

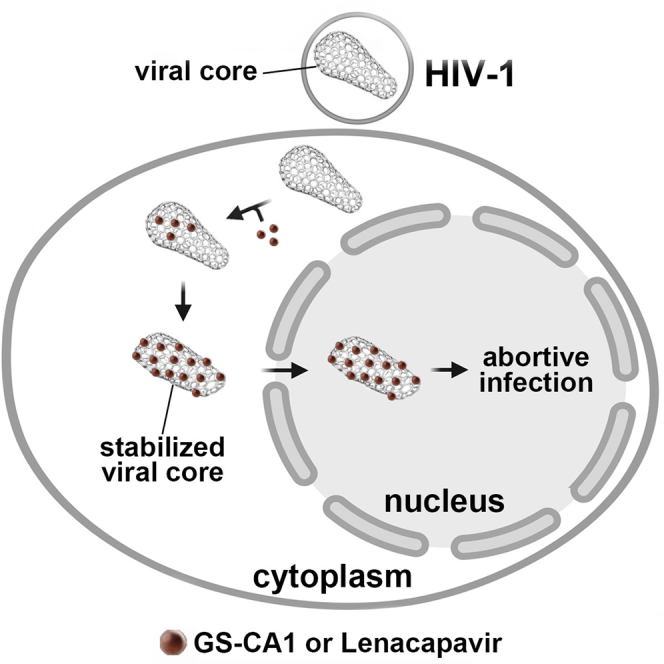
Summary
The HIV-1 capsid is the target for the antiviral drugs GS-CA1 and Lenacapavir (GS-6207). We investigated the mechanism by which GS-CA1 and GS-6207 inhibit HIV-1 infection. HIV-1 inhibition by GS-CA1 did not require CPSF6 in CD4+ T cells. Contrary to PF74 that accelerates uncoating of HIV-1, GS-CA1 and GS-6207 stabilized the core. GS-CA1, unlike PF74, allowed the core to enter the nucleus, which agrees with the fact that GS-CA1 inhibits infection after reverse transcription. Unlike PF74, GS-CA1 did not disaggregate preformed CPSF6 complexes in nuclear speckles, suggesting that PF74 and GS-CA1 have different mechanisms of action. GS-CA1 stabilized the HIV-1 core, possibly by inducing a conformational shift in the core; in agreement, HIV-1 cores bearing N74D regained their ability to bind CPSF6 in the presence of GS-CA1. We showed that GS-CA1 binds to the HIV-1 core, changes its conformation, stabilizes the core, and thereby prevents viral uncoating and infection.
Subject areas: Biological sciences, Immunology, Virology
Graphical abstract
Highlights
-
•
GS-CA1 and Lenacapavir (GS-6207) stabilizes the HIV-1 core during infection
-
•
GS-CA1/GS-6207 inhibit the interaction of the HIV-1 core with host factors
-
•
GS-CA1/GS-6207 do not disaggregate preformed CPSF6 complexes in nuclear speckles
-
•
GS-CA1/GS-6207 affects the dynamic surface of the HIV-1 core
Biological sciences; Immunology; Virology
Introduction
The HIV-1 capsid is critical throughout the HIV-1 life cycle; thus, it has become an important pharmacological target (Singh et al., 2019; Thenin-Houssier and Valente, 2016; Zhang et al., 2009). Capsid inhibitors, which range in size from peptides to small molecules, interact directly with the capsid to block HIV-1 infection. The peptide CAI (Sticht et al., 2005) and the small molecule CAP-1 (Kelly et al., 2007; Tang et al., 2003) bind to the capsid subunit protein and inhibit assembly in vitro. The small-molecule PF-3450074 (PF74) binds to HIV-1 core, prevents the binding of cleavage and polyadenylation specificity factor 6 (CPSF6) and Nup153 to the capsid (Buffone et al., 2018; Fricke et al., 2013b; Lee et al., 2010), inhibits entry of the core into the nucleus (Selyutina et al., 2020a), accelerates uncoating (Bhattacharya et al., 2014; Fricke et al., 2013b; Shi et al., 2011), inhibits integration (Balasubramaniam et al., 2019), and infection (Saito et al., 2016; Shi et al., 2011). Similar to PF74, the small molecule Bi-2 prevents the binding of CPSF6 to the core (Fricke et al., 2014a), inhibits entry of the core into the nucleus (Selyutina et al., 2020a), accelerates uncoating (Fricke et al., 2014a), and inhibits infection (Fricke et al., 2014a; Lamorte et al., 2013).
Two small molecules GS-CA1 and Lenacapavir (GS-6207, an analog of GS-CA1) bind to the capsid to potently inhibit HIV-1 replication in vitro and in vivo (Link et al., 2020; Yant et al., 2019). Structurally, GS-CA1 and PF74 utilize similar capsid amino acids to bind to the HIV-1 core (Bhattacharya et al., 2014; Link et al., 2020; Yant et al., 2019), and selection for drug resistance in vitro identified HIV-1 mutations that are either associated with PF74 resistance or are near the PF74 binding pocket in the capsid (Yant et al., 2019). Because GS-CA1 binds, in part, to the PF74 capsid binding pocket, this work investigates the mechanism by which GS-CA1 blocks HIV-1 infection and compares it to the inhibition mechanism used by PF74.
We determined that inhibition of HIV-1 by GS-CA1 and PF74 did not require CPSF6 expression in primary CD4+ T cells. Similar to PF74, GS-CA1 inhibited CPSF6 and Nup153 interaction with the HIV-1 core. A hallmark of PF74 is its ability to accelerate the uncoating of the HIV-1 core during infection, similar to the mechanism of rhesus tripartite motif protein 5α; however, GS-CA1 stabilized the core during infection. In contrast to PF74, which inhibits entry of the core into the nucleus, GS-CA1 allowed normal entry of the core into the nucleus, consistent with the fact that reverse transcription occurs in the presence of in GS-CA1. We found that GS-CA1 did not disaggregate preformed CPSF6 complexes in nuclear speckles, as PF74 does, suggesting a different mechanism of action for PF74. The stabilization of the HIV-1 core by GS-CA1 suggested that it induced a conformational change in the capsid. This is supported by our results showing that the capsid mutation N74D allows the binding of CPSF6 in the presence of GS-CA1. We showed that GS-CA1 bound to the HIV-1 core, changed the conformation of the capsid, and stabilized the core, thereby preventing uncoating and infection.
Results
Expression of CPSF6 in CD4+ T cells is not required for GS-CA1 and PF74 to block HIV-1 infection
The small molecule GS-CA1 binds to the capsid in a similar region to where the CPSF6 protein binds to the HIV-1 core suggesting that GS-CA1 interferes with CPSF6 binding to inhibit HIV-1 infection (Link et al., 2020; Yant et al., 2019). To determine the role of CPSF6 in GS-CA1 inhibition of HIV-1 infection, we administered increasing concentrations of GS-CA1 to CD4+ T cells that had been depleted for CPSF6 using CRISPR/Cas9. The effect of GS-CA1 treatment was measured by HIV-1NL4-3Δenv-GFP pseudotyped with VSV-G (HIV-1NL4-3-GFP) infection of the CPSF6-depleted CD4+ T cells. Cells depleted of CXCR4 using CRISPR/Cas9 or cells treated with a non-targeting RNA guide served as controls. Two guide RNAs, #5 and #6, depleted expression of CPSF6 in CD4+ T cells from two donors (Figure 1A); however, depletion of CPSF6 did not affect GS-CA1 inhibition of HIV-1NL4-3-GFP infection of CD4+ T cells (Figure 1B). Similarly, PF74 effectively inhibited HIV-1 infection in CPSF6-depleted CD4+ T cells (Figure 1B). Thus, CPSF6 expression is not required for GS-CA1 and PF74 inhibition of HIV-1 infection.
Figure 1.

GS-CA1 does not require the expression of CPSF6 in CD4+ T cells to inhibit HIV-1 infection
(A) Loss of CPSF6 expression using the CRISPR/Cas9 system in human primary CD4+ T cells from two different donors was determined by western blots using antibodies against CPSF6. Controls included gRNA against CXCR4 and a non-targeting gRNA. Expression of GAPDH was the control for equal loading.
(B) Human primary CD4+ T cells depleted for CPSF6 expression were infected with HIV-1NL4-3-GFP with the indicated concentrations of PF74, GS-CA1, or DMSO as a vehicle control. Infected GFP-positive cells were determined at 72 h post-infection. Experiments were performed in triplicates, and the mean infections ±SD are shown. Results for two different donors are shown.
GS-CA1 prevents the binding of CPSF6 to the HIV-1 core
Because GS-CA1 and PF74 utilize similar capsid amino acids to bind to the HIV-1 core (Bhattacharya et al., 2014; Link et al., 2020; Yant et al., 2019), and PF74 prevents CPSF6 from binding to the capsid (Fricke et al., 2013b; Lee et al., 2010), we determined whether GS-CA1 also prevents the binding of CPSF6 to the capsid. GS-CA1 prevented the interaction of CPSF6 with stabilized HIV-1 capsid tubes, which are core surrogates (Selyutina et al., 2018), as did the control PF74 (Figure 2A), which has previously been shown to prevent the binding of CPSF6 to the capsid. GS-CA1 at μM concentrations prevented the binding of CPSF6 to capsid, as does PF74; however, GS-CA1 prevented the binding of CPSF6 to the capsid more potently than did PF74. As shown in Figure 2, this is the only section of this work that uses μM concentrations of GS-CA1 and capsid (∼3 μM). Although GS-CA-1 strongly prevents binding of CPSF6 to capsid at 10 μM (Figure 2A), there is still a small fraction of CPSF6 that remains bound, which may be because of the fact that a 1:3 ratio of capsid:drug is not sufficient to completely prevent binding of CPSF6 to capsid.
Figure 2.

GS-CA1 prevents the binding of CPSF6 and Nup153 to the HIV-1 core
Human HEK293T cells were transfected with plasmids expressing CPSF6-FLAG (A) or Nup153-HA (B), and at 24 h post-transfection lysed cell extracts (INPUT) and stabilized wild-type HIV-1 capsid tubes were mixed with PF74 or GS-CA1 for 1 h before collection of the HIV-1 capsid tubes pellets by centrifugation. Pellets were washed two times and analyzed (BOUND). The input and bound fractions were analyzed by western blotting (a representative Western blot is shown) using anti-FLAG (A), anti-HA (B), or anti-p24 capsid antibodies. Experiments were repeated at least three times and a representative figure is shown. The amount of capsid relative to the total for three independent experiments is shown. Mean bound fractions relative to input ±SD are shown. ∗ indicates p value < 0.005, ∗∗ indicates p value < 0.001, ∗∗∗ indicates p value < 0.0005, as determined by using the unpaired t test.
Because GS-CA1 and PF74 prevented the interaction of CPSF6 with capsid, we also determined whether GS-CA1 prevented the binding of Nup153 to capsid, as does PF74 (Buffone et al., 2018). GS-CA1 inhibited the interaction of Nup153 with the capsid, but it was not more potent than PF74 in preventing the binding of Nup153 to the capsid (Figure 2B). These results showed that GS-CA1 and PF74 are similar in preventing the binding of cellular proteins CPSF6 and Nup153 to the HIV-1 core.
GS-CA1 stabilizes the HIV-1 core during infection
Because PF74 destabilizes the HIV-1 core during infection (Bhattacharya et al., 2014; Fricke et al., 2013b; Shi et al., 2011), and GS-CA1 shares some characteristics with PF74, we investigated the effect of GS-CA1 on HIV-1 core stability during infection using the fate of the capsid assay (Yang et al., 2014). Increasing concentrations of GS-CA1 resulted in increased amounts of pelletable capsid, indicating that it stabilized the capsid during infection in sharp contrast to the control, PF74, which destabilizes the core during infection (Figure 3A). This stabilization by GS-CA1 was similar to the effect observed by the overexpression of CPSF6 or MxB (Fricke et al., 2013b, 2014b). To correlate in vitro capsid stabilization with infection, we measured infection using the same conditions used for the fate of the capsid experiments and determined that the concentration of GS-CA1 that resulted in a gain of core stability coincided with the concentration of GS-CA1 that inhibited infection (Figure 3B). Thus, stabilization of the core may inhibit infection, as has been shown for MxB and NES-CPSF6 (Fricke et al., 2013b, 2014b). GS-6207, an analog of GS-CA1, also stabilized the HIV-1 core during infection (Figure 3C). GS-CA1 stabilization of the HIV-1 capsid was also demonstrated by using our capsid stabilization assay (Figure S1) (Fricke et al., 2013a; Fricke and Diaz-Griffero, 2016).
Figure 3.

GS-CA1 stabilizes the HIV-1 core during infection
(A) Human A549 cells were infected with HIV-1-GFP (pseudotyped with VSV-G) at MOI = 2 with PF74, GS-CA1, or DMSO as a vehicle control. After incubation for 16 h 37°C cells were harvested and processed using the fate of the capsid assay, as described in methods. INPUT, SOLUBLE, and PELLET fractions of lysed cells were analyzed by western blots using antibodies against the HIV-1 p24 capsid protein. Experiments were performed 3 times, and the mean pelletable capsids ±SD are shown.
(B) Human A549 cells were infected as described for (A) and infected GFP-positive cells were determined at 48 h post-infection. Infections were performed in triplicates, and the mean infections ±SD are shown.
(C) Human A549 cells were infected and analyzed as described for (A) with PF74, GS-6207, or DMSO as a vehicle control. Experiments were performed 3 times, and the mean pelletable capsids ±SD are shown.
(D) Human A549 cells were infected and analyzed as described for (A) and the PELLET fraction was also analyzed using antibodies against CPSF6. Experiments were performed 3 times, and the mean pelletable capsids ±SD are shown. ∗∗ indicates p value < 0.001, ∗∗∗ indicates p value < 0.0005, ∗∗∗∗ indicates p value < 0.0001, NS indicates not significant as determined by using the unpaired t test.
Structural analysis revealed that, unlike PF74, GS-CA1 interacts with capsid residues P38 and S41. To determine whether the interaction between GS-CA1 and residues P38 and S41 is important for stabilizing the core, we used the fate of the capsid assay for HIV-1 P38S, S41A, or P38S/S41A mutants testing various concentrations of GS-CA1 (Figure S2). Interestingly, a higher concentration of GS-CA1 was required to stabilize the core of viruses bearing the double capsid mutations P38S/S41A (Figure S2) than single mutations. These results revealed that contrary to PF74, which accelerates uncoating, GS-CA1 and GS-6207 prevent uncoating.
Using the capsid binding assay, we demonstrated that GS-CA1, like PF74, prevented the binding of CPSF6 to the capsid using μM concentrations of GS-CA1; however, we also wanted to know whether GS-CA1 dissociated CPSF6 from the HIV-1 core during infection using nM concentrations of GS-CA1, which are the concentrations sufficient to inhibit infection. Using the fate of the capsid (Fricke et al., 2014b), we determined whether GS-CA1 prevented endogenous CPSF6 from associating with the HIV-1 core during infection. Dissociation of CPSF6 from the core correlated with HIV-1 inhibition (Figure 3D), suggesting that GS-CA1 mediates the dissociation of CPSF6 from the core during infection when using nM concentrations of GS-CA1.
GS-CA1 does not inhibit the entry of the viral core into the nucleus
Because we have demonstrated that PF74 prevents the entry of the capsid into the nucleus (Selyutina and Diaz-Griffero, 2021; Selyutina et al., 2020a), based on a biochemical assay that tracks the presence of the capsid in the nucleus during infection, we also determined whether GS-CA1 prevented the entry of the capsid into the nucleus. We infected human A549 cells using HIV-1-GFP at a MOI = 2 for 8 h with GS-CA1. Cells were separated into nuclear and cytosolic fractions and analyzed for capsid protein (p24) by western blotting (Figure 4A). We determined the quality of fractionation using antibodies specific to the nuclear marker Nopp140 and the cytosolic marker tubulin (Figure 4A). As a control this assay utilizes PF74, which completely blocks the entry of capsid into the nucleus (Figure 4A). In contrast to PF74 (Selyutina and Diaz-Griffero, 2021; Selyutina et al., 2020a), GS-CA1 did not prevent the entry of capsid into the nucleus. Similar to MxB and CPSF6 (Selyutina et al., 2020a), GS-CA1 stabilization of the HIV-1 core during infection correlated with nuclear entry.
Figure 4.

GS-CA1 does not inhibit the entry of the viral core into the nucleus
(A and B) Human A549 cells were infected with wild-type HIV-1-GFP (pseudotyped with VSV-G) at MOI = 2 with PF74, GS-CA1, or DMSO as a vehicle control. (A) Capsids from the nuclear and cytosolic fractions of cells infected at 37°C for 8 h cells were determined using western blots with anti-p24 antibodies. Nopp140 and tubulin, determined using western blots with appropriate antibodies, were markers for nuclear and cytosolic content, respectively. Nuclear and cytosolic fractions of p24 were determined by western blots from three independent experiments (a representative Western blot is shown). The mean nuclear fractions relative to the cytosolic fractions ±SD are shown. ∗∗∗∗ indicates p value < 0.0001 as determined by using the unpaired t test. (B) Infected GFP-positive cells were determined at 48 h post-infection. Infections were performed in triplicates, and the mean infections ±SD are shown.
Although GS-CA1 and PF74 bind to a similar site in the HIV-1 capsid, their effect on the core during infection is remarkably different, with GS-CA1 allowing the transport of the core into the nucleus. Results from experiments that measured infection under the same conditions used for the nuclear import assay (Figure 4B) suggested that GS-CA1 inhibits HIV-1 infection after the core enters the nucleus. Because recent evidence suggested that HIV-1 reverse transcription occurs in the nucleus (Burdick et al., 2020; Dharan et al., 2020; Selyutina et al., 2020a), we determined whether GS-CA1 inhibits reverse transcription and the formation of 2-long terminal repeats (2-LTR) circles, which is considered an indirect measure of nuclear import. Although reverse transcription occurred at a concentration of GS-CA-1 that inhibited 98% of infection (5 nM), no 2-LTR circles were formed (Figure S3). Occurrence of reverse transcription in infected cells treated with GS-CA-1 is consistent with entry of the core into the nucleus, as has been shown for MxB and CPSF6 (Lee et al., 2010; Liu et al., 2013; Selyutina et al., 2020a). Contrary to PF74, which inhibits the entry of capsid into the nucleus, these results showed that GS-CA1 allowed entry of capsid into the nucleus.
PF74, but not GS-CA-1, results in disaggregation of preformed CPSF6 complexes in nuclear speckles caused by HIV-1 infection
We and others showed that HIV-1 infection induces the formation of CPSF6 aggregates that are colocalized with the nuclear speckle marker SC35 (Francis et al., 2020; Selyutina et al., 2020a), and found that PF74 prevented the formation of CPSF6 aggregates, as GS-CA1 did (Figure 5A). To determine the effect of PF74 and GS-CA1 on preformed CPSF6 aggregates, we infected human HeLa cells with HIV-1 until CPSF6 aggregated in nuclear speckles (18 h) and then treated the cells with PF74, GS-CA1, or solvent (DMSO) for 15 or 180 min. Although PF74 treatment for 15 or 180 min disaggregated CPSF6 from nuclear speckles, GS-CA1 did not affect the CPSF6 aggregates (Figure 5B). The addition of both drugs resulted in the disaggregation of preformed CPSF6 complexes in nuclear speckles (Figure 5C). These results indicated that PF74 can act on preformed CPSF6 complexes in nuclear speckles and trigger disaggregation but GS-CA1 cannot. Because PF74 and GS-CA1 had different effects, we added both drugs during infection and found that together they prevented the formation of CPSF6 in nuclear speckles (Figure 5C). Although both drugs bind to similar sites on the capsid, they have different mechanisms of inhibition.
Figure 5.

PF74, but not GS-CA1, disaggregates preformed CPSF6 complexes in nuclear speckles by HIV-1 infection
(A) HeLa cells were infected with wild-type HIV-1-Luc virus pseudotype with VSV-G in the presence of PF74, GS-CA1, or DMSO as a vehicle control. Cells were incubated for 18 h at 37°C. Mean nuclear speckles ±SD are shown.
(B) HeLa cells were infected with wild-type HIV-1-Luc virus, and 18 h post-infection were treated with PF74, GS-CA1, or DMSO as a vehicle control for 15 or 180 min. Mean nuclear speckles ±SD are shown.
(C) Left panels: HeLa cells were infected with wild-type HIV-1-Luc virus in the presence of PF74, GS-CA1, PF74 and GS-CA1, or DMSO as a vehicle control. Cells were incubated for 18 at 37°C. Right panels: HeLa cells were infected with wild-type HIV-1-Luc virus, and at 18 h post-infection PF74, GS-CA1, PF74 and GS-CA1, or DMSO as a vehicle control were added for 120 min (A–C). After drug treatment, cells were immunostained using specific antibodies directed against CPSF6 (green) and SC35 (red). Nuclei were counterstained with Hoeschst (DNA). Mean nuclear speckles ±SD are shown. Scale bars, 5 μM.
GS-CA1 changes the conformation of the capsid
The HIV-1 core is a higher-order structure with a dynamic surface that modulates its interaction with cellular factors (Gres et al., 2015; Yang et al., 2013). This dynamic surface is the direct result of the conformational plasticity of the monomers of capsid that affects the overall conformation of the assembled core, which is also known as core breathing. One possibility is that GS-CA1 binds to the core inducing a change of conformation that results in a more stable core. To determine whether GS-CA1 induces a conformational change that locks the core in a more stable conformation, we measured CPSF6 binding to wild-type and mutant (N74D) cores with GS-CA1 or PF74. We found that either GS-CA1 or PF74 prevented binding of CPSF6 to wild-type cores (Figure 6). In addition, CPSF6 bound less efficiently to cores with the N74D mutation compared to the wild type. Remarkably, while GS-CA1 restored the ability of cores with the N74D mutation to interact with CPSF6, PF74 did not affect the interaction of CPSF6 with N74D mutant cores. These results suggest that GS-CA1 changes the conformation of the HIV-1 core during infection, increasing core stability and modulating the ability of the core to interact with cellular factors.
Figure 6.

GS-CA1 changes the conformation of the capsid
Cell extracts of Human Jurkat cells (INPUT) were mixed for 1 h at room temperature with wild-type or mutant N74D HIV-1 capsid tubes in the presence of PF74, GS-CA1, or DMSO as a vehicle control. Stabilized HIV-1 capsid tubes were collected by centrifugation and washed two times (BOUND). The input and bound fractions were analyzed by western blotting using anti-CPSF6 and anti-p24 antibodies for three experiments (a representative Western blot is shown). Mean bound fractions relative to input ±SD are shown. ∗∗∗∗ indicates p value < 0.0001, NS indicates not significant as determined by using the unpaired t test.
Discussion
We investigated the mechanism by which GS-CA1 blocks HIV-1 infection by comparing it with PF74. GS-CA1 is a small molecule that, like PF74, prevents the binding of CPSF6 and Nup153 to the HIV-1 core. Although GS-CA1 might have inhibited HIV-1 infection via CPSF6, we found that depletion of CPSF6 expression in human primary CD4+ T cells did not change the ability of GS-CA1 to inhibit HIV-1 infection. These results suggested that CPSF6 is not necessary for the inhibitory effect of GS-CA1. Similarly, we found that expression of CPSF6 was not required for the ability of PF74 to inhibit HIV-1 infection.
Although we have reported that overexpression of MxB and NES-CPSF6 prevent uncoating (Fricke et al., 2013b, 2014b), here we report a small molecule that stabilizes the HIV-1 core, as determined by the fate of the capsid assay. These results are also in agreement with in vitro observations using isolated HIV-1 cores where the use of high concentrations (∼100 nM) of GS-CA1 increased core rigidity till a point that the core breaks open (Christensen et al., 2020). Consistent with our previous findings that a stabilized core undergoes reverse transcription (Fricke et al., 2013b, 2014b; Yang et al., 2013), we found that cores stabilized by GS-CA1 also undergo reverse transcription. Interestingly, when we tested reverse transcription at concentrations of GS-CA1 that inhibits 98–100% of infection (∼5 nM), we observed normal reverse transcription, but we did not observe the formation of 2-LTR circles; however, the use of higher concentrations of GS-CA1 can inhibit reverse transcription (Bester et al., 2020; Yant et al., 2019). Generation of 2-LTR circles was not observed when using ∼5 nM of GS-CA1 suggesting that GS-CA1 triggers a conformational change in the hexamer that stabilizes the entire core structure, which will prevent integration and formation of 2-LTR circles. In contrast to PF74, which accelerates uncoating, this work showed that GS-CA1 prevents HIV-1 uncoating. While this manuscript was under review, Bester and colleagues showed that GS-6207 stabilized the core during infection using an image-based assay (Bester et al., 2020).
The small molecules PF74 and Bi-2 prevent the entry of the HIV-1 core into the nucleus (Selyutina and Diaz-Griffero, 2021; Selyutina et al., 2020a), which are the control compounds for the assay that measures nuclear import of capsid in this manuscript. In contrast to PF74, we found that GS-CA1 at 5 nM did not affect the entry of the HIV-1 core into the nucleus, which also explains the normal occurrence of reverse transcription (Selyutina et al., 2020a). Thus, we found that upon GS-CA1 treatment, the core enters the nucleus and undergoes reverse transcription.
Our capsid nuclear import results do not agree with image-based results that showed GS-CA1 prevent the entry of capsid into the nucleus (Yant et al., 2019); however, this imaged-based assay differ from our assay in the observed phenotypes when using reverse transcription inhibitors. Contrary to our assay and the findings of others in which reverse transcription inhibitors did not affect the entry of capsid into the nucleus (Burdick et al., 2020; Dharan et al., 2020; Selyutina et al., 2020a), the Yant and colleagues assay showed that the reverse transcription inhibitor rilpivirine prevents the entry of capsid into the nucleus (Yant et al., 2019). These observations would imply that these assays are measuring different phenotypes. Our nuclear import assay does not show nuclear capsid when using agents such as PF74, BI-2, rhesus TRIM5α, and Owl monkey TRIMCyp (Selyutina and Diaz-Griffero, 2021; Selyutina et al., 2020a), which suggests that capsid does not adhere to the outer membrane of the nucleus in a nonspecific manner. One caveat of our nuclear import assay is that the nuclear fraction contains capsids that are in the nucleoplasm as well as capsids that are in the lumen of the nuclear pore. Considering this caveat, one possibility is that GS-CA1 stabilized cores are trapped in the lumen of the nuclear pore; therefore, stabilized cores do not reach the nucleoplasm.
We have shown that HIV-1 infection re-localizes CPSF6 to nuclear speckles that colocalize with the marker SC35 (Francis et al., 2020; Selyutina et al., 2020a). Although the contribution of these speckles to HIV-1 infection is unknown, compounds such as PF74 prevent the formation of CPSF6 accumulation in nuclear speckles during infection (Burdick et al., 2020; Dharan et al., 2020; Selyutina et al., 2020a). Interestingly, GS-CA1 added during infection also prevented CPSF6 enrichment in nuclear speckles. Surprisingly, PF74 disaggregated preformed CPSF6 complexes from nuclear speckles, suggesting that they are dynamic structures. Consistent with the role of GS-CA1 in stabilizing the HIV-1 capsid, GS-CA1 did not affect preformed CPSF6 complexes in nuclear speckles. These results indicate that GS-CA1 uses a different mechanism to inhibit HIV-1 infection.
GS-CA1 and GS-6207 stabilize the core during infection, which could be explained by a change in conformation of each monomeric capsid resulting in conformational change in the core. To indirectly measure conformational changes, we determined the effect of GS-CA1 on capsid interaction with CPSF6 and found that capsids with the N74D mutation recovered the ability to bind CPSF6 in the presence of GS-CA1. Therefore, these experiments suggest that GS-CA1 changes the conformation of N74D capsids, restoring their ability to interact with CPSF6. A second possibility is that GS-CA1 is binding capsid and CPSF6 simultaneously; however, (a) the binding of GS-CA1 and CPSF6 to capsid is mutually exclusive, and (b) there is no evidence in the literature suggesting that GS-CA-1 binds CPSF6. Although capsid assembled tubes are composed of hexameric rings of capsid, electron microscopy revealed that these tubes show a large heterogeneity of helical symmetry suggesting that the HIV-1 core can potentially acquire different conformations depending upon the conformation of the capsid protein (Li et al., 2000). This agrees with the concept of core breathing (Gres et al., 2015; Yang et al., 2013), which is likely to be modulated by the use of drugs and/or proteins that bind to the core. Based on the core breathing model, we think that the most likely explanation is that the binding of GS-CA1 to N74D capsid tubes is triggering a conformational change that rescues CPSF6 binding. Altogether these experiments suggest that GS-CA1 exerts its effect on the HIV-1 core by inducing a conformational change.
Limitations of the study
An important limitation of our nuclear import assay is that the nuclear fraction can possibly contain both proteins present in the nucleoplasm and proteins trapped in the lumen of the nuclear pore. Although this method was previously tested using a number of controls such as PF74, BI-2, and rhesus TRIM5α, which prevents the entry of capsid into the nucleus (Selyutina and Diaz-Griffero, 2021; Selyutina et al., 2020a), it is still possible that the nuclear fraction contains capsid complexes trapped in the lumen of the nuclear pore.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HIV-1 p24 (183-H12-5C) | NIH AIDS Reagent Program | Cat# 3537; RRID:AB_2832923 |
| Mouse monoclonal anti- CPSF6 (human) (3F11) | Novus | Cat# H00011052-M10; RRID:AB_2084564 |
| Mouse monoclonal anti-SC-35 (1SC-4F11) | Sigma-Aldrich | Cat# 04-1550; RRID:AB_11212756 |
| Mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Invitrogen (Thermo Fisher Scientific) | Cat# AM4300; RRID:AB_437392 |
| Rabbit polyclonal anti-alpha Tubulin | Invitrogen | Cat# PA5-29444; RRID:AB_2546920 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Mouse monoclonal anti-hemagglutinin (HA) | Sigma-Aldrich | Cat# H3663; RRID:AB_262051 |
| Rabbit polyclonal anti-Nopp140 (human) (RS8 serum) | (Kittur et al., 2007) | Gift of U. Thomas Meier, Albert Einstein College of Medicine |
| Goat anti-Mouse IRDye 680LT | LI-COR | Cat# 925-68020; RRID:AB_268782 |
| Goat anti-Rabbit IRDye 680LT | LI-COR | Cat# 925-68021; RRID:AB_2713919 |
| Goat anti-Mouse IRDye 800CW | LI-COR | Cat# 926-32210; RRID:AB_621842 |
| Goat anti-Rabbit IRDye 800CW | LI-COR | Cat# 925-32211; RRID:AB_621843 |
| Bacterial and virus strains | ||
| E. Coli DH5α competent cells | Zymo Research | Cat# T3007 |
| E. Coli One Shot BL21star (DE3) cells | Thermo Fisher Scientific | Cat# C601003 |
| HIV-1 NL4-3 | (Selyutina et al., 2020b) | NA |
| HIV-1-P38S | This paper | NA |
| HIV-1-S41A | This paper | NA |
| HIV-1-P38S/S41A | This paper | NA |
| Chemicals, peptides, and recombinant proteins | ||
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#: D2438; CAS: 67-68-5 |
| Difco LB Broth, Miller (Luria-Bertani) | Fisher Scientific | Cat#: BD244610 |
| Tris (TRIS(HYDROXYMETHYL)AMINOMETHANE) | Crystalgen | Cat#: 300-844-5000; CAS: 77-86-1 |
| cOmplete EDTA-free protease inhibitor cocktail | Millipore Sigma | Cat#: 11873580001 |
| Sodium Chloride (NaCl) | Crystalgen | Cat# 300-747-5000; CAS: 767-14-5 |
| β-Mercaptoethanol (BME) | Acros organics | Cat# 125470010; CAS: 60-24-2 |
| 2-(N-morpholino) ethanesulfonic acid (MES) | Calbiochem | Cat# 475893; CAS: 4432-31-9 |
| Magnesium Chloride (MgCl2) | Sigma-Aldrich | Cat# M2670; CAS: 7791-18-6 |
| Potassium Chloride (KCl) | Fisher Scientific | Cat# BP366-1; CAS: 7447-40-7 |
| Dithiothreitol (DTT) | VWR | Cat# 97061-340; CAS: 3483-12-3 |
| D-(+)-Sucrose | VWR | Cat# 97061-432; CAS: 57-50-1 |
| Dulbecco's Phosphate-Buffered Salt (PBS) Solution 1X | Corning | 21031CV |
| EDTA, pH 8.0, 0.5M | Corning | 46034CI |
| Paraformaldehyde (4% in PBS) | Boston BioProducts | Cat# BM-155 |
| PF74 | Sigma Aldrich | Cat# SML0835 |
| Nevirapine | NIH AIDS Reagent Program | Cat# 4666 |
| Triton X-100 | Sigma | Cat# T-9284 CAS: 9002-93-1 |
| Nonidet P40 Substitute (NP40) | Fluka Biochemika | Cat# 74385 CAS: 9016-45-9 |
| Glycerol | Millipore | Cat# 356350-1000ML CAS: 56-81-5 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Teknova | Cat# I3325 |
| Bovine Serum Albumin (BSA) (Fraction V) | Fisher bioreagents | Cat# BP1600-100 CAS: 9048-46-8 |
| Human IL-2 IS | Miltenyi Biotec | Cat# 130-097-743 |
| Pronase | Roche Diagnostics GmbH | Cat# 41844620 CAS: 9036-06-0 |
| Poly-L-Lysine (0.01% solution) | Sigma | Cat# P4707 |
| GS-CA1 | Generously provided by Stephen Yant (Gilead Sciences, CA) | |
| Lenacapavir | Generously provided by Stefan G. Sarafianos | |
| Critical commercial assays | ||
| QuikChange II site-directed mutagenesis kit | Agilent | Cat#: 200523 |
| QIAamp DNA Micro Kit | Qiagen | Cat# 56304 |
| Experimental models: Cell lines | ||
| Human: Jurkat | ATCC | TIB-152 |
| Human: 293T/17 | ATCC | CRL-11268 |
| Human: A549 | ATCC | CCL-185 |
| Human: HeLa | ATCC | CCL-2 |
| Oligonucleotides | ||
| All standard cloning primers for site-directed mutagenesis | Integrated DNA Technologies | NA |
| Late Reverse Transcripts (LTR) MH531: 5’-TGTGTGCCCGTCTGTTGTGT-3’ |
(Mbisa et al., 2009) | NA |
| Late Reverse Transcripts (LTR) MH532: 5’-GAGTCCTGCGTCGAGAGATC-3’ |
(Mbisa et al., 2009) | NA |
| Late Reverse Transcripts (LTR) Probe LRT-P: 5′ -(FAM)-CAGTGGCGCCCGA ACAGGGA-(TAMRA)-3’ |
(Mbisa et al., 2009) | NA |
| 2-LTR circles MH5355′-AACTAGGGAACC CACTGCTTAAG-3′ |
(Mbisa et al., 2009) | NA |
| 2-LTR circles MH5365′-TCCACAGATCAA GGATATCTTGTC-3′ |
(Mbisa et al., 2009) | NA |
| 2-LTR probe MH6035’-(FAM)-ACACTACTTG AAGCACTCAAG-GCAAGCTTT-(TAMRA)-3′ |
(Mbisa et al., 2009) | NA |
| CPSF6 crRNA, guide # 5 GGACCACATAGACATTTACG |
Dharmacon | CM-012334-05 |
| CPSF6 crRNA, guide # 6 ATATATTGGAAATCTAACAT |
Custom sequence | NA |
| TRIM5alpha crRNA, guide # 6 AAGAAGTCCATGCTAGACAA |
Custom sequence | NA |
| TRIM5alpha crRNA, guide # 7 GTTGATCATTGTGCACGCCA |
Custom sequence | NA |
| Recombinant DNA | ||
| pLPCX | Clontech | Cat# 631511 |
| pLPCX-CPSF6(1-358)-FLAG | Fricke et al., 2013b | NA |
| pLPCX-Nup153-HA | Buffone et al. (2018) | NA |
| Plasmid: Tat | Stremlau et al., (2006) | NA |
| Plasmid: Rev | Stremlau et al., (2006) | NA |
| Plasmid: VSVg | Stremlau et al., (2006) | NA |
| pVPack-GP | Agilent | Cat #: 217566 |
| pVPack-VSV-G | Agilent | Cat #: 217567 |
| pET-11a-p24-A14C/E45C | Selyutina et al., (2018) | NA |
| pET-11a-p24-A14C/E45C-N74D | Selyutina et al., (2018) | NA |
| Other | ||
| TURBO™ DNase (2 U/μL) | Invitrogen | Cat#AM2238 |
| Fetal Bovine Serum (FBS, heat-inactivated) | Gibco | 16140-071 |
| RPMI-1640 (high glucose) | Corning | MT10-017-CV |
| Penicillin–streptomycin (5 mg/mL) | Corning | MT10-040-CV |
| Sodium pyruvate (100x) | Corning | MT25-000-CI |
| HEPES (100x) | Fisher/HyClone | SH3023701 |
| Ficoll-Paque Plus | Cytiva | 17-1440-02 |
| In Vivo Ready™ Anti-Human CD3 (UCHT1) | Tonbo biosciences | 40-0038-U500 |
| Purified Anti-Human CD28 (CD28.2) | Tonbo biosciences | 70-0038-U100 |
| DMEM (high glucose) | Corning | MT10-017-CV |
| HiTrap Q HP (5 mL) | Cytiva | 17115401 |
| HiTrap SP FF (5 mL) | Cytiva | 17515701 |
| Centrifugal filter units Amicon Ultra-4 | Millipore | UFC801024 |
| SnakeSkin Dialysis tubing 10K MWCO | Thermo Scientific | 68100 |
| P3 Primary Cell 96-well Nucleofector kit | Lonza | V4SP-3096 |
| T cell Activation/Expansion Kit, human | Miltenyi Biotec | 130-091-441 |
| TaqMan Universal Probe Master Mix II, with UNG | Thermo Fischer Scientific | 4440038 |
| EasySep Human CD4+ T Cell Isolation Kit | Stemcell Technologies | Cat# 17952 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Felipe Diaz-Griffero (Felipe.Diaz-Griffero@einsteinmed.org).
Materials availability
This study generated the following new plasmids: HIV-1-P38S, HIV-1-S41A, HIV-1-P38S/S41A. Plasmids generated in this study are available upon request. Further information and requests for plasmids, resources, and reagents should be directed to and will be fulfilled by the lead contact Felipe Diaz-Griffero (Felipe.Diaz-Griffero@einsteinmed.org).
Experimental model and subject details
Bacterial strains
E. coli DH5α bacterial stain was used to generate new plasmids and E. Coli One Shot BL21star (DE3) bacterial strain was used for capsid protein expression. Bacteria were grown in Luria-Bertani (LB) medium at 37°C or at indicated temperatures.
Human cell lines
Human 293T/17, A549, and HeLa cells were grown in complete Dulbecco's modified Eagle's medium (DMEM) (DMEM, 1% penicillin-streptomycin, 10% fetal bovine serum [FBS]). Human Jurkat cells were grown in complete Roswell Park Memorial Institute (RPMI) medium (RPMI, 1% penicillin-streptomycin, 10% FBS). All cells were grown in cell culture incubator at 37°C and 5% CO2. All cell lines were obtained from the American type culture collection (ATCC).
Method details
Infection using HIV-1-GFP reporter viruses
Human immunodeficiency virus-1 (HIV-1) virus-like particles (wild type or with indicated mutations) expressing GFP or luciferase as a reporter gene and pseudotyped with VSV-G were used. Viruses were produced via transient transfection of 293T/17 cells as described (Diaz-Griffero et al., 2008). P24 levels of viral stocks were determined by Western blot and equal amounts of p24 were used to infect cells. Jurkat, A549, or human primary CD4+ T cells were infected in 24-well or 96-well plates (50,000/10,000 cells per well were used) and 48 h post infection the fraction of GFP-expressing cells was detected by flow cytometry using a BD FACSCelesta.
Capsid expression and purification
E. Coli One Shot BL21star (DE3) cells were transformed with pET-11a vector encoding HIV-1 capsid protein genes. In this work we used the capsid gene with A14C/E45C mutations. Capsid protein expression and purification was previously described (Selyutina et al., 2018). Bacteria were incubated at 30°C until reaching optical density A600 = 0.6–0.8 and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce protein expression. After overnight incubation at 18°C cells were collected by centrifugation (5,000 × g, 10 min, 4°C), resuspended in 40 mL of lysis buffer (100 mM β-mercaptoethanol, 50 mM Tris pH 8.0, Complete EDTA-free protease inhibitor tablets, 50 mM NaCl) and lysed by sonication (Qsonica microtip: 4420; A = 45; 2 min; 2 s on; 2 s off for 12 cycles). Cell lysate was centrifuged (40,000 × g, 20 min, 4°C) and to the resulting supernatant (39 mL) 13 mL of saturated ammonium sulfate with 100 mM β-mercaptoethanol was added. The mixture was incubated for 20 min at 4°C and then centrifuged (8,000 × g, 20 min, 4°C). Resulted pellet was resuspended in 30 mL of buffer A (100 mM β-mercaptoethanol, 25 mM 2-[N-morpholino] ethanesulfonic acid (MES) pH 6.5) and sonicated 2–3 times (Qsonica microtip: 4420; A = 45; 2 min; 1 s on; 2 s off). The protein mixture was dialyzed against buffer A (2 h, overnight, 2 h), sonicated, filtered, and diluted in 500 mL of buffer A. The sample was loaded on a HiTrap Q HP column and the flow-through subsequently was loaded on a HiTrap SP FF column. Elution was performed using 0–2 M NaCl gradient. Pooled fractions containing capsid protein were dialyzed against storage buffer (25 mM MES, 20 mM β-mercaptoethanol, 2 M NaCl). The capsid protein was concentrated to a final concentration of 20 mg/mL with a Centrifugal Filter Unit Amicon Ultra-4.
Assembly of stabilized HIV-1 capsid tubes
0.25 mL of purified monomeric capsid (20 mg/mL) were mixed with 0.75 mL of buffer 1 (50 mM Tris, pH 8.0, 100 mM β-mercaptoethanol, 1 M NaCl). The resulting mixture (1 mL of 5 mg/mL capsid protein) was dialyzed in SnakeSkin dialysis tubing against buffer 1 at 4°C for 8 h. After that it was dialyzed against buffer 1 without β-mercaptoethanol at 4°C for 8 h. Then the mixture was dialyzed against buffer 2 (40 mM NaCl, 20 mM Tris, pH 8.0) at 4°C for 8 h. Assembled capsid tubes were stored at 4°C.
Capsid binding assay
Human 293T/17 cells were transiently transfected with pLPCX plasmids encoding CPSF6-FLAG or Nup153-HA and after that incubated for 24 h at 37°C. Cell lysis was performed in 300 μL of capsid binding buffer per plate (CBB: 10 mM Tris, pH 8.0.10 mM KCl, 1.5 mM MgCl2). Alternatively, 10×106 Jurkat cells per point were centrifuged (300 × g, 5 min) and lysed in 300 μL of CBB. Cell lysates were incubated at 4°C for 15 min and subsequently centrifuged (21,000 × g, 15 min, 4°C). Supernatants were mixed with 5 μL of pre-assembled HIV-1 capsid tubes (5mg/mL) and incubated for 1 h at 25°C. Then the mixtures were centrifuged (21,000 × g, 2 min), supernatants were discarded and pellets were resuspended in 1 mL of CBB and centrifuged again as described before (wash step was repeated twice). Pellet fractions were resuspended in 50 μL of 1× Laemmli buffer. Samples were analyzed by western blotting using anti-p24, anti-FLAG, anti-HA, and anti-CPSF6 antibodies.
CA-NC stability assay
The assay was explained in detail Fricke and Diaz-Griffero (2016). In order to assemble HIV-1 CA-NC complexes 25 μL of monomeric CA-NC protein (20 mg/mL) were diluted two fold in buffer A (to get a final concentration of 10 mg/mL CA-NC protein, 50 mM Tris, pH 8.0, 2 mg/mL DNA oligo-[TG]50, 0.5 mM NaCl) and incubated at 4°C for at least 16 h. Then 5 μL of assembled CA-NC were mixed with 500 μL of either stabilization buffer (10mM Tris-HCl, pH 8.0, 2 mM MgCl2, 10 mM KCl, 0.5 mM DTT) or destabilization buffer (20 mM Tris-HCl, pH 8.0, 60 mM NaCl, 2 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1% NP-40, 1% glycerol) and incubated for 1 h at 25°C. Forty microliters of this mixture were mixed with 10 μL of 5x Laemmli buffer and used as input. The remaining mixture was layered onto 3 mL of sucrose cushion (70% sucrose, 1× PBS, 0.5 mM DTT) and centrifuged using SW55 rotor (Beckman) (100,000 × g, 1 h, 4°C). Then supernatant fraction was discarded and pellet fraction was resuspended in 50 μL of 1× Laemmli buffer. Samples were analyzed by western blotting using anti-p24 antibodies.
Fate of the capsid assay
The assay is explained in a detail (Yang et al., 2014; Stremlau et al., 2006). Human A549 cells were infected with HIV-1 virus-like particles in presence of the indicated concentrations of GS-CA1, Lenacapavir (GS-6207), or 10 μM PF74 or DMSO as control. Cells were incubated at 37°C overnight and then detached from the plate with 1 mL of Pronase per plate (7 mg/mL Pronase, DMEM media). Cells were collected in 50 mL tube and washed with ice-cold PBS. Cells were resuspended in 2 mL of buffer A (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 10 mM KCl). After incubation of cells for 20 min on ice they were lysed using a 7.0 mL Dounce homogenizer with pestle B. Cell lysates were centrifuged (7 min, 3,000 rpm, 4°C) and supernatants were layered onto 6 mL of sucrose cushion (50% sucrose, 1× PBS) and centrifuged using Beckman SW41 rotor (125,000 × g, 2 h, 4°C). Samples were analyzed by western blotting using anti-CPSF6 and anti-p24 antibodies.
CRISPR/Cas9 knockouts in primary CD4+ T cells
CRISPR/Cas9 knockouts in primary CD4+ T cells were performed as described (Hultquist et al., 2016). Briefly, crRNA and tracrRNA (Dharmacon) were diluted to 160 μM in resuspension buffer (10 mM Tris-HCl, pH 7.4, 150 mM KCl). To get gRNA:tracrRNA complexes 5 μL of crRNA was combined with 5 μL of tracrRNA and subsequently incubated for 30 min at 37°C. In order to form CRISPR-Cas9 ribonucleoprotein complexes (crRNPs) 10 μL of gRNA:tracrRNA complexes were gently mixed with 10 μL of 40 μM purified Cas9-NLS protein (UC-Berkeley Macrolab).
Peripheral blood mononuclear cells (PBMCs) were purified by Ficoll-Paque Plus density gradient centrifugation. Cells were washed with 1× PBS and suspended to 5 × 108 cells/mL in PBMCs resuspension buffer (1× PBS, 2 mM EDTA, 0.5% BSA). CD4+ T cells were purified from PBMCs using an EasySep Human CD4+ T cell Isolation Kit according to manufacturer's instructions. CD4+ T cells were suspended in complete RPMI medium supplemented with 20 IU/mL IL-2. To activate CD4+ T cells, bulk CD4+ T cells with 5 μg/mL soluble anti-CD28 were plated on anti-CD3 coated plates and incubated for 72 h at 37°C/5% CO2 before electroporation. Cell purity and activation were verified by CD4/CD25 immunostaining and flow cytometry.
For each electroporation reaction between 5 × 105 and 1 × 106 T cells were centrifuged (400 × g, 5 min), the cell pellet was resuspended in 20 μL of P3 electroporation buffer, then gently mixed with 3.5 μL of corresponding crRNP. 20 μL of cell suspension was placed into a 96-well electroporation cuvette for nucleofection with the 4D 96-well shuttle unit (Lonza) using pulse code EH-115. After electroporation, to each well 100 μL of complete RPMI medium without IL-2 was added, and cells were incubated for 30 min at 37°C. Then cells were transferred to a 96-well plate pre-filled with 100 μL of complete RPMI media with IL-2 at 40 U/mL (to make final concentration of 20 U/mL) and also containing anti-CD3/anti-CD2/anti-CD28 beads (T cell Activation/Expansion Kit, Miltenyi) at a 1:1 bead:cell ratio. Cells were incubated for 4 d. To check knock-out efficiency, cells were mixed and 50 μL cell aliquot were used to prepare cell lysate. It was analyzed by western blotting using antibodies against protein of interest.
Immunofluorescence microscopy
Human HeLa cells were seeded on poly-L-lysine–coated chamber slides (BD Biosciences) and regular chamber slides (Nalgene Nunc) and infected with HIV-1 virus-like particles in presence of indicated compounds. After that chamber slides were washed with PBS and incubated with paraformaldehyde (4% PFA, 1x PBS). Cells were incubated first with 0.1% Triton X-100 for 5 min for cell permeabilisation and then with blocking solution (3% BSA, 1× PBS) for 30 min to prevent non-specific binding. Samples were incubated with corresponding antibodies in blocking solution (anti-human CPSF6; anti-SC-35). Cells were washed with 1x PBS and then incubated with the corresponding secondary antibodies in blocking solution. Chamber slides were mounted using Gel Mount (Biomedia) and imaged using a Deltavision epifluorescent microscope system fitted with an automated stage (Applied Precision, Inc). Images were captured in z series on a CCD digital camera using a 63X lens. Out-of-focus images were digitally removed using the Softworks deconvolution software (Applied Precision, Inc).
Quantitative PCR to detect reverse transcription intermediates
Extraction of total DNA was performed using the QIAamp DNA micro kit (QIAGEN) (Butler, 2001 #48). One μL aliquot of total DNA (out of total 50 μL) was used to detect viral DNA forms of HIV-1 using quantitative PCR. Reactions were performed in a 20 μL volume (using 1× TaqMan Mix II). The amplification steps were: initial denaturation (95°C for 15 min), 40 cycles of amplification (95°C for 15 s, 58°C for 30 s, 72°C for 30 s), 4°C.
Quantification and statistical analysis
Quantification of the intensity of Western blot bands was determined using ImageJ. For all experiments, statistical, mean, and standard deviation (SD) values were calculated using GraphPad Prism 7.0c.
Acknowledgments
We thank the NIH AIDS repository for reagents. A.S., P.H., and F.D.-G. are supported by an NIH AI087390 grant to F.D.-G. Anti-Nopp140 antibodies were a kind gift from Dr. T. Meier. GS-CA1 was a donation of Stephen Yant (Gilead Sciences). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Author contributions
A.S., P.H., S.M., L.M.S., H.J.Y. designed, performed and analyzed experiments. J.F.H., KE.L., V.N.KR. designed the experiments. F.D.-G. designed experiments and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103593.
Supplemental information
Data and code availability
Unique standardized data types were not generated. All other unique data will be shared upon reasonable request to the lead contact.
This study did not generate unique original code.
Any additional information required to re-analyze the data reported in this paper is available upon reasonable request to the lead contact.
References
- Balasubramaniam M., Zhou J., Addai A., Martinez P., Pandhare J., Aiken C., Dash C. PF74 inhibits HIV-1 integration by altering the composition of the preintegration complex. J. Virol. 2019;93 doi: 10.1128/JVI.01741-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester S.M., Wei G., Zhao H., Adu-Ampratwum D., Iqbal N., Courouble V.V., Francis A.C., Annamalai A.S., Singh P.K., Shkriabai N., et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science. 2020;370:360–364. doi: 10.1126/science.abb4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A., Alam S.L., Fricke T., Zadrozny K., Sedzicki J., Taylor A.B., Demeler B., Pornillos O., Ganser-Pornillos B.K., Diaz-Griffero F., et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. U S A. 2014;111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffone C., Martinez-Lopez A., Fricke T., Opp S., Severgnini M., Cifola I., Petiti L., Frabetti S., Skorupka K., Zadrozny K.K., et al. Nup153 Unlocks the nuclear pore complex for HIV-1 nuclear translocation in nondividing cells. J. Virol. 2018;92 doi: 10.1128/JVI.00648-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick R.C., Li C., Munshi M., Rawson J.M.O., Nagashima K., Hu W.S., Pathak V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. U S A. 2020;117:5486–5493. doi: 10.1073/pnas.1920631117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen D.E., Ganser-Pornillos B.K., Johnson J.S., Pornillos O., Sundquist W.I. Reconstitution and visualization of HIV-1 capsid-dependent replication and integration in vitro. Science. 2020;370:eabc8420. doi: 10.1126/science.abc8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharan A., Bachmann N., Talley S., Zwikelmaier V., Campbell E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020;5:1088–1095. doi: 10.1038/s41564-020-0735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F., Perron M., McGee-Estrada K., Hanna R., Maillard P.V., Trono D., Sodroski J. A human TRIM5alpha B30.2/SPRY domain mutant gains the ability to restrict and prematurely uncoat B-tropic murine leukemia virus. Virology. 2008;378:233–242. doi: 10.1016/j.virol.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis A.C., Marin M., Singh P.K., Achuthan V., Prellberg M.J., Palermino-Rowland K., Lan S., Tedbury P.R., Sarafianos S.G., Engelman A.N., et al. Publisher correction: HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020;11:6165. doi: 10.1038/s41467-020-20152-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T., Brandariz-Nunez A., Wang X., Smith A.B., 3rd, Diaz-Griffero F. Human cytosolic extracts stabilize the HIV-1 core. J. Virol. 2013;87:10587–10597. doi: 10.1128/JVI.01705-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T., Buffone C., Opp S., Valle-Casuso J., Diaz-Griffero F. BI-2 destabilizes HIV-1 cores during infection and prevents binding of CPSF6 to the HIV-1 capsid. Retrovirology. 2014;11:120. doi: 10.1186/s12977-014-0120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T., Diaz-Griffero F. HIV-1 capsid stabilization assay. Methods Mol. Biol. 2016;1354:39–47. doi: 10.1007/978-1-4939-3046-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T., Valle-Casuso J.C., White T.E., Brandariz-Nunez A., Bosche W.J., Reszka N., Gorelick R., Diaz-Griffero F. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology. 2013;10:46. doi: 10.1186/1742-4690-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T., White T.E., Schulte B., de Souza Aranha Vieira D.A., Dharan A., Campbell E.M., Brandariz-Nunez A., Diaz-Griffero F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology. 2014;11:68. doi: 10.1186/s12977-014-0068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gres A.T., Kirby K.A., KewalRamani V.N., Tanner J.J., Pornillos O., Sarafianos S.G. Structural virology. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science. 2015;349:99–103. doi: 10.1126/science.aaa5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist J.F., Schumann K., Woo J.M., Manganaro L., McGregor M.J., Doudna J., Simon V., Krogan N.J., Marson A. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep. 2016;17:1438–1452. doi: 10.1016/j.celrep.2016.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B.N., Kyere S., Kinde I., Tang C., Howard B.R., Robinson H., Sundquist W.I., Summers M.F., Hill C.P. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 2007;373:355–366. doi: 10.1016/j.jmb.2007.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittur N., Zapantis G., Aubuchon M., Santoro N., Bazett-Jones D.P., Meier U.T. The nucleolar channel system of human endometrium is related to endoplasmic reticulum and R-rings. Mol. Biol. Cell. 2007;18:2296–2304. doi: 10.1091/mbc.E07-02-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte L., Titolo S., Lemke C.T., Goudreau N., Mercier J.F., Wardrop E., Shah V.B., von Schwedler U.K., Langelier C., Banik S.S., et al. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob. Agents Chemother. 2013;57:4622–4631. doi: 10.1128/AAC.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K., Ambrose Z., Martin T.D., Oztop I., Mulky A., Julias J.G., Vandegraaff N., Baumann J.G., Wang R., Yuen W., et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010;7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Hill C.P., Sundquist W.I., Finch J.T. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–413. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- Link J.O., Rhee M.S., Tse W.C., Zheng J., Somoza J.R., Rowe W., Begley R., Chiu A., Mulato A., Hansen D., et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature. 2020;584:614–618. doi: 10.1038/s41586-020-2443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Pan Q., Ding S., Qian J., Xu F., Zhou J., Cen S., Guo F., Liang C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe. 2013;14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- Mbisa J.L., Delviks-Frankenberry K.A., Thomas J.A., Gorelick R.J., Pathak V.K. Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol. Biol. 2009;485:55–72. doi: 10.1007/978-1-59745-170-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A., Ferhadian D., Sowd G.A., Serrao E., Shi J., Halambage U.D., Teng S., Soto J., Siddiqui M.A., Engelman A.N., et al. Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74. J. Virol. 2016;90:5808–5823. doi: 10.1128/JVI.03116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyutina A., Bulnes-Ramos A., Diaz-Griffero F. Binding of host factors to stabilized HIV-1 capsid tubes. Virology. 2018;523:1–5. doi: 10.1016/j.virol.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyutina A., Diaz-Griffero F. Biochemical detection of capsid in the nucleus during HIV-1 infection. STAR Protoc. 2021;2:100323. doi: 10.1016/j.xpro.2021.100323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyutina A., Persaud M., Lee K., KewalRamani V., Diaz-Griffero F. Nuclear import of the HIV-1 core precedes reverse transcription and uncoating. Cell Rep. 2020;32:108201. doi: 10.1016/j.celrep.2020.108201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyutina A., Persaud M., Simons L.M., Bulnes-Ramos A., Buffone C., Martinez-Lopez A., Scoca V., Di Nunzio F., Hiatt J., Marson A., et al. Cyclophilin a prevents HIV-1 restriction in lymphocytes by blocking human TRIM5alpha binding to the viral core. Cell Rep. 2020;30:3766–3777 e3766. doi: 10.1016/j.celrep.2020.02.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Zhou J., Shah V.B., Aiken C., Whitby K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 2011;85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K., Gallazzi F., Hill K.J., Burke D.H., Lange M.J., Quinn T.P., Neogi U., Sonnerborg A. GS-CA compounds: First-In-Class HIV-1 capsid inhibitors covering multiple grounds. Front Microbiol. 2019;10:1227. doi: 10.3389/fmicb.2019.01227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht J., Humbert M., Findlow S., Bodem J., Muller B., Dietrich U., Werner J., Krausslich H.G. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 2005;12:671–677. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- Stremlau M., Perron M., Lee M., Li Y., Song B., Javanbakht H., Diaz-Griffero F., Anderson D.J., Sundquist W.I., Sodroski J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U S A. 2006;103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C., Loeliger E., Kinde I., Kyere S., Mayo K., Barklis E., Sun Y., Huang M., Summers M.F. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 2003;327:1013–1020. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- Thenin-Houssier S., Valente S.T. HIV-1 capsid inhibitors as antiretroviral agents. Curr. HIV Res. 2016;14:270–282. doi: 10.2174/1570162x14999160224103555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Fricke T., Diaz-Griffero F. Inhibition of reverse transcriptase activity increases stability of the HIV-1 core. J. Virol. 2013;87:683–687. doi: 10.1128/JVI.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Luban J., Diaz-Griffero F. The fate of HIV-1 capsid: A biochemical assay for HIV-1 uncoating. Methods Mol. Biol. 2014;1087:29–36. doi: 10.1007/978-1-62703-670-2_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant S.R., Mulato A., Hansen D., Tse W.C., Niedziela-Majka A., Zhang J.R., Stepan G.J., Jin D., Wong M.H., Perreira J.M., et al. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019;25:1377–1384. doi: 10.1038/s41591-019-0560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Liu X., De Clercq E. Capsid (CA) protein as a novel drug target: recent progress in the research of HIV-1 CA inhibitors. Mini Rev. Med. Chem. 2009;9:510–518. doi: 10.2174/138955709787847958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unique standardized data types were not generated. All other unique data will be shared upon reasonable request to the lead contact.
This study did not generate unique original code.
Any additional information required to re-analyze the data reported in this paper is available upon reasonable request to the lead contact.