involved in the pathogenesis of AD and PD. In addition, Hansen et al., 2019,62 demonstrated the acetylation of CREB1 at K330 and K136, whereas Paz et al., 2014, demonstrated that sirtuin 1 directly downregulates the CREB transcriptional activity by binding and deacetylating CREB at K136, thereby reducing CREB interaction with CBP.102 Further, Lu et al., 2003, confirmed the acetylation of CREB1 at K91, K94, and K136 within the activation domain through CBP. However, they also concluded that a single mutation of the putative CBP acetylation sites has no significant effect on the transactivation potential of CREB.103 Thus, these pieces of evidence suggest the possibility of CREB1 acetylation and its binding with HDAC enzymes in the regulation of gene transcription.
Moreover, HINFP (AAH17234.1) consists of 27 potential acetylating lysine residues such as K6, K10, K31, K94, K96, K164, K174, K181, K185, K197, K213, K236, K256, K285, K294, K301, K330, K335, K346, K352, K366, K367, K371, K382, K439, K446, and K504, as observed in Supplementary Table 3. MuSite Deep predicted all 27 sites as potential acetylating lysine residues with no residue of high confidence score. However, five sites were predicted as potential acetylation sites with a moderate score as follows: K6, 0.318; K213, 0.311; K330, 0.420; K371, 0.354; and K382, 0.269. Thus, predicted acetylation sites were essential for triggering protein acetylation results in transcription initiation. Among the predicted acetylating lysine residues, HDAC6 (K6 and K330), HDAC1 (K382), and SIRT1 (K371) were important deacetylating residues involved in protein deacetylation, resulting in the progression of AD and PD. However, the binding score of HDAC6 (2.74 and 3.84) was predicted higher than HDAC1 (6.53) and SIRT1 (7.01). Similarly, PSKAcePred predicted 15 potential lysine acetylation sites, of which eight sites were predicted as potential lysine acetylation sites with a high confidence score: K31, 0.730; K96, 0.766; K174, 0.779; K330, 0.726; K335, 0.930; K367, 0.904; K371, 0.911; and K446, 0.719. Further, the HDAC enzyme prediction tool predicted that SIRT1 (K31, 6.51 and K371, 7.01), SIRT2 (K446, 0.95), SIRT7 (K174, 13.26), HDAC1 (K367, 4.23), and HDAC6 (K330, 3.84 and K335, 2.74) were crucial enzymes involved in the regulation of HINFP deacetylating activity. A comparative analysis of both the acetylation prediction tools and the type of deacetylating enzyme reflected that K330 and K371 were crucial proteins acetylating lysine residues with HDAC6 and SIRT1 as their interacting partners. However, the confidence score of SIRT1 is lower than that of HDAC6, while the confidence of K330 is higher than that of K371. Thus, it will be concluded that K330 interacts with HDAC6 to carry out HINFP deacetylation in AD and PD progression. Further, until now, no proteomic study has investigated the implementation of acetylation sites and HDAC binding residues in the activation domain of HINFP. However, mounting evidence suggests that HAT1, an acetyltransferase binding to HINFP promoters, has a specific stimulatory effect on H4 gene transcription. In addition, the authors concluded that HAT1 promotes the accumulation of newly synthesized H4 dimers without affecting the levels of histones embedded in the nucleosome.92 Another study concluded that HINFP forms a functional complex with NPAT that recruits the HAT cofactor TRRAP to facilitate the histone 4 acetylation at the PCSK9 promoter.91 Thus, this in silico analysis could be a milestone in providing an avenue for identifying crucial acetylation or deacetylation patterns of CREB1 and HINFP to minimize AD and PD progression (Table 4).
Table 4. List of Common Crucial Lysine Residues in CREB1 and HINFP.
| CREB1 | |
|---|---|
| lysine residue | interactor |
| K94 | SIRT7 |
| K292 | HDAC1 |
| K303 | SIRT7 |
| K304 | SIRT7 |
| HINFP | |
| K31 | SIRT1 |
| K174 | SIRT7 |
| K330 | HDAC6 |
| K335 | HDAC6 |
| K367 | HDAC1 |
| K371 | SIRT1 |
2.7. Glutamic Acid and Leucine Predominately Conserved Residues at Protein Acetylation Sites
The predicted protein acetylation sites were analyzed for the conserved lysine residues, which could be crucial for lysine selectivity and specificity for the acetylation process and binding of deacetylating enzymes. CREB1 has 15 potential lysine residues represented by 16220.2, while HINFP has 27 potential lysine residues represented by AAH17234.1. The multiple sequence alignment (MSA) analysis of predicted lysine residues for acetylation revealed the conservation of negatively charged glutamic acid (E) and neutrally charged leucine (L), methionine (M), valine (V), and glutamine (Q) in close association with the positively charged lysine residue, as shown in Figure 5A. These conserved residues provided a suitable environment and favorable conditions for associated potential lysine residues for the acetylation process, thus imparting lysine selectivity and specificity for the acetylation and deacetylation. However, further investigations are required to determine the potential of conserved residues in the acetylation and deacetylation processes of CREB1 and HINFP. However, glutamic acid is most prominent compared to other conserved amino acids as it decreases the overall positive charge of lysine and imparts a negative charge to the lysine site, which will promote acetylation and deacetylation reactions. For instance, Nguyen et al., 2016, concluded that glutamine triggers acetylation-dependent degradation of glutamine synthetase, whereas Son et al., 2020, demonstrated that leucine regulates autophagy through acetylation of the mTORC1.104,105 Moreover, the role of methionine involvement in lysine acetylation is not studied so far in AD and PD, but yet at the same time demonstrated the potential relationship between methionine and lysine acetylation in other neurological defects. For instance, Chiki et al., 2021, concluded that the presence of oxidation of methionine at position 8 and acetylation at K6 resulted in the dramatic inhibition of Httex1 fibrilization.106 Thus, these studies correlate with our results and suggest that glutamic acid (E), leucine (L), methionine (M), valine (V), and glutamine (Q) could be critical amino acid residues in acetylation and HDAC binding.
Figure 5.

(A) Multiple sequence analysis of potential acetylation/deacetylation lysine residues by taking 21 window sizes. 21 window size was taken by lysine at the center with ten amino acids on both sides. (B) Proposed action of mechanism or the signaling transduction pathway in CREB1- and HINFP-mediated neurodegeneration.
Further, structural information of CREB reveals that it consists of 11 exons and three isoforms that are produced through alternative splicing.107 Primary structure studies of CREB identified the presence of four functional domains, namely, Q1 basal transcriptional activity domain, kinase inducible domain, a glutamine-rich region, and basic region/leucine zipper domain.108 Thus, this relates to the importance of glutamine in the structural activity of CREB1. Similarly, structural information of HINFP confirms that the interaction of HINFP produced TF with methyl-CpG-binding protein-2, a component of the HDAC complex, and plays an important role in transcription repression. Sekimata et al., 2004, demonstrated that HINFP, through its DNA-binding activity, acts as a sequence-specific (conserved CGGAC core) transcriptional repressor,109 whereas Medina et al. concluded that the PSCR motif is required for the activation of histone H4 gene transcription and promotes its binding with DNA.110 Further, the study revealed the presence of acetylated H4 histone in the binding activity of HINFP to USF and GAL4-AH.111 In addition, a study concluded that lysine residues control the conformational dynamics of proteins.112 Thus, it is equally important to identify the structural features of CREB1 and HINFP that were involved in the acetylation mechanism. Thus, the potential and possible acetylation lysine residues were analyzed for their structural selectivity for lysine recognition pattern and potential deacetylating enzyme attachment, as discussed in Supplementary Table 4. The structural pattern of the putative deacetylating enzyme attachment binding to potential acetylation or deacetylation lysine residues revealed the presence of α-helix, strand, and coil region in the CREB1 and HINFP peptide. However, an in-depth analysis of the structural configuration of CREB1 and HINFP revealed that the helix region is predominant over the strand/coil region in the acetylation of CREB1. A study by Maltsev et al., 2012, concluded the involvement of the helical structure in the acetylation process, where acetylation increases α-helicity of the first six residues of α-synuclein.113 Similarly, the coil region is dominant over the helix/strand region in the potential lysine acetylation of HINFP. The results correlate with the study by Kulemzina et al., 2016, which concluded that lysine acetylation promotes interactions between Smc coiled coils that are required for cohesion ring assembly.114 Further, the results were analyzed precisely and revealed the involvement of structural selectivity in the acetylation and deacetylation of CREB1 and HINFP. In addition, the results also provide an avenue of helix and coil regions in the acetylation of predicted lysine residues of CREB1 and HINFP, respectively (Figure 5B). However, due to limited structural information of CREB1 and HINFP and the potential effect of acetylation on CREB1 and HINFP structural changes, these results need to be verified in vitro.
3. Materials and Methods
3.1. Data Collection and Identification of Differentially Expressed Genes (DEGs)
The microarray gene expression datasets for AD (GSE1297115 and GSE28146116) and PD (GSE7621117 and GSE19587118) were obtained from the NCBI-GEO database (https://www.ncbi.nlm.nih.gov/geo/),119 irrespective of the population. The datasets were analyzed in an R-environment for data normalization and data preprocessing. Further, Limma was used to identify DEGs in both AD and PD compared to controls. The p-value < 0.05 and ILog 2FcI > 1.1 was regarded as cutoff criteria to screen for significant DEGs. The significance for the selection of Log 2Fc is that the expression is slightly more than twice for both upregulated and downregulated genes. The BioMart data-mining tool (https://m.ensembl.org/info/data/biomart/index.html)120 was applied to convert probe symbols into gene symbols. Lastly, Venn analysis was performed using the online tool called Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/)121 to identify common DEGs from the four datasets.
3.2. Protein–Protein Interaction Network Analysis and Visualization
The interrelation between different DEGs of AD and PD was obtained from STRING database version 11.0 (https://string-db.org/).122 The search criteria in the STRING database are limited to a confidence score of 0.5. The obtained networks were imported into Cytoscape software version 3.8.0 (https://cytoscape.org/)123 for protein data integration, PPI network visualization, and PPI network analysis. Subsequently, node degree, number of edges, clustering coefficient, network homogeneity, shortest path length, and network density of AD and PD PPI networks were calculated.
3.3. PPI Network Clustering and Identification of HUB Proteins
The AD and PD networks were merged using a network merging tool of Cytoscape based on two methods, namely, network union and network intersection. Afterward, network clustering was performed through molecular complex detection (MCODE) (http://apps.cytoscape.org/apps/mcode)124 plugin of Cytoscape software. The clusters so formed were analyzed and visualized on different parameters such as the number of proteins (nodes) and physical interactions between them (Edges), network clustering coefficient, characteristics of path length, network centralization and homogeneity, and network density. The clusters of all PPI networks were statistically analyzed and ranked separately based on node degree. Lastly, the HUB proteins were identified using CytoHubba (http://apps.cytoscape.org/apps/cytohubba)125 through default parameters. Subsequently, the HUB proteins were mapped from all PPI network clusters individually, which include AD, PD, and AD–PD union PPI networks.
3.4. Functional Enrichment and Pathway Analysis of HUB Proteins
HUB protein overrepresentation was performed through the bioinformatics resource EnrichR (http://amp.pharm.mssm.edu/Enrichr/)126 and QuickGO (https://www.ebi.ac.uk/QuickGO/)127 to identify the molecular function, biological process, and cellular function. Further, pathway analysis of HUB proteins was carried out using freely accessible online databases and tools such as the REACTOME database (https://reactome.org/)128 and FunRich version 3.1.3 (http://funrich.org/).129 For statistical assessment of GO analysis and pathway analysis, a p-value less than 0.05 was considered significant, and a fold-enrichment value was considered. Here, the p-value reflects the chance of observing “n” number of genes in a gene list annotated to a specific term, whereas fold enrichment of a term was designated as overrepresented compared to the background, where overrepresentation is denoted as positive fold enrichment.
3.5. HUB Proteins–Transcription Factors (TFs) Interaction and Prediction of Protein Subcellular Localization
The subcellular localization of HUB genes was calculated to understand the mechanism of action of protein and its associated functions using CELLO version 2.5: subCELlular LOcalization predictor (http://cello.life.nctu.edu.tw/).130 To identify the TFs that control the HUB proteins at a transcriptional level, TF-target interactions were obtained from JASPAR version 8 (http://jaspar.genereg.net/)131 and an interaction network between TFs and HUB proteins was created using NetworkAnalyst tool version 3.0 (https://www.networkanalyst.ca/home.xhtml).132
3.6. Identification of Histone Lysine Signatures and Prediction of Protein Deacetylating Enzymes
Based on the previous experimental studies, it was evident that acetylation signatures were associated with the pathogenesis of AD and PD by altering gene expression patterns. Thus, we used the Epigenomics Roadmap CHIP-seq dataset, which is an inbuilt feature of EnrichR for their potential acetylation marks of HUB proteins. Moreover, acetylation sites in CREB1 and HINFP have been predicted through machine learning algorithm-based, freely accessible online tools such as MuSite Deep (https://www.musite.net/)133 and PSKAcePred (http://bioinfo.ncu.edu.cn/inquiries_PSKAcePred.aspx).134 Lastly, the type of deacetylating enzyme associated with CREB and HINFP was predicted with the help of a freely accessible online web server named Deep-PLA (http://deeppla.cancerbio.info/index.html).135
3.7. Prediction of Conserved Lysine Residues and Structural Features for HDAC’s Binding
The conserved sequence was predicted using a multiple sequence alignment (MSA) of 21 window size of lysine site residues that includes 10 residues on both the left and the right end and containing a lysine acetylating site in the middle for both CREB1 and HINFP using ClustalW MSA tool (https://www.genome.jp/tools-bin/clustalw).136 Additionally, the structural selectivity of lysine acetylating sites has been predicted with the help of PSIPRED: protein structure prediction server (http://bioinf.cs.ucl.ac.uk/psipred/).137 Subsequently, the secondary structure of the protein has been correlated with their respective protein acetylating sites.
4. Conclusions
In conclusion, the present study focuses on the crosstalk between AD and PD at the molecular level. Through this study, we identified the relationship between DEGs, HUB proteins, TFs, acetylation, and HDAC enzymes in the shared pathogenesis of AD and PD. Our findings highlighted the crucial role of CDC42, TUBB4B, and FGFR1 in the AD and PD crosstalk through Gap junction (TUBB4B), GnRH signaling pathway (CDC42), and Rap1 signaling pathway (CDC42 and FGFR1). In addition, the present study identified the potential TFs that regulate the expression of HUB proteins at the transcriptional level through biological network analysis. Our analysis identified FOXC1, GATA2, CREB1, FOXL1, NFIC, HINFP, and SREBF1 as potential TFs that regulate the activity of HUB proteins shared between AD and PD. Our bioinformatic analysis also revealed the effect of subcellular localization of HUB proteins and TFs in the AD and PD crosstalk. Lately, the study identified the 15 potential lysine residues and 27 potential lysine residues in CREB1 and HINFP, respectively. The study revealed that among 15 possible lysine residues of CREB1, only four lysine residues, namely, K91, K94, K136, and K330, had been studied in the past, while K123, K155, K285, K292, K303, K304, K305, K309, K323, K333, and K339 have been reported first time for their role in the acetylation process. Similarly, among HINFP, all 27 lysine residues have been reported for the first time. Further, the in silico analysis of CREB1 and HINFP revealed the importance of HDAC1 for its deacetylation activity at K292 of CREB1 and HDAC6 for its deacetylation activity at K330 of HINFP. This will provide a way to study the role of acetylation and HDAC enzymes in the transcriptional activity of CREB1 and HINFP in the AD and PD crosstalk. Further, the computational analysis identified the importance of negatively charged glutamic acid (E) and neutrally charged leucine (L), methionine (M), valine (V), and glutamine (Q) amino acid residues in the acetylation mechanism of CREB1 and HINFP in the AD and PD crosstalk. The study also highlighted the importance of the helix region over the strand/coil region in the acetylation of CREB1. Similarly, the coil region is dominant over the helix/strand region in the potential lysine acetylation of HINFP. Thus, this study highlighted the importance of two prominent biological pathways for the progression of AD and PD simultaneously, such as HDAC1-CREB1-TUBB4B/CDC42/CD44 and HDAC6-HINFP-TUBB4B/CDC42/CD44 (Figure 6). Further studies are required to generate the potential treatments targeting the abovementioned biological pathways to treat the adverse effects of AD and PD. Further, the current study is associated with some sort of limitation as the study uses only microarray data, which is not as comprehensive as transcriptomics data analysis. Thus, there is a growing need to simultaneously analyze the different types of AD and PD datasets, namely, microarray data, epigenetic data, and RNA data, to extract the novel biomarkers involved in disease pathology. Further, there should be a greater number of control as well as disease samples to conclude a general discussion. In addition, samples from different tissues could be more beneficial in understanding the molecular mechanism and role of HDAC in AD and PD simultaneously. In the current study, using Bioinformatics tools, we identified that CREB1 and HINFP are putative targets in the pathogenesis of AD and PD simultaneously; however, the different datasets with a large number of samples and wet lab experimentation are absolutely necessary to establish the molecular signature and validate the role of CREB1 and HINFP in AD and PD.
Figure 6.

Literature validation of the involvement of HDAC interaction with CREB1 and HINFP. HDAC inhibitors cause a decrease in HDAC activity, followed by the increased acetylation status of CREB1, and HINFP causes positive transcriptional regulation. Increased transcriptional activity causes an increase in the transcription of memory-associated genes, and Bcl-2 expression leads to an increase in cognitive function and memory function. The increased acetylation status of CREB1 and HINFP causes INPP5D and TUBB4B activation, which decreases neuronal cell death and leads to neuroprotection.
Acknowledgments
The authors thank the senior management of Delhi Technological University for constant support and encouragement.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05827.
Box plot, volcano plots, relative expression of HUB genes, list of HUB genes, putative lysine residues in CREB1 and HINFP, and secondary structure of acetylated lysine residues (PDF)
Author Contributions
P.K. and R.G. conceived and designed the manuscript. R.G. collected, analyzed, and critically evaluated these data. R.G. prepared the figures and tables. P.K and R.G. analyzed the entire data and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Luthra P. M. Paradigm of Protein Folding in Neurodegenerative Diseases. Protein Res. J. 2011, 2, 479. [Google Scholar]
- Nussbaum R. L.; Ellis C. E. Alzheimer’s Disease and Parkinson’s Disease. N. Engl. J. Med. 2003, 348, 1356–1364. 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- Kollmer M.; Close W.; Funk L.; Rasmussen J.; Bsoul A.; Schierhorn A.; Schmidt M.; Sigurdson C. J.; Jucker M.; Fändrich M. Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat. Commun. 2019, 10, 4760 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu X.-L.; Xiang Y.; Jin W.-S.; Wang J.; Shen L.-L.; Huang Z.-L.; Zhang K.; Liu Y.-H.; Zeng F.; Liu J.-H.; Sun H.-L.; Zhuang Z.-Q.; Chen S.-H.; Yao X.-Q.; Giunta B.; Shan Y.-C.; Tan J.; Chen X.-W.; Dong Z.-F.; Zhou H.-D.; Zhou X.-F.; Song W.; Wang Y.-J. Blood-Derived Amyloid-β Protein Induces Alzheimer’s Disease Pathologies. Mol. Psychiatry 2018, 23, 1948–1956. 10.1038/mp.2017.204. [DOI] [PubMed] [Google Scholar]
- Zheng T.; Zhang Z. Activated Microglia Facilitate the Transmission of α-Synuclein in Parkinson’s Disease. Neurochem. Int. 2021, 148, 105094 10.1016/J.NEUINT.2021.105094. [DOI] [PubMed] [Google Scholar]
- Chang C.-W.; Yang S.-Y.; Yang C.-C.; Chang C.-W.; Wu Y.-R. Plasma and Serum Alpha-Synuclein as a Biomarker of Diagnosis in Patients With Parkinson’s Disease. Front. Neurol. 2020, 10, 1388. 10.3389/FNEUR.2019.01388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si X.; Tian J.; Chen Y.; Yan Y.; Pu J.; Zhang B. Central Nervous System-Derived Exosomal Alpha-Synuclein in Serum May Be a Biomarker in Parkinson’s Disease. Neuroscience 2019, 413, 308–316. 10.1016/J.NEUROSCIENCE.2019.05.015. [DOI] [PubMed] [Google Scholar]
- Bao J.; Zheng L.; Zhang Q.; Li X.; Zhang X.; Li Z.; Bai X.; Zhang Z.; Huo W.; Zhao X.; Shang S.; Wang Q.; Zhang C.; Ji J. Deacetylation of TFEB Promotes Fibrillar Aβ Degradation by Upregulating Lysosomal Biogenesis in Microglia. Protein Cell 2016, 7, 417–433. 10.1007/s13238-016-0269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S. W.; Cho S. H.; Zhou Y.; Schroeder S.; Haroutunian V.; Seeley W. W.; Huang E. J.; Shen Y.; Masliah E.; Mukherjee C.; Meyers D.; Cole P. A.; Ott M.; Gan L. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca C.; Garamszegi S.; Eldick D.; Singer C.; Mash D. Altered SIRT4 Expression Is Associated with Lewy Body Pathology (P01.209). Neurology 2012, 78, P01.209. 10.1212/wnl.78.1_meetingabstracts.p01.209. [DOI] [Google Scholar]
- Choi H.; Kim H. J.; Yang J.; Chae S.; Lee W.; Chung S.; Kim J.; Choi H.; Song H.; Lee C. K.; Jun J. H.; Lee Y. J.; Lee K.; Kim S.; Sim H.; Il Choi Y.; Ryu K. H.; Park J.-C.; Lee D.; Han S.-H.; Hwang D.; Kyung J.; Mook-Jung I. Acetylation Changes Tau Interactome to Degrade Tau in Alzheimer’s Disease Animal and Organoid Models. Aging Cell 2020, 19, e13081 10.1111/ACEL.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Shi F.-X.; Li N.; Cao Y.; Lei Y.; Wang J.-Z.; Tian Q.; Zhou X.-W. AMPK Ameliorates Tau Acetylation and Memory Impairment Through Sirt1. Mol. Neurobiol. 2020, 57, 5011–5025. 10.1007/S12035-020-02079-X. [DOI] [PubMed] [Google Scholar]
- Fan F.; Li S.; Wen Z.; Ye Q.; Chen X.; Ye Q. Regulation of PGC-1α Mediated by Acetylation and Phosphorylation in MPP+ Induced Cell Model of Parkinson’s Disease. Aging 2020, 12, 9461. 10.18632/AGING.103219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Pi L.; Zhou S.; Li X.; Min F.; Wang S.; Liu Z.; Wu J. Activation of Sirtuin 1 Attenuates High Glucose-Induced Neuronal Apoptosis by Deacetylating P53. Front. Endocrinol. 2018, 9, 274. 10.3389/FENDO.2018.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhine-Diop S. M. S.; Niso-Santano M.; Rodríguez-Arribas M.; Gómez-Sánchez R.; Martínez-Chacón G.; Uribe-Carretero E.; Navarro-García J. A.; Ruiz-Hurtado G.; Aiastui A.; Cooper J. M.; López de Munaín A.; Bravo-San Pedro J. M.; González-Polo R. A.; Fuentes J. M. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2466–2481. 10.1007/S12035-018-1206-6. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Feng Y.; Guo Q.; Guo W.; Xu H.; Li X.; Yi F.; Guan Y.; Geng N.; Wang P.; Cao L.; O’Rourke B. P.; Jo J.; Kwon J.; Wang R.; Song X.; Lee I. H.; Cao L. SIRT1 Modulates Cell Cycle Progression by Regulating CHK2 Acetylation–phosphorylation. Cell Death Differ. 2020, 27, 482–496. 10.1038/s41418-019-0369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C.; Lee P. T.; Kao T. J.; Chou S. Y.; Su R. Y.; Lee Y. C.; Yeh S. H.; Liou J. P.; Hsu T. I.; Su T. P.; Chuang C. K.; Chang W. C.; Chuang J. Y. Upregulation of Znf179 Acetylation by SAHA Protects Cells against Oxidative Stress. Redox Biol. 2018, 19, 74–80. 10.1016/J.REDOX.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kim Y.; Liu T.; Hwang Y. J.; Hyeon S. J.; Im H.; Lee K.; Alvarez V. E.; McKee A. C.; Um S.-J.; Hur M.; Mook-Jung I.; Kowall N. W.; Ryu H. SIRT3 Deregulation Is Linked to Mitochondrial Dysfunction in Alzheimer’s Disease. Aging Cell 2018, 17, e12679 10.1111/ACEL.12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Wang S.; Yu L.; Jin J.; Ye X.; Liu Y.; Xu Y. HDAC3 Negatively Regulates Spatial Memory in a Mouse Model of Alzheimer’s Disease. Aging Cell 2017, 16, 1073–1082. 10.1111/ACEL.12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahady L.; Nadeem M.; Malek-Ahmadi M.; Chen K.; Perez S. E.; Mufson E. J. HDAC2 Dysregulation in the Nucleus Basalis of Meynert during the Progression of Alzheimer’s Disease. Neuropathol. Appl. Neurobiol. 2019, 45, 380–397. 10.1111/NAN.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.; Song S.; Park Y.; Kang S.; Seo H. HDAC Inhibition by Valproic Acid Induces Neuroprotection and Improvement of PD-like Behaviors in LRRK2 R1441G Transgenic Mice. Exp. Neurobiol. 2019, 28, 504. 10.5607/EN.2019.28.4.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzocchi M.; Goulding S. R.; Wyatt S. L.; Collins L. M.; Sullivan A. M.; O’Keeffe G. W. LMK235, a Small Molecule Inhibitor of HDAC4/5, Protects Dopaminergic Neurons against Neurotoxin- and α-Synuclein-Induced Degeneration in Cellular Models of Parkinson’s Disease. Mol. Cell. Neurosci. 2021, 115, 103642 10.1016/J.MCN.2021.103642. [DOI] [PubMed] [Google Scholar]
- Pilkington A. W.; Schupp J.; Nyman M.; Valentine S. J.; Smith D. M.; Legleiter J. Acetylation of Aβ 40 Alters Aggregation in the Presence and Absence of Lipid Membranes. ACS Chem. Neurosci. 2020, 11, 146–161. 10.1021/acschemneuro.9b00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Kumar P. Computational Analysis Indicates That PARP1 Acts as a Histone Deacetylases Interactor Sharing Common Lysine Residues for Acetylation, Ubiquitination, and SUMOylation in Alzheimer’s and Parkinson’s Disease. ACS Omega 2021, 6, 5739–5753. 10.1021/acsomega.0c06168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford K. M.; Leonard M. O.; Karhausen J.; Carey R.; Colgan S. P.; Taylor C. T. Small Ubiquitin-Related Modifier-1 Modification Mediates Resolution of CREB-Dependent Responses to Hypoxia. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 986. 10.1073/PNAS.0337412100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G.; Neptune M. A.; Depinho R. A. SUMO-1 Modification of Histone Deacetylase 1 (HDAC1) Modulates Its Biological Activities. J. Biol. Chem. 2002, 277, 23658–23663. 10.1074/JBC.M203690200. [DOI] [PubMed] [Google Scholar]
- Kirsh O.; Seeler J. S.; Pichler A.; Gast A.; Müller S.; Miska E.; Mathieu M.; Harel-Bellan A.; Kouzarides T.; Melchior F.; Dejean A. The SUMO E3 Ligase RanBP2 Promotes Modification of the HDAC4 Deacetylase. EMBO J. 2002, 21, 2682–2691. 10.1093/EMBOJ/21.11.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco S.; Leone L.; Barbati S. A.; Samengo D.; Piacentini R.; Maulucci G.; Toietta G.; Spinelli M.; McBurney M.; Pani G.; Grassi C. A CREB-Sirt1-Hes1 Circuitry Mediates Neural Stem Cell Response to Glucose Availability. Cell Rep. 2016, 14, 1195–1205. 10.1016/J.CELREP.2015.12.092. [DOI] [PubMed] [Google Scholar]
- Paz J. C.; Park S.; Phillips N.; Matsumura S.; Tsai W.-W.; Kasper L.; Brindle P. K.; Zhang G.; Zhou M.-M.; Wright P. E.; Montminy M. Combinatorial Regulation of a Signal-Dependent Activator by Phosphorylation and Acetylation. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 17116–17121. 10.1073/PNAS.1420389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider J.; Kotlyar M.; Saraon P.; Yao Z.; Jurisica I.; Stagljar I. Fundamentals of Protein Interaction Network Mapping. Mol. Syst. Biol. 2015, 11, 848. 10.15252/MSB.20156351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E.; Ozturk K.; Carter H. Integrating Molecular Networks with Genetic Variant Interpretation for Precision Medicine. Wiley Interdiscip. Rev.: Syst. Biol. Med. 2019, 11, e1443 10.1002/WSBM.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; Chang Q.; Peng W.; Zhong J.; Li Y. Network Embedding the Protein–Protein Interaction Network for Human Essential Genes Identification. Genes 2020, 11, 153. 10.3390/GENES11020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella D.; Marini S.; Vitali F.; Di Silvestre D.; Mauri G.; Bellazzi R. MTGO: PPI Network Analysis Via Topological and Functional Module Identification OPEN. Sci. Rep. 2018, 8, 5499 10.1038/s41598-018-23672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.-J.; Liao D.-L.; Chen C.-H.; Wang T.-Y.; Chen K.-C. Construction and Analysis of Protein-Protein Interaction Network of Heroin Use Disorder. Sci. Rep. 2019, 9, 4980 10.1038/s41598-019-41552-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel C. C.; Zimmer R. Inferring Topology from Clustering Coefficients in Protein-Protein Interaction Networks. BMC Bioinf. 2006, 7, 519. 10.1186/1471-2105-7-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K.; Bezakova I.; Bunimovich L.; Yi S. V. Path Lengths in Protein-Protein Interaction Networks and Biological Complexity. Proteomics 2011, 11, 1857–1867. 10.1002/PMIC.201000684. [DOI] [PubMed] [Google Scholar]
- Pavel A.; del Giudice G.; Federico A.; Di Lieto A.; Kinaret P. A. S.; Serra A.; Greco D. Integrated Network Analysis Reveals New Genes Suggesting COVID-19 Chronic Effects and Treatment. Briefings Bioinf. 2021, 22, 1430–1441. 10.1093/BIB/BBAA417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S.; Johnson J.; Faisal F. E.; Milenković T. From Homogeneous to Heterogeneous Network Alignment via Colored Graphlets. Sci. Rep. 2018, 8, 12524 10.1038/s41598-018-30831-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashtiani M.; Salehzadeh-Yazdi A.; Razaghi-Moghadam Z.; Hennig H.; Wolkenhauer O.; Mirzaie M.; Jafari M. A Systematic Survey of Centrality Measures for Protein-Protein Interaction Networks. BMC Syst. Biol. 2018, 12, 80. 10.1186/S12918-018-0598-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschützki D.; Schreiber F.. Comparison of Centralities for Biological Networks*.
- Zhou H.; Liu J.; Li J.; Duan W. A Density-Based Approach for Detecting Complexes in Weighted PPI Networks by Semantic Similarity. PLoS One 2017, 12, e0180570 10.1371/JOURNAL.PONE.0180570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NetworkAnalyzer Help, https://med.bioinf.mpi-inf.mpg.de/netanalyzer/help/2.5/ (accessed Aug 21, 2021).
- Rong Z.; Cheng B.; Zhong L.; Ye X.; Li X.; Jia L.; Li Y.; Shue F.; Wang N.; Cheng Y.; Huang X.; Liu C.-C.; Fryer J. D.; Wang X.; Zhang Y.; Zheng H. Activation of FAK/Rac1/Cdc42-GTPase Signaling Ameliorates Impaired Microglial Migration Response to Aβ42 in Triggering Receptor Expressed on Myeloid Cells 2 Loss-of-Function Murine Models. FASEB J. 2020, 34, 10984–10997. 10.1096/FJ.202000550RR. [DOI] [PubMed] [Google Scholar]
- Huang S.; Mao J.; Ding K.; Zhou Y.; Zeng X.; Yang W.; Wang P.; Zhao C.; Yao J.; Xia P.; Pei G. Polysaccharides from Ganoderma Lucidum Promote Cognitive Function and Neural Progenitor Proliferation in Mouse Model of Alzheimer’s Disease. Stem Cell Rep. 2017, 8, 84–94. 10.1016/J.STEMCR.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.; Kim D.; Ju S.; Shin S.; Cho I.; Park S.-H.; Grailhe R.; Lee C.; Kim Y. K. Glioblastoma-Secreted Soluble CD44 Activates Tau Pathology in the Brain. Exp. Mol. Med. 2018, 50, 1–11. 10.1038/s12276-017-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal M. L.; Boyle A. M.; Budge K. M.; Safadi F. F.; Richardson J. R. The Glycoprotein GPNMB Attenuates Astrocyte Inflammatory Responses through the CD44 Receptor. J. Neuroinflammation 2018, 15, 73. 10.1186/S12974-018-1100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrock K. K.; Sullivan P.; Martini-Stoica H.; Goldstein D. S.; Graham B. H.; Yamamoto S.; Bellen H. J.; Gibbs R. A.; Chen R.; D’Amelio M.; Stoica G. Pleiotropic Neuropathological and Biochemical Alterations Associated with Myo5a Mutation in a Rat Model. Brain Res. 2018, 1679, 155–170. 10.1016/J.BRAINRES.2017.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Cheng R.; Ye X.; Verbitsky M.; Kisselev S.; Mejia-Santana H.; Louis E. D.; Cote L. J.; Andrews H. F.; Waters C. H.; Ford B.; Fahn S.; Marder K.; Lee J. H.; Clark L. N. Increased Rate of Sporadic and Recurrent Rare Genic Copy Number Variants in Parkinson’s Disease among Ashkenazi Jews. Mol. Genet. Genomic Med. 2013, 1, 142. 10.1002/MGG3.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat G.; LaGrave D.; Millson A.; Herriges J.; Lamb A. N.; Matalon R. Xq11.1-11.2 Deletion Involving ARHGEF9 in a Girl with Autism Spectrum Disorder. Eur. J. Med. Genet. 2016, 59, 470–473. 10.1016/J.EJMG.2016.05.014. [DOI] [PubMed] [Google Scholar]
- Griffin K.; Bejoy J.; Song L.; Hua T.; Marzano M.; Jeske R.; Sang Q.-X. A.; Li Y. Human Stem Cell-Derived Aggregates of Forebrain Astroglia Respond to Amyloid Beta Oligomers. Tissue Eng., Part A 2020, 26, 527–542. 10.1089/ten.tea.2019.0227. [DOI] [PubMed] [Google Scholar]
- Qian G.; Ren Y.; Zuo Y.; Yuan Y.; Zhao P.; Wang X.; Cheng Q.; Liu J.; Zhang L.; Guo T.; Liu C.; Zheng H. Smurf1 Represses TNF-α Production through Ubiquitination and Destabilization of USP5. Biochem. Biophys. Res. Commun. 2016, 474, 491–496. 10.1016/J.BBRC.2016.04.135. [DOI] [PubMed] [Google Scholar]
- Tsai A. P.; Lin P. B. C.; Dong C.; Moutinho M.; Casali B. T.; Liu Y.; Lamb B. T.; Landreth G. E.; Oblak A. L.; Nho K. INPP5D Expression Is Associated with Risk for Alzheimer’s Disease and Induced by Plaque-Associated Microglia. Neurobiol. Dis. 2021, 153, 105303 10.1016/J.NBD.2021.105303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves I. M.; Lopes-Aguiar C.; Rossignoli M. T.; Ruggiero R. N.; Broggini A. C. S.; Bueno-Junior L. S.; Kandratavicius L.; Monteiro M. R.; Romcy-Pereira R. N.; Leite J. P. Chronic Nicotine Attenuates Behavioral and Synaptic Plasticity Impairments in a Streptozotocin Model of Alzheimer’s Disease. Neuroscience 2017, 353, 87–97. 10.1016/J.NEUROSCIENCE.2017.04.011. [DOI] [PubMed] [Google Scholar]
- Carvajal-Oliveros A.; Domínguez-Baleón C.; Zárate R. V.; Campusano J. M.; Narváez-Padilla V.; Reynaud E. Nicotine Suppresses Parkinson’s Disease like Phenotypes Induced by Synphilin-1 Overexpression in Drosophila Melanogaster by Increasing Tyrosine Hydroxylase and Dopamine Levels. Sci. Rep. 2021, 11, 9579 10.1038/s41598-021-88910-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Nagai T.; Ahammad R. U.; Kuroda K.; Nakamuta S.; Nakano T.; Yukinawa N.; Funahashi Y.; Yamahashi Y.; Amano M.; Yoshimoto J.; Yamada K.; Kaibuchi K. Balance between Dopamine and Adenosine Signals Regulates the PKA/Rap1 Pathway in Striatal Medium Spiny Neurons. Neurochem. Int. 2019, 122, 8–18. 10.1016/J.NEUINT.2018.10.008. [DOI] [PubMed] [Google Scholar]
- Kagawa Y.; Umaru B. A.; Shil S. K.; Hayasaka K.; Zama R.; Kobayashi Y.; Miyazaki H.; Kobayashi S.; Suzuki C.; Katori Y.; Abe T.; Owada Y. Mitochondrial Dysfunction in GnRH Neurons Impaired GnRH Production. Biochem. Biophys. Res. Commun. 2020, 530, 329–335. 10.1016/J.BBRC.2020.07.090. [DOI] [PubMed] [Google Scholar]
- Şişli H. B.; Hayal T. B.; Şenkal S.; Kıratlı B.; Sağraç D.; Seçkin S.; Özpolat M.; Şahin F.; Yılmaz B.; Doğan A. Apelin Receptor Signaling Protects GT1-7 GnRH Neurons Against Oxidative Stress In Vitro. Cell. Mol. Neurobiol. 2020, 10.1007/S10571-020-00968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadian E.; Eftekhari A.; Samiei M.; Maleki Dizaj S.; Vinken M. The Role and Therapeutic Potential of Connexins, Pannexins and Their Channels in Parkinson’s Disease. Cell. Signalling 2019, 58, 111–118. 10.1016/J.CELLSIG.2019.03.010. [DOI] [PubMed] [Google Scholar]
- Xing J.; Xu C. Role of Connexins in Neurodegenerative Diseases (Review). Mol. Med. Rep. 2021, 23, 1–6. 10.3892/MMR.2021.12034. [DOI] [PubMed] [Google Scholar]
- Angeli S.; Kousiappa I.; Stavrou M.; Sargiannidou I.; Georgiou E.; Papacostas S. S.; Kleopa K. A. Altered Expression of Glial Gap Junction Proteins Cx43, Cx30, and Cx47 in the 5XFAD Model of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 582934. 10.3389/FNINS.2020.582934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik M.; Vasan L.; Bose A.; Chowdhury S. D.; Sengupta N.; Sarma J. D. Amyloid-β Regulates Gap Junction Protein Connexin 43 Trafficking in Cultured Primary Astrocytes. J. Biol. Chem. 2020, 295, 15097–15111. 10.1074/JBC.RA120.013705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen B. K.; Gupta R.; Baldus L.; Lyon D.; Narita T.; Lammers M.; Choudhary C.; Weinert B. T. Analysis of Human Acetylation Stoichiometry Defines Mechanistic Constraints on Protein Regulation. Nat. Commun. 2019, 10, 1055 10.1038/s41467-019-09024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.; Li C.; Xiao S.; Tang Y.; Huang J.; Zhao S.; Li X.; Li J.; Zhang R.; Yu W. Acetylation Promotes TyrRS Nuclear Translocation to Prevent Oxidative Damage. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 687–692. 10.1073/PNAS.1608488114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Wu Y.; Li J.; Li D.; Jin Y.; Zhu P.; Liu Y.; Zhuang Y.; Yu S.; Cao W.; Wei H.; Wang X.; Han Y.; Chen G. JNK Activation-Mediated Nuclear SIRT1 Protein Suppression Contributes to Silica Nanoparticle-Induced Pulmonary Damage via P53 Acetylation and Cytoplasmic Localisation. Toxicology 2019, 423, 42–53. 10.1016/J.TOX.2019.05.003. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Asa S. L.; Ezzat S. Histone-Acetylated Control of Fibroblast Growth Factor Receptor 2 Intron 2 Polymorphisms and Isoform Splicing in Breast Cancer. Mol. Endocrinol. 2009, 23, 1397. 10.1210/ME.2009-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. L.; Wang M. J.; Chen J. Y. Acetylation and Activation of STAT3 Mediated by Nuclear Translocation of CD44. J. Cell Biol. 2009, 185, 949–957. 10.1083/JCB.200812060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Dong S.; Yao K.; Martinez M. F. Y. M.; Fleisher P. R.; Zhao Y.; Ma H.; Zhao J. Histone Acetyltransferase CBP Promotes Function of SCF FBXL19 Ubiquitin E3 Ligase by Acetylation and Stabilization of Its F-Box Protein Subunit. FASEB J. 2018, 32, 4284–4292. 10.1096/FJ.201701069R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójtowicz S.; Strosznajder A. K.; Jeżyna M.; Strosznajder J. B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. 10.1007/S11064-020-02993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis D. J.; Han S.; Schuurmans C. BHLH Transcription Factors in Neural Development, Disease, and Reprogramming. Brain Res. 2019, 1705, 48–65. 10.1016/J.BRAINRES.2018.03.013. [DOI] [PubMed] [Google Scholar]
- Yao Z.; Yang W.; Gao Z.; Jia P. Nicotinamide Mononucleotide Inhibits JNK Activation to Reverse Alzheimer Disease. Neurosci. Lett. 2017, 647, 133–140. 10.1016/J.NEULET.2017.03.027. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Lin Y.; Wang L.; Zhan H.; Luo X.; Zeng Y.; Wu W.; Zhang X.; Wang F. TREM2 Ameliorates Neuroinflammatory Response and Cognitive Impairment via PI3K/AKT/FoxO3a Signaling Pathway in Alzheimer’s Disease Mice. Aging 2020, 12, 20862. 10.18632/AGING.104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Wang R.; Hao Z.; Wang G.; Mu C.; Ding J.; Sun W.; Ren H. DJ-1 Regulates Tyrosine Hydroxylase Expression through CaMKKβ/CaMKIV/CREB1 Pathway in Vitro and in Vivo. J. Cell. Physiol. 2020, 235, 869–879. 10.1002/JCP.29000. [DOI] [PubMed] [Google Scholar]
- Marchese E.; Di Maria V.; Samengo D.; Pani G.; Michetti F.; Geloso M. C. Post-Natal Deletion of Neuronal CAMP Responsive-Element Binding (CREB)-1 Promotes Pro-Inflammatory Changes in the Mouse Hippocampus. Neurochem. Res. 2017, 42, 2230–2245. 10.1007/S11064-017-2233-9. [DOI] [PubMed] [Google Scholar]
- He T.; Shang J.; Gao C.; Guan X.; Chen Y.; Zhu L.; Zhang L.; Zhang C.; Zhang J.; Pang T. A Novel SIRT6 Activator Ameliorates Neuroinflammation and Ischemic Brain Injury via EZH2/FOXC1 Axis. Acta Pharm. Sin. B 2021, 11, 708–726. 10.1016/J.APSB.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelyanov A. K.; Lavrinova A. O.; Litusova E. M.; Knyazev N. A.; Kulabukhova D. G.; Garaeva L. A.; Milyukhina I. V.; Berkovich O. A.; Pchelina S. N. The Effect of Dopamine on Gene Expression of Alpha-Synuclein and Transcription Factors GATA-1, GATA-2, and ZSCAN21 in Parkinson’s Disease. Cell Tissue Biol. 2018, 12, 410–418. 10.1134/S1990519X18050024. [DOI] [Google Scholar]
- Zhang W.; Duan N.; Song T.; Li Z.; Zhang C.; Chen X. The Emerging Roles of Forkhead Box (FOX) Proteins in Osteosarcoma. J. Cancer 2017, 8, 1619. 10.7150/JCA.18778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M. R.; Islam T.; Turanli B.; Zaman T.; Faruquee H. M.; Rahman M. M.; Mollah M. N. H.; Nanda R. K.; Arga K. Y.; Gov E.; Moni M. A. Network-Based Approach to Identify Molecular Signatures and Therapeutic Agents in Alzheimer’s Disease. Comput. Biol. Chem. 2019, 78, 431–439. 10.1016/J.COMPBIOLCHEM.2018.12.011. [DOI] [PubMed] [Google Scholar]
- Jun G. R.; Chung J.; Mez J.; Barber R.; Beecham G. W.; Bennett D. A.; Buxbaum J. D.; Byrd G. S.; Carrasquillo M. M.; Crane P. K.; Cruchaga C.; Jager P.; De; Ertekin-Taner N.; Evans D.; Fallin M. D.; Foroud T. M.; Friedland R. P.; Goate A. M.; Graff-Radford N. R.; Hendrie H.; Hall K. S.; Hamilton-Nelson K. L.; Inzelberg R.; Kamboh M. I.; Kauwe J. S.; Kukull W. A.; Kunkle B. W.; Kuwano R.; Larson E. B.; Logue M. W.; Manly J. J.; Martin E. R.; Montine T. J.; Mukherjee S.; Naj A.; Reiman E. M.; Reitz C.; Sherva R.; George-Hyslop S. P. H.; Thornton T.; Younkin S. G.; Vardarajan B. N.; Wang L.-S.; Wendlund J. R.; Winslow A. R.; Consortium A. D. G.; Haines J.; Mayeux R.; Pericak-Vance M. A.; Schellenberg G.; Lunetta K. L.; Farrer L. A.; et al. Transethnic Genome-Wide Scan Identifies Novel Alzheimer Disease Loci. Alzheimer’s Dement. 2017, 13, 727. 10.1016/J.JALZ.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Zhao F.; Shen K.; Wang W.; Siedlak S. L.; Lee H.-G.; Phelix C. F.; Perry G.; Shen L.; Tang B.; Yan R.; Zhu X. The Sterol Regulatory Element-Binding Protein 2 Is Dysregulated by Tau Alterations in Alzheimer Disease. Brain Pathol. 2019, 29, 530–543. 10.1111/BPA.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Liu J. The Novel Function of HINFP as a Co-Activator in Sterol-Regulated Transcription of PCSK9 in HepG2 Cells. Biochem. J. 2012, 443, 757–768. 10.1042/BJ20111645. [DOI] [PubMed] [Google Scholar]
- Lou F.; Li M.; Liu N.; Li X.; Ren Y.; Luo X. The Polymorphism of SREBF1 Gene Rs11868035 G/A Is Associated with Susceptibility to Parkinson’s Disease in a Chinese Population. Int. J. Neurosci. 2019, 129, 660–665. 10.1080/00207454.2018.1526796. [DOI] [PubMed] [Google Scholar]
- Yuan X. Q.; Cao B.; Wu Y.; Chen Y. P.; Wei Q. Q.; Ou R. W.; Yang J.; Chen X. P.; Zhao B.; Song W.; Shang H. F. Association Analysis of SNP Rs11868035 in SREBF1 with Sporadic Parkinson’s Disease, Sporadic Amyotrophic Lateral Sclerosis and Multiple System Atrophy in a Chinese Population. Neurosci. Lett. 2018, 664, 128–132. 10.1016/J.NEULET.2017.11.015. [DOI] [PubMed] [Google Scholar]
- Pattaroni C.; Jacob C. Histone Methylation in the Nervous System: Functions and Dysfunctions. Mol. Neurobiol. 2013, 47, 740–756. 10.1007/S12035-012-8376-4. [DOI] [PubMed] [Google Scholar]
- Rahman M. R.; Islam T.; Zaman T.; Shahjaman M.; Karim M. R.; Huq F.; Quinn J. M. W.; Holsinger R. M. D.; Gov E.; Moni M. A. Identification of Molecular Signatures and Pathways to Identify Novel Therapeutic Targets in Alzheimer’s Disease: Insights from a Systems Biomedicine Perspective. Genomics 2020, 112, 1290–1299. 10.1016/J.YGENO.2019.07.018. [DOI] [PubMed] [Google Scholar]
- Zentner G. E.; Henikoff S. Regulation of Nucleosome Dynamics by Histone Modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- Ram O.; Goren A.; Amit I.; Shoresh N.; Yosef N.; Ernst J.; Kellis M.; Gymrek M.; Issner R.; Coyne M.; Durham T.; Zhang X.; Donaghey J.; Epstein C. B.; Regev A.; Bernstein B. E. Combinatorial Patterning of Chromatin Regulators Uncovered by Genome-Wide Location Analysis in Human Cells. Cell 2011, 147, 1628. 10.1016/J.CELL.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. Y.; Kalda A.; Yu L.; Ferrara J.; Zhu J.; Chen J. F. Additive Effects of Histone Deacetylase Inhibitors and Amphetamine on Histone H4 Acetylation, CAMP Responsive Element Binding Protein Phosphorylation and ΔFosB Expression in the Striatum and Locomotor Sensitization in Mice. Neuroscience 2008, 157, 644–655. 10.1016/J.NEUROSCIENCE.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Yildirim F.; Ji S.; Kronenberg G.; Barco A.; Olivares R.; Benito E.; Dirnagl U.; Gertz K.; Endres M.; Harms C.; Meisel A. Histone Acetylation and CREB Binding Protein Are Required for Neuronal Resistance against Ischemic Injury. PLoS One 2014, 9, e95465 10.1371/JOURNAL.PONE.0095465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.; Crossey E. L.; Zhang L.; Zucca S.; George O. L.; Valenzuela C. F.; Zhao X. Alcohol Exposure Decreases CREB Binding Protein Expression and Histone Acetylation in the Developing Cerebellum. PLoS One 2011, 6, e19351 10.1371/JOURNAL.PONE.0019351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass J. A.; Boyer M. E.; Pal S.; Wu J.; Weiss M. J.; Bresnick E. H. GATA-1-Dependent Transcriptional Repression of GATA-2 via Disruption of Positive Autoregulation and Domain-Wide Chromatin Remodeling. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8811–8816. 10.1073/PNAS.1432147100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Liu J. The Novel Function of HINFP as a Co-Activator in Sterol-Regulated Transcription of PCSK9 in HepG2 Cells. Biochem. J. 2012, 443, 757–768. 10.1042/BJ20111645. [DOI] [PubMed] [Google Scholar]
- Gruber J. J.; Geller B.; Lipchik A. M.; Chen J.; Salahudeen A. A.; Ram A. N.; Ford J. M.; Kuo C. J.; Snyder M. P. HAT1 Coordinates Histone Production and Acetylation via H4 Promoter Binding. Mol. Cell 2019, 75, 711–724.e5. 10.1016/J.MOLCEL.2019.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forma E.; Jóźwiak P.; Ciesielski P.; Zaczek A.; Starska K.; Bryś M.; Krześlak A. Impact of OGT Deregulation on EZH2 Target Genes FOXA1 and FOXC1 Expression in Breast Cancer Cells. PLoS One 2018, 13, e0198351 10.1371/JOURNAL.PONE.0198351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Wu X.; Liu Q.; Kong G.; Zhou J.; Jiang J.; Wu X.; Huang Z.; Su W.; Zhu Q. Ketogenic Metabolism Inhibits Histone Deacetylase (HDAC) and Reduces Oxidative Stress After Spinal Cord Injury in Rats. Neuroscience 2017, 366, 36–43. 10.1016/J.NEUROSCIENCE.2017.09.056. [DOI] [PubMed] [Google Scholar]
- Lee J.; Ko Y. U.; Chung Y.; Yun N.; Kim M.; Kim K.; Oh Y. J. The Acetylation of Cyclin-Dependent Kinase 5 at Lysine 33 Regulates Kinase Activity and Neurite Length in Hippocampal Neurons. Sci. Rep. 2018, 8, 13676 10.1038/s41598-018-31785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H.; Wang M.; Wang J.; Cao H.; Niu W.; Du L. Paeonol Attenuates Isoflurane Anesthesia-Induced Hippocampal Neurotoxicity via Modulation of JNK/ERK/P38MAPK Pathway and Regulates Histone Acetylation in Neonatal Rat. J. Matern.-Fetal. Neonat. Med. 2020, 33, 81–91. 10.1080/14767058.2018.1487396. [DOI] [PubMed] [Google Scholar]
- El-Naggar A. M.; Somasekharan S. P.; Wang Y.; Cheng H.; Negri G. L.; Pan M.; Wang X. Q.; Delaidelli A.; Rafn B.; Cran J.; Zhang F.; Zhang H.; Colborne S.; Gleave M.; Mandinova A.; Kedersha N.; Hughes C. S.; Surdez D.; Delattre O.; Wang Y.; Huntsman D. G.; Morin G. B.; Sorensen P. H. Class I HDAC Inhibitors Enhance YB-1 Acetylation and Oxidative Stress to Block Sarcoma Metastasis. EMBO Rep. 2019, 20, e48375 10.15252/EMBR.201948375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. D. W.; Chandru A.; Watson P. J.; Song Y.; Blades M.; Robertson N. S.; Jamieson A. G.; Schwabe J. W. R.; Cowley S. M. Histone Deacetylase (HDAC) 1 and 2 Complexes Regulate Both Histone Acetylation and Crotonylation in Vivo. Sci. Rep. 2018, 8, 14690 10.1038/s41598-018-32927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topuz R. D.; Gunduz O.; Tastekin E.; Karadag C. H. Effects of Hippocampal Histone Acetylation and HDAC Inhibition on Spatial Learning and Memory in the Morris Water Maze in Rats. Fundam. Clin. Pharmacol. 2020, 34, 222–228. 10.1111/FCP.12512. [DOI] [PubMed] [Google Scholar]
- Choi H.; Kim H. J.; Kim J.; Kim S.; Yang J.; Lee W.; Park Y.; Hyeon S. J.; Lee D.-S.; Ryu H.; Chung J.; Mook-Jung I. Increased Acetylation of Peroxiredoxin1 by HDAC6 Inhibition Leads to Recovery of Aβ-Induced Impaired Axonal Transport. Mol. Neurodegener. 2017, 12, 23. 10.1186/s13024-017-0164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S.-W.; Sohn P. D.; Li Y.; Devidze N.; Johnson J. R.; Krogan N. J.; Masliah E.; Mok S.-A.; Gestwicki J. E.; Gan L. SIRT1 Deacetylates Tau and Reduces Pathogenic Tau Spread in a Mouse Model of Tauopathy. J. Neurosci. 2018, 38, 3680–3688. 10.1523/JNEUROSCI.2369-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz J. C.; Park S.; Phillips N.; Matsumura S.; Tsai W.-W.; Kasper L.; Brindle P. K.; Zhang G.; Zhou M.-M.; Wright P. E.; Montminy M. Combinatorial Regulation of a Signal-Dependent Activator by Phosphorylation and Acetylation. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 17116. 10.1073/PNAS.1420389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q.; Hutchins A. E.; Doyle C. M.; Lundblad J. R.; Kwok R. P. S. Acetylation of CAMP-Responsive Element-Binding Protein (CREB) by CREB-Binding Protein Enhances CREB-Dependent Transcription. J. Biol. Chem. 2003, 278, 15727–15734. 10.1074/JBC.M300546200. [DOI] [PubMed] [Google Scholar]
- Van Nguyen T.; Lee J. E.; Sweredoski M. J.; Yang S. J.; Jeon S. J.; Harrison J. S.; Yim J. H.; Lee S. G.; Handa H.; Kuhlman B.; Jeong J. S.; Reitsma J. M.; Park C. S.; Hess S.; Deshaies R. J. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol. Cell 2016, 61, 809–820. 10.1016/J.MOLCEL.2016.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son S. M.; Park S. J.; Stamatakou E.; Vicinanza M.; Menzies F. M.; Rubinsztein D. C. Leucine Regulates Autophagy via Acetylation of the MTORC1 Component Raptor. Nat. Commun. 2020, 11, 3148 10.1038/s41467-020-16886-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiki A.; Zhang Z.; Rajasekhar K.; Abriata L. A.; Rostami I.; Krapp L. F.; Boudeffa D.; Dal Peraro M.; Lashuel H. A. Investigating Crosstalk Among PTMs Provides Novel Insight Into the Structural Basis Underlying the Differential Effects of Nt17 PTMs on Mutant Httex1 Aggregation. Front. Mol. Biosci. 2021, 8, 686086. 10.3389/FMOLB.2021.686086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiki T. Role of CAMP Response Element Binding Protein in Cardiovascular Remodeling. Arterioscler., Thromb., Vasc. Biol. 2006, 26, 449–455. 10.1161/01.ATV.0000196747.79349.D1. [DOI] [PubMed] [Google Scholar]
- Xu W.; Kasper L. H.; Lerach S.; Jeevan T.; Brindle P. K. Individual CREB-Target Genes Dictate Usage of Distinct CAMP-Responsive Coactivation Mechanisms. EMBO J. 2007, 26, 2890–2903. 10.1038/SJ.EMBOJ.7601734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimata M.; Homma Y. Sequence-Specific Transcriptional Repression by an MBD2-Interacting Zinc Finger Protein MIZF. Nucleic Acids Res. 2004, 32, 590–597. 10.1093/NAR/GKH249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R.; Buck T.; Zaidi S. K.; Miele-Chamberland A.; Lian J. B.; Stein J. L.; Van Wijnen A. J.; Stein G. S. The Histone Gene Cell Cycle Regulator HiNF-P Is a Unique Zinc Finger Transcription Factor with a Novel Conserved Auxiliary DNA-Binding Motif. Biochemistry 2008, 47, 11415–11423. 10.1021/BI800961D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettese-Dadey M.; Grant P. A.; Hebbes2′ T. R.; Crane-Robinson2 C.; David Allis4 C.; Workman5 J. L. Acetylation of Histone H4 Plays a Primary Role in Enhancing Transcription Factor Binding to Nucleosomal DNA in Vitro. EMBO J. 1996, 15, 2508–2518. 10.1002/j.1460-2075.1996.tb00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz I.; Nestler P.; Köppen S.; Delcea M. Lysine Residues Control the Conformational Dynamics of Beta 2-Glycoprotein I. Phys. Chem. Chem. Phys. 2018, 20, 26819–26829. 10.1039/C8CP03234C. [DOI] [PubMed] [Google Scholar]
- Maltsev A. S.; Ying J.; Bax A. Impact of N-Terminal Acetylation of α-Synuclein on Its Random Coil and Lipid Binding Properties. Biochemistry 2012, 51, 5004–5013. 10.1021/BI300642H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulemzina I.; Ang K.; Zhao X.; Teh J. T.; Verma V.; Suranthran S.; Chavda A. P.; Huber R. G.; Eisenhaber B.; Eisenhaber F.; Yan J.; Ivanov D. A Reversible Association between Smc Coiled Coils Is Regulated by Lysine Acetylation and Is Required for Cohesin Association with the DNA. Mol. Cell 2016, 63, 1044–1054. 10.1016/J.MOLCEL.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Landfield P. W.; Geddes J. W.; Chen K. C.; Blalock E. M.; Porter N. M.; Markesbery W. R. Incipient Alzheimer’s Disease: Microarray Correlation Analyses Reveal Major Transcriptional and Tumor Suppressor Responses. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 2173–2178. 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock E. M.; Buechel H. M.; Popovic J.; Geddes J. W.; Landfield P. W. Microarray Analyses of Laser-Captured Hippocampus Reveal Distinct Gray and White Matter Signatures Associated with Incipient Alzheimer’s Disease. J. Chem. Neuroanat. 2011, 42, 118–126. 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnick T. G.; Papapetropoulos S.; Mash D. C.; Ffrench-Mullen J.; Shehadeh L.; De Andrade M.; Henley J. R.; Rocca W. A.; Ahlskog J. E.; Maraganore D. M. A Genomic Pathway Approach to a Complex Disease: Axon Guidance and Parkinson Disease. PLoS Genet. 2007, 3, e98 10.1371/journal.pgen.0030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski N. M.; Ju S.; Verbitsky M.; Ross B.; Geddie M. L.; Rockenstein E.; Adame A.; Muhammad A.; Vonsattel J. P.; Ringe D.; Cote L.; Lindquist S.; Masliah E.; Petsko G. A.; Marder K.; Clark L. N.; Small S. A. Polyamine Pathway Contributes to the Pathogenesis of Parkinson Disease. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 16970–16975. 10.1073/pnas.1011751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D.; Haider S.; Durinck S.; Pandini L.; Provero P.; Allen J.; Arnaiz O.; Awedh M. H.; Baldock R.; Barbiera G.; Bardou P.; Beck T.; Blake A.; Bonierbale M.; Brookes A. J.; Bucci G.; Buetti I.; Burge S.; Cabau C.; Carlson J. W.; Chelala C.; Chrysostomou C.; Cittaro D.; Collin O.; Cordova R.; Cutts R. J.; Dassi E.; Di Genova A.; Djari A.; Esposito A.; Estrella H.; Eyras E.; Fernandez-Banet J.; Forbes S.; Free R. C.; Fujisawa T.; Gadaleta E.; Garcia-Manteiga J. M.; Goodstein D.; Gray K.; Guerra-Assunção J. A.; Haggarty B.; Han D. J.; Han B. W.; Harris T.; Harshbarger J.; Hastings R. K.; Hayes R. D.; Hoede C.; Hu S.; Hu Z. L.; Hutchins L.; Kan Z.; Kawaji H.; Keliet A.; Kerhornou A.; Kim S.; Kinsella R.; Klopp C.; Kong L.; Lawson D.; Lazarevic D.; Lee J. H.; Letellier T.; Li C. Y.; Lio P.; Liu C. J.; Luo J.; Maass A.; Mariette J.; Maurel T.; Merella S.; Mohamed A. M.; Moreews F.; Nabihoudine I.; Ndegwa N.; Noirot C.; Perez-Llamas C.; Primig M.; Quattrone A.; Quesneville H.; Rambaldi D.; Reecy J.; Riba M.; Rosanoff S.; Saddiq A. A.; Salas E.; Sallou O.; Shepherd R.; Simon R.; Sperling L.; Spooner W.; Staines D. M.; Steinbach D.; Stone K.; Stupka E.; Teague J. W.; Dayem Ullah A. Z.; Wang J.; Ware D.; Wong-Erasmus M.; Youens-Clark K.; Zadissa A.; Zhang S. J.; Kasprzyk A. The BioMart Community Portal: An Innovative Alternative to Large, Centralized Data Repositories. Nucleic Acids Res. 2015, 43, W589. 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros J. C.VENNY. An interactive tool for comparing lists with Venn Diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html.
- Szklarczyk D.; Gable A. L.; Lyon D.; Junge A.; Wyder S.; Huerta-Cepas J.; Simonovic M.; Doncheva N. T.; Morris J. H.; Bork P.; Jensen L. J.; Von Mering C. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L.; Gouy A.; Daub J. T.; Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Nucleic Acids Res. 2017, 45, e149. 10.1093/nar/gkx626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G. D.; Hogue C. W. V. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinf. 2003, 4, 2. 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C. H.; Chen S. H.; Wu H. H.; Ho C. W.; Ko M. T.; Lin C. Y. CytoHubba: Identifying Hub Objects and Sub-Networks from Complex Interactome. BMC Syst. Biol. 2014, 8, 41. 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M. V.; Jones M. R.; Rouillard A. D.; Fernandez N. F.; Duan Q.; Wang Z.; Koplev S.; Jenkins S. L.; Jagodnik K. M.; Lachmann A.; McDermott M. G.; Monteiro C. D.; Gundersen G. W.; Ma’ayan A. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer E. C.; Huntley R. P.; Barrell D. G.; Binns D.; Draghici S.; Camon E. B.; Hubank M.; Talmud P. J.; Apweiler R.; Lovering R. C. The Gene Ontology - Providing a Functional Role in Proteomic Studies. Proteomics 2008, 10.1002/pmic.200800002. [DOI] [Google Scholar]
- Jassal B.; Matthews L.; Viteri G.; Gong C.; Lorente P.; Fabregat A.; Sidiropoulos K.; Cook J.; Gillespie M.; Haw R.; Loney F.; May B.; Milacic M.; Rothfels K.; Sevilla C.; Shamovsky V.; Shorser S.; Varusai T.; Weiser J.; Wu G.; Stein L.; Hermjakob H.; D’Eustachio P. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan M.; Keerthikumar S.; Ang C. S.; Gangoda L.; Quek C. Y. J.; Williamson N. A.; Mouradov D.; Sieber O. M.; Simpson R. J.; Salim A.; Bacic A.; Hill A. F.; Stroud D. A.; Ryan M. T.; Agbinya J. I.; Mariadason J. M.; Burgess A. W.; Mathivanan S. FunRich: An Open Access Standalone Functional Enrichment and Interaction Network Analysis Tool. Proteomics 2015, 15, 2597. 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- Yu C. S.; Chen Y. C.; Lu C. H.; Hwang J. K. Prediction of Protein Subcellular Localization. Proteins 2006, 64, 643. 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- Bryne J. C.; Valen E.; Tang M. H. E.; Marstrand T.; Winther O.; Da piedade I.; Krogh A.; Lenhard B.; Sandelin A. JASPAR, the Open Access Database of Transcription Factor-Binding Profiles: New Content and Tools in the 2008 Update. Nucleic Acids Res. 2007, 36, D102–D106. 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G.; Soufan O.; Ewald J.; Hancock R. E. W.; Basu N.; Xia J. NetworkAnalyst 3.0: A Visual Analytics Platform for Comprehensive Gene Expression Profiling and Meta-Analysis. Nucleic Acids Res. 2019, 47, W234. 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Liu D.; Yuchi J.; He F.; Jiang Y.; Cai S.; Li J.; Xu D. MusiteDeep: A Deep-Learning Based Webserver for Protein Post-Translational Modification Site Prediction and Visualization. Nucleic Acids Res. 2020, 48, W140–W146. 10.1093/NAR/GKAA275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo S.-B.; Qiu J.-D.; Shi S.-P.; Sun X.-Y.; Huang S.-Y.; Chen X.; Liang R.-P. Position-Specific Analysis and Prediction for Protein Lysine Acetylation Based on Multiple Features. PLoS One 2012, 7, e49108 10.1371/JOURNAL.PONE.0049108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.; Zhang Q.; Liu Z.; Du Y.; Gao X.; Zhao Q.; Cheng H.; Li X.; Liu Z.-X. Deep Learning Based Prediction of Reversible HAT/HDAC-Specific Lysine Acetylation. Briefings Bioinf. 2020, 21, 1798–1805. 10.1093/BIB/BBZ107. [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Gibson T. J.; Higgins D. G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protoc. Bioinf. 2003, 151. 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- McGuffin L. J.; Bryson K.; Jones D. T. The PSIPRED Protein Structure Prediction Server. Bioinformatics 2000, 16, 404. 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.