

Abstract
Background
Sarcopenia with chronic kidney disease (CKD) progression is associated with life prognosis. Oxidative stress has attracted interest as a trigger for causing CKD‐related muscular atrophy. Advanced oxidation protein products (AOPPs), a uraemic toxin, are known to increase oxidative stress. However, the role of AOPPs on CKD‐induced muscle atrophy remains unclear.
Methods
In a retrospective case–control clinical study, we evaluated the relationship between serum AOPPs levels and muscle strength in haemodialysis patients with sarcopenia (n = 26, mean age ± SEM: 78.5 ± 1.4 years for male patients; n = 22, mean age ± SEM: 79.1 ± 1.5 for female patients), pre‐sarcopenia (n = 12, mean age ± SEM: 73.8 ± 2.0 years for male patients; n = 4, mean age ± SEM: 74.3 ± 4.1 for female patients) or without sarcopenia (n = 12, mean age ± SEM: 71.3 ± 1.6 years for male patients; n = 7, mean age ± SEM: 77.7 ± 1.6 for female ). The molecular mechanism responsible for the AOPPs‐induced muscle atrophy was investigated by using 5/6‐nephrectomized CKD mice, AOPPs‐overloaded mice, and C2C12 mouse myoblast cells.
Results
The haemodialysis patients with sarcopenia showed higher serum AOPPs levels as compared with the patients without sarcopenia. The serum AOPPs levels showed a negative correlation with grip strength (P < 0.01 for male patients, P < 0.01 for female patients) and skeletal muscle index (P < 0.01 for male patients). Serum AOPPs levels showed a positive correlation with cysteinylated albumin (Cys‐albumin), a marker of oxidative stress (r 2 = 0.398, P < 0.01). In the gastrocnemius of CKD mice, muscle AOPPs levels were also increased, and it showed a positive correlation with atrogin‐1 (r 2 = 0.538, P < 0.01) and myostatin expression (r 2 = 0.421, P < 0.05), but a negative correlation with PGC‐1α expression (r 2 = 0.405, P < 0.05). Using C2C12 cells, AOPPs increased atrogin‐1 and myostatin expression through the production of reactive oxygen species via CD36/NADPH oxidase pathway, and decreased myotube formation. AOPPs also induced mitochondrial dysfunction. In the AOPPs‐overloaded mice showed that decreasing running time and hanging time accompanied by increasing AOPPs levels and decreasing cross‐sectional area in gastrocnemius.
Conclusions
Advanced oxidation protein products contribute to CKD‐induced sarcopenia, suggesting that AOPPs or its downstream signalling pathway could be a therapeutic target for the treatment of CKD‐induced sarcopenia. Serum AOPPs or Cys‐albumin levels could be a new diagnostic marker for sarcopenia in CKD.
Keywords: Advanced oxidation protein products, Albumin, Biomarker, Chronic kidney disease, Muscle atrophy, Oxidative stress
Introduction
Patients with chronic kidney disease (CKD) also frequently have complications associated with muscle atrophy and muscle weakness (sarcopenia), and the onset of sarcopenia is associated with an increased risk of death in addition to being bedridden, as well as experiencing falls and fractures. The number of patients with sarcopenia increases with the progression of CKD. 1 In fact, the prevalence of sarcopenia in CKD ranges from 5% to 14% in the preservation period of CKD (eGFR category G3 to G5) and from 13% to 34% in the haemodialysis phase. It was also reported that a decrease in walking speed of 0.1 m/s increases mortality by 26% in patients with eGFR category G2 to G4. 2 Therefore, the prevention of CKD‐related sarcopenia is considered to be an urgent issue. Although the development of early diagnostic markers and therapies against CKD‐related sarcopenia are needed, the pathogenic mechanism responsible for CKD‐related sarcopenia remains unclear.
Oxidative stress has attracted attention as a trigger for CKD‐related muscular atrophy. Oxidative stress is caused by an imbalance between the oxidative damage derived from the reactive oxygen species (ROS) and the antioxidant defence system. It is known that oxidative stress increases as a function of age and in cases of chronic inflammation. 3 The ROS levels in human skeletal muscle are inversely correlated with cross‐sectional area. 4 In mouse myoblast cells (C2C12), an increase in the levels of intracellular ROS is associated with the development of muscular atrophy by increasing the expression of atrogin‐1 and myostatin, which are muscle atrophy‐related genes, and the increased ROS is also associated with a decrease in muscle strength accompanied by decreasing mitochondrial function. 5 , 6 , 7 We recently investigated the molecular mechanism of CKD‐related muscular atrophy by focusing on the levels of uraemic toxins that accumulate in the body with decreasing renal function. Among the small molecule uraemic toxins, indoxyl sulfate (IS) was taken up by skeletal muscle cells via an organic anion transporter. After being taken up, IS was involved in the development of muscle atrophy by enhancing ROS production through the activation of NADPH oxidase. 5 In addition, IS was also involved in the development of muscle weakness by impairing mitochondrial function. 6 , 8 Furthermore, we found that the administration of AST‐120 (oral carbon absorbent), which lowers serum IS concentrations, reversed muscle atrophy and mitochondrial function in CKD mice. 6
Advanced oxidation protein products (AOPPs) are products that are produced by the oxidation of serum proteins (mainly albumin). Increased ROS levels that occur in inflammatory conditions result in the production of hypochlorous acid (HOCl), via the action of neutrophil myeloperoxidase, resulting in the further oxidation of serum proteins. 9 , 10 , 11 , 12 Advanced oxidation protein products are also known to be one of the middle molecule uraemic toxins that are not removed by haemodialysis. The concentration of serum AOPPs in healthy subjects is about 20 to 30 μM, but this value increases to about 100 to 150 μM in CKD patients. 11 , 13 AOPPs are taken up by cells via CD36, a scavenger receptor. The AOPPs then further enhance ROS production by activating intracellular NADPH oxidase. Although AOPPs are involved in the development of renal tubular damage, chronic hepatitis, osteoporosis and Crohn's disease, 10 , 11 , 14 our current knowledge regarding the effect of AOPPs on skeletal muscle are limited. Taking these previous reports into consideration, we hypothesized that the increased levels of AOPPs in CKD patients may be closely associated with muscular atrophy.
The purpose of this study was to determine the involvement of AOPPs in CKD‐related muscle atrophy. We first carried out a retrospective case–control clinical study in which the relationship between serum AOPPs levels and muscle strength was evaluated in haemodialysis patients with or without sarcopenia. We then investigated the molecular mechanism of AOPPs‐induced muscle atrophy by using 5/6‐nephrectomized CKD mice, AOPPs‐overloaded mice and C2C12 mouse myoblast cells.
Materials and methods
Chemicals and materials
A commercial human serum albumin (HSA) solution was purchased from the Japan Blood Products Organization (Tokyo, Japan). Diphenylene iodonium (DPI), rabbit polyclonal anti‐mouse GAPDH antibody, anti‐laminin antibody produced in rabbits were purchased from Sigma‐Aldrich (St Louis, MO). The anti‐atrogin‐1 antibody was purchased from ECM biosciences (Versailles, KY, USA). Anti‐PGC‐1α (3G6) rabbit monoclonal antibody was purchased from Cell Signalling Technology (Danvers, MA, USA). The anti‐myogenin monoclonal antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The anti‐CD36/SR‐B3 antibody was purchased from Novus Biologicals (Centennial, CO, USA). The anti‐myosin heavy chain antibody was purchased from R&D systems (Minneapolis, MN, USA). The anti‐Akt antibody and phospho‐Akt (Ser473) rabbit monoclonal antibody were purchased from Cell Signaling Technology (Danvers, MA). ATP assay Kit was purchased from FUJIFILM Wako Chemicals (Tokyo, Japan). MitoRed, Hoechist33342 were purchased from Dojin Chemical (Kumamoto, Japan). Potassium iodide and chloramine T, acetic acid, N‐acetyl‐l‐cysteine (NAC) were purchased from Nacalai Tesque (Kyoto, Japan). 5‐(and 6)‐Chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate (CM‐H2DCFDA), Dulbecco's modified eagle medium (DMEM), Dulbecco's phosphate‐buffered saline (D‐PBS), MitoGreen were purchased from Invitrogen (Grand Island, NY). Block ace was purchased from DS Pharma Biomedical (Osaka, Japan). All methods were carried out in accordance with approved guidelines. All experimental protocols were approved by Kumamoto University.
Patients
This study enrolled 64 haemodialysis patients with sarcopenia (male, 38; female, 26, including pre‐sarcopenia) and 19 haemodialysis patients without sarcopenia (male, 12; female, 7) aged 65 years or older who were outpatients in the Department of Nephrology, Akebono Clinic, Kumamoto, Japan. Diagnosis criteria for sarcopenia was a walking speed of less than 1 m/s, or a grip strength of less than 25 kgf for male patients and less than 20 kgf for female patients, then a body mass index (BMI) value of less than 18.5 kg/m2 or a calf circumference of less than 30 cm in addition to skeletal muscle index (SMI) of less than 7.0 kg/m2 for male patients and less than 5.7 kg/m2 for female patients. Diagnosis criteria for pre‐sarcopenia was a BMI value of less than 18.5 kg/m2 or a calf circumference of less than 30 cm in addition to SMI of less than 7.0 kg/m2 for male patients and less than 5.7 kg/m2 for female although a walking speed of more than 1 m/s, or a grip strength of more than 25 kgf for male patients and more than 20 kgf for female patients.
This retrospective case–control study protocol was approved by the ethics committee of the Faculty of Life Sciences, Kumamoto University (Approval No. 1578). All of the subjects provided their written informed consent to participate in this study.
Advanced oxidation protein products level assay
Assays for AOPPs levels were performed as described in a previous report. 11 A 200 μL of samples diluted by 10‐fold in 67 mM phosphate buffer (pH 8.0) was added to a 96‐well plate. A 25 μL of 20% acetic acid and 10 μL of 1.16 M potassium iodide were added (dilutions were made with 67 mM phosphate buffer; pH 8.0). For the standard chloramine T solution, 25 μL of 20% acetic acid and 10 μL of 1.16 M potassium iodide were added, and a standard curve was prepared. The absorbance at 340 nm was measured by a microplate reader immediately after the solution was added.
Analysis of post‐translational modified albumin by electrospray ionization time‐of‐flight mass spectrometer
After collecting blood from the patient, 500 mM citrate buffer (pH 4.3) and blood were mixed at a ratio of 1:9. The resulting solution was centrifuged to obtain serum, and the serum was stored at −80°C until uses. A 5 μL volume of serum was added to 495 μL of 50 mM phosphate buffer (pH 6.0) to prepare a measurement sample. Solid‐phase extraction columns (Bond Elute‐C18 EWP 200 mg/3 cc, Varian) were initialized with 90% acetonitrile/0.1% formic acid and equilibrated with ultrapure water. The measurement sample was then added to the solid‐phase extraction column. To remove impurities, elution operation was carried out using 10% acetonitrile/0.1% formic acid. Finally, the column was eluted with 90% acetonitrile/0.1% formic acid to obtain the sample. A 2 μL aliquot of this eluate was measured with an electrospray ionization time‐of‐flight mass spectrometer (ESI‐TOF MS). The acetonitrile used in the experiment was LC grade.
Preparation of advanced oxidation protein products
Advanced oxidation protein products were prepared as described in a previous study. 15 HSA was defatted by treatment with activated carbon. 15 Defatted HSA (300 μM) was incubated with 100 mM chloramine T in 67 mM phosphate buffer (pH 8.0) for 1 h at 37°C. The oxidation reaction was stopped by dialysis with phosphate‐buffered saline (PBS). Finally, after the dialysis, the sample was freeze‐dried to prepare AOPPs. This AOPPs level was 205.6 μmol/g protein.
Animal experiments
All animals were purchased from Japan SLC (Shizuoka, Japan). All animals were freely provided with water and food and maintained in a room with controlled temperature (21–23°C) and a 12 h light/dark cycle (light 8 a.m.–8 p.m.). All animal experiments were conducted using procedures approved by the experimental animal ethics committee at Kumamoto University. 5/6‐Nephrectomized (Nx) mice were produced using a two‐step surgery using male ICR mice (5‐week‐old) according to previous reports. 5 , 6 In brief, 2/3 of the right kidney was removed, and 1 week later the left kidney was removed. In the AOPPs‐overloaded mice, AOPPs were intraperitoneally administered to 4‐week‐old male ICR mice daily for 7 weeks. As a control, PBS or defatted HSA (150 mg protein/kg/day: the same amount of protein as AOPPs) was also administered to male ICR mice (4‐week‐old) for 7 weeks.
Treadmill and wire‐hang‐test
An exercise test using a treadmill (MK‐680S, Muromachi Kikai, Tokyo, Japan) was performed to measure the muscle endurance of the mice. The experimental protocol was described in previous reports (incline: 10%, acceleration: 0.5 m/s2, speed: start from 10 m/min, run for 5 min, then an increase of 2 m/min every 2 min, time: until the mice remained on the electrical shocker plate for 30 s). 5 Mice to which AOPPs were administered every day for 7 weeks were used for this study. The running‐in was started 3 days before this experiment (incline: 0%, acceleration: 0.5 m/s2, speed: 14 m/min, time: 5 min). A wire‐hang‐test was used to evaluate agility and grip strength. Wire‐hang‐test was followed by the previous experimental protocols. 16 Measurements were performed a total of three times with an interval of 1 h for each individual.
Cell culture experiments
Mouse C2C12 myoblast cells were purchased from the RIKEN bioresource cell bank (Ibaraki, Japan). The culture medium was prepared by adding 10% deactivated foetal bovine serum (Capricorn Scientific, Ebsdorfergrund, Germany), antibiotic and antimycotic mixture to DMEM. Differentiation into myotube cells was performed by switching to a differentiation medium (DMEM supplemented with 2% heat‐inactivated horse serum; Sigma‐Aldrich) when myoblasts were 80% to 90% confluent.
Myotube formation assay
The measurement of myotubes was described in a previous report. 5 C2C12 cells were seeded on a 12‐well plate and adhered by culturing (overnight) at 37°C. The AOPPs (100 μM) were then added at the time of switching to the differentiation medium, and the cells were cultured for 7 days. The differentiation medium was changed every 24 h. On the 7th day, the cells were washed with D‐PBS and observed with a microscopy (Keyence, BZ‐X710 microscope, Osaka, Japan). Cells with a major axis of 40 μm or more of fused single cells were designated as myotube cells. The minor axis was measured with Image J software.
ROS measurements
Cells were seeded on 96‐well plates at 1.0 × 104 cell per well. After removing the culture solution, CM‐H2DCFDA was added and was allowed to become incorporated into cells by incubating at 37°C for 30 min. After removing the supernatant and adding D‐PBS or HSA or AOPPs, the fluorescence intensity was measured with a fluorescence plate reader (excitation/emission = 485 nm/535 nm, Spectra Fluor, TECAN). In the study of inhibitors, the cells were incubated with CM‐H2DCFDA for 30 min, then the supernatant was replaced with D‐PBS. After adding various inhibitors and treating for 30 min, AOPPs or HSA or D‐PBS was added in the presence of each inhibitor and the fluorescence intensity was measured in the same manner.
Quantitative RT‐PCR and mitochondrial DNA quantification
Total RNA was isolated from gastrocnemius or C2C12 cells using RNAiso Plus (Takara, Tokyo, Japan). The extracted RNA was determined for purity and concentration based on the absorbance at 260 and 280 nm. The master mix was used to prepare the cDNA and quantitative RT‐PCR measurements were then performed. The primers used for mRNA detection are shown in Supporting information, Table S1. The threshold cycle (Ct) values for each gene amplification were normalized by subtracting the Ct value calculated for GAPDH. Real‐time qPCR was performed for mitochondrial DNA‐encoded cytochrome c oxidase subunit II (Cox2) and nucleus‐encoded 18S ribosomal RNA genes. The ratio of Cox2 DNA copies to 18S rRNA represents the relative mitochondrial copy number.
Western blotting
Western blotting analyses were performed as previously described. 5 Total protein was extracted with RIPA buffer containing 1% nonidet P‐40,150 mM NaCl, 10 mM Tris–HCl (pH 7.4), 1% phosphatase inhibitor cocktail, and 1% protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). After centrifugation, the collected supernatant was mixed with a sample buffer containing 50 mM dithiothreitol and boiled at 100°C. The samples were then separated by 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene fluoride membrane, further immunoblotted with antibodies against myogenin, myosin heavy chain, myostatin, atrogin‐1 and PGC‐1α at 4°C for overnight. The membrane was then immunoblotted with a horseradish peroxidase‐conjugated secondary antibody at room temperature for 1 h. The intensity of each band was detected using LAS4000mini (GE Healthcare, UK Ltd, Backinghamshire, England) and quantified using ImageJ software. The densitometric intensity was normalized with GAPDH expression. Western blotting analyses for Akt and phospho‐Akt were also performed as previously described. 5
Mitochondrial staining
A flame‐sterilized cover glass was placed on a 6‐well plate and C2C12 myoblasts were seeded. After culturing at 37°C (overnight), the supernatant was removed. The cells were washed with D‐PBS and starved with serum‐free DMEM for 2 h. The medium was then changed to DMEM containing 10% FBS. At 3 h after adding the AOPPs, HSA or D‐PBS, the supernatant was removed; MitoRed or MitoGreen and Hoechist33342 in D‐PBS were added; and the mixture was then incubated for 20 min. After incubation, the cells were washed with D‐PBS and observed by the Keyence BZ‐X710 microscope (Keyence, Osaka, Japan).
Intracellular ATP assay
C2C12 cells were seeded on a 96‐well plate and cultured (overnight) at 37°C. ATP extraction was performed 6, 12, 24 hours after the addition of AOPPs, HSA, and D‐PBS. After removing the medium and washing with D‐PBS, 100 μL of ATP extraction reagent was added to each well. ATP was extracted from the cells with stirring at room temperature for 5 min. The extracted cell lysate was collected in a tube and transferred to an ice bath. Each 10 μL of sample was transferred to a luminescence measurement tube. After adding 100 μL of ATP luminescent reagent, the amount of luminescence was measured with a luminometer (Lumat3 LB9508, Berthold Technologies, DEU). The amount of ATP in each sample was calculated from the calibration curve and corrected by the protein concentration.
Measurement of oxygen consumption rate
C2C12 cells were seeded at 2.0 × 104 cells per well in a XF24‐well plate (Agilent Technologies, Santa Clara, CA, USA). After confirming cell growth and adhesion, the cells were treated with 100 μM AOPPs or HSA for 12 hours. The medium was switched from culture medium to analysis medium (Agilent Technologies) supplemented with 10 mM glucose, 1 mM pyruvate, and 2 mM l‐glutamine at 37 °C for 1 hour in a non‐CO2 incubator. The oxygen consumption rate (OCR) was analysed with a XF cell mitostress kit (Agilent Technologies, Santa Clara, CA) by sequential injection of 1.5 μM oligomycin A, 1.0 μM carbonyl cyanide‐4‐(trifluoromethoxy) phenylhydrazone (FCCP) and 0.5 μM rotenone. The basal respiration rate, ATP production, maximal respiration, and spare capacity was calculated by Seahorse XF software version 2.6.1.53.
Immunostaining of tissue sections
A 5 μm section of the frozen gastrocnemius was obtained. After washing with D‐PBS, blocking with 4% block ace in PBS at room temperature for 30 min. It was allowed to react overnight with an anti‐laminin antibody at 4 °C. After washing with D‐PBS, it was reacted with anti‐rabbit‐IgG‐Alexa546 at room temperature for 1 h under shading conditions. After encapsulation with a vector shield, it was observed using a microscope (Keyence, BZ‐X710 microscope, Osaka, Japan). The fluorescence of the skeletal muscle sections was quantified using BZ‐X Analyser.
Statistical analyses
The means for two group data were compared by the unpaired t test. The means for more than two groups were compared by one‐way ANOVA followed by Tukey's multiple comparison. Probability values of P < 0.05 or P < 0.01 were considered to be significant.
Results
Relationship between muscle strength and serum advanced oxidation protein products levels in haemodialysis patients
We investigated the relationship between muscle strength and serum AOPPs levels in haemodialysis patients without sarcopenia (19 patients) and with sarcopenia (48 patients)/with pre‐sarcopenia (16 patients) (Table 1). The haemodialysis in male patients with sarcopenia showed significantly higher serum AOPPs levels as compared with the patients without sarcopenia, and a higher tendency was found for female patients with sarcopenia (Table 1). The serum AOPPs levels showed a significant negative correlation with grip strength in both male and female patients (Figure 1A and 1B). Serum AOPPs levels also showed significant negative correlation with SMI in male and female patients (Figure 1C and 1D).
Table 1.
Characteristics of the participants in this study (n = 83)
| Male | Female | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sarcopenia (n = 26) | Pre‐sarcopenia (n = 12) | Non‐sarcopenia (n = 12) | P value (sarcopenia vs. non‐sarcopenia) | P value (sarcopenia vs. pre‐sarcopenia) | P value (pre‐sarcopenia vs. non‐sarcopenia) | Sarcopenia (n = 22) | Pre‐sarcopenia (n = 4) | Non‐sarcopenia (n = 7) | P value (sarcopenia vs. non‐sarcopenia) | P value (sarcopenia vs. pre‐sarcopenia) | P value (pre‐sarcopenia vs. non‐sarcopenia) | |
| Age (years) | 78.5 ± 1.4 | 73.8 ± 2.0 | 71.3 ± 1.6 | 0.01 | 0.13 | 0.65 | 79.1 ± 1.5 | 74.3 ± 4.1 | 77.7 ± 1.6 | 0.88 | 0.40 | 0.71 |
| Time on haemodialysis (years) | 9.0 ± 1.8 | 2.9 ± 0.8 | 2.7 ± 0.5 | 0.04 | 0.05 | 1.00 | 11.6 ± 3.9 | 9.5 ± 2.6 | 8.0 ± 2.1 | 0.86 | 0.97 | 0.99 |
| Body weight (kg) | 55.2 ± 1.9 | 62.1 ± 2.7 | 65.7 ± 2.0 | 0.006 | 0.09 | 0.62 | 44.5 ± 1.8 | 51.9 ± 1.0 | 56.0 ± 3.1 | 0.009 | 0.25 | 0.70 |
| Body mass index(kg/m2) | 21.7 ± 0.6 | 23.3 ± 0.8 | 23.9 ± 0.6 | 0.08 | 0.27 | 0.85 | 19.8 ± 0.6 | 24.0 ± 1.4 | 23.8 ± 1.3 | 0.03 | 0.07 | 1.00 |
| Skeletal mass index (kg/m2) | 6.0 ± 0.2 | 6.6 ± 0.2 | 7.4 ± 0.2 | < 0.001 | 0.06 | 0.04 | 4.8 ± 0.2 | 5.6 ± 0.6 | 5.3 ± 0.3 | 0.58 | 0.46 | 0.93 |
| Grip strength (kgf) | 19.9 ± 1.1 | 29.4 ± 1.4 | 32.8 ± 1.3 | < 0.001 | < 0.001 | 0.28 | 10.3 ± 1.0 | 12.6 ± 2.6 | 20.6 ± 0.8 | < 0.001 | 0.55 | 0.02 |
| Serum AOPPs level (μM) | 114.7 ± 4.6 | 90.0 ± 5.1 | 85.0 ± 6.0 | 0.001 | 0.007 | 0.85 | 88.2 ± 5.2 | 81.1 ± 17.5 | 68.3 ± 9.6 | 0.38 | 0.88 | 0.74 |
| Oxidized albumin (%) (Cys‐albumin) | 55.0 ± 1.5 | 52.9 ± 1.4 | 49.8 ± 1.4 | 0.07 | 0.63 | 0.47 | 51.6 ± 1.7 | 51.3 ± 3.1 | 42.5 ± 1.4 | 0.02 | 1.00 | 0.15 |
AOPPs, advanced oxidation protein products.
Figure 1.
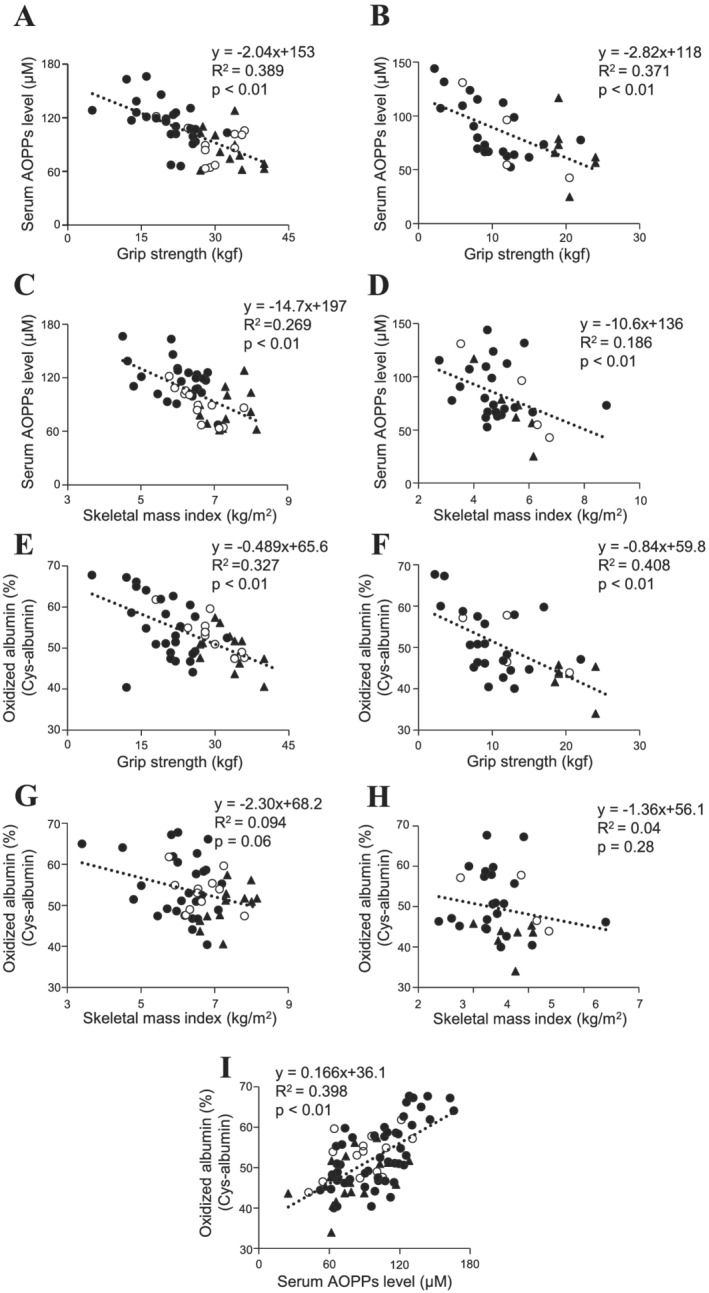
Relationship between muscle strength and serum advanced oxidation protein products (AOPPs) levels in patients undergoing haemodialysis. (A, B) Relationship between serum AOPPs levels and grip strength in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle), and without sarcopenia (closed triangle) (A: male, B: female). (C, D) Relationship between serum AOPPs levels and skeletal mass index (SMI) in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle), and without sarcopenia (closed triangle) (C: male, D: female). (E, F) Relationship between oxidized albumin (Cys‐albumin) and grip strength in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle), and without sarcopenia (closed triangle) (E: male, F: female). (G, H) Relationship between oxidized albumin (Cys‐albumin) and SMI in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle), and without sarcopenia (closed triangle) (G: male, H: female). (I) Relationship between serum AOPPs levels and oxidized albumin (Cys‐albumin) in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle), and without sarcopenia (closed triangle).
Because AOPPs are mainly derived from albumin, the correlation between oxidized albumin (cysteinylated albumin: Cys‐albumin) and grip strength or SMI was also investigated. A significant negative correlation between Cys‐albumin and grip strength was found in both male and female patients (Figure 1E and 1F). In addition, the Cys‐albumin levels tended to be correlated with SMI in male patients (Figure 1G), while no significant correlation with SMI was observed in female patients (Figure 1H). In fact, Cys‐albumin and serum AOPPs levels were significantly positively correlated (Figure 1I). These results suggest that serum AOPPs levels or Cys‐albumin has the potential to serve as a diagnostic biomarker for muscle weakness in haemodialysis patients, and AOPPs may contribute to the muscle weakness in haemodialysis patients.
Relationship between muscle advanced oxidation protein products levels and the expression of muscle atrophy‐related genes in 5/6‐nephrectomized mice
We next evaluated the relationship between AOPPs and muscle atrophy using 5/6‐nephrectomized (5/6‐Nx) CKD mice. At 8 weeks after 5/6‐Nx, the body weight was not significantly changed between the sham and CKD mice (Table 2). On the other hand, the weights of the gastrocnemius and soleus were significantly decreased in the CKD mice compared with those of the sham mice (Table 2). The AOPPs levels and the expression of muscle atrophy‐related genes in the gastrocnemius were also evaluated. As shown in Figure 2A, CKD mice showed significant increase in muscle AOPPs levels as compared with sham mice. Furthermore, muscle AOPPs levels showed a significant positive correlation with the expression of atrogin‐1 and myostatin (R 2 = 0.538, P < 0.01 for atrogin‐1; R 2 = 0.421, P < 0.05 for myostatin) (Figure 2B and 2C). Because exercise capacity is associated with mitochondrial function, 16 we evaluated the expression of PGC‐1α, a master regulator of mitochondrial biosynthesis, in the muscle. The muscle AOPPs levels and PGC‐1α mRNA expression in the gastrocnemius showed a significant negative correlation (R 2 = 0.405; P < 0.05) (Figure 2D). Protein expression of atrogin‐1 and myostatin were also increased in the muscle of CKD mice but PGC‐1α protein expression was not significantly changed in CKD mice (Figure 2E). These findings suggest that an increase in muscle AOPPs levels could cause the muscle atrophy and weakness that are observed in CKD mice.
Table 2.
Renal function, body weight and muscle weight profile for sham, 5/6‐nephrectomized mice (CKD mice)
| Sham | CKD | |
|---|---|---|
| BUN (mg/dL) | 23.3 ± 3.0 | 44.7 ± 2.2** |
| SCr (mg/dL) | 0.2 ± 0.05 | 0.3 ± 0.03 |
| Body weight (g) | 40.5 ± 3.1 | 36.9 ± 0.8 |
| Gastrocnemius (mg) | 144.7 ± 2.2 | 123.3 ± 3.0* |
| Tibialis anterior (mg) | 74.5 ± 0.8 | 71.8 ± 2.9 |
| Soleus (mg) | 12.3 ± 0.4 | 10.0 ± 0.2* |
BUN, blood urea nitrogen; CKD, chronic kidney disease; SCr, serum creatinine.
Data are expressed as the mean ± SEM.
P < 0.05.
P < 0.01 compared with sham
Figure 2.
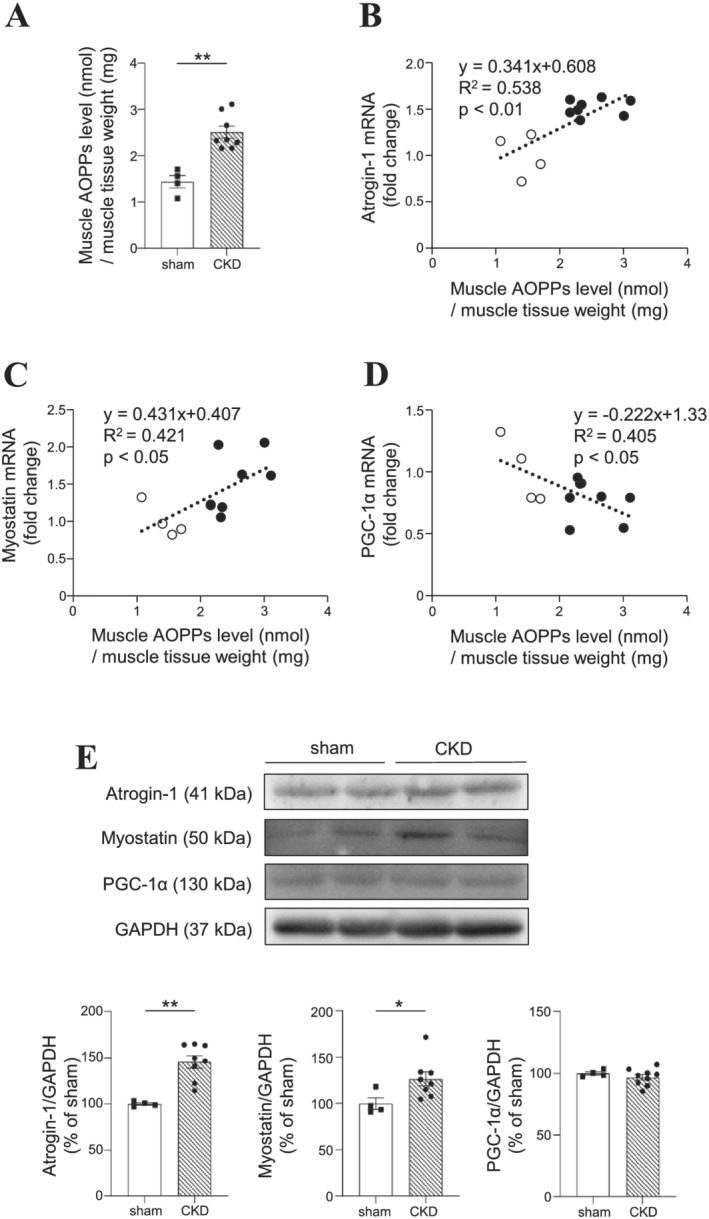
Relationship between muscle advanced oxidation protein products (AOPPs) levels and the expression of muscle atrophy‐related genes in 5/6‐nephrectomized chronic kidney disease (CKD) mice. (A) AOPPs levels in muscle and (B–D) the expression of muscle atrophy‐related genes in the gastrocnemius were measured at 8 weeks after 5/6‐nephrectomized. Relationship between muscle AOPPs levels and the mRNA expression of (B) atrogin‐1 (R 2 = 0.538, P < 0.01), (C) myostatin (R 2 = 0.421, P < 0.05), and (D) PGC‐1α (R 2 = 0.405, P < 0.05) in the gastrocnemius. The fold changes as compared with the average data from patients without sarcopenia were shown. (E) Western blot analyses of atrogin‐1, myostatin and PGC‐1α in the gastrocnemius of sham and CKD mice. Data are expressed the mean ± SEM (n = 4–8). **P < 0.01 compared with sham.
Molecular mechanism of advanced oxidation protein products‐induced muscle atrophy (C2C12 myoblast cells)
We investigated the molecular mechanism responsible for AOPPs‐induced muscle atrophy using C2C12 mouse myoblast cells. First, the expressions of atrogin‐1 and myostatin mRNA in C2C12 cells were evaluated at 48 h after incubating with a medium in which the medium contained 10% serum from haemodialysis patients [serum with low AOPPs levels (AOPPs levels < 80 μM: 56.6–73.2 μM) or serum with high AOPPs levels (AOPPs levels > 120 μM: 121–128.2 μM)]. In the presence of serum with high AOPPs levels, the expressions of atrogin‐1 and myostatin mRNA were significantly increased as compared with the serum with low AOPPs levels (Figure 3A and 3B), suggesting that the higher AOPPs levels could be involved in the increase of atrogin‐1 and myostatin mRNA in C2C12 cells. To evaluate the direct effect of AOPPs on muscle atrophy, AOPPs samples were prepared by oxidizing HSA (refer to Materials and methods) and C2C12 cells were then incubated with these AOPPs. As shown in Figure 3C and 3D, AOPPs caused an increase in the protein and mRNA expressions of atrogin‐1 and myostatin but unmodified HSA (the same protein concentration as that for the AOPPs) did not. An antioxidant, N‐acetylcysteine (NAC), suppressed the AOPPs‐induced expression of atrogin‐1 and myostatin. Under the same experimental conditions, the AOPPs had no effect on cell proliferation (data not shown). It was reported that AOPPs enhanced ROS production in renal tubular cells. 17 Other reports have also demonstrated that intracellular ROS production is involved in the increased expression of atrogin‐1 and myostatin. 5 Therefore, we investigated the effect of AOPPs on ROS production in C2C12 cells. As a result, the AOPPs increased ROS production in a dose‐dependent manner. A pre‐treatment with NAC and DPI, an NADPH oxidase inhibitor, significantly suppressed the AOPPs‐induced production of ROS (Figure 3E). Cao et al. reported that AOPPs are recognized by the scavenger receptor CD36 in proximal tubular epithelial cells. 18 In fact, in a previous study, we confirmed that C2C12 cells express CD36 by western blotting (data not shown). As shown in Figure 3E, AOPPs‐induced ROS production was significantly suppressed by the treatment with an anti‐CD36 neutralizing antibody, but not by the treatment with an isotype control IgG. These data suggest that AOPPs increased the expression of atrogin‐1 and myostatin mRNA through ROS production via CD36/NADPH oxidase pathway.
Figure 3.
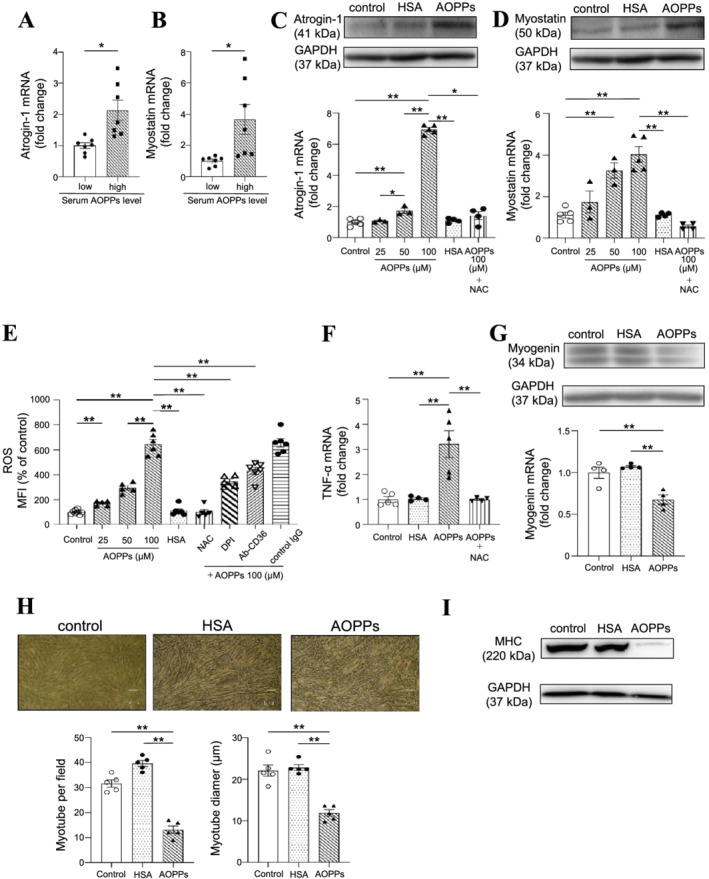
Molecular mechanism for the advanced oxidation protein products (AOPPs)‐induced muscle atrophy using C2C12 cells. (A, B) mRNA expression of atrogin‐1 and myostatin at 48 h after the addition of haemodialysis patients' serum [serum with low AOPPs levels (AOPPs levels < 80 μM: 56.6–73.2 μM) or serum with high AOPPs levels (AOPPs levels > 120 μM: 121–128.2 μM)] to C2C12 myoblasts cells was measured by real‐time qPCR. (C, D) Effect of AOPPs on protein and mRNA expression of atrogin‐1 and myostatin in C2C12 myoblasts cells at 48 h after the AOPPs treatment. N‐acetylcysteine: NAC (0.1 mM) was co‐treated with AOPPs. (E) Effect of AOPPs on ROS production in C2C12 myoblast cells. C2C12 myoblast cells were starved with serum‐free medium for 2 h and then treated with CM‐H2DCFDA in Dulbecco's phosphate‐buffered saline (D‐PBS) for 30 min. After removal of the D‐PBS, the cells were treated with AOPPs and incubated for 90 min. Fluorescence intensity was measured at an excitation wavelength of 485 nm and at an emission wavelength of 535 nm. In the inhibitor experiments, CM‐H2DCFDA in D‐PBS was removed and the inhibitor was then added. After incubating for 30 min, the cells were treated with AOPPs and incubated for 90 min in the presence of each inhibitor. NAC (0.1 mM), NADPH oxidase inhibitors [diphenyleneiodonium chloride: DPI (10 μM)] and CD36 neutralizing antibodies (4 mg/mL) were used. (F) Effect of AOPPs on the mRNA expression of TNF‐α in C2C12 myoblasts cells at 48 h after treatment with 100 μM AOPPs. (G) Effect of AOPPs on myogenin protein and mRNA expression at 48 h after 100 μM AOPPs treatment. (H) Effect of AOPPs on C2C12 myoblast cell tubular formation. C2C12 myoblast cells were seeded on a 6‐well plate and cultured overnight. After cell adhesion, the cultured medium was changed to a differentiation medium containing with 100 μM AOPPs or human serum albumin (HSA) and cultured for 7 days. The cell density in each treatment group was the same before changing the culture medium to a differentiation medium. Cell morphology was observed by microscopy and the number of myotube cells and myotube diameter evaluated using the ImageJ. (I) Effect of AOPPs on myotube formation was evaluated using myosin heavy chain protein expression. Data are expressed the mean ± SEM (n = 3–7). *P < 0.05, **P < 0.01.
It has been reported that TNF‐α expression is increased during muscle atrophy, 19 and that TNF‐α is involved in the increased secretion of myostatin from muscle tissue. 20 Furthermore, in chondrocytes, AOPPs were found to increase TNF‐α expression by activating NADPH oxidase/MAPK signals. 21 Therefore, we evaluated the ability of AOPPs to induce the expression of TNF‐α in C2C12 cells. As shown in Figure 3F, AOPPs significantly increased the expression of TNF‐α mRNA, and the increase in TNF‐α expression was suppressed by the presence of NAC, demonstrating that AOPPs‐induced ROS production is involved in the increase in TNF‐α expression.
We also evaluated the effect of AOPPs on the expression of myogenin, one of the genes responsible for myogenesis, in C2C12 cells. As shown in Figure 3G, AOPPs significantly decreased the myogenin protein level and mRNA expression. AOPPs also significantly decreased myotube formation (both the number of myotubes and myotube diameter) compared with control or the HSA treatment (Figure 3H). In addition, the levels of the myosin heavy chain (MHC) protein, which is a marker for the differentiation of skeletal muscle, was decreased in the presence of AOPPs (Figure 3I).
Effect of advanced oxidation protein products on mitochondria (C2C12 myoblast cells)
To evaluate the effect of AOPPs on mitochondria, we first evaluated PGC‐1α expression in C2C12 cells. Incubation with serum containing high AOPPs levels (121–128.2 μM) showed a significant decrease in PGC‐1α mRNA expression in C2C12 cells as compared with the serum containing low AOPPs levels (56.6–73.2 μM) (Figure 4A). Similarly, AOPPs also significantly decreased the expression of PGC‐1α protein and mRNA compared with the control (non‐treated group) or the HSA‐treated group (Figure 4B). The number of mitochondria was evaluated by MitoGreen staining, and the mitochondrial membrane potential was measured as an index of mitochondrial activity by MitoRed staining. As a result, AOPPs significantly reduced the number of mitochondria and mitochondrial membrane potential in C2C12 cells compared to the control or HSA‐treated group (Figure 4C). In addition, we found that mitochondrial DNA copies number (Figure 4D) and cellular ATP levels were decreased in the AOPPs‐treated group. Mitochondrial oxygen consumption rate (OCR) (basal respiration, ATP production and maximal respiration) was also decreased in the AOPPs‐treated group (Figure 4F).
Figure 4.
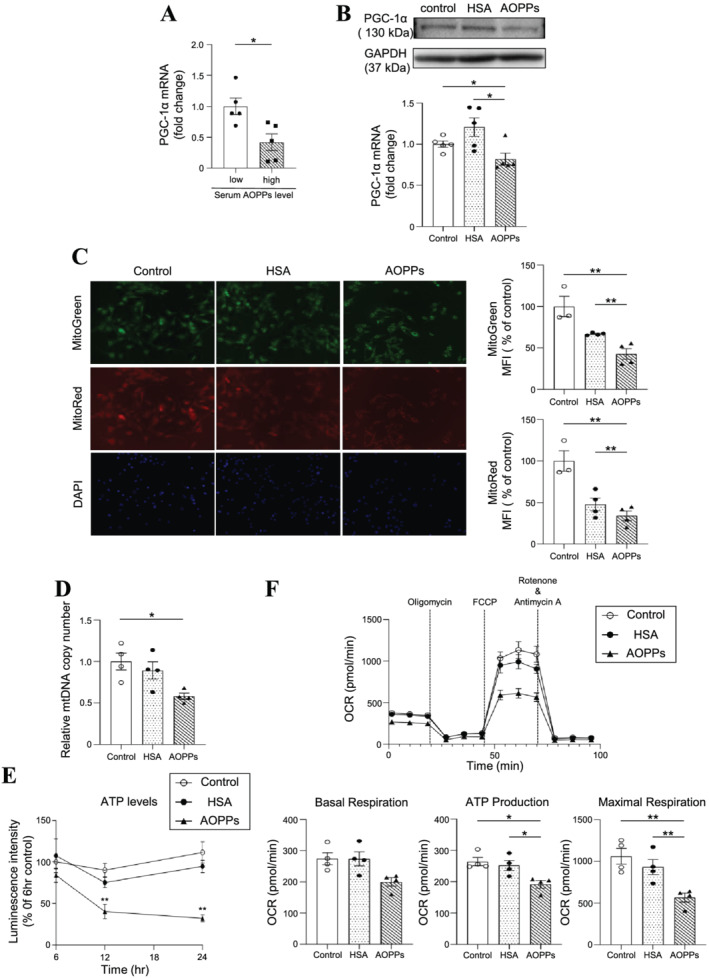
Effect of advanced oxidation protein products (AOPPs) on mitochondria in C2C12 myoblast cells. (A) mRNA expression of PGC‐1α at 3 h after the addition of serum from a haemodialysis patient [serum with low AOPPs levels (AOPPs levels < 80 μM: 56.6–73.2 μM) or serum with high AOPPs levels (AOPPs levels > 120 μM: 121–128.2 μM)] to C2C12 myoblasts was measured by real‐time qPCR. (B) The mRNA expression of PGC‐1α at 3 h after treatment with 100 μM AOPPs was measured by real‐time qPCR. (C) The number of mitochondria of C2C12 myoblasts at 3 h after treatment with 100 μM AOPPs was evaluated using MitoGreen. Mitochondrial membrane potential of C2C12 myoblasts at 3 h after AOPPs treatment was evaluated by MitoRed. (D) Mitochondrial DNA (mtDNA) copy number at 12 h after treatment with 100 μM AOPPs in C2C12 myoblasts, (E) intracellular ATP level after treatment with 100 μM AOPPs in C2C12 myoblasts, and (F) mitochondrial oxygen consumption rate (OCR) was determined at 12 h after treatment with 100 μM AOPPs in C2C12 myoblasts. Representative time course data for OCR and aggregate data were shown. Data are expressed the mean ± SEM (n = 3–6). *P < 0.05, **P < 0.01.
Effect of advanced oxidation protein products on skeletal muscle (advanced oxidation protein products‐overloaded mice)
Lastly, we investigated the effect of AOPPs on skeletal muscle in vivo using AOPPs‐overloaded mice (Figure 5A). At 7 weeks after the daily administration of AOPPs, no significant decrease in renal function was observed as evidenced by the finding that BUN and SCr levels were not increased (Table 3). Although body weight was not affected by the administration of the AOPPs, a significant decrease in the weight of the gastrocnemius was observed (Table 3). The weight of the tibialis anterior and soleus tended to decrease in AOPPs‐overloaded mice.
Figure 5.
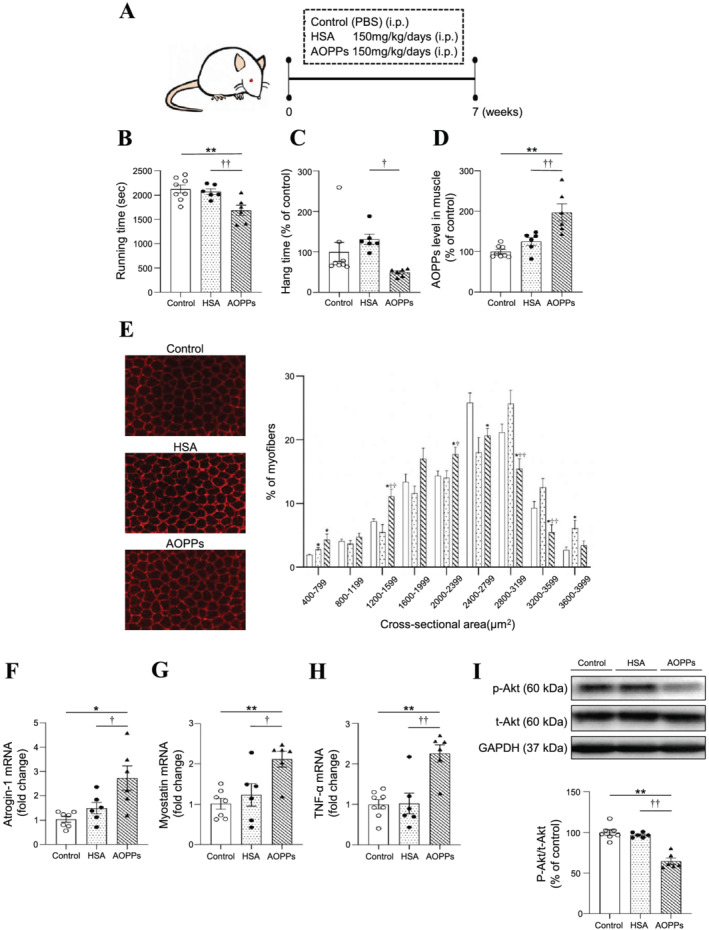
In vivo effect of advanced oxidation protein products (AOPPs) on skeletal muscle using AOPPs‐overloaded mice. (A) AOPPs‐overloaded mice were created by intraperitoneally administering AOPPs at a dosage of 150 mg protein/kg/day for 7 weeks. In the comparison group, phosphate‐buffered saline (PBS) and human serum albumin (HSA) (150 mg protein/kg/days) were intraperitoneally administered. (B) Muscle endurance at 6 weeks after the administration of AOPPs was determined using a treadmill. (C) Grip strength at 6 weeks after AOPPs administration was determined using a wire‐hang‐test. (D) Muscle AOPPs levels in the gastrocnemius muscle was measured at 7 weeks after the administration of AOPPs administration. (E) Cryosections of gastrocnemius muscle were immune‐stained with anti‐laminin antibody to assess myofibre size. (F–H) After AOPPs administration for 7 weeks, the mRNA expression of (F) atrogin‐1, (G) myostatin, and (H) TNF‐α in gastrocnemius muscle were measured by real‐time qPCR. (I) Phosphorylated‐Akt (p‐Akt) was detected by western blotting. Data are expressed the mean ± SEM (n = 6–8). *P < 0.05, **P < 0.01; † P < 0.05, †† P < 0.01 compared with HSA.
Table 3.
Renal function, body weight and muscle weight profile for control, HSA‐overloaded mice or AOPPs‐overloaded mice
| Control | HSA | AOPPs | |
|---|---|---|---|
| BUN (mg/dL) | 25.7 ± 1.7 | 27.7 ± 0.9 | 27.9 ± 2.2 |
| SCr (mg/dL) | 0.20 ± 0.02 | 0.18 ± 0.03 | 0.22 ± 0.03 |
| Body weight (g) | 39.6 ± 1.0 | 40.3 ± 0.6 | 40.5 ± 1.4 |
| Gastrocnemius (mg) | 199.7 ± 6.3 | 215.1 ± 3.7** | 168.7 ± 7.3** , †† |
| Tibialis anterior (mg) | 78.5 ± 1.7 | 74.7 ± 6.2 | 71.5 ± 4.9 |
| Soleus (mg) | 13.5 ± 1.7 | 12.1 ± 0.9 | 11.1 ± 1.2 |
AOPPs, advanced oxidation protein products; BUN, blood urea nitrogen; HSA, human serum albumin; SCr, serum creatinine.
Data are expressed as the mean ± SEM.
P < 0.01 compared with control.
P < 0.01 compared with HSA.
In the treadmill and wire‐hang‐tests, the AOPPs‐treated mice showed a significant decrease in muscle endurance as compared to the control or HSA‐treated mice (Figure 5B and 5C). At that time, the AOPPs levels in the gastrocnemius were increased by about two‐fold in the AOPPs‐treated group (Figure 5D). In addition, the muscle tissue of the AOPPs‐treated group showed a significant decrease in cross‐sectional area (Figure 5E), an increase in atrogin‐1, myostatin, and TNF‐α mRNA (Figure 5F, 5G and 5H). AOPPs‐overload also decreased MHC formation (Figure S1). These in vivo data support the results using C2C12 myoblast cells. Regarding protein synthesis, muscle phosphorylated‐Akt (p‐Akt) was detected. AOPPs‐overload suppressed muscle p‐Akt (Figure 5I). On the other hands, in C2C12 myoblast cells, AOPPs‐treatment (~48 h) did not affect p‐Akt levels (data not shown) but it suppressed myogenin expression (Figure 3G), suggesting AOPPs also negatively affected muscle protein synthesis.
Discussion
Although sarcopenia with CKD progression is associated with a poor life expectancy, the molecular mechanism responsible for CKD‐induced sarcopenia remains unclear. The findings of our in vitro and animal studies indicate that AOPPs, a uraemic toxin, induced the muscle atrophy‐related genes such as atrogin‐1/myostatin through CD36/NADPH oxidase/ROS pathway. In addition, AOPPs induced mitochondrial dysfunction. In dialysis patients, serum AOPPs levels were inversely correlated with muscle strength (grip strength) and muscle mass (SMI), suggesting that the increased AOPPs are involved in the decrease in muscle strength and muscle mass in humans. The serum AOPPs levels also showed a positive correlation with oxidized albumin (Cys‐albumin). These results suggest that the increased levels of AOPPs could contribute to the sarcopenia in dialysis patients and also suggest that serum AOPPs levels and Cys‐albumin measurement could be useful biomarkers in the diagnosis of sarcopenia in these patients.
It has been reported that the AOPPs‐induced production of ROS is involved in the pathogenesis and progression of chronic inflammatory diseases including CKD. In fact, AOPPs have been reported to be involved in the development of osteoporosis by enhancing the expression of TNF‐α via NADPH oxidase/ROS/p38MAPK signalling in chondrocytes. 21 In intestinal epithelial cells, AOPPs were found to induce an epithelial‐mesenchymal transition by enhancing ROS production via the PKC/NADPH oxidase pathway. 22 In addition, Iwao et al., using renal tubular epithelial cells, reported that the intracellular uptake of AOPPs via CD36 was the first step in intracellular ROS production. 10 However, information concerning the effects of AOPPs on skeletal muscle in CKD was lacking. In this study, we confirmed the expression of CD36 in C2C12 cells (data not shown). In addition, the expression of NADPH oxidase was also reported in skeletal muscle. 21 In our studies using a CD36 neutralizing antibody and an NADPH oxidase inhibitor, we found that the intracellular uptake of AOPPs via CD36 and the increase in ROS production associated with the activation of NADPH oxidase both contribute to the development of muscle atrophy (Figure 3).
In general, it is known that muscle atrophy involves an enhancement in the proteolytic system and the suppression of the protein synthetic system. It was reported that the proteolytic system was enhanced in haemodialysis patients. 23 Zhang et al., in a previous study, reported that serum myostatin levels were negatively correlated with muscle weight in CKD model mice. 24 It is also known that myostatin and TNF‐α induce the expression of genes that are involved in proteolysis such as atrogin‐1 and MuRF1 via NF‐κB signalling in cells. 25 In this study, AOPPs were found to increase the expression of atrogin‐1, myostatin and TNF‐α expression, and to suppress the expression of myogenin, demonstrating that increased proteolytic activity could be involved in AOPPs‐induced muscle atrophy. These effects of AOPPs were significantly suppressed by the presence of NAC. We therefore conclude that the intracellular ROS production induced by AOPPs triggers the induction of muscle atrophy.
During muscle atrophy, it is known that the distribution density of mitochondria in muscle cells is reduced by about 20%. 26 Regarding CKD‐induced muscular atrophy, it was also reported that the amount of mitochondria in skeletal muscle decreases due to decreased expression of PGC‐1α and TFAM, which are involved in mitochondrial biosynthesis and an increase in the expression of BNIP3L and PINK1, which are involved in mitochondrial degradation. 27 Su et al. also reported that the activation of autophagy induced mitochondrial dysfunction when uraemic serum was added to cultured myotube cells. 28 In this study, AOPPs were observed to decrease mitochondrial mass and activity in addition to causing a decrease in PGC‐1α expression. Xiao et al. recently reported that AOPPs cause a change in mitochondrial morphology via the CD36/PKC pathway in tubular epithelial cells, which leads to an enhanced production in mitochondrial ROS. 29 Therefore, in skeletal muscle, AOPPs may also affect mitochondrial morphology and may be involved in the exacerbation of oxidative stress by increasing mitochondrial‐derived ROS production. Further study regarding this will be needed in the future.
Regarding the treatment of AOPPs‐induced muscular atrophy, the inhibition of the activin (ActRIIB) receptor, a myostatin receptor, using follistatin and bimagrumab, could be a therapeutic strategy for suppressing the proteolytic system. 30 In fact, AOPPs‐induced muscular atrophy was found to be associated with increased myostatin expression. In addition, this study revealed that AOPPs reduced the expression of PGC‐1α expression. DPP‐4 inhibitors are known to promote mitochondrial production by increasing the expression of PGC‐1α. 31 Interestingly, teneligliptin, a DPP‐4 inhibitor, has also been reported to reduce CD36 expression in mouse peritoneal macrophages and in human macrophage cell lines. 29 In fact, using CKD mice, we previously reported that teneligliptin suppresses CKD‐induced muscle weakness. 6 Thus, DDP‐4 inhibitors may have the potential to be effective in suppressing the progression of muscle atrophy from two aspects, enhancing mitochondrial biosynthesis in skeletal muscle and suppressing the uptake of AOPPs via CD36. The results reported in this study serve to clarify that ROS is involved in the muscle atrophy caused by AOPPs. We speculate that ROS produced by AOPPs may further activate neutrophil myeloperoxidase, thus creating a vicious cycle that further increases the production of AOPPs. Therefore, anti‐oxidative and inflammatory modulator such as Nrf2‐activator intended to suppress the production of AOPPs would also be expected to be a potential future therapeutic strategy. In addition, unbiased approach such as RNA‐Seq analysis may be needed in the future study to confirm the aberrant effects of AOPPs on skeletal muscle.
Advanced oxidation protein products have been reported to be correlated with increased serum creatinine, urinary N‐acetyl‐β‐d‐glucosaminidase activity and advanced glycation end products and decreased eGFR in diabetic patients. 29 AOPPs have also been reported to be a risk factor for coronary artery disease. 32 In addition, the fact that AOPPs are correlated with lumbar bone mineral density and bone turnover markers (BALP, TACP5b) suggests that AOPPs might be useful as a marker for postmenopausal osteoporosis. 33 On the other hand, oxidized albumin (Cys‐albumin) is an oxidative modification of the free cysteine residue present at position 34 in HSA. We previously reported that Cys‐albumin was increased in patients with chronic hepatitis, diabetes mellitus, and CKD. 34 , 35 , 36 , 37 In this study, serum Cys‐albumin also showed an inverse correlation with grip strength (male and female patients) and SMI (male patients) of dialysis patients, and serum AOPPs and Cys‐albumin showed a positive correlation, suggesting that serum AOPPs and Cys‐albumin could be a useful marker for the diagnosis of sarcopenia in such patients. But it still remained unclear that Cys‐Albumin itself could be a ‘culprit’ causing muscle wasting in CKD so far. The C‐reactive protein (CRP)/albumin ratio was recently reported to be a possible index of sarcopenia associated with dialysis and cancer. 38 In this study, the CRP/albumin ratios were significantly correlated with only male grip strength (Figure S2). Therefore, the use of AOPPs and Cys‐albumin as a sarcopenia marker should be compared with existing markers such as the CRP/albumin ratio in a future study. Our data also showed that, in female patients, serum AOPPs, Cys‐albumin and CRP/albumin were not significantly correlated with SMI. These results may be due to the fact that the female patients enrolled in this study showed lower AOPPs levels, Cys‐albumin and CRP/albumin ratios compared with male patients. In addition, as Table 1 showed that the age with sarcopenia was significantly higher than that with non‐sarcopenia (only in male patients). Therefore, we could not exclude the contribution of ageing‐induced sarcopenia as a baseline. Further investigation will clearly be needed to clarify these points in the future.
Conclusions
In this study, we found that AOPPs are involved in the development of CKD‐induced sarcopenia through CD36/NADPH oxidase/ROS pathway, and AOPPs also contribute to mitochondrial dysfunction. These results suggest that AOPPs or a related downstream signalling pathway could be a therapeutic target for the CKD‐induced sarcopenia. In addition, we also found that serum AOPPs or Cys‐albumin could be a new diagnostic marker for sarcopenia pathology in CKD.
Conflict of interest
The authors have no potential conflicts of interest to disclose.
Funding
This work was supported by a grant from JSPS KAKENHI (grant number JP20H03406).
Supporting information
Figure S1. Effect of AOPPs‐overload on the protein expression of (A) myosin heavy chain (MHC), (B) PGC‐1α, (C) myostatin and (D) atrogin‐1 in gastrocnemius muscle of mice were determined by Western blotting. AOPPs overloaded to ICR mice for 7 weeks, Data are expressed the mean ± SEM (n = 6 ~ 8). *p < 0.05, **p < 0.01; †p < 0.05, ††p < 0.01 compared with control or HSA.
Figure S2. Relationship between muscle strength and C‐reactive protein (CRP)/albumin ratio in patients who are undergoing haemodialysis. (A, B) The relationship between CRP/albumin ratio and grip strength in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle) and without sarcopenia (closed triangle) (A: male, B: female). (C, D) The relationship between CRP/albumin ratio and skeletal mass index in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle) and without sarcopenia (closed triangle) (C: male, D: female).
Table S1. The primers used for mRNA detection.
Acknowledgements
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. 39
Kato H., Watanabe H., Imafuku T., Arimura N., Fujita I., Noguchi I., Tanaka S., Nakano T., Tokumaru K., Enoki Y., Maeda H., Hino S., Tanaka M., Matsushita K., Fukagawa M., and Maruyama T. (2021) Advanced oxidation protein products contribute to chronic kidney disease‐induced muscle atrophy by inducing oxidative stress via CD36/NADPH oxidase pathway, Journal of Cachexia, Sarcopenia and Muscle, 12, 1832–1847, 10.1002/jcsm.12786
Hiromasa Kato and Hiroshi Watanabe contributed equally to the study.
Contributor Information
Hiroshi Watanabe, Email: hnabe@kumamoto-u.ac.jp.
Toru Maruyama, Email: tomaru@gpo.kumamoto-u.ac.jp.
References
- 1. Moon SJ, Kim TH, Yoon SY, Chung JH, Hwang HJ. Relationship between stage of chronic kidney disease and sarcopenia in korean aged 40 years and older using the Korea National Health and Nutrition Examination Surveys (KNHANES IV‐2, 3, and V‐1, 2), 2008–2011. PLoS ONE 2015;10:e0130740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roshanravan B, Robinson‐Cohen C, Patel KV, Ayers E, Littman AJ, de Boer IH, et al. Association between physical performance and all‐cause mortality in CKD. J Am Soc Nephrol 2013;24:822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Faitg J, Reynaud O, Leduc‐Gaudet JP, Gouspillou G. Skeletal muscle aging and mitochondrial dysfunction: an update. Med Sci (Paris) 2017;33:955–962. [DOI] [PubMed] [Google Scholar]
- 4. Dalla Libera L, Ravara B, Gobbo V, Tarricone E, Vitadello M, Biolo G, et al. A transient antioxidant stress response accompanies the onset of disuse atrophy in human skeletal muscle. J Appl Physiol (1985) 2009;107:549–557. [DOI] [PubMed] [Google Scholar]
- 5. Enoki Y, Watanabe H, Arake R, Sugimoto R, Imafuku T, Tominaga Y, et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress‐mediated expression of myostatin and atrogin‐1. Sci Rep 2016;6:32084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, et al. Potential therapeutic interventions for chronic kidney disease‐associated sarcopenia via indoxyl sulfate‐induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle 2017;8:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abrigo J, Simon F, Cabrera D, Vilos C, Cabello‐Verrugio C. Mitochondrial dysfunction in skeletal muscle pathologies. Curr Protein Pept Sci 2019;20:536–546. [DOI] [PubMed] [Google Scholar]
- 8. Watanabe H, Enoki Y, Maruyama T. Sarcopenia in chronic kidney disease: factors, mechanisms, and therapeutic interventions. Biol Pharm Bull 2019;42:1437–1445. [DOI] [PubMed] [Google Scholar]
- 9. Iwao Y, Anraku M, Hiraike M, Kawai K, Nakajou K, Kai T, et al. The structural and pharmacokinetic properties of oxidized human serum albumin, advanced oxidation protein products (AOPP). Drug Metab Pharmacokinet 2006;21:140–146. [DOI] [PubMed] [Google Scholar]
- 10. Iwao Y, Nakajou K, Nagai R, Kitamura K, Anraku M, Maruyama T, et al. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am J Physiol Renal Physiol 2008;295:F1871–F1880. [DOI] [PubMed] [Google Scholar]
- 11. Witko‐Sarsat V, Friedlander M, Nguyen Khoa T, Capeillère‐Blandin C, Nguyen AT, Canteloup S, et al. Advanced oxidation protein products as novel mediators of inflammation and monocyte activation in chronic renal failure. J Immunol 1998;161:2524–2532. [PubMed] [Google Scholar]
- 12. Witko‐Sarsat V, Gausson V, Descamps‐Latscha B. Are advanced oxidation protein products potential uremic toxins? Kidney Int Suppl 2003;63:S11–S14. [DOI] [PubMed] [Google Scholar]
- 13. Witko‐Sarsat V, Friedlander M, Capeillère‐Blandin C, Nguyen‐Khoa T, Nguyen AT, Zingraff J, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 1996;49:1304–1313. [DOI] [PubMed] [Google Scholar]
- 14. Zhu SY, Zhuang JS, Wu Q, Liu ZY, Liao CR, Luo SG, et al. Advanced oxidation protein products induce pre‐osteoblast apoptosis through a nicotinamide adenine dinucleotide phosphate oxidase‐dependent, mitogen‐activated protein kinases‐mediated intrinsic apoptosis pathway. Aging Cell 2018;17:e12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imafuku T, Watanabe H, Satoh T, Matsuzaka T, Inazumi T, Kato H, et al. Advanced oxidation protein products contribute to renal tubulopathy via perturbation of renal fatty acids. Kidney 360 2020;1:781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Melchior B, Mittapalli GK, Lai C, Duong‐Polk K, Stewart J, Güner B, et al. Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: a potential therapeutic for Alzheimer's disease. Aging Cell 2019;18:e13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao W, Hou FF, Nie J. AOPPs and the progression of kidney disease. Kidney Int Suppl 2011;2014:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao W, Xu J, Zhou ZM, Wang GB, Hou FF, Nie J. Advanced oxidation protein products activate intrarenal renin–angiotensin system via a CD36‐mediated, redox‐dependent pathway. Antioxid Redox Signal 2013;18:19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Léger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS‐3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res 2008;11:163–75B. [DOI] [PubMed] [Google Scholar]
- 20. Rodriguez J, Fernández‐Verdejo R, Pierre N, Priem F, Francaux M. Endurance training attenuates catabolic signals induced by TNF‐α in muscle of mice. Med Sci Sports Exerc 2016;48:227–234. [DOI] [PubMed] [Google Scholar]
- 21. Liao CR, Wang SN, Zhu SY, Wang YQ, Li ZZ, Liu ZY, et al. Advanced oxidation protein products increase TNF‐α and IL‐1β expression in chondrocytes via NADPH oxidase 4 and accelerate cartilage degeneration in osteoarthritis progression. Redox Biol 2020;28:101306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu X, Sun S, Xie F, Ma J, Tang J, He S, et al. Advanced oxidation protein products induce epithelial‐mesenchymal transition of intestinal epithelial cells via a PKC δ‐mediated, redox‐dependent signaling pathway. Antioxid Redox Signal 2017;27:37–56. [DOI] [PubMed] [Google Scholar]
- 23. Mitch WE. Malnutrition: a frequent misdiagnosis for hemodialysis patients. J Clin Invest 2002;110:437–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 2011;25:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hong Y, Lee JH, Jeong KW, Choi CS, Jun HS. Amelioration of muscle wasting by glucagon‐like peptide‐1 receptor agonist in muscle atrophy. J Cachexia Sarcopenia Muscle 2019;10:903–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peterson CM, Johannsen DL, Ravussin E. Skeletal muscle mitochondria and aging: a review. J Aging Res 2012;2012:194821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yazdi PG, Moradi H, Yang JY, Wang PH, Vaziri ND. Skeletal muscle mitochondrial depletion and dysfunction in chronic kidney disease. Int J Clin Exp Med 2013;6:532–539. [PMC free article] [PubMed] [Google Scholar]
- 28. Su Z, Klein JD, Du J, Franch HA, Zhang L, Hassounah F, et al. Chronic kidney disease induces autophagy leading to dysfunction of mitochondria in skeletal muscle. Am J Physiol Renal Physiol 2017;312:F1128–F1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Xu L, Hou X, Geng J, Tian J, Liu X, et al. Advanced oxidation protein products aggravate tubulointerstitial fibrosis through protein kinase C‐dependent mitochondrial injury in early diabetic nephropathy. Antioxid Redox Signal 2019;30:1162–1185. [DOI] [PubMed] [Google Scholar]
- 30. Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 2013;45:2333–2347. [DOI] [PubMed] [Google Scholar]
- 31. Hansotia T, Maida A, Flock G, Yamada Y, Tsukiyama K, Seino Y, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 2007;117:143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaneda H, Taguchi J, Ogasawara K, Aizawa T, Ohno M. Increased level of advanced oxidation protein products in patients with coronary artery disease. Atherosclerosis 2002;162:221–225. [DOI] [PubMed] [Google Scholar]
- 33. Wu Q, Zhong ZM, Pan Y, Zeng JH, Zheng S, Zhu SY, et al. Advanced oxidation protein products as a novel marker of oxidative stress in postmenopausal osteoporosis. Med Sci Monit 2015;21:2428–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watanabe H, Imafuku T, Otagiri M, Maruyama T. Clinical implications associated with the posttranslational modification‐induced functional impairment of albumin in oxidative stress‐related diseases. J Pharm Sci 2017;106:2195–2203. [DOI] [PubMed] [Google Scholar]
- 35. Nagumo K, Tanaka M, Chuang VT, Setoyama H, Watanabe H, Yamada N, et al. Cys34‐cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress‐related chronic diseases. PLoS ONE 2014;9:e85216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Setoyama H, Tanaka M, Nagumo K, Naoe H, Watanabe T, Yoshimaru Y, et al. Oral branched‐chain amino acid granules improve structure and function of human serum albumin in cirrhotic patients. J Gastroenterol 2017;52:754–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Imafuku T, Watanabe H, Oniki K, Yoshida A, Kato H, Nakano T, et al. Cysteinylated albumin as a potential biomarker for the progression of kidney disease in patients with type 2 diabetes. Diabetes Care 2021;26:dc203003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wong TC, Su HY, Chen YT, Wu PY, Chen HH, Chen TH, et al. Ratio of C‐reactive protein to albumin predicts muscle mass in adult patients undergoing hemodialysis. PLoS ONE 2016;11:e0165403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of AOPPs‐overload on the protein expression of (A) myosin heavy chain (MHC), (B) PGC‐1α, (C) myostatin and (D) atrogin‐1 in gastrocnemius muscle of mice were determined by Western blotting. AOPPs overloaded to ICR mice for 7 weeks, Data are expressed the mean ± SEM (n = 6 ~ 8). *p < 0.05, **p < 0.01; †p < 0.05, ††p < 0.01 compared with control or HSA.
Figure S2. Relationship between muscle strength and C‐reactive protein (CRP)/albumin ratio in patients who are undergoing haemodialysis. (A, B) The relationship between CRP/albumin ratio and grip strength in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle) and without sarcopenia (closed triangle) (A: male, B: female). (C, D) The relationship between CRP/albumin ratio and skeletal mass index in dialysis patients with sarcopenia (closed circle), pre‐sarcopenia (open circle) and without sarcopenia (closed triangle) (C: male, D: female).
Table S1. The primers used for mRNA detection.