

Abstract
Background:
Recent studies revealed that human and experimental alcohol-related liver disease (ALD) is robustly associated with dysregulation of bile acid homeostasis which may in turn modulates disease severity. Pharmacological agents targeting bile acid metabolism and signaling may be potential therapeutics for treating ALD.
Methods:
The potential beneficial effects of a gut restricted apical sodium-dependent bile acid transporter (ASBT) inhibitor are studied in a chronic-plus-binge ALD mouse model.
Results:
Here we report that blocking intestinal bile acid re-absorption by a gut restricted ASBT inhibitor GSK2330672 attenuated hepatic steatosis and liver injury in a chronic-plus-binge ALD mouse model. Alcohol feeding is associated with intestinal bile acid accumulation but paradoxically impaired ileal farnesoid x receptor (FXR) function, and repressed hepatic cholesterol 7α-hydrolase (CYP7A1) expression despite decreased hepatic small heterodimer partner (SHP) and ileal fibroblast growth factor 15 (FGF15) expression. ASBT inhibitor treatment decreases intestinal bile acid accumulation and increases hepatic CYP7A1 expression, but further decreases ileal FXR activity. Alcohol feeding induces serum bile acid concentration that strongly correlate with liver injury marker. However, alcohol-induced serum bile acid elevation is not due to intrahepatic bile acid accumulation but is strongly and positively associated with hepatic multidrug resistance-associated protein 3 (MRP4) and MRP4 induction but poorly associated with sodium-taurocholate co-transporting peptide (NTCP) expression. ASBT inhibitor treatment decreases serum bile acid concentration without affecting hepatocyte basolateral bile acid uptake and efflux transporters.
Conclusion:
We report that ASBT inhibitor treatment corrects alcohol-induced bile acid dysregulation and attenuates liver injury in experimental ALD.
Keywords: alcohol-related liver disease, ASBT, bile acid, liver injury, CYP7A1
Introduction
Alcohol-related liver disease (ALD) is a major cause of liver cirrhosis (Gao and Bataller, 2011). Currently, clinical ALD is mainly managed by abstinence, corticosteroids and nutritional support. There is still an unmet need for identifying new serological markers and therapeutic strategies for diagnosis and treatment of ALD. Recently, dysregulation of bile acid homeostasis in ALD has been increasingly recognized. It has been reported that ALD patients showed reduced bile acid synthesis and elevated serum bile acids that correlated with disease severity (Brandl et al., 2018). Consistently, alcohol feeding results in elevated serum bile acids in mice (Hartmann et al., 2018). Furthermore, intestinal farnesoid x receptor (FXR) has been shown to play a protective role in experimental mouse models of ALD (Hartmann et al., 2018, Huang et al., 2020). Currently, the underlying causes of bile acid dysregulation in ALD are still incompletely understood. Furthermore, the potential therapeutic benefits of targeting bile acid metabolism and signaling in ALD require further investigation.
Bile acids circulate between the liver and small intestine in a process termed the enterohepatic circulation where bile acids activate the nuclear receptor FXR to feedback inhibit cholesterol 7α-hydroxylase (CYP7A1) gene which encodes the rate-limiting enzyme in hepatic bile acid synthesis (Goodwin et al., 2000, Inagaki et al., 2005). In the small intestine, the majority of the bile acids are re-absorbed at the terminal ileum by the apical sodium-dependent bile acid transporter (ASBT) into the enterocytes (Dawson et al., 2003), while the organic solute transporter α (OSTα)/OSTβ heterodimer effluxes bile acids into the portal circulation (Rao et al., 2008). Blocking intestinal bile acid re-absorption by ASBT inhibitors promotes fecal bile acid loss and decreases total bile acid pool, which results in increased de novo bile acid synthesis. Studies have shown that gut restricted ASBT inhibitors decrease hepatic fat and cholesterol accumulation in experimental fatty liver models (Rao et al., 2016, Wang et al., 2020), and alleviate liver injury in mouse models of cholestasis (Baghdasaryan et al., 2015, Miethke et al., 2015). Given that ALD is associated with both hepatic fat accumulation and dysregulation of bile acid homeostasis, we investigated the potential therapeutic benefits of a gut restricted ASBT inhibitor GSK2330672 (GSK672) in an acute on chronic Gao-Binge ALD mouse model (Bertola et al., 2013).
Experimental Procedure
Reagents.
GSK2330672 was purchased from MedChem Express (Monmouth Junction, NJ). Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) assay kits, total cholesterol assay kit and triglyceride (TG) assay kit were purchased from Pointe Scientific (Canton. MI). Bile acid assay kit was purchased from Diazyme Laboratories (Poway, CA). Oil Red O was purchased from Sigma (St. Louis, MO).
Animal experiments.
Male 12 weeks old C57BL/6J mice were purchased from the Jackson Lab (Bar Harbor, ME). Mice were housed in micro-isolator cages (2 mice/cage) under 7 am - 7 pm light cycle and 7 pm - 7 am dark cycle. Ethanol feeding was carried out as previously reported (Bertola et al., 2013). Briefly, mice were first acclimated to the Lieber-DeCarli liquid diet (F1259SP; Bio-serv, Flemington, NJ) for 5 days. Mice were then fed ethanol (Decon, King of Prussia, PA)–containing (5%, v/v) liquid diet (F1258SP; Bio-serv) supplemented with maltose dextrin (Bio-serv) for 10 days or calorie- and volume-matched control liquid diet. On the last day, mice were orally gavaged with ethanol (5 g/kg). GSK672 was prepared in methylcellulose and administered via oral gavage at 4 mg/kg/day for the duration of the ethanol feeding. Vehicle was administered to the control group. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals.” All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center.
Triglyceride, cholesterol and bile acid measurement.
Lipids were extracted from liver tissues in a mixture of chloroform: methanol (2:1; v: v), dried under nitrogen, and resuspended in isopropanol containing 1% triton X-100. Total TG and cholesterol were measured with assay kits. For bile acid measurement, bile acids were extracted from liver, whole gallbladder bile, whole small intestine with content, and dried feces in 90% ethanol. Total serum and fecal bile acid amount was measured with an assay kit. Bile acids in liver, gallbladder bile and small intestine extract was measured by an LC-MS method.
Histological analysis.
Liver tissues were fixed in 4% paraformaldehyde and paraffin embedded. Hematoxylin and Eosin (H&E) was performed with an automated stainer. Oil Red O stock solution was prepared by dissolving 2 g Oil Red O in 400 mL isopropanol. A working solution was prepared by diluting three parts Oil Red O stock solution with two parts water, followed by filtering. Frozen tissues were sectioned and fixed in 10% formalin for 5 minutes. Slides were stained with freshly prepared Oil Red O working solution for 15 minutes and rinsed with 60% isopropanol before mounting in an aqueous mounting medium.
Real-time PCR.
Liver and distal ileum total RNA was purified with Trizol (Sigma-Aldrich, St. Louis, MO). Reverse transcription was performed with Oligo dT primer and SuperScript III reverse transcriptase (Thermo Fisher Scientific, Grand Island, NY). Real-time PCR was performed on a Bio-Rad CFX384 Real-time PCR system with iQ SYBR Green Supermix (Bio-rad, Hercules, CA). The comparative CT (Ct) method was used to calculate the relative mRNA expression and the results were expressed as 2 −ΔΔCt with the control group arbitrarily set as “1”.
Statistical analysis.
Results are expressed as mean ± SEM. Student’s t-test or two-way ANOVA and Tukey post hoc test was used to calculate the p value. A p < 0.05 was considered statistically significant.
Results
Gut-restricted ASBT inhibitor reduces liver fat accumulation and serum transaminase in alcohol-fed mice.
To investigate the effects of alcohol feeding and ASBT inhibitor treatment on bile acid metabolism and ALD development, mice were subjected to chronic-plus-binge alcohol feeding and daily treatment with a gut-restricted ASBT inhibitor GSK672. Alcohol feeding caused a significant reduction in body weight, while GSK672 did not affect body weight in either the control diet group (CD) or the alcohol diet (ED) group (Fig 1A). Alcohol feeding did not alter liver weight, but significantly increased liver weight: body weight (LW:BW) ratio due to reduced body weight (Fig 1B). Interestingly, GSK672 decreased serum transaminases in the alcohol-fed mice (Fig 1C–D). Furthermore, GSK672 decreased alcohol-induced hepatic steatosis and caused a significant reduction in hepatic TG and cholesterol content (Fig 1E–H). Alcohol feeding did not induce hepatic sterol regulatory element-binding protein 1c (SREBP-1c) or lipogenic genes sterol CoA desaturase 1 (SCD1) and fatty acid synthetase (FASN), but repressed hepatic fatty acid oxidation genes peroxisomal proliferator-activated receptor α (PPAR α), carnitine palmityoltransferase 1 (CPT1) and PPARγ-coactivator 1α (PGC1α) (Fig 1I). Reduced hepatic SREBP1c gene expression in ALD is consistent with a previous report (Zhang et al., 2018), suggesting that alcohol-induced hepatic steatosis may be independent of upregulation of hepatic lipogenic gene network. GSK672 did not affect the expression of the lipogenic genes nor the fatty acid oxidation. Alcohol-fed mice also showed increased plasma TG concentration, which tended to be lower in the GSK672-treated group (p=0.2) (Fig 1J). Plasma cholesterol concentration was similar between all groups (Fig 1K). In summary, these results suggest that gut restricted ASBT inhibitor is effective in attenuating alcohol feeding-induced hepatic steatosis and liver injury, and these beneficial effects may be independent of regulation of the lipogenic and fatty acid oxidation gene network.
Figure 1. GSK672 treatment decreases hepatic steatosis and injury in the alcohol-fed mice.
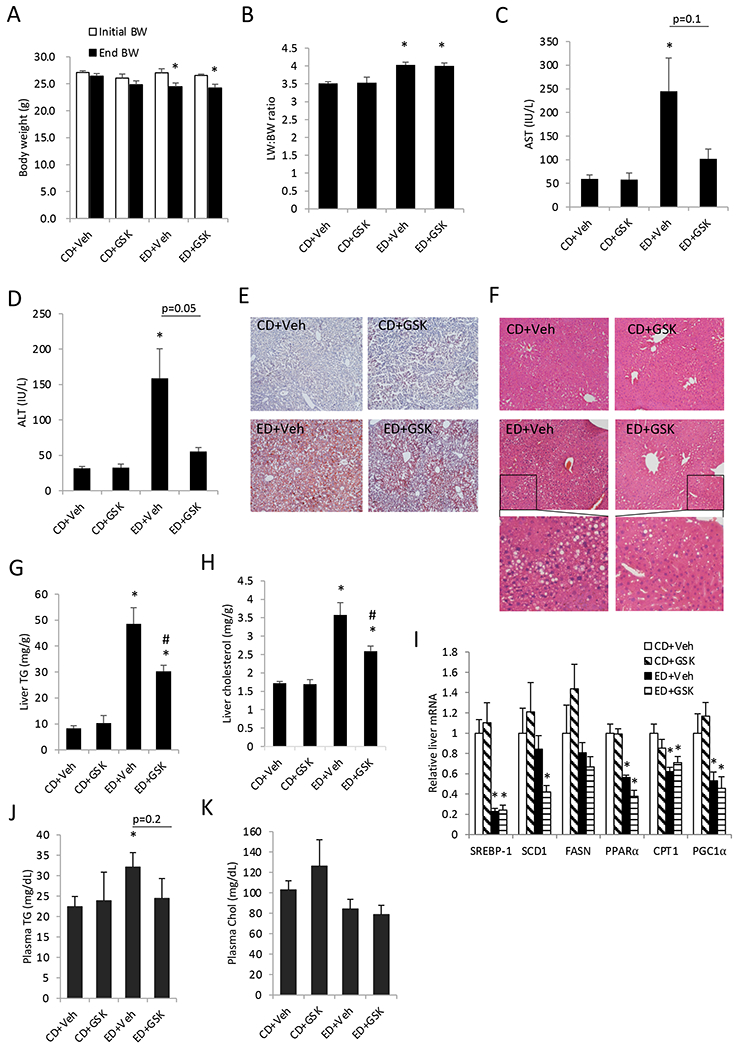
Male C57BL/6J mice were fed control diet (CD) or alcohol diet (ED) and treated with vehicle (Veh) or GSK672 (4 mg/kg/day) as described in the “Materials and Methods”. A. Body weight before and after alcohol feeding. B. Liver weight (LW): body weight (BW) ratio. C. serum AST concentration. D. Serum ALT concentration. E. Representative liver Oil Red O staining. F. Representative liver H&E staining. G. Liver triglyceride (TG) content. H. Liver total cholesterol content. I. Liver mRNA expression. J. Plasma TG concentration. K. Plasma total cholesterol concentration. All results are expressed as mean±SEM (n=5-6). “*”, vs. CD+Veh. “#”, vs. ED+Veh.
GSK672 treatment prevents alcohol-induced intestinal bile acid accumulation and serum bile acid elevation.
Analysis of bile acid metabolism revealed that alcohol feeding did not alter fecal bile acid excretion or hepatic bile acid content (Fig 2A–B), but significantly decreased gallbladder bile acid content and increased small intestinal bile acid content (Fig 2C–D). GSK672 treatment increased fecal bile acid loss (Fig 2A), and decreased gallbladder and small intestinal bile acid content (Fig 2C–D). Furthermore, serum bile acid concentration was elevated in the alcohol-fed mice (p=0.07) and was reduced upon GSK672 treatment (p=0.15) (Fig 2E). Given the relatively large variation of serum bile acid concentration in the alcohol-fed group, we further analyzed the association between serum bile acid levels and serum AST levels in this group of mice. The results showed that serum bile acid concentration was strongly and positively associated with serum AST in alcohol-fed mice (Fig 2F), suggesting liver injury-associated serum bile acid elevation in the alcohol-fed mice.
Figure 2. GSK672 treatment decreases small intestinal and serum bile acid concentration in the alcohol-fed mice.
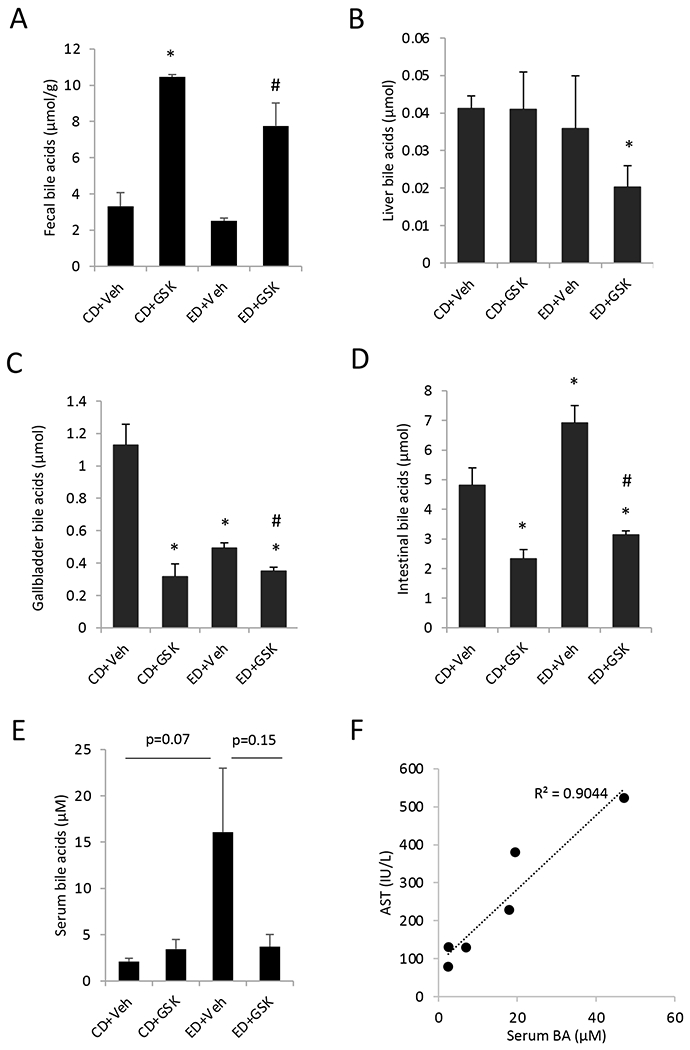
Male C57BL/6J mice were fed control diet (CD) or alcohol diet (ED) and treated with vehicle (Veh) or GSK672 (4 mg/kg/day) as described in the “Materials and Methods”. A. Fecal bile acids. Feces were collected from 3 cages per experimental condition (2 mice/cage). B-D. Total bile acids in liver, gallbladder and small intestine. The values are total bile acid content in the whole liver, gallbladder, and whole small intestine with its content. E. Serum bile acid concentration. Body weight before and after alcohol feeding. F. Correlation between serum AST and serum bile acids in the ED+Veh group. All results are expressed as mean±SEM (n=5-6). “*”, vs. CD+Veh. “#”, vs. ED+Veh.
Gene expression analysis revealed that alcohol feeding led to significantly reduced OSTα, OSTβ and ileal bile acid-binding protein (I-BABP) expression and unaltered ASBT or FXR expression in the distal ileum (Fig 3A). Interestingly, fibroblast growth factor 15 (FGF15), which is induced by FXR to mediate hepatic CYP7A1 inhibition, was strongly repressed in the distal ileum of the alcohol-fed mice (Fig 3A). Upon GSK672 treatment, these FXR target genes were further decreased (Fig 3A), which correlated with decreased small intestinal bile acid content (Fig 2D). Unexpectedly, hepatic CYP7A1 and sterol 12α-hydroxylase (CYP8B1) mRNA expression was significantly repressed in the alcohol-fed mice, which cannot be explained by reduced ileal FGF15 and hepatic small heterodimer partner (SHP) expression or unaltered intrahepatic bile acid content (Fig 2B, 3A, 3B). As expected, GSK672 treatment increased hepatic CYP7A1 in the CD group and the ED group (Fig 3B). Such GSK672-mediated changes were less pronounced in the alcohol-fed mice possibly due to significantly altered baseline expression of these genes (Fig 3B). Taken together, these results suggest that alcohol feeding is associated with significantly impaired FXR activity in the distal ileum despite elevated small intestinal bile acid content in alcohol-fed mice. GSK672 treatment decreases intestinal and serum bile acid levels without restoring ileal FXR activity.
Figure 3. Alcohol feeding is associated with impaired ileal FXR target gene expression and reduced hepatic CYP7A1 gene expression.
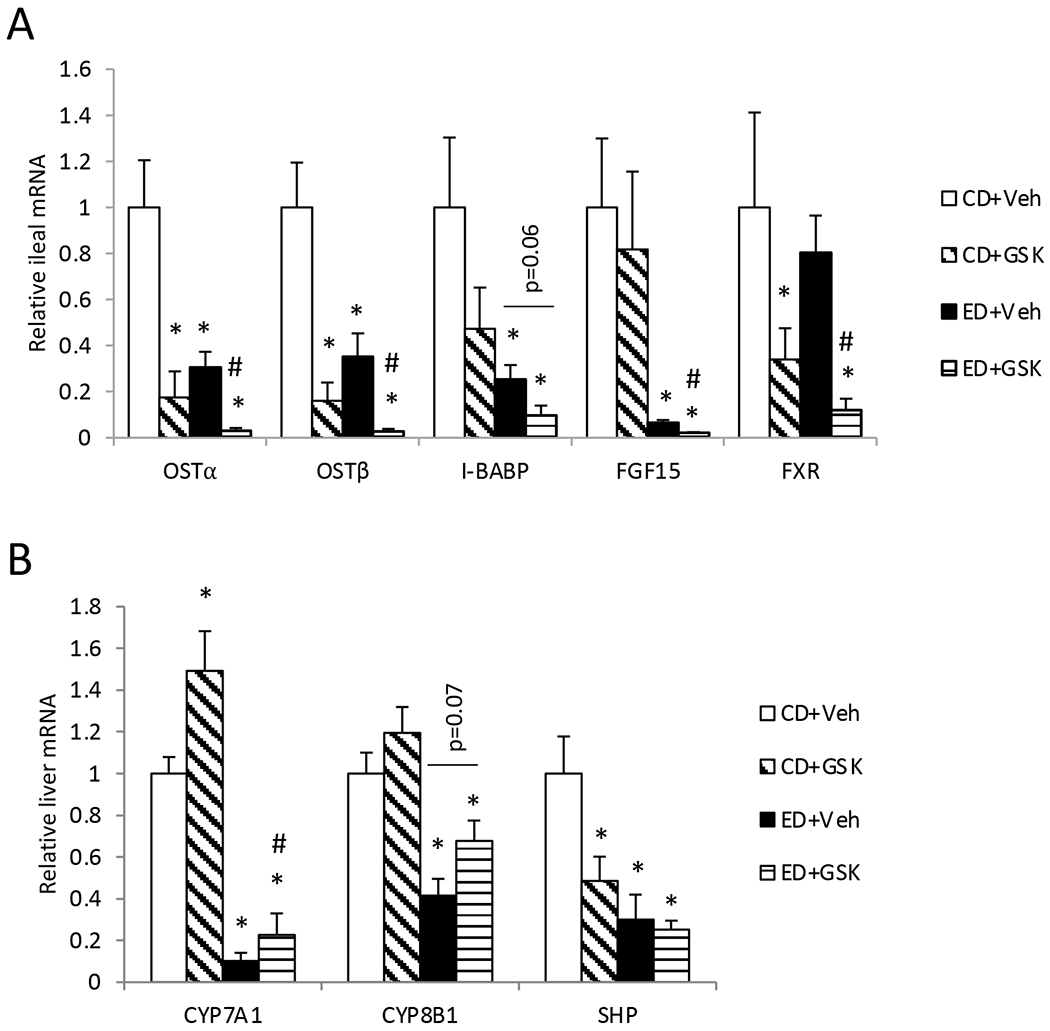
Male C57BL/6J mice were fed control diet (CD) or alcohol diet (ED) and treated with vehicle (Veh) or GSK672 (4 mg/kg/day) as described in the “Materials and Methods”. A. Ileal mRNA expression. A ~1 cm segment of the distal ileum was used for mRNA expression measurement by real-time PCR. B. Liver mRNA expression. All results are expressed as mean±SEM (n=5-6). “*”, vs. CD+Veh. “#”, vs. ED+Veh.
Hepatic MRP3 and MRP4 induction contributes to elevated serum bile acid levels in alcohol-fed mice.
Due to the lack of intrahepatic bile acid accumulation to explain serum bile acid elevation in alcohol-fed mice, we further measured hepatic expression of bile acid transporters. We found that bile salt export pump (BSEP), the apical bile acid efflux transporter, was not altered in the alcohol-fed mice (Fig 4A). In contrast, basolateral bile acid uptake transporter sodium-taurocholate co-transporting peptide (NTCP) was down-regulated by ~50% in the alcohol-fed mice (Fig 4B). Organic anion transporting polypeptide 1A1 (OATP1A1) and OATP1B2, which mediate basolateral bile acid uptake in mice (Slijepcevic et al., 2017), were also downregulated in alcohol-fed mice (Fig 4C–D). Interestingly, the basolateral bile acid efflux transporter multidrug resistance-associated protein 3 (MRP3) and MRP4 were induced in the alcohol-fed mice (Fig 4E–F).
Figure 4. Basolateral bile acid efflux transporters MRP3 and MRP4 are induced in the alcohol-fed mice.
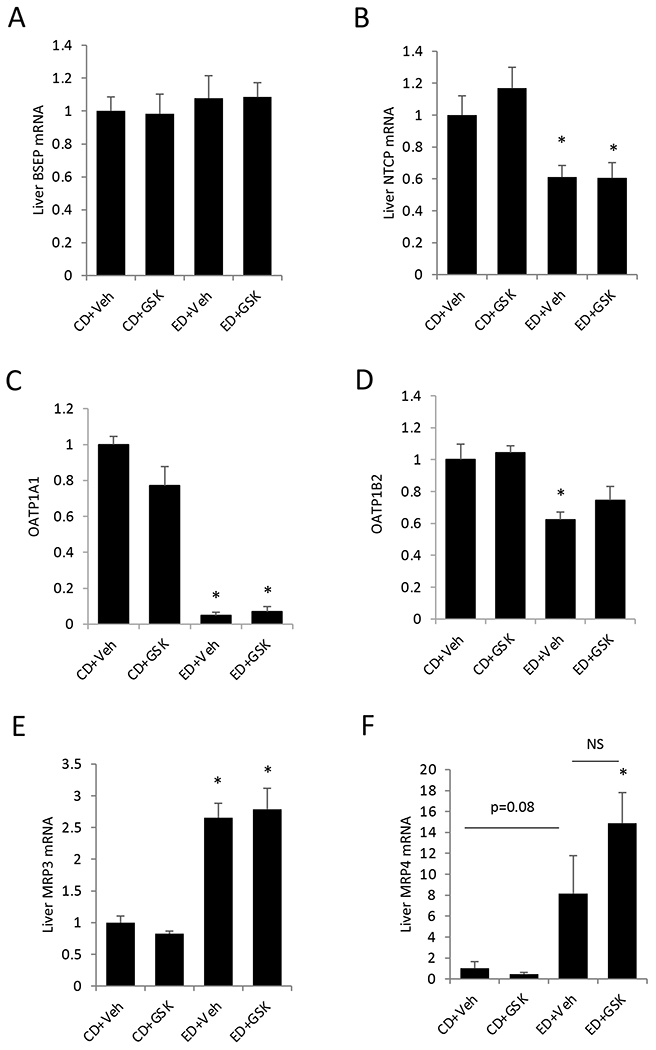
Male C57BL/6J mice were fed control diet (CD) or alcohol diet (ED) and treated with vehicle (Veh) or GSK672 (4 mg/kg/day) as described in the “Materials and Methods”. Relative liver mRNA expression is expressed as mean±SEM (n=5-6). “*”, vs. CD+Veh. “#”, vs. ED+Veh. NS. not significant.
Given the close association of serum bile acids and serum AST in the alcohol-fed mice, we further determined the association of MRP3 and MRP4 expression with serum bile acids and serum AST. Interestingly, we found that serum bile acids are positively correlated with hepatic MRP3 and MRP4 expression (Fig 5A–B), which further suggest that induction of MRP3 and MRP4 act as the underlying cause of serum bile acid elevation upon alcohol feeding. Furthermore, positive and strong association between MRP3 and MRP4 expression and serum AST was also observed in the alcohol-fed mice (Fig 5C–D). In contrast, serum bile acids and AST showed poor association with hepatic NTCP expression despite downregulated NTCP expression in the alcohol-fed mice (Fig 5E–F). Taken together, these results suggest that hepatic MRP3 and MRP4 induction mediates serum bile acid elevation in the alcohol-fed mice.
Figure 5. Hepatic MRP3 and MRP4 expression strongly and positively correlates with serum bile acid concentration and serum AST in the alcohol-fed mice.
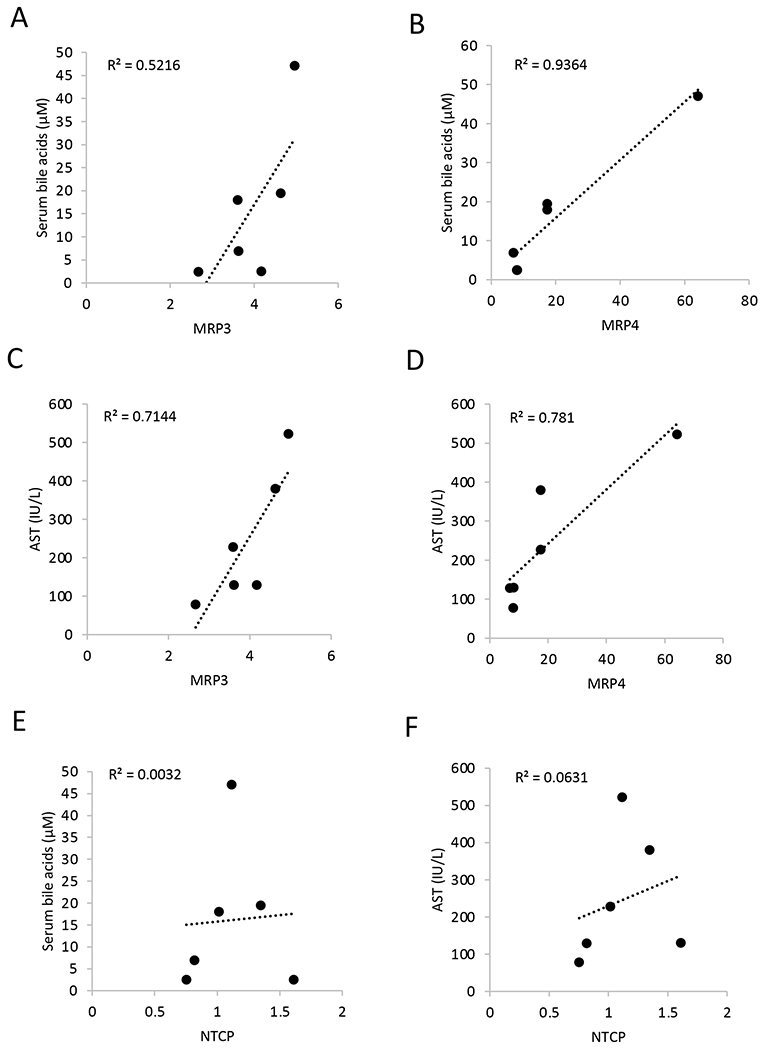
Male C57BL/6J mice were fed alcohol diet (ED) and treated with vehicle (Veh) as described in the “Materials and Methods”. Correlation between relative liver MRP3, MRP4, and NTCP mRNA expression and serum bile acid concentration and serum AST concentration were illustrated. (n=6)
GSK672 treatment increases deoxycholic acid (DCA) abundance and bile acid pool hydrophobicity.
Given the significant impact of alcohol feeding on bile acid metabolism and FXR activity, we further analyzed the effects of alcohol feeding and GSK672 on biliary and small intestinal bile acid composition. We found that although alcohol feeding significantly increased bile acid pool and altered bile acid metabolizing genes, the composition of the biliary bile acids was generally unaltered in the alcohol-fed mice (Fig 6A–B). Biliary bile acid pool consisted of less than 1% of unconjugated bile acids (not shown), which was not further analyzed. In the small intestine, alcohol feeding did not cause a significant shift in the hydrophobic to hydrophilic bile acid abundance (Fig 6C–D). Compared to the biliary bile acid pool, small intestinal bile acid pool consisted of ~10% unconjugated bile acids (Fig 6C–E), which may reflect the presence of bile acid metabolizing bacteria in the small intestine. Alcohol feeding did not alter the ratio of total conjugated bile acids and unconjugated bile acids (Fig 6F). Interestingly, GSK672 treatment significantly decreased biliary tauro-muricholic acid (T-MCA) abundance and increased tauro-DCA content without altering tauro-cholic acid (T-CA) content (Fig 6A–B). In the small intestine, GSK672 treatment also decreased T-MCA content and increased both T-DCA and DCA content (Fig 6C). As a result, GSK672 treatment increased the unconjugated bile acid abundance in the small intestinal bile acid pool (Fig 6F). These changes suggest that GSK672 treatment decreased total bile acid pool but retained more hydrophobic bile acids in the enterohepatic circulation.
Figure 6. GSK672 treatment increases DCA abundance in biliary bile acid pool and small intestine bile acid pool.
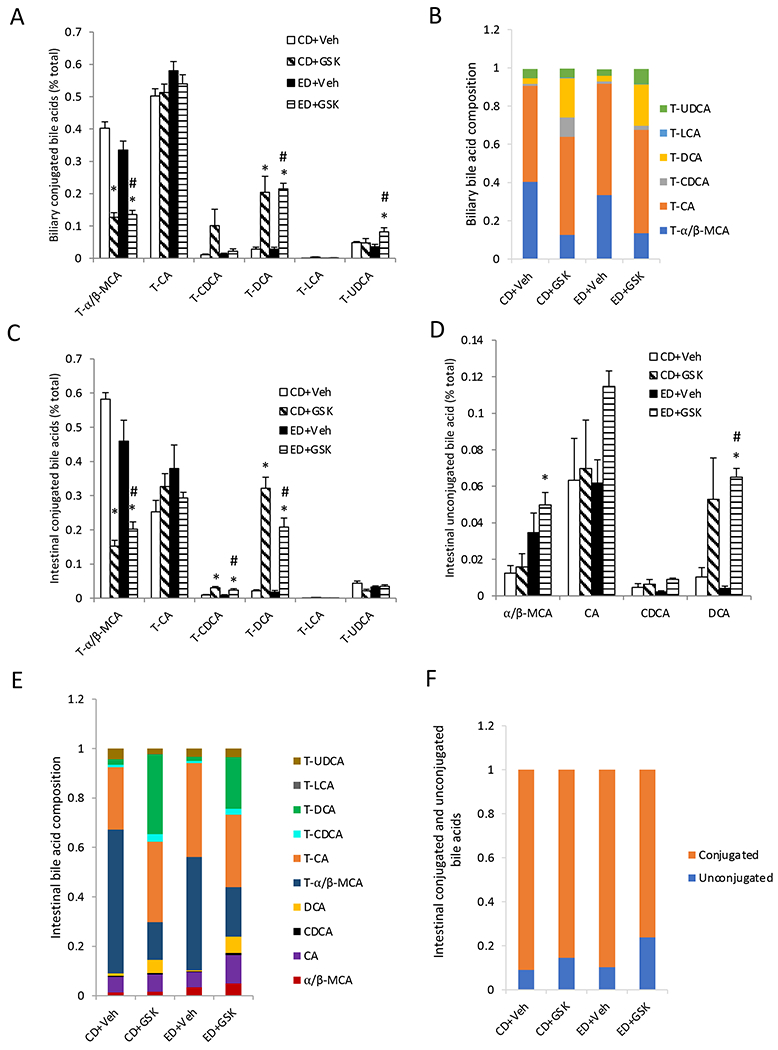
Male C57BL/6J mice were fed control diet (CD) or alcohol diet (ED) and treated with vehicle (Veh) or GSK672 (4 mg/kg/day) as described in the “Materials and Methods”. Results are expressed as mean±SEM (n=5-6). “*”, vs. CD+Veh. “#”, vs. ED+Veh.
Discussion
A major finding from this study is that treatment of a gut restricted ASBT inhibitor GSK672 effectively attenuates steatosis and liver injury in a chronic-plus-binge (Gao-Binge) ALD mouse model. These findings not only suggest that blocking intestinal bile acid reabsorption may be a potential therapeutic approach for ALD treatment, but also provides new mechanistic insights on the relationship between bile acid dysregulation and ALD. We found that alcohol feeding significantly increased small intestinal bile acid content. Such bile acid changes are consistent with findings from several other studies conducted in mouse models (Donepudi et al., 2018, Huang et al., 2020, Hartmann et al., 2018). Interestingly, we found that FXR target genes involved in bile acid transport OSTα/OSTβ and I-BABP were paradoxically reduced in the distal ileum upon alcohol feeding. When bile acid levels increase in small intestine, FXR induces OSTα/OSTβ and I-BABP and represses ASBT expression in order to decrease enterocyte bile acid uptake and promote enterocyte bile acid efflux (Dawson et al., 2009). The lack of such adaptive responses to higher intestinal bile acid content suggests that alcohol consumption also impaired ileal FXR function in mice. Currently, how dysregulation of intestinal bile acid homeostasis and impaired ileal FXR signaling contribute to the pathogenesis of ALD is incompletely understood. It has been reported that intestinal FXR deletion, but not hepatocyte FXR deletion, exacerbates alcoholic liver disease in mice (Zhang et al., 2018, Huang et al., 2020), suggesting that impaired intestinal FXR function plays a pathogenic role in ALD. Intestinal FXR is required to induce genes involved in preventing ileal bacteria overgrowth and intestinal epithelial injury (Inagaki et al., 2006). Decreased intestinal FXR function down-regulates OSTα and OSTβ, which could slow trans-intestinal bile acid transport to contribute to elevated bile acid abundance in the small intestine of the alcohol-fed mice. These changes could further exacerbate alcohol-induced gut epithelial barrier dysfunction (Szabo, 2015, Ferrebee et al., 2018). In this study, we showed that GSK672 treatment prevented small intestinal bile acid accumulation but further decreased ileal FXR target gene expression in the alcohol-fed mice. Therefore, this pharmacological model helps separate the effect of ileal FXR activity and intestinal bile acid pool and provides new evidence that decreasing intestinal bile acid accumulation without restoring ileal FXR activity can be beneficial in ALD. Blocking ileal bile acid re-absorption is expected to have metabolic impacts beyond the small intestine, and the mechanisms that contribute to improved ALD remain to be further delineated in future studies.
The robust observation of serum bile acid elevation and its close correlation with alcohol-induced liver injury marker in mice and humans suggest that serum bile acid elevation is a characteristic feature and potential serological marker of ALD. We found that alcohol feeding did not cause intrahepatic bile acid accumulation, indicating that elevated serum bile acid concentration was unlikely a direct result of cholestatic changes in the livers of the alcohol-fed mice. In contrast, we found that hepatic NTCP and OATPs were significantly down-regulated and MRP3 and MRP4 were induced in the alcohol-fed mice. Notably, the expression levels of MRP3 and MRP4 showed a strong and positive correlation with serum bile acid concentration, suggesting that MRP3 and MRP4 induction may play an important role in serum bile acid elevation in alcohol-fed mice. In contrast, NTCP did not show a negative correlation with serum bile acid levels despite being downregulated in ALD livers. Patients with alcoholic hepatitis also showed increased hepatic MRP4 expression (Brandl et al., 2018), suggesting that activation of basolateral bile acid efflux rather than the presence of cholestasis may significantly contribute to serum bile acid elevation in ALD patients. In normal physiology, hepatic first pass extraction of portal bile acids is highly efficient, resulting in significantly lower systemic bile acid concentration. Therefore, modest changes in basolateral bile acid uptake/excretion may result in significant elevation of bile acid concentration in the systemic circulation. This may explain the elevated circulating bile acids in the absence of intrahepatic bile acid accumulation in alcohol-fed mice. In addition, gallbladder bile acid content was significantly lower and small intestinal bile acid content was significantly higher in alcohol-fed mice. Increased small intestinal bile acid content was reported in both chronic alcohol-fed mice and chronic-plus-binge ALD mice (Hartmann et al., 2018), suggesting that such change is not likely a result of acute alcohol binge. One possibility is that alcohol metabolism in the gut, via mechanisms remain to be determined, slows transintestinal bile acid re-uptake because ileal I-BABP, OSTα and OSTβ expression was significantly decreased. Reduced ileal bile acid recycling and decreased hepatic basolateral bile acid uptake may further cause decreased trans-hepatic bile acid secretion into the bile, which may explain decreased gallbladder bile acid content without intrahepatic bile acid accumulation in the alcohol-fed mice. Reduced ileal FGF15 may also decrease gallbladder refilling in alcohol-fed mice to contribute to such changes (Choi et al., 2006). However, it should be noted that these tissue bile acid changes were measured at a steady state. Experiments that directly determine biliary bile acid secretion rate and ileal bile acid uptake kinetics are needed to better define the alcohol impact on bile acid metabolism and transport. Currently, the mechanisms underlying MRP3 and MRP4 induction in ALD is not well understood. MRP3 and MRP4 are well-established target genes of PXR (Teng et al., 2003, Zollner et al., 2007). It is possible that injury-associated metabolites may activate PXR to induce MRP3 and MRP4, resulting in a close correlation between serum bile acids and transaminase in the absence of hepatic bile acid accumulation. However, PXR is known to have very broad ligand specificity. ALD-associated hepatic metabolites that cause PXR activation remain to be defined. It should be noted that GSK672 decreased serum bile acid concentration without affecting hepatic MRP3 and MRP4 expression, suggesting that the effect may be due to blocked intestinal bile acid re-absorption.
Despite the significantly improved hepatic steatosis and liver injury upon GSK672 treatment, the molecular mechanisms mediating these benefits are still not well understood. Although the metabolic impact of ASBT inhibitor has not been studied in experimental ALD previously, the effects of ASBT inhibitors have been investigated in the settings of diabetes and fatty liver diseases whereby ASBT inhibitors improve hepatic organelle function, lipid catabolism, and insulin sensitivity (Rao et al., 2016, Wang et al., 2020, Wu et al., 2013). However, given the different disease etiology, how ASBT inhibitor impacts hepatic lipid metabolism in ALD remains to be investigated. In addition, the potential effects of GSK672 on gut microbiome and gut barrier function have not been explored. Compared to small intestine, large intestine has much higher bacteria abundance. Alcohol consumption in both humans and mice results in dysbiosis in the large intestine, which contributes to increased gut permeability and endotoxin translocation to the liver and systemic circulation (Szabo, 2015, Parlesak et al., 2000, Bala et al., 2014). Bile acid concentration is lower in the large intestine where bacterial enzymes deconjugate bile acids to produce secondary bile acids. Bile acids are bacteriostatic agents that play a key role in limiting gut bacteria growth (Inagaki et al., 2006, Hofmann and Eckmann, 2006). This effect is likely more prominent in the terminal ileum than in the large intestine because of the significantly higher ileal bile acid concentration. However, GSK672 treatment not only decreases small intestinal bile acids but also increases bile acid load in the large intestine. Furthermore, GSK672 significantly increased intestinal conjugated and unconjugated DCA, which possesses strong inhibitory activity of bacteria growth among different bile acid species (Watanabe et al., 2017). Although gut microbiome is not investigated in this study, it is tempting to speculate that ileal ASBT inhibition and subsequently increased bile acid load in the large intestine may exert a negative effect on bacteria overgrowth and possibly alter gut microbiome. Given that the gut-liver axis plays a critical role in the pathogenesis of ALD, the potential impact and implications of ASBT inhibition on gut microbiome remain to be investigated in future study.
Hepatic CYP7A1 was strongly inhibited upon alcohol feeding although alcohol-fed mice showed lower hepatic SHP and intestinal FGF15 and no apparent intrahepatic bile acid accumulation. Furthermore, intestinal FGF15 as well as other FXR target genes OSTs and I-BABP were paradoxically reduced upon alcohol feeding in the presence of elevated bile acids in the small intestine. These findings suggest that alcohol feeding may somehow disrupt the tight bile acid feedback regulation of bile acid synthesis via FXR independent mechanisms that remain to be identified. Human ALD patients show lower serum C4 levels (Brandl et al., 2018), which is indicative of hepatic CYP7A1 downregulation. ALD is associated with hepatic inflammation and inflammatory cytokines are known inhibitors of hepatic CYP7A1 expression (Miyake et al., 2000). However, it has also been shown that chronic alcohol feeding for 8 weeks without alcohol binge before tissue collection resulted in increased hepatic CYP7A1 expression and decreased ileal FGF15 expression in mice (Hartmann et al., 2018). This may imply the possibility that the strong CYP7A1 downregulation in our model may be an acute effect of alcohol binge that temporarily dissociates hepatic CYP7A1 expression from hepatic and ileal FXR activity, while hepatic CYP7A1 expression may be elevated during chronic alcohol feeding that negatively correlates with reduced hepatic and ileal FXR signaling. Given that mice were sacrificed 9 hours after the last alcohol binge while controls were gavaged with isocaloric maltose dextrin that is rapidly digested and absorbed as glucose, differential nutrient status during this period may also cause altered hepatic CYP7A1 expression because CYP7A1 expression is highly sensitive to rapid nutrient and insulin stimulation (Li et al., 2006, Li et al., 2012). Because bile acid homeostasis is significantly disrupted in ALD and circulating bile acid concentration closely correlates with liver injury marker, mechanisms altering hepatic CYP7A1 expression in various ALD models may be further investigated in future studies.
CYP7A1 deficient mice were more susceptible to alcohol-induced liver injury, while CYP7A1 transgenic mice were protected against alcohol-induced liver injury (Donepudi et al., 2018), suggesting that the strong downregulation of hepatic CYP7A1 in the alcohol-fed mice may play a pathogenic role in ALD. This defect was partially corrected by GSK672 treatment in the alcohol-fed mice. Furthermore, GSK672 also significantly increased DCA abundance in the bile acid pool. This is possibly a result of increased CYP8B1 expression that produces more CA, which is subsequently converted to DCA by gut bacterial enzymes. The potential contribution of altered gut microbiome to increased DCA synthesis remains to be determined. Alcohol feeding did not significantly shift the hydrophobic and hydrophilic bile acid abundance except a modest but non-significant decrease of T-MCA and increased T-CA in the small intestine bile acid pool. However, given the significant differences between human and mouse bile acid pool composition, the significance of bile acid composition changes observed in mice may not be extrapolated to humans.
In summary, our study demonstrates that inhibition of ileal ASBT is effective in attenuating hepatic steatosis and liver injury in an experimental ALD mouse model. These hepatic improvements are associated with decreased intestinal bile acid accumulation and lower serum bile acid concentration. ASBT inhibitors have been under active basic and clinical investigation for treating cholestasis and fatty liver disease (Rao et al., 2016, Newsome et al., 2020, Baghdasaryan et al., 2016). ASBT inhibitor treatment is generally well-tolerated in humans (Tiessen et al., 2018). However, a recent clinical study showed that ASBT inhibitor monotherapy did not offer therapeutic benefits in NASH patients (Newsome et al., 2020). Whether ASBT inhibitor offers clinical benefits in ALD patients remains to be determined. In experimental ALD, targeting FXR or FGF19 signaling has also been reported to attenuate ALD (Hartmann et al., 2018, Iracheta-Vellve et al., 2018). Future studies focusing on delineating the causative relationship between bile acid metabolism and alcohol-induced liver injury may improve our understanding of the role of bile acids in the pathogenesis of ALD and the potential of targeting bile acid metabolism and signaling for ALD treatment.
Financial support:
This study is supported in part by NIH grant 1R01 DK117965-01A1 (T.L.) and U01 AA024733, R37 AA020518, R01 DK102142, and R01 AG072895 (W.X.D).
Abbreviations
- ALD
Alcohol-related liver disease
- SREBP-1c
Sterol regulatory element-binding protein 1c
- SCD1
Sterol CoA desaturase 1
- FASN
Fatty acid synthetase (FASN)
- PPAR a
Peroxisomal proliferator-activated receptor a
- CPT1
Carnitine palmityoltransferase 1
- PGC1a
PPARg-coactivator 1a
- FXR
farnesoid x receptor
- ASBT
apical sodium-dependent bile acid transporter
- OSTα
organic solute transporter α
- OSTβ
organic solute transporter β
- GSK672
GSK2330672
- AST
Aspartate aminotransferase
- ALT
alanine aminotransferase
- CYP7A1
cholesterol 7α-hydroxylase
- FGF15
fibroblast growth factor 15
- I-BABP
ileal bile acid-binding protein
- CYP8B1
sterol 12α-hydroxylase
- SHP
small heterodimer partner
- BSEP
bile salt export pump
- NTCP
sodium-taurocholate co-transporting peptide
- MRP3
multidrug resistance-associated protein 3
- MRP4
multidrug resistance-associated protein 4
- PXR
pregnane x receptor
- DCA
deoxycholic acid
- T-MCA
tauro-muricholic acid
- T-CA
tauro-cholic acid
- OATP
organic-anion-transporting polypeptide
Reference
- BAGHDASARYAN A, FUCHS CD, OSTERREICHER CH, LEMBERGER UJ, HALILBASIC E, PAHLMAN I, GRAFFNER H, KRONES E, FICKERT P, WAHLSTROM A, STAHLMAN M, PAUMGARTNER G, MARSCHALL HU & TRAUNER M 2015. Inhibition of Intestinal Bile Acid Absorption Improves Cholestatic Liver and Bile Duct Injury in a Mouse Model of Sclerosing Cholangitis. J Hepatol. [DOI] [PubMed] [Google Scholar]
- BAGHDASARYAN A, FUCHS CD, OSTERREICHER CH, LEMBERGER UJ, HALILBASIC E, PAHLMAN I, GRAFFNER H, KRONES E, FICKERT P, WAHLSTROM A, STAHLMAN M, PAUMGARTNER G, MARSCHALL HU & TRAUNER M 2016. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol, 64, 674–81. [DOI] [PubMed] [Google Scholar]
- BALA S, MARCOS M, GATTU A, CATALANO D & SZABO G 2014. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One, 9, e96864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERTOLA A, MATHEWS S, KI SH, WANG H & GAO B 2013. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc, 8, 627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRANDL K, HARTMANN P, JIH LJ, PIZZO DP, ARGEMI J, VENTURA-COTS M, COULTER S, LIDDLE C, LING L, ROSSI SJ, DEPAOLI AM, LOOMBA R, MEHAL WZ, FOUTS DE, LUCEY MR, BOSQUES-PADILLA F, MATHURIN P, LOUVET A, GARCIA-TSAO G, VERNA EC, ABRALDES JG, BROWN RS JR., VARGAS V, ALTAMIRANO J, CABALLERIA J, SHAWCROSS D, STARKEL P, HO SB, BATALLER R & SCHNABL B 2018. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol, 69, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI M, MOSCHETTA A, BOOKOUT AL, PENG L, UMETANI M, HOLMSTROM SR, SUINO-POWELL K, XU HE, RICHARDSON JA, GERARD RD, MANGELSDORF DJ & KLIEWER SA 2006. Identification of a hormonal basis for gallbladder filling. Nat Med, 12, 1253–5. [DOI] [PubMed] [Google Scholar]
- DAWSON PA, HAYWOOD J, CRADDOCK AL, WILSON M, TIETJEN M, KLUCKMAN K, MAEDA N & PARKS JS 2003. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem, 278, 33920–7. [DOI] [PubMed] [Google Scholar]
- DAWSON PA, LAN T & RAO A 2009. Bile acid transporters. J Lipid Res, 50, 2340–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONEPUDI AC, FERRELL JM, BOEHME S, CHOI HS & CHIANG JYL 2018. Deficiency of cholesterol 7alpha-hydroxylase in bile acid synthesis exacerbates alcohol-induced liver injury in mice. Hepatol Commun, 2, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERREBEE CB, LI J, HAYWOOD J, PACHURA K, ROBINSON BS, HINRICHS BH, JONES RM, RAO A & DAWSON PA 2018. Organic Solute Transporter alpha-beta Protects Ileal Enterocytes From Bile Acid-Induced Injury. Cell Mol Gastroenterol Hepatol, 5, 499–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO B & BATALLER R 2011. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology, 141, 1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOODWIN B, JONES SA, PRICE RR, WATSON MA, MCKEE DD, MOORE LB, GALARDI C, WILSON JG, LEWIS MC, ROTH ME, MALONEY PR, WILLSON TM & KLIEWER SA 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell, 6, 517–26. [DOI] [PubMed] [Google Scholar]
- HARTMANN P, HOCHRATH K, HORVATH A, CHEN P, SEEBAUER CT, LLORENTE C, WANG L, ALNOUTI Y, FOUTS DE, STARKEL P, LOOMBA R, COULTER S, LIDDLE C, YU RT, LING L, ROSSI SJ, DEPAOLI AM, DOWNES M, EVANS RM, BRENNER DA & SCHNABL B 2018. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology, 67, 2150–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFMANN AF & ECKMANN L 2006. How bile acids confer gut mucosal protection against bacteria. Proc Natl Acad Sci U S A, 103, 4333–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG M, KONG B, ZHANG M, RIZZOLO D, ARMSTRONG LE, SCHUMACHER JD, CHOW MD, LEE YH, JOSEPH LB, STOFAN M, ZHANG L & GUO GL 2020. Enhanced alcoholic liver disease in mice with intestine-specific farnesoid X receptor deficiency. Lab Invest, 100, 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INAGAKI T, CHOI M, MOSCHETTA A, PENG L, CUMMINS CL, MCDONALD JG, LUO G, JONES SA, GOODWIN B, RICHARDSON JA, GERARD RD, REPA JJ, MANGELSDORF DJ & KLIEWER SA 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab, 2, 217–25. [DOI] [PubMed] [Google Scholar]
- INAGAKI T, MOSCHETTA A, LEE YK, PENG L, ZHAO G, DOWNES M, YU RT, SHELTON JM, RICHARDSON JA, REPA JJ, MANGELSDORF DJ & KLIEWER SA 2006. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A, 103, 3920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IRACHETA-VELLVE A, CALENDA CD, PETRASEK J, AMBADE A, KODYS K, ADORINI L & SZABO G 2018. FXR and TGR5 Agonists Ameliorate Liver Injury, Steatosis, and Inflammation After Binge or Prolonged Alcohol Feeding in Mice. Hepatol Commun, 2, 1379–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI T, FRANCL JM, BOEHME S, OCHOA A, ZHANG Y, KLAASSEN CD, ERICKSON SK & CHIANG JY 2012. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem, 287, 1861–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI T, KONG X, OWSLEY E, ELLIS E, STROM S & CHIANG JY 2006. Insulin regulation of cholesterol 7alpha-hydroxylase expression in human hepatocytes: roles of forkhead box O1 and sterol regulatory element-binding protein 1c. J Biol Chem, 281, 28745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIETHKE AG, ZHANG W, SIMMONS J, TAYLOR AE, SHI T, SHANMUKHAPPA SK, KARNS R, WHITE S, JEGGA AG, LAGES CS, NKININ S, KELLER BT & SETCHELL KD 2015. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAKE JH, WANG SL & DAVIS RA 2000. Bile acid induction of cytokine expression by macrophages correlates with repression of hepatic cholesterol 7alpha-hydroxylase. J Biol Chem, 275, 21805–8. [DOI] [PubMed] [Google Scholar]
- NEWSOME PN, PALMER M, FREILICH B, SHEIKH MY, SHEIKH A, SARLES H, HERRING R, MANTRY P, KAYALI Z, HASSANEIN T, LEE HM, AITHAL GP & VOLIXIBAT IN ADULTS STUDY, G. 2020. Volixibat in adults with non-alcoholic steatohepatitis: 24-week interim analysis from a randomized, phase II study. J Hepatol, 73, 231–240. [DOI] [PubMed] [Google Scholar]
- PARLESAK A, SCHAFER C, SCHUTZ T, BODE JC & BODE C 2000. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol, 32, 742–7. [DOI] [PubMed] [Google Scholar]
- RAO A, HAYWOOD J, CRADDOCK AL, BELINSKY MG, KRUH GD & DAWSON PA 2008. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A, 105, 3891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO A, KOSTERS A, MELLS JE, ZHANG W, SETCHELL KD, AMANSO AM, WYNN GM, XU T, KELLER BT, YIN H, BANTON S, JONES DP, WU H, DAWSON PA & KARPEN SJ 2016. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med, 8, 357ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLIJEPCEVIC D, ROSCAM ABBING RLP, KATAFUCHI T, BLANK A, DONKERS JM, VAN HOPPE S, DE WAART DR, TOLENAARS D, VAN DER MEER JHM, WILDENBERG M, BEUERS U, OUDE ELFERINK RPJ, SCHINKEL AH & VAN DE GRAAF SFJ 2017. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology, 66, 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABO G 2015. Gut-liver axis in alcoholic liver disease. Gastroenterology, 148, 30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TENG S, JEKERLE V & PIQUETTE-MILLER M 2003. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab Dispos, 31, 1296–9. [DOI] [PubMed] [Google Scholar]
- TIESSEN RG, KENNEDY CA, KELLER BT, LEVIN N, ACEVEDO L, GEDULIN B, VAN VLIET AA, DORENBAUM A & PALMER M 2018. Safety, tolerability and pharmacodynamics of apical sodium-dependent bile acid transporter inhibition with volixibat in healthy adults and patients with type 2 diabetes mellitus: a randomised placebo-controlled trial. BMC Gastroenterol, 18, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y, GUNEWARDENA S, LI F, MATYE DJ, CHEN C, CHAO X, JUNG T, ZHANG Y, CZERWINSKI M, NI HM, DING WX & LI T 2020. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis. Nat Commun, 11, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATANABE M, FUKIYA S & YOKOTA A 2017. Comprehensive evaluation of the bactericidal activities of free bile acids in the large intestine of humans and rodents. J Lipid Res, 58, 1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU Y, AQUINO CJ, COWAN DJ, ANDERSON DL, AMBROSO JL, BISHOP MJ, BOROS EE, CHEN L, CUNNINGHAM A, DOBBINS RL, FELDMAN PL, HARSTON LT, KALDOR IW, KLEIN R, LIANG X, MCINTYRE MS, MERRILL CL, PATTERSON KM, PRESCOTT JS, RAY JS, ROLLER SG, YAO X, YOUNG A, YUEN J & COLLINS JL 2013. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem, 56, 5094–114. [DOI] [PubMed] [Google Scholar]
- ZHANG M, KONG B, HUANG M, WAN R, ARMSTRONG LE, SCHUMACHER JD, RIZZOLO D, CHOW MD, LEE YH & GUO GL 2018. FXR deletion in hepatocytes does not affect the severity of alcoholic liver disease in mice. Dig Liver Dis, 50, 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZOLLNER G, WAGNER M, FICKERT P, SILBERT D, GUMHOLD J, ZATLOUKAL K, DENK H & TRAUNER M 2007. Expression of bile acid synthesis and detoxification enzymes and the alternative bile acid efflux pump MRP4 in patients with primary biliary cirrhosis. Liver Int, 27, 920–9. [DOI] [PubMed] [Google Scholar]