

Abstract
We investigated the prevalence of coronaviruses in 44 bats from four families in northeastern Eswatini using high-throughput sequencing of fecal samples. We found evidence of coronaviruses in 18% of the bats. We recovered full or near-full-length genomes from two bat species: Chaerephon pumilus and Afronycteris nana, as well as additional coronavirus genome fragments from C. pumilus, Epomophorus wahlbergi, Mops condylurus, and Scotophilus dinganii. All bats from which we detected coronaviruses were captured leaving buildings or near human settlements, demonstrating the importance of continued surveillance of coronaviruses in bats to better understand the prevalence, diversity, and potential risks for spillover.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10393-021-01567-3.
Keywords: Chiroptera, alphacoronavirus, betacoronavirus, emerging infectious diseases, zoonotic disease, human–wildlife interface
Introduction
Coronaviruses are a family of zoonotic viruses comprised of four genera, two of which, alpha- and betacoronaviruses, have an evolutionary origin in bats, while gamma- and deltacoronaviruses, originate in birds (Graham et al. 2013). Coronaviruses have since radiated to a variety of hosts (Drexler et al. 2014). Notably, in humans, coronaviruses have caused COVID-19 (Zhou et al. 2020; Gorbalenya et al. 2020), Severe Acute Respiratory Syndrome (SARS) (Marra et al. 2003; Li et al. 2005), and Middle East Respiratory Syndrome (MERS) (Memish et al. 2013). While recent studies have increased our knowledge of coronavirus diversity and ecology, large gaps in sampling mean there are probably still many undiscovered species and strains in bats (Anthony et al. 2013, 2017).
Southern Africa has a diverse bat community (Monadjem et al. 2020b) that appears to host many coronaviruses, including strains phylogenetically close to MERS-CoV (Geldenhuys et al. 2013, 2018; Ithete et al. 2013), although studies are still limited (Markotter et al. 2020). Globally, the diversity and distribution of coronaviruses in bats makes it likely that future transmission of these pathogens to humans or other animal species will occur (Woo et al. 2009; Anthony et al. 2017). Although there are no known cases of coronavirus spillover in Africa thus far (Markotter et al. 2020), this could occur where bat species come into frequent, close contact with humans or domestic animals (Monadjem 1998; Fenton et al. 2004; Jacobs and Barclay 2009; Noer et al. 2012; Monadjem et al. 2020b).
Therefore, we investigated the prevalence of coronaviruses in bats belonging to eight species from four families (Pteropodidae: Epomophorus wahlbergi; Emballonuridae: Taphozous mauritianus; Molossidae: Chaerephon pumilus, Mops condylurus, and Mops midas; and Vespertilionidae: Afronycteris nana, Scotophilus dinganii, and Scotophilus viridis). These species are all widely distributed and abundant across southeastern Africa and are commonly found in or near human settlements in northeast Eswatini (Monadjem et al. 2020b, 2021; Shapiro et al. 2020). We subjected fecal samples to virion enrichment followed by RNA sequencing to noninvasively investigate the prevalence and types of coronavirus in the bats of this region. We used this approach to recover whole coronavirus genomes and thus more reliably characterize them (Drexler et al. 2014; De Sabato et al. 2019). This method also allowed us to detect both known and unknown coronaviruses regardless of the specific sequences or genomic region present in samples.
We captured bats at eight sites in northeast Eswatini (Fig. 1) from December 2013–May 2014 using mist-nets and/or a harp trap. Taxonomy follows Monadjem et al. (2010, 2020b, 2020a). To aid in the identification of species, we measured forearm length of each captured bat with calipers to the nearest 0.1 mm and mass to the nearest 0.5 g with a spring balance. Captured bats were placed individually in cloth holding bags for the deposition of feces. We trapped, handled, and released bats in accordance with a permit from the Eswatini National Trust Commission and University of Florida Institutional Animal Care and Use Committee approval (Protocol #201,508,751).
Figure 1.
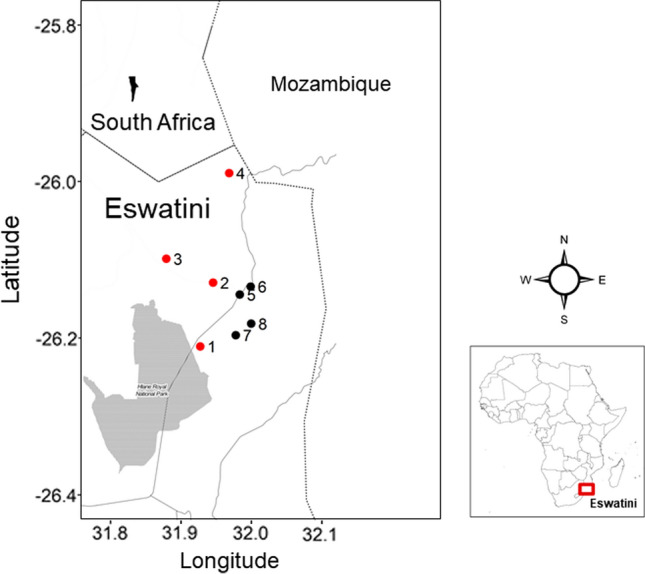
Map of study region. Site numbers indicate where bats were captured and are referenced in Table 2. Sites from which coronaviruses were detected in bats are marked in red, while coronaviruses were not detected in bats captured from sites marked in black. The area shaded in gray is Hlane National Park. Solid lines indicate national borders and dotted lines indicate roads.
Fecal samples from insectivorous species were desiccated and preserved with silica gel (Sigma-Aldrich), while samples from the frugivorous Epomophorus wahlbergi were placed in RNALater (Thermo Fisher Scientific) because due to their wet condition they could not be properly dried. Samples were stored at − 10 °C until the end of the field season (May 2014), then transferred to − 80 °C.
Frozen fecal samples were vortexed vigorously in 600 µl of PBS with beads from the PowerFecal kit (MoBio) for 1 min and incubated at room temperature for 10 min. Following incubation, samples were vortexed for 1 min, then centrifuged at 2500 × g for 3 min. The supernatant was then filtered and the flow-through nuclease-treated following Jensen et al. (2015). Viral nucleic acids were subsequently extracted using Roche High Pure Viral RNA kit (Roche) according to the manufacturer’s guidelines after which 1 µl RNase Out (Invitrogen) was added to the final RNA extract (Jensen et al. 2015; Hansen et al. 2015).
Forty-four RNA libraries were produced, each one from an individual fecal sample, using ScriptSeq v2 RNA-seq library preparation kit (Epicentre, Illumina), according to the manufacturer’s guidelines. Samples were DNase-treated with Promega DNase for 30 min at 37 °C and purified on RNeasy MinElute columns (Qiagen). Seven or eight individually and uniquely single-indexed sequencing libraries were pooled together in equimolar ratios for sequencing with paired-end reads of 100 bp (PE100) on an Illumina Hiseq 2000 platform. The library from one sample (Bat50) was resequenced individually on one lane of PE100 on an Illumina Hiseq 2000 platform.
Reads with overlapping sections of sequences were assembled into longer contiguous sequences (contigs) using Ray Meta v2.2.0 with default settings (Boisvert et al. 2012). The contigs were searched for coronaviruses using megablast and BLASTn on the NCBI Nucleotide collection (nt) database (Altschul et al. 1990, 1997) and by mapping against NCBI’s nr database using DIAMOND (Buchfink et al. 2014).
De novo assembly with an alternative assembler was attempted on the eight coronavirus-positive samples using MEGAHIT v1.1.1 (Li et al. 2015) with the following parameters: minimum contig length = 100, minimum kmer size = 15, maximum kmer size = 101, increment of kmer size of each iteration = 2. To search for potential coronavirus genomes, the 20 longest contigs from each assembly were selected and analyzed using BLASTn on the nt/nr databases, which resulted in the identification of longer coronavirus contigs spanning and extending shorter contigs already identified. Further assembly was attempted on the combined set of contigs using Geneious v.11 software (https://www.geneious.com/), resulting in full or near-full genomes for four bats. Reads were mapped back to the genomes using bowtie2 (Langmead and Salzberg 2012) to correct ambiguous bases.
We also mapped all the sequenced reads from individual samples back to the coronavirus contigs using bowtie2 (v2.2.9) (Langmead and Salzberg 2012). We did this in order to confirm which samples the sequences came from and identify any potential cases of bleed over (the misidentification of the sample from which each sequence read originated) following Kircher et al. (2012) and Jensen et al. (2015).
We identified three full-length and one partial alphacoronavirus genomes from four individual bats (accession numbers OL807608, OL807609, OL807610, OL807611; Supplementary File 1). Three of these were isolated from the species Chaerephon pumilus: two of the full genomes (from Bat143 and Bat151; 27,956 nt and 28,061 nt respectively) and one partial genome (Bat180; 20,826 nt). The best hit using BLASTn for all three of these coronavirus genomes was Chaerephon bat coronavirus/Kenya/KY22/2006 from Kenya (Tong et al. 2009). When aligned in Geneious, all three were 86–87% identical to this species. Pairwise identity for the ORF1ab gene was 97.1–97.2%, indicating these coronaviruses likely belong to the same species as Chaerephon bat coronavirus/Kenya/KY22/2006 based on the coronavirus species demarcation criterion of the International Committee on Taxonomy of Viruses (Lefkowitz et al. 2018; ICTV 2019). In a bootstrapped maximum likelihood tree using RAxML based on full coronavirus genomes following De Sabato et al. (2019), all three Chaerephon pumilus coronavirus genomes clustered together as a sister clade to Chaerephon bat coronavirus/Kenya/KY22/2006 (Fig. 2). This coronavirus may be widespread within the bat genus Chaerephon across Africa. Other coronaviruses have been found in bats of the same species or genera that are geographically distant, sometimes across continents (Drexler et al. 2014) and could indicate connectivity between bat populations across their distribution.
Figure 2.

Maximum likelihood phylogeny of coronaviruses (CoVs) based on full genomes, including reference genomes and the four full-length and partial genomes from this study, which are labelled in red and can be retrieved under accession numbers OL807608, OL807609, OL807610, OL807611. Stars indicate branches with 100% bootstrap support.
When aligned to each other in Geneious, the three Chaerephon pumilus coronavirus genomes were 98.8% identical. The full genomes from Bat143 and Bat151 were slightly more similar to each other (99.4%) than to the partial genome from Bat180 (98.3 – 98.6%). All three genomes were confirmed using real-time PCR using strain-specific primers and fluorescently labeled TaqMan probe designed with Primer3 software in Geneious based on the coronavirus sequences from these three bats (Untergasser et al. 2012) (Supplementary Fig. 1, Supplementary File 2).
We detected a third full-length coronavirus genome from the bat species Afronycteris nana (Bat77; 26,977 nt) that likely represents a newly described alphacoronavirus. Its best hit in BLASTn was Alphacoronavirus Bat-CoV/P.kuhlii/Italy/206645-41/2011 isolated from Pipistrellus kuhli in Italy (De Sabato et al. 2019). When aligned to Alphacoronavirus Bat-CoV/P.kuhlii/Italy/206645-41/2011 in Geneious, pairwise identity was 76.4% across the full genome and 77.6% across the ORF1ab region. In our phylogeny, this coronavirus was sister to Alphacoronavirus Bat-CoV/P.kuhlii/Italy/206645-41/2011 (Fig. 2). Afronycteris and Pipistrellus are closely related pipistrelle-like bats in the subfamily Vespertilioninae (Vespertilionidae), albeit in different tribes (Vespertilionini and Pipistrellini, respectively) (Monadjem et al. 2020a); thus it is not unexpected that the coronaviruses from these genera would be relatively similar.
In addition to these full- and near-full-length genomes, we identified 75 shorter coronavirus genome fragments ranging from 103 to 5241 nt from five bats belonging to the species: Chaerephon pumilus (Bat180), Mops condylurus (Bat166), Scotophilus dinganii (Bat167), and Epomophorus wahlbergi (Bat50, Bat76) (accession numbers OM000306–OM000380; Supplementary File 3). Only alphacoronavirus sequences were isolated from molossids (Chaerephon pumilus and Mops condylurus) and vespertilionids (Scotophilus dinganii). The sequences detected in the fruit bat Epomophorus wahlbergi were either betacoronaviruses (the same genus as SARS-CoV-2 (Zhou et al. 2020; Gorbalenya et al. 2020), SARS-CoV-1 (Li et al. 2005), and MERS-CoV (Memish et al. 2014)), or unclassified coronaviruses (Tables 1, and 2). These sequences were short, ranging from 106–517 nt (mostly < 200 nt), with 71–93% percent identity to previously described coronaviruses. They appear most closely related to coronaviruses sequenced from other pteropodid fruit bats, particularly Eidolon helvum in Kenya (Tong et al. 2009), Cameroon (Yinda et al. 2018), and Nigeria (Leopardi et al. 2016), and to a lesser extent Rousettus leschenaultii in southern China (Woo et al. 2007). None appeared particularly closely related to any human betacoronavirus pathogens. Targeted PCR of specific genes, such as the RNA-dependent RNA polymerase (RdRp), which is widely used in studies of animal coronaviruses (Drexler et al. 2014), could provide further information about specific lineages of coronaviruses, including the betacoronaviruses. However, lack of material prevents such endeavors at this time.
Table 1.
Table summarizing coronavirus detection in bats in northeast Eswatini.
| Family | Species | Number of captured individuals | Detected CoV (no. of samples) | Proportion of CoV-positive individuals (%) |
|---|---|---|---|---|
| Pteropodidae | Epomophorus wahlbergi | 9 | 2 | 15 |
| Emballonuridae | Taphozous mauritianus | 2 | 0 | 0 |
| Molossidae | Chaerephon pumilus | 18 | 3 | 17 |
| Mops condylurus | 7 | 1 | 14 | |
| Mops midas | 1 | 0 | 0 | |
| Vespertilionidae | Afronycteris nana | 1 | 1 | 100 |
| Scotophilus dinganii | 3 | 1 | 33 | |
| Scotophilus viridis | 3 | 0 | 0 | |
| Total | 44 | 8 | 18 |
Table 2.
Coronaviruses detected in individual bats.
| Family | Species | Bat ID | Capture Sitea | Number CoV Contigs | Contig length (nt) |
|---|---|---|---|---|---|
| Pteropodidae | Epomophorus wahlbergi | 50 | 1 | 36 | 105–517 |
| 76 | 6 | 106–176 | |||
| Molossidae | Chaerephon pumilus | 143 | 3 | 1 | 27,956 |
| 151 | 3 | 1 | 28,061 | ||
| 180 | 4 | 6 | 110–20,826 | ||
| Mops condylurus | 166 | 4 | 24 | 249–4121 | |
| Vespertilionidae | Afronycteris nana | 77 | 2 | 1 | 26,977 |
| Scotophilus dinganii | 167 | 4 | 4 | 103–148 |
aCapture site numbers correspond to site numbers Fig. 1.
In conclusion, from a sample of 44 bats in Eswatini, we detected both alpha- and betacoronaviruses. All eight bats from which coronaviruses were detected were captured leaving roosts in houses, churches, or within human settlements. More research is necessary to determine whether any of these detected coronaviruses could be a concern for the health of humans or livestock. Limiting direct contact with these bats or their feces might possibly aid in preventing future emerging infectious diseases, while continued monitoring may shed light on the diversity and ecology of coronaviruses.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank Hervé Echecolonea, Phumlile Simelane, Mduduzi Ngwenya, and Zanele Dlamini for assistance in the field and Mandla Motsa, Smart Shabangu, Tal Fineberg the staff at Mbuluzi Game Reserve, Thea Litschka-Koen, Clifton Koen, Nick Jackson, and Kim Roques and All-Out Africa, for help with logistics. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1315138 (J.T.S), a National Science Foundation Graduate Research Opportunities Worldwide grant (J.T.S.), Innovation Fund Denmark (The Genome Denmark platform, grant no. 019-2011-2), a Student Research Grant from Bat Conservation International (J.T.S), a University of Florida Biodiversity Institute Fellowship (J.T.S), the Zuckerman STEM Leadership Program (J.T.S), and an NIH Grant (1R01GM114362) (N.D.N). Initial phylogenetic analyses used the Extreme Science and Engineering Discovery Environment (XSEDE) resources, which is supported by National Science Foundation grant number ACI-1053575. XSEDE resources were provided by project allocation TG-ASC160034.
References
- Altschul S, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Khan SA, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JAK, Daszak P, Lipkin WI. A strategy to estimate unknown viral diversity in mammals. mBio. 2013 doi: 10.1128/mBio.00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony SJ, Johnson CK, Greig DJ, Kramer S, Che X, Wells H, Hicks AL, Joly DO, Wolfe ND, Daszak P, Karesh W, Lipkin WI, Morse SS, Mazet JAK, Goldstein T. Global patterns in coronavirus diversity. Virus Evolution. 2017;3:1814–1820. doi: 10.1093/ve/vex012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert S, Raymond F, Godzaridis É, Laviolette F, Corbeil J, Wold B, Myers R, Brenner S, McPherson J, Mardis E, Compeau P, Pevzner P, Tesler G, Flicek P, Birney E, Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G, Miller J, Koren S, Sutton G, Salzberg S, Treangen T, Salzberg S, Lorenz P, Eck J, Scholz M, Lo C, Chain P, Schoenfeld T, Patterson M, Richardson P, Wommack K, Young M, Mead D, Varin T, Lovejoy C, Jungblut A, Vincent W, Corbeil J, Varin T, Lovejoy C, Jungblut A, Vincent W, Corbeil J, Narasingarao P, Podell S, Ugalde J, Brochier-Armanet C, Emerson J, Brocks J, Heidelberg K, Banfield J, Allen E, Tringe S, Mering C von, Kobayashi A, Salamov A, Chen K, Chang H, Podar M, Short J, Mathur E, Detter J, Bork P, Hugenholtz P, Rubin E, Tyson G, Chapman J, Hugenholtz P, Allen E, Ram R, Richardson P, Solovyev V, Rubin E, Rokhsar D, Banfield J, Cho I, Blaser M, Gill S, Pop M, Deboy R, Eckburg P, Turnbaugh P, Samuel B, Gordon J, Relman D, Fraser-Liggett C, Nelson K, Qin J, Li R, Raes J, Arumugam M, Burgdorf K, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende D, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J, Hansen T, Paslier D Le, Linneberg A, Nielsen H, Pelletier E, Renault P, Arumugam M, Raes J, Pelletier E, Paslier D Le, Yamada T, Mende D, Fernandes G, Tap J, Bruls T, Batto J, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen H, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Consortium T, Schloss P, Handelsman J, Liu B, Gibbons T, Ghodsi M, Pop M, Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C, McDonald D, Price M, Goodrich J, Nawrocki E, DeSantis T, Probst A, Andersen G, Knight R, Hugenholtz P, Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, Harris M, Hill D, Issel-Tarver L, Kasarskis A, Lewis S, Matese J, Richardson J, Ringwald M, Rubin G, Sherlock G, Simpson J, Wong K, Jackman S, Schein J, Jones S, Birol I, Boisvert S, Laviolette F, Corbeil J, Schatz M, Langmead B, Salzberg S, Huson D, Mitra S, Ruscheweyh H, Weber N, Schuster S, Meyer F, Paarmann D, D M, Dixon P, Caporaso J, Kuczynski J, Stombaugh J, Bittinger K, Bushman F, Costello E, Fierer N, Pena A, Goodrich J, Gordon J, Huttley G, Kelley S, Knights D, Koenig J, Ley R, Lozupone C, McDonald D, Muegge B, Pirrung M, Reeder J, Sevinsky J, Turnbaugh P, Walters W, Widmann J, Yatsunenko T, Zaneveld J, Knight R, Krause L, Diaz N, Goesmann A, Kelley S, Nattkemper T, Rohwer F, Edwards R, Stoye J, Brady A, Salzberg S, Namiki T, Hachiya T, Tanaka H, Sakakibara Y, Peng Y, Leung H, Yiu S, Chin F, Laserson J, Jojic V, Koller D, Wu G, Chen J, Hoffmann C, Bittinger K, Chen Y, Keilbaugh S, Bewtra M, Knights D, Walters W, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman F, Lewis J, Pevzner P, Tang H, Waterman M, Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg S, Schadt E, Linderman M, Sorenson J, Lee L, Nolan G, Benson D, Boguski M, Lipman D, Ostell J, Kulikova T, Aldebert P, Althorpe N, Baker W, Bates K, Browne P, Broek A van den, Cochrane G, Duggan K, Eberhardt R, Faruque N, Garcia-Pastor M, Harte N, Kanz C, Leinonen R, Lin Q, Lombard V, Lopez R, Mancuso R, McHale M, Nardone F, Silventoinen V, Stoehr P, Stoesser G, Ann M, Tzouvara K, Vaughan R, Wu D, Zhu W, Apweiler R, Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R, Gabriel E, Fagg G, Bosilca G, Angskun T, Dongarra J, Squyres J, Sahay V, Kambadur P, Barrett B, Lumsdaine A, Castain R, Daniel D, Graham R, Woodall T, Gabriel E, Fagg G, Bosilca G, Angskun T, Dongarra J, Squyres J, Sahay V, Kambadur P, Barrett B, Lumsdaine A, Castain R, Daniel D, Graham R, Woodall T, Gropp W (2012) Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biology 10.1186/gb-2012-13-12-r122 [DOI] [PMC free article] [PubMed]
- Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature Methods. 2014;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- De Sabato L, Lelli D, Faccin F, Canziani S, Di Bartolo I, Vaccari G, Moreno A. Full genome characterization of two novel Alpha-coronavirus species from Italian bats. Virus Research. 2019;260:60–66. doi: 10.1016/j.virusres.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Research. 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton MB, Jacobs DS, Richardson EJ, Taylor PJ, White W. Individual signatures in the frequency-modulated sweep calls of African large-eared, free-tailed bats Otomops martiensseni (Chiroptera: Molossidae) J Zool. 2004;262:11–19. doi: 10.1017/S095283690300431X. [DOI] [Google Scholar]
- Geldenhuys M, Mortlock M, Weyer J, Bezuidt O, Seamark ECJ, Kearney T, Gleasner C, Erkkila TH, Cui H, Markotter W. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLOS ONE. 2018;13:e0194527. doi: 10.1371/journal.pone.0194527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldenhuys M, Weyer J, Nel LH, Markotter W. Coronaviruses in South African bats. Vector-Borne and Zoonotic Diseases. 2013;13:516–519. doi: 10.1089/vbz.2012.1101. [DOI] [PubMed] [Google Scholar]
- Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, Haagmans BL, Lauber C, Leontovich AM, Neuman BW, Penzar D, Perlman S, Poon LLM, Samborskiy DV, Sidorov IA, Sola I, Ziebuhr J. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RL, Donaldson EF, Baric RS. A decade after SARS : Strategies for controlling emerging coronaviruses. Nature Reviews Microbiology. 2013;11:836–848. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TA, Fridholm H, Frøslev TG, Kjartansdóttir KR, Willerslev E, Nielsen LP, Hansen AJ. New type of papillomavirus and novel circular single stranded DNA virus discovered in urban Rattus norvegicus using circular DNA enrichment and metagenomics. PLoS ONE. 2015;10:e0141952. doi: 10.1371/journal.pone.0141952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICTV (2019) (International Committee on Taxonomy of Viruses). Virus Taxonomy: 2019 Release. https://talk.ictvonline.org/taxonomy/. Accessed 1 Sep 2020
- Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, Drosten C, Drexler JF, Preiser W. Close relative of human Middle East Respiratory Syndrome coronavirus in bat, South Africa. Emerging Infectious Diseases. 2013;19:1697–1699. doi: 10.3201/eid1910.130946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs DS, Barclay RMR. Niche Differentiation in Two Sympatric Sibling Bat Species, Scotophilus dinganii and Scotophilus mhlanganii. Journal of Mammalogy. 2009;90:879–887. doi: 10.1644/08-MAMM-A-235.1. [DOI] [Google Scholar]
- Jensen RH, Mollerup S, Mourier T, Hansen TA, Fridholm H, Nielsen LP, Willerslev E, Hansen AJ, Vinner L. Target-dependent enrichment of virions determines the reduction of high-throughput sequencing in virus discovery. PLoS ONE. 2015;10:e0122636. doi: 10.1371/journal.pone.0122636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Research. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz EJ, Dempsey DM, Hendrickson RC, Orton RJ, Siddell SG, Smith DB. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV) Nucleic Acids Research. 2018;46:D708–D717. doi: 10.1093/nar/gkx932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopardi S, Oluwayelu D, Meseko C, Marciano S, Tassoni L, Bakarey S, Monne I, Cattoli G, De Benedictis P. The close genetic relationship of lineage D Betacoronavirus from Nigerian and Kenyan straw-colored fruit bats (Eidolon helvum) is consistent with the existence of a single epidemiological unit across sub-Saharan Africa. Virus Genes. 2016;52:573–577. doi: 10.1007/s11262-016-1331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- Li WD, Shi ZL, Yu M, Ren WZ, Smith C, Epstein JH, Wang HZ, Crameri G, Hu ZH, Zhang HJ, Zhang JH, McEachern J, Field H, Daszak P, Eaton BT, Zhang SY, Wang LF. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Markotter W, Coertse J, De Vries L, Geldenhuys M, Mortlock M. Bat-borne viruses in Africa: a critical review. Journal of Zoology. 2020;311:77–98. doi: 10.1111/jzo.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra MA, Jones SJM, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Memish ZA, Cotten M, Meyer B, Watson SJ, Alsahafi AJ, Al Rabeeah AA, Corman VM, Sieberg A, Makhdoom HQ, Assiri A, Al Masri M, Aldabbagh S, Bosch B-J, Beer M, Müller MA, Kellam P, Drosten C. Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013. Emerging Infectious Diseases. 2014;20:1012–1015. doi: 10.3201/eid2006.140402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, AlHakeem R, Durosinloun A, Al Asmari M, Islam A, Kapoor A, Briese T, Daszak P, Al Rabeeah AA, Lipkin WI. Middle East Respiratory Syndrome coronavirus in bats, Saudi Arabia. Emerging Infectious Diseases. 2013;19:1819–1823. doi: 10.3201/eid1911.131172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monadjem A (1998) Mammals of Swaziland. The Conservation Trust of Swaziland and Big Game Parks, Mbabane, Swaziland
- Monadjem A, Demos TC, Dalton DL, Webala PW, Musila S, Kerbis Peterhans JC, Patterson BD. A revision of pipistrelle-like bats (Mammalia: Chiroptera: Vespertilionidae) in East Africa with the description of new genera and species. Zoological Journal of the Linnean Society. 2020 doi: 10.1093/zoolinnean/zlaa087. [DOI] [Google Scholar]
- Monadjem A, Simelane F, Shapiro JT, Gumbi BC, Mamba ML, Sibiya MD, Lukhele SM, Mahlaba TAM. Using species distribution models to gauge the completeness of the bat checklist of Eswatini. European Journal of Wildlife Research. 2021;67:1–10. doi: 10.1007/s10344-021-01463-9. [DOI] [Google Scholar]
- Monadjem A, Taylor PJ, Cotterill FPD, Schoeman MC. Bats of Southern and Central Africa A Biogeographic and Taxonomic Synthesis. 2. Johannesburg: Wits University Press; 2020. [Google Scholar]
- Monadjem A, Taylor PJ, Cotterill FPD, Schoeman MC. Bats of Southern and Central Africa: A Biogeographic and Taxonomic Synthesis. Johannesburg: Wits University Press; 2010. [Google Scholar]
- Noer CL, Dabelsteen T, Bohmann K, Monadjem A, Monadiem A. Molossid bats in an African agro-ecosystem select sugarcane fields as foraging habitat. African Zoology. 2012;47:1–11. doi: 10.3377/004.047.0120. [DOI] [Google Scholar]
- Shapiro JT, Monadjem A, Röder T, McCleery RA. Response of bat activity to land cover and land use in savannas is scale-, season-, and guild-specific. Biological Conservation. 2020;241:108245. doi: 10.1016/j.biocon.2019.108245. [DOI] [Google Scholar]
- Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerging Infectious Diseases. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3- New capabilities and interfaces. Nucleic Acids Research. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo PCY, Lau SKP, Huang Y, Yuen K-Y. Coronavirus diversity, phylogeny and interspecies jumping. Experimental Biology and Medicine. 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- Woo PCY, Wang M, Lau SKP, Xu H, Poon RWS, Guo R, Wong BHL, Gao K, Tsoi H-w, Huang Y, Li KSM, Lam CSF, Chan K-h, Zheng B-j, Yuen K-y. Comparative Analysis of Twelve Genomes of Three Novel Group 2c and Group 2d Coronaviruses Reveals Unique Group and Subgroup Features. Journal of Virology. 2007;81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yinda CK, Ghogomu SM, Conceição-Neto N, Beller L, Deboutte W, Vanhulle E, Maes P, Van Ranst M, Matthijnssens J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evolution. 2018 doi: 10.1093/ve/vey008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Lou Yang X, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Di Jiang R, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.