

Abstract
Copper is an essential trace element for human biology where its metal dyshomeostasis accounts for an increased level of serum copper, which accelerates protein aggregation. Protein aggregation is a notable feature for many neurodegenerative disorders. Herein, we report an experimental study using two model proteins, bovine serum albumin (BSA) and human serum albumin (HSA), to elucidate the mechanistic pathway by which serum albumins get converted from a fully folded globular protein to a fibril and an amorphous aggregate upon interaction with copper. Steady-state fluorescence, time-resolved fluorescence studies, and Raman spectroscopy were used to monitor the unfolding of serum albumin with increasing copper concentrations. Steady-state fluorescence studies have revealed that the fluorescence quenching of BSA/HSA by Cu(II) has occurred through a static quenching mechanism, and we have evaluated both the quenching constants individually. The binding constants of BSA–Cu(II) and HSA–Cu(II) were found to be 2.42 × 104 and 0.05 × 104 M–1, respectively. Further nanoscale morphological changes of BSA mediated by oligomers to fibril and HSA to amorphous aggregate formation were studied using atomic force microscopy. This aggregation process correlates with the Stern–Volmer plots in the absence of discernible lag phase. Raman spectroscopy results obtained are in good agreement with the increase in antiparallel β-sheet structures formed during the aggregation of BSA in the presence of Cu(II) ions. However, an increase in α-helical fractions is observed for the amorphous aggregate formed from HSA.
Introduction
Proteins are the building blocks of life that possess diverse functions owing to proper folding and supramolecular assembly. Failure in normal protein folding machinery accounts for protein aggregation in human body.1−3 Proteinopathies ranging from neurodegeneration to type 2 diabetes and systemic amyloidosis result from protein aggregation.4 Metal ions, pH, temperature, and protein concentration are few factors which accelerate protein aggregation.5 During the aggregation process, proteins undergo a number of conformational and intermolecular rearrangements with the formation of small clusters to amyloid fibrils and amyloid plaques.6 Such species marked their presence in many of the neurodegenerative disorders. Each of the intermediate species formed is said to possess standard features such as the intermolecular β-sheet core, as seen in the case of amyloid fibrils.7 Studies on intermediate species suggest that the toxicity of intermediate species (e.g., oligomers) is more than that of the final fibrils.8 Therefore, it is necessary to analyze the possible mechanism of production and growth of protein clusters formed during the on/off pathway during protein aggregation.
In vitro studies help to follow the mechanism of protein aggregation with the help of different techniques which are sensitive to the different types of structuring in proteins. The study of the structural organization of protein aggregates uses a wide variety of combined spectroscopic and microscopic techniques such as fluorescence spectroscopy, time-correlated single-photon counting measurements (TCSPC), Raman spectroscopy, and atomic force microscopy (AFM) imaging technique. The intrinsic fluorescence of Trp residues is highly sensitive to the changes in its microenvironment. Therefore, a change in the intensity of intrinsic Trp fluorescence suggests an alteration in the microenvironment with the addition of a quencher molecule. Therefore, a strong binding occurs between the protein and quencher molecule. Thus, fluorescence spectroscopy is a valuable tool to measure the changes in the fluorescence intensity, spectral shifts, and lifetime of fluorophores during protein aggregation. Raman spectroscopy measures the Raman scattered radiation associated with secondary structural reorganization in proteins. Moreover, it can differentiate between parallel and antiparallel β-sheets as well as β-sheet versus amyloid formation. AFM images reveal the morphology of micro- and nanodimensional protein structures.7−10
Protein aggregation does not occur at normal physiological conditions, but some trigger factors can induce aggregation. Metal ion concentration is one among the trigger factors that promotes protein aggregation the most. Dyshomeostasis and generation of reactive oxygen species by metal ions promote protein misfolding and aggregation.11 Recent research has revealed the role of transition metals such as Fe, Cu, and Zn in the aggregation of proteins associated with neurodegenerative disorders.12,13 Among the metals, the transition metal Cu(II) can bind with the disease-related proteins such as amyloid β (Aβ), α-synuclein (AS), and Tau (TP) with high affinity.14−17 Besides the involvement of Cu(II) in protein aggregation diseases, it is essential for homeostasis in the human body. Copper has various functions in applications such as energy production, neuropeptide activation, iron metabolism, and neurotransmitter synthesis.18 A copper concentration of about 50–80 mg is required for healthy adults and concentrations above this are toxic. An adequate amount of copper reaches the human body through food supplements and water. Excess copper exposures occur through consumption of acidic food cooked in uncoated copper cookware, consumption of excess copper containing drinking water, copper pipes, birth controls, dietary supplements, breathing air or dust holding copper, use of copper salt topical creams, in farming as pesticides, leather industry, and from other environmental sources. Due to its multiple entry points, the copper concentration in the human body increases in tissues, leading to copper toxicity and its associated protein aggregation diseases.19,20 Copper complexation and reactive oxygen species production are the major pathways by which Cu(II) brings about severe aggregation in proteins.20 Also, the copper concentrations and pH may vary the propensity of binding and pathway of aggregation in proteins.21−23
Therefore, protein aggregation and amyloid formation are considered to be the key issues for various proteinopathies. To put light on this issue, a systematic study is necessary to outline a relationship between serum albumin aggregation in the presence of Cu(II) ions at physiological pH. We have chosen two serum albumins, human serum albumin (HSA) and a model protein bovine serum albumin (BSA), for the present study. For in vitro studies, serum albumins act as exemplar proteins by playing a carrier role in several bioactive molecules and metals that reversibly bind to it. BSA has 80% sequence homology and 76% structural similarity with HSA. The present investigation was carried out at physiological pH and with micromolar copper concentrations. A combined use of spectroscopic and microscopic techniques is used to monitor the aggregation process.
Results and Discussion
Binding Mechanism Analysis of Cu(II) Ions with Serum Albumins
Fluorescence spectroscopy is a widely used tool to study the interaction of ligands with proteins; hence, it is used to measure the extent of binding to protein. Intrinsic fluorescence of proteins is due to the presence of three amino acid residues: tryptophan, tyrosine, and phenylalanine. Among the amino acid residues, Trp is the dominant source for intrinsic fluorescence in protein. Particularly in the case of serum albumins, Tyr and Trp residues act as the probe to study protein fluorescence. Fluorescence quenching is the phenomena by which the intrinsic fluorescence of fluorophores is decreased by the interaction with a quencher. A variety of molecular interactions such as excited-state reactions, molecular rearrangements, energy transfer, ground-state complex formation, and collisional quenching can result in fluorescence quenching.26 The fluorescence quenching mechanism is broadly classified under the following categories: dynamic/collisional, static, and combined static–dynamic quenching mechanisms. Dynamic quenching results when the excited-state fluorophore molecule is deactivated upon contact with a quencher molecule in solution and returns to the ground state by a diffusive encounter with the quencher. The formation of a nonfluorescent ground-state complex between the fluorophore and quencher occurs when the mechanism of quenching is static.27 Static and dynamic quenching can be differentiated by their temperature dependence. The quenching rate constant is found to be decreased with increasing temperature for dynamic/collisional quenching, while the reverse effect is observed for static quenching. In all the cases, a molecular contact between a fluorophore and quencher is essential for fluorescence quenching to occur.28
The present study deals with the binding interaction of Cu(II) ions to BSA and HSA. The intrinsic fluorescence spectra of BSA and HSA in tris-HCl were recorded by keeping their concentration constant (10 μM) and with an incremental concentration of Cu(II) ions. The results are depicted in Figure 1a,b.
Figure 1.
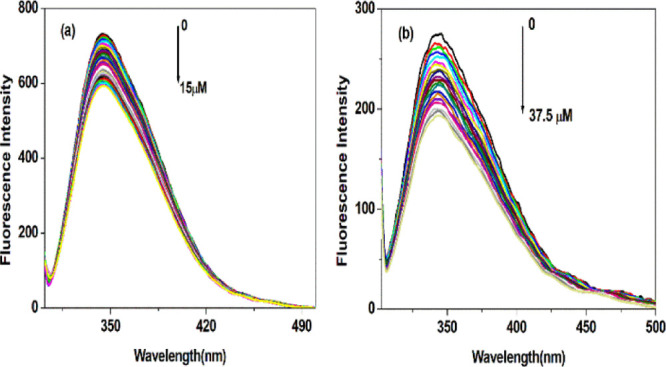
(a) Fluorescence spectra showing quenching of intrinsic fluorescence of BSA and (b) HSA with an increase in Cu(II) ion concentration.
From the experiments, it is clear that Cu(II) ions have no fluorescence under experimental conditions. BSA and HSA have a maximum fluorescence emission in the wavelengths of 344 and 345.5 nm, respectively, upon excitation at 295 nm. The stepwise increase in Cu(II) concentration decreases the fluorescence intensities of BSA and HSA. This observation immediately elucidates that a strong interaction occurs between BSA and HSA with Cu(II) ions. The maximum emission wavelength for BSA has a red shift from 344 to 345 nm, while a blue shift from 345.5 to 344 nm is observed for HSA when the wavelength of excitation is kept at 295 nm. This result indicates that Cu(II) ions have quenched the intrinsic fluorescence of both serum albumins with a change in the microenvironment of fluorophores. Cu(II) has increased the polarity around the microenvironment of BSA. However, a symptomatic blue shift is observed with an increase in hydrophobicity in the microenvironment of HSA. Literature reports support that a shift in the emission spectrum to shorter wavelengths with a progressive increase in quencher concentrations is mainly attributed to the fluorescence quenching of Trp residues buried deep inside the protein’s hydrophobic core.29,30 Also, it is interesting to note that the fluorescence intensities of BSA and HSA decreased to approximately 19.15 and 29.93%, respectively, with the addition of Cu(II) at pH 7.4.
To further elucidate/understand the fluorescence quenching mechanism, the fluorescence data obtained were analyzed with the help of the Stern–Volmer equation.
| 1 |
where Fo and F represent the fluorescence intensities in the absence and presence of Cu(II) ions, Ksv is the Stern–Volmer quenching constant, and its value is obtained from the linear regression of a plot of Fo/F versus [Q]. Kq is the quenching rate constant of the biomolecule.
| 2 |
τ0 is the average lifetime of the fluorophore without the quencher (τ0 = 2.80 × 10–9 s) and [Q] is the concentration of Cu(II) ions.31,32
The Stern–Volmer plots of BSA and HSA under varying concentrations of Cu(II) ions are presented in Figure 2.
Figure 2.
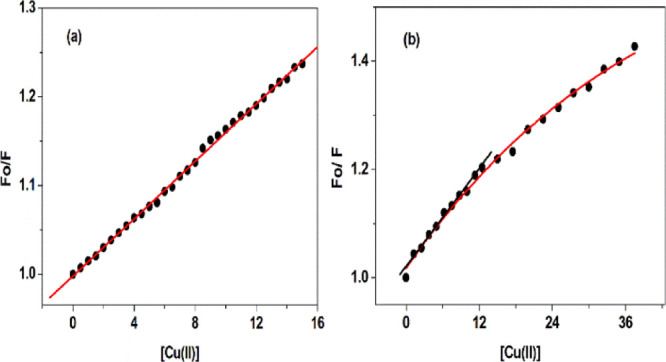
(a) Corrected Stern–Volmer plots of BSA and (b) HSA under varying concentrations of Cu(II) ions at 298 K.
The Stern–Volmer plots for the BSA–Cu(II) system in Figure 2a shows a good linear relationship, which indicates a single quenching mechanism (static or dynamic) or the presence of a single class of fluorophore. BSA has two tryptophan residues with intrinsic fluorescence in a distinct environment. Trp-212 located within a hydrophobic pocket of the protein and Trp-134 located on the surface of the molecule. Here, the quenching of tryptophan fluorescence present in both hydrophilic and hydrophobic pockets is equally affected, that is, Cu(II) does not have a preferential site of binding in BSA. This is the reason why linear Stern–Volmer plots are observed even at increasing copper concentrations.33 The Ksv and Kq values calculated for the BSA–Cu(II) complex are listed in Table 1.
Table 1. Quenching Constant and Binding Parameter of the BSA/HSA–Cu(II) Ion System at 298 K.
| Ksv (M–1) | Kq (M–1 s–1) | n1 | Kb1 (M–1) | ΔG1 (kJ mol–1) | |
|---|---|---|---|---|---|
| BSA | 1.62 × 104 | 1.62 × 1012 | 1.04 | 2.42 × 104 | –25.01 |
| HSA | 1.52 × 104 | 1.52 × 1012 | 0.70 | 0.05 × 104 | –15.64 |
Thus, the probable mechanism of quenching is by complex formation, namely, static quenching in the case of the BSA–Cu(II) system. This prediction is possible from the verification of bimolecular quenching constant values calculated. Diffusion-controlled quenching typically results in Kq values approximately 1 × 1010 M–1 s–1. However, the values of Kq obtained here are 160-fold higher, which suggests a complex formation between BSA and Cu(II) (static mechanism).34 Static quenching in the case of BSA interaction with other ligands was reported earlier. For instance, the interaction of BSA with tannic acid and its derivatives resulted in static quenching. Bimolecular quenching constant values of several orders of magnitude higher than the maximum value required for diffusion-controlled quenching were observed.35 The Stern–Volmer plots of HSA in Figure 2b revealed that up to a concentration of 12.50 μM, the Stern Volmer plots remain linear, and after that, a clear negative deviation is observed. The negative deviation observed for quenching of Trp fluorescence in HSA is due to polar or charged quenchers.30,36 Here, in the linear range, linear fitting of the experimental data was used to calculate the Stern–Volmer constants and quenching rate constants, and the results have been tabulated in Table 1. In the present case, the quenching rate constant of HSA fluorescence induced by Cu(II) is 1.52 × 1012 M–1 s–1, which is higher than that reported in the case of the collisional quenching process. This clearly suggests the mechanism of quenching to be static. Further confirmation for static mechanism of quenching in the case of both serum albumins is understood from the UV–vis spectra and fluorescence lifetime measurements.
Further using the fluorescence data, the binding parameters of Cu(II) ions to BSA and HSA are calculated using the following equation:
| 3 |
where Fo and F represent the fluorescence intensities of BSA and HSA in the absence and presence of Cu(II) ions. Kb and n represent the binding constant and number of binding sites, respectively. The thermodynamics of binding is estimated by calculating the free energy of binding (ΔGo) using eq 4.37
| 4 |
The binding data (Kb, n and ΔGo) obtained at 298 K are summarized in Table 1.
As can been seen from Figure S1, the plots of log[Fo – F/F] versus log Q displayed a straight line in the case of BSA and HSA. Meanwhile, from the data of Kb value, BSA is said to have a strong interaction with Cu(II) and the value of n is approximately equal to 1, indicating that there is only one class of binding site for Cu(II) in BSA. However, the Kb value for HSA is of the same order as that of BSA but having a lower value compared to BSA. This value suggests that an interaction has occurred between Cu(II) and HSA also. The binding site is lower than the one in the case of HSA–Cu(II), which implied that Cu(II) is only partially bound to HSA. The binding process is all spontaneous for both serum albumins as is evident from the negative sign of ΔGo values.38,39
Fluorescence Lifetime Measurement
To further confirm whether the interaction of Cu(II) ions with BSA and HSA has occurred by a static/dynamic quenching mechanism, time-resolved fluorescence (TRF) decay of BSA and HSA before and after the addition of Cu(II) ions in tris-HCl was measured at pH 7.4 and 298 K using the TCSPC method. The results are depicted in Figure 3.
Figure 3.
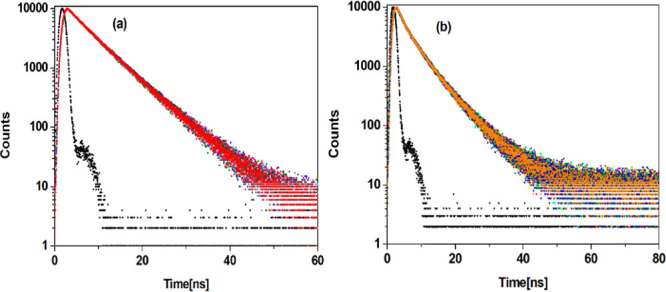
TRF decay of (a) BSA–Cu(II) and (b) HSA–Cu(II) systems at pH 7.4 and 298 K.
TCSPC serves as a complementary method for the measurement of fluorescence intensity, which provides a direct information about the conformational heterogeneity of proteins during the protein–ligand interaction. Moreover, it can be used to differentiate between static and dynamic quenching mechanisms. In static mechanism, the ground-state complex formed has no influence on the lifetime of the uncomplexed fluorophore. The excited-state lifetime of the fluorophore decreases in dynamic quenching because it is a rate process entirely dependent on the excited-state population of the fluorophore.40 In TRF, the excited-state lifetime of a fluorophore is highly dependent on the structure and conformation of the fluorophore, and its concentration-independent nature is an added advantage of TRF measurements.41 The samples were excited at 295 nm using a picosecond diode laser, and the corresponding fluorescence decay profiles are noted in the absence and presence of Cu(II) ions at their respective emission maximum wavelengths. The decay curves are fitted to biexponential and multiexponential functions using the equation
| 5 |
where τ1, τ2, and τ3 are the lifetime components and α1, α2, and α3 are the associated fractional intensities. In TRF, intrinsic fluorescence of Trp residues in BSA and HSA is considered. Trp can exist as rotamers or rotational isomers presented in Figure 4, which are responsible for its multiexponential decay profiles in the solution state. Fleming and his coworkers42 studied on the decay profiles of Trp residues in an aqueous solution and reported the presence of three conformers for Trp residues at physiological pH.
Figure 4.

Rotational conformational isomers of Trp in BSA and HSA.
The relative population of various conformers in the excited state contributes to the Trp lifetime. Among the three conformers, 4c is rather stable, and its conversion to either 4a or 4b form is difficult in nanosecond time scale. In aqueous solution, Trp undergoes multiexponential decays, and conformer 4c contributes to a shorter lifetime component. The longer lifetime component arises from the rapid interconversion of 4a and 4b conformers. Fluorescence quenching occurs as a result of fluorophore–quencher interaction. As a result of quenching, the planarity of the indole ring in Trp is distorted; therefore, a change in the microenvironment occurs, followed by a decrease in lifetime, and sometimes a change in contribution is observed from the rotameric lifetime.42
In the present case, BSA shows biexponential decay due to the presence of two tryptophan residues (Trp-134 and Trp-212)24 with an average lifetime of 6.18 ns with lifetime components of 3.51 and 6.94 ns. Thus, the shorter lifetime component (τ1 = 3.51 ns) is due to the Trp-212 residue which is located in the hydrophobic pocket of BSA and the longer lifetime component (τ2 = 6.94 ns) is due to the presence of Trp-134 on the surface of BSA.43 With increasing concentration of Cu(II) ions, the fluorescence decay curve remains biexponential. A decrease in the average lifetime from 6.18 to 5.98 ns and a marginal decrease in the two lifetime components (τ1 and τ2) are also observed. The lifetime parameters have been tabulated in Table S1. Moreover, the relative amplitudes of longer lifetime components (τ2) show a slight decrease (77.86–76.26%), which is due to the augmented interconversion from 4c to 4a or 4b conformers. The rotation of indole ring in the Trp moiety is facilitated in the excited state due to the enhanced planarity of the ring. This will contribute to the interconversion of the Trp rotamer to a more stable conformer 4c, which is clear from the increasing amplitude of shorter lifetime component (τ1) (22.14–23.74%). Thus, both Trp residues are actively involved in binding with BSA but with a higher binding affinity of Cu(II) toward Trp-134 residues.44 The percentage reduction in average lifetime values in the presence of 15 μM Cu(II) is 3.24%, which suggests that the mechanism of quenching is static in nature. The calculated time-resolved parameter very well agrees with the results from steady-state fluorescence measurements. Thus, it can be concluded that in static quenching, the fluorescence lifetime is unaffected because the fluorescence observed is only from fluorophores that are not interacting with quencher molecules.32
Similarly, Figure 3b shows the fluorescence decay profile of HSA with increasing concentrations of Cu(II) ions. All the decay parameters including average lifetimes (τm) and their associated fractional intensities are summarized in Table S2. HSA with one Trp-214 residue decay curves is fitted to a sum of three exponentials. The fluorescence lifetimes (and their amplitudes in parenthesis) obtained for HSA are τ1 = 9.99 ns (9.13%), τ2 = 3.33 ns (44.28%), and τ3 = 7.33 ns (46.59%) with an average lifetime of 5.80 ns. Similar multiple decay components have also been observed in the case of proteins containing single Trp residues. Previous investigators also reported that the TRF decay of Trp-214 in HSA has heterogeneous lifetimes.43,45,46 Studies of Wahl and Auchet also resolved the decay of Trp-214 into three exponentials having lifetimes of 12.08 ns (5.70%), 6.17 ns (84.80%), and 1.50 ns (9.50%) on excitation at 295 nm.47 In our study, it has been observed that the relative amplitude of the three lifetime components is significantly altered. The fluorescence decay of HSA after its interaction with Cu(II) was further determined to fit well to a multiexponential model with three components. A change in the mean fluorescence lifetime from 5.80 to 5.67 ns (0–5 μM) and 4.94–4.89 ns (7.50–37.50 μM) is observed. The percentage reduction in the average lifetime values in the presence of 5 μM Cu(II) is about 2.24 and 1.01% in the presence of 37.50 μM Cu(II). This observation further supports static quenching. The pattern of variation in the average lifetime of Trp on interaction with Cu(II) followed the same trend as observed in steady-state fluorescence measurements. A combined effect of quenching and conformational transition of Trp induced by Cu(II) has decreased the average lifetime.
Thus, time-resolved studies help to corroborate the results of steady-state fluorescence measurements and strengthen the fact that introduction of Cu(II) to serum albumins results in the formation of Cu(II)–serum albumin complexes.
Conformational Investigation
UV–Vis Absorption Spectra Studies
UV–vis absorption spectra are a unique tool to identify the structural changes and to confirm the complex formation between the protein and quencher. The absorbance spectra of the protein in the absence and presence of varying copper ion concentrations are presented in Figure S2. The π–π* transition of aromatic amino acid residues such as Trp, Tyr, and Phe accounts for the absorption around 280 nm.48,49 With the increasing concentration of Cu(II) ions, hyperchromicity is observed in both the absorbance spectra of serum albumins at 279 nm, which is an indication for the complex formation between serum albumin and Cu(II) ions.50 The dynamic quenching process only affects the excited-state lifetime of a fluorophore, and no change in the absorbance spectrum is observed.51 Therefore, we get further confirmation that the fluorescence quenching of both serum albumins proceeds through a static quenching mechanism.
Conformational Studies Using Synchronous Fluorescence
Alteration in the microenvironment around the fluorophores (Tyr and Trp) of BSA and HSA was further explored by synchronous fluorescence spectra. The presence of tryptophan and tyrosine accounts for the fluorescence in BSA and HSA. According to Miller52 in synchronous fluorescence spectra, the difference between excitation and emission wavelength (Δλ = λemis – λexc) spectra reflects a spectrum of different nature of a chromophore, that is, Δλ = 60 nm gives information regarding any changes in the tryptophan residues of BSA and HSA, whereas Δλ = 15 nm gives characteristics of tyrosine residues. Synchronous fluorescence spectra of BSA and HSA with various concentrations of Cu(II) ions were recorded at Δλ = 60 and 15 nm. Figures S3 and S4 display the effect of the increasing concentration of Cu(II) ions on the synchronous fluorescence spectra of BSA and HSA when the scanning interval Δλ was fixed at 60 and 15 nm, respectively.
Upon the addition of Cu(II) ions to BSA, the fluorescence intensity of both tryptophan and tyrosine was decreased, but the maximum emission wavelength of Trp at Δλ = 60 nm displays an obvious red shift from 281.5 to 282 nm, which suggests that the hydrophobicity decreased and the polarity increased around the Trp residue.53,54 However, in the case of HSA, the fluorescence intensity of both the tryptophan and tyrosine was decreased, but the emission wavelength of tryptophan residues is slightly blue-shifted from 281.5 to 280.5 nm, which suggests that the hydrophobicity increased and the polarity decreased around the Trp residue with the increasing concentration of Cu(II) ions. At the same time, no change in emission wavelength for tyrosine is observed. It suggests that the interaction of Cu(II) ions with BSA and HSA affects the conformation of tryptophan microregion only.55
Influence of Cu(II) Ions on Oligomerization, Aggregation, and Amyloid Fibril Formation in the Case of Serum Albumins
The Cu(II) ion concentration-dependent nanoscale morphological changes of serum albumins in aqueous solution were investigated using tapping mode AFM and compared with that of pure proteins. Before the addition of Cu(II) ions, BSA contained homogeneously distributed globular molecules that have no resemblance to any form of aggregates or elongated fibrils with a height distribution of about 3 nm as shown in Figure 5a,b. This is due to the predominant α-helical nature of serum albumins, which lacks the property to self-aggregate and results in the formation of amyloid fibrils.56,57 This large helical structure of serum albumins is confirmed from its Raman spectroscopic data presented in Table 2. The 17 disulfide bonds and 1 unpaired cysteine (Cys 34) residue in BSA enhance its higher-order association.58 Moreover, the aggregation of proteins starts from a partially denatured state and favorable under conditions of low pH, high temperatures, and chemical denaturants.59,60 Also, the presence of hydrogen bonding and hydrophobic interactions associated with the secondary structure of protein accounts for the aggregation in proteins.56 In this case, BSA fibrillation proceeds without a lag phase, as it is evident from the Stern–Volmer plots. However, in usual cases, protein aggregation and fibril formation proceed through a series of three steps: (1) nucleation—formation of a stable nuclei; (2) growth—elongation of nuclei to fibrils; and (3) precipitation—floccule formation. The above ambient temperature nucleation occurs, but fibril formation proceeds even at ambient and low temperatures. Later, it was modified that fibril formation has taken place through a nucleation-dependent elongation mechanism, which involves nucleation, elongation, and equilibrium phases. The nucleation step involves the assembly of monomers to form an organized structure called nucleus, which serves as a precursor for fibril formation. This nucleus forming step is also referred to as the lag phase, which is different for each protein. It is a time-consuming step since the formation of the initial nucleus is energetically difficult. Next, the addition of monomers to nucleus results in the formation of oligomers, which corresponds to an exponential/elongation/polymerization phase during fibrillation. In the last stage, oligomers were elongated to fibrils, and thus, an equilibrium stage is reached. Proteins such as Aβ-peptides, α-synuclein, follow the above-described model of protein fibrillation.56,61 There are reports on the fibrillation of serum albumins proceeding without a lag phase.58 One such example is observed in the case of BSA aggregation under neutral conditions and at elevated temperatures. Fibrils with β-sheet-rich fractions were formed, and the aggregation proceeds without a lag phase with a linear dependence on protein concentration. In our studies too, BSA fibrillation in the presence of Cu(II) proceeds without a lag phase. The absence of lag phase in BSA fibrillation suggests that nucleus is rapidly formed under the influence of Cu(II) ions.
Figure 5.

AFM topographical images of BSA in the absence and presence of Cu(II) ions. (a,b) Free BSA and the respective height profile at physiological pH. (c,d) Formation of oligomers after introducing 3 μM Cu(II) and the corresponding height profiles. (e–j) Dimer, trimer, and tetramer formation after introducing 5, 9, and 11 μM Cu(II) ions and the corresponding height profiles. (k) Protofibril formation after introducing 15 μM Cu(II). (l,m) Fibril formation after a longtime incubation period of 10 and 30 days.
Table 2. Percentage Analysis of Different Secondary Structural Elements Obtained after Deconvolution of the Amide I (1600–1700 cm–1) Region of BSA and with the Addition of Varying Concentrations of Cu(II) (pH 7.4).
| BSA 7.4 | +5 μM Cu(II) | +7 μM Cu(II) | +15 μM Cu(II) | |
|---|---|---|---|---|
| β-sheet | 13.47 | 19.06 | 22.34 | |
| α-helix | 28.33 | 14.12 | 9.02 | 6.36 |
| random coils | 25.69 | 13.08 | 21.46 | |
| β-turns | 21.28 | 12.39 | 27.96 | 25.50 |
| antiparallel β | 11.23 | 22.97 | 22.97 | 25.50 |
With increasing Cu(II) concentration from 0 to 15 μM, visible morphological changes from globular oligomers to fibrillar formation were observed. The globular oligomers upon mutual interaction get converted to dimers, trimers, and tetramers of oligomers and finally to elongated bead-like protofibrils as shown in Figure 5c. Initially, oligomers appeared as globular-shaped particles from 3 μM concentration of Cu(II) with a height distribution of about 1–3.50 nm. Further, its transformation to a dimer, trimer, and tetramer is observed until a concentration of about 11 μM Cu(II). In the concentration range of 13–15 μM, protofibril formation takes place. Curly bead-like structures of protofibrils arise from the attractive interaction between spherical protein aggregates arising from an increased exposure of hydrophobic residues. The bead-like structures get elongated through mutual interactions and converted to protofibrils. Then, through a longer period of incubation, mature fibrils were formed from 9 to 15 μM Cu(II) concentrations as shown in Figure 5l,m. Through the lateral association or lateral fusion of protofibrils or by the addition of protein oligomers to the growing end of protofibrils, mature fibrils get formed. The mechanism of aggregation here reported is said to have a close similarity to Aβ1–40 fibrillization reported by Seremetal.62 Aβ1–40 fibrillation proceeds through a two-step polymerization process, nucleation and elongation. In the two-step process, monomeric amyloid proteins self-aggregate to generate spherical shaped oligomers as seed particles, which then converted to protofilaments. These seed-shaped particles are formed at the early stage of aggregation. The protofilaments get elongated through the monomer addition to the ends of protofilaments. Then, two or three protofilaments associated to form protofibrils; from intertwining protofibrils, fibrils are formed. Through a series of addition steps such as self-association, annealing of protofibrils ends, and lateral association of protofibrils contributes to elongation branching and intertwining of fibrils. Hydrophobic and hydrogen bonding interactions between the accessible faces of Aβ peptides are the factors which accelerate the rate of fibril formation.
Thus, Cu(II) ions can accelerate the fibrillation process in BSA by an enhancement of self-assembly process of protofibrils. These morphological changes are mainly attributed to the increased exposure of hydrophobic residues to Cu(II) ions, giving rise to these more elongated bead-like structures. This elongation possibly involves a conformational conversion in proteins.
Similar to BSA, HSA also lacks the property to self-aggregate, resulting in the formation of amyloid fibrils. Because HSA is a major α-helix protein in its native form at pH 7.4, whose structure is supported with the help of intermolecular interactions such as hydrogen bonds. Raman spectroscopy data well supported this fact presented in Table S4. The 17 disulfide bonds and 1 free SH group facilitate higher-order association in HSA.63 From AFM images, it is clear that free HSA molecules exist as well-separated ellipsoidal particles with a height distribution of around 1–5 nm (Figure 6a,b). The extent of protein aggregation and their morphologies also get varied in HSA under the influence of Cu(II) ions. In the presence of 3.75 μM Cu(II) ions, HSA monomers undergo self-association into noncovalently linked oligomers as shown in Figure 6c,d, with a height distribution of ∼1.60 nm. This oligomer formation is observed up to a concentration of 37.50 μM. The oligomers formed are transformed into amorphous aggregates at concentrations above 7.50 μM Cu(II) after an incubation period of 10 days as shown in Figure 6e–i. Oligomeric species of different sizes were observed in the aggregation pathway. Smaller, larger, and elongated oligomeric species were also observed. An increase in β-sheet fractions with a proportional increase in Cu(II) ion concentration is in agreement with the Raman spectroscopy data (Table S4) because oligomeric species are rich in β-sheet fractions, and these nonfibrillar oligomers have been suggested as pathogenic species.
Figure 6.

AFM topographical images of HSA in the absence and presence of Cu(II) ions. (a,b) Free HSA and the respective height profile at physiological pH. (c,d) Formation of oligomers after introducing 3.75 μM Cu(II) and the corresponding height profiles. (e–i) Amorphous aggregate formation after an incubation period of 10 days with the introduction of 7.50, 11.30, 17.50, 32.50, and 37.50 μM Cu(II) ions.
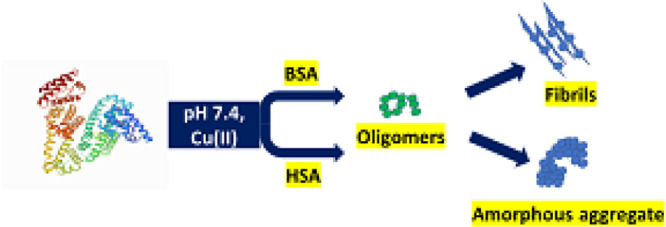
Large multidomain proteins such as BSA and HSA have the tendency to form propagation—a competent nucleus-like structure such as oligomers. HSA forms these oligomeric species by classical coagulation or downhill polymerization. In downhill polymerization, protein aggregation starts without the need of a multimeric nucleus, and it is independent of monomer concentration also referred to as classical coagulation. As it is evident from the Stern–Volmer plots, no discernible lag phase is involved in downhill polymerization. The absence of lag phase is strongly correlated with the mechanism of aggregation to be as a downhill process64 because Cu(II)-induced aggregation in HSA is characterized by a sigmoidal growth curve comprising a growth phase and saturation phase. The nonexistence of lag phase in protein aggregation is also a characteristic property of amyloidogenesis in proteins such as acylphosphatase and β-2 microglobulin. With further increase in Cu(II) concentration, oligomeric species is changed to amorphous aggregates with a larger α-helix and unordered conformation contents confirmed from Raman spectroscopic data. An amorphous aggregate is formed by the addition of monomers to the growing clump of the aggregated protein. This type of amorphous aggregation formation was reported in the case of HSA fibrillation at physiological pH and at higher salt concentrations.65 Up to 50 mM NaCl, fibril formation takes place rapidly due to the increased hydrophobicity arising as a result of screening of repulsive electrostatic interactions between proteins. However, at a concentration above 50 mM NaCl, bundles of short fibrils along with amorphous aggregates with larger α-helix fractions are observed. This different behavior is due to the shielding of intermolecular electrostatic forces between protein molecules, leading to an enhancement in solution hydrophobicity promoting random aggregation. The nonstructured aggregates of tau protein in Alzheimer’s disease (AD), AS-unstructured aggregates called as Lewis body in Parkinson’s disease, and Aβ peptides in AD coexist as amorphous aggregates and amyloid fibrils in neurodegenerative disorders.66 The overall mechanistic pathway for the formation of amyloid fibrils from BSA and amorphous aggregate from HSA is presented in Scheme 1.
Scheme 1. Mechanistic Pathway of BSA and HSA Aggregation on Exposure to Cu(II) at Physiological pH.

Delineating the Secondary Structural Changes of Serum Albumins during Aggregation at the Molecular Level Using Raman Spectroscopy
The secondary structural changes during the sequential process of protein aggregation were monitored with the help of Raman spectroscopy. The Raman spectra of BSA in the range of 800–1800 cm–1 at physiological pH in the absence and presence of Cu(II) are shown in Figure 7a.
Figure 7.

Raman spectra of BSA and the BSA–Cu(II) complex at physiological pH. (a) Raman spectra of BSA alone and in the presence of different concentrations of Cu(II) (800–1800 cm–1). (b) Amide I (1600–1700 cm–1) region. (c,d) Amide I region of BSA alone and with the addition of 15 μM Cu(II) deconvoluted displaying an increase in antiparallel β-sheet conformation.
Backbone amide group markers such as amide I and amide III were monitored and analyzed for the shift in intensity and band positions of serum albumins as a function of aggregation under the influence of chemical stress induced by Cu(II) ions. Deconvolution of both amide I and amide III regions gives α-helix, β-sheet, and random coils as secondary structural components. The Raman spectrum obtained for BSA prior to the exposure of Cu(II) displayed a broad amide I band at 1648 cm–1 derived from α-helical conformations (Figure 7b). Deconvolution of the amide I band followed by percentage analysis yielded the secondary structural components with a high content of α-helical structure (28.33%), random coils (25.69%), β-turn (21.28%), and with a minor contribution from β-sheet (13.47%) and antiparallel β (11.23%) (Figure 7c). With an exposure of 5 μM Cu(II) ions, the amide I band gets shifted to 1639 cm–1 with an increase in antiparallel β-sheet conformation (22.97%), β-turn (12.39%), followed by a decrease in α-helical (14.12%) content. Further addition of 7 μM Cu(II) ions showed an increase in antiparallel β-sheet (22.97%) and β-turn (27.96%) at the expense of a decrease in α-helix (9.02%) content. Further addition of 15 μM Cu(II) showed that the amide I band is shifted to 1641 cm–1 (Figure 7d). The secondary structural analysis in Table 2 revealed that antiparallel β-sheet fractions have increased to a larger extent at the expense of α-helix with increasing concentrations of Cu(II).
In the aggregation process from native to fibril formation, the oligomeric species is found to be rich in antiparallel β-sheet fractions and is potentially cytotoxic too. Prefibrillar species such as oligomers on infusing onto the left ventricles of brain induced significant impairment in learning and memory functions, whereas fibrils do not bring about such effects. Small size and high surface hydrophobicity account for the cytotoxicity of amyloid oligomers. For small oligomers of Aβ and tau proteins, cytotoxicity increases with oligomer size, whereas a large Aβ oligomer has decreased cytotoxicity with increasing size. Besides size, the structural features of oligomers also account for their cytotoxicity. Both spheroidal and annular oligomers are cytotoxic, which can efficiently bind with brain-derived membrane fractions than fibrils. Membrane permeabilization and calcium dyshomeostasis are the main toxicity mechanism associated with amyloid oligomers.67 FTIR studies of Aβ42 peptides by Ha et al. reported the presence of cross-β structures in Aβ42 aggregates in the presence of Fe3+ ions, and these cross-β structures are characteristics of amyloid oligomers.68 Through this β-sheet formation, the binding of Fe3+ to Aβ peptides takes place and accelerates the formation of fibrillar amyloid plaques. Also, pH is an important environmental factor that controls Aβ aggregation. At normal physiological pH, Fe3+ promotes fibril formation but on lowering the pH to 6 or 4.6 caused reduction in Fe3+-induced fibrillar aggregates. Also, Rivas-Arancibia et al. using Raman spectroscopy studied the influence of oxidative stress on amyloid fibril formation in the case of amyloid β 1–42 (Aβ 1–42) in the brain associated with AD.69 In their study, a group of rats were administered small doses of ozone for a period of up to 90 days, followed by evaluating the conformational structure of Aβ 1–42 present in the dentate gyrus of each animal using Raman spectroscopy. Between 30 and 60 days of ozone exposure, the Raman band at 1671 cm–1 associated with the β-sheet component begins to appear in the spectrum with pronounced intensity. After 60 days of ozone exposure, it was observed that at the expense of α-helix conformation, the quantity of β-sheet conformation of Aβ 1–42 has increased, along with β-turn and unordered structures, which is an indication for the effect of oxidative stress on secondary structure alterations of proteins associated with various neurological disorders. The analysis of the amide III region supported the information obtained from the amide I band as shown in Figure S5 and Table S3.
However, in the case of HSA, the amide I band is observed at 1638 cm–1, and the percentage compositions of secondary structural elements are 31.63% for α-helix, 16.34% for β-turns, and 15.94% for the antiparallel β-sheet, as shown in Figure S6. The addition of 3.75 μM Cu(II) has shifted the amide I band to 1639 cm–1. The secondary structural elements have further decreased in α-helix fraction (14.04%) with a corresponding increment in antiparallel β-sheet, and random coil fractions are presented in Table S4. The increase in antiparallel β-sheet fractions accounts for the formation of oligomers. Further addition of 12.50 μM Cu(II) shifts the amide I band to 1646 cm–1 with an increase in α-helix fraction (15.19%). On increasing the concentration to 37.50 μM, the amide I band is shifted to 1645 cm–1 with a corresponding increment in α-helix (27.19%) fractions, which is in agreement with the amorphous aggregate formed during the aggregation process. Juárez et al. reported a similar case in the FTIR spectra of HSA and amorphous aggregate with an increase in α-helix conformation at physiological pH and with increasing salt concentrations.65 The intensity of the amide I band is decreased up to 50 mM NaCl and at physiological pH. The appearance of well-defined peaks at 1625 and 1693 cm–1 suggests the existence of antiparallel β-sheet fractions. A further increase in NaCl concentration decreased the intensity of the peak at 1625 cm–1, which points out a lower antiparallel β-sheet conformation, followed by an increase in α-helix fractions at larger salt concentrations. A shift in peak positions with varying contents of secondary structural elements clearly reflects an alteration in secondary structural elements for both serum albumins.
Conclusions
Our results revealed that protein aggregation has taken place through different mechanistic pathways for both serum albumins under the influence of Cu(II) ions. BSA undergoes a series of transformations from globular shape to oligomer, dimer, trimer, protofibril, and finally to fibril formation. The presence of Cu(II) has accelerated the formation of nucleus required for aggregation. The protein aggregation pathway observed here for BSA has close similarity to Aβ1–40 amyloid fibril formation, while for HSA, a downhill polymerization mechanism is observed. Stern–Volmer plots obtained are also in agreement with the observed mechanism with the absence of discernible lag phase. Oligomers of varying sizes are formed in the pathway to amorphous aggregate formation in the case of HSA. The formation of these toxic intermediates formed during the protein aggregation process is further confirmed from the secondary structure elemental analysis of amide I region in Raman spectra. The study using two model proteins is very helpful for a better understanding of molecular mechanisms of disease-associated amyloidogenesis.
Experimental Section
Materials
BSA and HSA were procured from Sigma-Aldrich and used without further purification. Tris(hydroxymethyl)aminomethane and copper sulfate pentahydrate were purchased from Merck India and used as received. All the chemicals and reagents used in the study were of analytical grade. Stock solutions of BSA and HSA both having concentrations of 10 μM were prepared in tris-HCl buffer of pH 7.4. The pH of the buffer solution was measured using a Metler pH Meter. The aqueous solution of copper sulfate pentahydrate was prepared. Milli-Q water was used for the preparation of all solutions and buffers.
Methods
UV–Vis Spectroscopy
A UV-1700 Shimadzu UV–vis spectrophotometer was used to record the absorption spectrum of BSA and HSA in the absence and presence of varying concentrations of CuSO4·5H2O. The Quartz cell with a path length of 1 cm was used, and the wavelength range was set from 200 to 800 nm. The concentrations of BSA and HSA were kept fixed as 10 μM, and the concentration of copper sulfate pentahydrate was varied for recording the absorption spectrum. The baseline correction was done using the same buffer.
Steady-State Fluorescence Measurements
The fluorescence emission spectrum was recorded on a LS55 (PerkinElmer) Fluorescence Spectrometer having a 20 kW continuous powered high-pressure Xe lamp as the excitation source and an R928 photomultiplier as the photodetector. The fluorescence spectrum was recorded at an excitation wavelength of 295 nm, which excites Trp residues only. The maximum fluorescence emission was recorded in the wavelength range of 300–500 nm for BSA and 290–500 nm for HSA. Excitation and emission bandwidths were all set at 5 nm. A 10 μM protein solution prepared in tris-HCl buffer of pH 7.4 and with varying concentrations of copper sulfate pentahydrate was used to record the emission spectrum of both proteins at 298 K.24
Synchronous Fluorescence Measurements
Synchronous fluorescence measurements were recorded with the same instruments, and the concentrations of the protein solution and copper sulfate pentahydrate were the same as used for fluorescence emission spectrum measurements. The spectra were recorded by simultaneously scanning between emission and excitation wavelengths. The wavelength interval between excitation and emission wavelengths was set at Δλ = 60 nm and Δλ = 15 nm, respectively. The intervals of Δλ = 60 nm and Δλ = 15 nm were chosen to study the changes in the microenvironment around the Trp and Tyr residues of the selected proteins.24
TRF Measurements
The TRF spectra of BSA and HSA in the absence and presence of copper sulfate pentahydrate of varying concentrations were recorded by using a single-photon counting spectrometer equipped with a pulsed nanosecond LED excitation source at 295 nm (HORIBA Fluorolog Jobin Yvon spectrometer) and at 298 K. The fluorescence lifetime data were measured by setting peak count to 10,000. The excitation and emission slits were all set at 5 nm for all the experiments. Analysis of the fluorescence lifetime decay profile was performed using the DAS 6 software attached to the system. The goodness of fit was determined from the χ2 values.24
AFM and Raman Spectroscopy
AFM measurements were made to observe the morphological changes happened to the protein after interaction with the metal ion. The measurements were made in a confocal Raman microscope coupled with an AFM [WITecALPHA 300RA Germany] instrument. In this study, measurements were carried out in the noncontact mode using a silicon tip of 75 kHz resonant frequency, 2.8 N/m force constant, and a radius of less than 8 nm. The samples were prepared on a fresh mica sheet, and it is dried under a gentle stream of nitrogen gas and subjected to AFM study. The AFM images obtained were processed and analyzed using the WiTec Project 4 program.
For Raman spectroscopy studies, a confocal Raman microscopy system equipped with a 532 nm DPSS laser, a maximum power of 42 mW focused on the sample, coupled to a microscope equipped with a 100× /0.9 DIC Zeiss (EC Epiplan—Neofluar) objective, a spectrometer (UHTS 300, focal length 300 mm, with a 1800 gmm–1 grating), and a CCD camera were used for the experiments. Solution phase spectra were acquired for serum albumins. Before measurements, a system calibration was performed using silicon wafer in order to check the standard band position and intensity. The Raman spectra were recorded in the spectral range of 800–1800 cm–1. Each spectrum consists of three accumulations with an integration time of 100 s. All the spectra acquired were preprocessed for baseline correction and cosmic ray removal, and each set of spectra was averaged using the WITec Project 4 program. The baseline-corrected and smoothened Raman spectra were plotted using Origin 6 software. Deconvolution of amide I band was also performed using Origin 6.25
Acknowledgments
The authors acknowledge the Inter University Instrumentation Centre (IUIC) and Sophisticated Analytical Instrument Facility (SAIF), Mahatma Gandhi University, Kottayam, for providing the instrumental facilities. They also acknowledge the technical support of WITec Team India.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05119.
Double log graph of the Stern–Volmer equation for BSA and HSA at pH 7.4 and 298 K; fluorescence lifetime parameters of BSA in the presence of Cu(II) ions; fluorescence lifetime parameters of HSA in the presence of Cu(II) ions; UV–vis absorbance spectra of BSA and HSA in the presence of increasing concentrations of Cu(II) ions at pH 7.4 and 298 K; synchronous fluorescence spectra of BSA and HSA in the presence of increasing concentrations of Cu(II); deconvoluted amide III region of BSA alone and with the addition of 15 μM Cu(II) ion displaying an increment in antiparallel β-sheet and random clew conformations; percentage analysis of different secondary structural elements obtained after deconvolution of the amide III (1200–300 cm–1) band of BSA and with the addition of varying concentrations of Cu(II) ions, pH 7.4; Raman spectra of HSA and the HSA–Cu(II) complex at physiological pH; and percentage analysis of different secondary structural elements obtained after deconvolution of the amide I (1600–1700 cm–1) region of HSA and with the addition of varying concentrations of Cu(II) ions at pH 7.4 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Simpson L. W.; Good T. A.; Leach J. B. Protein Folding and Assembly in Confined Environments: Implications for Protein Aggregation in Hydrogels and Tissues. Biotechnol. Adv. 2020, 42, 107573. 10.1016/j.biotechadv.2020.107573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsi T. N.; Athar T.; Parveen R.; Fatima S. A Review on Protein Misfolding, Aggregation and Strategies to Prevent Related Ailments. Int. J. Biol. Macromol. 2017, 105, 993–1000. 10.1016/j.ijbiomac.2017.07.116. [DOI] [PubMed] [Google Scholar]
- Kalmar B.; Greensmith L. Cellular Chaperones as Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neurosci. 2017, 10, 251. 10.3389/fnmol.2017.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantoulea K.; Louros N.; Rousseau F.; Schymkowitz J. Heterotypic Interactions in Amyloid Function and Disease. FEBS J. 2021, 1–22. 10.1111/febs.15719. [DOI] [PubMed] [Google Scholar]
- Alam P.; Siddiqi K.; Chturvedi S. K.; Khan R. H. Protein Aggregation: From Background to Inhibition Strategies. Int. J. Biol. Macromol. 2017, 103, 208–219. 10.1016/j.ijbiomac.2017.05.048. [DOI] [PubMed] [Google Scholar]
- Adamcik J.; Mezzenga R. Amyloid Polymorphism in the Protein Folding and Aggregation Energy Landscape. Angew. Chem., Int. Ed. 2018, 57, 8370–8382. 10.1002/anie.201713416. [DOI] [PubMed] [Google Scholar]
- Giugliarelli A.; Tarpani L.; Latterini L.; Morresi A.; Paolantoni M.; Sassi P. Spectroscopic and Microscopic Studies of Aggregation and Fibrillation of Lysozyme in Water/Ethanol Solutions. J. Phys. Chem. B 2015, 119, 13009–13017. 10.1021/acs.jpcb.5b07487. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Peptide Fibrillization. Angew. Chem., Int. Ed. 2007, 46, 8128–8147. 10.1002/anie.200700861. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Kurouski D. Structural Characterization of Individual α-Synuclein Oligomers Formed at Different Stages of Protein Aggregation by Atomic Force Microscopy-Infrared Spectroscopy. Anal. Chem. 2020, 92, 6806–6810. 10.1021/acs.analchem.0c00593. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Mishra S.; Khan E. Emerging Methods for Structural Analysis of Protein Aggregation. Protein Pept. Lett. 2017, 24, 331–339. 10.2174/0929866524666170206123150. [DOI] [PubMed] [Google Scholar]
- Giampietro R.; Spinelli F.; Contino M.; Colabufo N. A. The Pivotal Role of Copper in Neurodegeneration: A New Strategy for the Therapy of Neurodegenerative Disorders. Mol. Pharm. 2018, 15, 808–820. 10.1021/acs.molpharmaceut.7b00841. [DOI] [PubMed] [Google Scholar]
- Kim A.; Lim S.; Kim Y. K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. 10.3390/ijms19010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorlet P.; Hureau C. Copper Complexation by Peptides Implicated in Neurodegenerative Diseases. Electron Paramagn. Reson. Spectrosc. Appl. 2020, 83–108. 10.1007/978-3-030-39668-8_4. [DOI] [Google Scholar]
- Prakash A.; Dhaliwal G. K.; Kumar P.; Majeed A. B. A. Brain Biometals and Alzheimer’s Disease–Boon or Bane?. Int. J. Neurosci. 2017, 127, 99–108. 10.3109/00207454.2016.1174118. [DOI] [PubMed] [Google Scholar]
- Poulson B. G.; Szczepski K.; Lachowicz J. I.; Jaremko L.; Emwas A. H.; Jaremko M. Aggregation of Biologically Important Peptides and Proteins: Inhibition or Acceleration Depending on Protein and Metal Ion Concentrations. RSC Adv. 2019, 10, 215–227. 10.1039/c9ra09350h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaglia M.; Bubacco L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis so Relevant?. Biomolecules 2020, 10, 195. 10.3390/biom10020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamás M. J.; Sharma S. K.; Ibstedt S.; Jacobson T.; Christen P. Heavy Metals and Metalloids as a Cause for Protein Misfolding and Aggregation. Biomolecules 2014, 4, 252. 10.3390/biom4010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt B.; Schaumlöffel D.; Schwerdtle T. Subcellular Localization of Copper—Cellular Bioimaging with Focus on Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 2341. 10.3390/ijms21072341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.; Aschner M. Commonalities between Copper Neurotoxicity and Alzheimer’s Disease. Toxics 2021, 9, 1–11. 10.3390/toxics9010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadi S.; Zhu S.; Sharma R.; Wilson D. J.; Kraatz H. B. Interaction of Metal Ions with Tau Protein. The Case for a Metal-Mediated Tau Aggregation. J. Inorg. Biochem. 2019, 194, 44. 10.1016/j.jinorgbio.2019.02.007. [DOI] [PubMed] [Google Scholar]
- Okita Y.; Rcom-H’cheo-Gauthier A. N.; Goulding M.; Chung R. S.; Faller P.; Pountney D. L. Metallothionein, Copper and Alpha-Synuclein in Alpha-Synucleinopathies. Front. Neurosci. 2017, 11, 114. 10.3389/fnins.2017.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasia R. M.; Bertoncini C. W.; Marsh D.; Hoyer W.; Cherny D.; Zweckstetter M.; Griesinger C.; Jovin T. M.; Fernandez C. O. Structural Characterization of Copper(II) Binding to α-Synuclein: Insights into the Bioinorganic Chemistry of Parkinson’s Disease. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4294–4299. 10.1073/pnas.0407881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung Y. H.; Bush A. I.; Cherny R. A. Copper in the Brain and Alzheimer’s Disease. J. Biol. Inorg Chem. 2010, 15, 61–76. 10.1007/s00775-009-0600-y. [DOI] [PubMed] [Google Scholar]
- Mathew M.; Sreedhanya S.; Manoj P.; Aravindakumar C. T.; Aravind U. K. Exploring the Interaction of Bisphenol-S with Serum Albumins: A Better or Worse Alternative for Bisphenol A?. J. Phys. Chem. B 2014, 118, 3832–3843. 10.1021/jp500404u. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Neumann O.; Wang H.; Yuwono V. M.; Barhoumi A.; Perham M.; Hartgerink J. D.; Wittung-Stafshede P.; Halas N. J. Gold Nanoparticles Can Induce the Formation of Protein-Based Aggregates at Physiological PH. Nano Lett. 2009, 9, 666–671. 10.1021/nl803054h. [DOI] [PubMed] [Google Scholar]
- Shahabadi N.; Maghsudi M. Binding Studies of a New Copper (II) Complex Containing Mixed Aliphatic and Aromatic Dinitrogen Ligands with Bovine Serum Albumin Using Different Instrumental Methods. J. Mol. Struct. 2009, 150, 186. 10.1016/j.jbiotec.2010.08.485. [DOI] [Google Scholar]
- Mariam J.; Dongre P. M.; Kothari D. C. Study of Interaction of Silver Nanoparticles with Bovine Serum Albumin Using Fluorescence Spectroscopy. J. Fluoresc. 2011, 21, 2193–2199. 10.1007/s10895-011-0922-3. [DOI] [PubMed] [Google Scholar]
- Gao H.; Lei L.; Liu J.; Kong Q.; Chen X.; Hu Z. The Study on the Interaction between Human Serum Albumin and a New Reagent with Antitumour Activity by Spectrophotometric Methods. J. Photochem. Photobiol., A 2004, 167, 213–221. 10.1016/j.jphotochem.2004.05.017. [DOI] [Google Scholar]
- Li J.; Feng H.; Liu R.; Ding G.; Si H.; He W.; Sun Z. The Computational and Experimental Studies on a 1, 2, 3-Triazole Compound and Its Special Binding to Three Kinds of Blood Proteins. J. Biomol. Struct. Dyn. 2020, 38, 1185–1196. 10.1080/07391102.2019.1598498. [DOI] [PubMed] [Google Scholar]
- Dahiya V.; Chaubey B.; Dhaharwal A. K.; Pal S. Solvent-Dependent Binding Interactions of the Organophosphate Pesticide, Chlorpyrifos (CPF), and Its Metabolite, 3,5,6-Trichloro-2-Pyridinol (TCPy), with Bovine Serum Albumin (BSA): A Comparative Fluorescence Quenching Analysis. Pestic. Biochem. Physiol. 2017, 139, 92–100. 10.1016/j.pestbp.2017.04.011. [DOI] [PubMed] [Google Scholar]
- Gao W.; Li N.; Chen G.; Xu Y.; Chen Y.; Hu S.; Hu Z. Binding Studies of Costunolide and Dehydrocostuslactone with HSA by Spectroscopy and Atomic Force Microscopy. J. Lumin. 2011, 131, 2063–2071. 10.1016/j.jlumin.2011.04.041. [DOI] [Google Scholar]
- Lakowicz J. R.; Weber G. Quenching of Fluorescence by Oxygen. a Probe for Structural Fluctuations in Macromolecules. Biochemistry 1973, 12, 4161–4170. 10.1021/bi00745a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Chen M.; Jiang L.; Song L. New Insight into Molecular Interaction of Heavy Metal Pollutant-Cadmium(II) with Human Serum Albumin. Environ. Sci. Pollut. Res. 2014, 21, 6994. 10.1007/s11356-014-2610-8. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Wang G.; Lu X.; Lv J.; Xu M.; Zhang W. Molecular Mechanism of Interaction between Norfloxacin and Trypsin Studied by Molecular Spectroscopy and Modeling. Spectrochim. Acta, Part A 2010, 75, 261–266. 10.1016/j.saa.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Labieniec M.; Gabryelak T. Interactions of Tannic Acid and Its Derivatives (Ellagic and Gallic Acid) with Calf Thymus DNA and Bovine Serum Albumin Using Spectroscopic Method. J. Photochem. Photobiol., B 2006, 82, 72–78. 10.1016/j.jphotobiol.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Faridbod F.; Ganjali M. R.; Larijani B.; Riahi S.; Saboury A. A.; Hosseini M.; Norouzi P.; Pillip C. Interaction Study of Pioglitazone with Albumin by Fluorescence Spectroscopy and Molecular Docking. Spectrochim. Acta, Part A 2011, 78, 96–101. 10.1016/j.saa.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Najaran A.; Divsalar A.; Saboury A. A.; Roodbari N. H. Probing the Interaction of Newly Synthesized Pt(II) Complex on Human Serum Albumin Using Competitive Binding Site Markers. J. Fluoresc. 2019, 29, 827–835. 10.1007/s10895-019-02383-3. [DOI] [PubMed] [Google Scholar]
- Xie X.; Wang Z.; Zhou X.; Wang X.; Chen X. Study on the Interaction of Phthalate Esters to Human Serum Albumin by Steady-State and Time-Resolved Fluorescence and Circular Dichroism Spectroscopy. J. Hazard. Mater. 2011, 192, 1291. 10.1016/j.jhazmat.2011.06.038. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Huang C.-r.; Jiang M.; Zhu Y.-y.; Wang J.; Chen J.; Shi J.-h. Binding Interaction of Atorvastatin with Bovine Serum Albumin: Spectroscopic Methods and Molecular Docking. Spectrochim. Acta, Part A 2016, 156, 155–163. 10.1016/j.saa.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Ma X.; He J.; Sun Q.; Li Y.; Li H. Binding Properties of Drospirenone with Human Serum Albumin and Lysozyme in Vitro. Spectrochim. Acta, Part A 2016, 153, 612–618. 10.1016/j.saa.2015.09.017. [DOI] [PubMed] [Google Scholar]
- Millan S.; Satish L.; Kesh S.; Chaudhary Y. S.; Sahoo H. Interaction of Lysozyme with Rhodamine B: A Combined Analysis of Spectroscopic & Molecular Docking. J. Photochem. Photobiol., B 2016, 162, 248–257. 10.1016/j.jphotobiol.2016.06.047. [DOI] [PubMed] [Google Scholar]
- Petrich J. W.; Chang M. C.; McDonald D. B.; Fleming G. R. On the Origin of Nonexponential Fluorescence Decay in Tryptophan and Its Derivatives. J. Am. Chem. Soc. 1983, 105, 3824. 10.1021/ja00350a014. [DOI] [Google Scholar]
- Ghosh N.; Mondal R.; Mukherjee S. Hydrophobicity Is the Governing Factor in the Interaction of Human Serum Albumin with Bile Salts. Langmuir 2015, 31, 1095–1104. 10.1021/la504270a. [DOI] [PubMed] [Google Scholar]
- Anand U.; Kurup L.; Mukherjee S. Deciphering the Role of PH in the Binding of Ciprofloxacin Hydrochloride to Bovine Serum Albumin. Phys. Chem. Chem. Phys. 2012, 14, 4250–4258. 10.1039/c2cp00001f. [DOI] [PubMed] [Google Scholar]
- Kamal A. J.; Behere D. V. Spectroscopic Studies on Human Serum Albumin and Methemalbumin: Optical, Steady-State, and Picosecond Time-Resolved Fluorescence Studies, and Kinetics of Substrate Oxidation by Methemalbumin. J. Biol. Inorg Chem. 2002, 7, 273–283. 10.1007/s007750100294. [DOI] [PubMed] [Google Scholar]
- Abou-Zied O. K.; Al-Shihi O. I. K. Characterization of Subdomain HA Binding Site of Human Serum Albumin in Its Native, Unfolded, and Refolded States Using Small Molecular Probes. J. Am. Chem. Soc. 2008, 130, 10793–10801. 10.1021/ja8031289. [DOI] [PubMed] [Google Scholar]
- She M.; Dong W. J.; Umeda P. K.; Cheung H. C. Time-Resolved Fluorescence Study of the Single Tryptophans of Engineered Skeletal Muscle Troponin C. Biophys. J. 1997, 73, 1042–1055. 10.1016/s0006-3495(97)78137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Wang S. Binding of Glutathione and Melatonin to Human Serum Albumin: A Comparative Study. Colloids Surf., B 2015, 125, 96–103. 10.1016/j.colsurfb.2014.11.023. [DOI] [PubMed] [Google Scholar]
- Liu X.; Ling Z.; Zhou X.; Ahmad F.; Zhou Y. Comprehensive Spectroscopic Probing the Interaction and Conformation Impairment of Bovine Serum Albumin (BSA) by Herbicide Butachlor. J. Photochem. Photobiol., B 2016, 162, 332–339. 10.1016/j.jphotobiol.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Shamsi A.; Ahmed A.; Bano B. Probing the Interaction of Anticancer Drug Temsirolimus with Human Serum Albumin: Molecular Docking and Spectroscopic Insight. J. Biomol. Struct. Dyn. 2018, 36, 1479–1489. 10.1080/07391102.2017.1326320. [DOI] [PubMed] [Google Scholar]
- Wang C. Comparative Studies on the Interaction of Rhodamine B with Bovine Serum Albumin Using Fluorescence Method and Synchronous Fluorescence Method. J. Chem. Environ. Biol. Eng. 2018, 2, 17. 10.11648/j.jcebe.20180201.14. [DOI] [Google Scholar]
- Llyod J. B. F. Synchronized Fluorescence. Nat. Phys. Sci. 1971, 231, 64–65. 10.1038/physci231064a0. [DOI] [Google Scholar]
- Wang Y.; Zhang G.; Wang L. Potential Toxicity of Phthalic Acid Esters Plasticizer: Interaction of Dimethyl Phthalate with Trypsin in Vitro. J. Agric. Food Chem. 2015, 63, 75–84. 10.1021/jf5046359. [DOI] [PubMed] [Google Scholar]
- Paramaguru G.; Kathiravan A.; Selvaraj S.; Venuvanalingam P.; Renganathan R. Interaction of Anthraquinone Dyes with Lysozyme: Evidences from Spectroscopic and Docking Studies. J. Hazard. Mater. 2010, 175, 985. 10.1016/j.jhazmat.2009.10.107. [DOI] [PubMed] [Google Scholar]
- Cheng Z.-J.; Zhao H.-M.; Xu Q.-Y.; Liu R. Investigation of the Interaction between Indigotin and Two Serum Albumins by Spectroscopic Approaches. J. Pharm. Anal. 2013, 3, 257–269. 10.1016/j.jpha.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi M.; Kalhor H. R.; Laurent S.; Lynch I. Protein Fibrillation and Nanoparticle Interactions: Opportunities and Challenges. Nanoscale 2013, 5, 2570–2588. 10.1039/c3nr33193h. [DOI] [PubMed] [Google Scholar]
- Lacerda S. H. D. P.; Park J. J.; Meuse C.; Pristinski D.; Becker M. L.; Karim A.; Douglas J. F. Interaction of Gold Nanoparticles with Common Human Blood Proteins. ACS Nano 2010, 4, 365–379. 10.1021/nn9011187. [DOI] [PubMed] [Google Scholar]
- Holm N. K.; Jespersen S. K.; Thomassen L. V.; Wolff T. Y.; Sehgal P.; Thomsen L. A.; Christiansen G.; Andersen C. B.; Knudsen A. D.; Otzen D. E. Aggregation and Fibrillation of Bovine Serum Albumin. Biochim. Biophys. Acta, Proteins Proteomics 2007, 1774, 1128–1138. 10.1016/j.bbapap.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Khan A. Y.; Suresh Kumar G. Exploring the Binding Interaction of Potent Anticancer Drug Topotecan with Human Serum Albumin: Spectroscopic, Calorimetric and Fibrillation Study. J. Biomol. Struct. Dyn. 2018, 36, 2463–2473. 10.1080/07391102.2017.1359671. [DOI] [PubMed] [Google Scholar]
- Deng Z. J.; Liang M.; Toth I.; Monteiro M. J.; Minchin R. F. Molecular Interaction of Poly(Acrylic Acid) Gold Nanoparticles with Human Fibrinogen. ACS Nano 2012, 6, 8962–8969. 10.1021/nn3029953. [DOI] [PubMed] [Google Scholar]
- Nielsen L.; Khurana R.; Coats A.; Frokjaer S.; Brange J.; Vyas S.; Uversky V. N.; Fink A. L. Effect of Environmental Factors on the Kinetics of Insulin Fibril Formation: Elucidation of the Molecular Mechanism. Biochemistry 2001, 40, 6036–6046. 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- Serem W. K.; Bett C. K.; Ngunjiri J. N.; Garno J. C. Studies of the Growth, Evolution, and Self-Aggregation of β-Amyloid Fibrils Using Tapping-Mode Atomic Force Microscopy. Microsc. Res. Tech. 2011, 74, 699–708. 10.1002/jemt.20940. [DOI] [PubMed] [Google Scholar]
- Barbosa S.; Taboada P.; Mosquera V.. Fibrillation and Polymorphism of Human Serum Albumin; Elsevier, 2013. [Google Scholar]
- Siddiqi M. K.; Alam P.; Chaturvedi S. K.; Shahein Y. E.; Khan R. H. Mechanisms of Protein Aggregation and Inhibition. Front. Biosci. 2017, 9, 1–20. [DOI] [PubMed] [Google Scholar]
- Juárez J.; López S. G.; Cambón A.; Taboada P.; Mosquera V. Influence of Electrostatic Interactions on the Fibrillation Process of Human Serum Albumin. J. Phys. Chem.B 2009, 113, 10521–10529. 10.1021/jp902224d. [DOI] [PubMed] [Google Scholar]
- Pedersen J. T.; Heegaard N. H. H. Analysis of Protein Aggregation in Neurodegenerative Disease. Anal. Chem. 2013, 85, 4215–4227. 10.1021/ac400023c. [DOI] [PubMed] [Google Scholar]
- Siddiqi M. K.; Malik S.; Majid N.; Alam P.; Khan R. H.. Cytotoxic Species in Amyloid-Associated Diseases: Oligomers or Mature Fibrils; Elsevier Ltd, 2019; Vol. 118. [DOI] [PubMed] [Google Scholar]
- Ha C.; Ryu J.; Park C. B. Metal Ions Differentially Influence the Aggregation and Deposition of Alzheimer’s β-Amyloid on a Solid Template. Biochemistry 2007, 46, 6118–6125. 10.1021/bi7000032. [DOI] [PubMed] [Google Scholar]
- Rivas-Arancibia S.; Rodríguez-Martínez E.; Badillo-Ramírez I.; López-González U.; Saniger J. M. Structural Changes of Amyloid Beta in Hippocampus of Rats Exposed to Ozone: A Raman Spectroscopy Study. Front. Mol. Neurosci. 2017, 10, 137. 10.3389/fnmol.2017.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.