

Summary
Gene replacements through homologous recombination (HR) have been extensively used for functional genomic studies. However, the general efficiency of HR repair can be low in filamentous fungi and the process laborious. Here, we provide a detailed protocol for efficient gene editing by inserting donor DNA into a region of interest following Cas12a ribonucleoprotein (RNP)-mediated DNA double-strand break. We demonstrate this protocol using Magnaporthe oryzae (synonym of Pyricularia oryzae), a model plant pathogenic fungus that is used to study plant-fungal interactions.
For complete details on the use and execution of this protocol, please refer to Huang et al. (2021).
Subject areas: Genomics, Microbiology, Model Organisms, Plant sciences, Molecular Biology, CRISPR, Biotechnology and bioengineering
Graphical abstract
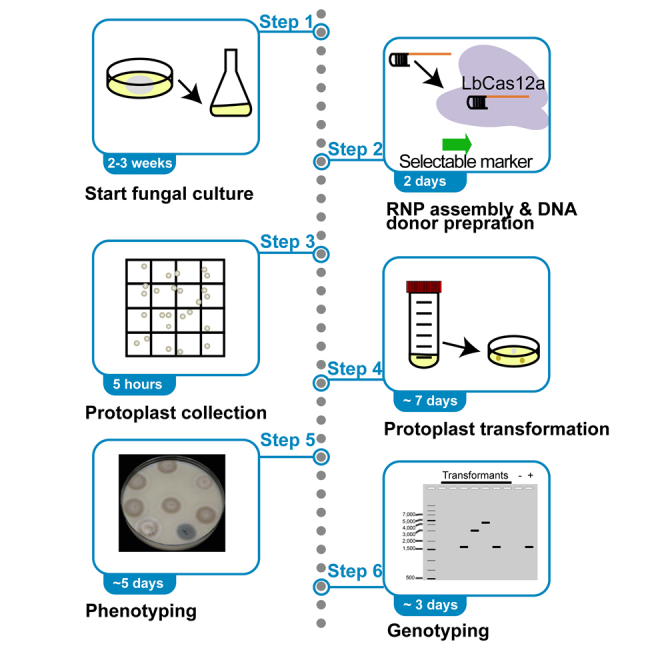
Highlights
-
•
Protocol for designing and performing Cas12a genome editing in a filamentous fungus
-
•
Donor DNA integration provides efficient transformation and selection
-
•
The donor DNA does not require homologous sequence to the targeted locus
-
•
The transformation and genotyping approach identifies many types of DNA mutations
Gene replacements through homologous recombination (HR) have been extensively used for functional genomic studies. However, the general efficiency of HR repair can be low in filamentous fungi and the process laborious. Here, we provide a detailed protocol for efficient gene editing by inserting donor DNA into a region of interest following Cas12a ribonucleoprotein (RNP)-mediated DNA double-strand break. We demonstrate this protocol using Magnaporthe oryzae (synonym of Pyricularia oryzae), a model plant pathogenic fungus that is used to study plant-fungal interactions.
Before you begin
CRITICAL: To avoid contamination, all the steps involving fungal protoplasts and cultures require a sterile work environment, such as a biosafety cabinet. This includes using sterilized materials and tools.
Fungal culture
Timing: 1–3 weeks
-
1.
Begin your starter culture on plates from a stock. For M. oryzae, transfer isolate from filter paper stock to oatmeal agar (OTA) using a sterilized toothpick.
-
2.
Incubate the culture in a growth chamber at 25°C under light for 1–2 weeks.
-
3.
Cut 5–8 small mycelial plugs (1–2 mm side length) from the OTA plate with a sterilized toothpick. Move the mycelial plugs into 100 mL liquid complete medium (CM) and grow at 28°C, 120 rpm for approximately 2–4 days in the dark.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Oatmeal Agar | BD | Cat#DF0552-17-3 |
| Sucrose | Fisher Scientific | Cat#S5-12 |
| Yeast extract | BD | Cat#21750 |
| Casamino acid | VWR | Cat#J851-500G |
| Yeast Nitrogen Base Without Amino Acids and Ammonium Sulfate | Fisher Scientific | Cat#DF0335-15-9 |
| Ammonium nitrate | Sigma-Aldrich | Cat#221244-500G |
| L- (+) - Asparagine | Fisher Scientific | Cat#AAB2147322 |
| Glucose | Sigma-Aldrich | Cat#G8270-100G |
| Agarose, low gelling temperature | Sigma-Aldrich | Cat#A9414-5G |
| di-sodium hydrogen phosphate | Sigma-Aldrich | Cat#1065851000 |
| Bacteriological Agar | IBI Scientific | Cat#IB49171 |
| Hygromycin B in solution | Corning | Cat#45000-806 |
| G418 solution | VWR | Cat#97064-358 |
| Bialaphos/glufosinate ammonium | Crescent Chemical | Cat#C140300 |
| FK506 | LC Laboratories | Cat#NC0876958 |
| Agarose | VWR | Cat#0710-500G |
| LbCas12a protein | New England BioLabs | Cat#M0653T |
| Lysing Enzymes from Trichoderma harzianum | Sigma-Aldrich | Cat#L1412-10G |
| Sodium chloride | Fisher Scientific | Cat#S271-3 |
| PEG4000 | FLUKA | Cat#81240 |
| Potassium chloride | Sigma-Aldrich | Cat#P-4504 |
| Chloroform | Sigma-Aldrich | Cat#C2432-1L |
| Isopropanol | Fisher Scientific | Cat#A416-4 |
| Ethanol | Fisher Scientific | Cat#BP2818-500 |
| Tris-HCI, pH 8.0 | Thermo Fisher Scientific | Cat#AM9856 |
| Calcium chloride | Sigma-Aldrich | Cat#C8106-1KG |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Fisher Scientific | Cat#SM1331 |
| Critical commercial assays | ||
| Phusion® High-Fidelity DNA Polymerase | New England BioLabs | Cat#M0530L |
| Q5® High-Fidelity DNA Polymerases | New England BioLabs | Cat#M0491L |
| Taq DNA Polymerase with Standard Taq Buffer | New England BioLabs | Cat#M0273L |
| HiScribe™ T7 High Yield RNA Synthesis Kit | New England BioLabs | Cat#E2040S |
| Monarch® RNA Cleanup Kit | New England BioLabs | Cat#T2050L |
| DNase I (RNase-free) | New England BioLabs | Cat#M0303S |
| Deoxynucleotide (dNTP) Solution Mix | New England BioLabs | Cat#N0447L |
| Wizard SV Gel and PCR Clean-Up system | Promega | Cat#A9282 |
| Experimental models:Organisms/strains | ||
| M. oryzae field isolate O-137 | Dr. Barbara Valent (Kansas State Univ) | N/A |
| Oligonucleotides | ||
| Please see the oligos used in this protocol in Table S1 | Huang et al. (2021) | N/A |
| Recombinant DNA | ||
| pFGL821 | Addgene | Cat#58223 |
| pFGL921 | Zhang et al. (2021) | N/A |
| pBV9/pSM324 | Kankanala et al. (2007) | N/A |
| Software and algorithms | ||
| BLAST | FungiDB | https://fungidb.org/fungidb/app/search/transcript/UnifiedBlast |
| Other | ||
| Membrane Filter, 0.22 μm pore size | Millex | Cat#SLGVM33RS |
| Plastic syringes | Thermo Fisher Scientific | Cat#S7510-10 |
| Toothpick | Fisher Scientific | Cat#S24554 |
| Hemocytometer | Fisher Scientific | Cat#0267151B |
| Miracloth | Calbiochem | Cat#475855-1R |
| 61/2 in Disks Non Gauze Milk Filter | KenAG | N/A |
| Water, Molecular Biology Grade | VWR | Cat#VWRL0201-0500 |
| 150 × 15 mm petri dishes | Fisher Scientific | Cat#FB0875714 |
| 60 × 15 mm Petri dishes | MIDSCI | Cat#901 |
| 100 × 15 mm Petri dishes | MIDSCI | Cat#900 |
| 50 mL falcon tubes | MIDSCI | Cat#C50B |
| 1.5 mL microcentrifuge tubes | USA scientific | Cat#1615-5500 |
| 0.2 mL PCR tubes | MIDSCI | Cat#AVTW-F |
| Funnels | Fisher Scientific | Cat#FB6015865 |
| 125 mL flask | PYREX | N/A |
| 200 mL flask | PYREX | N/A |
| Parafilm | Sigma-Aldrich | Cat#P7793-1EA |
| NanoDrop spectrophotometer | Thermo Fisher Scientific | N/A |
| 2.0 mL impact resistant screw cap tube | USA scientific | Cat #1420-9600 |
| 1.0 mm Silica Beads | BioSpec | Cat #11079110z |
| Bead ruptor elite-Bead mill homogenizer | OMNI international | Cat#19-2141E |
| Centrifuge | Eppendorf | Cat#5415D |
| Centrifuge | Beckman Coulter | Cat#Allegra 25R |
| Filter paper | N/A | N/A |
Materials and equipment
Prepare medium and stock solution
Oatmeal agar (OTA)
Dissolve 72.5 g commercial OTA powder in 1 L of distilled water, mix well. Heat to near boiling using a microwave for 1–2 min. Autoclave at 121°C for 20 min in liquid cycle. Pour the sterilized OTA in a 100 × 15 mm petri dish after autoclave, store at room temperature (∼28°C). The plate can be stored for up to 2 months.
Alternatives: Microwave can be replaced with a water bath for pre-heating.
Complete medium (CM)
| Reagent | Final concentration | Amount |
|---|---|---|
| Sucrose | 10 g/L | 10 g |
| Yeast extract | 6 g/L | 6 g |
| Casamino acid | 6 g/L | 6 g |
| Agar | 20 g/L | 20 g |
| ddH2O | n/a | up to 1000 mL |
| Total | n/a | 1000 mL |
Autoclave at 121°C for 20 min in liquid cycle. Agar is not required for liquid CM.
Store at room temperature up to 6 months.
TB3 medium (TB3)
| Reagent | Final concentration | Amount |
|---|---|---|
| Sucrose | 200 g/L | 200 g |
| Yeast extract | 6 g/L | 6 g |
| Casamino acid | 6 g/L | 6 g |
| Agar | 15 g/L | 15 g |
| ddH2O | n/a | up to 1000 mL |
| Total | n/a | 1000 mL |
Autoclave at 121°C for 20 min in liquid cycle. Agar is not required for liquid TB3.
Store at room temperature up to 6 months.
Defined complex medium (DCM)
| Reagent | Final concentration | Amount |
|---|---|---|
| Yeast nitrogen base without amino acids and ammonium sulphate | 1.7 g/L | 1.7 g |
| Ammonium nitrate | 1 g/L | 1 g |
| Asparagine | 2 g/L | 2 g |
| Glucose | 10 g/L | 10 g |
| Agarose, low gelling temperature | 20 g/L | 20 g |
| ddH2O | n/a | up to 1000 mL |
| Total | n/a | 1000 mL |
Adjust pH to 6 with di-sodium hydrogen phosphate (Na2HPO4). Autoclave at 121°C for 15 min in liquid cycle. Store at room temperature up to 6 months.
1×STC solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Sucrose | 20% w/v | 80 g |
| 1M Tris-Hcl pH = 8.0 | 50 mM | 20 mL |
| CaCl2. 2H2O | 50 mM | 2.94 g |
| ddH2O | n/a | up to 400 mL |
| Total | n/a | 400 mL |
Autoclave at 121°C for 20 min in liquid cycle. Store at 4°C for up to 6 months
PTC solution
| Reagent | Final concentration | Amount |
|---|---|---|
| PEG4000 | 60% w/v | 60 g |
| 1× STC solution | n/a | up to 100 mL |
| Total | n/a | 100 mL |
Autoclave at 121°C for 20 min in liquid cycle. Store at room temperature for up to 6 months.
If the solution is not completely dissolved before you use, microwave PTC solution to help it dissolve.
0.7 M NaCl solution
Dissolve 16.36 g NaCl in 400mL of distilled water. Autoclave at 121°C for 20 min in liquid cycle. Store at room temperature for up to 6 months.
Protoplast lysing solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Lysing Enzymes from Trichoderma harzianum | 10 mg/mL | 0.3 g |
| 0.7M NaCl solution | n/a | up to 30 mL |
| Total | n/a | 30 mL |
Filter sterilize using 0.22 μm filter. This solution needs to be freshly prepared on the date when you are ready to perform protoplast preparation.
1 M KCl solution
Dissolve 29.8 g KCl in 400 mL of distilled water. Autoclave at 121°C for 20 min in liquid cycle. Store at room temperature for up to 6 months.
Vector usage
We amplified the coding sequence for hygromycin, g418 and Bialaphos from the vectors pFGL821, pFGL921 and pBV9/pSM324 respectively. Additional details can be found in the Note of step 9. Other vectors or templates can additionally be used to amplify other genes of interest.
Step-by-step method details
Design oligos for LbCas12a guide RNA
This step is required to design a guide RNA that will direct the Cas12a nuclease to a specific site for editing. To demonstrate CRISPR-Cas12a editing in M. oryzae, we mutated the trihydroxynaphthalene reductase encoding gene termed BUF1 (MGG_02252), which is involved in fungal melanin biosynthesis (Chumley and Valent, 1990). The buf1 deletion mutant showed buff color (i.e., tan/orange) compared with the gray/black mycelial color in the wild-type strain. In this step, we describe the procedure to design the Cas12a guide RNA targeting BUF1.
-
1.
Download the sequence of gene of interest from FungiDB (https://fungidb.org/fungidb/app). For the BUF1 gene, download the sequence for the gene MGG_02252.
-
2.
Search for the LbCas12a PAM sequences (5′-TTTV-3′) in your gene of interest. Select a 23 bp DNA sequence downstream (3′ end) of PAM as a targeting sequence (Figure 1). In the case of BUF1, we designed two spacer/targeting sequences.
Figure 1.
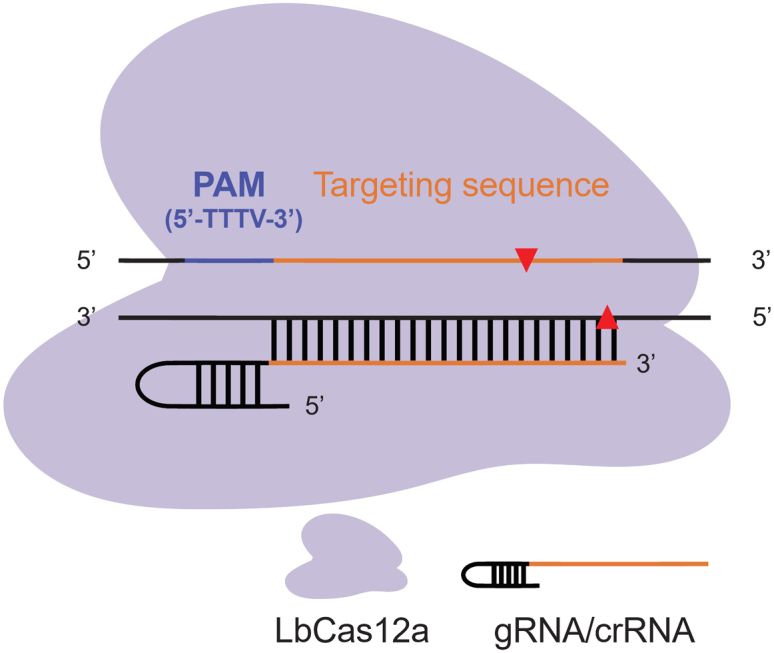
Schematic diagram of Cas12a cutting
Blue line highlights the PAM sequence, the orange line indicates the designed targeting sequence. Arrows indicate the cutting site (17th base after PAM in the non-targeted strand and 22nd base after PAM in the targeted strand) of Cas12a, which is represented by the purple shape. The gRNA contains a hairpin at the 5′ end to facilitate Cas12a interaction.
BUF1-targeting sequence#1: TTTCTCACCTTCAGCTCACTTCCCCAA
BUF1-targeting sequence#2: TTTACCATCAACACCCGTGGCCAGTTC
First four bold letters are the PAM, the remaining 23 bp are the targeting sequences.
Note: For LbCas12a guide designing, the targeting sequences must be on the same strand of PAM (‘5-TTTV-3’). V indicates A, C or G, but not T. Previous research reported that targeting sequences with GC-content (∼40%–60%) provides the highest editing activity for Cas12a (Kim et al., 2017). We advise avoiding targeting sequences with GC content <30% or >70% (Kim et al., 2017).
-
3.Check the off-target effect by blasting the designed targeting sequences within FungiDB database (https://fungidb.org/fungidb/app/search/transcript/UnifiedBlast)
-
a.Choose ‘genome’ as Target Data Type
-
b.Choose ‘blastn’ as BLAST Program
-
c.Choose ‘Pyricularia oryzae 70-15’ as Target organism
-
d.Input 27 bp sequences (4 bp PAM + 23 bp targeting sequences)
-
e.If the blast result shows the other loci match first 4 bp PAM and at least 16 bp targeting sequences after PAM perfectly, it may cause off-target editing. It is best to discard this potential guide sequence. Otherwise, move forward with the targeting sequence design.
-
a.
Note:Pyricularia oryzae is a synonym of Magnaporthe oryzae.
-
4.Design oligos for preparing the DNA template for in vitro guide RNA synthesis. Two oligos are designed to construct the DNA template.
-
a.Universal forward oligo including the T7 promoter and LbCas12a mature direct repeat (Figure 2).
-
b.Target specific reverse oligo including the reverse complemented sequences of LbCas12a mature direct repeat and the 23 bp targeting sequences you designed for your gene of interest (Figure 2). Oligos designed for two guides targeting the BUF1 locus, termed BUF1-guide1 and BUF1-guide2, are provided as examples (Figure 2).
-
a.
Note: Do not include PAM sequence in your target reverse oligo.
Note: Various online tools have been developed for genome-wide CRISPR target design. Many are dependent on the organism of interest and must be checked for suitability.
Optional: Commercial guide RNA synthesis kits, compatible with different Cas proteins, are available as an alternative option for preparing gRNA. For example, the NEB EnGen® sgRNA Synthesis Kit for the S. pyogenes SpCas9 protein can be used for making the corresponding gRNA (NEB, Cat#E3322V).
Figure 2.

Schematic examples of oligo design
In vitro guide RNA synthesis and RNP assembly
Following guide RNA design, this step is required to synthesize and associate the guide RNA with the Cas12a protein. This results in a competent RNP ready for transformation.
-
5.
Generate DNA template for in vitro guide RNA synthesis. Set up the below PCR reaction with Phusion® High-Fidelity DNA Polymerase.
| Reagent | Amount |
|---|---|
| 5× Phusion HF Buffer | 4 μL |
| 10 mM dNTPs | 0.8 μL |
| Universal forward primer (100 μM) | 0.4 μL |
| Target specific reverse primer (100 μM) | 0.4 μL |
| Phusion DNA Polymerase | 0.2 μL |
| Molecular water | 14.2 μL |
| Total | 20 μL |
PCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 30 s | 1 |
| Denaturation | 95°C | 10 s | 35 cycles |
| Annealing | 57°C | 10 s | |
| Extension | 72°C | 10 s | |
| Final extension | 72°C | 2 min | 1 |
| Hold | 4°C | 2 min | |
Optional: Ordering the top and bottom oligos with full sequences (i.e., T7 promoter, LbCas12a mature direct repeat and 23 bp targeting sequences), and annealing them as the DNA template would serve as an alternative option to avoid this PCR step.
-
6.
Guide RNA in vitro transcription. Set up the transcription as below by using HiScribe™ T7 High Yield RNA Synthesis Kit (NEB, Cat#E2040S).
| Reagent | Amount |
|---|---|
| 10× Reaction Buffer | 2 μL |
| ATP (100 mM) | 2 μL |
| GTP (100 mM) | 2 μL |
| CTP (100 mM) | 2 μL |
| UTP (100 mM) | 2 μL |
| Prepared template DNA from step 5 | 8 μL |
| T7 RNA Polymerase Mix | 2 μL |
| Total | 20 μL |
Run the reaction in a thermocycler with 37°C for 8–18 h.
Note: Based on our experience, the yield should be at least 50 μg RNA from this step.
-
7.Purification of synthetic guide RNA
-
a.Remove the DNA template by adding 1 μL (2 units) RNase-free DNase to the guide RNA reaction and incubate for 15 min at 37°C in a thermocycler.
-
b.Purify the DNase treated guide RNA through Monarch® RNA Cleanup Kit (NEB Cat#T2050L) followed the manufacturer's protocol (https://www.neb.com/protocols/2018/06/28/monarch-rna-cleanup-kit-protocol).
-
c.Quantify the RNA quality by NanoDrop spectrophotometer. A 260/280 ratio of 2.0–2.2 following guide RNA transcription and purification indicates successful synthesis.
-
d.Use guide RNA immediately for RNP assembly or store at −80°C for up to 2 months.
-
a.
-
8.
Assemble the RNP for one reaction in a 0.2 mL PCR tube as indicated below.
| Reagent | Final concentration | Amount |
|---|---|---|
| LbCas12a protein (100 μM) | ∼ 5 ug/20 μL | 0.34 μL |
| gRNA | ∼ 0.5 ug/20 μL | x μl |
| NEBuffer 2.1 Reaction Buffer (10×) | n/a | 2 μL |
| Molecular water | n/a | up to (17.66-x) μl |
| Total | n/a | 20 μL |
The amount of gRNA depends on the concentration of gRNA (e.g., if the concentration is 1000 ng/μL, then use x = 0.5 μL for the reaction).
Incubate the above reaction for 15 min at 25°C using a thermocycler.
Note: Add the LbCas12a protein last to avoid protein precipitation (Fernandez et al., 2018). Assemble the RNP immediately before fungal protoplast transformation.
Donor DNA preparation
This protocol uses donor DNA for selection, which is integrated at the Cas12a DNA double-strand break site. This step is used to amplify and purify the donor DNA prior to transformation.
-
9.
Amplify the no-homology HYG DNA donor as indicated below
| Reagent | Amount |
|---|---|
| 5× Phusion HF Buffer | 10 μL |
| 10 mM dNTPs | 1 μL |
| no-homology HYG DNA forward primer (10 μM) | 2.5 μL |
| no-homology HYG DNA reverse primer (10 μM) | 2.5 μL |
| Phusion DNA Polymerase | 0.5 μL |
| pFGL821 (∼5 ng/μL) | 1 μL |
| Molecular water | 32.5 μL |
| Total | 50 μL |
PCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 30 cycles |
| Annealing | 57°C | 30 s | |
| Extension | 72°C | 1 min | |
| Final extension | 72°C | 10 min | 1 |
| Hold | 4°C | 2min | |
Note: Expected PCR amplification size is 1,466 bp. Each fungal protoplast transformation requires ∼3 μg donor DNA in total. Adjust the number of above PCR reactions based on the total number of transformations you will run. We amplified the HYG DNA donor by using pFGL821 plasmid as DNA template. Other selectable markers (e.g., G418 and Bialaphos) could be used. We amplified G418 coding sequence from plasmid pFGL921 and Bialaphos coding DNA from plasmid pBV9/pSM324 (Kankanala et al., 2007; Zhang et al., 2021). If the gene being edited can be directly used for selection, such as editing FKBP12 (Huang et al., 2021) or URA3/URA5 as has been shown in Fusarium oxysporum for instance (Wang et al., 2018), the inclusion of donor DNA in the experiment is not necessary.
Note: Selection based on URA3/URA5 has not been established in M. oryzae, and preliminary experiments might be needed to confirm selection based on URA3/URA5 in M. oryzae.
-
10.
Check the amplification quality by running 2 μL of PCR product on a 1% agarose gel. Successful amplification should yield a single product of the expected size.
-
11.
Purify the PCR reaction through Wizard SV Gel and PCR Clean-Up system (Promega). The detailed purification step follows manufacturer's protocols (https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/101/wizard-sv-gel-and-pcr-clean-up-system-protocol.pdf?la=en). If there are more products from the amplification product as indicated by step 10, gel purification can be used to isolate the product of interest by using the same Wizard SV Gel and PCR Clean-Up system (Promega).
-
12.
Quantify the DNA quality by NanoDrop spectrophotometer. Donor concentration near 200 ng/μL–350 ng/μL is preferred.
Note: Store the DNA donor in −20°C freezer until future use.
M. oryzae protoplast preparation
This step is required to generate the necessary M. oryzae cells to be used for transformation. The protoplasts are obtained by degrading the fungal cell wall, and allows more efficient RNP and nucleic acid transfer.
-
13.
Prepare the protoplast lysing solution freshly as described in “prepare medium and stock solution” section.
-
14.
Collect the mycelia from sub-cultured liquid CM through 2-layers of 61/2 in Disks Non Gauze Milk Filter papers and funnel. Wash the collected mycelia with a 5 mL 1×STC solution.
-
15.
Dry the collected mycelia with extra filter paper, transfer the dry mycelia to 100 mL flask with 25–30 mL protoplast lysing solution. Shake dry mycelia containing the flask at 30°C, 70–80 rpm, for 2.5–3 h under dark to digest the fungal cell wall and release protoplasts.
Note: When transferring the mycelium, manually separate the mass so it is not a single large mass. This will increase the accessibility to the lysing solution.
-
16.
Check the digestion progress and protoplast quality using a hemocytometer under a dissecting microscope. Successful digestion should release many healthy protoplasts, while if many undigested mycelia are observed, extend the digestion time. However, excessive digestion can also cause protoplast collapse (Figure 3).
-
17.
Collect the protoplast by filtering the above lysing reaction containing the fungal protoplasts, through 2-layer miracloth into pre-chilled, 50 mL falcon tube. Wash the lysing debris left in the miracloth with 1×STC again to collect all the protoplasts.
-
18.
Centrifuge the 50mL falcon tube with protoplast at 4°C, 5,000 rpm (i.e., 3,829 g in Beckman Coulter-Allegra 25R centrifuge) for 10 min.
Note: A visible pellet containing protoplasts should be seen.
-
19.
Discard the supernatant into a biohazard trash container. Gently resuspend the protoplast using 5–10 mL 1×STC by pipetting or flicking.
-
20.
Repeat step 18.
-
21.
Discard the supernatant into a biohazard trash container, and gently resuspend the protoplast in 1mL 1×STC. Quantify the protoplast concentration using a hemocytometer. Add additional 1×STC to adjust the final concentration of protoplasts to 8 × 106 to 5 × 107 protoplasts/mL. Aliquot 200–250 μL protoplast solution to individual 50 mL falcon tubes for further transformation.
Optional: The protoplasts can be frozen and stored by adding 7% v/v DMSO into the protoplast solution and freezing at −80°C. We recommend performing the protoplast transformation immediately after preparation for best results, but the frozen cultures can still be used for successful editing.
Note: This protocol is strain independent, and we have used it to make protoplasts for various isolates including O-137 and Guy11 (Bao et al., 2017).
Figure 3.

Different protoplast status after lysing enzyme digestion
(A–C) (A) An example of healthy protoplasts, (B) undigested mycelium, and (C) collapsed protoplasts each highlighted by black circles. Scale Bars = 50 μm
M. oryzae protoplast transformation
This step transfers the assembled RNP and donor DNA into the fungal cell to allow editing and donor DNA integration.
-
22.
Slowly add 20 μL assembled RNP and 3 μg of no-homology HYG DNA donor into the 50 mL falcon tube containing the aliquoted protoplast. Mix the solution by flicking gently. Place the tube at room temperature for 20–25 min.
Note: If your gene of interest can be used as selection directly, DNA donor is not required in step 22 For example, we have generated fkbp12 (FK506 receptor) mutants by using the selection of drug FK506 without DNA donor successfully (Huang et al., 2021).
-
23.
Slowly add 1,000 μL PTC solution to the 50 mL falcon tube. Mix the solution by flicking gently. Place the tube at room temperature for 20–25 min.
-
24.
Slowly add 5 mL liquid TB3 medium into the 50 mL falcon tube. Mix the solution by flicking gently. Cover the 50 mL falcon tube lid with parafilm.
-
25.
Shake 50 mL falcon tube at 28°C, 90 rpm overnight (12–20 h) in the dark.
-
26.
Next day, unseal the 50 mL falcon tube and add melting TB3 medium (near 40 mL for one transformation, 50°C–60°C) with final concentration of 100 μg/mL hygromycin solution. Mix gently, then pour the mixture (overnight protoplast culture + medium) into 150 × 15 mm petri-dish. Wait for about 20 min until complete solidification.
Note: Protoplasts are fragile and require gentle handling. Other antibiotics corresponding to a different no-homology DNA donor can also be used. For example, we have also used G418 at a final concentration: 300 μg/mL. Bialaphos also can be used at a final concentration: 25 μg/mL in DCM instead of TB3 medium.
-
27.
After the first selection layer has completely solidified, pour a second layer of melting TB3 medium (near 50 mL) with final concentration of 200 μg/mL hygromycin solution into the 150 × 15 mm petri-dish above the first layer. Wait for about 20 min until complete solidification.
Note: For G418 selection, the final concentration for the second layer is 600 μg/mL. Bialaphos also can be used at a final concentration: 100 μg/mL in DCM instead of TB3 medium.
-
28.
Seal the 150 × 15 mm petri-dish with parafilm and incubate the plate upside down at 28°C for 5–7 days under dark.
Note: Transformants using M. oryzae isolate O-137 can typically be seen and transferred at 5 days, while those from isolate Guy11 typically take 7 days. Other isolates will have to be empirically determined.
-
29.
Pick the transformants growing in the top layer for further phenotyping and genotyping. This can be done using a sterile toothpick to move a piece of mycelium to a new plate containing the selection.
-
30.
For BUF1 deletion identification, we cultured the transformants to OTA to check the mycelial color change. For other genes of interest, transformants can be moved to CM with corresponding appropriate selection: 200 μg/mL hygromycin or 500 μg/mL G418. If you use bialaphos selection (final concentration: 100 μg/mL), select DCM instead of CM for screening.
Quick fungal DNA extraction for genotyping
DNA is extracted from transformants in order to genotype putative DNA edits.
-
31.
Prepare the screw cap tubes with 1.0 mm beads, 400 μL 1M KCL and 400 μL chloroform (added in hood).
-
32.
Add fungal plug (near 1 cm diameter) into the above prepared tubes. (Performed in the biosafety cabinet hood)
-
33.
Put the above tubes in a homogenizer with power 4 × 6 cycles (30 s each cycle) to disrupt the fungal material.
-
34.
Centrifuge 13,200 rpm (i.e.,16,100 g in Eppendorf 5415D centrifuge) × 10 min
-
35.
Transfer 200 μL supernatant into new labeled tubes
-
36.
Add 120 μL isopropanol in the tube, mix well to precipitate the DNA for at least 10 min (added in hood).
-
37.
Centrifuge 13,200 rpm (i.e.,16,100 g in Eppendorf 5415D centrifuge) × 10 min.
-
38.
Discard the supernatant and wash the pellet with 500 μL 75% ethanol.
-
39.
Centrifuge 13,200 rpm (i.e.,16,100 g in Eppendorf 5415D centrifuge) × 10 min.
-
40.
Discard the ethanol and air dry for 10 min.
-
41.
Add 50 μL sterilized water to dissolve the DNA pellet.
Note: We prefer to use 1 μL of the dissolved DNA as template for PCR amplification. The DNA extracted by this quick extraction protocol is sufficient for PCR genotyping but should not be considered adequate for other uses such as next-generation sequencing or southern blot.
PCR genotyping
Using the extracted DNA, PCR is used to determine if the gene of interest has been mutated by amplifying multiple regions at the target locus. This allows the researcher to understand and identify a range of possible DNA mutations, including INDELS, donor DNA insertions, and large deletions.
-
42.
We prefer to design three pairs of primer for genotyping; one primer pair amplifies within the gene of interest (∼1–2 kb for PCR amplification, the RNP targeting sequence should be included in this amplifying region), and the other two primer pairs amplifying the 5′ and 3′ regions of the targeted sequence (∼ 0.5 kb for PCR amplification, the reverse primer for 5′ upstream amplification and the forward primer for 3′ downstream amplification should locate closely (1–200 bp) to the start codon and stop codon of the gene of interest) (Figure 4).
-
43.
Perform genotyping PCR followed the below program for the gene of interest. We used the BUF1 gene as an example.
| Reagent | Amount |
|---|---|
| 5× Q5 reaction buffer | 5 μL |
| 10mM dNTPs | 0.5 μL |
| BUF1 gene region forward primer (10 μM) | 1.25 μL |
| BUF1 gene region reverse primer (10 μM) | 1.25 μL |
| Q5 High-Fidelity DNA Polymerase | 0.25 μL |
| DNA extracted from quick extraction protocol | 1 μL |
| Molecular water | 15.75 μL |
| Total | 25 μL |
PCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 30 cycles |
| Annealing | 65°C | 30 s | |
| Extension | 72°C | 2 min | |
| Final extension | 72°C | 2 min | 1 |
| Hold | 4°C | 2 min | |
Note: The expected amplification size for wild-type is 1,648 bp, the size for mutants might vary based on the mutation pattern (Huang et al., 2021).
-
44.
Perform genotyping PCR for 5′upstream, 3′ downstream of BUF1 and ACTIN (loading control) with below program
| Reagent | Amount |
|---|---|
| 10× standard Taq reaction buffer | 2.5 μL |
| 10mM dNTPs | 0.5 μL |
| BUF1 5′ upstream or 3' downstream or ACTIN forward primer (10 μM) | 0.5 μL |
| BUF1 5′ upstream or 3' downstream or ACTIN Reverse primer (10 μM) | 0.5 μL |
| Taq DNA Polymerase | 0.125 μL |
| DNA extracted from quick extraction protocol | 1 μL |
| Molecular water | 19.875 μL |
| Total | 25 μL |
PCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 30 s | 1 |
| Denaturation | 95°C | 10 s | 30 cycles |
| Annealing | See below note | 30 s | |
| Extension | 72°C | 35 s | |
| Final extension | 68°C | 5 min | 1 |
| Hold | 4°C | 2 min | |
Note: Annealing temperature for BUF1 5′ upstream or downstream is 54°C, while it requires 57°C for ACTIN. The expected amplification size for BUF1 upstream, downstream and ACTIN is 311 bp, 458 bp and 503 bp, respectively.
Figure 4.

Schematic illustration of PCR primer pairs used for genotyping
Expected outcomes
Gene disruption mutants can be generated by our method with an editing efficiency from ∼50% to 100% at BUF1 locus (Figure 5A). Genotyping results suggested that large-scale insertion and deletions are common in the buf1 mutants (Figure 5B). Please see more detailed analysis and discussion in the original manuscript (Huang et al., 2021).
Figure 5.

Representative phenotyping and genotyping outcomes at BUF1 locus with BUF1-guide1 RNP and no-homology HYG DNA donor
(A) Δbuf1 showed the buff/orange mycelial color in OTA. CP641 (buf-) derived from O-137 is the positive strain for buff mycelia. O-137 (BUF+) is the wild-type strain used in this assay.
(B) DNA extracted from the transformants in (A) were used for genotyping with BUF1, BU1 5′upstream, BUF1 3′ downstream and Actin primer pairs (loading control). – and + indicated the water (negative control) and O-137 genomic DNA (positive control) used in PCR reaction.
Limitations
Due to locus-dependent and physiological variables (e.g., gRNA efficiency, chromatin feature, sub-telomere/telomere location, and cell cycle), gene-editing efficiency can vary between loci and experiments. (Ceccaldi et al., 2016; Gisler et al., 2019; Huang et al., 2021; Schep et al., 2021; Xue and Greene, 2021). The use of two separate RNPs, targeting DNA regions within proximity to the desired location of editing may increase editing efficiency if single RNP transformation does not generate the desired mutation.
Troubleshooting
Problem 1
Cannot get the DNA template for T7 in vitro transcription from step 5.
Potential solution
Double check the oligo concentration you used in your system, it requires 100 μM for oligos, which is 10× higher than the normal PCR reaction.
Confirm the complementary sequences shared between universal forward primer and target specific reverse primer.
Problem 2
gRNA quality and concentration are low from step 6–7.
Potential solution
A low yield of gRNA could result from the following:
Inefficient gRNA synthesis
Run DNA template before T7 in vitro transcription in 2% agarose gel to confirm the integrity of the initial DNA template. A single, strong signal near 66 bp is expected.
Exactly follow the T7 in vitro transcription step described in this protocol (step 6). Mix all the reaction components well by pipetting before starting the reaction. Extend the reaction time to 18 h for increased gRNA yield.
Inefficient gRNA purification
Follow the purification step described in Monarch® RNA Cleanup Kit (NEB Cat#T2050L), store the gRNA correctly prior to usage. For this, if we cannot immediately use the gRNA following purification, we store at −80°C for storage.
Problem 3
Poor quality of protoplast from step 21.
Potential solution
Avoid using the melanized mycelia for lysing enzyme digestion.
Prepare the lysing enzyme freshly.
Don’t over digest (> 4 h) the fungi.
Make sure you use miracloth rather than other filter materials in the filtering step after protoplasts are released.
Gently mix the protoplasts before hemocytometer quantification.
Keep released protoplast on the ice until you start the transformation.
Problem 4
Buff phenotype is not visibly obvious from step 30.
Potential solution
The buff phenotype is most visibly pronounced when M. oryzae is grown on OTA or RPA media. We found that plating the buff strains on CM produces less clear visible color differences when compared to the wild-type strain.
Problem 5
Low gene editing efficiency from step 30.
Potential solution
Many factors contribute to genome editing efficiency.
Guide design, if you are new for designing CRISPR gRNA, do an in vitro digestion assay followed the attached protocol (https://international.neb.com/protocols/2017/12/19/in-vitro-digestion-of-dna-with-engen-lba-cas12a-cpf1-neb-m0653), it will tell you whether your designed gRNA is compatible with Cas12a protein and the cutting efficiency.
Fungal protoplast, use freshly prepared protoplast for transformation. We found that freshly prepared protoplast with correct concentration always showed better transformation efficiency than frozen stored protoplast.
Temperature sensitivity, it has been shown before that LbCas12a is a temperature sensitive enzyme, with best editing efficiency at ∼28 or higher °C (Malzahn et al., 2019). Therefore, it is necessary to maintain your lab temperature around 28°C when you perform Cas12a transformation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David E. Cook, decook@ksu.edu.
Materials availability
M. oryzae wild-type strains and derived mutants within this study are available upon reasonable request and may require permit.
Acknowledgments
The authors would like to thank Dr. Barbara Valent and Melinda Dalby (Kansas State University) for sharing the resources and fungal isolates, and David Rowe (Kansas State University) for critically reading prior to manuscript submission. The research was funded by the United State Department of Agriculture–National Institute of Food and Agriculture (USDA-NIFA) (award no. 2018-67013-28492) and the National Science Foundation Division of Molecular and Cellular Biosciences–Systems and Synthetic Biology (award no. 1936800) to D.E.C. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
J.H. executed the experiments and wrote the first draft of the manuscript. D.E.C. supervised the study and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.101072.
Contributor Information
Jun Huang, Email: jhuang90@ksu.edu.
David E. Cook, Email: decook@ksu.edu.
Supplemental information
Data and code availability
This study did not generate any unique datasets or code.
References
- Bao J., Chen M., Zhong Z., Tang W., Lin L., Zhang X., Jiang H., Zhang D., Miao C., Tang H., et al. PacBio sequencing reveals transposable elements as a key contributor to genomic plasticity and virulence variation in Magnaporthe oryzae. Mol. Plant. 2017;10:1465–1468. doi: 10.1016/j.molp.2017.08.008. [DOI] [PubMed] [Google Scholar]
- Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumley F.G., Valent B. Genetic analysis of melanin-deficient, nonpathogenic mutants of Magnaporthe grisea. Mol. Plant Microbe Interact. 1990;3:135–143. [Google Scholar]
- Dean R.A., Talbot N.J., Ebbole D.J., Farman M.L., Mitchell T.K., Orbach M.J., Thon M., Kulkarni R., Xu J.R., Pan H., et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature. 2005;434:980–986. doi: 10.1038/nature03449. [DOI] [PubMed] [Google Scholar]
- Fernandez J.P., Vejnar C.E., Giraldez A.J., Rouet R., Moreno-Mateos M.A. Optimized CRISPR-Cpf1 system for genome editing in zebrafish. Methods. 2018;150:11–18. doi: 10.1016/j.ymeth.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisler S., Goncalves J.P., Akhtar W., de Jong J., Pindyurin A.V., Wessels L.F.A., van Lohuizen M. Multiplexed Cas9 targeting reveals genomic location effects and gRNA-based staggered breaks influencing mutation efficiency. Nat. Commun. 2019;10:1598. doi: 10.1038/s41467-019-09551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Rowe D., Zhang W., Suelter T., Valent B., Cook D. CRISPR-Cas12a induced DNA double-strand breaks are repaired by locus-dependent and error-prone pathways in a fungal pathogen. bioRxiv. 2021 doi: 10.1101/2021.09.08.459484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankanala P., Czymmek K., Valent B. Roles for rice membrane dynamics and plasmodesmata during biotrophic invasion by the blast fungus. Plant Cell. 2007;19:706–724. doi: 10.1105/tpc.106.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.K., Song M., Lee J., Menon A.V., Jung S., Kang Y.M., Choi J.W., Woo E., Koh H.C., Nam J.W., et al. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat. Methods. 2017;14:153–159. doi: 10.1038/nmeth.4104. [DOI] [PubMed] [Google Scholar]
- Malzahn A.A., Tang X., Lee K., Ren Q., Sretenovic S., Zhang Y., Chen H., Kang M., Bao Y., Zheng X., et al. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 2019;17:9. doi: 10.1186/s12915-019-0629-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep R., Brinkman E.K., Leemans C., Vergara X., van der Weide R.H., Morris B., van Schaik T., Manzo S.G., Peric-Hupkes D., van den Berg J., et al. Impact of chromatin context on Cas9-induced DNA double-strand break repair pathway balance. Mol. Cell. 2021;81:2216–2230.e10. doi: 10.1016/j.molcel.2021.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Cobine P.A., Coleman J.J. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genet. Biol. 2018;117:21–29. doi: 10.1016/j.fgb.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C., Greene E.C. DNA repair pathway choices in CRISPR-Cas9-mediated genome editing. Trends Genet. 2021;37:639–656. doi: 10.1016/j.tig.2021.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Huang J., Cook D.E. Histone modification dynamics at H3K27 are associated with altered transcription of in planta induced genes in Magnaporthe oryzae. PLoS Genet. 2021;17:e1009376. doi: 10.1371/journal.pgen.1009376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.