

Abstract
Background
Although mounting evidence indicates that insulin resistance (IR) co‐occurs with mitochondrial dysfunction in skeletal muscle, there is no clear causal link between mitochondrial dysfunction and IR pathogenesis. In this study, the exact role of mitochondria in IR development was determined.
Methods
Six‐week‐old C57BL/6 mice were fed a high‐fat diet for 2 weeks to induce acute IR or for 24 weeks to induce chronic IR (n = 8 per group). To characterize mitochondrial function, we measured citrate synthase activity, ATP content, mitochondrial DNA (mtDNA) content, and oxygen consumption rate in gastrocnemius and liver tissues. We intraperitoneally administered mitochondrial division inhibitor 1 (mdivi‐1) to mice with acute IR and measured mitochondrial adaptive responses such as mitophagy, mitochondrial unfolded protein response (UPRmt), and oxidative stress (n = 6 per group).
Results
Acute IR occurred coincidently with impaired mitochondrial function, including reduced citrate synthase activity (−37.8%, P < 0.01), ATP production (−88.0%, P < 0.01), mtDNA (−53.1%, P < 0.01), and mitochondrial respiration (−52.2% for maximal respiration, P < 0.05) in skeletal muscle but not in liver. Administration of mdivi‐1 attenuated IR development by increasing mitochondrial function (+58.5% for mtDNA content, P < 0.01; 4.06 ± 0.69 to 5.84 ± 0.95 pmol/min/mg for citrate synthase activity, P < 0.05; 13.06 ± 0.70 to 34.87 ± 0.70 pmol/min/g for maximal respiration, P < 0.001). Western blot analysis showed acute IR resulted in increased autophagy (mitophagy) and UPRmt induction in muscle tissue. This adaptive response was inhibited by mdivi‐1, which reduced the mitochondrial oxidative stress of skeletal muscle during acute IR.
Conclusions
Acute IR induced mitochondrial oxidative stress that impaired mitochondrial function in skeletal muscle. Improving mitochondrial function has important potential for treating acute IR.
Keywords: Insulin resistance, Mitochondria, Oxidative stress, Skeletal muscle, Mitophagy
Introduction
Insulin maintains glucose homeostasis by stimulating glucose uptake in peripheral tissues, primarily adipose and skeletal muscle (SKM), and reducing gluconeogenesis in liver. Insulin resistance (IR) is characterized by the inability of tissues to respond appropriately to insulin. 1 Intake of foods high in fat and sugar can cause caloric excess that drives obesity, IR, and impaired insulin signalling. IR is an important risk factor for type 2 diabetes, coronary heart disease, and non‐alcoholic fatty liver disease. 2 IR is also accompanied by reduced mitochondrial content of SKM. 3 Because SKM accounts for 60–70% of insulin‐stimulated glucose uptake in the whole body, 4 IR in SKM is a major obstacle to normoglycaemia. 5
Mitochondria are dynamic organelles that contribute to oxidative stress mediation, cellular redox state, and cell signalling. Because mitochondria are central to cellular energy metabolism, they play major roles in metabolic disorders, such as reduced ATP synthesis, reactive oxygen species (ROS) generation, and apoptotic cell death with a high‐fat diet (HFD). 6 The preliminary evidence linking mitochondrial dysfunction to IR comes from studies of obese and insulin‐resistant individuals who had defective lipid metabolism and lower SKM mitochondria oxidative capacity than healthy lean controls. 7 Mitochondrial capacity for oxidative metabolism is lower in insulin‐responsive tissues of patients. 3 , 8 First‐degree relatives of individuals with T2DM have decreased mitochondrial respiration, suggesting that mitochondrial dysfunction precedes T2DM onset. 9 Moreover, compromised mitochondria can promote diabetes pathogenesis, especially in cases of hereditary mitochondrial disease. However, the role of mitochondria in IR development is still controversial, because mitochondrial dysfunction does not always occur with IR. 10 , 11
Mitochondria constantly undergo fusion and fission, which establish a dynamic mitochondrial network. The physiological importance of mitochondrial dynamics for the regulation of mitochondrial function has been reported in neurodegenerative disease and cancer. 2 , 3 In the case of IR, mitochondrial fission is increased in SKM from diet‐induced obese mice. 4 Moreover, the inhibition of mitochondrial fission improves muscle insulin signalling and systemic insulin sensitivity, which suggest that aberrant mitochondrial fission is casually associated with mitochondrial dysfunction and IR.
To clarify the relationship between mitochondrial dysfunction and IR severity in SKM, we compared insulin sensitivity and mitochondrial function of mice fed a 2‐week HFD with those of mice fed a 24‐week HFD (HFD‐S and HFD‐L, respectively). Also, to determine the role of mitochondrial plasticity in the development acute IR, we treated HFD‐S mice with mdivi‐1, a mitochondrial fission inhibitor, and measured insulin sensitivity and mitochondrial function.
Materials and methods
Animals
Five‐week‐old male C57BL/6 mice were obtained from Orient Bio (Seongnam, Korea). After a week of acclimation, mice were fed a control diet (CD) or a 60% high‐fat diet long‐term for 24 weeks (HFD‐L). For short‐term HFD feeding (HFD‐S), we fed mice a CD for 22 weeks and then a 60% HFD for 2 weeks. On the day of sacrifice, we compared HFD‐S and HFD‐L mice with age matched CD mice. Mitochondrial division inhibitor 1 (mdivi‐1) is a quinazolinone derivative that selectively inhibits dynamin‐related protein 1 (DRP1)‐dependent mitochondrial fission and was purchased from Sigma‐Aldrich (St. Louis, MO). Mdivi‐1 was dissolved in DMSO and diluted with sterile water for intraperitoneal injection into mice. We injected HFD‐S mice with 50 mg/kg body weight of mdivi‐1 twice, 24 and 6 h before sacrifice.
Blood measurements
We collected blood, centrifuged samples at 3600 g for 20 min, and stored at −80°C until use. Serum glucose levels were measured with a commercial kit (Shinyang Chemical Co., Busan, Korea). Enzyme‐linked immunosorbent assay kits were used to measure serum insulin levels (ALPCO Diagnostics, Salem, NH, USA). We calculated homeostatic model assessment‐IR (HOMA‐IR) from fasting glucose and insulin levels.
Glucose tolerance and insulin sensitivity test
We performed intraperitoneal glucose tolerance tests (IPGTT) and intraperitoneal insulin tolerance tests (IPITT) to measure IR. Prior to each test, mice were fasted for 4 h, a baseline blood sample was taken from their tail, and each mouse intraperitoneally received either a 2 g d‐glucose/kg body weight (bw) or 0.75 U insulin/kg bw for the IPGTT or the IPITT, respectively. We collected tail blood samples at 15, 30, 60, 90, and 120 min after injection and analysed immediately for glucose content using the Accu‐Check Performa Instrument (Roche Diagnostics, Basel, Switzerland).
Hyperinsulinaemic‐euglycaemic clamp
We performed a hyperinsulinaemic‐euglycaemic clamp as previously described. 12 Briefly, after overnight fasting, we performed a 2 h hyperinsulinaemic‐euglycaemic clamp with a 900 pmol/kg priming dose and a continuous infusion of human regular insulin (Novolin, Denmark) at 15 pmol/kg/min, and infused 20% glucose at variable rates to maintain constant glucose concentrations of 5–6 mM. We estimated insulin‐stimulated rates of whole‐body glucose uptake using a continuous infusion of [3‐3H] glucose (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) through the clamps (0.1 μCi/min). To estimate insulin‐stimulated glucose uptake in individual tissues, we administered 2‐deoxy‐d‐[1‐14C] glucose (2‐[14C]DG; PerkinElmer Life and Analytical Sciences) as a bolus (10 μCi) 75 min after starting clamps. We calculated rates of insulin‐stimulated whole‐body glucose uptake by dividing [3H] glucose infusion rates [disintegrations per minute (dpm/min)] by specific activities of plasma glucose (dpm/μmol) during the final 30 min of the clamp. Hepatic glucose production during clamps was determined by subtracting the glucose infusion rate from the whole‐body glucose uptake rate. We calculated glucose uptake in SKM from the plasma 2‐[14C]DG profile, which was fitted with a double exponential or linear curve using MLAB (Civilized Software, Silver Spring, MD, USA) and tissue 2‐[14C]DG‐6‐P content.
Mitochondria analysis
Protein extraction was performed in RIPA buffer (Thermo Fisher Scientific, Rockford, IL, USA). Citrate synthase activity was measured in the protein fraction with a spectrophotometer using a commercial kit (Sigma‐Aldrich, St. Louis, MO, USA) and adjusted by protein content. We determined ATP content using an ATP luminescence detection assay system (ATPlite, PerkinElmer, Waltham, MA, USA). The mitochondrial fraction was isolated using the Mitochondrial Isolation Kit for Tissue (Thermo Fisher Scientific) and used for the measurement of complex I activity using a Complex І Enzyme Activity Assay Kit (Abcam, Cambridge, UK). To measure mitochondria content, we extracted total DNA from muscle using phenol/chloroform/isoamyl alcohol (25:24:1) followed by ethanol precipitation. The content of mtDNA was calculated using real‐time quantitative PCR (qRT‐PCR) by measuring the threshold cycle ratio (ΔCt) of a mitochondrial‐encoded gene (Cox5, forward 5′‐ACTATACTACTACTAACAGACCG‐3′, reverse 5′‐GGTTCTTTTTTTCCGGAGTA‐3′) versus a nuclear‐encoded gene (Cyclophilin A, forward 5′‐ACACGCCATAATGGCACTGG‐3′, reverse 5′‐CAGTCTTGGCAGTGCAGAT‐3′).
Quantitative real time‐polymerase chain reaction (qRT‐PCR) assay
We extracted total RNAs from muscle using RNeasy Fibrous Tissue Mini Kit (Qiagen, Germantown Rd, MD, USA) and from liver tissue using NucleoSpin® RNA Plus kit (Macherey‐Nagel, Bethlehem, PA, USA). The ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan) was used to synthesize cDNA. We performed qRT‐PCR with SYBR Green Master Mix (Toyobo) and a StepOne™ PCR System (Applied Biosystems, Foster City, CA, USA). Primer sequences are listed in Supporting Information, Table S1.
Bioenergetics of extensor digitorum longus (EDL) myofibre and isolated muscle mitochondria
To isolate EDL myofibres, we dissected the EDL muscle and digested with DMEM supplemented with collagenase II and 1% antibiotics for 1 h. We gently triturated EDL muscle through a fire‐polished glass pipette and transferred intact single fibres to new medium. The oxygen consumption rate (OCR) was measured in EDL muscles using a previously described method with minor modifications. 13 Briefly, collagenase‐digested single muscle fibres were seeded into XF24 wells and the OCR was measured with a Seahorse XF24 Analyzer (Agilent, Santa Clara, CA, USA). After basal OCR measurement, we injected mitochondrial inhibitor, oligomycin (1 μM), a mitochondrial uncoupler, carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (FCCP; 400 nM), pyruvate (10 mM), and rotenone to inhibit the mitochondrial complex. Next, we performed a bicinchoninic acid (BCA) assay to determine the concentration of muscle protein and normalized to total protein content. The respiratory O2 consumption in the mitochondrial fraction isolated from gastrocnemius or liver tissues was measured as previously described. 14 For the electron flow experiment, we used 10 μM pyruvate, 2 mM malate, and 4 μM FCCP as substrate. The final concentrations were 2 μM rotenone, 10 mM succinate, 4 μM antimycin A, and 100 mM ascorbate with 0.2 mM TMPD. Rotenone and antimycin A were used to inhibit complex I and complex III respiration. The flow of electrons through each complex (I–IV) was determined using specific substrates: pyruvate and malate for complex I, succinate for complex II, and ascorbate/TMPD for complex IV. Coupling experiments (rates of state2, state3, state4O, and state3 μ) were performed using 80 μM palmitoyl‐carnitine and 0.5 mM malate as substrate and oligomycin as a coupling agent. FCCP was used as an uncoupling agent to evaluate the effect of HFD feeding on maximal respiration rate. The final concentrations were 4 mM ADP, 10 μM oligomycin, 10 μM FCCP, and 10 μM antimycin A.
Western blot analysis
We extracted protein fractions with RIPA buffer (Thermo Fisher Scientific, Rockford, IL, USA) containing protease and phosphatase inhibitors (Thermo Fisher Scientific), separated 20 μg of protein from each sample by 12% SDS‐PAGE, and electrophoretically transferred the protein to a polyvinylidene difluoride membrane. Blocked membranes were incubated overnight at 4°C with primary antibodies, then incubated with horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse Ig. Immunoreactive bands were visualized by enhanced chemiluminescence solution (Thermo Scientific). Band quantitative analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Antibodies against T‐OXPHOS (ab110413, 1:1000) (Abcam), Mfn1 (sc‐50330, 1:1000), Mfn2 (sc‐100560, 1:1000), Opa1 (sc‐30573, 1:1000), Fis1 (PA1–41082, 1:1000), DRP1 (#5391, 1:1000), p‐DRP1 (#3455, 1:1000), PGC1α (ST1202, 1:1000), PGC1β (sc‐373771, 1:1000), PPARδ (PA5–29678, 1:1000), p70 S6 Kinase (#2708, 1:1000), p‐p70 S6 Kinase [Thr389 (#9205, 1:1000)], LC3 (#2775, 1:1000), p62 (BD, 610832, 1:1000), Atg5 (#12994, 1:1000), p‐S556‐ULK1 (OABF01248, 1:1000)], p‐S757‐ULK1 (#6888, 1:1000)], PINK1 (#6946, 1:1000), Parkin (ab15954, 1:1000), HSP25 (ADI‐SPA‐801‐D, 1:1000), TNX2 (#14907, 1:1000), CLPP (ab124822, 1:1000), 4‐HNE (ab46545, 1:1000), β‐actin (sc‐47778, 1:1000), and VDAC (#4661, 1:1000) were used. β‐actin and VDAC were used as reference proteins for total protein and mitochondrial fraction, respectively.
Liver lipid measurement
For liver histology, we fixed liver tissues with 10% formaldehyde solution, embedded in paraffin, and stained with haematoxylin–eosin. Hepatic lipids were extracted according to the conventional Foch method. 15 Total triglyceride (TG) and total cholesterol (TC) were measured using commercial kits (Shinyang Chemical Co., Busan, Korea) according to manufacturer protocols.
Transmission electron microscopy (TEM)
We studied the ultrastructural characteristics of mitochondria using TEM. First, gastrocnemius and liver tissues were fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M PBS (pH 7.4) at 4°C for 12 h and then post‐fixed with 1% osmium tetroxide for 1 h. The tissues were embedded in pure Epon 812 mixture after dehydration in an ethanol series and infiltration in a propylene oxide : epon mixture series. Ultra‐thin sections (~70 nm) were cut with a model MT‐X ultramicrotome (RMC, Tucson, AZ, USA) and stained with 2% uranyl acetate and 0.2% lead citrate. We used a Cryo‐TEM (JEM‐1400 Plus, 120 kV) and Bio‐HVEM (JEM‐1000BEF, 1000 kV) (Jeol, Japan). The percentage of the surface area was calculated, and the mean percentage of the surface area for each animal was used to determine the overall mean for each group.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 8 software (San Diego, CA, USA). We used one‐way ANOVA to compare quantitative data among groups and the Bonferroni post hoc test to adjust for multiple comparisons (P < 0.05).
Results
HFD feeding induces IR development
To examine whether the length of time on a HFD affects IR development, we fed mice a HFD for 2 weeks to induce acute IR and 24 weeks to induce chronic IR. We found that increases in mouse body weight and metabolic organ weight were dependent on the HFD feeding period (Figure S1a, b). Serum cholesterol and high‐density lipid (HDL)‐cholesterol significantly increased on a HFD (P < 0.01) (Table S2).
Both HFD‐S and HFD‐L significantly increased fasting blood glucose, insulin levels, and HOMA‐IR (P < 0.01) (Figure 1A–C). Fasting blood glucose and HOMA‐IR further increased with a longer HFD feeding period. We found HFD feeding induced glucose intolerance and IR, according to glucose tolerance and insulin sensitivity tests (Figure 1D,E). However, length of feeding period did not make a difference.
Figure 1.
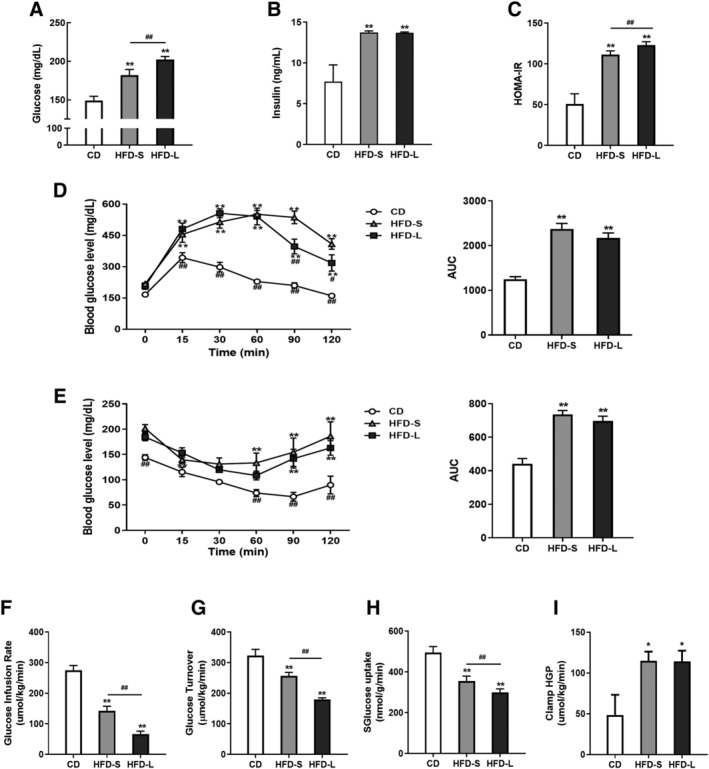
Induction of insulin resistance (IR) with short‐term and long‐term high‐fat diets (HFDs) in C56BL/6 mice. Fasting glucose levels (A), plasma insulin levels (B), and HOMA‐IR (C) of control diet (CD), 2 weeks of HFD (HFD‐S)‐, and 24 weeks of HFD (HFD‐L)‐fed mice. (D) Glucose tolerance test (left) with area under curve (AUC, right). (E) Insulin sensitivity test (left) with AUC (right). (F) Glucose infusion rate and (G) glucose turnover measured by hyperinsulinaemic‐euglycaemic clamp. (H) Impaired muscle glucose uptake measured by 2‐[14C]deoxyglucose (2‐DG) uptake. (I) Hepatic glucose production (HGP) during clamp. Values expressed as means ± SEM (n = 8). *P < 0.05, **P < 0.01, significantly different from CD. Mean differences detected by one‐way ANOVA test. # P < 0.05, ## P < 0.01, significant difference between HFD‐S and HFD‐L. Mean differences detected by Student's t‐test.
HFD changed mRNA expression levels of glucose 6‐phosphatase, phosphoenolpyruvate carboxykinase, and phosphofructokinase in liver and SKM, but the length of the HFD feeding period made no difference (Figure S1c). Measurements of fatty acid oxidation (FAO)‐related genes by qRT‐PCR demonstrated that the HFD‐evoked upregulation of FAO was only observed in HFD‐L mouse SKM (Figure S1d). However, increases in hepatic FAO‐related genes were observed in both HFD‐S and HFD‐L mice, with greater upregulation in HFD‐S mice than in HFD‐L mice (Figure S1e). An increase in FAO was induced by the activation of AMPK, which promotes a metabolic switch from glucose oxidation to FAO 16 in SKM (Figure S1f). A HFD impaired insulin signalling, and the effect was more severe in HFD‐L mice (Figure S1g).
To better understand the effect of a HFD on whole‐body and tissue‐specific glucose metabolism, we performed hyperinsulinaemic‐euglycaemic clamp studies. HFD feeding resulted in progressive decreases in glucose infusion rate and glucose turnover, indicating systemic IR (Figure 1F, G). We also observed a gradual decrease of glucose uptake in the SKM with a HFD (Figure 1H). In contrast, the increased clamp hepatic glucose production caused by HFD‐S persisted in the HFD‐L group (Figure 1I). These findings suggest that short‐term HFD is sufficient to cause systemic IR and that incremental IR progression can be observed after 24 weeks of HFD feeding.
Acute IR impairs mitochondrial function in SKM
As reduced mitochondrial capacity in SKM is suggested to underlie IR development, 3 , 17 , 18 we measured the mitochondrial function of gastrocnemius muscle in HFD‐S and HFD‐L mice. We found that HFD significantly decreased citrate synthase activity, ATP production, and mtDNA content (P < 0.01). The decreases were significantly greater for acute IR than chronic IR (P < 0.05) (Figure 2A–C). Mitochondrial biogenesis‐related genes were upregulated by HFD (Figure 2D)
Figure 2.
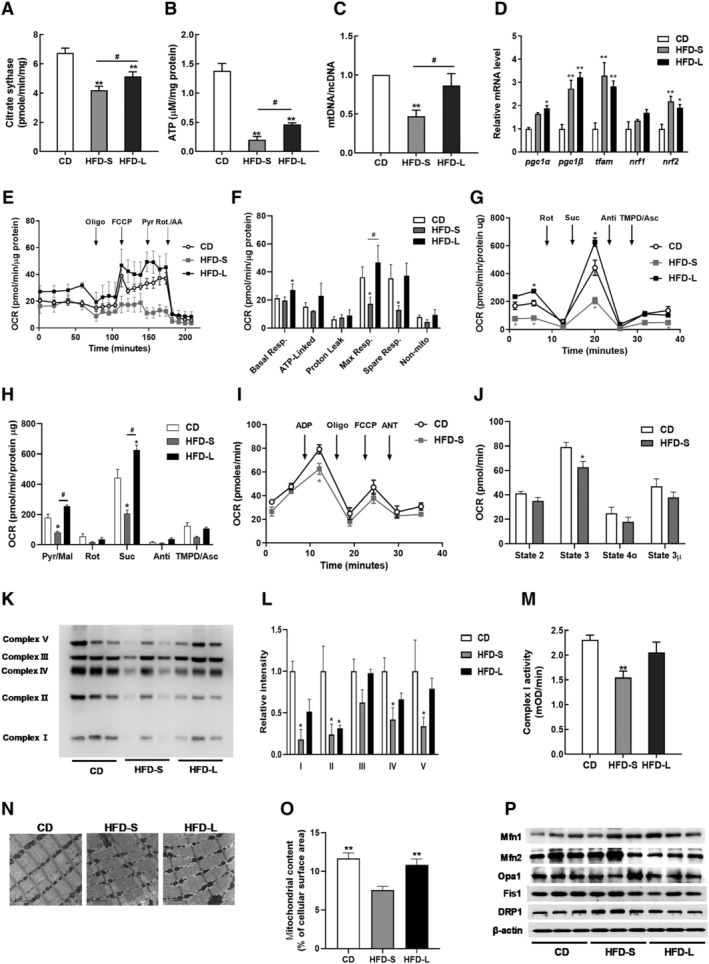
Impairment of skeletal mitochondrial function in acute IR. (A) Citrate synthase activity in gastrocnemius muscles from CD, HFD‐S, and HFD‐L groups. (B) Total ATP content in gastrocnemius muscles normalized to CD mice. (C) The mitochondrial DNA (mtDNA) content calculated as the ratio of Cox5 to Cyclophilin A DNA levels measured by quantitative PCR in gastrocnemius muscles. (D) Relative gene expressions for mitochondrial biogenesis and mtDNA replication measured by qRT‐PCR. (E) Representative oxygen consumption rate (OCR) curves, showing the response for each agent in isolated EDL myofibres. (F) Mitochondrial respiration by OCR quantification. (G) Complex‐dependent respiration determined by electron flow assay in mitochondrial proteins. (H) OCR quantification to measure complex‐dependent respiration. (I) Respiratory coupling determined by mitochondrial coupling assay. (J) OCR quantification to measure mitochondrial respiratory coupling. (K) Expression of OXPHOS Complexes in gastrocnemius muscles. (L) Quantification of OXPHOS complex expression by densitometry analysis. (M) Complex I activity in gastrocnemius muscles. (N) Representative mitochondrial morphology imaged by transmission electron microscopy (TEM). (O) Quantitative analyses of images to measure the mitochondrial surface area (n = 5). (P) Immunoblot analyses of proteins related to mitochondrial dynamics. Values expressed as means ± SEM (n = 8). *P < 0.05, **P < 0.01, significantly different from CD. Mean differences detected by ANOVA test. # P < 0.05, ## P < 0.01, significant difference between HFD‐S and HFD‐L. Mean differences detected by Student's t‐test.
To measure the mitochondrial function, we performed extracellular flux analysis in EDL muscle fibres. In brief, we measured basal OCR and OCR after sequential exposure to the ATP synthase inhibitor oligomycin A, the uncoupler FCCP, pyruvate, and electron transport chain inhibitors antimycin A/rotenone (Figure 2E). Basal respiration did not change with HFD‐S but increased with HFD‐L (Figure 2F). HFD‐S significantly decreased the FCCP‐evoked maximal respiration (P < 0.05), whereas HFD‐L had no effect (Figure 2F).
Next, we examined how SKM mitochondria responded to HFD‐induced IR. We isolated the mitochondrial fraction from SKM and performed an electron flow assay by monitoring the flow of electrons from complex I to complex IV in the presence of FCCP, an uncoupler that carries protons away through the inner membrane without forming a proton gradient. Therefore, this system does not produce ATP. Defects in a specific complex are determined by a decrease or increase in oxygen consumption when a complex‐specific substrate is provided. We used rotenone and antimycin A to inhibit complex I and complex III respiration. The flow of electrons through each complex (I–IV) was determined using pyruvate and malate for complex I, succinate for complex II, and ascorbate/TMPD for complex IV. As shown in Figure 2G, H, complex I and complex II respiration significantly decreased with HFD‐S (P < 0.05). Interestingly, HFD‐L did not affect complex I‐dependent respiration and significantly increased complex II‐dependent respiration relative to a CD. HFD had no effect on complex IV respiration.
Mitochondrial coupling is the process by which electron flow generates proton motive force via proton extrusion. To investigate mitochondrial electron flow in HFD‐S induced IR, we performed a mitochondrial coupling assay of SKM from mice with CD or HFD‐S. We determined the effect of HFD on basal respiration (state 2), phosphorylating respiration in presence of ADP (state 3), resting respirations in presence of oligomycin (state 4o), maximal respiration in presence of FCCP (state 3 μ), and the response to antimycin A (Figure 2I). We found that state 3 OCR significantly decreased with HFD‐S (Figure 2J, P < 0.05).
We measured the expression levels of electron transport chain proteins and found that expressions of complexes I, II, IV, and V decreased with HFD‐S (Figure 2K, L). With HFD‐L, only complex II expression decreased. HFD‐S also reduced complex I enzyme activity (Figure 2M). In addition, TEM showed a decrease in matrix density, loss of cristae structure, and smaller and shorter mitochondria in HFD‐S mice (Figure 2N). Quantitative morphometric analyses revealed a significant decrease in the mitochondrial content of HFD‐S SKM (P < 0.01) (Figure 2O). Western blot analysis of key mitochondrial components showed increased DRP1 expression in HFD‐S mice (Figure 2P). Taken together, these results indicate that that HFD‐S, but not HFD‐L, damages mitochondrial function in SKM.
Short‐term and long‐term HFD have variable effects on hepatic mitochondrial function
HFD‐induced hepatic steatosis and lipid accumulation increased as the HFD duration increased (Figure 3A, B). TC content was significantly higher with HFD‐L than HFD‐S (Figure 3C). However, increased triglyceride levels did not depend on HFD duration (Figure 3D).
Figure 3.
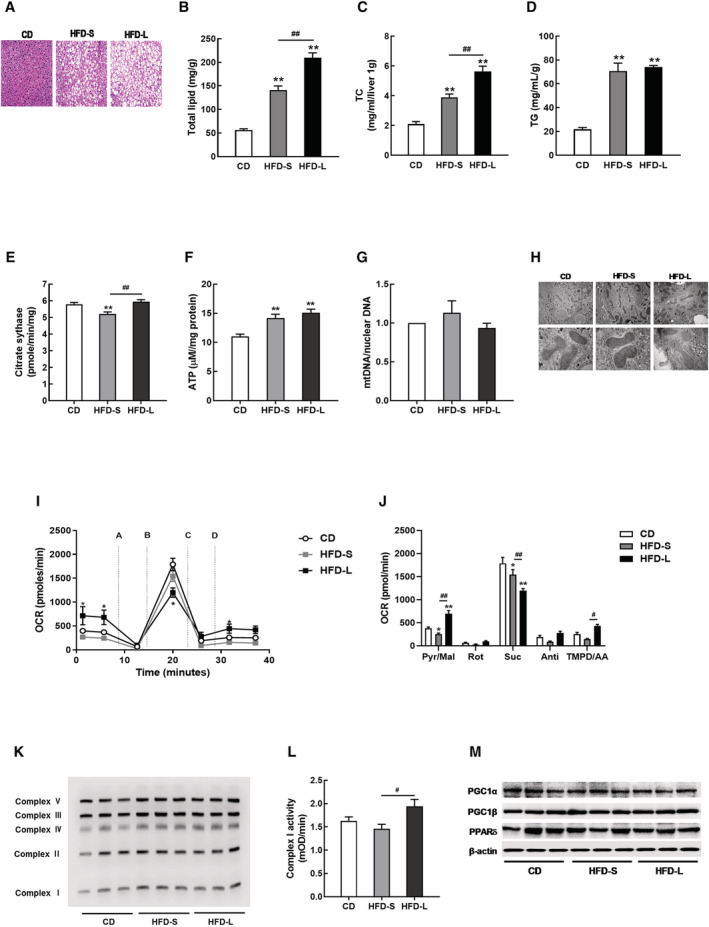
Hepatic mitochondrial dysfunction was not observed in acute IR. (A) Representative haematoxylin and eosin (H&E)‐liver sections from CD, HFD‐S, and HFD‐L mice. Hepatic lipid profiles, including total lipid content (B), total cholesterol (TC, C), and triglyceride (TG, D). (E) Citrate synthase activity in liver tissues from CD, HFD‐S, and HFD‐L groups. (F) Total ATP content in liver tissues. (G) The mitochondrial DNA (mtDNA) content calculated as the ratio of COX5 to Cyclophilin A DNA levels measured by quantitative PCR in liver tissues. (H) Mitochondrial morphology imaged by transmission electron microscopy (TEM). (I) Complex‐dependent respiration determined by electron flow assay in mitochondrial proteins. (J) OCR quantification to measure complex‐dependent respiration. (K) OXPHOS complex expression in liver tissues. (L) Complex I activity in liver tissues. (M) Expressions of PGC1 and PPARδ measured by western blot.
Next, we measured mitochondrial function in the livers of HFD‐S and HFD‐L mice. Only HFD‐S decreased citrate synthase activity (Figure 3E). HFD increased ATP content in liver, but we found no differences in mtDNA content or mitochondrial ultrastructure (Figure 3F–H).
The mitochondrial coupling assay showed that HFD‐S decreased complex I‐ and complex II‐dependent respirations in liver tissues (Figure 3I, J). In contrast to HFD‐S, HFD‐L significantly increased complex I‐ and complex IV‐dependent respirations (P < 0.01 and P < 0.05, respectively) and significantly decreased complex II‐dependent respiration in liver tissues (P < 0.01).
HFD did not affect complex I protein expression or activity (Figure 3K, L). Protein expressions of PGC1α/β and PPARδ did not change in steatotic liver tissues (Figure 3M).
Mdivi‐1 improves acute IR by recovering mitochondrial function
As described above, we found that acute IR induced by HFD‐S impaired mitochondrial dynamics and mitochondrial function in SKM. Inhibiting mitochondrial fission is known to improve insulin signalling in muscle. 19 Similarly, we found that glucose uptake and mitochondrial respiration improved in PA‐induced insulin‐resistant C2C12 cells treated with mdivi‐1 (Figure S2a–c). Thus, we hypothesized that improved mitochondrial function could attenuate the development of acute IR caused by HFD. To test this hypothesis, we administrated mdivi‐1 to HFD‐S mice and performed the glucose tolerance test (Figure 4A). Mdivi‐1 treatment decreased fasting blood glucose levels and improved glucose intolerance (Figure 4B–D). However, mdivi‐1 treatment did not change body weight (Figure S3a) or blood lipid profile (Table S3). Aspartate aminotransferase and alanine aminotransferase analyses demonstrated that mdivi‐1 treatment did not result in hepatotoxicity (Table S3).
Figure 4.
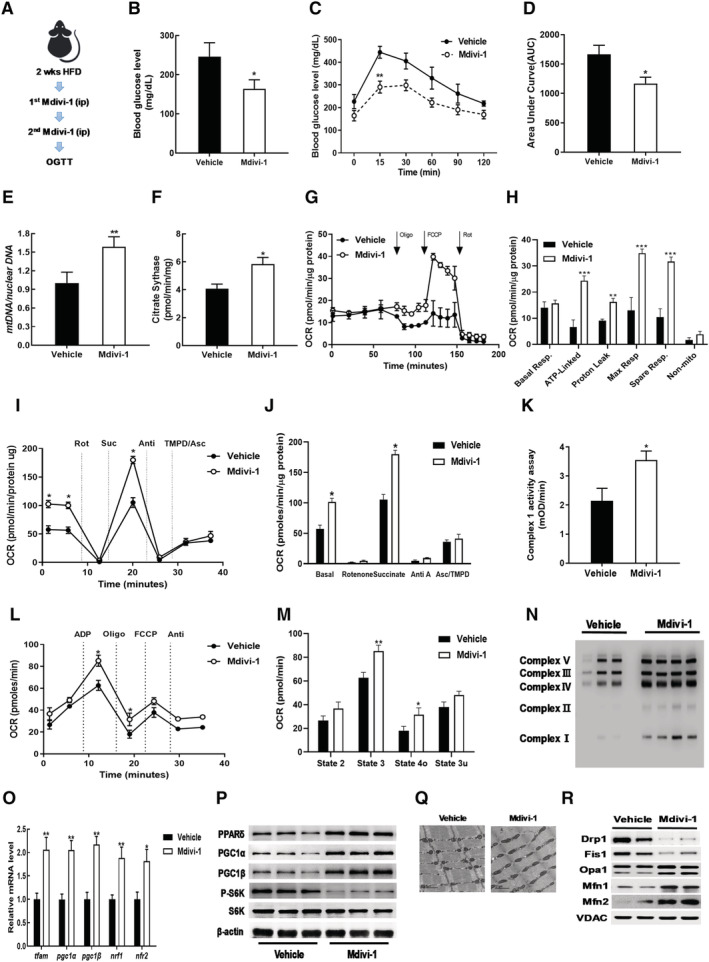
Mdivi‐1 administration improved acute IR via increased mitochondrial function. (A) Workflow for mdivi‐1 injections. (B) Blood glucose levels in plasma from vehicle DMSO or mdivi‐1 administrated HFD‐S mice. (C) Glucose tolerance test result. (D) AUC of glucose tolerance test result. (E) Mitochondrial DNA content in gastrocnemius muscles of mdivi‐1 treated mice versus normalized vehicle. (F) Citrate synthase activity in gastrocnemius muscles of mdivi‐1 group normalized to vehicle. (G) OCR measurements in EDL myofibres. (H) Analysis of mitochondrial respiration by OCR quantification. (I) Electron flow assay in isolated mitochondrial fraction of gastrocnemius muscles. (J) OCR quantification to measure complex‐dependent respiration. (K) Complex I activity in gastrocnemius muscles. (L) Mitochondrial respiratory coupling assay in isolated mitochondrial fraction of gastrocnemius muscles. (M) Quantification of respiratory coupling. (N) Expression of OXPHOS complex in gastrocnemius muscles. (O) Relative gene expressions for mitochondrial biogenesis and mtDNA replication measured by qRT‐PCR. (P) Western blot analysis for expression of PPARδ, PGC1α/β, S6K, and p‐S6K in gastrocnemius muscles. (Q) Mitochondrial morphology imaged by transmission electron microscopy (TEM). (R) Immunoblot analyses of proteins related to mitochondrial dynamics. Values expressed as means ± SEM and mean differences detected by Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001 versus Vehicle, n = 5.
Upon further examination of mitochondrial function, we found that mdivi‐1 treatment significantly increased mtDNA content (P < 0.01) and citrate synthase activity (P < 0.05) (Figure 4E, F). We performed extracellular flux analysis in isolated EDL myofibres and found that mdivi‐1 treatment significantly increased maximal respiration (**P < 0.001) (Figure 4G, H). An electron flow assay showed that both complex I‐dependent and complex II‐dependent respiration increased in the gastrocnemius of mdivi‐1 treated mice (Figure 4I, J). Mdivi‐1 treatment also increased complex I enzyme activity (Figure 4K).
Mitochondrial coupling assay confirmed that mdivi‐1 treatment increased state 3 and state 4o respirations (Figure 4L, M). Western blot analysis showed that mdivi‐1 treatment increased protein expression of oxidative phosphorylation (OXPHOS) complex subunits (Figure 4N).
To determine how mdivi‐1 treatment improved mitochondrial dysfunction induced by HFD‐S, we measured mitochondrial biogenesis related factors and observed that mdivi‐1 significantly upregulated transcriptional regulators of mitochondrial biogenesis, including pgc1, nrf1/2, and tfam (Figure 4O). PPARδ activation improves glucose metabolism and insulin sensitivity by inducing FAO, which is dependent on PGC1α and PGC1β. 20 Additionally, activation of PPARδ and PGC1α increases mitochondrial gene expression and function. 21 , 22 We found that mdivi‐1 treatment increased expression of PPARδ and PPARγ coactivator 1 (PGC1) proteins in gastrocnemius tissue (Figure 4P). Mdivi‐1 treatment also decreased phosphorylation of protein S6 kinase.
We next observed mitochondrial morphology via TEM and found that muscle treated with mdivi‐1 had a higher frequency of elongated mitochondria than vehicle‐treated muscle (Figure 4Q). Consistent with mitochondrial morphology, there were lower levels of proteins involved in mitochondrial fission (DRP1 and Fis1), whereas mdivi‐1 treatment increased levels of fusion‐associated factors such as optic atrophy 1 (Opa1) and mitofusins 1 and 2 (Mfn1/2) (Figure 4R). These results indicate that mdivi‐1 inhibition of mitochondrial fission improved acute IR induced by HFD‐S by improving mitochondrial function.
Mdivi‐1 decreases mitophagy and mitochondrial unfolded protein response (UPRmt) caused by acute IR
To address the cause mitochondrial dysfunction in SKM after HFD‐S, we measured autophagy‐related markers. As shown in Figure 5A, HFD‐S and HFD‐L increased LC3‐II and decreased p62/SQSTM1, a protein degraded by autophagy. This finding indicated increased autophagic flux during IR. We also observed increased Atg5 with HFD‐S and HFD‐L. Collectively, these results suggest the induction of canonical autophagy with HFD.
Figure 5.
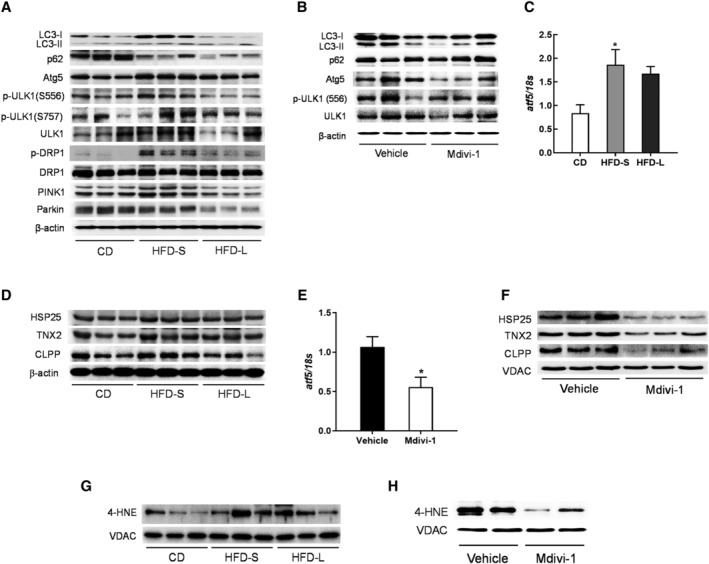
Mitochondrial oxidative stress is crucial for mitochondrial dysfunction in acute IR. (A) Western blot analysis for the expression of autophagy and mitophagy related markers in gastrocnemius tissues from CD, HFD‐S, and HFD‐L mice. (B) Western blot analysis of autophagy‐related markers in gastrocnemius muscles from vehicle DMSO or mdivi‐1 administrated HFD‐S mice. (C) qRT‐PCR analysis for ATF5 expression in gastrocnemius muscles from CD, HFD‐S, and HFD‐L mice (n = 8). (D) Western blot analysis of UPRmt targets in gastrocnemius muscles. (E) qRT‐PCR analysis for ATF5 expression in gastrocnemius muscles from vehicle DMSO or mdivi‐1 administrated HFD‐S mice (n = 5). (F) Western blot analysis of UPRmt targets in gastrocnemius muscles. (G) Western blot analysis of 4‐HNE in gastrocnemius muscles from CD, HFD‐S, and HFD‐L mice. (H) Western blot analysis of 4‐HNE in gastrocnemius muscles from vehicle or mdivi‐1 treated mice.
ULK1 contains various phosphorylation sites, including S757 for ULK1 inactivation by mammalian target of rapamycin complex (mTOR) 1 and S556 for ULK1 activation by AMPK. 23 We observed that ULK1 phosphorylation on S556 increased with HFD‐S but not HFD‐L (Figure 5A). A HFD did not affect S757 phosphorylation on ULK1. Mitophagy is the autophagic turnover of damaged mitochondria that maintains mitochondrial integrity. 24 DRP1‐dependent mitochondrial fission is required to initiate mitophagy. 25 HFD‐S increased DPR‐1 phosphorylation and PTEN‐induced kinase 1 (PINK1) expression (Figure 5A), suggesting induction of mitophagy by HFD‐S. However, HFD‐L decreased PINK1 and Parkin expression. In contrast, mdivi‐1 treatment effectively reversed changes in the protein expression of autophagy markers. We observed mdivi‐1 treatment decreased LC‐II accumulation, Atg5 expression, and ULK1 phosphorylation (Figure 5B). These results suggest that autophagy induction by HFD‐S resulted from an adaptive response to mitochondrial damage.
UPRmt is a conserved adaptive transcriptional stress response that promotes recovery of mitochondrial function. 26 We examined whether UPRmt was involved in the perturbation of mitochondrial function with HFD‐S. We measured mRNA expression of ATF5, a transcription factor that mediates mammalian UPRmt, 27 in SKM tissues and found that HFD‐S induced ATF5 upregulation (Figure 5C). Then we investigated UPRmt activation by measuring UPRmt biomarkers that are downstream targets of ATF5 (Figure 5D). Similar to ATF5 findings, we found upregulation of HSP25, TNX2, and CLPP with HFD‐S and HFD‐L, though the increase was greater with HFD‐S than HFD‐L. Conversely, mdivi‐1 treatment decreased expression of ATF5 and UPRmt biomarkers (Figure 5E,F). Overall, these data indicate that mitochondrial dysfunction during IR induced UPRmt.
Finally, we investigated how oxidative stress affected mitochondrial dysfunction caused by HFD. ROS are an important mediator of metabolic dysfunction. 28 To address whether oxidative stress‐induced lipid peroxidation is associated with mitochondrial dysfunction in HFD‐S, we measured expression of 4‐hydroxynonenal (4‐HNE) expression in the mitochondrial fraction. Derived from lipid hydroperoxides, 4‐HNE inhibits mitochondrial function by the formation of protein adducts. 29 We found that HFD‐S, but not HFD‐L, increased mitochondrial 4‐HNE (Figure 5G). This finding indicates that HFD‐S induced oxidative stress of mitochondria. Interestingly, mdivi‐1 treatment decreased 4‐HNE expression (Figure 5H). In conclusion, these results showed that mitochondrial oxidative stress is crucial to induce mitochondrial dysfunction which is associated with HFD‐S and subsequent IR.
Discussion
Considerable evidence indicates that impaired OXPHOS is associated with IR development and metabolic disease. 30 However, it is not clear whether impaired mitochondrial oxidative activity causes IR or vice versa. In this study, we show that impaired mitochondrial function causes acute IR induced by HFD‐S. Furthermore, improved mitochondrial function attenuates glucose intolerance in acute IR. Therefore, impaired mitochondrial function contributes to development of acute IR in mice.
The primary evidence linking mitochondrial dysfunction to IR comes from a study of obese and IR individuals who had reduced SKM mitochondrial oxidative capacity. 31 Further, diabetes‐prone individuals have decreased mitochondrial respiration. 32 Previously, a comparison of mice fed a HFD for 2 weeks with those fed a HFD for 12–16 weeks suggested that glucose intolerance by a long‐term HFD resulted from inflammation in white adipose tissue and SKM. 33 However, short‐term HFD‐fed mice had no changes in inflammatory markers; thus, the cause of IR development due to short‐term HFD remains unknown. Thus, we hypothesized that mitochondrial dysfunction is involved in acute IR development. To prove this hypothesis, we induced IR by feeding mice a HFD for 2 or 24 weeks to induce acute IR or chronic IR, respectively, and compared mitochondrial function in SKM and liver. Consistent with a previous study, 34 2 weeks of HFD was sufficient to induce IR, though IR severity increased with longer HFD duration.
Mitochondria are the major site of fatty acid β‐oxidation and accumulation of hepatic fatty acid can lead to hepatic IR. 35 Thus, mitochondrial dysfunction is involved in the development hepatic fatty acid‐induced IR. We found that hepatic lipid content increased as HFD duration increased. However, mitochondrial dysfunction did not increase as HFD duration increased. Moreover, HFD increased ATP synthesis. Collectively, mitochondrial dysfunction was not observed in the liver under conditions of IR. The reason for this uncoupling is unclear, but it has been postulated that mitochondrial dysfunction is not a prerequisite for liver steatosis. 36
Because SKM is responsible for most insulin‐stimulated glucose uptake in the body, dysregulation of SKM metabolism has a profound effect on systemic glucose homeostasis and insulin sensitivity. 37 In acute IR, a decrease in mitochondrial content was confirmed by measurements of mitochondrial DNA content and mitochondrial surface volume. The mtDNA copy number can be determined by the mtDNA/nuclear DNA (ncDNA) ratio by qRT‐PCR. 38 This analysis must be carefully considered under conditions of muscle atrophy where the nuclear density is increased. 39 We and another group demonstrated that a HFD causes muscle atrophy. 40 , 41 In the current study, however, the decrease of muscle mtDNA content under acute IR conditions was negligible because 2 weeks of HFD feeding was insufficient to induce muscle atrophy.
Mitochondrial bioenergetics can be used to understand real‐time cellular metabolism in EDL myofibres 42 and isolated mitochondria. 14 We found that SKM of HFD‐S mice had lower maximal respiration and spare respiratory capacity. Electron flow and mitochondrial coupling assays using isolated mitochondria confirmed that HFD‐S mice had lower mitochondrial respiration than controls. Conversely, HFD‐L mice had increased OCR in myofibres and mitochondria isolated from muscle, suggesting that a chronic HFD increases mitochondrial function in SKM to offset an oversupply of fatty acids. Among the biochemical measurements for mitochondrial function, cardiolipin content, citrate synthase activity, and complex I activities are the biomarkers that exhibit the strongest association with mitochondrial content, whereas complex IV activity is strongly associated with OXPHOS capacity in human SKM. 43 We observed greater mitochondrial dysfunction in SKM from HFD‐S mice than from HFD‐L mice, as shown by citrate synthase activity, ATP content, and mtDNA content. Measurements of mitochondrial bioenergetics using the Seahorse XF24 Analyzer also showed similar results. These findings provide a strong rationale for the involvement of mitochondrial dysfunction in the development of acute IR.
Mitochondrial dynamics is the balance of fission and fusion events. Mitochondrial fusion is controlled by Mfn1, Mfn2, and Opa1, whereas mitochondrial fission is controlled by DRP1 and mitochondrial fission factor. 44 Dysfunctional mitochondrial dynamics is associated with various diseases. 45 Mitochondrial fission is increased in obese mice; inhibiting mitochondrial fission by mdivi‐1 improves muscle insulin signalling and systemic insulin sensitivity. 19 Consistent with this information, we observed that mitochondrial fission increased as DRP1 expression increased in the muscles of HFD‐S mice. Mdivi‐1, a putative DRP1 inhibitor, inhibits palmitate‐evoked IR by reducing mitochondrial fragmentation. 19 Recently, mdivi‐1 was shown to attenuate IR induced by 4 weeks of HFD supplementation in mice. 46 We also found that mdivi‐1 diminished the reduction in glucose uptake caused by fatty acids and also increased OCR in C2C12 cells. Additionally, mdivi‐1 treatment reduced glucose intolerance by increasing mitochondrial function in HFD‐S mice. Mitochondrial dysfunction in HFD‐S mice suggests that boosting mitochondrial function may be a therapeutic strategy for IR.
Malfunction of autophagy has been linked to the incidence of type 2 diabetes; the activation of autophagy has been shown to increase insulin sensitivity in insulin‐responsive tissues such as muscle and liver in mice. 47 In our study, short‐term HFD‐induced autophagy, as shown by increased LC3‐II and Atg5 expression and decreased p62 expression. These findings indicate that the accumulation of dysfunctional mitochondria induces autophagy. Mitochondrial damage induces stress responses and adaptations, such as mitophagy and UPRmt. 48 Mitophagy is a highly selective type of autophagy that selectively degrades damaged mitochondria to ensure the overall quality of mitochondria. 24 Mitophagy is regulated by Parkin and PINK1 in mammals, and mutations in these two genes are associated with Parkinson's disease. 24 Mitophagy can occur via Parkin‐independent mechanisms, such as those activated by receptor‐mediated mitophagy. Dysfunctional mitochondrial are specifically recognized by receptors, such as FUDNC1, NIX, BNIP3, PHB2, NLRX1, and BCL2L13, which connect mitochondria with LC3‐II in the autophagosomal membrane. 49 , 50 DRP1 is involved in the regulation of autophagy and is activated by phosphorylation on Ser616. 51 In acute IR, DRP1 phosphorylation and increased PINK1 expression induce mitophagy. However, autophagy induction decreases after mdivi‐1 treatment. These findings suggest that mitophagy can result from acute IR‐evoked mitochondrial damage.
UPRmt is an adaptive transcriptional response activated by misfolded protein in the mitochondria. 52 UPRmt is regulated by ATFS‐1 in C. elegans or ATF‐5 in mammals, which has both a mitochondrial targeting sequence and a nuclear localization sequence. 53 Under homeostatic conditions, ATF‐5 is imported into the mitochondrial matrix and degraded by the protease. 54 However, under mitochondrial stress, mitochondrial import of ATFS‐1 decreases and nuclear accumulation of ATFS‐1 increases. Thus, UPRmt activation occurs when import efficiency of the mitochondrial network decreases. Impairment of OXPHOS genes, such as complex III or IV, also activates the UPRmt. 55 We found that HFD‐S caused upregulation of ATF5 and an increase in UPRmt related markers in muscle tissue, suggesting short‐term HFD‐induced UPRmt in SKM tissue. Similar to mitophagy, mdivi‐1 treatment reduced UPRmt. Our results suggest that mitochondrial damage from HFD‐S induced mitophagy and UPRmt as stress responses in muscle tissue.
Compared with HFD‐S, HFD‐L exerts less damage to mitochondrial function in SKM tissues, such as increased mitochondrial biogenesis, ATP production, complex I activity, and mitochondrial respiration. The reason for this phenomenon is unclear, but it may be that increased autophagy and the induction of UPRmt may affect mitochondrial function because we observed increased autophagy (autophagic flux and the phosphorylation of ULK‐1 and DRP1) and UPRmt (induction of ATF5 and its targets) in SKM from mice with chronic IR.
Excessive ROS production is a key trigger and regulator of IR. 56 , 57 Mitochondria are regarded as the major site of ROS production, predominantly in electron transport chain complexes I and III. 58 ROS produced during oxidative phosphorylation primarily trigger oxidative damage to mitochondria and other macromolecules, such as DNA, proteins, and lipids. 59 Cell membrane phospholipids, including linoleic acid and arachidonic acid, are major targets for 4‐HNE, a destructive by‐product formed in mitochondria by cardiolipin oxidation. 60 , 61 Besides membrane lipids, proteins and DNA are the principal targets of 4‐HNE attack. 62 Thus, 4‐HNE is a critical biomarker for oxidative stress. We showed that HFD‐S increased 4‐HNE levels in the mitochondrial fraction of muscle tissue, indicating that mitochondrial oxidative stress is induced by short‐term HFD. Similarly, a short‐term high‐calorie diet is known to increase mitochondrial oxidative stress and expression of OXPHOS enzyme in brain. 63 Mitochondrial oxidative stress inhibits insulin signalling and interferes with acetyl‐CoA oxidation, consequently resulting in IR. 56 We saw that mdivi‐1 treatment reduced 4‐HNE levels as IR decreased, suggesting that mitochondrial oxidative stress leads to acute IR development in SKM.
In conclusion, we show that short‐term HFD‐induced acute mitochondrial dysfunction in SKM, indicating that mitochondrial dysfunction is a prerequisite for acute IR. In HFD‐S mice with acute IR, oxidative stress‐induced mitochondrial damage and led to mitochondrial dysfunction. Increased mitochondrial activity reduced glucose intolerance in acute IR. In conclusion, mitochondrial dysfunction in SKM plays a major role in acute IR development in mice.
Author contributions
J.A. and T.Y.H. designed the experiments. J.A., H.L., and F.N. performed the cell and animal studies. C.H.J. conducted western blots. S.Y.P. performed the clamp experiment. H.Y.H. conducted the TEM image analysis.
Conflict of interest
There are no conflict of interests to declare.
Supporting information
Figure S1. Effect of HFD feeding period on body weights, organs weights, glucose/fat metabolism‐related genes, and insulin signaling.
Figure S2. Mdivi‐1 increased glucose uptake and mitochondrial respiration in FA‐evoked insulin resistant C2C12 cells.
Figure S3. Mdivi‐1 administration increases mitochondrial biogenesis and complex I activity.
Table S1. Primer sequences used for qRT‐PCR.
Table S2. Serum lipid levels.
Table S3. Serum biochemical analysis.
Acknowledgements
This work was supported by a grant from the Korea Food Research Institute (E0210101). The authors certify compliance with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 64 All experimental procedures performed on the mice were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Korea Food Research Institute (KFRI‐M‐14002).
Lee H., Ha T. Y., Jung C. H., Nirmala F. S., Park S.‐Y., Huh Y. H., and Ahn J. (2021) Mitochondrial dysfunction in skeletal muscle contributes to the development of acute insulin resistance in mice, Journal of Cachexia, Sarcopenia and Muscle, 12, 1925–1939, 10.1002/jcsm.12794
References
- 1. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 2016;126:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006;55:S9–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin‐resistant offspring of patients with type 2 diabetes. N Engl J Med 2004;350:664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khalyfa A, Wang Y, Zhang SX, Qiao Z, Abdelkarim A, Gozal D. Sleep fragmentation in mice induces nicotinamide adenine dinucleotide phosphate oxidase 2‐dependent mobilization, proliferation, and differentiation of adipocyte progenitors in visceral white adipose tissue. Sleep 2014;37:999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caro JF, Dohm LG, Pories WJ, Sinha MK. Cellular alterations in liver, skeletal muscle, and adipose tissue responsible for insulin resistance in obesity and type II diabetes. Diabetes Metab Rev 1989;5:665–689. [DOI] [PubMed] [Google Scholar]
- 6. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res 2008;102:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 1999;277:E1130–E1141. [DOI] [PubMed] [Google Scholar]
- 8. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387. [DOI] [PubMed] [Google Scholar]
- 9. Phielix E, Meex R, Ouwens DM, Sparks L, Hoeks J, Schaart G, et al. High oxidative capacity due to chronic exercise training attenuates lipid‐induced insulin resistance. Diabetes 2012;61:2472–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holloszy JO. Skeletal muscle "mitochondrial deficiency" does not mediate insulin resistance. Am J Clin Nutr 2009;89:463S–466S. [DOI] [PubMed] [Google Scholar]
- 11. Schiff M, Loublier S, Coulibaly A, Benit P, de Baulny HO, Rustin P. Mitochondria and diabetes mellitus: untangling a conflictive relationship? J Inherit Metab Dis 2009;32:684–698. [DOI] [PubMed] [Google Scholar]
- 12. Kwon MJ, Ju TJ, Heo JY, Kim YW, Kim JY, Won KC, et al. Deficiency of clusterin exacerbates high‐fat diet‐induced insulin resistance in male mice. Endocrinology 2014;155:2089–2101. [DOI] [PubMed] [Google Scholar]
- 13. Schuh RA, Jackson KC, Khairallah RJ, Ward CW, Spangenburg EE. Measuring mitochondrial respiration in intact single muscle fibers. Am J Physiol Regul Integr Comp Physiol 2012;302:R712–R719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 2011;6:e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497–509. [PubMed] [Google Scholar]
- 16. Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang Cc C, Itani SI, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl‐CoA carboxylase inhibition and AMP‐activated protein kinase activation. Proc Natl Acad Sci U S A 2002;99:16309–16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Short KR, Nair KS, Stump CS. Impaired mitochondrial activity and insulin‐resistant offspring of patients with type 2 diabetes. N Engl J Med 2004;350:2419–2421. [DOI] [PubMed] [Google Scholar]
- 18. Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005;54:8–14. [DOI] [PubMed] [Google Scholar]
- 19. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 2012;32:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleiner S, Nguyen‐Tran V, Bare O, Huang X, Spiegelman B, Wu Z. PPAR{delta} agonism activates fatty acid oxidation via PGC‐1{alpha} but does not increase mitochondrial gene expression and function. J Biol Chem 2009;284:18624–18633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga‐Ocampo CR, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol 2004;2:e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vega RB, Huss JM, Kelly DP. The coactivator PGC‐1 cooperates with peroxisome proliferator‐activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 2000;20:1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fonseca TB, Sanchez‐Guerrero A, Milosevic I, Raimundo N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019;570:E34–E42. [DOI] [PubMed] [Google Scholar]
- 26. Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS‐1 promotes OXPHOS recovery during the UPR (mt). Mol Cell 2015;58:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 2016;26:2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sci 2016;148:183–193. [DOI] [PubMed] [Google Scholar]
- 29. Humphries KM, Yoo Y, Szweda LI. Inhibition of NADH‐linked mitochondrial respiration by 4‐hydroxy‐2‐nonenal. Biochemistry 1998;37:552–557. [DOI] [PubMed] [Google Scholar]
- 30. Sergi D, Naumovski N, Heilbronn LK, Abeywardena M, O'Callaghan N, Lionetti L, et al. Mitochondrial (dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front Physiol 2019;10:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950. [DOI] [PubMed] [Google Scholar]
- 32. Phielix E, Schrauwen‐Hinderling VB, Mensink M, Lenaers E, Meex R, Hoeks J, et al. Lower intrinsic ADP‐stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008;57:2943–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams LM, Campbell FM, Drew JE, Koch C, Hoggard N, Rees WD, et al. The development of diet‐induced obesity and glucose intolerance in C57BL/6 mice on a high‐fat diet consists of distinct phases. PLoS One. 2014;9:e106159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raher MJ, Thibault HB, Buys ES, Kuruppu D, Shimizu N, Brownell AL, et al. A short duration of high‐fat diet induces insulin resistance and predisposes to adverse left ventricular remodeling after pressure overload. Am J Physiol Heart Circ Physiol 2008;295:H2495–H2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. A possible link between hepatic mitochondrial dysfunction and diet‐induced insulin resistance. Eur J Nutr 2016;55:1–6. [DOI] [PubMed] [Google Scholar]
- 36. Franko A, von Kleist‐Retzow JC, Neschen S, Wu M, Schommers P, Bose M, et al. Liver adapts mitochondrial function to insulin resistant and diabetic states in mice. J Hepatol 2014;60:816–823. [DOI] [PubMed] [Google Scholar]
- 37. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A 2007;104:12587–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Venegas V, Wang J, Dimmock D, Wong LJ. Real‐time quantitative PCR analysis of mitochondrial DNA content. Curr Protoc Hum Genet 2011. [DOI] [PubMed] [Google Scholar]
- 39. Gundersen K, Bruusgaard JC. Nuclear domains during muscle atrophy: nuclei lost or paradigm lost? J Physiol 2008;586:2675–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abrigo J, Rivera JC, Aravena J, Cabrera D, Simon F, Ezquer F, et al. High fat diet‐induced skeletal muscle wasting is decreased by mesenchymal stem cells administration: implications on oxidative stress, ubiquitin proteasome pathway activation, and myonuclear apoptosis. Oxid Med Cell Longev 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoo A, Jang YJ, Ahn J, Jung CH, Seo HD, Ha TY. Chrysanthemi Zawadskii var. latilobum attenuates obesity‐induced skeletal muscle atrophy via regulation of PRMTs in skeletal muscle of mice. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li R, Steyn FJ, Stout MB, Lee K, Cully TR, Calderon JC, et al. Development of a high‐throughput method for real‐time assessment of cellular metabolism in intact long skeletal muscle fibre bundles. J Physiol 2016;594:7197–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 2012;590:3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet 2009;18:R169–R176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol 2015;33:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kugler BA, Deng W, Duguay AL, Garcia JP, Anderson MC, Nguyen PD, et al. Pharmacological inhibition of dynamin‐related protein 1 attenuates skeletal muscle insulin resistance in obesity. Physiol Rep 2021;9:e14808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamamoto S, Kuramoto K, Wang N, Situ X, Priyadarshini M, Zhang W, et al. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep 2018;23:3286–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pellegrino MW, Haynes CM. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol 2015;13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol 2019;20:433–446. [DOI] [PubMed] [Google Scholar]
- 50. Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age‐associated diseases. Front Cell Dev Biol 2020;8:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin JR, Shen WL, Yan C, Gao PJ. Downregulation of dynamin‐related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler Thromb Vasc Biol 2015;35:1413–1422. [DOI] [PubMed] [Google Scholar]
- 52. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 2002;21:4411–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF‐1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans . Mol Cell 2010;37:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science 2012;337:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment‐specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 2004;117:4055–4066. [DOI] [PubMed] [Google Scholar]
- 56. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006;440:944–948. [DOI] [PubMed] [Google Scholar]
- 57. Dong K, Ni H, Wu M, Tang Z, Halim M, Shi D. ROS‐mediated glucose metabolic reprogram induces insulin resistance in type 2 diabetes. Biochem Biophys Res Commun 2016;476:204–211. [DOI] [PubMed] [Google Scholar]
- 58. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hu F, Liu F. Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal 2011;23:1528–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uchida K. 4‐Hydroxy‐2‐nonenal: a product and mediator of oxidative stress. Prog Lipid Res 2003;42:318–343. [DOI] [PubMed] [Google Scholar]
- 61. Liu W, Porter NA, Schneider C, Brash AR, Yin H. Formation of 4‐hydroxynonenal from cardiolipin oxidation: intramolecular peroxyl radical addition and decomposition. Free Radic Biol Med 2011;50:166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao Y, Miriyala S, Miao L, Mitov M, Schnell D, Dhar SK, et al. Redox proteomic identification of HNE‐bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment. Free Radic Biol Med 2014;72:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Corral‐Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet 1992;2:324–329. [DOI] [PubMed] [Google Scholar]
- 64. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of HFD feeding period on body weights, organs weights, glucose/fat metabolism‐related genes, and insulin signaling.
Figure S2. Mdivi‐1 increased glucose uptake and mitochondrial respiration in FA‐evoked insulin resistant C2C12 cells.
Figure S3. Mdivi‐1 administration increases mitochondrial biogenesis and complex I activity.
Table S1. Primer sequences used for qRT‐PCR.
Table S2. Serum lipid levels.
Table S3. Serum biochemical analysis.