

Abstract
Background
Skeletal muscle as a metabolic consumer determines systemic energy homeostasis by regulating myofibre type conversion and muscle mass control. Perturbation of the skeletal muscle metabolism elevates the risk of a variety of diseases including metabolic disorders. However, the regulatory pathways and molecules are not completely understood. The discovery of relevant responsible molecules and the associated network could be an attractive strategy to overcome diseases associated with muscle problems.
Methods
An initial screening using quantitative trait locus analysis enabled us to extract a set of genes including ubiquitin‐specific proteases21 (USP21) (r = 0.738; P = 0.004) as potential targets associated with fasting blood glucose content. Given tight regulation of the ubiquitination status of proteins in muscle, we focused on USP21 and generated whole‐body (KO) and skeletal muscle‐specific USP21 knockout (MKO) mice. Transcriptomics, proteomics, and lipidomics assays in combination with various in vivo and in vitro experiments were performed to understand the functions of USP21 and underlying mechanisms. A high‐fat diet (60%)‐fed mouse model and diabetic patient‐derived samples were utilized to assess the effects of USP21 on energy metabolism in skeletal muscle.
Results
USP21 was highly expressed in both human and mouse skeletal muscle, and controlled skeletal muscle oxidative capacity and fuel consumption. USP21‐KO or USP21‐MKO significantly promoted oxidative fibre type changes (Δ36.6% or Δ47.2%), muscle mass increase (Δ13.8% to Δ22.8%), and energy expenditure through mitochondrial biogenesis, fatty acid oxidation, and UCP2/3 induction (P < 0.05 or P < 0.01). Consistently, cold exposure repressed USP21 expression in mouse skeletal muscle (Δ55.3%), whereas loss of USP21 increased thermogenesis (+1.37°C or +0.84°C; P < 0.01). Mechanistically, USP21 deubiquitinated DNA‐PKcs and ACLY, which led to AMPK inhibition. Consequently, USP21 ablation diminished diet‐induced obesity (WT vs. USP21‐KO, Δ8.02 g, 17.1%, P < 0.01; litter vs. USP21‐MKO, Δ3.48 g, 7.7%, P < 0.05) and insulin resistance. These findings were corroborated in a skeletal muscle‐specific gene KO mouse model. USP21 was induced in skeletal muscle of a diabetic patient (1.94‐fold), which was reciprocally changed to p‐AMPK (0.30‐fold).
Conclusions
The outcomes of this research provide novel information as to how USP21 in skeletal muscle contributes to systemic energy homeostasis, demonstrating USP21 as a key molecule in the regulation of myofibre type switch, muscle mass control, mitochondrial function, and heat generation and, thus, implicating the potential of this molecule and its downstream substrates network as targets for the treatment and/or prevention of muscle dysfunction and the associated metabolic diseases.
Keywords: USP21, Skeletal muscle, Oxidative fibre type, Muscle mass control, Obesity, Diabetes
Introduction
Skeletal muscle is a major determinant of systemic energy homeostasis due to its large mass and high rate of fuel consumption. 1 Catabolism of lipids from myocellular triglycerides and circulating fatty acids (FAs) is enhanced in oxidative fibres to meet fuel demand. Perturbation of skeletal muscle metabolism may thus elevate the risk of metabolic diseases, 2 which is attributed to muscle fibre type composition and muscle mass. 3 In addition, progressive defect of muscle mass and/or quality during aging is linked to low energy expenditure and fat accumulation in the body, 4 implying the importance of skeletal muscle function in systemic fuel catabolism. Moreover, individuals suffering from muscle disorders such as sarcopenia and cachexia exhibit abnormal muscle wasting caused by elevated protein breakdown in association with metabolic disturbances. 4 , 5
Because patients with metabolic diseases have fewer oxidative myofibre, the recomposition of myofibre to oxidative phenotype and related mitochondrial function are closely linked to protection from obesity and diabetes. 3 , 6 Currently, the myofibre type conversion can be accomplished principally by endurance exercise. So, identifying the molecules responsible for regulating myofibre type composition, muscle mass, and energy expenditure may have a profound impact to overcome muscle dysfunction and metabolic diseases. Nonetheless, the basis for myofibre phenotype and energy expenditure control, particularly in the aspect of quality controlling and regulatory systems of protein ubiquitination, remains largely unknown.
In skeletal muscle, the balance between protein synthesis and degradation needs to be tightly regulated to properly maintain organ function and homeostasis. 5 Protein ubiquitination is one of the most common post‐translational modifications that affect biological functions through protein stability and molecular trafficking. 7 Ubiquitin modifications are counter‐balanced by the opposing actions of E3 ligases and deubiquitinases. 7 Ubiquitin‐specific proteases (USPs) belong to the largest group of the deubiquitinase family that play roles in fine‐tuning the ubiquitination status of targets and their fates, 8 suggestive of their roles in quality control of skeletal muscle and energy metabolism. In our quantitative trait locus analysis of fasting glucose levels, we encountered the association of a set of genes with glucose metabolism and focused on USP21 as a potential target for the regulation of systemic energy homeostasis.
Here, we report the identification of USP21 and its substrates network as new targets for muscle quality control, fibre type composition, and thermogenesis in skeletal muscle. Our findings indicate that ablation of USP21 enhances energy expenditure by promoting oxidative myofibre type switch, mitochondrial biogenesis and fuel oxidation, uncoupler induction, and thermogenesis. Leveraging RNA‐sequencing (RNA‐seq), proteomics, and lipidomics, our studies reveal DNA‐dependent protein kinase, catalytic subunit (DNA‐PKcs) and adenosine triphosphate (ATP) citrate lyase (ACLY) to be substrates of USP21 that control AMP‐activated protein kinase (AMPK) activity. As a functional outcome, USP21 deficiency decreases obesity and diabetes induced by a high‐fat diet (HFD) feeding. Additionally, we found USP21 up‐regulation in the skeletal muscle of a diabetic patient and diet‐induced obese mice, and its down‐regulation in animals subjected to exercise or cold conditions. Our findings demonstrate a novel rheostatic role of USP21 in balancing fuel storage and energy expenditure in skeletal muscle, providing alternative ways for therapeutic targeting in muscle dysfunction and the associated metabolic diseases.
Methods
Cell lines and cell culture
Mouse C2C12 cells and human HEK 293 cells were obtained from American Type Culture Collection. C2C12 cells were grown in Dulbecco's modified Eagle's medium (DMEM, GIBCO, 11995‐065) supplemented with 10% foetal bovine serum (FBS, GIBCO, 26140‐079) and 100 units/mL penicillin and 100 μg/mL streptomycin (Hyclone, SV30010). Differentiation medium (DMEM containing 2% horse serum, GIBCO, 16050‐122) was used to differentiate confluent C2C12 cells to myotubes. All studies were performed using differentiated myotubes. Human female HEK293 cells were maintained in DMEM (Hyclone, SH.30243.01) with MEM NEAA (GIBCO, 11140‐050), containing 10% FBS and 100 μg/mL penicillin/streptomycin. The cells before passage 20 were used.
Animal studies
All animal studies were approved by the Institutional Animal Care and Use Committee of Seoul National University or of the Center of Animal Care and Use at Lee Gil Ya Cancer and Diabetes Institute, Gachon University, and were conducted under the guidelines. Mice were maintained in a specific pathogen‐free barrier area at ambient temperature (22 ± 2°C) and humidity (55 ± 10%), on a 12/12 h light–dark cycle (7 a.m. to 7 p.m.) and fed rodent normal chow diet (ND, Purina lab, 38057) ad libitum unless otherwise stated. Usp21 tm1a/tm1a mice were purchased from Infrafrontier. Protamine‐Flpe mice were generously provided by Dr SH Lee, Seoul National University, Korea. Muscle creatine kinase (Mck)‐Cre mice were purchased from the Jackson Laboratory. Usp21 tm1a/tm1a mice were used to examine USP21‐knockout (KO) phenotype. Protamine‐Flpe mice were crossed with Usp21 tm1a/tm1a mice to generate control littermates (Usp21 tm1c/tm1c ) for the skeletal muscle‐specific USP21‐KO (USP21‐MKO; Usp21 tm1d/tm1d ), which were bred with Mck‐Cre mice to produce the USP21‐MKO mice. All mice used were male and backcrossed to C57BL/6 background. Validation of the line was assessed by genotyping genomic DNA from pups isolated from an ear punch of two‐weeks‐old pups using the Maxime™ polymerase chain reaction (PCR) PreMix (i‐Taq) (iNtRON Bio, 25026). PCR primer pairs for genotyping are listed in Supporting information, Table S2. Animal group sizes were determined by our experiences or similar previous studies. Age‐matched and weight‐matched mice were randomly sub‐grouped and were subjected to study, but the experimenters were not blinded to the genotype of the mice because of the experimental design. For cold tolerance tests, USP21‐KO [or wild type (WT)] mice and USP21‐MKO (or litter) mice were placed in individual cages at 4°C. Rectal temperature measurements were performed using a rectal thermometer (Physitemp, BAT‐7001H) to monitor core temperatures for the indicated times. For the fasting model, WT and USP21‐KO mice were fed ad libitum and then fasted for 24 h. For an insulin injection study, overnight fasted mice were intraperitoneally injected with insulin [0.75 U/kg body weight (BW), Humalog, Lilly] and subsequently sacrificed 15 min afterward. For a diet‐induced obesity model, WT and USP21‐KO mice or litter and USP21‐MKO mice were subjected to ad libitum feeding of either an normal diet (ND) or an HFD with 60% kcal fat (Research Diets, D12492). Blood was taken for serum biochemical analyses, and excised tissues were dissected and processed for histopathological analyses, and protein and RNA quantifications.
Human study
The study was approved by the Institutional Review Board (IRB) of Seoul National University. Human samples were purchased from Genetex (GTX24366 and GTX21805), which sources tissue products from select laboratories that have an established IRB complaint to 45 CFR part 46, 21 CFR part 56.
RNA isolation and quantitative real‐time PCR assays
Total RNA was extracted using Trizol (Thermo Fisher Scientific, 15596‐018) and was reverse‐transcribed to cDNA according to the previously published procedures. 9 The cDNA was amplified by qRT‐PCR under the manufacturer's instructions using a SYBR Premix Ex Taq II kit (Takara, RR420). Gene expression was assessed by quantitative real‐time (qRT)‐PCR analysis using the primers shown in Table S3.
Electroporation‐mediated gene transfer
The experiment was performed as previously described, 9 with some modifications as detailed here. Under anaesthesia, tibialis anterior muscle was injected with hyaluronidase (40 μg). After 3 h, 50 μg plasmid vector encoding for USP21 (OriGene, RC210554) was injected into one tibialis anterior muscle, and pCMV (an empty plasmid) was used for mock transfection to the contralateral muscle. Electric pulses (50 ms, 1 Hz, 100 V, eight times) were delivered immediately to the injected tibialis anterior muscle using a square‐pulse stimulator (Harvard Apparatus, ECM830) through a Tweezertrode electrode (Harvard Apparatus, 45‐0165) on the medial and lateral sides of the tibialis anterior muscle so that the electrical field was perpendicular to the long axis of the myofibre. Mice were sacrificed to isolate muscle 7 days after injection.
Transmission electron microscopy
Soleus muscle was fixed in Karnovsky's fixative immediately after isolation. Samples were then post‐fixed by 1% osmium tetraoxide in 0.1 M sodium cacodylate buffer. After overnight incubation with 0.5% uranyl acetate, the samples were dehydrated in ethanol and propylene oxide and were polymerized in Spurr's resin at 70°C. The observation was performed by the National Instrumentation Center for Environmental Management at Seoul National University. The images were taken using a JEM‐1010 transmission electron microscope (JEOL, Japan).
Histological analysis
Succinate dehydrogenase staining
Cryosections of tibialis anterior muscles were cut from the mid‐belly region of the snap‐frozen muscle. After drying for 5 min at room temperature, histochemical staining of succinate dehydrogenase (SDH) activity was performed with incubation of the sections for 30 min in a 0.2 M sodium phosphate buffer solution containing 0.6 mM nitrotetrazolium blue chloride (Sigma, N6876) and 50 mM sodium succinate. The sections were then rinsed three times for 30 s each in physiological saline, for 5 min in 15% ethanol and mounted.
Immunohistochemistry
Skeletal muscle samples were fixed in 4% formaldehyde, embedded in paraffin, cut into 10‐μm sections, and were mounted on slides. Skeletal muscle sections were immunostained with antibodies directed against USP21 (1:200, ab38864 and 1:100, NBP2‐75701) and phosphorylated‐AMPKα (p‐AMPKα; 1:50, Cell Signaling, 2535). Skeletal muscle samples were fixed and immediately frozen with liquid nitrogen‐cooled isopentane after dissection and were sectioned on a cryostat microtome for immunofluorescence. Immunofluorescence staining was performed using antibodies specific to myosin heavy chain 1 (MyHC1; 1:50, DSHB BA‐F8), MyHC2a (1:100, DSHB SC‐71) or MHC2b (1:200, DSHB BF‐F3).
Oil red O staining
Cryosections of snap‐frozen muscles were stained with Oil Red O stain kit (ScyTek, ORK‐1‐IFU) for fat analysis.
Haematoxylin and eosin staining
Liver, epididymal fat, and brown adipose tissue (BAT) were fixed in 10% formalin, embedded in paraffin, cut into 4 μm thick sections and were mounted on slides. Tissue sections were stained with haematoxylin and eosin for morphology analysis.
Ex vivo fatty acid uptake and oxidation
Fatty acid uptake
Soleus muscle was removed from mice of each genotype and immediately soaked in DMEM supplemented with 0.5 μCi/mL [9,10‐3H]‐palmitate (PerkinElmer, NET043‐1000 μCi) in the presence of 2% BSA and 0.2 mM cold palmitate (Sigma, P9767). After 30 min incubation at 30°C, tissues were washed thoroughly and lysed with 50°C 1 M KOH solution for 1 h. Samples were then used for scintillation counting.
Fatty acid oxidation
Soleus muscle, liver, white adipose tissue (WAT), and BAT were freshly removed from mice and were placed in flasks fitted with centre wells to trap 14CO2. Tissues were incubated in 3 mL Krebs‐Ringer phosphate buffer with 2 μCi of [1‐14C] oleic acid (PerkinElmer) and cold oleic acid (0.6 mM final concentration, Sigma) in complex with bovine serum albumin for 60 min (soleus muscle and WAT) or 30 min (liver and BAT) at 37°C. Then, 1 mL of 0.5 N sulfuric acid was injected into the media to stop the FA oxidation. Flasks were maintained at 50°C for 3 h to release and evaporation of 14CO2 from the media to NaOH solution in the centre well. After 3 h‐incubation, contents of the centre well were transferred to scintillation fluid and counted radioactivity with liquid scintillation counter (Tri‐Carb 3110 TR, PerkinElmer).
Cellular respiration assays
The cellular oxygen consumption rate (OCR) was measured using the Seahorse XFp Extracellular Flux Analyzer (Agilent Technology) according to the manufacturer's instruction. Cells were washed with XF assay media (XF base medium containing 1 mM sodium pyruvate, 25 mM glucose, and 4 mM glutamine) and placed into non‐CO2 incubator maintained at 37°C for 30 min before the experiment. In addition to basal respiration, uncoupled, and maximal mitochondrial respiration rates were determined by sequential additions of 1 μM oligomycin, 2 μM carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP), and 0.5 μM rotenone plus antimycin A, respectively.
Immunoblottings
A standard protocol was used for immunoblotting of protein lysates. In brief, protein lysates were resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) and were transferred onto an NC membrane (GE Healthcare, BA85), followed by immunoblotting with the primary and HRP‐conjugated secondary antibodies according to the previously published procedures. 10 For ACLY transfer, 0.45 mm PVDF membrane (Thermo Fisher Scientific, 88518) was used. Antibodies to USP21 (ab171028) and DNA‐PKcs (ab32566) were supplied from Abcam and were used in 1:5000 dilutions. Anti‐DNA‐PKcs was purchased from Santa Cruz Biotechnology (sc‐390849 and sc‐390495, Santa Cruz Biotechnology) and used in 1:1000 dilution. Antibodies to p‐AMPKα (2535), p‐acetyl‐CoA carboxylase (p‐ACC) (3661), AMPKα (2532), ACC (3662), ACLY (4332), FLAG‐tag (8146), p‐protein kinase B (p‐Akt) (Ser473) (9271), p‐Akt (Thr308) (9275), Akt (4685), p‐serine/threonine kinase 11 (p‐LKB1) (3482), LKB1 (3050), and p‐S6 ribosomal protein (p‐S6) (Ser235/236) (2211) were provided from Cell Signaling and used in 1:10 000 dilution. Anti‐S6 was purchased from Genetex (GTX113542). Anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (60004‐1‐Ig) from Proteintech was used in 1:10 000 dilution. Anti‐β‐actin antibody (A5441) and anti‐ubiquitin (Ub) antibody (U0508) were obtained from Sigma‐Aldrich, whereas horseradish peroxidase‐conjugated goat anti‐rabbit (G21234) and goat anti‐mouse immunoglobulin G (G21040) were from Invitrogen.
Immunoprecipitation and immunoblot assays
Protein lysates were centrifuged at 10 000 g for 10 min at 4°C. The supernatant fraction was pre‐cleaned using Protein G Agarose beads (Millipore, 16‐266) and was incubated with specific primary antibodies as indicated with gentle rotating at 4°C. Protein G beads were added to the solution followed by incubation for 90 min with gentle rotating at 4°C. The beads were then collected by centrifugation and washed four times with cell lysis buffer. Proteins were re‐suspended with 2 X sample dilution buffer and boiled for 2 min and then subjected to immunoblottings.
Transfection of plasmids and siRNAs
The USP21 expression clone was purchased from OriGene (RC210554). The specific base substitution was made through PCR, followed by Gibson assembly. Mutagenic primers flanking Cys221 residue of human USP21 (5′‐AAA CAC GGC CTT CCT GAA TGC TGT GCT‐3′ and 5′‐CAG GAA GGC CGT GTT TCC CAG GTT TCG‐3′) were used for alanine substitution. The resulting vector expressing C221A mutant was verified by Sanger sequencing. In the USP21 or USP21 (C221A) overexpression experiment, the empty plasmid, pCMV, was used for mock transfection. For gene knockdown assays, scrambled control siRNA was made by Cosmogenetech (A06001), and SMARTpool (a mixture of four siRNAs) for mouse USP21 (L‐063216‐01‐0005), DNA‐PKcs (L‐040958‐00‐0005), and ACLY (L‐040092‐01‐0005) was obtained from Dharmacon. C2C12 cells, seeded in 60 pi dish, were transfected with 3 μg of indicated plasmid DNA and/or 200 pmoles of siRNA using FuGENE HD Reagent (Promega, E231A) for 24 h, and were differentiated to myotubes for 48 h. Otherwise, C2C12 cells were transfected with plasmid DNA and/or siRNA using electroporator (Lonza, Amaxa NucleofectorII) according to the supplier's instructions, followed by differentiation to myotube for 48 h. HEK293 cells were seeded in 6‐well plates and incubated with 1 μg of the indicated plasmid DNA and FuGENE HD Reagent for 24 h. For the detection of ubiquitination, the cells were treated with 10 μΜ MG132 for 6 h after co‐transfection with USP21 (or pCMV) and ubiquitin plasmids (1 μg each).
Analysis of mouse metabolic status
Fat and lean body masses were measured by 1H minispec system (Bruker Optik, LF90II) in mice. To measure whole‐body energy metabolism in HFD‐fed mice, a comprehensive laboratory animal monitoring system (Columbus Instruments, CLAMS) was used for 3 days (2 days of acclimation followed by 1 day of measurement).
Glucose tolerance test and insulin tolerance test
For glucose tolerance test, mice fed with an ND or an HFD (14 weeks‐fed in WT or USP21‐KO mice; 9 weeks‐fed in litter or USP21‐MKO mice) were fasted overnight (16 h) and injected intraperitoneally with a glucose solution (2 g/kg BW in WT and USP21‐KO mice; 1.5 g/kg BW in litter and USP21‐MKO mice). Blood glucose concentrations were measured before and 15, 30, 60, and 90 min after glucose injection. For insulin tolerance test, mice fed an ND or an HFD for 10 weeks were morning‐fasted and injected intraperitoneally with insulin at 0.75 IU/kg BW to 1.5 IU/kg BW. Blood glucose concentrations were monitored before and 30, 60, and 90 min after insulin injection.
Treadmill running
Prior to the tests, mice were trained by running at a speed of 10 m/min, 10 min for 2 days to acquire running skills. For treadmill running tests, the inclination angle was set at 5%. The running speed was set at 10 m/min for 30 min and increased by increments of 2 m/min every 10 min, up to 16 m/min, or exhausted for the measurement of total running time and distance. Mice were determined to be exhausted and were excluded if they resisted running and rested for more than 20 s.
RNA‐sequencing analysis
For RNA‐seq analysis, gastrocnemius muscle from the mice of litter and USP21‐MKO were used (n = 3 each). Total RNA concentration was calculated by Quant‐IT RiboGreen (Thermo Fisher Scientific, R11490). To assess the integrity of the total RNA, samples were run on the Tape Station RNA screen tape (Agilent Technologies, 5067‐5576). Only high‐quality RNA preparations, with RIN N7.0, were used for RNA library construction. A library was independently prepared with 1 μg of total RNA for each sample by Illumina TruSeq Stranded mRNA Sample Prep Kit (Illumina, Inc., RS‐122‐2101). The first step in the workflow involved purifying the poly‐A containing mRNA molecules using poly‐T‐attached magnetic beads. Following purification, the mRNA was fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments were copied into first strand cDNA using SuperScript II reverse transcriptase (Invitrogen, 18064014) and random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I, RNase H, and dUTP. These cDNA fragments then went through an end repair process, the addition of a single ‘A’ base, and then ligation of the adapters. The products were then purified and enriched with PCR to create the final cDNA library. The libraries were quantified using KAPA Library Quantification kits for Illumina Sequencing platforms according to the qRT‐PCR Quantification Protocol Guide (Kapa Biosystems, KK4854) and qualified using the TapeStation D1000 ScreenTape (Agilent Technologies, 5067‐5582). Indexed libraries were submitted to an Illumina NovaSeq 6000 (Illumina, Inc.), and the paired‐end (2 × 100 bp) sequencing was performed by the Macrogen Incorporated. For RNA‐seq analysis, the raw ‘fragments per kilobase million’ (FPKM) values were processed and normalized by logarithm and quantile normalization method. Differentially expressed genes (DEGs) by USP21‐MKO were identified using independent t test: DEGs were selected as the genes with P values under 0.05 with over 1.5× fold‐change. Heatmap and hierarchical correlation analysis of the DEGs were analysed using R software 3.5.2. Statistically enriched signalling pathways of clustered DEGs were ranked and categorized according to gene ontology (GO) using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway using KEGG database (http://www.genome.jp/kegg/). Gene interaction network was visualized using Cytoscape 3.7.2 software (http://www.cytoscape.org/). The raw transcriptomics data have been deposited in the Gene Expression Omnibus (GEO) under the accession code GSE159558.
Liquid chromatography–mass spectrometry analyses
ATP, AMP/ATP, and ADP/ATP ratio detection
Metabolite extraction of skeletal muscle from WT and USP21‐KO mice was performed as previously described, 11 , 12 with some modifications as detailed here (n = 6 each). In brief, the frozen tissue samples were cut into pieces approximately 30 mg each, and transferred to tissue grinders (BioMasher II, Kimble, Japan). After homogenization with 300 μL of cold methanol/chloroform (2:1), the samples were vortexed‐mixed for 30 s and submerged for 1 min in liquid N2, followed by thawing on ice for 2–3 min. The vortex–submerge–thaw step was repeated three times. The samples were then added 200 μL of cool water and 200 μL of cold chloroform and centrifuged at 15 800 g for 20 min at 4°C. The supernatant was dried using a vacuum concentrator (Vision Scientific, VS‐802 Centra‐vac) and the extract was dissolved in 40 μL of water/acetonitrile (1:1) before injection to liquid chromatography–mass spectrometry (LC–MS). The middle layer was collected and dissolved in 3 M guanidine‐HCl solution for bicinchoninic acid protein assay. All steps were executed on ice. Chromatographic separation was performed on a ZIC‐HILIC column (100 × 2.1 mm, 3.5 μm, 200 Å, SeQuant, Merck Millipore) using an Agilent 1200 Infinity Series liquid chromatography system. The column temperature was set at 40°C with a flow rate of 0.2 mL/min. A 2 μL of analytes was eluted with a mobile phase composed of 5 mM ammonium acetate in water (A) and acetonitrile (B). Gradient conditions were as follows: 0–30 min gradient 5–70% A, 30–40 min isocratic 70% A, and 40.1–50 min isocratic 5% A. Mass spectrometry experiments were performed on a Q‐TOF (Agilent Technologies, 6530 Accurate‐Mass) equipped with electrospray ionization sources. Data were acquired in a positive mode and the scan range was 100–1000 m/z. Raw data were converted as MZdata.xml form and processed using MZmine 2.20 (mzmine.github.io) as previously described. 13 Briefly, chromatograms were built, and peaks were recognized using local minimum search function, and the ion intensities, matching m/z, and retention time were grouped into peak lists. All peaks were aligned and normalized by protein concentrations acquired from bicinchoninic acid test. Identification of analytes was completed using house‐library. Student's t test was performed for evaluation of target analytes.
LC–MS/MS analysis of USP21‐interacting proteins
HEK293 cells overexpressing FLAG‐tagged USP21 (or pCMV) were subjected to SDS‐PAGE. Bands of control group and USP21‐overexpressed group were cut from the SDS‐PAGE gel and were in‐gel digested with trypsin at 37°C overnight. The samples were subsequently resolved by online reversed‐phase chromatography for each run using Easy‐nLC II (Thermo Fisher Scientific). An autosampler was used to load the peptide solutions into a peptide trap EASY‐column (100 μm inner diameter, 2 cm length, 5 μm particle size, C18‐AQ) and peptides were then separated on an analytical EASY‐Column (75 μm inner diameter, 10 cm length, 3 μm particle size, C18‐AQ). The mobile phases were composed of 100% water (A) and 100% acetonitrile (B), and each contained 0.1% formic acid. The voltage applied to produce the electrospray was 1.9 kV, at a flow rate of 300 nL/min. During the chromatographic separation, the MS/MS scans were acquired in LTQ Orbitrap Velos mass spectrometer equipped with a CID source [1 full MS (Orbitrap) + 20 MS2 (LTQ)]. The MS data scanned from 300–2000 m/z were acquired in Orbitrap at a resolution of 100 000 for each MS/MS measurement. Fragmented ions were excluded for 180 s. Then, the datasets generated by LTQ‐Orbitrap were analysed using the Sorcerer 2, (Sage‐N Research, version 4.0) and Scaffold 4 Q + S (Proteome Software Inc., version 4.8.7) by Biocon. Peptide identifications were accepted if they could be established at a greater than protein and peptide threshold of 90% probability by the Scaffold local false discovery rate algorithm. These were accepted if they contained at least two identified unique peptides.
Lipid profiling
Snap frozen tissue samples were weighed and homogenized in 4 volumes of ice‐cold water using a TissueLyser II (Qiagen). For lipid profiling of mouse samples, 10 μL of tissue homogenate was extracted in 190 μL of isopropanol containing 0.2 ng/μL 1,2‐dilauroyl‐sn‐glycero‐3‐phosphocholine, vortexed, and centrifuged (10 000 g, room temperature, 10 min), and the supernatants were stored at −80°C until analysis. A 2 μL of extracted mouse tissue or plasma was separated by reversed phase chromatography using an Acquity UPLC BEH C8 column (Waters); mobile Phase A: 10 mM ammonium acetate in 95% water/5% methanol/0.1% formic acid; mobile phase B: 0.1% formic acid in methanol. The column was eluted isocratically with 20% mobile Phase B for 1 min, followed by a linear gradient to 80% mobile Phase B over 2 min, a linear gradient to 100% mobile Phase B over 7 min, then 3 min at 100% mobile Phase B. MS data were acquired with a Q Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific) in the positive ion mode using electrospray ionization and full scan MS over m/z 200–1100. 14 Raw data were integrated and visually inspected using TraceFinder 3.3 software (Thermo Fisher Scientific). For each lipid analyte, the first number denotes the total number of carbons in the lipid acyl chain(s), and the second number (after the colon) does the total number of double bonds in the lipid acyl chain(s).
Statistical analysis
Two‐tailed Student's t tests or one‐way analysis of variance tests (ANOVA) followed by Tukey's honestly significant difference or least significant difference multiple comparisons correction were performed to assess the significance of differences among treatment groups using Sigma Plot 10.0 (Systat Software) or SPSS 25 (IBM). Data are expressed as the mean ± standard error of the mean (SEM). The criterion for statistical significance was set at P < 0.05 or P < 0.01. Coefficients of correlation (r) were determined by the Pearson's or Spearman's correlation methods.
Results
Ablation of USP21 promotes oxidative fibre phenotype in skeletal muscle
In an effort to find new target molecules related to energy metabolism, we first used a quantitative trait locus analysis approach to determine genetic associations with fasting blood glucose levels in mice. The Usp21 gene located on chromosome 1 was identified as a candidate gene most significantly affecting fasting glucose phenotype variations, implicating this locus in energy homeostasis (r = 0.738, P = 0.004; Figure 1A). To estimate the distribution of USP21 expression in human tissues, we analysed a public database and found that among different metabolic organs, USP21 was predominantly expressed in skeletal muscle (GSE43346) (Figure 1B). Similarly, in an analysis of mouse samples, USP21 was highly expressed in soleus muscle as well as BAT and heart (Figure 1C) and was also expressed in muscles with high glycolytic myofibre content (i.e. gastrocnemius, tibialis anterior, and extensor digitorum longus) ( S1 ). With the aim of identifying the role of USP21 in fuel catabolism in skeletal muscle, we generated Usp21‐mutant mouse strains using the KO‐first strategy. 15 The Usp21 tm1a/tm1a allele (hereafter referred to as USP21‐KO) was first utilized to assess the phenotype of the whole‐body KO mouse. Later, the Usp21 tm1d/tm1d allele (hereafter referred to as USP21‐MKO) was created to ascertain the effects of the skeletal muscle‐specific deletion of Usp21 on phenotypic changes (Figure 1D). Immunoblottings and qRT‐PCR assays in combination with genotyping verified the silencing of Usp21 in skeletal muscle (Figure 1E). USP21‐KO mice exhibited no change in BW compared with WT animals. Interestingly, however, the soleus or tibialis anterior muscle‐to‐body weight ratio (%) was higher in USP21‐KO mice and USP21‐MKO mice than respective control (Δ13.8% to Δ22.8%; P < 0.05 or P < 0.01; Figure 1F and 1G). These results raise the possibility that USP21 plays a role in skeletal muscle function and energy metabolism.
Figure 1.
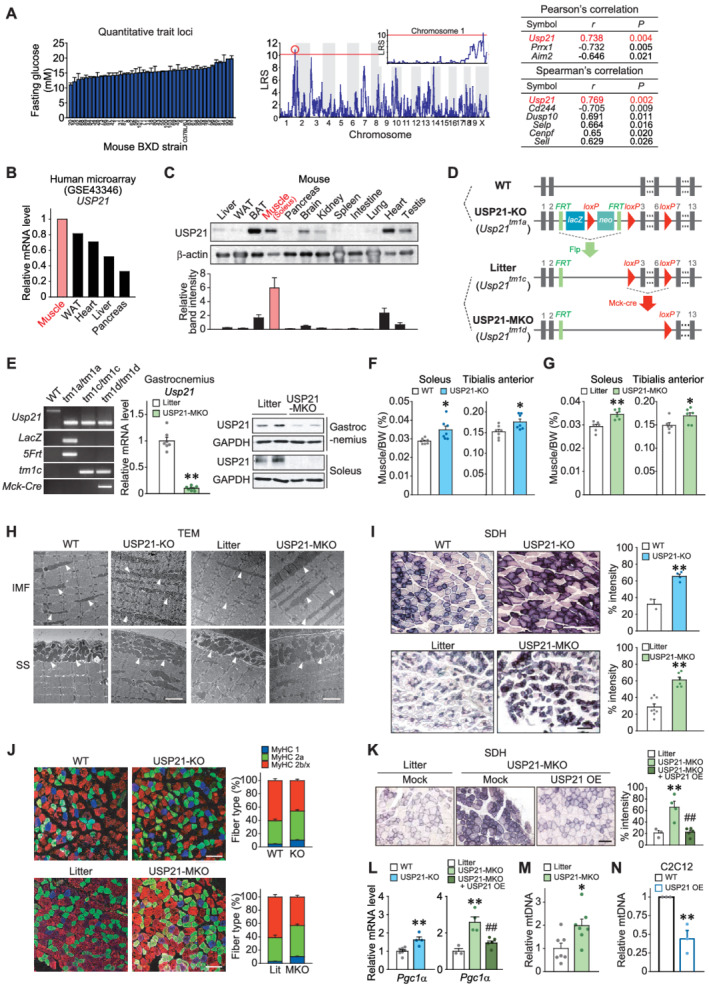
Ablation of USP21 in skeletal muscle increases muscle mass and oxidative fibre type switch. (A) Quantitative trait loci analysis obtained from glucose levels in mice fasted for 4 h (BXD_12925) (left). Quantitative loci where likelihood ratio statistics (LRS) over 10 in chromosome 1 was analysed (middle). Top ranks of Pearson's and Spearman's correlation analyses are listed (right). (B) USP21 transcript levels in a microarray GEO dataset (GSE43346) available in the public domain from major human tissues indicated. (C) USP21 protein levels in different tissues. Immunoblottings were performed for a variety of tissues obtained from 18‐week‐old C57BL/6 mice fasted overnight prior to sacrifice (n = 3). A representative blot was shown. WAT, white adipose tissue; BAT, brown adipose tissue. (D) Generation of whole‐body knockout (KO) mice and skeletal muscle‐specific Usp21 knockout (MKO) mice models. Wild type (WT) and Usp21 tm1a/tm1a mice were used to assess the phenotype of systemic KO of USP21. Control littermate (Usp21 tm1c/tm1c ) mice for the skeletal muscle‐specific KO mice (Usp21 tm1d/tm1d ; MKO) were generated by breeding Usp21 tm1a/tm1a mice with protamine‐Flpe mice. Usp21 tm1c/tm1c mice were crossed with muscle creatine kinase (Mck)‐Cre mice to produce USP21‐MKO mice. (E) PCR assays for respective genotypes using genomic DNAs extracted from the tails of mice (left). qRT‐PCR assays for Usp21 in the gastrocnemius muscle of USP21‐MKO mice (middle) (n = 7, 8 each). Immunoblottings for USP21 in gastrocnemius and soleus muscle of USP21‐MKO mice (right) (n = 2 each). (F,G) Muscle weights of indicated genotypes. (F) 18‐week‐old WT or USP21‐KO mice (n = 7 each). (G) 25‐week‐old litter or USP21‐MKO mice (n = 6 each). BW, body weight. (H) Representative transmission electron micrographs (TEM) of longitudinal sections of soleus muscle. Arrows indicate intermyofibrillar (IMF) or subsarcolemmal (SS) mitochondria (n = 3 each). Scale bars: 2 μm. (I) Representative succinate dehydrogenase (SDH) activity in tibialis anterior muscle and histological quantification of staining intensity (n = 2–8 each). Scale bars: 200 μm. (J) Representative immunofluorescence images of red gastrocnemius muscle. Immunostainings were performed in 12‐week‐old WT or USP21‐KO mice, and litter or USP21‐MKO mice using specific antibodies for MyHC 1 or MyHC 2a or MyHC 2b/2x (n = 7, 7, 3, 5 each), and myofibre types of each genotype were quantified. Scale bar: 100 μm. (K) Representative SDH activity in tibialis anterior muscle and histological quantification of staining intensity (n = 3–5 each). Scale bars: 200 μm. (L) qRT‐PCR assays for peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (Pgc1α) in gastrocnemius muscle of WT or USP21‐KO mice (n = 7, 4 each), and litter or USP21‐MKO mice (n = 3–5 each). (Μ,Ν) Relative mitochondrial DNA (mtDNA) copy number in gastrocnemius muscle of litter or USP21‐MKO mice (M) (n = 7 each) and C2C12 myotubes with Usp21 overexpression (OE) or mock (N) (n = 3 each). mtDNA copy number represents the ratio of mtDNA to nuclear DNA measured by qRT‐PCR. Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by Student's t test (E–G,I,L–N). **P < 0.01 for WT vs. USP21‐MKO mice; ##P < 0.01 for USP21‐MKO vs. USP21‐MKO mice plus USP21 overexpression by one‐way ANOVA with Tukey's multiple comparisons correction (K,L).
To understand the role of USP21 in skeletal muscle physiology, we then assessed the effects of USP21‐KO and USP21‐MKO on mitochondrial oxidative capacity and myofibre type composition. Electron microscopic analyses showed larger‐size and higher density intermyofibrillar and subsarcolemmal mitochondria in skeletal muscle of USP21‐KO mice and USP21‐MKO mice (Figure 1H). SDH activity assays verified a significant increase in mitochondrial oxidation in USP21‐deficient animals (2.05‐fold or 2.14‐fold, P < 0.01; Figure 1I). Moreover, an increase in the number of myosin heavy chain subtypes of Type 1 or 2a oxidative fibres along with a reciprocal decrease in Type 2b or 2x glycolytic fibres demonstrated switching of myofibre types to a more oxidative phenotype as a result of gene deletion (Δ36.6% or Δ47.2%; P < 0.05 or P < 0.01; Figure 1J). To corroborate the bona fide regulatory effect of USP21 abrogation on mitochondrial function, we studied the effect of USP21 overexpression in the skeletal muscle of USP21‐MKO mice using an in vivo gene delivery technique. The electroporation‐mediated forced expression of USP21 in the tibialis anterior prevented an increase in SDH activity (P < 0.01; Figure S2A and 1K). Consistently, the transcript levels of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (Pgc‐1α) as the mitochondrial biogenesis and oxidative fibre marker were greater in both USP21‐KO mice and USP21‐MKO mice, which was prevented by USP21 overexpression (P < 0.01; Figure 1L). Consistently, USP21 deficiency increased mitochondrial DNA (mtDNA) content in gastrocnemius muscle of USP21‐MKO mice (Δ98.2%, P < 0.05; Figure 1M). USP21 overexpression in C2C12 myotubes showed the opposite effect (Δ56.1%, P < 0.01; Figure 1N). These results support the notion that USP21 deficiency promotes myofibre type change to oxidative phenotype with muscle mass increase.
Skeletal muscle ablation of USP21 increases fatty acid oxidation and thermogenesis
Given the changes in mitochondrial capacity induced by USP21 modulations, we assessed the effects of USP21 on fuel consumption. Lipid profiling enabled us to substantiate decreases in not only overall fat content but also key lipids including diacylglycerol (DAG), triacylglycerols (TAGs, 11), cholesterol ester (CE), lysophosphatidylcholines (LPCs, 7), phosphatidylcholines (PCs, 7), and sphingomyelins (SMs, 10) in skeletal muscle (P < 0.05 or P < 0.01; Figure 2A). Correspondingly, the transcript levels of genes encoding for proteins representing fatty acid oxidation, FA uptake and utilization were all higher in the KO animals (P < 0.05 or P < 0.01; Figure 2B), indicating an increase in overall fuel consumption. Consistently, the rate of palmitate uptake by skeletal muscle was significantly higher in the KO animals (1.34‐fold, P < 0.05; Figure 2C). Induction of the genes involved in lipid oxidation was also found in USP21‐MKO mice, which was completely prohibited by the electroporation‐mediated delivery of USP21 (P < 0.05 or P < 0.01; Figure 2D). Respiratory exchange ratios were lower in USP21‐KO mice, supporting the concept that USP21 deficiency promotes FA oxidation (Δ8.8%, P < 0.05; Figure 2E). Although a decrease in Usp21 was observed in the heart muscle of USP21‐MKO mice (Figure S3A), the change was comparable with that in skeletal muscle. In addition, the transcript levels of genes involved in mitochondrial function were comparable with controls, as was histology (Figure S3B and S3C), supportive of the prominent phenotype changes in skeletal muscle.
Figure 2.
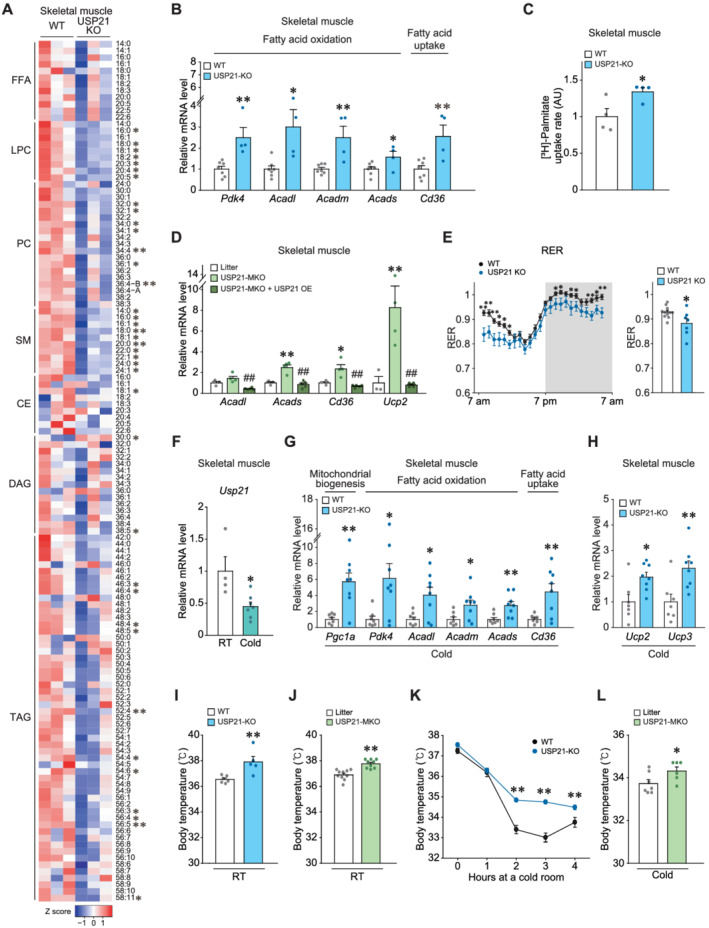
USP21 ablation in skeletal muscle promotes fatty acid oxidation and thermogenesis switch. (A) Heatmaps for lipids profiling in tibialis anterior muscle of 12‐week‐old WT and USP21‐KO mice as analysed by liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) (n = 3 each). The first number of analyte indicates the number of carbons in the lipid acyl chain and the number after the colon indicates the number of double bonds in the lipid acyl chain(s). Lipids peak intensities with independent Student's t test (*P < 0.05, **P < 0.01) were presented as heatmap according to the Z score. FFA, free fatty acid; LPC, lysophosphatidylcholine; SM, sphingomyelin; PC, phosphatidylcholine; CE, cholesterol ester; DAG, diacylglycerol; TAG, triacylglycerol. (B) qRT‐PCR assays for the genes associated with mitochondrial biogenesis, fatty acid (FA) oxidation, and FA uptake in gastrocnemius muscle of indicated genotypes (n = 7, 4 each). (C) Ex vivo FA uptake assays. Soleus muscle was freshly removed from mice of indicated genotypes, incubated in a medium containing [3H]‐palmitic acid for 30 min, and lysed for scintillation counting (n = 4 each). AU, arbitrary unit. (D) Quantitative real‐time (qRT)‐PCR assays for the genes representing mitochondrial functions in gastrocnemius muscle of indicated genotypes (n = 3–5 each). (E) In vivo energy balance of respiratory exchange ratios (RERs) were analysed in mice housed in individual metabolic cages for 24 h (12 weeks, n = 12, 7 each). (F) qRT‐PCR assays for Usp21 in the gastrocnemius muscle of 13‐week‐old mice housed individually in a room of ambient temperature (RT) or a 4°C cold room for 6 h (n = 7, 4 each). (G,H) Transcript levels in gastrocnemius muscle of 13‐week‐old WT or USP21‐KO mice exposed to 4°C temperature for 6 h (n = 7, 8 each). (G) qRT‐PCR assays for those representing mitochondrial biogenesis, oxidation, and fatty acid uptake. (H) qRT‐PCR assays for Ucp2 and Ucp3 representing mitochondrial uncoupling. (I–L) Core body temperatures of mice were monitored using a rectal thermometer. (I) The 13‐week‐old WT or USP21‐KO mice at RT (n = 6, 5 each). (J) The 12‐week‐old litter and USP21‐MKO mice at RT (n = 10, 9 each). (K) The 11‐week‐old WT and USP21‐KO mice were acutely exposed to 4°C temperature for the indicated times (n = 9, 13 each). (L) The 13‐week‐old litter and USP21‐MKO mice were exposed to 4 °C temperature for 6 h (n = 7, 6 each). Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by Student's t test (B, C, E–L). *P < 0.05, **P < 0.01 for WT vs. USP21‐MKO mice; ##P < 0.01 for USP21‐MKO vs. USP21‐MKO mice plus USP21 overexpression by one‐way ANOVA with Tukey's multiple comparisons correction (D).
Because mitochondria play an important role in thermogenic response, we next assessed USP21 levels upon cold exposure condition. In mice exposed to a low temperature (4°C) for 6 h, Usp21 transcript levels were significantly lowered in skeletal muscle (Δ55.3%, P < 0.05; Figure 2F). The expression levels of genes responsible for mitochondrial biogenesis, and FA oxidation and uptake were raised in USP21‐KO mice (P < 0.05 or P < 0.01; Figure 2G). Of the mitochondrial uncoupling proteins (UCPs), the transcript levels of Ucp2 and Ucp3, which participate in thermogenesis and FA oxidation in skeletal muscle, 16 were significantly elevated in USP21‐KO mice subjected to 6 h cold stress in comparison with WT controls (P < 0.05 or P < 0.01; Figure 2H). Consistently, both USP21‐KO and USP21‐MKO displayed meaningful increases in body temperature compared with the respective controls at ambient temperature (22 ± 2°C) (+1.37°C or +0.84°C; P < 0.01; Figure 2I and 2J). In addition, when the mice were subjected to cold stress at 4°C for 1 to 4 h, core body temperature was maintained at the higher level in USP21‐KO mice than WT mice (P < 0.01; Figure 2K). USP21‐MKO mice showed comparable outcomes (+0.59 °C, P < 0.05; Figure 2L), strengthening the role of USP21 in fuel oxidation and thermogenesis. In BAT, Ucp1 levels were decreased in USP21‐KO mice, whereas other genes related to mitochondrial biogenesis and fat oxidation were unaffected (Figure S4A). Also, both sarco/endoplasmic reticulum Ca 2+ ‐ATPase (Serca) and sarcolipin transcript levels, possibly associated with non‐shivering thermogenesis, were unaltered (Figure S4B).
Next, we examined the relationship between USP21 transcript levels and exercise. From the analysis of human skeletal muscle microarray datasets available in the public domain, we found that exercise training significantly resulted in lower USP21 expression compared with sedentary conditions (Figure S5A). In an animal model, exercise training also significantly down‐regulated Usp21 expression (Figure S5B). Interestingly, however, we could not find differences in exercise capacity between the control mice and Usp21‐deficient mice (Figure S5C and S5D). Collectively, these results indicate that USP21 regulates genes related to FA oxidation in skeletal muscle and thermogenesis.
USP21 ablation promotes uncoupler induction, mitochondrial oxygen consumption, and ATP depletion, persistently activating AMPK
To understand the increased mitochondrial capacity and thermogenesis that occurs with USP21 ablation in greater depth, we performed RNA‐seq analysis of skeletal muscle and found marked changes in groups of DEGs linked to mitochondrial function. In the GO analysis, terms related to lipid and nutrient metabolic processes, mitochondrial uncoupler activity, and the mitochondrial inner membrane were identified (P < 0.05; Figure 3A). The DEGs assigned to GO terms related to uncoupling, mitochondrial oxidative phosphorylation, and fuel oxidation are shown as a heatmap (Figure 3B). Forced expression of USP21 consistently lowered the basal, uncoupled, and mitochondrial maximal OCR in C2C12 myotubes (Figure 3C), supporting the regulatory role of USP21 in the mitochondrial respiration. In addition, USP21‐KO mice exhibited significantly lower ATP (Δ68.7%; P < 0.05) levels in skeletal muscle with a tendency towards increased AMP/ATP and ADP/ATP ratios (Figure 3D). These data show the functional effects of USP21 ablation on uncoupler genes, mitochondrial oxygen consumption, and cellular ATP content.
Figure 3.
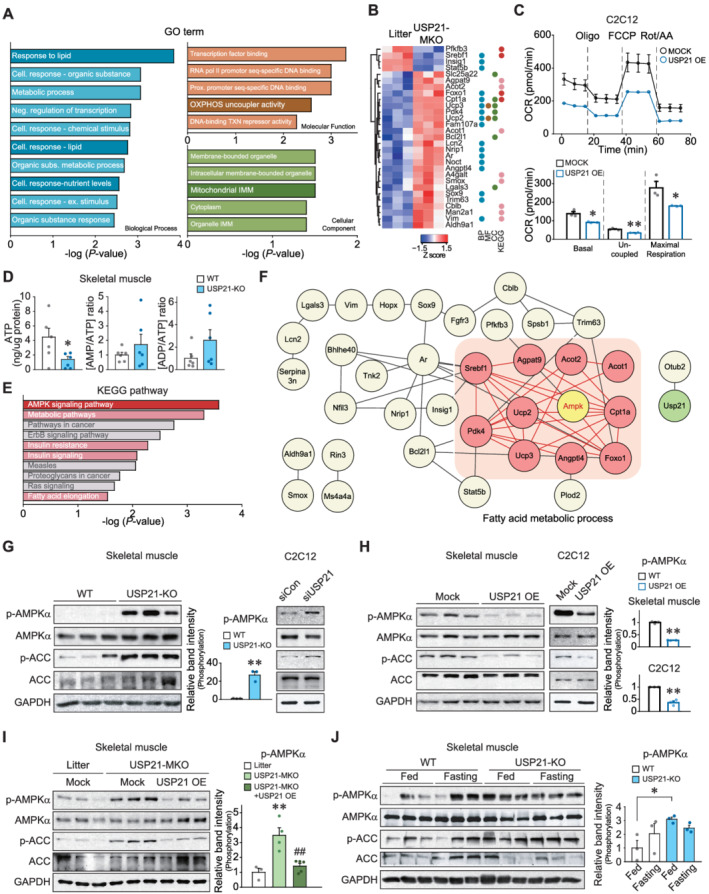
USP21 ablation induces AMPK activation. (A,B) RNA‐seq analyses using gastrocnemius muscle of 12‐week‐old WT and USP21‐MKO mice (n = 3 each). Significantly up‐regulated or down‐regulated genes by skeletal muscle‐specific Usp21 deletion. (A) Results of gene ontology (GO) terms. GO terms enrichment were analysed using g:Profiler. GO terms relevant to mitochondrial homeostasis were presented as dark coloured bar graph. (B) Hierarchically clustered differentially expressed genes (DEGs) were presented as heatmaps according to the Z score (blue, underexpression; red, overexpression). The DEGs corresponding to GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were marked as the same coloured circles with relevant bar graphs. BP, biological process; CC, cellular component; MF, molecular function; Cell, cellular; Neg, negative; Subs, substances; Ex, external; Prox, proximal, OXPHOS, oxidative phosphorylation; TXN, transcription; and IMM, inner membrane. (C) Respiration assays using an XFp Extracellular Flux Analyzer. The oxygen consumption rate (OCR) in C2C12 myotubes with USP21 overexpression (or mock). Real‐time triplicate readings (upper) and mitochondrial respiration rates of basal, uncoupled, maximal respiration (lower) are shown (three points each). The data were normalized to the cell numbers. Oligo, oligomycin; FCCP, carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone; Rot, rotenone; and AA, antimycin A. (D) Adenosine triphosphate (ATP) concentrations, AMP/ATP, and ADP/ATP ratios in tibialis anterior muscle of WT and USP21‐KO mice (n = 6 each). (E) KEGG pathway analysis using KEGG pathway database derived from RNA‐seq as in Panel (A). Energy metabolism related pathways are marked as red and pink bar graphs. (F) Core network analysis associated with the AMPK pathway derived from RNA‐seq dataset as in Panel (A). AMPK‐associated neighbour genes, whose expression was assigned to FA metabolic process and affected by USP21‐MKO, are shown as pink coloured circles. The network was analysed using STRING, Cytoscape plugin Genemania, and Cluster ONE. (G–J) Immunoblottings for phosphorylated‐AMPKα and phosphorylated‐ACC in skeletal muscle or C2C12 myotubes. (G) Gastrocnemius muscle of 13‐week‐old WT or USP21‐KO mice (n = 3 each, left) and C2C12 myotubes transfected with Usp21‐specific siRNA (or mock) (right). The cell‐based data were confirmed in a separate experiment. (H) Tibialis anterior muscle of 12‐week‐old WT mice 7 days after electroporation‐mediated USP21 gene delivery (or mock) (n = 3 each, left). C2C12 cells were differentiated to myotubes after transfection with USP21 or mock for 24 h (n = 3 each, right). (I) Tibialis anterior muscle 7 days after electroporation‐mediated gene delivery. Each mouse of the indicated genotype received a control vector in one limb, and USP21‐MKO mice received a plasmid expressing USP21 in the contralateral limb (n = 3–5 each). (J) Soleus muscle from mice fed ad libitum or fasted for 24 h (n = 3 each). Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by Student's t‐test (C,D,G,H,J). **P < 0.01 for WT vs. USP21‐MKO mice; ##P < 0.01 for USP21‐MKO vs. USP21‐MKO mice plus USP21 overexpression by one‐way ANOVA with Tukey's multiple comparisons correction ANOVA (I).
Given the changes in the OCR in mitochondria via USP21 modulation, we wanted to determine whether loss of cellular ATP affects AMPK activation. As expected, analyses using KEGG, a database that integrates systemic functional information used for interpreting the comprehensive functions of biological systems, 17 allowed us to identify the AMPK pathway as being the most exclusively changed pathway in skeletal muscle (Figure 3B and 3E). In the clustering analyses, genes functionally related to FA metabolic process were differentially expressed (Figure 3F). Consistently, USP21‐KO mice displayed profound increases in the phosphorylation of AMPK and its substrate ACC (P < 0.01; Figure 3G, left). Silencing of the Usp21 gene using siRNA showed the same effect in C2C12 myotubes (Figures 3G, right, and S2B). Likewise, USP21 overexpression repressed the phosphorylation of AMPK and ACC in both skeletal muscle and C2C12 myotubes (P < 0.01; Figures 3H and S2C). AMPK was not activated in the liver of USP21‐KO or USP21‐MKO mice, nor the heart of USP21‐MKO mice (Figure S6A and S6B). Additionally, LKB1, an upstream effector of AMPK, 18 was not activated in the skeletal muscle of Usp21‐deficient mice (Figure S6C). We further confirmed that the in vivo electroporation‐mediated delivery of USP21 almost completely prevented AMPK activation elicited by USP21‐MKO (P < 0.01; Figure 3I).
AMPK is activated during fasting as a metabolic adaptation to limited glucose availability. 19 To physiologically validate the link between USP21 and AMPK, we compared the effects of feeding and fasting on AMPK in skeletal muscle. As expected, fasting of WT animals for 24 h enhanced the phosphorylation of AMPK and ACC. Ablation of USP21 similarly activated AMPK phosphorylation, even in animals provided food, and the phosphorylation was not further enhanced by fasting (P < 0.05; Figure 3J), indicative of the fasting‐mimic effect of USP21 ablation. Together, these results demonstrate the link between USP21 modulation and mitochondrial oxygen consumption and the resultant AMPK activation reflecting the loss of ATP.
USP21 interacts with DNA‐PKcs and ACLY as novel substrates
Subsequently, we sought to identify USP21 substrates using proteomic approaches. USP21 immunoprecipitation, in‐gel digestion, and proteomic analyses identified a total of 95 binding partners in HEK293 cells overexpressing FLAG‐tagged USP21 (Figure 4A, left, and Table S1). From the top 18 differential binding proteins showing two‐fold or more increases in abundance with greater than five spectral counts (Figure 4A, right), we narrowed our focus to DNA‐PKcs, the protein with the highest spectral count in association with AMPK. ACLY was chosen as another target because of its role in lipid anabolism. In our analysis of the GO biological process terms, we found that binding partners involved in cellular metabolic processes were enriched and, further, that DNA‐PKcs and ACLY might be connected to USP21 and each other (FDR < 0.05; Figures 4B, S7A, and S7B). Because a link between USP21 and either DNA‐PKcs or ACLY has not previously been described, we hypothesized that USP21 may use DNA‐PKcs and ACLY as substrates for metabolic processing.
Figure 4.
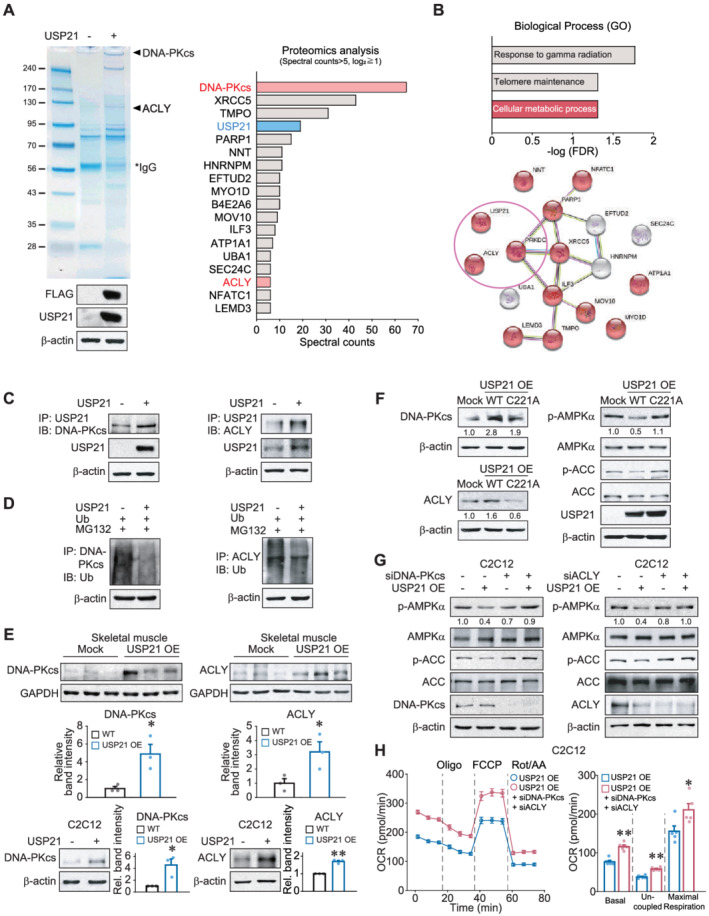
USP21 interacts with DNA‐PKcs and ACLY as substrates. (A) Liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) analysis for USP21 interacting proteins. Mock or USP21‐overexpressing HEK293 cells were used for SDS‐PAGE, and the bands of SDS‐PAGE were subjected to LC–MS/MS analysis. Top differentially interacting proteins with USP21 were listed (spectral counts >5, fold change ≧2). IgG, immunoglobulin G. (B) Gene ontology terms and the cluster obtained using STRING bioinformatics database from top differentially interacted proteins. FDR, false discovery rate. (C) Immunoprecipitation and immunoblotting assays. Immunoblottings for DNA‐PKcs or ACLY were performed on USP21 immunoprecipitates from HEK293 cells transfected with USP21 (or mock) for 24 h. The data were confirmed in a separate experiment. IP, immunoprecipitation; IB, immunoblotting. (D) The effect of USP21 overexpression on the ubiquitination of DNA‐PKcs and ACLY. HEK293 cells were treated with 10 μM MG132 for 6 h after transfection with the plasmid encoding for ubiquitin in combination with USP21‐expressing plasmid (or mock) for 12 h. The data were confirmed in a separate experiment. Ub, ubiquitin.(E) Immunoblottings for DNA‐PKcs and ACLY in skeletal muscle and C2C12 myotubes. Tibialis anterior muscle from 12‐week‐old WT mice 7 days after electroporation‐mediated USP21 (or mock) gene delivery (n = 3 each, upper). C2C12 cells were differentiated to myotubes after transfection with USP21 (or mock) for 24 h (n = 3 each, lower). (F) Immunoblottings for DNA‐PKcs, ACLY, phosphorylated‐AMPKα, and phosphorylated‐ACC in HEK293A cells transfected with mock or USP21 or its C221A mutant for 24 h. The data were confirmed in a separate experiment. (G) Immunoblottings for phosphorylated‐AMPKα and phosphorylated‐ACC in C2C12 myotubes. C2C12 cells were differentiated to myotubes after co‐transfection with USP21 (or mock) and siRNA of Dna‐pkcs or Acly for 24 h. The data were confirmed in a separate experiment. (H) Respiration assays using an XFp Extracellular Flux Analyzer. Real‐time readings of OCR (left) and mitochondrial respiration rates of basal, uncoupled, and maximal respiration (right) are shown (n = 5 each, triplicates per each experiment). C2C12 cells were differentiated to myotubes after co‐transfection of siDNA‐PKcs and siACLY with USP21 for 24 h. The data were normalized to the cell numbers. Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by Student's t test (E,H).
To assess the bona fide interaction of USP21 with potential targets, we conducted immunoprecipitation and immunoblotting assays and found that USP21 indeed binds with DNA‐PKcs and ACLY (Figure 4C). Because USP21 is a deubiquitinase, we confirmed the effect of USP21 overexpression on the ubiquitination status of DNA‐PKcs and ACLY, verified via the attenuation of the band intensities (Figure 4D). We then corroborated the effects of USP21 on the identified substrates by performing additional electroporation experiments using mouse skeletal muscle and confirmed that the forced expression of USP21 increased DNA‐PKcs and ACLY levels, supporting the ability of USP21 to stabilize the proteins (P < 0.05; Figure 4E, upper). This outcome was further verified in C2C12 myotubes (P < 0.05 or P < 0.01; Figure 4E, lower). The lack of changes in the levels of DNA‐PKcs, ACLY, p‐AMPK, and p‐ACC by overexpression of USP21 C221A mutant lacking deubiquitinase activity strengthens its enzymatic role for the identified substrates (Figure 4F). In addition, siRNA‐mediated silencing of either Dna‐pkcs or Acly prevented USP21 from inhibiting AMPK activity in C2C12 myotubes (Figure 4G). Moreover, co‐transfection of siDNA‐PKcs and siACLY with USP21 exhibited a greater OCR than that of USP21 alone, confirmative of their effects on energy consumption (P < 0.05 or P < 0.01; Figure 4H). These results show that USP21 uses DNA‐PKcs and ACLY as substrates for the regulation of AMPK‐mediated catabolism.
Ablation of USP21 prevents obesity through increased energy expenditure
Having identified the effects of USP21 ablation on fibre type switching, mitochondrial fuel consumption in skeletal muscle, and thermogenesis, we wanted to verify whether USP21‐KO has any beneficial effect on obesity. Interestingly, USP21‐KO mice resisted HFD‐induced BW gain (Δ8.02 g, 17.1%, P < 0.01; Figure 5A) as well as an increase in fat mass (Δ21.5%, P < 0.01; Figure 5B) compared with WT mice. By contrast, no differences were observed in USP21‐KO mice fed an ND. In an analysis using an animal metabolism‐monitoring system, both energy expenditure and maximal oxygen consumption were higher in USP21‐KO mice over the time periods examined (Δ6.8% or Δ6.3%; P < 0.05; Figure 5C and 5D). Food intake and locomotor activities were comparable with WT controls (Figure 5E and S8A). USP21‐KO mice fed an ND also showed no changes in food intake (Figure S8B). In addition, energy expenditure tended to increase at certain times in the USP21‐KO mice, but the differences were not statistically significant (Figure S8C).
Figure 5.
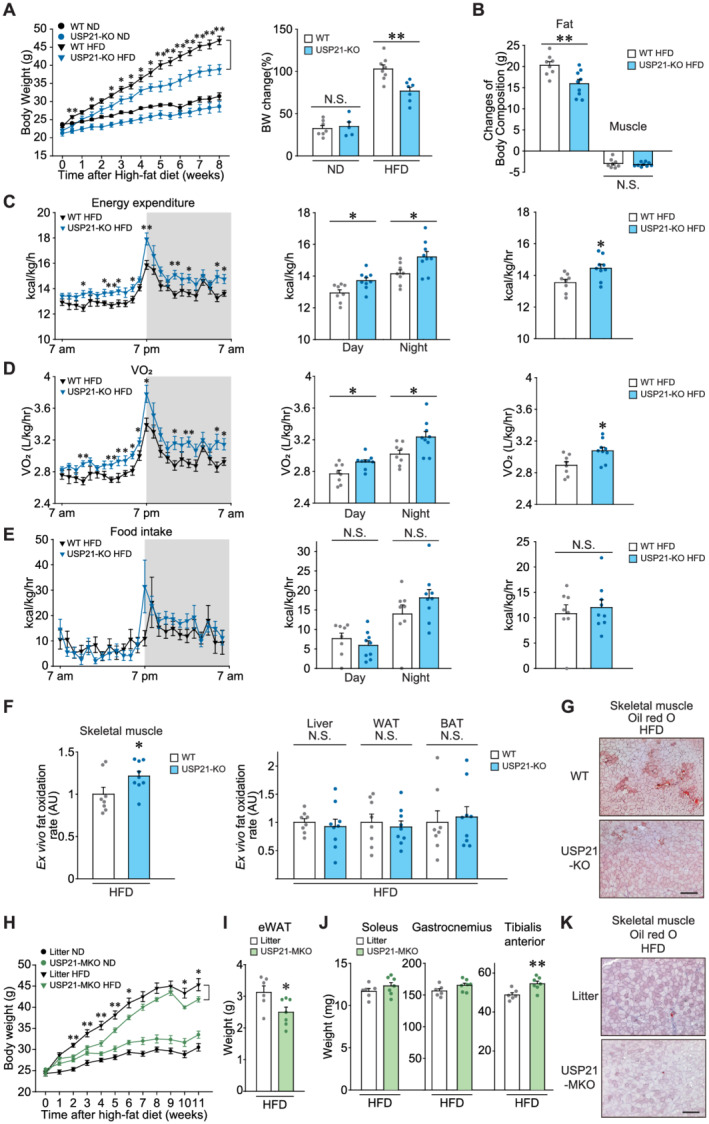
USP21 ablation increases energy expenditure, protecting mice from diet‐induced obesity. (A,B) WT or USP21‐KO mice fed a normal diet (ND) or a high‐fat diet (HFD). (A) BW gains and per cent changes (8 weeks HFD‐fed, n = 5–8/group). (B) Fat and lean muscle mass changes before and after 7 weeks of HFD‐feeding as assessed using nuclear magnetic resonance (n = 8, 9 each). (C–E) In vivo energy balance. Energy expenditure (C), maximal oxygen consumption (VO2) (D), and food intake (E) were analysed in mice housed in individual metabolic cages for 24 h (7 weeks HFD‐fed, n = 8, 9 each). (F) Ex vivo FA oxidation assays in soleus muscle, liver, WAT, and BAT of indicated genotype (11 weeks HFD‐fed, n = 8, 9 each). Tissues quickly isolated from mice of indicated genotypes were incubated in a medium containing [14C]‐oleic acid for 60 min (soleus muscle and WAT) or 30 min (liver and BAT), and the released 14CO2 was lysed for scintillation counting. AU, arbitrary unit. (G) Oil red O staining of tibialis anterior muscle (n = 5, 9 each). Scale bar: 200 μm. (H–K) Litter or USP21‐MKO mice fed an ND (n = 6 each) or an HFD (n = 6, 7 each) for 11 weeks. After HFD feeding, the mice were fasted overnight and then sacrificed. (H) BW gains. (I) Epididymal fat (eWAT) weights. (J) Skeletal muscle weights. (K) Oil red O staining of tibialis anterior muscle (n = 3 each). Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by Student's t test (A–F,H–J). N.S., not significant.
In the analyses of ex vivo FA oxidation rates, HFD‐fed USP21‐KO mice displayed a significantly higher rate of FA oxidation in skeletal muscle, but not in liver, WAT, and BAT, compared with HFD‐fed WT controls (Δ21.5%, P < 0.05; Figure 5F). The HFD‐induced accumulation of intramyocellular lipid droplets was also lower in the KO animals (Figure 5G). We additionally employed USP21‐MKO mice and corroborated the inhibitory effects of USP21 ablation on the BW gain and epididymal fat mass increase caused by HFD feeding (P < 0.05; Figure 5H and 5I). USP21‐MKO mice showed an increase in tibialis anterior muscle mass with a decrease in fat accumulation (Figure 5J and 5K). These results support the conclusion that ablation of USP21 promotes fat catabolism in skeletal muscle and enhances systemic energy expenditure.
Skeletal muscle ablation of USP21 protects other metabolic organs from HFD feeding
We further examined changes in the metabolic phenotypes of other organs in mice fed an HFD. USP21‐KO and USP21‐MKO mice fed an HFD for 11 weeks had significantly attenuated increases in liver weight compared with WT or litter mice (P < 0.05 or P < 0.01; Figure 6A). USP21‐KO and USP21‐MKO mice fed on ND showed no difference compared with control animals. Consistently, liver injury and steatosis were diminished in the USP21‐KO and USP21‐MKO mice fed an HFD, as evidenced by changes in serum alanine transaminase activities (P < 0.05 or P < 0.01) and histopathological analyses (Figure 6B and 6C).
Figure 6.
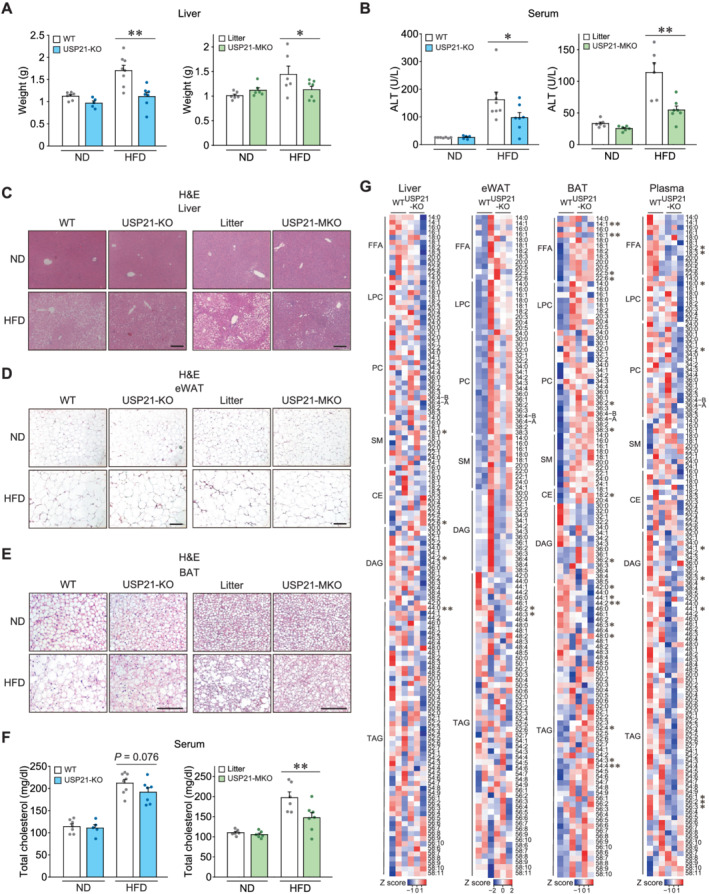
Loss of USP21 protects other metabolic organs from diet‐induced obesity. (A–F) WT or USP21‐KO mice (n = 5–8 each); or litter or USP21‐MKO mice (n = 6–7 each) were fed an ND or an HFD for 11 weeks. (A) Liver weights. (B) Serum ALT activities. (C–E) Haematoxylin and eosin (H&E)‐stained images of liver (C), epididymal fat (D), and BAT (E) of indicated genotypes. Scale bar: 200 μm. (F) Serum cholesterol contents. ALT, alanine transaminase. (G) Heatmaps for lipids profiling in the indicated tissues and plasma of 12‐week‐old WT and USP21‐KO mice as assessed by liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) (n = 3 each). The first number of analyte indicates the number of carbons in the lipid acyl chain and the number after the colon indicates the number of double bonds in the lipid acyl chain(s). Lipids peak intensities with independent Student's t test (*P < 0.05, **P < 0.01) were presented as heatmaps according to the Z score. Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by one‐way ANOVA with least significant difference multiple comparisons correction (A,B,F).
In addition, fat accumulation and adipose hypertrophy in WAT and BAT were also attenuated in USP21‐KO mice fed an HFD (Figure 6D, left, and 6E, left). Similarly, the inhibitory effect of USP21 ablation on adipose tissue was pronounced in the USP21‐MKO mice (Figure 6D, right, and 6E, right). These results support the notion that a deficiency in USP21 in skeletal muscle contributes to decreases in fat accumulation in other metabolic organs, including the liver, WAT, and BAT. Consistent with these changes, USP21‐MKO mice displayed a decrease in total blood cholesterol content (P < 0.01; Figure 6F), verifying the systemic effect of USP21 modulation on lipid catabolism. Furthermore, when we employed lipidomic approaches to study the metabolic tissues and plasma of USP21‐KO animals, the results were consistent with the changes in skeletal muscle lipid profiles. Loss of USP21 caused a decrease in DAG (C34:2) in the liver, TAGs (C46:2, C46:3) in WAT, and free fatty acids (FFAs) (C14:1**, 16:1**) and TAGs (C42:0, C44:1, C44:2**, C46:3) in BAT. Moreover, the contents of FFAs (C18:2, C18:3), LPC (C16:0), PC (C32:2), DAGs (C34:1, C36:3), and TAGs (C44:1, C56:1, 56:2. 56:3) were significantly decreased in the plasma of the KO animals (P < 0.05 or P < 0.01; Figure 6G). These results show that USP21 ablation systemically benefits lipid catabolism during the high‐fat dietary challenge.
USP21 ablation ameliorates HFD‐induced hyperglycaemia
Metabolic dysfunction in skeletal muscle deregulates systemic glucose metabolism and induces hyperglycaemia. 2 To better understand the association between USP21 levels in skeletal muscle and glucose homeostasis, we assessed USP21 levels in the skeletal muscle of a type 2 diabetic patient. From the immunochemical assay, USP21 level was shown to be higher, while phosphorylated‐AMPK was reciprocally lower, in the patient with diabetes than the non‐diabetic control (1.94‐fold or 0.30‐fold; Figure 7A and 7B). Similarly, HFD‐fed mice displayed induction of USP21 in skeletal muscle, as supported by the outcomes of immunohistochemistry, immunoblotting, and qRT‐PCR assays (P < 0.05 or P < 0.01; Figure 7C–7E).
Figure 7.
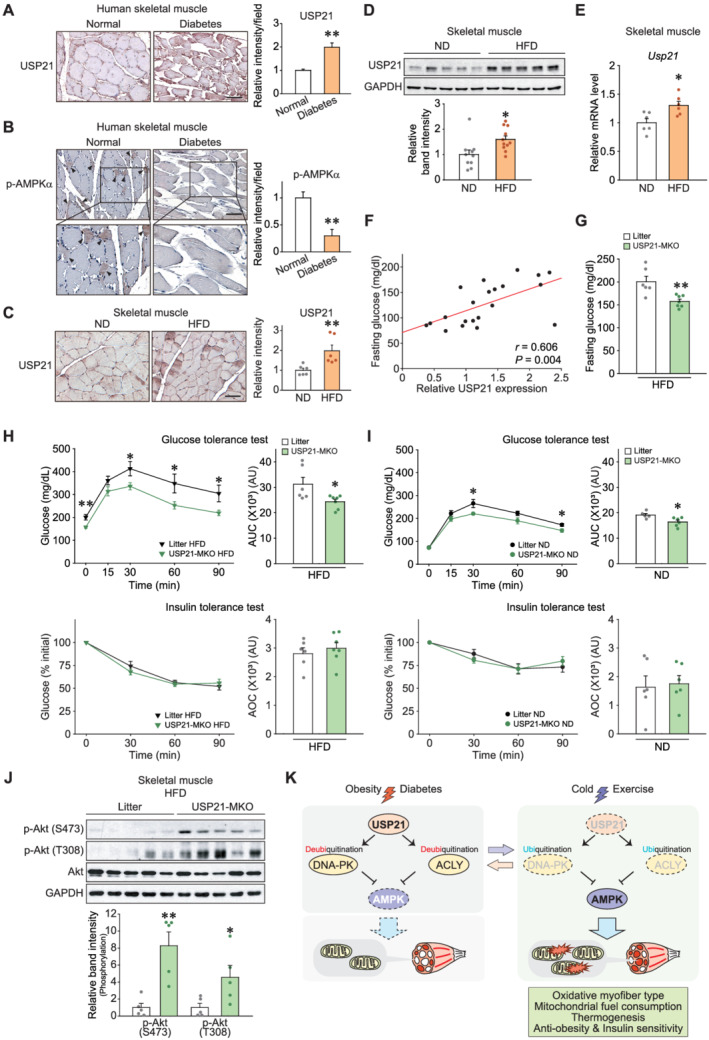
Skeletal muscle‐specific ablation of Usp21 ameliorates insulin resistance. (A,B) Immunostainings for USP21 and phosphorylated‐AMPKα in the skeletal muscle of a healthy subject or a diabetic patient (five images per each sample). (B) Arrowheads indicate phosphorylated‐AMPK signals. Scale bar: 100 μm. (C–F) WT or USP21‐KO mice were fed on an ND or an HFD. (C) Immunostainings for USP21 in tibialis anterior muscle (11 weeks fed, n = 6 each). Scale bar: 100 μm. (D) Immunoblottings for USP21 in soleus muscle (18 weeks fed, n = 10, 11 each). A representative blot was shown. (E) Quantitative real‐time (qRT)‐PCR assays for Usp21 in gastrocnemius muscle of mice (11 weeks fed, n = 6 each). (F) Correlation between USP21 levels in soleus muscle from Panel (D) and fasting blood glucose contents in ND‐fed or HFD‐fed mice (8 weeks fed, n = 21). Data were analysed by Pearson correlations. (G) Fasting blood glucose contents in litter or USP21‐MKO mice fed an HFD for 9 weeks, as assessed at the starting point of glucose tolerance test (GTT) (n = 6, 7 each). (H) GTT (glucose gavage; 1.5 g/kg BW, 9 weeks HFD‐fed) and insulin tolerance tests (ITT, insulin injection; 1.5 IU/kg BW, 10 weeks HFD‐fed, % of initial) in litter or USP21‐MKO mice (n = 6, 7 each). (I) GTT (glucose gavage; 1.5 g/kg BW) and ITT (insulin injection; 1 IU/kg BW, % of initial) in litter or USP21‐MKO mice fed an ND (n = 5–6 each). (J) Immunoblottings for phospho‐Akt (Ser473 or Thr308) and Akt in soleus muscle of litter or USP21‐MKO mice fed an HFD for 11 weeks (n = 5 each). (K) Schematic diagrams illustrating as to how USP21 and its target molecules regulate fuel metabolism in skeletal muscle. Values are expressed as means ± standard error of the mean (SEM). *P < 0.05, **P < 0.01, by Student's t test (A–E,G–J). AOC; area over the curve; AU, arbitrary unit; AUC, area under the curve; IU, insulin unit.
USP21 levels in skeletal muscle positively correlated with fasting blood glucose levels (r = 0.606, P = 0.004, Figure 7F). Moreover, the skeletal muscle‐specific KO of Usp21 significantly attenuated the hyperglycaemia induced via HFD feeding for 9 weeks (Δ21.5%, P < 0.01, Figure 7G). In addition, systemic glucose intolerance was relieved in animals fed an HFD or an ND, as assessed by glucose tolerance test (Δ22.1% or Δ14.3%; P < 0.05; Figure 7H, upper, and 7I, upper). Blood glucose levels were lower in USP21‐MKO mice fed an HFD compared with those in litter, as assessed by insulin tolerance test (Δ19.2%, P < 0.05; Figure S9A). When glucose levels were normalized to the initial glucose content (i.e. zero‐time), the relative glucose contents and area over the curve of blood glucose were comparable with controls (Figure 7H, lower, and 7I, lower). The levels of phospho‐Akt at Ser473 and Thr308 representing insulin sensitivity were both increased in skeletal muscle of USP21‐MKO mice fed an HFD compared with controls (P < 0.05 or P < 0.01; Figure 7J). Similar outcomes were obtained in all experiments using USP21‐KO mice (Figure S9B–S9D). These data provide strong evidence that USP21 ablation in skeletal muscle ameliorates HFD‐induced hyperglycaemia and improved insulin sensitivity.
Discussion
Reprogramming of myofibres to oxidative phenotype through endurance exercise training changes overall metabolic capacity. 20 In the present study, bioinformatic and experimental analyses demonstrated down‐regulation and up‐regulation of USP21 by exercise and diabetes, respectively, suggestive of its role in skeletal muscle pathophysiology. The outcomes of this study accomplished using USP21 modulation, comprehensive RNA‐seq, and lipidomics elaborate USP21‐mediated myofibre phenotypic change, muscle mass control, mitochondrial biogenesis, and fuel burning. As the first functional impact, the effects of USP21 ablation on obesity and diabetes control were discovered using two different genetic animal models, supporting the hypothesis that USP21 in skeletal muscle indeed plays a role in systemic energy metabolism. Another interesting phenotype change is the regulation of thermogenesis and tolerance to cold stress by USP21 ablation, indicative of fuel consumption and temperature control. The phenotypic changes combined with the repression of USP21 in skeletal muscle through exercise or cold stress strengthen its impact on physical adaptations to external stresses.
The role of skeletal muscle in systemic metabolism has been shown in human studies, which demonstrates that oxidative myofibres and muscle heat generation confer protection from obesity and related disorders. 3 , 21 , 22 A key finding is that the ablation of USP21 reduces BW gains in animals fed an HFD. Moreover, USP21 levels were elevated in skeletal muscle under metabolic overload conditions. Therefore, it would be reasonable to assume that enhanced energy metabolism in skeletal muscle systemically helps to prevent metabolic dysregulation. FA oxidation is coupled to an increased fuel expenditure, whereas its reduction causes fat accumulation. In our studies, HFD‐induced fat accumulation was ameliorated in other organs, supporting the inference that USP21 modulation in skeletal muscle and the resultant changes in myofibre type conversion, muscle mass, and energy expenditure benefit the whole body.
Our findings consistently identified USP21 as a regulator of lipid oxidation as shown by changes in FA oxidation and skeletal muscle lipid content, particularly DAG and TAG contents in the KO model. This idea was supported by the outcomes of RNA‐seq analysis, gene clustering, and lipidomics. Changes in oxidative capacity along with the dysregulated mitochondrial capacity of muscle correlate with an accumulation of intramyocellular lipids. 23 , 24 Mitochondria are critical organelles for the control of metabolic status of skeletal muscle, and thus, the mitochondrial processes need to be dynamically and tightly regulated by diverse pathways. Another important finding of our study is that loss of USP21 enhanced mitochondrial respiration, which concurred with the results that USP21‐KO and USP21‐MKO mice showed augmented mitochondrial oxidative capacity and expression of the responsible genes. The idea that mitochondrial respiration is increased by USP21 ablation was fortified by our results on SDH activity, thermogenesis, up‐regulation of Ucp2 and Ucp3, and myosin heavy chain subtype analyses. Hence, USP21 appears to serve as a switch controlling fuel catabolism through dynamic regulation of mitochondria. Moreover, transmission electron microscopy and mtDNA assay also support the notion that overall mitochondrial contents are increased by USP21 ablation. Hence, increased respiratory activity and mitochondrial content may coordinately contribute to the effect of USP21 deficiency on fuel oxidation in skeletal muscle.
Changes in fuel oxidation in peripheral organs are considered a primary contributor to systemic glucose homeostasis. 23 , 24 In particular, fuel oxidation in skeletal muscle is indispensable in maintaining systemic glucose homeostasis. Indeed, muscle glucose intolerance precedes hepatic glucose imbalance, contributing to excess fat accumulation and metabolic disorders by loading ingested glucose and FAs to different organs, 25 and thus, glucose intolerance in skeletal muscle promotes obesity. 26 , 27 Consistently, the skeletal muscle‐specific ablation of USP21 in our study had a beneficial effect on systemic insulin sensitivity. The liver takes up FA from the bloodstream and converts it to TAG for storage or very low‐density lipoprotein secretion. Adipose tissue is the main storage organ for TAG and the uptake of FA from lipoprotein in the bloodstream. Skeletal muscle absorbs FA from the bloodstream to use as an energy source. 28 Our results showing reductions in neutral lipids in the liver and WAT and reductions in FFAs and several neutral lipids in plasma support the idea that high lipid consumption by skeletal muscle reduces lipid transport from liver to adipose tissue and promotes a shift from lipid storage to utilization, contributing to glucose homeostasis.
Uncoupling is an essential process for heat production in skeletal muscle and BAT. 21 In our research, the thermogenic effect in the skeletal muscle of the USP21‐KO animals seemed to be linked at least in part to mitochondrial uncoupling activity. The results of our RNA‐seq dataset analysis, the first publicly disseminated global gene expression database obtained using the USP21‐MKO model, support the ability of USP21 ablation to control thermogenesis through an increase in oxidative uncoupling activity accompanying Ucp2/3 up‐regulation. In particular, UCP3 seems to promote FA oxidation, decrease of BW gain, and would thus prevent obesity and diabetes, 16 being consistent with our findings. Moreover, a large mass of skeletal muscle and its contribution to metabolism render UCP2 (ubiquitously expressed) and UCP3 (preferentially expressed in muscles) significant players in thermogenesis and the regulation of energy metabolism. 16 By contrast, USP21‐KO animals showed no increase in Ucp1 in BAT in response to cold exposure, implying that the thermogenic effect of USP21 deficiency was derived from skeletal muscle. In addition, BAT shows restricted UCP1 expression in humans except in the neonatal stages. 21 Together, it is highly likely that skeletal muscle thermogenesis by USP21 ablation results from increased mitochondrial biogenesis in oxidative fibres and mitochondrial uncoupling mediated, at least in part, by USP2/3, as also supported by the observations of enhanced FA catabolic processes.
Emerging evidence indicates the importance of the ubiquitination status of proteins in diverse biological functions. USPs control target protein stability by inhibiting ubiquitin‐mediated proteasomal degradation. The regulation of metabolism by several USPs and their substrates has been studied; USP10, USP4, and USP18 inhibit nonalcoholic steatohepatitis, 29 , 30 , 31 whereas USP14 aggravates nonalcoholic steatohepatitis. 32 USP17 affects vascular disease 33 and USP19 promotes muscle wasting. 34 The present study described the ability of USP21 to regulate the post‐translational modification of substrates involved in fuel consumption and energy homeostasis. Our study suggests persistent activation of AMPK, a central kinase for cellular metabolism, as a robust downstream event of USP21 modulation, both in cells and animals. Through the study, comprehensive proteome assays and the examination of the ubiquitination status of neighbour molecules led us to discover the substrates DNA‐PKcs and ACLY. The role of E3 ligases in DNA‐PKcs and ACLY ubiquitination has been studied, 35 , 36 , 37 but the ubiquitinase for these molecules was unknown. Although DNA‐PKcs is known to play a role in DNA double‐stranded break responses, 38 recent studies suggest that it has other functions as well. 39 , 40 , 41 The induction of DNA‐PK decreased AMPK activity and mitochondrial function in aged muscle, whereas the inhibition of DNA‐PK alleviated physical decline and improved insulin sensitivity, 39 being consistent with our study. DNA‐PKcs may phosphorylate upstream stimulatory factor‐1 for the transcriptional activation of FA synthase 40 and promote alcohol‐related liver disease. 41 ACLY is an enzyme responsible for lipid biosynthesis. 42 In chondrocytes, ACLY inhibition activates AMPK. 43 Identification of USP21 as an upstream regulator of DNA‐PKcs and ACLY which signal through AMPK integrates previous findings and suggests a newly described metabolic pathway.
The AMPK senses the ATP status of the cell to maintain energy balance by fine‐tuning anabolic and catabolic signalling. In response to energy demand, AMPK acts as a signal integration platform by phosphorylating proteins necessary for adaptation to energy availability. 44 Despite the importance of AMPK and its upstream activators, 45 the inhibitory regulator of AMPK for mitochondrial functions has been elusive. The marked and persistent activation of AMPK in skeletal muscle by the loss of USP21 was substantiated by our KEGG analysis, and in vivo and in vitro experiments. ATP consumption increases the AMP/ATP and ADP/ATP ratios, thereby activating AMPK. 44 , 46 Here, we showed there was a significant decrease in ATP in skeletal muscle tissue because of USP21 ablation, which indicates that there was a change in the mitochondrial membrane potential, reflecting uncoupling under the conditions of fuel oxidation. Consequently, the lack of an increase in the endurance exercise capacity via USP21 deficiency may also reflect ATP depletion. This idea was strengthened by the persistent activation of AMPK, even during augmented mitochondrial biogenesis. The lack of statistical significance in the AMP/ATP and ADP/ATP ratios may have been due to the dynamic circuitry of the nucleotides. FA oxidation in skeletal muscle and myofibre switching to an oxidative type may represent the adaptation of muscles to a lack of ATP maintenance. In this setting, our findings showing USP21‐dependent AMPK modulation through DNA‐PKcs and ACLY render a new metabolic pathway for the control of energy expenditure. An increase in UCP2/3 expression by AMPK 47 , 48 and by genetic ablation of USP21 in our study strengthens the idea that the identified molecules may also affect mitochondrial uncoupling and fuel oxidation.
Previously, we showed Gα13‐NFATc1 signalling pathway as a regulatory axis for oxidative fibre switch, but not muscle mass control nor thermogenesis. 9 The findings in the present study expand our knowledge on oxidative myofibre type conversion, muscle mass control, and thermogenic physiology involved in energy expenditure and demonstrate the impact of USP21 and its substrates network in skeletal muscle as a key metabolic sensory axis for this event through the regulation of systemic fuel storage and consumption (Figure 7K) and suggest further investigations into the inhibitors of USP21 or its downstream targets, which may provide an opportunity to develop new therapeutic strategies to treat patients suffering from muscle problems and the associated metabolic diseases.
Conflict of interest
The authors have declared that they have no competing interests.
Funding
We thank Dr SH Lee (Seoul National University, Seoul, South Korea) for providing the protamine‐Flpe mice. This work was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (2017K1A1A2004511 to S. G. K.; 2014M3A9D5A01073886 to C. S. C.; 2018R1A3B1052328 to S. P.; 2021R1C1C1013323 and 2021R1A4A5033289 to J. H. K.) and by Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health and Welfare (HR14C0001 to C. S. C.). This work was supported by the Dongguk University Research Fund of 2020.
Supporting information
Figure S1. USP21 expression levels in different skeletal muscles. Immunoblottings were done for skeletal muscles excised from 15‐week‐old C57BL/6 mice morning fasted prior to sacrifice (n = 3). A representative blot was shown. Sol, soleus; GC, gastrocnemius; TA, tibialis anterior; and EDL, extensor digitorum longus.
Figure S2. Validation of USP21 modulations. (A) qRT‐PCR assays for Usp21 in tibialis anterior muscle of 12‐week‐old litter or USP21‐MKO mice with electric shocks for Usp21 (or mock) gene delivery. Mice were subjected to analyses 7 days after electroporation (n = 3–5 each). Values are expressed as means ± SEM. **P < 0.01 for WT versus USP21‐MKO mice; ##P < 0.01 for USP21‐MKO versus USP21‐MKO mice plus USP21 overexpression by Student's t‐test. (B, C) Immunoblottings for USP21 in C2C12 myotubes or skeletal muscle. (B) C2C12 cells were differentiated to myotubes after transfection with Usp21‐specific siRNA (or mock) for 24 h. (C) Tibialis anterior muscle of 12‐week‐old WT mice 7 days after electroporation‐mediated mock or Usp21 gene delivery (n = 3 each, left). C2C12 cells were differentiated to myotubes after transfection with Usp21 plasmid (or mock) for 24 h (right).
Figure S3. Analysis of heart muscle in USP21 MKO mice. (A) qRT‐PCR assays for Usp21 in the liver and heart of litter and USP21‐MKO mice (n = 3 each). (B) H&E‐stained images of heart muscle of litter and USP21‐MKO mice (n = 3 each). Scale bar: 1000 μm. (C) qRT‐PCR assays for genes associated with mitochondrial function in heart of litter and USP21‐MKO mice (n = 3 each). Values are expressed as means ± SEM. **P < 0.01, by Student's t‐test. N.S., not significant.
Figure S4. Expression of thermogenic genes in USP21‐KO mice exposed to cold stress. (A, B) 13‐week‐old mice were housed individually in a room of ambient temperature (RT) or a 4 °C cold room for 6 h (n = 5–8 each). (A) qRT‐PCR assays for Ucp1, Pgc1a, and Cd36 in BAT. (B) qRT‐PCR assays for Serca1, Serca2, and Sarcolipin in skeletal muscle. Values are expressed as means ± SEM. *P < 0.05, by one‐way ANOVA with Tukey's multiple comparisons correction. N.S., not significant.
Figure S5. Endurance exercise capacity tests. (A) Relative USP21 transcript levels in human skeletal muscle of sedentary or trained individuals (n = 10 each). Data were extracted from GSE48278 and GSE9103 available in the public domain. (B) Immunoblottings for USP21 in gastrocnemius muscle of WT mice sedentary or exercised until exhaustion (n = 5 each). (C, D) Endurance capacities. (C) 10‐week‐old USP21‐KO mice (n = 20, 22 each) or (D) 11‐week‐old USP21‐MKO mice (n = 11, 9 each) as compared with its littermate mice (left). Percentage of mice running until exhaustion, running duration, and distance in endurance capacity tests (right). Values are expressed as means ± SEM. **P < 0.01, by Student's t‐test. N.S., not significant.
Figure S6. Levels of phosphorylated and total forms of AMPKα, ACC, and LKB1 in USP21‐deficient mice. (A) Immunoblottings for phosphorylated AMPKα and phosphorylated ACC in liver homogenates of 13‐week‐old WT or USP21‐KO mice (n = 3 each). (B) Immunoblottings for phosphorylated AMPKα and phosphorylated ACC in liver and heart homogenates of 12‐week‐old WT or USP21‐MKO mice (n = 3 each). (C) Immunoblottings for phosphorylated LKB1 and LKB1 in homogenates of gastrocnemius muscle in 13‐week‐old WT or USP21‐KO mice (n = 3 each).
Figure S7. LC–MS/MS analyses for the proteins interacting with USP21. (A) Amino acid sequences of DNA‐PKcs and ACLY. The number of spectral counts for DNA‐PKcs and ACLY sequences are 65 and 6, respectively. Analysed peptide sequences of each spectral count were marked as yellow. Unique amino acids for each protein were marked as green. (B) Shown are schematic illustrations and their corresponding spectra for DNA‐PKcs [(R)CGAALAGHQLR(G)] and ACLY [(R)SGGmSNELNNIISR(T)].
Figure S8. Locomotor activity, food intake, and energy expenditure in USP21‐KO mice. (A) Locomotor activities of WT or USP21‐KO mice fed a HFD for 7 weeks (n = 8, 9 each). (B, C) Food intake (B), and energy expenditure (C) profiles of WT or USP21‐KO mice fed a ND. Values are expressed as means ± SEM. *P < 0.05, **P < 0.01, by Student's t‐test.
Figure S9. Insulin sensitivity of USP21‐KO mice. (A) ITT (1.5 IU/kg BW, 10 weeks HFD‐fed; 1 insulin IU/kg BW, ND‐fed) in litter or USP21‐MKO mice (n = 6–7 each) from Figure 7G and 7H. (B) GTT (14 weeks HFD‐fed) and ITT (9 weeks HFD‐fed; mg/dL or % of initial) in WT or USP21‐KO mice (n = 6–8 each). (C) GTT and ITT (mg/dL or % of initial) in WT or USP21‐KO mice fed a ND (n = 5–8 each). (D) Immunoblottings for insulin sensitivity markers in soleus muscle of WT or USP21‐KO mice injected with insulin (0.75 U/kg BW; i.p., 15 min) (n = 2–3 each). For B and C, glucose solution (2 g/kg BW); insulin injection (0.75 IU/kg BW). Values are expressed as means ± SEM. *P < 0.05, **P < 0.01, Student's t‐test (A‐C). AUC, area under the curve; AOC, area over the curve; AU, arbitrary unit; and IU, insulin unit.
Table S2. Primers used in genotyping (mouse).
Table S3. Primers used for qRT‐PCR assays (mouse).
Table S1. Lists of top differentially interacting proteins with USP21 by LC–MS/MS analysis. Top proteins differentially interacting with USP21 and showing increases in abundance with over 5 spectral counts in LC–MS/MS analysis were listed in separate supplemental information.
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. 49
Kim A., Koo J. H., Jin X., Kim W., Park S.‐Y., Park S., Rhee E. P., Choi C. S., and Kim S. G. (2021) Ablation of USP21 in skeletal muscle promotes oxidative fibre phenotype, inhibiting obesity and type 2 diabetes, Journal of Cachexia, Sarcopenia and Muscle, 12, 1669–1689, 10.1002/jcsm.12777.
References
- 1. Ye J, Medzhitov R. Control strategies in systemic metabolism. Nat Metab 2019;1:947–957. [DOI] [PubMed] [Google Scholar]
- 2. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009;32:S157–S163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanner CJ, Barakat HA, Dohm GL, Pories WJ, MacDonald KG, Cunningham PR, et al. Muscle fiber type is associated with obesity and weight loss. Am J Physiol Endocrinol Metab 2002;282:E1191–E1196. [DOI] [PubMed] [Google Scholar]
- 4. Batsis JA, Villareal DT. Sarcopenic obesity in older adults: aetiology, epidemiology and treatment strategies. Nat Rev Endocrinol 2018;14:513–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowen TS, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle 2015;6:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He J, Watkins S, Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes 2001;50:817–823. [DOI] [PubMed] [Google Scholar]
- 7. Swatek KN, Komander D. Ubiquitin modifications. Cell Res 2016;26:399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009;10:550–563. [DOI] [PubMed] [Google Scholar]
- 9. Koo JH, Kim TH, Park SY, Joo MS, Han CY, Choi CS, et al. Gα13 ablation reprograms myofibers to oxidative phenotype and enhances whole‐body metabolism. J Clin Invest 2017;127:3845–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim TH, Koo JH, Heo MJ, Han CY, Kim YI, Park SY, et al. Overproduction of inter‐α‐trypsin inhibitor heavy chain 1 after loss of Gα(13) in liver exacerbates systemic insulin resistance in mice. Sci Transl Med 2019;11:eaan4735. [DOI] [PubMed] [Google Scholar]
- 11. Yoon DW, Kwon HN, Jin X, Kim JK, Lee SK, Park S, et al. Untargeted metabolomics analysis of rat hippocampus subjected to sleep fragmentation. Brain Res Bull 2019;153:74–83. [DOI] [PubMed] [Google Scholar]
- 12. Yanes O, Clark J, Wong DM, Patti GJ, Sánchez‐Ruiz A, Benton HP, et al. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol 2010;6:411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wen H, Yang HJ, An YJ, Kim JM, Lee DH, Jin X, et al. Enhanced phase II detoxification contributes to beneficial effects of dietary restriction as revealed by multi‐platform metabolomics studies. Mol Cell Proteomics 2013;12:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim W, Deik A, Gonzalez C, Gonzalez ME, Fu F, Ferrari M, et al. Polyunsaturated fatty acid desaturation is a mechanism for glycolytic NAD(+) recycling. Cell Metab 2019;29 4:856–70.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, et al. A conditional knockout resource for the genome‐wide study of mouse gene function. Nature 2011;474:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab 2005;2:85–93. [DOI] [PubMed] [Google Scholar]
- 17. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lage R, Diéguez C, Vidal‐Puig A, López M. AMPK: a metabolic gauge regulating whole‐body energy homeostasis. Trends Mol Med 2008;14:539–549. [DOI] [PubMed] [Google Scholar]
- 19. Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 2010;11:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 2013;17:162–184. [DOI] [PubMed] [Google Scholar]
- 21. Betz MJ, Enerbäck S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat Rev Endocrinol 2018;14:77–87. [DOI] [PubMed] [Google Scholar]
- 22. Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov 2010;9:465–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcelloni C, et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H–13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999;48 8:1600–1606. [DOI] [PubMed] [Google Scholar]
- 24. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950. [DOI] [PubMed] [Google Scholar]
- 25. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 2016;126:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim JK, Zisman A, Fillmore JJ, Peroni OD, Kotani K, Perret P, et al. Glucose toxicity and the development of diabetes in mice with muscle‐specific inactivation of GLUT4. J Clin Invest 2001;108:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Finck BN, Bernal‐Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, et al. A potential link between muscle peroxisome proliferator‐ activated receptor‐alpha signaling and obesity‐related diabetes. Cell Metab 2005;1:133–144. [DOI] [PubMed] [Google Scholar]
- 28. Shi Y, Burn P. Lipid metabolic enzymes: emerging drug targets for the treatment of obesity. Nat Rev Drug Discov 2004;3:695–710. [DOI] [PubMed] [Google Scholar]
- 29. Luo P, Qin C, Zhu L, Fang C, Zhang Y, Zhang H, et al. Ubiquitin‐specific peptidase 10 (USP10) inhibits hepatic steatosis, insulin resistance, and inflammation through Sirt6. Hepatology 2018;68:1786–1803. [DOI] [PubMed] [Google Scholar]
- 30. Zhao Y, Wang F, Gao L, Xu L, Tong R, Lin N, et al. Ubiquitin‐specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease in mice. Hepatology 2018;68:897–917. [DOI] [PubMed] [Google Scholar]
- 31. An S, Zhao LP, Shen LJ, Wang S, Zhang K, Qi Y, et al. USP18 protects against hepatic steatosis and insulin resistance through its deubiquitinating activity. Hepatology 2017;66:1866–1884. [DOI] [PubMed] [Google Scholar]
- 32. Liu B, Jiang S, Li M, Xiong X, Zhu M, Li D, et al. Proteome‐wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat Commun 2018;9:4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akoumianakis I, Sanna F, Margaritis M, Badi I, Akawi N, Herdman L, et al. Adipose tissue‐derived WNT5A regulates vascular redox signaling in obesity via USP17/RAC1‐mediated activation of NADPH oxidases. Sci Transl Med 2019;11:eaav5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bédard N, Jammoul S, Moore T, Wykes L, Hallauer PL, Hastings KE, et al. Inactivation of the ubiquitin‐specific protease 19 deubiquitinating enzyme protects against muscle wasting. FASEB J 2015;29:3889–3898. [DOI] [PubMed] [Google Scholar]
- 35. Ho SR, Mahanic CS, Lee YJ, Lin WC. RNF144A, an E3 ubiquitin ligase for DNA‐PKcs, promotes apoptosis during DNA damage. Proc Natl Acad Sci USA 2014;111:E2646–E2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang C, Liu J, Huang G, Zhao Y, Yue X, Wu H, et al. Cullin3‐KLHL25 ubiquitin ligase targets ACLY for degradation to inhibit lipid synthesis and tumor progression. Genes Dev 2016;30:1956–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li K, Zhang K, Wang H, Wu Y, Chen N, Chen J, et al. Hrd1‐mediated ACLY ubiquitination alleviate NAFLD in db/db mice. Metabolism 2020;114:154349. [DOI] [PubMed] [Google Scholar]
- 38. Blackford AN, Jackson SP. ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 2017;66:801–817. [DOI] [PubMed] [Google Scholar]
- 39. Park SJ, Gavrilova O, Brown AL, Soto JE, Bremner S, Kim J, et al. DNA‐PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metab 2017;25:1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA‐PK for the metabolic gene regulation in response to insulin. Cell 2009;136:1056–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA‐PKcs promotes alcohol‐related liver disease by activating Drp1‐related mitochondrial fission and repressing FUNDC1‐required mitophagy. Signal Transduct Target Ther 2019;4:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pinkosky SL, Groot PHE, Lalwani ND, Steinberg GR. Targeting ATP‐citrate lyase in hyperlipidemia and metabolic disorders. Trends Mol Med 2017;23:1047–1063. [DOI] [PubMed] [Google Scholar]
- 43. Chen LY, Lotz M, Terkeltaub R, Liu‐Bryan R. Modulation of matrix metabolism by ATP‐citrate lyase in articular chondrocytes. J Biol Chem 2018;293:12259–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kjøbsted R, Hingst JR, Fentz J, Foretz M, Sanz MN, Pehmøller C, et al. AMPK in skeletal muscle function and metabolism. FASEB J 2018;32:1741–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab 2018;27:299–313. [DOI] [PubMed] [Google Scholar]
- 46. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011;13:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Putman CT, Kiricsi M, Pearcey J, MacLean IM, Bamford JA, Murdoch GK, et al. AMPK activation increases uncoupling protein‐3 expression and mitochondrial enzyme activities in rat muscle without fibre type transitions. J Physiol 2003;551:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang J, Liu W, Doycheva DM, Gamdzyk M, Lu W, Tang J, et al. Ghrelin attenuates oxidative stress and neuronal apoptosis via GHSR‐1α/AMPK/Sirt1/PGC‐1α/UCP2 pathway in a rat model of neonatal HIE. Free Radic Biol Med 2019;141:322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. USP21 expression levels in different skeletal muscles. Immunoblottings were done for skeletal muscles excised from 15‐week‐old C57BL/6 mice morning fasted prior to sacrifice (n = 3). A representative blot was shown. Sol, soleus; GC, gastrocnemius; TA, tibialis anterior; and EDL, extensor digitorum longus.
Figure S2. Validation of USP21 modulations. (A) qRT‐PCR assays for Usp21 in tibialis anterior muscle of 12‐week‐old litter or USP21‐MKO mice with electric shocks for Usp21 (or mock) gene delivery. Mice were subjected to analyses 7 days after electroporation (n = 3–5 each). Values are expressed as means ± SEM. **P < 0.01 for WT versus USP21‐MKO mice; ##P < 0.01 for USP21‐MKO versus USP21‐MKO mice plus USP21 overexpression by Student's t‐test. (B, C) Immunoblottings for USP21 in C2C12 myotubes or skeletal muscle. (B) C2C12 cells were differentiated to myotubes after transfection with Usp21‐specific siRNA (or mock) for 24 h. (C) Tibialis anterior muscle of 12‐week‐old WT mice 7 days after electroporation‐mediated mock or Usp21 gene delivery (n = 3 each, left). C2C12 cells were differentiated to myotubes after transfection with Usp21 plasmid (or mock) for 24 h (right).
Figure S3. Analysis of heart muscle in USP21 MKO mice. (A) qRT‐PCR assays for Usp21 in the liver and heart of litter and USP21‐MKO mice (n = 3 each). (B) H&E‐stained images of heart muscle of litter and USP21‐MKO mice (n = 3 each). Scale bar: 1000 μm. (C) qRT‐PCR assays for genes associated with mitochondrial function in heart of litter and USP21‐MKO mice (n = 3 each). Values are expressed as means ± SEM. **P < 0.01, by Student's t‐test. N.S., not significant.
Figure S4. Expression of thermogenic genes in USP21‐KO mice exposed to cold stress. (A, B) 13‐week‐old mice were housed individually in a room of ambient temperature (RT) or a 4 °C cold room for 6 h (n = 5–8 each). (A) qRT‐PCR assays for Ucp1, Pgc1a, and Cd36 in BAT. (B) qRT‐PCR assays for Serca1, Serca2, and Sarcolipin in skeletal muscle. Values are expressed as means ± SEM. *P < 0.05, by one‐way ANOVA with Tukey's multiple comparisons correction. N.S., not significant.
Figure S5. Endurance exercise capacity tests. (A) Relative USP21 transcript levels in human skeletal muscle of sedentary or trained individuals (n = 10 each). Data were extracted from GSE48278 and GSE9103 available in the public domain. (B) Immunoblottings for USP21 in gastrocnemius muscle of WT mice sedentary or exercised until exhaustion (n = 5 each). (C, D) Endurance capacities. (C) 10‐week‐old USP21‐KO mice (n = 20, 22 each) or (D) 11‐week‐old USP21‐MKO mice (n = 11, 9 each) as compared with its littermate mice (left). Percentage of mice running until exhaustion, running duration, and distance in endurance capacity tests (right). Values are expressed as means ± SEM. **P < 0.01, by Student's t‐test. N.S., not significant.
Figure S6. Levels of phosphorylated and total forms of AMPKα, ACC, and LKB1 in USP21‐deficient mice. (A) Immunoblottings for phosphorylated AMPKα and phosphorylated ACC in liver homogenates of 13‐week‐old WT or USP21‐KO mice (n = 3 each). (B) Immunoblottings for phosphorylated AMPKα and phosphorylated ACC in liver and heart homogenates of 12‐week‐old WT or USP21‐MKO mice (n = 3 each). (C) Immunoblottings for phosphorylated LKB1 and LKB1 in homogenates of gastrocnemius muscle in 13‐week‐old WT or USP21‐KO mice (n = 3 each).
Figure S7. LC–MS/MS analyses for the proteins interacting with USP21. (A) Amino acid sequences of DNA‐PKcs and ACLY. The number of spectral counts for DNA‐PKcs and ACLY sequences are 65 and 6, respectively. Analysed peptide sequences of each spectral count were marked as yellow. Unique amino acids for each protein were marked as green. (B) Shown are schematic illustrations and their corresponding spectra for DNA‐PKcs [(R)CGAALAGHQLR(G)] and ACLY [(R)SGGmSNELNNIISR(T)].
Figure S8. Locomotor activity, food intake, and energy expenditure in USP21‐KO mice. (A) Locomotor activities of WT or USP21‐KO mice fed a HFD for 7 weeks (n = 8, 9 each). (B, C) Food intake (B), and energy expenditure (C) profiles of WT or USP21‐KO mice fed a ND. Values are expressed as means ± SEM. *P < 0.05, **P < 0.01, by Student's t‐test.
Figure S9. Insulin sensitivity of USP21‐KO mice. (A) ITT (1.5 IU/kg BW, 10 weeks HFD‐fed; 1 insulin IU/kg BW, ND‐fed) in litter or USP21‐MKO mice (n = 6–7 each) from Figure 7G and 7H. (B) GTT (14 weeks HFD‐fed) and ITT (9 weeks HFD‐fed; mg/dL or % of initial) in WT or USP21‐KO mice (n = 6–8 each). (C) GTT and ITT (mg/dL or % of initial) in WT or USP21‐KO mice fed a ND (n = 5–8 each). (D) Immunoblottings for insulin sensitivity markers in soleus muscle of WT or USP21‐KO mice injected with insulin (0.75 U/kg BW; i.p., 15 min) (n = 2–3 each). For B and C, glucose solution (2 g/kg BW); insulin injection (0.75 IU/kg BW). Values are expressed as means ± SEM. *P < 0.05, **P < 0.01, Student's t‐test (A‐C). AUC, area under the curve; AOC, area over the curve; AU, arbitrary unit; and IU, insulin unit.
Table S2. Primers used in genotyping (mouse).
Table S3. Primers used for qRT‐PCR assays (mouse).
Table S1. Lists of top differentially interacting proteins with USP21 by LC–MS/MS analysis. Top proteins differentially interacting with USP21 and showing increases in abundance with over 5 spectral counts in LC–MS/MS analysis were listed in separate supplemental information.