

Abstract
Mitochondria dysfunction occurs in the aging brain as well as in several neurodegenerative disorders and predisposes neuronal cells to enhanced sensitivity to neurotoxins. 3-nitropropionic acid (3-NP) is a naturally occurring plant and fungal neurotoxin that causes neurodegeneration predominantly in the striatum by irreversibly inhibiting the tricarboxylic acid respiratory chain enzyme, succinate dehydrogenase (SDH), the main constituent of the mitochondria respiratory chain complex II. Significantly, although 3-NP-induced inhibition of SDH occurs in all brain regions, neurodegeneration occurs primarily and almost exclusively in the striatum for reasons still not understood.
In rodents, 3-NP-induced striatal neurodegeneration depends on the strain background suggesting that genetic differences among genotypes modulate toxicant variability and mechanisms that underlie 3-NP-induced neuronal cell death. Using the large BXD family of recombinant inbred (RI) strains we demonstrate that variants in Ccnd1 - the gene encoding cyclin D1 - of the DBA/2J parent underlie the resistance to 3-NP-induced striatal neurodegeneration. In contrast, the Ccnd1 variant inherited from the widely used C57BL/6J parental strain confers sensitivity. Given that cellular stress triggers induction of cyclin D1 expression followed by cell-cycle re-entry and consequent neuronal cell death, we sought to determine if the C57BL/6J and DBA/2J Ccnd1 variants are differentially modulated in response to 3-NP. We confirm that 3-NP induces cyclin D1 expression in striatal neuronal cells of C57BL/6J, but this response is blunted in the DBA/2J. We further show that striatal-specific alternative processing of a highly conserved 3’UTR negative regulatory region of Ccnd1 co-segregates with the C57BL/6J parental Ccnd1 allele in BXD strains and that its differential processing accounts for sensitivity or resistance to 3-NP. Our results indicate that naturally occurring Ccnd1 variants may play a role in the variability observed in neurodegenerative disorders involving mitochondria complex II dysfunction and point to cyclin D1 as a possible therapeutic target.
Keywords: 3-nitropropionic acid, mouse, neurodegeneration, striatum, BXD, cell cycle, Cyclin D1
1. Introduction
Mitochondrial respiratory chain complex II dysfunction is associated with neuronal degeneration in several diseases, including Huntington’s disease (HD) and multiple system atrophy (MSA), and is likely to play a pivotal role in the progression of these disorders (Reiner et al., 2011; Stefanova and Wenning, 2016; Arun et al., 2016). Livestock poisoning by either plants or fungi containing 3-nitropropionic acid (3-NP), a naturally occurring neurotoxin that irreversibly inhibits succinate dehydrogenase (SDH), the main constituent of the mitochondrial respiratory chain complex II (Alston et al., 1977; Coles et al., 1979) has been extensively documented (Hamilton et al., 2000). Outbreaks of human poisonings have also been reported, and similar to what has been observed in animals, 3-NP ingestion by humans leads to severe clinical features resembling HD, as well as significant neuronal cell death majorly limited to the striatum (Lui et al., 1989, Ludolph et al., 1991; Hamilton et al., 2000; Birkelund et al., 2021).
In animal models, systemic administration of 3-NP leads to extensive neuronal cell death, almost exclusively in the striatum. Chronic or acute administration of 3-NP in rodents and primates has been extensively used to reproduce clinical features of HD, closely recapitulating the motor, biochemical and pathological features of the disorder (Brouillet et al., 1995; Palfi et al., 1996; Brouillet et al., 2005; Bonsi et al., 2006; Túnez et al., 2010), while the first models of MSA that fully recapitulate the disorder, were generated by 3-NP administration in mice transgenic for oligodendrocyte-specific overexpression of alpha-synuclein, underscoring the possible role of complex II dysfunction in the disease (Stefanova et al., 2005; Ubhi et al., 2009; Kuzdas-Wood et al., 2014, Foti et al., 2019).
Despite the extensive body of work in animal and cellular models, the mechanisms leading to 3-NP-induced neuronal cell death are still poorly understood. 3-NP inhibition of SDH leads to impaired mitochondrial bioenergetics, oxidative stress, and loss of ATP (Alston et al., 1977; Coles et al., 1979), triggering a cascade of intracellular events that ultimately result in cell death (Túnez et al., 2010). However, although 3-NP-induced inhibition of SDH occurs in all brain regions, neurodegeneration occurs primarily in the striatum (Brouillet et al., 1998; Bizat et al., 2003; Bonsi et al., 2006). The underlying basis for preferential striatal vulnerability to 3-NP is currently unknown.
In both rats and mice 3-NP-induced striatal neurodegeneration depends on the strain employed (Alexi et al., 1998; Brouillet et al., 2005; Schauwecker et al., 2005), suggesting that genetic differences between specific strains interact and modify the mechanisms that underlie the toxin-induced neuronal cell death. So far, attempts to identify genetic factors that influence 3-NP-induced neurotoxicity consisted in empirically testing the function of a battery of genes/factors, most of them already known to be involved in either mitochondria function, or neurodegenerative disorders, and in particular in HD. Unfortunately, this approach was not fruitful since inactivation of the examined genes either had no effect (Hanbury et al., 2003; Junyent et al., 2012), or led to moderate increase or decrease in 3-NP toxicity in wild-type mice in a C57BL/6 (B6) genetic background (Mealer et al., 2013; Glat et al., 2016). Similarly, overexpression of genes known to be involved in neuroprotection were also shown to partially protect against 3-NP toxicity (Martínez-Turrillas et al., 2012; Jovicic et al., 2013). Neither of these approaches however is likely to unveil naturally occurring genetic variants that could explain strain-specific resistance or susceptibility, since either inactivation or overexpression of the identified genes is not only out of the normal range of natural variation but also often interfere with unrelated pathways.
Forward genetics using linkage analyses provides an unbiased strategy to screen for relevant susceptibility and resistance genes in vivo. Recombinant inbred (RI) strains have been an invaluable resource for genetic mapping of Mendelian quantitative traits in the mouse over the past several decades. Conventional mouse RI strains are developed by crossing two inbred parental strains and sequentially mating the resulting siblings for over 20 generations to ensure that they are at least 99% inbred (Williams et al., 2001; Mulligan et al., 2017). For our study, the RI strains utilized, are derived from a cross (X) of the parental C57BL/6J (B6) and DBA/2J (D2) strains, therefore referred to as BXDs. This panel of strains has been densely genotyped and the genomes of both parental strains have been sequenced at a high level of genomic coverage. Over 6 million sequence variants segregating between B6 and D2 and among the BXD set of strains have now been identified, including nearly 12,000 missense mutations, over 11,000 small insertions and deletions in coding regions, and nearly 16,000 copy number variants spanning one or more exons (Wang et al., 2016; Ashbrook et al., 2021). In addition, gene expression has been profiled in multiple brain regions, including whole brain and striatum for the parental strains and BXD strains using traditional microarray platforms and next generation RNA sequencing. These expression resources allow for the identification of genes whose expression is controlled by a sequence variant within or near the cognate gene — a cis expression quantitative trait locus (eQTL) (Schadt et al., 2003; Chesler et al., 2005). The success of using BXDs for high throughput genetic screening is underscored by recent endeavors such as the identification of specific genes and gene variants that confer immunological resistance to Chlamydia (Myiari et al., 2007; Myiari et al., 2012), modulate cognitive aging (Neuner et al., 2016), extend longevity (Merkwirth et al., 2016) or influence cognition in Alzheimer’s disease (Heuer et al., 2020).
Using sub-acute 3-NP administration, we have found that while 3-NP treatment induces extensive striatal lesions in the BXD parental B6 strain, the parental D2 strain is completely resistant, with no signs of striatal damage. By analyzing striatal integrity/neuronal cell death in a panel of BXD strains treated with 3-NP, we found that resistance to 3-NP is a highly heritable trait that maps to a 1.2 Mb region in Chromosome 7. Among the possible genes, we identified Ccnd1, the gene encoding cyclin D1, as a potential major modulator of 3-NP induced striatal neurodegeneration.
2. Materials and Methods
2.1. Animals
C57BL/6J (B6), DBA/2J (D2) and BXD recombinant inbred strains were bred and maintained in a pathogen-free facility at UTHSC. Mice were maintained on a 12hr light/dark cycle, fed standard chow (Harlan Teklad 7912) and provided with water and food ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee at University of Tennessee Health Science Center (Memphis, TN) and are in accordance with the National Institutes of Health guide for the care and use of Laboratory animals.
2.2. 3-nitropropionic acid administration
3-nitropropionic acid (Sigma N5636) was prepared fresh weekly in phosphate-buffered saline at a concentration of 5 mg/ml and stored at 4 °C. Mice were injected intraperitoneally (IP) with 50 mg/kg 3-NP twice a day for a period of 3 to 5 days, leading to cumulative doses of 300 to 500 mg of 3-NP.
2.3. Assay for succinate dehydrogenase (SDH) activity
SDH activity was determined as described (Ahmad et al., 2018) with some modifications. Briefly, cerebral cortex, striatum and whole brain from two-month-old untreated and 3-NP treated D2 and B6 female mice were homogenized in ice-cold isolation buffer (10 mM Tris pH 7.4, 1 mM EDTA, 320 mM sucrose). The homogenate was then centrifuged at 1,300 g for 10 min at 4 °C. The post-nuclear supernatant was used for SDH determinations. Protein concentrations were determined using the Bradford method. For SDH activity, each reaction mixture contained 100 μg of protein sample in phosphate buffer containing 3 mM ferricyanide (FeIII) as electron acceptor. The reaction was initiated by addition of 50 mM succinate and SDH activity was determined by calculating the rate of decrease in OD at 420 nm, which corresponds to the rate of potassium ferrocyanide (FeII) formation. As controls, protein extracts were incubated in the same reaction buffer in the absence of succinate or in the presence of 50 mM malonate, an SDH inhibitor.
2.4. Histological analyses
Brains were dissected and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for one week; incubated for 24 hours at 4 °C in PBS containing 0.25 M sucrose, 0.2 M glycine; dehydrated; cleared with toluene; and embedded in paraffin. Paraffin blocks were sectioned at 7 μm, mounted in superfrost slides (Fisher) and stained with haematoxylin and eosin (H&E).
2.5. Immunohistochemistry
Slides were deparaffinized, rehydrated, and incubated with 0.3% H202 in methanol for 20 min to quench endogenous peroxidase. Sections were then washed with PBS, blocked for 1 hr with 4% BSA, 0.2% Triton X-100 in PBS, and incubated at 4 °C overnight with primary rabbit polyclonal anti-DARPP-32 (AB 1656, Chemicon) or rabbit monoclonal anti-cyclin D1 (MA5–14512, Invitrogen), in 0.4% BSA; 0.2% Triton X-100 in PBS. For the detection of cyclin D1, antigen retrieval was performed by microwaving the slides for 15 min in a microwave safe dish containing antigen-unmasking solution (H-3300, Vector), according to manufacturer’s instructions. In all cases, following primary antibody incubation, slides were washed three times with PBS, and primary antibody detection was carried out using the Vector ABC Elite kit according to manufacturer’s instructions, followed by incubation with DAB brown substrate (BD Biosciences). Slides immunostained for cyclin D1 were counterstained with hematoxylin QS (H-3404, Vector).
2.6. Counts of Cyclin D1 positive neurons
Cyclin D1 positive neurons were quantified by counting the total number of neurons and number of cyclin D1-stained nuclei in cross sections of the striatum of untreated and 3-NP-treated D2 and B6 mice (n = 3 for each strain and condition). For each case, four images per striatal area of both hemispheres from at least two independently stained forebrain coronal sections were captured using 10X objective and divided into quadrants. Neuronal counts were performed using Image J, as described (Dietrich et al., 2017).
2.7. Quantitative Trait Loci mapping
Quantitative trait locus (QTL) mapping was performed using the GeneNetwork web resource (www.genenetwork.org). Interval mapping was performed by genotype regression against trait values in combination with whole genome permutation to determine the significance threshold for the association between genotype and trait expression. A permutation test establishes genome-wide significance criteria of p <0.05. Three-month old females (n = 2 or 3 per strain) were treated with 3-NP as described above, always paired with at least one sensitive age-matched B6 female as a positive control. Mice were weighed daily during the course of the 3-NP treatment and assessed daily for behavioral changes. Sensitivity or resistance to 3-NP was scored by histological analyses: presence of lesion with overt neuronal cell loss was scored as sensitive (−1) while lack of lesion with no overt signs of neuronal cell loss was scored as resistant (+1).
2.8. Gene expression data
Gene expression data of B6, D2 and their BXD progeny strains were derived from the open access GeneNetwork and GeneNetwork2 databases (gn1.genenetwork.org; gn2.genenetwork.org). Gene expression data for the mouse Ccnd1 mRNA encompassing the last coding exon/proximal 3’UTR and mid-to distal portions of its 3’UTR in striatum, hippocampus, prefrontal cortex and whole brain were derived from the HBP Rosen Striatum M430v2 PDNN clean, Hippocampus Consortium M430v2 (Jun06) RMA, VCU BXD PFC Sal M430 2.0(Dec06) RMA, and INIA Brain mRNA M430 RMA databases, respectively.
Gene expression data was restricted to probe sets that showed 100% gene specificity, which was verified using the Basic Local Alignment Search Tool (BLAST).
Log2 values were converted to normal scale values prior to ratio determination and graphing.
2.9. Semi-quantitative RT-PCR
Semi-quantitative RT-PCR was performed as described (Dietrich et al., 2011). Briefly, brains were dissected and total RNA was isolated from cortex and striatum of 3 month-old B6 (n=3) and D2 (n=3) female mice using Trizol reagent (GIBCO-BRL), according to the manufacturer’s instructions. For reverse transcription, samples were first treated with RQ1 DNAse (to remove DNA contaminants), 1μg total RQ1 DNAse-treated RNA was annealed with random hexamers and first strand cDNA synthesis was carried out using SuperScript IV First Strand Synthesis System (Invitrogen). For each pair of primers, 1.5μl of each RT reaction (equivalent to 75 ng of starting mRNA) was used for semi-quantitative PCR amplification. All PCR reactions were carried out in the same conditions: 45 sec denaturation at 94 °C, 45 sec annealing at 61 °C, and 1 min extension at 72 °C using the pairs of primers specific for Ccnd1: C3 5’-TCCTCAACGACCGGGTGCTG-3’ and C4 5’-GGAAGCGGTCCAGGTAGTTCA-3’, which amplify a 200 bp product, and MD1 5’-GAGGAAAATTAGGGGACTCCAAA-3’ and MD4 5’-GGCATCTGTAAATACACTCTATGA-3’, which amplify a 388 bp product.
For internal control, 18S rRNA was PCR amplified as above, using the primers 18Sfor 5’-GGTGGTGGTGCATGGCCGTT-3’ and 18Srev 5’-GCAGCCCCGGACATCTAAGG-3’ which amplify a 200 bp product.
PCR reactions products were fractionated in 2% agarose gels and stained with ethidium bromide. Images were captured in high definition and band intensities were analyzed by Image J.
2.10. MicroRNA binding sites
To identify the location of putative microRNA (miR) binding sites in the Ccnd1 mRNA 3’UTR, we used miRDB (mirdb.org) an online database for miR target prediction. The whole sequence of the mouse Ccnd1 mRNA 3’UTR (2676bp) was screened for potential miR binding sites, and the locations of sites with target scores above 85 were then annotated.
2.11. Statistics
Data presented are shown as mean ± SD. Data were derived from multiple independent experiments from distinct mice. Animal studies were performed without blinding of the investigator. No statistical method was used to predetermine sample size, but sample size was based on preliminary data and previous publications. In all experiments the differences were considered significant when p <0.05. Open access software was used in most cases. Student’s t-test was used for comparison of two groups.
3. Results
Although a large fraction of 3-NP studies has been done in rats, several studies have demonstrated that in both, rats and mice, 3-NP striatal neurotoxicity depends on the strain employed (Schauweker, 2005; Brouilllet et al., 2005). Taking this observation into account, we decided to investigate whether the BXD parental strains, B6 and D2, display differences in susceptibility to 3-NP toxicity, which, if so, would justify the continuation of the analyses using the BXD family.
3.1. 3-NP induces striatal neurodegeneration in B6 but not in the D2 strain
Two- to three-month-old D2 (n = 7) and B6 (n = 7) females were injected intraperitoneally (IP) with 50 mg/kg 3-NP twice a day and their behavior and weight monitored daily. After 3 to 4 days of systemic 3-NP administration (total cumulative dose of 300–400 mg/kg), all B6 females became runted, displayed reduced locomotive activity and a head tilt. In contrast, D2 females injected twice daily for a period of 5 consecutive days (total cumulative dose of 500 mg/kg) did not exhibit any abnormal behavior. Females from both strains exhibited a comparable weight loss of about 10% of their initial weight (Fig. S1A), indicating that both strains metabolized the drug similarly. Notably, striatal SDH activity was reduced at a similar rate in both strains, attaining almost complete inhibition of SDH activity after a cumulative 3-NP dose of 200 mg/kg (Fig. S1B), providing direct evidence that differences observed in the two strains are not due to differences in the bioavailability of 3-NP. All mice were sacrificed a few hours after the last injection, and brains were collected and processed for histological analyses. Analyses of unstained and H&E-stained coronal paraffin sections revealed extensive striatal lesions in the brains of all B6 females while no signs of lesions were observed in the brains of D2 females (Fig. 1A-D). Immunohistochemistry for DARPP-32 confirmed extensive medium spiny neuronal cell loss in the striatum of the B6 mice while most neurons were spared in the D2 strain (Fig. 1E, 1F). Importantly, similar results were obtained with age-matched males of each strain (n = 3), suggesting that the mechanism that confers sensitivity in B6 strain and resistance in D2 strain is conserved between males and females.
Fig. 1.
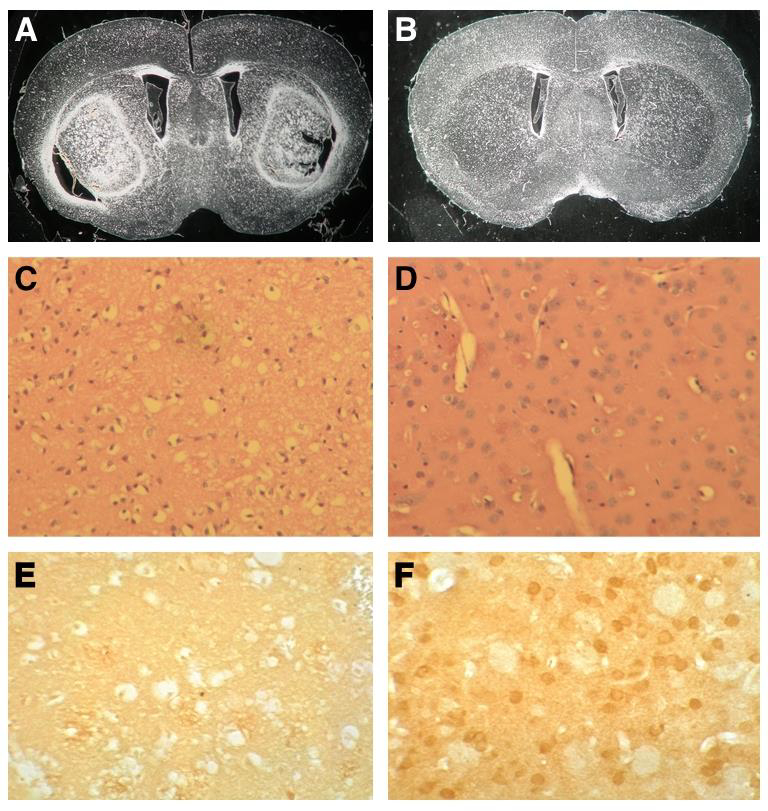
The D2 strain is resistant to 3-NP-induced striatal neurodegeneration. Dark field images of unstained coronal paraffin sections of B6 (A) and D2 (B) brains. Note that large striatal lesions are seen in B6 but not in D2. H&E-stained coronal paraffin sections spanning the striatum of B6 (C) and D2 (D). Note the extensive neuronal cell loss in B6 striatum. (E-F) Immunohistochemistry for DARPP-32. Extensive medium spiny neuronal cell loss is demonstrated by high magnification (20X) in B6 (E), while neurons are preserved in D2 (F).
To ascertain whether this differential response to 3-NP is maintained in adulthood, 12–15-month-old females and males of each strain were subjected to the same experimental procedure. Remarkably, adult B6 females (n = 7) and males (n = 4) exhibited an even more substantial sensitivity to 3-NP, with extensive striatal damage observed with cumulative doses as low as 200 mg/kg, while no lesions were observed in age-matched D2 females (n = 3) or males (n = 3) with cumulative doses as high as 500 mg/kg.
These data indicated that there is a clear phenotype distinction between the two strains, regardless of sex and age, and suggested that the use of BXDs for genetic screen to identify modifiers of 3-NP toxicity would likely lead to the identification of loci that confer resistance to 3-NP-induced striatal neurodegeneration.
3.2. Forward genetics identifies a 1.2 Mb interval in chromosome 7 that confers resistance to 3-NP neurotoxicity
In order to map potential neuroprotective loci, three-month-old females from 18 randomly selected BXD strains (n = 2 to 3 per strain) were IP injected with 50 mg/kg 3-NP twice a day for 5 consecutive days. For each experimental batch, at least one B6 female was also injected in parallel, as a positive control. Similar to the parental strains, all BXD strains exhibited comparable weight loss during the course of 3-NP administration, but variable behavior at endpoint, ranging from no behavioral abnormalities even with a cumulative dose of 500 mg/kg to severe compromise including runted body, reduced locomotive activity and a head tilt with doses as low as 300 mg/kg. Mice were sacrificed at day 5, and brains processed for paraffin embedding. Histological analyses of coronal brain sections spanning the striatal region revealed that 14 of the 18 strains showed no signs of neuronal loss, recapitulating the resistant phenotype of the parental D2 strain while the other 4 exhibited an identical outcome as the susceptible parental B6 strain, that is, a visible lesion characterized by extensive neuronal cell loss (Fig. 2) and (Fig. S2).
Fig. 2.

Resistance to 3-NP-induced striatal neurodegeneration is a heritable trait. (A-J) Representative images of coronal brain sections of sensitive (A,C,E,G,I) and resistant (B,D,F,H,J) BXD strains. (A-B) Dark field images of unstained coronal paraffin sections of the sensitive BXD 102 (A) and resistant BXD 60 (B) strains. (C-J) High magnification of H&E-stained coronal paraffin sections of the striatum from the sensitive BXD strains, BXD 102 (C), BXD 100 (E), BXD 43 (G), BXD 24 (I) and from the resistant BXD strains, BXD 60 (D), BXD 103 (F), BXD 27 (H), BXD 40 (J). Note the extensive cell death in the sensitive BXD strains and the preservation of neuronal integrity in the resistant BXD strains.
The heritability of the resistance trait allowed us to identify one strong QTL linked to 3-NP resistance in Chromosome 7, with a significant likelihood ratio statistic (LRS) score at 144.1–145.3 Mb interval (Fig. 3). This region contains 12 known genes (Table 1), and literature search pointed to Ccnd1 (the gene encoding cyclin D1) as the strongest candidate for differential 3-NP sensitivity in parental and BXD strains since aberrant cell cycle re-entry, marked by increased expression of cyclin D1, is a hallmark of neurodegeneration (Park et al., 2000; Akashiba et al., 2008; Liu et al., 2010; Folch et al., 2012; Pelegri et al.,2008; Frade and Ovejero-Benito, 2015).
Fig. 3.

Recombinant inbred strains identify a 1.2 Mb resistance locus in chromosome 7. Eighteen BXD strains were treated with 3-NP and monitored for the presence or absence of striatal lesions. Results were analyzed by WebQTL software (www.genenetwork.org) and interval mapping are shown as a blue line representing the LRS score across the entire mouse genome. A single significant LRS is present in chromosome 7. All data are accessible through the GeneNetwork web site (www.genenetwork.org; trait identification (ID) BXD 15959).
Table 1.
The 1.2-Mb QTL on chromosome 7 contains 12 known genes
| Symbol | Description | Mb Start |
|---|---|---|
| Shank2 | SH3/ankyrin domain gene 2 | 144.175519 |
| Cttn | cortactin | 144.435723 |
| Fadd | Fas-associated via death domain | 144.578322 |
| Tmem16a | transmembrane protein 16A | 144.588662 |
| Fgf3 | fibroblast growth factor 3 | 144.838611 |
| Fgf4 | fibroblast growth factor 4 | 144.861385 |
| Fgf15 | fibroblast growth factor 15 | 144.896531 |
| Oraov1 | oral cancer overexpressed 1 | 144.915193 |
| Ccnd1 | cyclin D1 | 144.929930 |
| Tpcn2 | two pore segment channel 2 | 145.253922 |
| Mrgprf | MAS-related GPR, member F | 145.300908 |
| Mrgprd | MAS-related GPR, member D | 145.314834 |
Significantly, haplotype analyses revealed that all the BXD strains carrying the D2 Ccnd1 allele were resistant to 3-NP-induced striatal neurodegeneration, while the BXD strains sensitive to 3-NP all carried the B6 Ccnd1 allele.
3.3. 3-NP induces Cyclin D1 expression in striatal neurons of the B6 strain
To evaluate the genetic differences between the two strains we first identified single nucleotide polymorphisms (SNPs) between the B6 and D2 Ccnd1 alleles (ENSMUSG00000070348) using a bioinformatics approach. This analysis revealed the presence of 39 sequence variants in the Ccnd1 allele between the two strains, most of them located either in introns or in the regulatory 3’UTR of the Ccnd1 gene, while the only variant located in the coding region is predicted to be silent. These observations suggested that differences in regulation of Ccnd1 mRNA expression, stability or translation could be the underlying basis for the differential sensitivity/resistance to 3-NP between the two strains.
It has been shown that in vitro and in vivo exposure to 3-NP leads to increased cyclin D1 expression in striatal neurons, and this induction precedes neuronal apoptotic cell death (Akashiba et al., 2008; Pelegri et al., 2008). Because most SNPs are located in the 3’UTR regulatory region of Ccnd1, we sought to determine if differential regulation of cyclin D1 in response to 3-NP could be the underlying cause of differential resistance/sensitivity between the two strains. To test this hypothesis, we performed immunohistochemistry for cyclin D1 in untreated and 3-NP treated B6 and D2 brain coronal sections spanning the striatal region, prior to overt neurodegeneration. Our analyses revealed a significant 3.5-fold increase in the number of cyclin D1 positive neuronal nuclei in the B6 striatum compared to the D2 strain in response to 3-NP administration (Fig. 4).
Fig. 4.
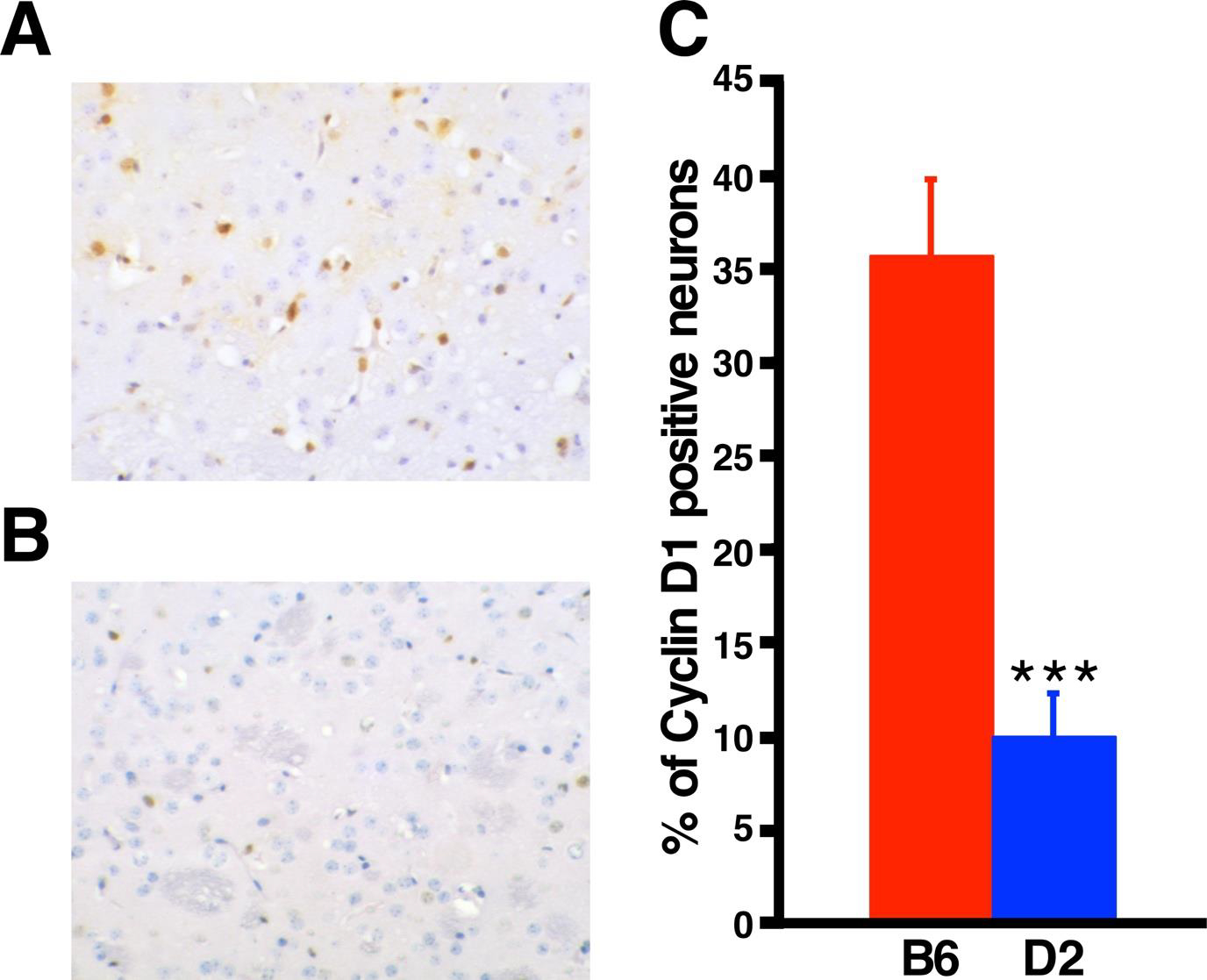
3-NP induces cyclin D1 expression in B6 but not in D2 striatal neurons. Representative immunohistochemistry for cyclin D1 in B6 (A) and D2 striatum (B). Positive signal is brown and negative nuclei are stained blue with Hematoxylin QS. (B) Graph representing percentage of cyclin D1 positive neurons in 3-NP-treated B6 (n = 4) and D2 (n = 4) striatum. Data are expressed as mean ± SD. ***p <0.001.
3.4. Ccnd1 mRNA 3’UTR is alternatively processed in the B6 striatum
To gain further insight into the possible mechanisms underlying the differential cyclin D1 induction, we obtained the data regarding Ccnd1 mRNA expression in the striatum of B6 and D2 strains from databases at GeneNetwork2 (gn2.genenetwork.org, see materials and methods) to determine whether basal levels of Ccnd1 expression differ between the two strains. Intriguingly, we found that Ccnd1 mRNA probes that recognized the last exon, proximal 3’UTR and mid 3’UTR showed similar striatal expression between B6 is and D2. In contrast, Ccnd1 mRNA probes that recognized the mid-to-distal portion of the 3’UTR showed a dramatic difference in striatal expression between B6 and D2, with this region demonstrating ~70% less abundance in B6 relative to D2 (Fig. 5). This observation prompted us to investigate if this phenomenon occurs in other brain regions as well. For this, we obtained the available data regarding Ccnd1 mRNA last exon and mid-to-distal 3’UTR expression in cerebral cortex, hippocampus, and whole brain of B6 and D2 strains from databases at GeneNetwork (gn2.genenetwork.org, see materials and methods). As shown in Fig. 5, in the cortex and hippocampus the mid-to-distal portion of the 3’UTR is marginally less represented in B6 Ccnd1 transcripts than in D2, and this difference is even lower in other brain regions, raising the possibility that Ccnd1 mRNA transcripts undergo differential post-transcriptional alternative processing preferentially in the striatum. The Affymetrix data was further confirmed by semi-quantitative RT-PCR in cortex and striatum mRNA derived from B6 and D2 mice, using pairs of primers specific for the Ccnd1 coding region and mid-to-distal 3’UTR (Fig. 6).
Fig. 5.
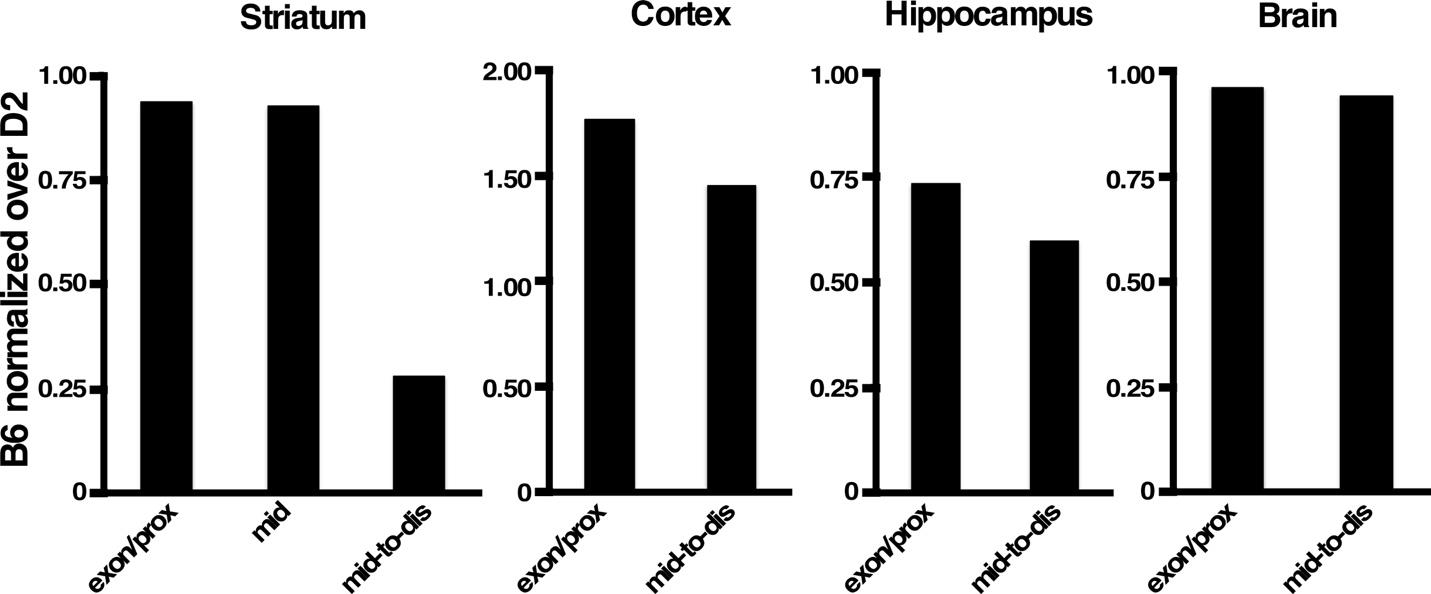
The mid-to-distal portion of Ccnd1 3’UTR mRNA is alternatively processed in B6 striatum. Affymetrix values of Ccnd1 expression (last exon, mid and mid-to-distal 3’UTR) in Striatum, Cortex, Hippocampus and whole brain in B6 mice were normalized over the corresponding Affymetrix values of D2. Exon/prox = exon 5 and proximal 3’UTR; mid = mid 3’UTR, mid-to-dis = mid-to-distal 3’UTR.
Fig. 6.
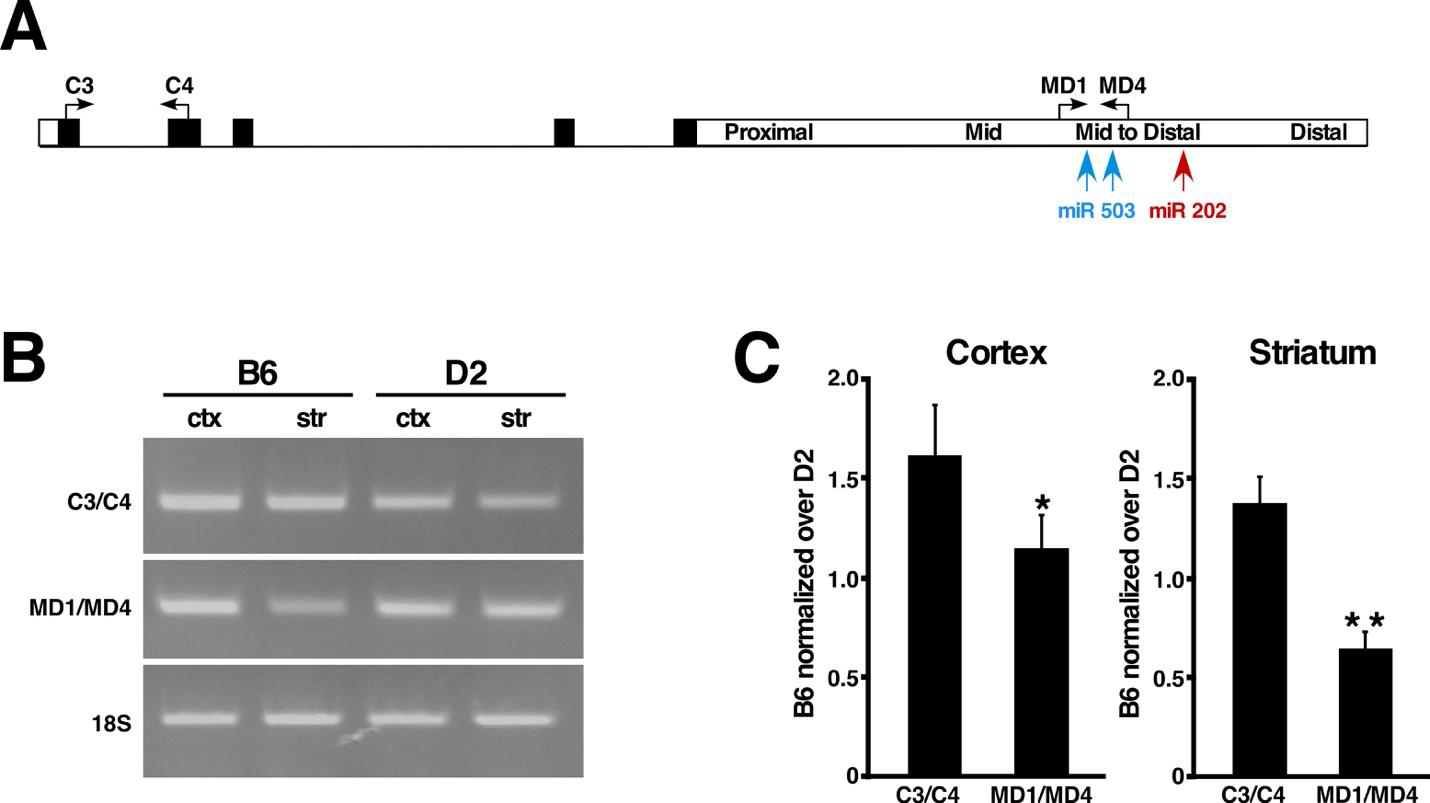
Semi-quantitative RT-PCR confirms preferential alternative processing of the mid-to-distal 3’UTR in B6 striatum. (A) Schematic representation of the Ccnd1 full length transcript. The positions and orientation of the oligonucleotides used for semi-quantitative RT-PCR are indicated by arrows. Black rectangles represent exons and white rectangle represent the 3’UTR. The positions of miR 503 and miR 202 are indicated in blue and red respectively. (B) Representative image of semi-quantitative RT-PCR of B6 and D2 Ccnd1 mRNAs from cortex (ctx) and striatum (str). 18S amplification was used as internal control. (C) Band intensities of semi-quantitative RT-PCR from B6 and D2 quantified by Image J and normalized over 18S band intensity. The resulting values of B6 were then normalized over D2 values. Data are expressed as mean ± SD. *p <0.01; **p <0.005.
3.5. The mid-to-distal Ccnd1 mRNA 3’UTR contains highly conserved negative regulatory elements
The mechanisms involved in the regulation of cyclin D1 expression have mostly been studied and defined in the context of cancer. In human cancer cell lines it is well established that expression of cyclin D1 is negatively regulated by small non-coding microRNAs (miRs) that bind to specific sites located in the mid-to-distal portion of the CCND1 mRNA 3’UTR (Sander et al., 2005; Jiang et al., 2009), raising the possibility that this regulatory function might be conserved in the mouse Ccnd1 mid-to-distal 3’UTR and play a role in regulating cyclin D1 expression in response to stress in neuronal cells. For this, we first performed sequence alignment between the mouse Ccnd1 3’UTR and human CCND1 3’UTR. Sequence alignment between the human and mouse 3’UTR revealed a high degree of homology (78%) between the mouse mid-to-distal portion and the corresponding portion of the human 3’UTR, while flanking sequences share less than 30% homology (Fig. S3). To investigate the distribution of miR binding sites along the 3’UTR of the mouse Ccnd1 gene, we then performed in silico analysis using the miRDB-MicroRNA target prediction website (www.mirdb.org). We found that binding sites for miR 503–5p, and miR 202–3p, which have been shown to negatively regulate the expression of human cyclin D1 and decrease cell proliferation in cancer cell lines (Jiang et al., 2016; Yi et al., 2016; Peng et al., 2019), are conserved in the mouse and are present exclusively in the mid-to-distal portion of the mouse Ccnd1 3’UTR (Fig. 6A, Fig. S3). Altogether, these observations suggest that the lack of negative regulatory elements in most of B6 Ccnd1 transcripts may be responsible for the high levels of 3-NP-induced cyclin D1 protein expression, while retention of negative regulatory elements in D2 Ccnd1 transcripts is sufficient to blunt the induction of cyclin D1 protein expression in response to 3-NP (Fig. 4).
3.6. Alternative processing of the mid-to-distal 3’UTR in the striatum is dictated by the parental origin of the Ccnd1 allele
To investigate whether the differential striatal alternative processing of the mid-to-distal 3’UTR is a heritable trait and if it is regulated in cis or in trans, we obtained the Affymetrix Ccnd1 expression data of 30 random BXD strains (see materials and methods) and organized the data according to the parental origin of their Ccnd1 alleles. Strikingly, similar to the B6 parental strain, all the BXD strains that carry the B6 Ccnd1 allele also display a significant relative reduced expression of the mid-to-distal 3’UTR specifically in the striatum while this region is mostly retained in other brain regions (Fig. 7). Likewise, similar to the parental D2 strain, the mid-to-distal Ccnd1 3’UTR is mostly retained in the striatum and other brain regions of all the BXD strains that carry the D2 Ccnd1 allele (Fig. 7). These data strongly indicate that cis-elements present in the Ccnd1 alleles are responsible for differential post-transcriptional processing of the mid-to-distal portion of the 3’UTR in the striatum.
Fig. 7.

Alternative processing of the mid-to-distal 3’UTR in the striatum co-segregates with the parental B6 Ccnd1 allele in BXDs. The graphs represent Affymetrix probe values of mid-to-distal 3’UTR over the last exon (exon 5) of Ccnd1 coding region derived from 30 BXDs, 18 carrying the parental D2 Ccnd1 allele (blue) and 12 carrying the parental B6 Ccnd1 allele (red). The data was organized and graphed according to the parental origin of the Ccnd1 allele.
In conclusion, our results strongly suggest that the D2 Ccnd1 variant underlies resistance to 3-NP-induced striatal neurodegeneration while the B6 Ccnd1 variant confers sensitivity and point to striatal-specific alternative post-transcriptional processing as a potential mechanism underlying strain and brain region selective 3-NP-induced neurotoxicity.
4. Discussion
In humans and animals, 3-NP exposure results in widespread impairment of energy metabolism throughout the brain but only the basal ganglia, and in particular the striatum (caudate and putamen) undergo selective neurodegeneration (Bizat et al., 2003; Brouillet et al., 2005; Bonsi et al., 2006). The mechanisms underlying regional selective neuronal cell loss are still poorly understood. Over the years, several hypotheses have been proposed, but the most widely accepted explanation is that glutamatergic input from the cortex predisposes striatal neurons to excitotoxic and oxidative stress damage, and that their GABAergic nature also contributes to their increased sensitivity (Alexi et al., 1998; Brouillet et al., 2005). Significantly though, in vitro studies have shown that Wistar rats’ striatal neurons are intrinsically more sensitive than cortical neurons to 3-NP-induced cell death (Akashiba et al., 2008), ruling out that glutamatergic input is required for their increased 3-NP sensitivity and suggesting instead that cell autonomous mechanisms may be important contributing factors. Consistent with this assumption, though 3-NP exposure in vitro induces cell death in both striatal and cortical neurons, the pathways leading to cell death differ between the two neuronal cell types. Notably, 3-NP exposure induces up-regulation of cyclin D1 and cell cycle-re-entry in striatal neurons but not in cortical neurons and pharmacological inhibition of cyclin D-dependent kinases 4 and 6 (CDK4/6) significantly reduces 3-NP-induced cell death in striatal neurons but has little effect in cortical neurons (Park et al., 2000; Akashiba et al., 2008). Though these results point to an important role of cyclin D1 and cell cycle re-entry in 3-NP-induced striatal neuronal cell death in vitro, the mechanisms leading to differential 3-NP-induced cyclin D1 upregulation in striatal neurons in vivo (Pelegri et al., 2008; Duran-Vilaregut et al., 2011), as well as its role in 3-NP-induced striatal neurodegeneration remain to be elucidated.
In this study we have shown that, contrary to the commonly used B6 mouse strain, the D2 strain is highly resistant to 3-NP-induced striatal neurodegeneration. This trait is highly heritable, and using unbiased forward genetics, we have found that a genetic variant located in a 1.2 Mb region in Chromosome 7 accounts for the differences in sensitivity between the two strains. In view of the literature linking cyclin D1, cell cycle re-entry and 3-NP-induced neurodegeneration, out of the 12 possible genes we identified Ccnd1 (the gene encoding cyclin D1) as a strong candidate. Our analyses showed that 3-NP induces cyclin D1 in B6 striatal neurons but not in D2 and that cis-elements present in the B6 Ccnd1 allele lead to striatal-specific post-transcriptional alternative processing of the Ccnd1 mRNA regulatory 3’UTR.
The mechanisms of regulation of cyclin D1 expression have so far been studied mostly in the context of cancer. CCND1 represents the second most frequently amplified locus in the human cancer genome (Beroukhim et al., 2010). Interestingly, in most tumors that exhibit elevated cyclin D1 levels, two types of transcripts are present due to alternative post-transcriptional processing and polyadenylation, with the long transcript containing the full-length 3’untranslated region (3’UTR) of the CCND1 mRNA and the short transcript lacking most of the mid and distal 3’UTR region (Xiong et al., 1991; Mayr and Bartel, 2009). Not only do cancer cells expressing predominantly the short transcript exhibit higher rates of proliferation than those expressing mostly the long transcript (Sander et al., 2005; Slotta-Huspenina et al., 2012), but genetic editing of the 3’UTR favoring preferential expression of the short transcript leads to acceleration of cell cycle re-entry and high rates of cell proliferation (Sander et al., 2005; Wang et al., 2018). Negative regulation of cyclin D1 protein levels is mediated through binding of small non-coding RNAs, miRs, to the mid/distal 3’UTR of the CCND1 long transcript, thereby reducing mRNA translation (Deshpande et al., 2009). Several miR binding sites have been identified in the mid-to-distal CCND1 3’ UTR (Deshpande et al., 2009; Jiang et al., 2009), and the essential negative regulatory function of some of them have been confirmed experimentally. For instance, it has been demonstrated that miR 503 and miR 202 bind to the mid-to-distal region of the CCND1 3’UTR, and that overexpression of either miR 503 or miR 202 reduce cyclin D1 protein levels in cancer cells and halt cell proliferation, while their downregulation increases cell proliferation (Long et al., 2015; Jiang et al., 2017; Qi et al., 2018; Jiang et al., 2016; Yi et al., 2016; Peng et al., 2019). While the mechanisms leading to reduced retention of the mid-to-distal 3’UTR of Ccnd1 transcripts in B6 striatal neuronal cells have yet to be established, the high degree of sequence conservation between the mouse and human mid-to-distal portion, including the miR binding sites, suggest that the function of this region is also conserved in the mouse, implying that in B6 Ccnd1 transcripts are likely to be more stable in the striatum, which could explain the increased cyclin D1 expression seen in B6 striatum in response to 3-NP.
In dividing cells, the first stage of the cell cycle, the G1 phase, is initiated by increased levels of members of the cyclin D family, followed by activation of CDKs and phosphorylation of the tumor suppressor protein retinoblastoma, which in turn leads to activation of the E2 promoter-binding factor (E2F) family of transcription factors. Active E2F in turn induces transcription of various genes involved in cell cycle, ultimately resulting in mitosis in dividing cells. In vitro and in vivo, increased cyclin D1 expression in striatal neurons in response to 3-NP leads to activation of cell cycle re-entry pathway (Park et al., 2000; Akashiba et al., 2008; Pelegri et al., 2008). However, in contrast to most cell types, in which cell cycle re-entry leads to cell proliferation, in neuronal cell cycle re-entry instead leads to apoptosis. This has been demonstrated in vitro, in several paradigms, including excitotoxicity, oxidative stress and DNA damage (Stefanis et al., 1999; Kruman et al., 2004; Zhang et al., 2020; Park et al., 1997; Park et al., 1998; Akashiba et al., 2008), as well as in vivo in mouse models of traumatic brain injury (Hilton et al., 2008; Kabadi et al., 2012a, Kabadi et al., 2012b; Kabadi and Faden, 2014). Notably, inhibition of cell cycle re-entry in all these paradigms exerts significant neuroprotective effect. It is likely therefore that the absence of cyclin D1 induction in the D2 strain prevents cell cycle re-entry, and this appears to be sufficient to prevent 3-NP induced neurodegeneration in this strain.
While the mechanisms leading to cell cycle re-entry and consequent apoptosis following 3-NP exposure are still far from being elucidated, the current knowledge points to 3-NP-induced oxidative stress and oxidative DNA damage as the initial trigger. One of the first and main intracellular effects of 3-NP is the generation of reactive oxygen species (ROS), including hydrogen peroxide (H2O2), superoxide anion and hydroxyl radicals, which are known to cause oxidative DNA damage (Liot et al., 2009). Notably, both oxidative damage and neuronal cell loss induced by 3-NP are reduced by antioxidants (Pedraza-Chaverri et al., 2009; Colle et al., 2009; Gao et al., 2015), while mutations in SDH subunits were shown to cause genomic instability (Owens et al., 2012). The mechanisms by which DNA damage elicits cell cycle re-entry in neurons is still not clear. One hypothesis is that post-mitotic neurons re-enter the cell cycle as part of their DNA repair response (Kim and Tsai, 2009; Tomashevski et al., 2010; Zhang et al., 2020). In support of this hypothesis, inhibition of cell cycle re-entry decreases DNA double strand break repair in neurons exposed to DNA damaging agents (Schwartz et al., 2007; Tomashevski et al., 2010). It is currently not known however why in some circumstances neuronal cell cycle re-entry leads to apoptotic cell death. One appealing possibility is that when DNA damage exceeds a certain threshold, due to the decreased baseline levels of DNA repair proteins in neurons, cell-cycle checkpoint responses are hindered and instead of DNA being repaired leading to cell cycle progression, neuronal cells enter the apoptotic pathway (Kim and Tsai, 2009; Schwartz et al., 2007).
Although our analyses indicate that strain-specific Ccnd1 genetic polymorphisms account for resistance or susceptibility to 3-NP-induced striatal neurodegeneration in mice, in terms of translatability it would be of great importance to determine if similar genetic modifiers of complex II deficiency-induced neurodegeneration also exist in humans. Genetically inherited complex II deficiency is rare, but even within a limited number of patients the age of onset and range of neurological abnormalities vary extensively despite similar reductions in enzymatic activity, with most patients displaying lesions in the basal ganglia in infancy while others display adult-onset cerebellar atrophy with no involvement of the basal ganglia (Jain-Ghai et al., 2013). Significantly, though the neurological findings vary between individuals, they are strikingly similar between family members (Jain-Ghai et al., 2013), suggesting that epigenetic factors may play an essential role in the age of onset and pathology caused by complex II deficiency. Likewise, the well documented outbreaks of human poisonings with 3-NP also resulted in variable neurological involvement, with bilateral basal ganglia lesions and consequent permanent dystonia observed in only about 50% of the cases (Peraica et al., 1999), further strengthening the likelihood that as in mice genetic factors play an essential role in conferring resistance to 3-NP-induced striatal lesions in humans. Whether polymorphisms in the CCND1 gene play a role in these differential outcomes or not remain to be determined. Genetic polymorphisms in CCND1 exist in the human population, with some polymorphisms linked to increased susceptibility to cancer or increased malignancy (Diehl, 2002; Knudsen et al., 2006; Wang et al., 2014), and others predicted to alter cyclin D1 function (Aftab et al., 2021), however their potential role in neurodegeneration has not been evaluated so far.
Our findings may also have implications to the pathology of Huntington’s Disease (HD). HD is an autosomal dominant neurodegenerative disorder caused by expansion of a CAG tract in the amino terminal portion of the protein Huntingtin (Walker, 2007). Although several genetic models of the disease have been generated over the past decades, none of them genetic models recapitulate the extensive striatal neuronal cell death of HD. In contrast, 3-NP models of HD recapitulate not only HD neuropathology but also biochemical and phenotypic manifestations and have been extensively used to investigate mechanisms of HD (Túnez et al., 2010). Importantly, biochemical studies in HD brain tissues have shown a consistent deficit in mitochondria complex II in the caudate and putamen, associated with 50–70% reduction in SDH activity (Gu et al., 1996; Browne et a., 1997). Importantly, restoration of complex II function abolishes cell death in genetic cellular models of HD, indicating that complex II deficits play a significant role in the disease pathology (Benchoua et a., 2006). Interestingly, evidence of cell cycle re-entry has been shown in cellular and animal models of HD, including elevated levels of cyclin D1 (Gines et al., 2003), cdk4 (Fernandez-Fernandez et al., 2011), cyclin B1, and activated cdk5 (Liu et al., 2015; Paoletti et al., 2008). Significantly, evidence of cell cycle re-entry, including elevated levels of cyclin D1, cdk4, E2F-1, and cyclin E (Pelegri et al., 2008; Fernandez-Fernandez et al., 2011) is also seen in HD patients’ striata. The possible direct role of cyclin D1 and cell cycle re-entry in HD striatal neuronal cell death has not been investigated so far. However, epigenetic factors are known to modulate the age-of-onset and the progression of striatal neuronal cell death in HD (Reiner et al., 2011; Gusella et al., 2014) and increased expression of cell cycle-related genes is positively associated with the severity of HD in mice (Langfelder et al., 2016), suggesting that genetic polymorphisms in CCND1 or other cell cycle-related genes might modulate HD neuropathology.
In summary, our study suggests that preventing 3-NP-induced cyclin D1 expression might be sufficient to promote resistance to 3-NP-induced striatal neurodegeneration, making cyclin D1 and its downstream pathway possible therapeutic targets for neurodegenerative disorders in which mitochondrial complex II is compromised. These results can potentially be extended to other neurodegenerative disorders as well, since increased cyclin D1 and evidence of cell cycle re-entry is also a hallmark of amyotrophic lateral sclerosis, Alzheimer’s disease, and Parkinson’s disease (Liu et al., 2010; Folch et al., 2012; Frade and Ovejero-Benito, 2015). Further understanding of the mechanisms underlying regulation of expression of cyclin D1 in response to cellular stress and its role in different neuronal cell types may open new avenues for novel therapeutic strategies for different neurodegenerative disorders.
Supplementary Material
Highlights.
3-nitropropionic acid (3-NP) induces striatal lesions in C57BL/6J (B6) but not in DBA/2J (D2) mice
Forward genetics identified Ccnd1 (cyclin D1) as a strong candidate for strain-specific 3-NP response
3-NP induces cyclin D1 expression in B6 striatum but not in D2
Ccnd1 mRNA’s 3’UTR is alternatively processed in B6 striatum
Alternative B6 striatal-specific 3’UTR processing is predicted to increase cyclin D1 levels
Acknowledgements
Rachel Cox was an undergraduate Summer Research Scholar in the College of Graduate Health Sciences at the University of Tennessee Health Sciences Center.
We would like to thank members of the Dragatsis-Dietrich’s laboratory for valuable discussions and technical help.
Funding
This work was supported by an intramural New Grant award from the UTHSC Office of Research, by a CITG seeding Funding (PD and ID) and by an NIH/NIEHS R21ES028429 grant (PD, MM and ID).
Abbreviations
- 3-NP
3-nitropropionic acid
- B6
C57BL/6J
- D2
DBA/2J
- HD
Huntington’s disease
- MSA
multiple system atrophy
- DARPP-32
dopamine and cyclic-AMP-regulated phosphoprotein of molecular weight 32,000
- Ccnd1
cyclin D1
- RI
recombinant inbred
- QTL
quantitative trait locus
- miR
microRNA
- H&E
haematoxylin and eosin
- IP
intraperitoneally
- CDK
cyclin D-dependent kinase
- SDH
succinate dehydrogenase
Footnotes
Credit author statement
All authors have read, edited, and approved the manuscript. The authors contributed to the study as follows: PD, SA and RC performed experiments. PD, MKM, DGA and RWW performed in silico analyses. MKM and RWW provided animal models. PD and ID analyzed the data and performed statistical analyses. PD and ID conceptualized the study, drafted, edited, and finalized the manuscript. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Declaration of competing interests
The authors declare no competing financial interests.
Supplementary data
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aftab A, Khan R, Shah W, Azhar M, Unar A, Jafar Hussain HM, Waqas A, 2021. Computational analysis of Cyclin D1 gene SNPs and association with breast cancer. Biosci. Rep. 41, BSR20202269. 10.1042/BSR20202269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad F, Alamoudi W, Haque S, Salahuddin M, Alsamman K, 2018. Simple, reliable, and time-efficient colorimetric method for the assessment of mitochondrial function and toxicity. Bosn. J. Basic. Med. Sci. 18, 367–374. 10.17305/bjbms.2018.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashiba H, Ikegaya Y, Nishiyama N, Matsuki N, 2008. Differential involvement of cell cycle reactivation between striatal and cortical neurons in cell death induced by 3-nitropropionic acid. J. Biol. Chem. 283, 6594–6606. 10.1074/jbc.M707730200. [DOI] [PubMed] [Google Scholar]
- Alexi T, Hughes PE, Faull RL, Williams CE, 1998. 3-Nitropropionic acid’s lethal triplet: cooperative pathways of neurodegeneration. Neuroreport. 9, R57–64. 10.1097/00001756-199808030-00001. [DOI] [PubMed] [Google Scholar]
- Alston TA, Mela L, Bright HJ, 1977. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. U. S. A. 74, 3767–3771. 10.1073/pnas.74.9.3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun S, Liu L, Donmez G, 2016. Mitochondrial biology and neurodegenerative diseases. Curr. Neuropharmacol. 14, 143–154. 10.2174/1570159×13666150703154541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, Lutz CM, Valenzuela A, Bohl CJ, Ingels JF, McCarty MS, Centeno AG, Hager R, Auwerx J, Lu L, Williams RW, 2021. A platform for experimental precision medicine: the extended BXD mouse family. Cell. Syst. 13, S2405–4712(20)30503–2. 10.1016/j.cels.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Déglon N, Brouillet E, 2006. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol. Biol. Cell. 17, 1652–1663. 10.1091/mbc.e05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G et al. , 2010. The landscape of somatic copy-number alteration across human cancers. Nature. 463, 899–905. 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkelund T, Johansen RF, Illum DG, Dyrskog SE, Østergaard JA, Falconer TM, Andersen C, Fridholm H, Overballe-Petersen S, Jensen JS, 2021. Fatal 3-nitropropionic acid poisoning after consuming coconut water. Emerg. Infect. Dis. 27, 278–280. 10.3201/eid2701.202222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizat N, Hermel JM, Humbert S, Jacquard C, Créminon C, Escartin C, Saudou F, Krajewski S, Hantraye P, Brouillet E, 2003. In vivo calpain/caspase cross-talk during 3-nitropropionic acid-induced strital degeneration: implication of a calpain-mediated cleavage of active caspase-3. J. Biol. Chem. 278, 43245–43253. 10.1074/jbc.M305057200. [DOI] [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, Martella G, Sciamanna G, Tolu M, Calabresi P, Bernardi G, Pisani A, 2006. Mitochondrial toxins in Basal Ganglia disorders: from animal models to therapeutic strategies. Curr. Neuropharmacol. 4, 69–75. 10.2174/157015906775203039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Guyot MC, Mittoux V, Altairac S, Condé F, Palfi S, Hantraye P, 1998. Partial inhibition of brain succinate dehydrogenase by 3-nitropropiomnic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 70, 794–805. 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF, 1995. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. U. S. A. 92, 7105–7109. 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Jacquard C, Bizat N, Blum D, 2005. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 95, 1521–1540. 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF, 1997. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann. Neurol. 41, 646–653. 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, Hsu HC, Mountz JD, Baldwin NE, Langston MA, Threadgill DW, Manly KF, Williams RW, 2005. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat. Genet. 37, 233–242. 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- Coles CJ, Edmondson DE, Singer TP, 1979. Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem. 254, 5161–5167. [PubMed] [Google Scholar]
- Colle D, Santos DB, de Souza V, Lopes MW, Leal RB, de Souza Brocardo P, Farina M, 2019. Sodium selenite protects from 3-nitropropionic acid-induced oxidative stress in cultured primary cortical neurons. Mol Biol Rep. 46, 751–762. 10.1007/s11033-018-4531-y. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Pastore A, Deshpande AJ, Zimmermann Y, Hutter G, Weinkauf M, Buske C, Hiddemann W, Dreyling M, 2009. 3’UTR mediated regulation of the cyclin D1 proto-oncogene. Cell Cycle 8, 3592–3600. 10.4161/cc.8.21.9993 [DOI] [PubMed] [Google Scholar]
- Diehl JA, 2002. Cycling to cancer with Cyclin D1. Cancer Biol Ther. 1, 226–231. 10.4161/cbt.72. [DOI] [PubMed] [Google Scholar]
- Dietrich P, Johnson IM, Alli S, Dragatsis I, 2017. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain homeostasis. PLoS Genet. 13, e1006846. 10.1371/journal.pgen.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich P, Yue J, E. S., Dragatsis I, 2011. Deletion of exon 20 of the Familial Dysautonomia gene Ikbkap mice cause developmental delay, cardiovascular defects, and early embryonic lethality. PLoS One. 6, e27015. 10.1371/journal.pone.0027015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Vilaregut J, Manich G, del Valle J, Pallàs M, Camins A, Pelegrí C, Vilaplana J, 2011. Neuronal apoptosis in the striatum of rats treated with 3-nitropropionic acid is not triggered by cell-cycle re-entry. Neurotoxicology. 32, 734–741. 10.1016/j.neuro.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez MR, Ferrer I, Lucas JJ, 2011. Impaired ATF6a processing, decreased Rheb and neuronal cell cycle re-entry in Huntington’s disease. Neurobiol Dis. 41, 23–32. 10.1016/j.nbd.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Folch J, Junyent F, Verdaguer E, Auladell C, Pizarro JG, Beas-Zarate C, Pallàs M, Camins A, 2012. Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotox Res. 22, 195–207. 10.1007/s12640-011-9277-4. [DOI] [PubMed] [Google Scholar]
- Foti SC, Hargreaves I, Carrington S, Kiely AP, Houlden H, Holton JL, 2019. Cerebral mitochondrial electron transport chain dysfunction in multiple systematrophy and Parkinson’s disease. Sci Rep. 9, 6559. 10.1038/s41598-019-42902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Ovejero-Benito MC, 2015. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle. 14, 712–720. 10.1080/15384101.2015.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Chu SF, Li JP, Zhang Z, Yan JQ, Wen ZL, Xia CY, Mou Z, Wang ZZ, He WB, Guo XF, Wei GN, Chen NH, 2015. Protopanaxtriol protects against 3-nitropropionic acid-induced oxidative stress in a rat model of Huntington’s disease. Acta Pharmacol Sin. 36, 311–322. 10.1038/aps.2014.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME, 2003. Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington’s disease knock-in striatal cells. J Biol Chem. 278, 50514–50522. 10.1074/jbc.M309348200. [DOI] [PubMed] [Google Scholar]
- Glat MJ, Ben-Zur T, Barhum Y, Offen D, 2016. Neuroprotective effect of a DJ-1 based peptide in a toxin induced mouse model of multiple system atrophy. PLoS One.11, e0148170. 10.1371/journal.pone.0148170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH, 1996. Mitochondrtial defect in Huntington’s disease caudate nucleus. Ann Neurol. 39, 385–389. 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME, Lee JM, 2014. Genetic modifiers of Huntington’s disease. Mov Disord. 2014 29, 1359–1365. 10.1002/mds.26001. [DOI] [PubMed] [Google Scholar]
- Hamilton BF, Gould DH, Gustine DL. (2000). “History of 3-Nitropropionic acid – Occurrence and Role in Human and Animal Disease”, in Mitochondrial Inhibitors and Neurodegenerative Disorders, part of the series Contemporary Neuroscience, Sanberg, Nishino, Borlongan eds. p 21–33. [Google Scholar]
- Hanbury R, Chen EY, Wuu J, Kordower JH, 2003. Knockout of p75NTR does not alter the viability of striatal neurons following a metabolic or excitotoxic injury. J Mol Neurosci. 20, 93–102. 10.1385/JMN:20:2:93. [DOI] [PubMed] [Google Scholar]
- Heuer SE, Neuner SM, Hadad N, O’Connell KMS, Williams RW, Philip VM, Gaiteri C, Kaczorowski CC, 2020. Identifying the molecular systems that influence cognitive resilience to Alzheimer’s disease in genetically diverse mice. Learn Mem. 27, 355–371. 10.1101/lm.051839.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton GD, Stoica BA, Byrnes KR, Faden AI, 2008. Roscovitine reduces neuronal loss, glial activation and neurologic deficits after brain trauma. J Cereb Blood Flow Metab. 28, 1845–1859. 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, Raiman J, 2013. Complex II deficiency – a case report and review of the literature. Am J Med Genet A. 161A, 285–294. 10.1002/ajmg.a.35714. [DOI] [PubMed] [Google Scholar]
- Jiang J, Huang J, Wang XR, Quan YH, 2016. MicroRNA-202 induces cell cycle arrest and apoptosis in lung cancer cells through targeting cyclin D1. Eur Rev Med Pharmacol Sci. 20, 2278–2284. [PubMed] [Google Scholar]
- Jiang L, Zhao Z, Zheng L, Xue L, Zhan Q, Song Y, 2017. Downregulation of miR-503 promotes ESCC Cell proliferation, migration, and invasion by targeting Cyclin D1. Genomics Proteomics Bioinformatics. 15, 208–217. 10.1016/j.gpb.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Feng MG, Mo YY, 2009. Systematic validation of predicted microRNAs for cyclin D1. BMC Cancer. 9, 194. 10.1186/1471-2407-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovicic A, Zaldivar Jolissaint JF, Moser R, Silva Santos Mde F, Luthi-Carter R, 2013. MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS One 8,e54222. 10.1371/journal.pone.0054222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junyent F, de Lemos L, Verdaguer E, Pallàs M, Folch J, Beas-Zárate C, Camins A, Auladell C, 2012. Lack of Jun-N-terminal kinase 3 (JNK3) does not protect against neurodegeneration induced by 3-nitropropionic acid. Neuropathol Appl Neurobiol. 38, 311–321. 10.1111/j.1365-2990.2011.01214.x. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Faden AI, 2014. Selective CDK inhibitors: promising candidates for future clinical traumatic brain injury trials. Neural Regen Res. 9, 1578–1580. 10.4103/1673-5374.141779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Loane DJ, Byrnes KR, Hanscom M, Cabatbat RM, Tan MT, Faden AI, 2012a. Cyclin D1 gene ablation confers neuroprotection in traumatic brain injury. J Neurotrauma. 29, 813–827. 10.1089/neu.2011.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Byrnes KR, Hanscom M, Loane DJ, Faden AI 2012b. Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J Cereb Blood Flow Metab. 32, 137–149. 10.1038/jcbfm.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Tsai LH, 2009. Linking cell cycle re-entry and DNA damage in neurodegeneration. Ann N Y Acad Sci. 1170, 674–679. 10.1111/j.1749-6632.2009.04105.x. [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Diehl JA, Haiman CA, Knudsen ES, 2006. Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene. 25, 1620–1628. 10.1038/sj.onc.1209371. [DOI] [PubMed] [Google Scholar]
- Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R Jr., Gorospe M, Mattson MP, 2004. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 41, 549–561. 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- Kuzdas-Wood D, Stefanova N, Jellinger KA, Seppi K, Schlossmacher MG, Poewe W, Wenning GK, 2014. Towards translational therapies for multiple system atrophy. Prog Neurobiol. 118, 19–35. 10.1016/j.pneurobio.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, Deverasetty S, Tebbe A, Schaab C, Lavery DJ, Howland D, Kwak S, Botas J, Aaronson JS, Rosinski J, Coppola G, Horvath S, Yang XW, 2016. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 19, 623–633. 10.1038/nn.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E, 2009. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 16, 899–909. 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DZ, Ander BP, Sharp FR, 2010. Cell cycle inhibition without disruption of neurogenesis is a strategy for treatment of central nervous system diseases. Neurobiol Dis. 37, 549–557. 10.1016/j.nbd.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, James Shen CK, Marsh JL (2015). Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntingon’s disease. Hum Mol Genet. 24,1602–1616. 10.1093/hmg/ddu574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Ou C, Xia H, Zhu Y, Liu D, 2015. MIR-503 inhibited cell proliferation of human breast cancer cells by suppressing CCND1 expression. Tumour Biol. 36, 8697–8702. 10.1007/s13277-015-3623-8. [DOI] [PubMed] [Google Scholar]
- Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M, 1991. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci. 18, 492–498. 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- Lui XJ, Hu WJ, Wang YH et al. , 1989. Studies on the mycology and mycotoxins in an outbreak of deteriorated sugar cane poisoning. Chin Prev Med. 23, 345–348. [Google Scholar]
- Martínez-Turrillas R, Puerta E, Chowdhury D, Marco S, Watanabe M, Aguirre N, Pérez-Otaño I, 2012. The NMDA receptor subunit GluN3A protects against 3-nitropropionic-induced striatal lesions via inhibition of calpain activation. Neurobiol Dis. 48, 290–298. [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP, 2009. Widespread shortening of 3’UTR by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 138, 673–684. 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealer RG, Subramaniam S, Snyder SH, 2013. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of Huntington’s disease. J Neurosci. 33, 4206–4210. 10.1523/JNEUROSCI.3730-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, Murillo V, Wolff SC, Shaw RJ, Auwerx J, Dillin A, 2016. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 165, 1209–1223. 10.1016/j.cell.2016.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyairi I, Tatireddigari VR, Mahdi OS, Rose LA, Belland RJ, Lu L, Williams RW, Byrne GI, 2007. The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J Immunol. 179,1814–1824. [DOI] [PubMed] [Google Scholar]
- Miyairi I, Ziebarth J, Laxton JD, Wang X, van Rooijen N, Williams RW, Lu L, Byrne GI, Cui Y, 2012. Host genetics and Chlamydia disease: prediction and validation of disease severity mechanisms. PLoS One. 7, e33781. 10.1371/journal.pone.0033781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Mozhui K, Prins P, Williams RW, 2017. GeneNetwork: A toolbox for systems genetics. Methods Mol Biol. 1488, 75–120. 10.1007/978-1-4939-6427-7_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner SM, Garfinkel BP, Wilmott LA, Ignatowska-Jankowska BM, Citri A, Orly J, Lu L, Overall RW, Mulligan MK, Kempermann G, Williams RW, O’Connell KM, Kaczorowski C,C., 2016. Systems genetics identifies Hp1bp3 as a novel modulator of cognitive aging. Neurobiol Aging. 46, 58–67. 10.1016/j.neurobiolaging.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens KM, Aykin-Burns N,. Dayal D, Coleman MC, Domann FE, Spitz DR, 2012. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2(−) and H2O2. Free Radic Biol Med. 52, 160–166. 10.1016/j.freeradbiomed.2011.10.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P, 1996. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 16, 3019–3025. 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Vila I, Rifé M, Lizcano JM, Alberch J, Ginés SJ (2008). Dopaminergic and glutamatergic signaling crosstalk in Huntington’s disease neurodegeneration: the role of p25/cycli-dependent kinase 5. Neurosci. 28, 10090–10101. 10.1523/JNEUROSCI.3237-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA, 1997. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 17, 8975–8983. 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA, 1998. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 143, 457–467. 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Obeidat A, Giovanni A, Greene LA, 2000. Cell cycle regulators in neuronal death evoked by excitotoxic stress: implications for neurodegeneration and its treatment. Neurobiol Aging. 21, 771–781. 10.1016/s0197-4580(00)00220-7. [DOI] [PubMed] [Google Scholar]
- Pedraza-Chaverrí J, Reyes-Fermín LM, Nolasco-Amaya EG, Orozco-Ibarra M, Medina-Campos ON, González-Cuahutencos O, Rivero-Cruz I, Mata R, 2009. ROS scavenging capacity and neuroprotective effect of alpha-mangostin against 3-nitropropionic acid in cerebellar granule neurons. Exp Toxicol Pathol. 61, 491–501. 10.1016/j.etp.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Pelegrí C, Duran-Vilaregut J, del Valle J, Crespo-Biel N, Ferrer I, Pallàs M, Camins A, Vilaplana J, 2008. Cell cycle activation in striatal neurons from Huntington’s disease patients and rats treated with 3-nitropropionic acid. Int J Dev Neurosci. 26, 665–671. 10.1016/j.ijdevneu.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Peng J, Chen XL, Cheng HZ, Xu ZY, Wang H, Shi ZZ, Liu J, Ning XG, Peng H, 2019. Silencing of KCNK15-AS1 inhibits lung cancer cell proliferation via upregulation of miR-202 and miR-370. Oncol Lett. 18, 5968–5976. 10.3892/ol.2019.10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraica M, Radić B, Lucić A, Pavlović M, 1999. Toxic effects of mycotoxins in humans, Bull World Health Organ. 77, 754–766. [PMC free article] [PubMed] [Google Scholar]
- Qi H, Wen B, Wu Q, Cheng W, Lou J, Wei J, Huang J, Yao X, Weng G, 2018. Long noncoding RNA SNHG7 accelerates prostate cancer proliferation and cell cycle progression through cyclin D1 by sponging miR-503. Biomed Pharmacother. 102, 326–332. 10.1016/j.biopha.2018.03.011. [DOI] [PubMed] [Google Scholar]
- Reiner A, Dragatsis I, Dietrich P, 2011. Genetics and neuropathology of Huntington’s disease. Int Rev Neurobiol. 98, 325–372. 10.1016/B978-0-12-381328-2.00014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander B, Flygare J, Porwit-Macdonald A, Smith CI, Emanuelsson E, Kimby E, Liden J, Christensson B, 2005. Mantle cell lymphomas with low levels of cyclin D1 long mRNA transcripts are highly proliferative and can be discriminated by elevated cyclin A2 and cyclin B1. Int J Cancer. 117, 418–430. 10.1002/ijc.21166. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, 2005. Susceptibility to excitotoxic and metabolic striatal neurodegeneration in the mouse is genotype dependent. Brain Res. 1040, 112–120. 10.1016/j.brainres.2005.01.067. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, Linsley PS, Mao M, Stoughton RB, Friend SH, 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature. 422, 297–302. 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Schwartz EI, Smilenov LB, Price MA, Osredkar T, Baker RA, Ghosh S, Shi FD, Vollmer TL, Lencinas A, Stearns DM, Gorospe M, Kruman II, 2007. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle. 6, 318–329. 10.4161/cc.6.3.3752. [DOI] [PubMed] [Google Scholar]
- Slotta-Huspenina J, Koch I, de Leval L, Keller G, Klier M, Bink K, Kremer M, Raffeld M, Fend F, Quintanilla-Martinez L, 2012. The impact of cyclin D1 mRNA isoforms, morphology and p53 in mantle cell lymphoma: p53 alterations and blastoid morphology are strong predictors of a high proliferation index. Haematologica. 97, 1422–1430. 10.3324/haematol.2011.055715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova N, Reindl M, Neumann M, Haass C, Poewe W, Kahle P,J,, Wenning GK, 2005. Oxidative stress in transgenic mice with oligodendroglial aplpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am J Pathol. 166, 869–876. 10.1016/s0002-9440(10)62307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova N, Wenning GK, 2016. Review: Multiple system atrophy: emerging targets for interventional therapies. Neuropathol Appl Neurobiol. 42, 20–32. 10.1111/nan.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Park DS, Friedman WJ, Greene LA, 1999. Caspase-dependent and - independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 19, 6235–6247. 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomashevski A, Webster DR, Grammas P, Gorospe M, Kruman II, 2010. Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ. 17, 1189–1198. 10.1038/cdd.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Túnez I, Tasset I, Pérez-De La Cruz V, Santamaría A, 2010. 3-nitropropionic acid as a tool to study the mechanisms involved in Huntington’s Disease: past, present and future. Molecules. 15, 878–916. 10.3390/molecules15020878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi K, Lee PH, Adame A, Inglis C, Mante M, Rockenstein E, Stefanova N, Wenning GK, Masliah E, 2009. Mitochondrial inhibitor 3-nitropropionic acid enhances oxidative modification of alpha-synuclein in a transgenic mouse model of multiple system atrophy. J Neurosci Res. 87, 2728–2739. 10.1002/jnr.22089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO, 2007. Huntington’s disease. Lancet. 369, 218–228. 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Wang L, Jiao Y, Wang Y, Zhang M, Gu W, 2016. Self-confirmation and ascertainment of the candidate genomic regions of complex trait loci – a none-experimental solution. PLoS One.11, e0153676. 10.1371/journal.pone.0153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, He G, Hou M, Chen L, Chen S, Xu A, Fu Y, 2018. Cell cycle regulation by alternative polyadenylation of CCND1. Sci Rep 8, 6824–6831. 10.1038/s41598-018-25141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ma S, 2014. Risk factors for etiology and prognosis of mantle cell lymphoma. Expert Rev Hematol. 7, 233–243. 10.1586/17474086.2014.889561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L, 2001. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2, RESEARCH0046. 10.1186/gb-2001-2-11-research0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Connolly T, Futcher B, Beach D, 1991. Human D-type cyclin. Cell. 65, 691–699. 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- Yi Y, Li H, Lv Q, Wu K, Zhang W, Zhang J, Zhu D, Liu Q, Zhang W, 2016. miR-202 inhibits the progression of human cervical cancer through inhibition of cyclin D1. Oncotarget. 7, 72067–72075. 10.18632/oncotarget.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Song X, Herrup K, 2020. Context-dependent functions of E2F1: cell-cycle, cell death, and DNA damage repair in cortical neurons. Mol Neurobiol. 57, 2377–2390. 10.1007/s12035-020-01887-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.