

Abstract
Background:
High-risk pediatric acute myeloid leukemia confers a poor prognosis, and alternative strategies are needed to improve outcomes. We hypothesized that intensifying induction on the AAML1031 clinical trial would improve outcomes compared to the predecessor trial AAML0531.
Methods:
Patients on AAML0531 received cytarabine (1600 mg/m2)/daunorubicin (150 mg/m2)/etoposide (ADE) for induction II and patients on AAML1031 received mitoxantrone (48 mg/m2)/cytarabine (8000 mg/m2) (MA). Stem cell transplant (SCT) conditioning included busulfan/cyclophosphamide on AAML0531, whereas AAML1031 used busulfan/fludarabine and liberalized donor eligibility. Patients were included in this analysis if they met high-risk criteria common to the two trials by cytogenics or poor disease response after induction I ADE.
Results:
MA provided no benefit over ADE at: induction II response (complete response [CR]: 64% vs. 62%, p = .87; measurable residual disease [MRD]+: 57% vs. 46%, p = .34); or intensification I response (CR: 79% vs. 94%, p = .27; MRD+: 27% vs. 20%, p = 1.0). When considered with altered SCT approach, MA did not improve 5-year disease-free survival (24% ± 9% vs. 18% ± 15%, p = .63) or 5-year overall survival (35% ± 10% vs. 38% ± 18%, p = .66). MA was associated with slower neutrophil recovery (median 34 vs. 27 days, p = .007) and platelet recovery (median 29 vs. 24.5 days, p = .04) and longer hospital stay (32 vs. 28 days, p = .01) during induction II.
Conclusion:
Intensification of induction II did not improve treatment response or survival, but did increase toxicity and resource utilization. Alternative strategies are urgently needed to improve outcomes for pediatric patients with high-risk acute myeloid leukemia (trials registered at clinicaltrials.gov NCT01371981, NCT00372593).
Keywords: acute leukemia, Children’s Oncology Group, induction, mitoxantrone, myeloid leukemia, pediatric oncology
1 ∣. INTRODUCTION
Outcomes for children with acute myeloid leukemia (AML) have improved primarily because of intensification of therapy1-3 and progress in supportive care,4,5 but relapse rates remain high. For patients with high-risk disease based on cytogenetics or poor response to initial therapy, 3-year disease-free survival (DFS) is as low as 20%–30%, even with the inclusion of stem cell transplant (SCT) in first remission.6-9 For these patients, intensification of remission induction through prolonging chemotherapy exposure and increasing drug dose may improve initial leukemia response and long-term postremission disease control.1,2 However, intensified induction approaches may also increase treatment-related morbidity and mortality.
The inclusion of high-dose cytarabine to intensify consolidation has been shown to reduce relapse and prolong survival.10-16 However, intensification of induction courses of therapy with high-dose cytarabine has yielded mixed results relative to treatment outcomes.17-19 Alternatively, prior studies have suggested there may be benefit from alternative anthracycline or anthracenediones in induction for pediatric patients.5,15,16
In an effort to improve outcomes for high-risk AML, the Children’s Oncology Group (COG) AAML1031 trial intensified induction II chemotherapy by switching from daunorubicin to mitoxantrone, and escalating cytarabine exposure compared to the predecessor trial.20,21 This provided an opportunity to evaluate the impact of induction intensification using the predecessor trial, AAML0531, as a comparator. In a protocol described aim, high-risk patients who received cytarabine/mitoxantrone (MA) as induction II on AAML1031 were compared to a similar cohort who received cytarabine/daunorubicin/etoposide (ADE) as induction II on AAML0531. AAML1031 also modified SCT by liberalizing donor source and utilizing a different conditioning regimen. Here, we describe the impact of these two changes on survival. We also examine the impact of induction II intensification on disease response pretransplant and induction II toxicities, costs, and resource utilization. We hypothesized that these changes would be associated with improved survival but would also confer increased toxicity.
2 ∣. METHODS
2.1 ∣. Study population
The source population was patients treated on either COGAAML0531 or AAML1031 who survived and remained on study to the beginning of the second cycle of chemotherapy (termed Induction II). The entry criteria for these trials included newly diagnosed de novo AML patients aged 0–30 years old. Patients with secondary AML, acute promyelocytic leukemia, and bone marrow failure syndromes were excluded. The National Cancer Institute central institutional review board (IRB) and IRB at each enrolling institution approved both studies with patients and families providing consent and assent as appropriate. The trials were conducted in accordance with the Declaration of Helsinki. Both trials were registered at clinicaltrials.gov NCT01371981, NCT00372593.
These studies had identical backbones for cycles 1 and 3, and then diverged in their approach (Figure 1,Table S1). On AAML0531, patients were randomized to receive or not receive gemtuzumab ozogamicin (GO) during cycle 1. Patients were allocated to high-risk therapy based on cytogenetics or >15% blasts by morphology after induction I chemotherapy. Multidimensional flow cytometry measurable residual disease (MRD) was analyzed, but investigators were blinded to the result. High-risk patients then received two more cycles of chemotherapy, and then proceeded to related or unrelated allogeneic SCT with busulfan/cyclophosphamide, or two additional cycles of chemotherapy if there was no suitable donor.20 On AAML1031, patients were randomized to receive or not receive bortezomib on days 1, 4, and 8 of each cycle of chemotherapy. Risk stratification occurred after cycle 1, and poor response to therapy was defined as MRD ≥0.1%. On AAML1031, after two additional cycles of chemotherapy, high-risk patients underwent SCT with busulfan/fludarabine and haploidentical donors were also permitted in addition to matched related and unrelated donors. Those without an appropriate donor received a single additional cycle of chemotherapy.
FIGURE 1.
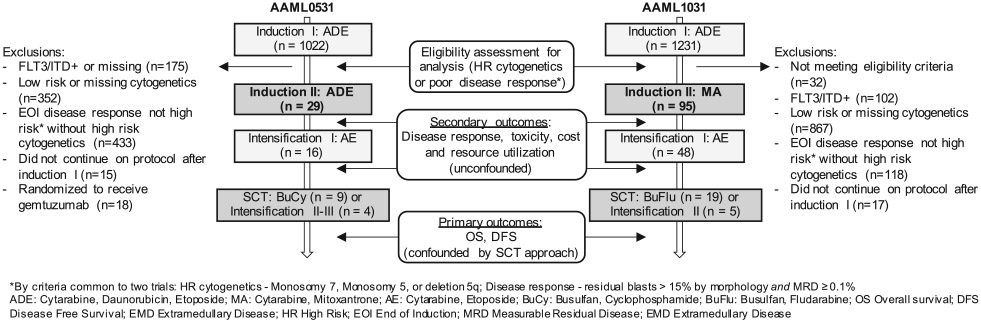
Schematic of therapy for high-risk patients on AAML0531 and AAML1031. Patients were included for analysis if they met high-risk criteria common to both trials: cytogenetics (7-, 5-, del5q), or poor disease response requiring morphological blasts >15%, and measurable residual disease (MRD) ≥0.1%. Exclusions and timing of outcome ascertainment are shown
The definition of “high risk” was discrepant between trials. To create a uniform population for this analysis, high risk was defined based on criteria common to the two trials: (a) adverse cytogenetics (monosomy 7, monosomy 5, or deletion 5q); or (b) poor disease response to Induction 1 ADE. The inclusion criteria for poor disease response required patients to satisfy both studies’ criteria: residual blasts >15% by morphology, the definition used on AAML053120; and ≥0.1% MRD, the definition used on the AAML1031 trial.21,22
Patients with high allelic ratio FLT3/ITD+ were excluded. In addition, patients randomized to the experimental arm of AAML0531 (standard therapy + GO) were excluded to mitigate confounding as GO was found to improve EFS and GO was not included in AAML1031.9 AAML1031 randomization of bortezomib receipt did not modify survival so all patients were included in the primary analysis.21
2.2 ∣. Exposure
On COG AAML1031, induction II consisted of cytarabine 1000 mg/m2/dose every 12 hours on days 1–4 (cumulative dose 8000 mg/m2) and mitoxantrone 12 mg/m2/dose on days 3–6. Induction II on AAML0531 included cytarabine 100 mg/m2/dose every 12 hours on days 1–8 (cumulative dose 1600 mg/m2), daunorubicin 50 mg/m2/dose on days 1, 3, and 5, and etoposide 100 mg/m2/dose on days 1–5. The AAML1031 approach equated to 50% more cytarabine exposure (20 vs. 13.6 g/m2) and 15%–78% more anthracycline exposure (342–534 vs. 300 mg/m2 doxorubicin equivalents) over the first three cycles of chemotherapy.
2.3 ∣. Outcomes
Patients were observed from the start of induction II through last available follow-up. The primary protocol-described outcomes were DFS and OS. DFS was defined as the time from end of induction I to induction failure, relapse, secondary malignancy, or death. OS was defined as time from end of induction I to death. We anticipated that DFS and OS would be confounded due to differences in approach to SCT between the two trials. Therefore, we included secondary outcomes proximal to transplant to evaluate treatment response: complete remission (CR)/CR incomplete recovery (CRi) rate and MRD at the end of induction II and intensification I; and median change in MRD between end of induction I and end of induction II chemotherapy. Secondary outcomes to evaluate toxicity included hematologic toxicity, infections, course length, hospital stay, and intensive care unit (ICU) days. Additional resource utilization and cost outcomes were examined in a subset of patients treated at hospitals contributing to the Pediatric Health Information System (PHIS), as described previously.23
2.4 ∣. Covariates
Demographic information including sex, age at diagnosis, race, Hispanic ethnicity, central nervous system disease, end induction marrow response, and cytogenetics were obtained from the COG study databases for AAML0531 and AAML1031.
2.5 ∣. Analysis
Patient characteristics and treatment responses were compared by study using chi-square test, or, in the event of sparse data, Fisher’s exact test. Medians for continuous characteristics were compared using the Mann–Whitney test. The Kaplan–Meier method was used to compare DFS and OS from end of induction I for patients who continued on protocol; patients were censored at date of last known contact. Multivariable cox models were constructed to adjust for baseline variables that were unbalanced between the two trials. Inpatient treatment costs were summarized as means, and crude cost ratios were estimated from a general linear model with gamma distribution to compare costs between studies. Poisson regression was used to compare resource utilization rates with resource days as the outcome, inpatient days as the offset, and Pearson scale adjustment for overdispersion. These methodologies have been described in detail previously.24,25 Sensitivity analyses were performed to compare survival and toxicities between the standard and experimental arms of AAML1031 to determine if the inclusion of bortezomib as an experimental agent was associated with differential outcomes. Data from AAML0531 and AAML1031 were current as of December 31, 2019. All analyses were performed using SAS (version 9.4, SAS Institute, Inc., Cary, NC).
3 ∣. RESULTS
A total of 124 patients were treated on AAML0531 (n = 29) or AAML1031 (n = 95) and met criteria for inclusion in the current analyses (Figure 1). Patient characteristics are shown in Table 1 and the distribution of high-risk features differed between trials. Patients on AAML1031 had a higher disease burden at the end of induction I by MRD (25% vs. 16%, p = .027) and morphology (median blast percentage 30% vs. 16.5%, p = .097). More patients on AAML0531 had monosomy 5/del5q compared to AAML1031 (34.5% vs. 10.6%, p = .002). Following intensification I, 13 of 29 (44.8%) patients on AAML0531 continued on protocol-directed therapy, including nine of 13 who proceeded to SCT. On AAML1031, 24 of 95 (25.3%) patients continued on protocol-directed therapy and 19 of 24 underwent SCT.
TABLE 1.
Characteristics of high-riska AML study population
| AAML0531 (arm A) | AAML1031 | p-Value | |
|---|---|---|---|
| Total, N | 29 | 95 | |
| Sex | |||
| Male | 16 (55.2%) | 55 (57.9%) | .795 |
| Female | 13 (44.8%) | 40 (42.1%) | |
| Age at diagnosis, years | |||
| Median (range) | 9.95 (0.88–29.8) | 9.03 (1.08–24.7) | .549 |
| 0–1 | 6 (20.7%) | 14 (14.7%) | .564 |
| 2–10 | 11 (37.9%) | 45 (47.4%) | .371 |
| 11–20 | 11 (37.9%) | 32 (33.7%) | .674 |
| ≥21 | 1 (3.4%) | 4 (4.2%) | 1.000 |
| Race | |||
| American Indian or Alaskan Native | 0 (0.0%) | 2 (2.5%) | 1.000 |
| Asian | 1 (3.7%) | 3 (3.8%) | 1.000 |
| Black or African American | 8 (29.6%) | 15 (18.8%) | .234 |
| White | 18 (66.7%) | 60 (75%) | .400 |
| Unknown | 2 (6.9%) | 15 (15.8%) | |
| Ethnicity | |||
| Hispanic or Latino | 6 (21.4%) | 20 (22.0%) | .951 |
| Not Hispanic or Latino | 22 (78.6%) | 71 (78.0%) | |
| Unknown | 1 (3.4%) | 4 (4.3%) | |
| CNS disease classification at study entry | |||
| CNS1 | 25 (86.2%) | 71 (75.5%) | .258 |
| CNS2 | 3 (10.3%) | 16 (17.0%) | .559 |
| CNS3 | 1 (3.4%) | 6 (6.4%) | 1.000 |
| Unknown | 0 (0.0%) | 2 (2.1%) | |
| Adverse cytogeneticsb | |||
| Monosomy 7 | 9 (31.0%) | 19 (20.2%) | .224 |
| Monosomy 5/del5q | 10 (34.5%) | 10 (10.6%) | .002 |
| End induction I marrow response | |||
| Median MRD% (range) | 16 (0–73) | 25 (0–92) | .027 |
| Median blast percentage by morphology (range) | 16.5 (0–89) | 30 (0–89) | .097 |
| Criteria for high-risk definition | |||
| Cytogenetics | 15 (51.7%) | 23 (24.2%) | .005 |
| Disease response | 11 (37.9%) | 64 (67.4%) | .005 |
| Both | 3 (10.3%) | 8 (8.4%) | .718 |
| Patients proceeding to transplant, n | 9 | 19 | |
| Matched sibling | 1(11.1%) | 3 (15.8%) | |
| Unrelated | 8 (88.9%) | 16 (84.2%) | 1.000 |
Note: Data presented as n (%) unless otherwise noted.
Abbreviations: AML, acute myeloid leukemia; CNS, central nervous system; MRD, measurable residual disease.
High risk based on criteria mutual to both studies.
Specific cytogenetics unknown for two patients.
3.1 ∣. Comparison of survival and disease response
Figure 2 presents the Kaplan–Meier estimates for DFS and OS from end of induction I. High-risk patients treated on AAML1031 did not have different 5-year DFS (24.2% ± 8.8% vs. 18.3% ± 14.7%, p = .632) or OS (34.6% ± 10.1% vs. 37.9% ± 18.0%, p = .658) compared to AAML0531. In the sensitivity analysis that considered the two arms of AAML1031 separately, Kaplan–Meier estimates of DFS and OS were similar in all three groups. In this group, 5-year DFS on AAML1031 arm A was 28.7% ± 13.5% and on arm B was 20.0% ± 11.3%, and 5-year OS on AAML1031 was 36.6% ± 15.2% and 32.3% ± 13.5% on arms A and B, respectively. The corresponding survival curves are shown in Figure S1.
FIGURE 2.

Kaplan–Meier curves for overall survival (A) and disease-free survival (B) comparing high-risk patients treated on AAML0531 arm A and AAML1031
Similarly, there was no improvement in treatment response at end of induction II as a result of inclusion of MA (Table 2). On AAML1031, 63.7% of patients who received MA as induction II achieved a CR/CRi compared to 62.1% of patients who received ADE as induction II on AAML053 (p = .871). At the end of induction II, there were no statistically significant differences in the proportion with positive MRD ≥0.1% or median MRD between the two trials. Intensification I treatment was identical between the two trials, and end-intensification I disease evaluation also did not reveal any statistically significant differences between the studies. While patients in the AAML1031 cohort had higher mean MRD at the start of induction II, there was no significant difference in the median percentage change of MRD from induction I to either end induction II (−12.7 vs. −18.0, p = .788), or end intensification I (−22.4 vs. −15.8, p = .676).
TABLE 2.
Disease response
| AAML0531 (arm A) | AAML1031 | p-Value | |
|---|---|---|---|
| Induction II | |||
| Total, N | 29 | 95 | |
| Response | |||
| Clinical remission | 18 (62.1%) | 58 (63.7%) | |
| Refractory/relapse | 11 (37.9%) | 33 (36.3%) | 0.871 |
| Death | 0 (0.0%) | 0 (0.0%) | |
| Not evaluable | 0 | 4 | |
| MRD evaluation | |||
| Median MRD (range) | 0.01 (0.0–46.0) | 0.4 (0.0–75.0) | .426 |
| MRD positive (>0.1) | 10 (45.5%) | 41 (56.9%) | .344 |
| Unknown MRD | 7 (24.1%) | 23 (24.2%) | |
| Median MRD change from end induction I (range) | −18.0 (−39.2, −3.02) | −12.7 (−78.9,32) | .788 |
| Intensification I | |||
| Total, N | 16 | 48 | |
| Response | |||
| Clinical remission | 15 (93.8%) | 38 (79.2%) | .265 |
| Relapse | 1 (6.3%) | 9 (18.8%) | .429 |
| Death | 0 (0.0%) | 1 (2.1%) | 1.000 |
| MRD evaluation | |||
| Median MRD (range) | 0.0 (0.0–4.4) | 0.0 (0.0–15.0) | .523 |
| MRD positive (>0.1) | 1 (20.0%) | 8 (26.7%) | 1.000 |
| Unknown MRD | 11 (68.8%) | 18 (37.5%) | |
| Median MRD change from end induction I (range) | −15.8 (−18.0, −13.6) | −22.4 (−68.0, 0.2) | .676 |
Note: Data presented as N (%) unless otherwise noted.
Abbreviation: MRD, measurable residual disease.
As the proportion of patients who met the high-risk definition by cytogenetics or end induction I disease response differed between the two trials (Table 1), we performed a post hoc multivariable Cox analysis to evaluate if survival was confounded by these variables. This analysis did not identify a difference between DFS and OS on the two trials: DFS-adjusted HR 0.67, 95% CI 0.41–1.10 and OS-adjusted HR 0.95, 95% CI 0.56–1.64. The full multivariable model is shown in Table 3. To further explore this, we performed stratified analyses in subgroups of patients who were high risk by cytogenetics (n = 49) and by disease response (n = 86). The end induction I disease response or OS estimates were similar to the overall estimates and did not demonstrate a difference by induction II regimen in either strata (Table S2). The point estimates did suggest a potential benefit of MA compared to ADE relative to DFS for patients who were high risk by disease response (DFS 0% vs. 16.7%, p = .154).
TABLE 3.
Multivariable Cox analyses for disease-free survival and overall survival from end of induction 1
| Disease-free survival |
Overall survival |
||||||
|---|---|---|---|---|---|---|---|
| N | HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Study | |||||||
| AAML0531 arm A | 29 | 1 | 1 | ||||
| AAML1031 | 95 | 0.67 | 0.41–1.10 | .112 | 0.95 | 0.56–1.64 | .873 |
| High-risk cytogenetics | |||||||
| None | 75 | 1 | 1 | ||||
| Present | 49 | 1.51 | 0.77–2.98 | .235 | 2.64 | 1.32–5.28 | .006 |
| End induction MRD | |||||||
| Negative | 38 | 1 | 1 | ||||
| Positive | 86 | 3.69 | 1.70–7.99 | .009 | 3.59 | 1.63–7.94 | .002 |
Abbreviations: HR, hazard ratio; MRD, measurable residual disease.
3.2 ∣. Comparison of toxicity
Patients on AAML1031 who received MA had a significantly lower probability of neutrophil and platelet recovery and significantly longer median time to recovery of both cell lines in induction II (Table 4). In addition, patients on AAML1031 had more median hospital days during induction II (32 [range 5–72] vs. 28 [range 1–49] days, p = .013). Hematologic toxicity, course length, and hospital days in intensification I were not statistically significantly different between the two trials (Table 4). The proportion of patients requiring ICU care was similar in both courses. In addition, the proportion of patients who experienced microbiologically documented infectious toxicity in induction II and intensification I did not differ between the two trials (Table 4). Importantly, there was no evidence of increased hematological toxicity or course length among AAML1031 patients allocated to bortezomib versus not (Table S3) nor was it differential by end induction I disease status (Table S4).
TABLE 4.
Hematologic and infectious toxicity, hospital days, and course length
| AAML0531 (arm A) | AAML1031 | p-Value | |
|---|---|---|---|
| Induction II | |||
| Total, N | 29 | 95 | |
| Neutrophil recovery (ANC >500 cells/ml) | |||
| Patients recovered | 23 (79.3%) | 50 (52.6%) | .011 |
| Days to recovery, median (range) | 27 (22–58) | 34 (19–73) | .007 |
| Platelet recovery (platelets >50,000/ml) | |||
| Patients recovered | 24 (82.8%) | 57 (60.0%) | .024 |
| Days to recovery, median (range) | 24.5 (11–62) | 29 (14–63) | .040 |
| Days in course, median (range) | 35 (21–69) | 40 (16–115) | .007 |
| Hospital days, median (range) | 28 (1–49) | 32 (5–72) | .013 |
| ICU days, median (range) | 4 (1–28) | 4 (1–54) | .859 |
| Bacterial infection | 8 (27.6%) | 27 (28.4%) | .930 |
| Viridans group streptococcus | 3 (10.3%) | 14 (14.7%) | .760 |
| Gram negative bacilli | 4 (13.8%) | 6 (6.3%) | .241 |
| Fungal infection | 0 (0.0%) | 3 (3.2%) | 1.000 |
| Intensification I | |||
| Total, N | 16 | 48 | |
| Neutrophil recovery (ANC >500 cells/ml) | |||
| Patients recovered | 12 (75.0%) | 37 (77.1%) | 1.000 |
| Days to recovery, median (range) | 25 (19–33) | 28 (21–49) | .052 |
| Platelet recovery (platelets >50,000/ml) | |||
| Patients recovered | 10 (62.5%) | 35 (72.9%) | .530 |
| Days to recovery, median (range) | 23 (15–30) | 28(19–55) | .011 |
| Days in course, median (range) | 38.5 (1–73) | 45.5 (25–66) | .121 |
| Hospital days, median (range) | 23 (1–38) | 48 (5–66) | .067 |
| ICU days, median (range) | 2 (1–3) | 11.5 (2–20) | .088 |
| Bacterial infection | 5 (31.3%) | 20 (41.7%) | .460 |
| Viridans group streptococcus | 1 (6.3%) | 8 (16.7%) | .430 |
| Gram-negative bacilli | 1 (6.3%) | 10 (20.8%) | .265 |
| Fungal infection | 0 (0.0%) | 0 (0.0%) |
Note: Data presented as n (%) unless otherwise noted.
Abbreviation: ANC, absolute neutrophil count.
3.3 ∣. Comparisons of resource utilization and cost
There were 9 and 27 patients from AAML0531 and AAML1031, respectively, identified in PHIS. These patients were representative of the larger cohorts of patients included in these analyses (Table S5). The induction II costs did not differ significantly between trials when total course cost was compared (cost ratio 1.41 [95% CI 0.86–2.29], p = .17) or cost per day was compared (cost ratio 1.26 [95% CI 0.92–1.74], p = .15) (Table S6). Rates of specific resource utilization did not differ between trials after accounting for inpatient days (Table S7).
4 ∣. DISCUSSION
The COG trial AAML1031 modified therapy for patients with high-risk AML by intensifying induction II chemotherapy with MA (mitoxantrone and cytarabine 8000 mg/m2), using busulfan/fludarabine as SCT conditioning and liberalizing donor eligibility. The data comparing AAML0531 and AAML1031 outcomes do not demonstrate a survival benefit for pediatric patients with high-risk AML as a result of those changes, although the relative contribution of each change cannot be parsed. These two trials demonstrated equivalent rates of remission induction after induction II, an outcome that was not confounded by changes in the SCT approach between the two trials. As remission induction is highly correlated with survival in AML,22,26-28 together these data suggest that there is no short- or long-term benefit especially as a result of intensified induction II chemotherapy. Although the chemotherapy intensification did not improve outcomes, it was associated with additional hematologic toxicity and longer time to course completion.
Compared to a backbone of cytarabine and daunorubicin, attempts have been made to intensify induction in order to more effectively induce or maintain remissions, including increasing cytarabine exposure, using alternative anthracycline agents, or incorporating additional chemotherapeutic agents. To date, the evidence supporting any of these approaches has been limited.
Prior studies have compared low-dose cytarabine (100–200 mg/m2) to intermediate and high-dose cytarabine (1000–3000 mg/m2 BID) on backbones that vary the number of induction cycles, additional agents, and methods to evaluate treatment response. A comparison of successive AML Berlin–Frankfurt–Münster (BFM) group studies reported that an additional cycle containing cytarabine (3000 mg/m2) and mitoxantrone in induction was associated with improved survival in high-risk16 and cytogenetically favorable RUNX1-RUNX1T129 AMLs compared to patients treated on the predecessor trial. In addition, the AML-12 study performed by the European Organization for Research and Treatment of Cancer and Research and Gruppo Italiano Malattie Ematologiche dell’ Adulto randomized young adults to induction with high- (total 24,000 mg/m2) or low-dose cytarabine (total 1000 mg/m2). Intensified cytarabine was associated with superior CR rate and 6-year OS for patients younger than 46 years.19 The authors hypothesized that the survival benefit was due to improvements in supportive care and the availability of more intensive postremission strategies. In contrast, three analyses, Pediatric Oncology Group 9421, St. Jude AML02, and Medical Research Council (MRC) AML-12, did not show a benefit to intensified induction, with similar CR rates, EFS, and OS between treatment approaches.17,18,30
Prior pediatric studies have also examined the utility of incorporating alternative anthracycline or anthracenediones in induction.5,15,16 As above, the use of mitoxantrone in combination with an extra cycle of high-dose cytarabine improved outcomes on the BFM 93 trial.16 Similarly, results of the MRC AML12 trial suggested that substituting mitoxantrone for daunorubin during induction improved DFS. However, this survival benefit was offset by increased treatment-related mortality.5 Importantly, in adults, dose intensification of anthracyclines during induction has not consistently improved response rates and overall survival.31-34
These analyses do suggest that MA is associated with increased hematologic toxicity as evidenced by longer time to both neutrophil and platelet recovery in induction II. This effect may persist even beyond induction II, as our analyses suggest increased hematologic toxicity after identical consolidation I chemotherapy as well. The increase in hospital days for patients treated with MA, likely as a result of delayed count recovery, has important implications for health services delivery.25,35 In addition, these analyses did not capture late effects of treatment, but the use of mitoxantrone on AAML1031 may confer an increased risk of long-term cardiotoxicity compared to daunorubicin.36-38
This observational study is limited by its use of historic controls, leading to potential misclassification and confounding.39,40 Figure 1 highlights the limitations of such a study, demonstrating the small proportion of patients that met inclusion criteria for these analyses and imbalance in patient and disease charateristics between studies. Risk of misclassification in this analysis was mitigated through the stringent inclusion criteria employed for inclusion on these analyses and the requirement that patients meet high-risk criteria common to both trials. With regards to confounding, unobserved differences in supportive care and SCT strategies would be expected to bias in favor of AAML1031, the more recent clinical cohort,4,6,7,39,40 so an absence of improved outcomes on the later trial further reinforces that intensified induction may not confer a survival advantage. Furthermore, the post hoc analysis that controlled for important unbalanced clinical criteria between the trials did not substantially change the response rate or survial estimates. Finally, this study was limited by its modest sample size, particularly among patients who continued on protocol-defined SCT, which may have limited our ability to detect a true association in either direction.
In conclusion, data from AAML0531 and AAML1031 do not suggest a disease response or overall survival benefit from higher dose cytarabine and mitoxantrone in induction II in pediatric high-risk AML in the context of COG chemotherapy, even in the modern era of supportive care and broader transplant criteria. Despite the acknowledged limitations to this study, these are the best data available to guide selection of COG backbone chemotherapy for future high-risk clinical trials, and importantly these data provide a clear signal of increased hematologic toxicity associated with MA. In the context of increased toxicity and an absent signal of efficacy, the next-generation COG Phase III trial, AAML1831, has reverted to the prior backbone and now uses low-dose cytarabine with daunorubicin as induction II for high-risk AML patients. Unfortunately, these analyses underscore that despite intensified therapy in both the induction and postremission phases, survival for children with high-risk AML remains unacceptably poor. Alternative approaches that incorporate rationally designed targeted therapies are urgently needed to achieve cure.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the patients and families who participated in the AAML0531 and AAML1031 clinical trials. This research was supported by the COG NCTN Network Group Operations Centres (5U10CA180886-07) and NCTN Statistics & Data Center Grant (2U10CA180899-07).
Abbreviations:
- ADE
cytarabine/daunorubicin/etoposide
- AML
acute myeloid leukemia
- BFM
Berlin–Frankfurt–Münster
- COG
Children’s Oncology Group
- CR
complete remission
- DFS
disease-free survival
- GO
gemtuzumab ozogamicin
- ICU
intensive care unit
- IRB
institutional review board
- MA
mitoxantrone/cytarabine
- MRC
Medical Research Council
- MRD
measurable residual disease
- PHIS
Pediatric Health Information System
- SCT
stem cell transplant
Footnotes
CONFLICT OF INTEREST
Michael Loken and Lisa Eidenschink Brodersen are employees of Hematologics Inc, which performed the MRD-based flow cytometry assays used in this clinical trial.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Prior presentations: Findings presented in part as an oral presentation at the annual meeting of the American Society of Clinical Oncology June 2019 (https://doi.org/10.1200/JCO.2019.37.15_suppl.10002; Journal of Clinical Oncology (May 20, 2019) 37(15_suppl):10002).
REFERENCES
- 1.Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015;33(27):2949–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woods WG, Kobrinsky N, Buckley JD, et al. Timed-sequential induction therapy improves postremission outcome in acute myeloid leukemia: a report from the Children’s Cancer Group. Blood. 1996;87(12):4979–4989. [PubMed] [Google Scholar]
- 3.Castellino SM, Alonzo TA, Buxton A, Gold S, Lange BJ, Woods WG. Outcomes in childhood AML in the absence of transplantation in first remission–Children’s Cancer Group (CCG) studies 2891 and CCG 213. Pediatr Blood Cancer. 2008;50(1):9–16. [DOI] [PubMed] [Google Scholar]
- 4.Sung L, Aplenc R, Alonzo TA, Gerbing RB, Lehrnbecher T, Gamis AS. Effectiveness of supportive care measures to reduce infections in pediatric AML: a report from the Children’s Oncology Group. Blood. 2013;121(18):3573–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibson BE, Webb DK, Howman AJ, et al. Results of a randomized trial in children with acute myeloid leukaemia: Medical Research Council AML12 trial. Br J Haematol. 2011;155(3):366–376. [DOI] [PubMed] [Google Scholar]
- 6.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29(5):551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore AS, Kearns PR, Knapper S, Pearson AD, Zwaan CM. Novel therapies for children with acute myeloid leukaemia. Leukemia. 2013;27(7):1451–1460. [DOI] [PubMed] [Google Scholar]
- 8.Nunes AL, Paes CA, Murao M, Viana MB, De Oliveira BM. Cytogenetic abnormalities, WHO classification, and evolution of children and adolescents with acute myeloid leukemia. Hematol Transfus Cell Ther. 2019;41(3):236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol. 2014;32(27):3021–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med. 1994;331(14):896–903. [DOI] [PubMed] [Google Scholar]
- 11.Mayer RJ, Schiffer CA, Peterson BA, et al. Intensive postremission therapy in adults with acute nonlymphocytic leukemia with ara-C by continuous infusion or bolus administration: preliminary results of a CALGB phase I study. Semin Oncol. 1985;12(2 Suppl 3): 84–90. [PubMed] [Google Scholar]
- 12.Woods WG, Ruymann FB, Lampkin BC, et al. The role of timing of high-dose cytosine arabinoside intensification and of maintenance therapy in the treatment of children with acute nonlymphocytic leukemia. Cancer. 1990;66(6):1106–1113. [DOI] [PubMed] [Google Scholar]
- 13.Wells RJ, Woods WG, Lampkin BC, et al. Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Children’s Cancer Group. J Clin Oncol. 1993;11(3):538–545. [DOI] [PubMed] [Google Scholar]
- 14.Creutzig U, Berthold F, Boos J, et al. Improved treatment results in children with AML: results of study AML-BFM 93. Klin Padiatr. 2001;213(4):175–185. [DOI] [PubMed] [Google Scholar]
- 15.Creutzig U, Ritter J, Zimmermann M, et al. Idarubicin improves blast cell clearance during induction therapy in children with AML: results of study AML-BFM 93. AML-BFM Study Group. Leukemia. 2001;15(3):348–354. [DOI] [PubMed] [Google Scholar]
- 16.Creutzig U, Ritter J, Zimmermann M,et al. Improved treatment results in high-risk pediatric acute myeloid leukemia patients after intensification with high-dose cytarabine and mitoxantrone: results of Study Acute Myeloid Leukemia-Berlin-Frankfurt-Munster 93. J Clin Oncol. 2001;19(10):2705–2713. [DOI] [PubMed] [Google Scholar]
- 17.Rubnitz JE, Inaba H, Dahl G, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11(6):543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becton D, Dahl GV, Ravindranath Y, et al. Randomized use of cyclosporin A (CsA) to modulate P-glycoprotein in children with AML in remission: Pediatric Oncology Group Study 9421. Blood. 2006;107(4):1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willemze R, Suciu S, Meloni G, et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12 trial. J Clin Oncol. 2014;32(3):219–228. [DOI] [PubMed] [Google Scholar]
- 20.Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol. 2014;32(27):3021–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aplenc R, Meshinchi S, Sung L, et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the Children’s Oncology Group. Haematologica. 2020;105(7):1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loken MR, Alonzo TA, Pardo L, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children’s Oncology Group. Blood. 2012;120(8):1581–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aplenc R, Fisher BT, Huang YS, et al. Merging of the National Cancer Institute-funded cooperative oncology group data with an administrative data source to develop a more effective platform for clinical trial analysis and comparative effectiveness research: a report from the Children’s Oncology Group. Pharmacoepidemiol Drug Saf. 2012;21(Suppl 2):37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Getz KD, Li Y, Alonzo TA, et al. Comparison of in-patient costs for children treated on the AAML0531 clinical trial: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(10):1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kavcic M, Fisher BT, Li Y, et al. Induction mortality and resource utilization in children treated for acute myeloid leukemia at free-standing pediatric hospitals in the United States. Cancer. 2013;119(10):1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agarwal SK, Mangal N, Menon RM, Freise KJ, Salem AH. Response rates as predictors of overall survival: a meta-analysis of acute myeloid leukemia trials. J Cancer. 2017;8(9):1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheatley K, Burnett AK, Goldstone AH, et al. A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. United Kingdom Medical Research Council’s Adult and Childhood Leukaemia Working Parties. Br J Haematol. 1999;107(1):69–79. [DOI] [PubMed] [Google Scholar]
- 28.Short NJ, Zhou S, Fu C, et al. Association of measurable residual disease with survival outcomes in patients with acute myeloid leukemia: a systematic review and meta-analysis. JAMA Oncol. 2020;6(12):1890–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Creutzig U, Zimmermann M, Bourquin JP, et al. Second induction with high-dose cytarabine and mitoxantrone: different impact on pediatric AML patients with t(8;21) and with inv(16). Blood. 2011;118(20):5409–5415. [DOI] [PubMed] [Google Scholar]
- 30.Burnett AK, Hills RK, Milligan DW, et al. Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol. 2010;28(4):586–595. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRIAML17 trial in 1206 patients. Blood. 2015;125(25):3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood. 2011;118(14):3832–3841. [DOI] [PubMed] [Google Scholar]
- 34.Ohtake S, Miyawaki S, Fujita H, et al. Randomized study of induction therapy comparing standard-dose idarubicin with high-dose daunorubicin in adult patients with previously untreated acute myeloid leukemia: the JALS GAML201 study. Blood. 2011;117(8):2358–2365. [DOI] [PubMed] [Google Scholar]
- 35.Getz KD, Miller TP, Seif AE, et al. A comparison of resource utilization following chemotherapy for acute myeloid leukemia in children discharged versus children that remain hospitalized during neutropenia. Cancer Med. 2015;4(9):1356–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipshultz SE, Adams MJ, Colan SD, et al. Long-term cardiovascular toxicity in children, adolescents, and young adults who receive cancer therapy: pathophysiology, course, monitoring, management, prevention, and research directions: a scientific statement from the American Heart Association. Circulation. 2013;128(17):1927–1995. [DOI] [PubMed] [Google Scholar]
- 37.Armenian SH, Hudson MM, Mulder RL, et al. Recommendations for cardiomyopathy surveillance for survivors of childhood cancer: a report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. 2015;16(3):e123–e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feijen EAM, Leisenring WM, Stratton KL, et al. Derivation of anthracycline and anthraquinone equivalence ratios to doxorubicin for late-onset cardiotoxicity. JAMA Oncol. 2019;5(6):864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaynon PS. Historical controls? Haematologica. 2017;102(3):e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wheatley K SAB–a promising new treatment to improve remission rates in AML in the elderly? Br J Haematol. 2002;118(2):432–433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.